 Woosuk Choi
Woosuk Choi Gee W. Lau
Gee W. Lau- Department of Pathobiology, University of Illinois at Urbana-Champaign, Urbana, IL, United States
Forkhead box (FOX) proteins are transcriptional factors that regulate various cellular processes. This minireview provides an overview of FOXA2 functions, with a special emphasis on the regulation airway mucus homeostasis in both healthy and diseased lungs. FOXA2 plays crucial roles during lung morphogenesis, surfactant protein production, goblet cell differentiation and mucin expression. In healthy airways, FOXA2 exerts a tight control over goblet cell development and mucin biosynthesis. However, in diseased airways, microbial infections and proinflammatory responses deplete FOXA2 expression, resulting in uncontrolled goblet cell hyperplasia and metaplasia, mucus hypersecretion, and impaired mucociliary clearance of pathogens. Furthermore, accumulated mucus clogs the airways and creates a niche environment for persistent microbial colonization and infection, leading to acute exacerbation and deterioration of pulmonary function in patients with chronic lung diseases. Various studies have shown that FOXA2 inhibition is mediated through induction of antagonistic EGFR and IL-13R-STAT6 signaling pathways as well as through posttranslational modifications induced by microbial infections. An improved understanding of how bacterial pathogens inactivate FOXA2 may pave the way for developing therapeutics that preserve the protein's function, which in turn, will improve the mucus status and mucociliary clearance of pathogens, reduce microbial-mediated acute exacerbation and restore lung function in patients with chronic lung diseases.
Introduction
The Forkhead (fox) genes encode evolutionarily-conserved transcriptional regulators characterized by a winged-helix DNA-binding domain (DBD), called the forkhead box. Members of the FOX family have divergent roles, including embryonic development, cell survival, proliferation, differentiation, and energy homeostasis (1). Initially discovered in Drosophila melanogaster, a mutation in the fox gene generated a homeotic transformation of foregut into head that originates the nomenclature “forked head” (2). Over 100 FOX members have been identified, with 50 human fox genes cataloged into 19 subgroups from FOXA to FOXS (3). The FOXA subclass was the first discovered in mammals (4).
Structure and Function Of Foxa2
FOXA1, FOXA2, and FOXA3 were originally identified in rat liver, the so-called hepatocyte nuclear factor 3 (HNF3)-α, -β, and -γ, respectively (4). The conserved domains of FOXA2 were originally analyzed (2), and the DBD was found to be the most evolutionarily conserved among the FOXA members, with an unique AKT/PKB phosphorylation site at the threonine (Thr)156 (5). The DBD of FOXA is structurally similar to H1/H5 linker histones capable of unwinding chromatin, which enables the recruitment of other transcriptional cofactors to the promoter (6, 7). The C-terminus of FOXA interacts with histones H3/H4 within nucleosome to support the opening of chromatin by the DBD (8). FOXA members regulate the expression of target genes by displacing histones from chromatin and serving as transcription factors at the enhancer region of the promotor (9, 10).
Modulation Of Foxa2 Activity by Post-Translational Modifications
FOXA transcription factors are abundantly expressed in liver and regulate metabolic homeostasis (4). During hypoglycemia, low plasma insulin activates FOXA2 to elevate the transcription of genes encoding metabolic enzymes involved in fatty acid oxidation and ketogenesis, supplying energy for gluconeogenesis and maintaining brain function, respectively. However, excess blood glucose elevates plasma insulin, which inactivates FOXA2 and decreases the expression of enzymes involved in gluconeogenesis (11, 12). This is supported by studies that show inactivation of hepatic FOXA2 in hyperinsulinemic ob/ob and db/db mice and in diet-induced obese mice (13). Insulin promotes nuclear export of FOXA2 through AKT-mediated phosphorylation at Thr156 (Figures 1A,B) (5). Cells expressing the phosphorylation-deficient FOXA2-T156A are unresponsive to insulin-induced AKT, resulting in constitutive nuclear localization. Interestingly, DNA-binding ability of FOXA2 is not altered by insulin and phosphorylation-deficiency, suggesting that phosphorylation at Thr156 does not regulate transcriptional activity. Further study indicates that in the presence of insulin, phosphorylation at Thr156 inactivates transcriptional function and induces nuclear exclusion of FOXA2. Nuclear export of FOXA2 is dependent upon nuclear export factor CRM1, which recognizes the leucin-rich NES consensus sequence LX2,3(L/I/V/F/M)X2,3LX(L/I) within amino acids 106-111 (LSPSLS) of FOXA2 (14). In contrast, others have reported that FOXA2 is constitutively localized to the nucleus, independent of the metabolic conditions (15, 16). The aforementioned discrepancies might be caused by distinct experimental conditions, including differences in transgenic obese mouse strains, feeding conditions, and immortalized hepatic cell lines. Subsequently, it was revealed that FOXA2 function could be modulated by IKKα-mediated phosphorylation on Ser107/111 (Figure 1A) (17). Both Thr156 and Ser107/111 residues are located within the nuclear localization signal domain and the nuclear export signal domain, respectively (Figure 1A), implying that phosphorylation of these residues dictates subcellular localization of FOXA2 (14).
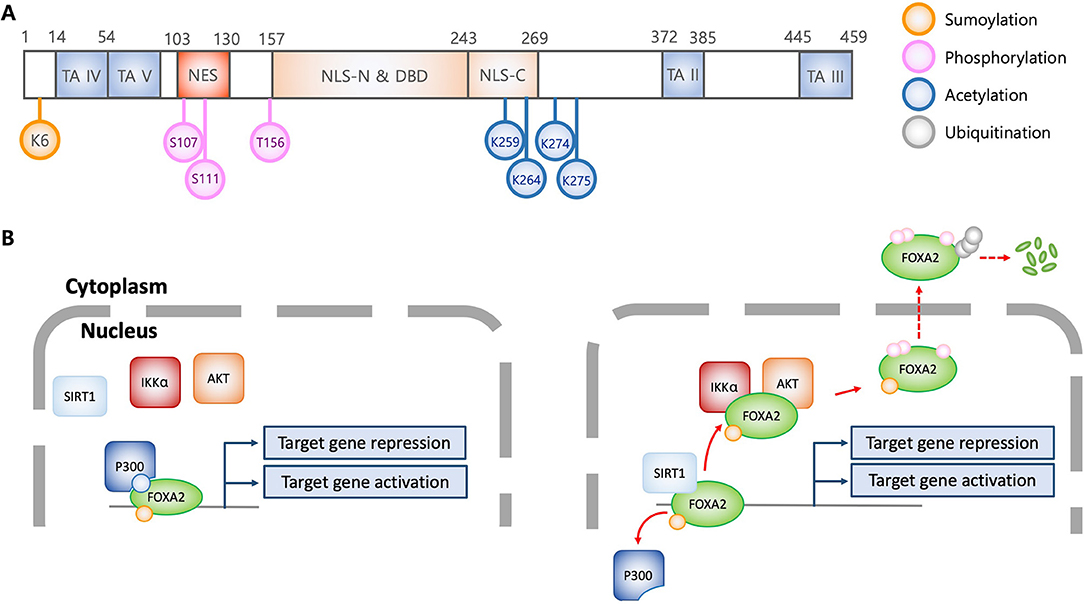
Figure 1. Structural and functional characteristics of FOXA2 in association with posttranslational modification. (A) Schematic diagram of FOXA2 (adapted from J Biol Chem 2009, 284:24816-24; and Mol Cell Biol 1992, 12:3723-3732). Diagram shows functional domains of FOXA2. Colorized circles indicate post-translationally modified amino acid residues which alter FOXA2 functions. TA, transactivation domain; NES, nuclear export signal; NLS, Nuclear localization signal; DBD, DNA binding domain; K, lysine; S, serine; T, threonine. (B) Regulation of transcriptional activity and stability of FOXA2 by posttranslational modifications. Acetylation by P300 allows FOXA2 to be functionally active. In contrast, SIRT1 deacetylates FOXA2, leading to nuclear export via AKT and IKKα-mediated phosphorylation, and subsequently, ubiquitination and degradation. Sumoylation enhances the stability of nuclear FOXA2, which is diminished by ubiquitination, resulting in FOXA2 degradation.
Acetylation and deacetylation also compete to modulate the transcriptional activity of FOXA2. Insulin induces the SIRT1 deacetylase to deacetylate Lys259 on FOXA2 (Figure 1A), which attenuates target gene expression and increases export from the nuclei in hepatocytes. In contrast, glucagon induces FOXA2 acetylation through P300 acetyltransferase, which leads to nuclear accumulation and increased expression of target genes (12). The acetylation-deficient FOXA2-K259R has attenuated DNA binding ability, and is sequestered to the cytoplasm for degradation. As discussed above, FOXA2-T156A is sequestered in the nucleus; however, hepatic cells transfected with FOXA2-T156A-K259R exhibit similar phenotype to those transfected with FOXA2-K259R, suggesting that acetylation/deacetylation diminish phosphorylation event, and therefore, confer a dominant phenotype (12). Interestingly, it has been reported that during nutrient-deprivation, deacetylation by SIRT1 only attenuates FOXA2's transcriptional activity but not its nuclear export for degradation by the proteasome (11).
Sumoylation and ubiquitination also compete for maintenance and degradation of FOXA2 (18) Sumoylation of the Lys6 by PIAS stabilizes FOXA2's transcriptional activity. In contrast, polyubiquitination facilitates FOXA2 degradation by proteasome. Sumoylation-resistance FOXA2-K6R is more susceptible to ubiquitination and rapid degradation, suggesting that additional lysine residues might be ubiquitinated. Notably, although sumoylation increases both stability and transcriptional activity of FOXA2, nuclear localization is not affected.
Role of Foxa2 in Lung Development and Homeostasis
FOXA2 is required for lung development. During mouse embryogenesis, FOXA2 is initially expressed in the primitive streak and in the node on embryonic day 6.5 (E6.5), inducing gastrulation. By E7.5, FOXA2 is highly expressed in both mesoderm and endoderm, and thereafter, persistently expressed throughout development and in adult endoderm-derived tissues, including the lung (19). After E10-E11, lung morphogenesis is facilitated by the spatiotemporal expression of FOXA2 restricted to respiratory epithelium. By E12.5, mouse embryo develops conducting airways and alveolar epithelial cells, and forms lung buds during late embryonic development (20). In contrast, FOXA2 null mouse embryo shows lethality on E11-E12, with severe defects in all three germ layers before the initiation of lung morphogenesis (21). The conditional loss of FOXA2 on E12.5 disrupts branching formation and airway epithelial cell differentiation, resulting in the dilation of distal airways. In addition, postnatal (PN) lungs of foxa1 null allele and lung epithelium-specific foxa2 depleted mice (foxa1−/−/foxa2Δ/Δ) exhibit regressed formation of alveolar and peripheral lung saccules by PN day 3 (PN3), and extensive airspace enlargement with mucin glycoprotein overexpression by PN10–PN20 (20).
In postnatal lungs, FOXA2 is constitutively expressed in subsets of respiratory epithelial cells and transcriptional controlled genes encoding club cell specific protein CC10 (22), surfactant proteins (SP) (23), thyroid transcription factor-1 (TTF-1) (24), and mucins MUC5AC and MUC5B (25, 26). Pulmonary surfactant is composed of 90% phospholipids and 10% proteins, including SP-A, SP-B, SP-C, and SP-D secreted by the type II alveolar cells and non-ciliated terminal bronchiolar Club cells. Together with phospholipids, SP-B and SP-C provide critical surface tension-lowering properties that reduce the work of breathing and maintain airspace patency. FOXA2Δ/Δ mice show significantly reduced SP-B expression with deteriorating respiratory distress syndrome (27). In contrast to upregulation of CC10, SFTPB, and TTF-1, FOXA2 represses the transcription of mucin genes. Conditional deletion of FOXA2 in the mouse respiratory epithelium causes airspace enlargement, goblet cell hyperplasia, increased mucin expression and neutrophil infiltration (25). Collectively, the aforementioned studies indicate crucial roles of FOXA2 in regulating embryonic lung development and postnatal lung homeostasis.
Foxa2 and Mucus Homeostasis
The apical surface of healthy airways is covered by the airway surface liquid (ASL) composed of mucin glycoproteins, antimicrobial peptides and proteins, innate immune cells, signaling molecules, and enzymes (28). ASL is bilayer, with the periciliary layer sandwiched between the top mucus gel layer and the bottom airway epithelium, forming the “gel-on-brush” structure (29) (Figure 2A). The periciliary layer is filled with hydrogel that provides space for ciliary beating and supports mucociliary clearance. Within the mobile mucus layer, MUC5AC and MUC5B are the predominant mucins that provide viscosity and gel-forming properties to mucus, trapping inhaled pathogens and irritants. Mucins also keep moisture in the airway epithelium (30), which help to maintain the periciliary layer and mucociliary clearance. MUC5AC is produced by the goblet cells while MUC5B is expressed in submucosal glands of trachea and bronchi, and in surface secretory cells throughout the airway down to the level of preterminal bronchioles (31, 32). MUC5AC-overexpressing mice are more resistant to PR8/H1N1 influenza virus (33). Similarly, MUC5B-deficient mice are more susceptible to lung infection with increased mortality caused by ensuing bacteremia (34).
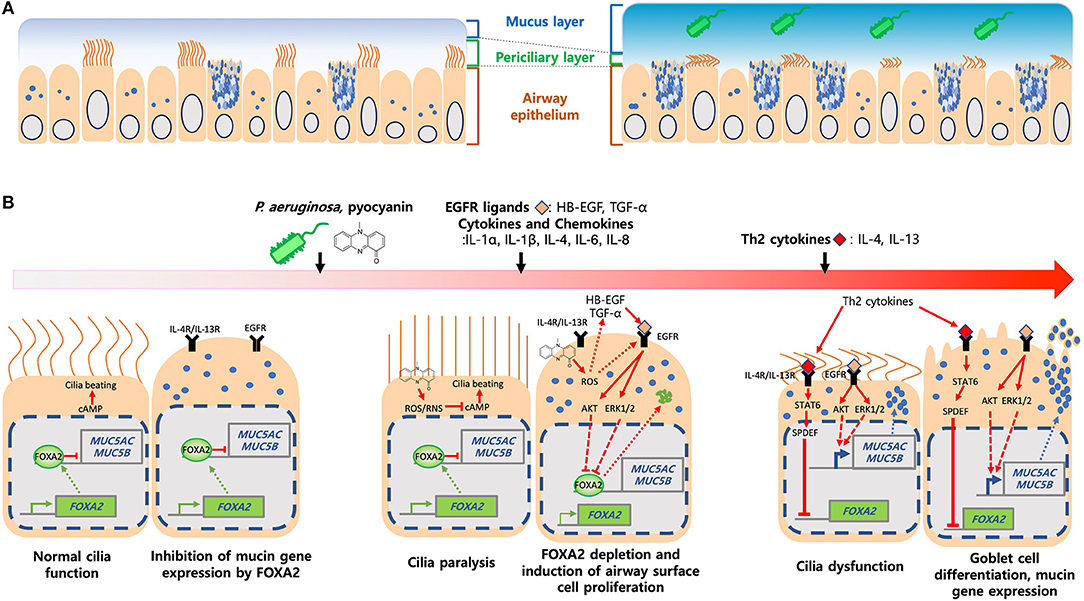
Figure 2. Induction of mucus hypersecretion by P. aeruginosa pyocyanin. (A) Composition of mucus layers in heathy and diseased airways. (B) P. aeruginosa and its virulence factors, especially pyocyanin, stimulate excessive ROS, chemokines and cytokines and, ligands that activate IL-4R/IL-13R-STAT6-SPDEF, and EGFR-AKT/ERK1/2-mediated signaling pathways. Both kinase cascades converged to inhibit FOXA2, resulting in over proliferation and differentiation of airway epithelial (ciliated and club) cells to mucus-secreting goblet cells. Excessive mucus and failure in the clearance exacerbates airway obstruction and microbial colonization and infection.
FOXA2 deficiency causes pulmonary eosinophilia, recruitment of inflammatory immune cells and upregulation of IL-4, IL-13, IL-33, CCL-17, and CCL-20 that promote Th2 cell differentiation, goblet cell hyperplasia and metaplasia, and mucus hypersecretion (25, 35). These suggest that FOXA2 regulates airway mucus homeostasis by counteracting the effects of IL-4 and IL-13 (36, 37). Metaplastic effects of IL-4 and IL-13 are mediated through the STAT6 (35) and its downstream effector SPDEF (25, 38, 39). MUC5Ac and MUC5B expression is dependent on SPDEF (38, 40). IL-13-stimulated airway epithelial cells show decreased FOXA2 transcript, a process mediated by SPDEF (41). Conditional induction of SPDEF within transgenic mouse airways downregulates the foxa2 gene, resulting in goblet cell hyperplasia (39). These findings indicate that FOXA2 and SPDEF compete to regulate the expression of MUC5AC and MUC5B.
Th2 cytokines also amplify the mucus-inducing EGFR signaling, which is highly activated in cystic fibrosis (CF), chronic obstructive pulmonary disease (COPD) and asthma (42, 43). In response to IL-4, IL-5, and IL-13, bronchial epithelial and immune cells produce ligands (EGF, TGF-α, amphiregulin) that induce EGFR in an autocrine manner (41, 44–46), which subsequently, activates the downstream cRAF-MEK-ERK and PI3K-AKT signaling cascades that inhibit FOXA2 and increase MUC5AC and MUC5B production (47). Interestingly, these pathways have distinct effects, with PI3K-AKT augments cell proliferation while cRAF-MEK-ERK directly enhances goblet cell metaplasia and mucin synthesis (48).
Notch signaling regulates cell-cell communication and differentiation of airway basal cells into secretory and ciliated cells (49). Both Notch1 and Notch2 are required for goblet cell development (50, 51). Interestingly, Notch ligands promotes goblet cell metaplasia independent of the IL-13R-STAT6 axis. It is unknown if Notch induction depletes FOXA2 expression during goblet cell development in diseased airways.
Interestingly, MUC5B gene transcription is differentially-regulated by FOXA2. Inhibition of FOXA2 by bacterial pathogens elevates MUC5B expression (26, 52–54). In contrast, FOXA2 positively regulates MUC5B expression in both idiopathic pulmonary fibrosis (55) and asthma (56), most likely caused by polymorphism in the MUC5B promoter (57). Additionally, inhibition of FOXA2 by IL-13 results in different inhibitory kinetics on MUC5B expression in the air-liquid interface (ALI) culture of airway cells vs. in mouse lungs (41), suggesting that additional mediators may modulate mucin production in an intact lung. Collectively, these findings suggest that regulatory activity of FOXA2 on MUC5B may vary, depending on disease context and additional interacting factors.
Foxa2 Inactivation by Respiratory Bacterial Pathogens
As previously discussed, excessive mucus causes airway obstruction, narrowing and airflow limitation in chronic lung diseases. Significantly, FOXA2 expression is depleted in airways of patients with bronchopulmonary dysplasia, bronchiectasis (25), and asthma (58). Cigarette smoking, the most important etiologic agent in COPD, directly suppress FOXA2 expression (59). Accumulated mucus allows microbes to thrive, resulting in persistent inflammation, acute exacerbation (60), and lung function impairment (61), with increased morbidity, and mortality (62). Among the bacterial pathogens, Staphylococcus aureus and Pseudomonas aeruginosa (PA) are the most important in young CF patients, but in adulthood, PA predominates (63). Chlamydophila pneumoniae and Mycoplasma pneumoniae are the most important in asthma induction and acute exacerbation (64). Streptococcus pneumoniae, Haemophilus influenza, and Moraxella catarrhalis are the most common in COPD. However, in advanced stages of COPD, PA, M. pneumoniae, H. parainfluenzae, and Klebsiella pneumoniae predominate. Acquisition of PA increases episodes of acute exacerbation, especially in COPD patients who received antibiotics and those who require mechanical ventilation. Significantly, a subset of these patients becomes chronically-infected with PA (65).
For the remainder of this review, we will focus on the FOXA2 inactivation by PA (Figures 2A,B). PA forms biofilms in the mucus-rich environments and becomes more resistant to antibiotics and phagocytic clearance. PA virulence factors, including pyocyanin (26, 52, 66, 67), LPS (68), flagellin (69, 70), alginate (71) and protease (72) induce mucus overproduction.
Among these aforementioned virulence factors, the tricyclic phenazine pyocyanin, is the most robust mucus inducer (73). Pyocyanin is zwittwerionic, which ionizes at physiological pH, penetrates cell membrane, and increases both intracellular reactive oxygen species (ROS) and nitrogen species (RNS) (74–76). Redox cycling of pyocyanin with intracellular electron donors and acceptors generates ROS/RNS (76, 77). Persistent oxidative stress causes dysfunction of ion pumps, antioxidant proteins and cellular reducing agents, resulting in cytotoxicity (78, 79).
Pyocyanin is important for lung infections (80) and recoverable at 0.1 mM concentrations from both COPD and CF sputa (81). Additionally, the levels of pyocyanin within sputa negatively-correlates with the function of CF lungs (82). Pyocyanin interferes with ciliary beating and mucus transport (81), induces bronchoconstriction (83), and decreases mucus velocity (84, 85). Mouse lungs chronically-exposed to pyocyanin develop goblet cell hyperplasia and metaplasia, peribronchial fibrosis, and alveolar airspace destruction, accompanied by polarization from initially a Th1 response toward a Th2 response dominated by IL-4 and IL-13 secreted by activated macrophages and CD4+ T cells, with concomitant influx of neutrophils (52, 66). Many of these pathological features resemble the airways of FOXA2-deficient mice (25). Further studies in the both primary and immortalized human airway cells and in mice demonstrate that pyocyanin depletes FOXA2 expression by activating antagonistic EGFR-PI3K-AKT, EGFR-MEK-ERK and IL-13R-STAT6-SPDEF pathways, resulting in goblet cell hyperplasia and metaplasia and excessive mucins (52, 66). Additionally, pyocyanin activates EGFR directly through ROS or indirectly by inducing the release of proinflammatory cytokines and EGFR ligands from airway cells (86). ROS/RNS generated by pyocyanin post-translationally modify FOXA2 and reduce its binding affinity to the MUC5B promoter. Glutathione restores the expression of FOXA2, which inhibits the transcription of MUC5AC and MUC5B (26). Collectively, these results suggest that pyocyanin inactivates FOXA2 through EGFR-PI3K-AKT, EGFR-MEK-ERK, and IL-13R-STAT6-SPDEF signaling, and post-translational modification of FOXA2.
As for the remaining PA virulence factors, LPS activates the Src-dependent Ras-p38MAPK-pp90rsk pathway, leading to mucin overproduction (68). Flagellum binds asialoGM1 and induces a signaling cascade, leading to cleavage of PIP2 by PLC, formation of IP3, Ca2+ mobilization, phosphorylation of ERK1/2, and finally, transcription of the MUC2 gene (70). Flagellum also activates mucin biosynthesis through the NF-κB induced by TLR5-IL-8 signaling (69). The mechanisms underlying mucin induction by exoproteases (72), alginate (71), and their association to FOXA2 inactivation remain uncharacterized.
FOXA2 appears to be an evolutionally-conserved target of inactivation. Besides PA, we have shown that M. pneumoniae inactivates FOXA2 by inducing both STAT3-STAT6 and EGFR signaling, resulting in overexpression of airway mucins (53). Additional evidences are found in canine species. Because of genetic predisposition and exposure to environmental pollutants and infectious agents, older dogs, especially of smaller breeds, develop lung diseases similar to those in humans. Our recent study in dogs with COPD and chronic bronchitis indicate that infection by PA and Bordetella bronchiseptica, and by viral-bacterial combination activate the antagonistic STAT6 and EGFR signaling to inhibit FOXA2, resulting in goblet cell hyperplasia and metaplasia and mucus hypersecretion (16).
Summary
Although many aspects of mucus biology have been explored as therapeutic targets, few drugs are available because of inefficacy and adverse effects (87). Despite its importance, FOXA2 has not been targeted for the development of novel mucoregulators, perhaps due to the complexity in gene regulation, post-translational modifications, and toxicity associated with its regulation of multiple cellular processes. Recently, the peptide ADEL (Ala-Asp-Glu-Leu) was found to relieve bacterial-mediated inflammation and improve lung function while boosting FOXA2 expression (88). Additionally, GLP-1 analogs were shown to reduce mortality and improve lung function in mice with acute obstructive lung disease (89). Our latest results indicate that the GLP-1 analog Exenatide restores FOXA2-regulated airway mucus homeostasis through the GLP1R-PKA-PPARγ-phosphatase signaling, by dephosphorylating key kinases within both STAT6 and EGFR cascades (90). Some factors need to be considered before repurposing the GLP-1 analogs. Systemically-administered GLP-1 analogs could suppress appetite (91). Because patients with muco-obstructive diseases (e.g., CF, COPD) commonly experience inappetence, and in the case of CF patients, nutrient malabsorption (92, 93), GLP-1 analogs may adversely impact the health status of these patients. To minimize systemic toxicity, direct aerosolization should be considered. Also, co-prescribing steroids and appetite stimulants with GLP-1 analogs may boost positive outcome. Finally, detailed mechanistic characterization of how these drugs restore FOXA2 function may lead to new methods of controlling excessive mucus, lowering bacterial burden and improving the quality of life in patients with chronic lung diseases.
Author Contributions
WC, SC, and GL co-wrote the manuscript.
Funding
This work was supported in part by the American Lung Association DeSouza Research Award (DS-192835-N), the NIH (HL090699; HL142626A1), the University of Illinois Research Board (RB18060), and the Companion Animal Grant Program (James Harkness Fund) to GL. The funders had no role in study design, data collection, and interpretation, or the decision to submit the work for publication.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Carlsson P, Mahlapuu M. Forkhead transcription factors: key players in development and metabolism. Dev Biol. (2002) 250:1–23. doi: 10.1006/dbio.2002.0780
2. Costa R, Grayson D, Darnell J. Multiple hepatocyte-enriched nuclear factors function in the regulation of transthyretin and alpha 1-antitrypsin genes. Mol Cell Biol. (1989) 9:1415–25. doi: 10.1128/MCB.9.4.1415
3. Kaestner KH, Knöchel W, Martínez DE. Unified nomenclature for the winged helix/forkhead transcription factors. Genes Dev. (2000) 14:142–6. doi: 10.1101/gad.14.2.142
4. Lai E, Prezioso VR, Smith E, Litvin O, Costa RH, Darnell J. HNF-3A, a hepatocyte-enriched transcription factor of novel structure is regulated transcriptionally. Genes Dev. (1990) 4:1427–36. doi: 10.1101/gad.4.8.1427
5. Wolfrum C, Besser D, Luca E, Stoffel M. Insulin regulates the activity of forkhead transcription factor Hnf-3 /Foxa-2 by Akt-mediated phosphorylation and nuclear/cytosolic localization. Proc Natl Acad Sci. USA. (2003) 100:11624–9. doi: 10.1073/pnas.1931483100
6. Clark KL, Halay ED, Lai E, Burley SK. Co-crystal structure of the HNF-3/fork head DNA-recognition motif resembles histone H5. Nature. (1993) 364:412. doi: 10.1038/364412a0
7. Zaret K, Caravaca J, Tulin A, Sekiya T. Nuclear mobility and mitotic chromosome binding similarities between pioneer transcription factor FoxA and linker histone H1. Cold Spring Harbor Symp Quant Biol. (2010) 219–26. doi: 10.1101/sqb.2010.75.061
8. Cirillo LA, Lin FR, Cuesta I, Friedman D, Jarnik M, Zaret KS. Opening of compacted chromatin by early developmental transcription factors HNF3 (FoxA) and GATA-4. Mol Cell. (2002) 9:279–89. doi: 10.1016/S1097-2765(02)00459-8
9. Zaret KS, Carroll JS. Pioneer transcription factors: establishing competence for gene expression. Genes Dev. (2011) 25:2227–41. doi: 10.1101/gad.176826.111
10. Iwafuchi-Doi M, Zaret KS. Pioneer transcription factors in cell reprogramming. Genes Dev. (2014) 28:2679–92. doi: 10.1101/gad.253443.114
11. Westerheide SD, Van Gent R, Di Sanza C, Van Den Broek NJF, Fleskens V, Veenstra A, et al. SIRT1 mediates FOXA2 breakdown by deacetylation in a nutrient-dependent manner. PLoS ONE. (2014) 9:e98438. doi: 10.1371/journal.pone.0098438
12. Von meyenn F, Porstmann T, Gasser E, Selevsek N, Schmidt A, Aebersold R, et al. Glucagon-induced acetylation of Foxa2 regulates hepatic lipid metabolism. Cell Metab. (2013) 17:436–47. doi: 10.1016/j.cmet.2013.01.014
13. Wolfrum C, Shih DQ, Kuwajima S, Norris AW, Kahn CR, Stoffel M. Role of Foxa-2 in adipocyte metabolism and differentiation. J Clin Invest. (2003) 112:345–56. doi: 10.1172/JCI18698
14. Howell JJ, Stoffel M. Nuclear export-independent inhibition of Foxa2 by insulin. J Biol Chem. (2009) 284:24816–24. doi: 10.1074/jbc.M109.042135
15. Steneberg P, Rubins N, Bartoov-Shifman R, Walker MD, Edlund H. The FFA receptor GPR40 links hyperinsulinemia, hepatic steatosis, and impaired glucose homeostasis in mouse. Cell Metab. (2005) 1:245–58. doi: 10.1016/j.cmet.2005.03.007
16. Zhang L, Rubins NE, Ahima RS, Greenbaum LE, Kaestner KH. Foxa2 integrates the transcriptional response of the hepatocyte to fasting. Cell Metab. (2005) 2:141–8. doi: 10.1016/j.cmet.2005.07.002
17. Liu M, Lee D-F, Chen C-T, Yen C-J, Li L-Y, Lee H-J, et al. IKKα activation of NOTCH links tumorigenesis via FOXA2 suppression. Mol Cell. (2012) 45:171–84. doi: 10.1016/j.molcel.2011.11.018
18. Belaguli NS, Zhang M, Brunicardi FC, Berger DH. Forkhead box protein A2 (FOXA2) protein stability and activity are regulated by sumoylation. PLoS ONE. (2012) 7:e48019. doi: 10.1371/journal.pone.0048019
19. Burtscher I, Lickert H. Foxa2 regulates polarity and epithelialization in the endoderm germ layer of the mouse embryo. Development. (2009) 136:1029–38. doi: 10.1242/dev.028415
20. Wan H, Dingle S, Xu Y, Besnard V, Kaestner KH, Ang S-L, et al. Compensatory roles of Foxa1 and Foxa2 during lung morphogenesis. J Biol Chem. (2005) 280:13809–16. doi: 10.1074/jbc.M414122200
21. Weinstein DC, I Altaba AR, Chen WS, Hoodless P, Prezioso VR, Jessell TM, et al. The winged-helix transcription factor HNF-3β is required for notochord development in the mouse embryo. Cell. (1994) 78:575–88. doi: 10.1016/0092-8674(94)90523-1
22. Chang A, Ramsay P, Zhao B, Park M, Magdaleno S, Reardon MJ, et al. Physiological regulation of uteroglobin/CCSP expression. Ann N Y Acad Sci. (2000) 923:181–92. doi: 10.1111/j.1749-6632.2000.tb05529.x
23. Bohinski RJ, Di Lauro R, Whitsett JA. The lung-specific surfactant protein B gene promoter is a target for thyroid transcription factor 1 and hepatocyte nuclear factor 3, indicating common factors for organ-specific gene expression along the foregut axis. Mol Cell Biol. (1994) 14:5671–81. doi: 10.1128/MCB.14.9.5671
24. Ikeda K, Shaw-White JR, Wert SE, Whitsett JA. Hepatocyte nuclear factor 3 activates transcription of thyroid transcription factor 1 in respiratory epithelial cells. Mol Cell Biol. (1996) 16:3626–36. doi: 10.1128/MCB.16.7.3626
25. Wan H, Kaestner KH, Ang S-L, Ikegami M, Finkelman FD, Stahlman MT, et al. Foxa2 regulates alveolarization and goblet cell hyperplasia. Development. (2004) 131:953–64. doi: 10.1242/dev.00966
26. Hao Y, Kuang Z, Xu Y, Walling BE, Lau GW. Pyocyanin-induced mucin production is associated with redox modification of FOXA2. Respirat Res. (2013) 14:82. doi: 10.1186/1465-9921-14-82
27. Wan H, Xu Y, Ikegami M, Stahlman MT, Kaestner KH, Ang S-L, et al. Foxa2 is required for transition to air breathing at birth. Proc Natl Acad Sci USA. (2004) 101:14449–54. doi: 10.1073/pnas.0404424101
28. Kato A, Schleimer RP. Beyond inflammation: airway epithelial cells are at the interface of innate and adaptive immunity. Curr Opin Immunol. (2007) 19:711–20. doi: 10.1016/j.coi.2007.08.004
29. Button B, Cai L-H, Ehre C, Kesimer M, Hill DB, Sheehan JK, et al. A periciliary brush promotes the lung health by separating the mucus layer from airway epithelia. Science. (2012) 337:937–41. doi: 10.1126/science.1223012
30. Sheehan JK, Kesimer M, Pickles R. Innate immunity and mucus structure and function. Novartis Foundation Symp. (2006) 155:155–66. doi: 10.1002/9780470035399.ch13
31. Reid CJ, Gould S, Harris A. Developmental expression of mucin genes in the human respiratory tract. Am J Respirat Cell Mol Biol. (1997) 17:592–8. doi: 10.1165/ajrcmb.17.5.2798
32. Buisine M-P, Devisme L, Copin M-C, Durand-Réville M, Gosselin B, Aubert J-P, et al. Developmental mucin gene expression in the human respiratory tract. Am J Respirat Cell Mol Biol. (1999) 20:209–18. doi: 10.1165/ajrcmb.20.2.3259
33. Ehre C, Worthington EN, Liesman RM, Grubb BR, Barbier D, O'neal WK, et al. Overexpressing mouse model demonstrates the protective role of Muc5ac in the lungs. Proc Natl Acad Sci USA. (2012) 109:16528–33. doi: 10.1073/pnas.1206552109
34. Roy MG, Livraghi-Butrico A, Fletcher AA, Mcelwee MM, Evans SE, Boerner RM, et al. Muc5b is required for airway defence. Nature. (2014) 505:412–6. doi: 10.1038/nature12807
35. Chen G, Wan H, Luo F, Zhang L, Xu Y, Lewkowich I, et al. Foxa2 programs Th2 cell-mediated innate immunity in the developing lung. J Immunol. (2010) 184:6133–41. doi: 10.4049/jimmunol.1000223
36. Jain-Vora S, Wert SE, Temann U-A, Rankin JA, Whitsett JA. Interleukin-4 alters epithelial cell differentiation and surfactant homeostasis in the postnatal mouse lung. Am J Respirat Cell Mol Biol. (1997) 17:541–51. doi: 10.1165/ajrcmb.17.5.2883
37. Kuperman DA, Huang X, Koth LL, Chang GH, Dolganov GM, Zhu Z, et al. Direct effects of interleukin-13 on epithelial cells cause airway hyperreactivity and mucus overproduction in asthma. Nat Med. (2002) 8:885. doi: 10.1038/nm734
38. Park K-S, Korfhagen TR, Bruno MD, Kitzmiller JA, Wan H, Wert SE, et al. SPDEF regulates goblet cell hyperplasia in the airway epithelium. J Clin Invest. (2007) 117:978–88. doi: 10.1172/JCI29176
39. Chen G, Korfhagen TR, Xu Y, Kitzmiller J, Wert SE, Maeda Y, et al. SPDEF is required for mouse pulmonary goblet cell differentiation and regulates a network of genes associated with mucus production. J Clin Invest. (2009) 119:2914–24. doi: 10.1172/JCI39731
40. Chen G, Volmer AS, Wilkinson KJ, Deng Y, Jones LC, Yu D, et al. Role of Spdef in the regulation of Muc5b expression in the airways of naive and mucoobstructed mice. Am J Respirat Cell Mol Biol. (2018) 59:383–96. doi: 10.1165/rcmb.2017-0127OC
41. Zhen G, Park SW, Nguyenvu LT, Rodriguez MW, Barbeau R, Paquet AC, et al. IL-13 and epidermal growth factor receptor have critical but distinct roles in epithelial cell mucin production. Am J Respirat Cell Mol Biol. (2007) 36:244–53. doi: 10.1165/rcmb.2006-0180OC
42. Burgel P, Nadel J. Epidermal growth factor receptor-mediated innate immune responses and their roles in airway diseases. Eur Respirat J. (2008) 32:1068–81. doi: 10.1183/09031936.00172007
43. Lai HY, Rogers DF. Mucus hypersecretion in asthma: intracellular signalling pathways as targets for pharmacotherapy. Curr Opin Allergy Clin Immunol. (2010) 10:67–76. doi: 10.1097/ACI.0b013e328334643a
44. Booth BW, Adler KB, Bonner JC, Tournier F, Martin LD. Interleukin-13 induces proliferation of human airway epithelial cells in vitro via a mechanism mediated by transforming growth factor-α. Am J Respirat Cell Mol Biol. (2001) 25:739–43. doi: 10.1165/ajrcmb.25.6.4659
45. Burgel PR, Lazarus SC, Tam DCW, Ueki IF, Atabai K, Birch M, et al. Human eosinophils induce mucin production in airway epithelial cells via epidermal growth factor receptor activation. J Immunol. (2001) 167:5948–54. doi: 10.4049/jimmunol.167.10.5948
46. Okumura S, Sagara H, Fukuda T, Saito H, Okayama Y. FcεRI-mediated amphiregulin production by human mast cells increases mucin gene expression in epithelial cells. J Allergy Clin Immunol. (2005) 115:272–9. doi: 10.1016/j.jaci.2004.10.004
47. Jorissen RN, Walker F, Pouliot N, Garrett TP, Ward CW, Burgess AW. Epidermal growth factor receptor: mechanisms of activation and signalling. EGF Receptor Family. (2003) 284:33–55. doi: 10.1016/B978-012160281-9/50004-9
48. Tyner JW, Kim EY, Ide K, Pelletier MR, Roswit WT, Morton JD, et al. Blocking airway mucous cell metaplasia by inhibiting EGFR antiapoptosis and IL-13 transdifferentiation signals. J Clin Invest. (2006) 116:309–21. doi: 10.1172/JCI25167
49. Boucherat O, Morissette MC, Provencher S, Bonnet S, Maltais F. Bridging lung development with chronic obstructive pulmonary disease. Relevance of developmental pathways in chronic obstructive pulmonary disease pathogenesis. Am J Respirat Crit Care Med. (2016) 193:362–75. doi: 10.1164/rccm.201508-1518PP
50. Guseh JS, Bores SA, Stanger BZ, Zhou Q, Anderson WJ, Melton DA, et al. Notch signaling promotes airway mucous metaplasia and inhibits alveolar development. Development. (2009) 136:1751–9. doi: 10.1242/dev.029249
51. Danahay H, Pessotti AD, Coote J, Montgomery BE, Xia D, Wilson A, et al. Notch2 is required for inflammatory cytokine-driven goblet cell metaplasia in the lung. Cell Reports. (2015) 10:239–52. doi: 10.1016/j.celrep.2014.12.017
52. Hao Y, Kuang Z, Walling BE, Bhatia S, Sivaguru M, Chen Y, et al. Pseudomonas aeruginosa pyocyanin causes airway goblet cell hyperplasia and metaplasia and mucus hypersecretion by inactivating the transcriptional factor FoxA2. Cell Microbiol. (2012) 14:401–15. doi: 10.1111/j.1462-5822.2011.01727.x
53. Hao Y, Kuang Z, Jing J, Miao J, Mei LY, Lee RJ, et al. Mycoplasma pneumoniae modulates STAT3-STAT6/EGFR-FOXA2 signaling to induce overexpression of airway mucins. Infect Immunity. (2014) 82:5246–55. doi: 10.1128/IAI.01989-14
54. Choi W, Yang AX, Waltenburg MA, Choe S, Steiner M, Radwan A, et al. FOXA2 depletion leads to mucus hypersecretion in canine airways with respiratory diseases. Cell Microbiol. (2018) 21:e12957. doi: 10.1111/cmi.12957
55. Helling BA, Gerber AN, Kadiyala V, Sasse SK, Pedersen BS, Sparks L, et al. Regulation of MUC5B expression in idiopathic pulmonary fibrosis. Am J Respirat Cell Mol Biol. (2017) 57:91–9. doi: 10.1165/rcmb.2017-0046OC
56. Lachowicz-Scroggins ME, Finkbeiner WE, Gordon E, Yuan S, Zlock L, Bhakta NR, et al. Corticosteroid and long-acting ß-agonist therapy reduces epithelial goblet cell metaplasia. Clin Exp Allergy. (2017) 47:1534–45. doi: 10.1111/cea.13015
57. Seibold MA, Wise AL, Speer MC, Steele MP, Brown KK, Loyd JE, et al. A common MUC5B promoter polymorphism and pulmonary fibrosis. N Engl J Med. (2011) 364:1503–12. doi: 10.1056/NEJMoa1013660
58. Park SW, Verhaeghe C, Nguyenvu LT, Barbeau R, Eisley CJ, Nakagami Y, et al. Distinct roles of FOXA2 and FOXA3 in allergic airway disease and asthma. Am J Respirat Crit Care Med. (2009) 180:603–10. doi: 10.1164/rccm.200811-1768OC
59. Du C, Lu J, Zhou L, Wu B, Zhou F, Gu L, et al. MAPK/FoxA2-mediated cigarette smoke-induced squamous metaplasia of bronchial epithelial cells. Int J Chronic Obstruct Pulmonary Dis. (2017) 12:3341. doi: 10.2147/COPD.S143279
60. Tunney MM, Einarsson GG, Wei L, Drain M, Klem ER, Cardwell C, et al. Lung microbiota and bacterial abundance in patients with bronchiectasis when clinically stable and during exacerbation. Am J Respirat Crit Care Med. (2013) 187:1118–26. doi: 10.1164/rccm.201210-1937OC
61. Wilson C, Jones P, O'leary C, Hansell D, Cole P, Wilson R. Effect of sputum bacteriology on the quality of life of patients with bronchiectasis. Eur Respirat J. (1997) 10:1754–60. doi: 10.1183/09031936.97.10081754
62. Kreuter M, Cottin V. The threat in chronic lung diseases: acute exacerbations. Eur Respiratory Soc. (2017) 26:170075. doi: 10.1183/16000617.0075-2017
63. Riquelme SA, Ahn D, Prince A. Pseudomonas aeruginosa and Klebsiella pneumoniae adaptation to innate immune clearance mechanisms in the lung. J Innate Immunity. (2018) 10:442–54. doi: 10.1159/000487515
64. Resiliac J, Grayson MH. Epidemiology of infections and development of asthma. Immunol Allergy Clin. (2019) 39:297–307. doi: 10.1016/j.iac.2019.03.001
65. Döring G, Parameswaran IG, Murphy TF. Differential adaptation of microbial pathogens to airways of patients with cystic fibrosis and chronic obstructive pulmonary disease. FEMS Microbiol Rev. (2011) 35:124–46. doi: 10.1111/j.1574-6976.2010.00237.x
66. Caldwell CC, Chen Y, Goetzmann HS, Hao Y, Borchers MT, Hassett DJ, et al. Pseudomonas aeruginosa exotoxin pyocyanin causes cystic fibrosis airway pathogenesis. Am J Pathol. (2009) 175:2473–88. doi: 10.2353/ajpath.2009.090166
67. Xu Y, Duan C, Kuang Z, Hao Y, Jeffries JL, Lau GW. Pseudomonas aeruginosa pyocyanin activates NRF2-ARE-mediated transcriptional response via the ROS-EGFR-PI3K-AKT/MEK-ERK MAP kinase signaling in pulmonary epithelial cells. PLoS ONE. (2013) 8:e72528. doi: 10.1371/journal.pone.0072528
68. Li JD, Feng W, Gallup M, Kim JH, Gum J, Kim Y, et al. Activation of NF- B via a Src-dependent Ras-MAPK-pp90rsk pathway is required for Pseudomonas aeruginosa-induced mucin overproduction in epithelial cells. Proc Natl Acad Sci USA. (1998) 95:5718–23. doi: 10.1073/pnas.95.10.5718
69. Mohamed FB, Garcia-Verdugo I, Medina M, Balloy V, Chignard M, Ramphal R, et al. A crucial role of Flagellin in the induction of airway mucus production by Pseudomonas aeruginosa. PLoS ONE. (2012) 7:e39888. doi: 10.1371/journal.pone.0039888
70. Mcnamara N, Khong A, Mckemy D, Caterina M, Boyer J, Julius D, et al. ATP transduces signals from ASGM1, a glycolipid that functions as a bacterial receptor. Proc Natl Acad Sci USA. (2001) 98:9086–91. doi: 10.1073/pnas.161290898
71. Kishioka C, Okamoto K, Hassett DJ, De Mello D, Rubin BK. Pseudomonas aeruginosa alginate is a potent secretagogue in the isolated ferret trachea. Pediatric Pulmonol. (1999) 27:174–9. doi: 10.1002/(SICI)1099-0496(199903)27:3<174::AID-PPUL4>3.0.CO;2-A
72. Klinger JD, Tandler B, Liedtke C, Boat T. Proteinases of Pseudomonas aeruginosa evoke mucin release by tracheal epithelium. J Clin Invest. (1984) 74:1669–78. doi: 10.1172/JCI111583
73. Jeffries JL, Jia J, Choi W, Choe S, Miao J, Xu Y, et al. Pseudomonas aeruginosa pyocyanin modulates mucin glycosylation with sialyl-Lewisx to increase binding to airway epithelial cells. Mucosal Immunol. (2015) 9:1039–50. doi: 10.1038/mi.2015.119
74. Ran H, Hassett DJ, Lau GW. Human targets of Pseudomonas aeruginosa pyocyanin. Proc Natl Acad Sci USA. (2003) 100:14315–20. doi: 10.1073/pnas.2332354100
75. Gardner PR. Superoxide production by the mycobacterial and pseudomonad quinoid pigments phthiocol and pyocyanine in human lung cells. Arch Biochem Biophy. (1996) 333:267–74. doi: 10.1006/abbi.1996.0390
76. O'malley YQ, Reszka KJ, Spitz DR, Denning GM, Britigan BE. Pseudomonas aeruginosa pyocyanin directly oxidizes glutathione and decreases its levels in airway epithelial cells. Am J Physiol. (2004) 287:L94–103. doi: 10.1152/ajplung.00025.2004
77. Lau GW, Hassett DJ, Ran H, Kong F. The role of pyocyanin in Pseudomonas aeruginosa infection. Trends Mol Med. (2004) 10:599–606. doi: 10.1016/j.molmed.2004.10.002
78. O'malley YQ, Reszka KJ, Rasmussen GT, Abdalla MY, Denning GM, Britigan BE. The Pseudomonas secretory product pyocyanin inhibits catalase activity in human lung epithelial cells. Am J Physiol. (2003) 285:L1077–86. doi: 10.1152/ajplung.00198.2003
79. Schwarzer C, Fischer H, Kim E-J, Barber KJ, Mills AD, Kurth MJ, et al. Oxidative stress caused by pyocyanin impairs CFTR Cl- transport in human bronchial epithelial cells. Free Rad Biol Med. (2008) 45:1653–62. doi: 10.1016/j.freeradbiomed.2008.09.011
80. Lau GW, Ran H, Kong F, Hassett DJ, Mavrodi D. Pseudomonas aeruginosa pyocyanin is critical for lung infection in mice. Infect Immunity. (2004) 72:4275–8. doi: 10.1128/IAI.72.7.4275-4278.2004
81. Wilson R, Sykes D, Watson D, Rutman A, Taylor G, Cole P. Measurement of Pseudomonas aeruginosa phenazine pigments in sputum and assessment of their contribution to sputum sol toxicity for respiratory epithelium. Infect Immunity. (1988) 56:2515–7. doi: 10.1128/IAI.56.9.2515-2517.1988
82. Hunter RC, Klepac-Ceraj V, Lorenzi MM, Grotzinger H, Martin TR, Newman DK. Phenazine content in the cystic fibrosis respiratory tract negatively correlates with lung function and microbial complexity. Am J Respirat Cell Mol Biol. (2012) 47:738–45. doi: 10.1165/rcmb.2012-0088OC
83. Forteza R, Lauredo IT, Burch R, Abraham WM. Extracellular metabolites of Pseudomonas aeruginosa produce bronchoconstriction by different mechanisms. Am J Respirat Crit Care Med. (1994) 149:687–93. doi: 10.1164/ajrccm.149.3.8118638
84. Munro N, Barker A, Rutman A, Taylor G, Watson D, Mcdonald-Gibson W, et al. Effect of pyocyanin and 1-hydroxyphenazine on in vivo tracheal mucus velocity. J Appl Physiol. (1989) 67:316–23. doi: 10.1152/jappl.1989.67.1.316
85. Dormehl I, Ras G, Taylor G, Hugo N. Effect of Pseudomonas aeruginosa-derived pyocyanin and 1-hydroxyphenazine on pulmonary mucociliary clearance monitored scintigraphically in the baboon model. Int J Rad Appl Instrument B Nucl Med Biol. (1991) 18:455–9. doi: 10.1016/0883-2897(91)90105-T
86. Rada B, Gardina P, Myers TG, Leto TL. Reactive oxygen species mediate inflammatory cytokine release and EGFR-dependent mucin secretion in airway epithelial cells exposed to Pseudomonas pyocyanin. Mucos Immunol. (2011) 4:158–71. doi: 10.1038/mi.2010.62
87. Vallath S, Hynds RE, Succony L, Janes SM, Giangreco A. Targeting EGFR signalling in chronic lung disease: therapeutic challenges and opportunities. Eur Respiratory Soc. (2014) 44:513–22. doi: 10.1183/09031936.00146413
88. Khavinson VK, Tendler S, Vanyushin B, Kasyanenko N, Kvetnoy I, Linkova N, et al. Peptide regulation of gene expression and protein synthesis in bronchial epithelium. Lung. (2014) 192:781–91. doi: 10.1007/s00408-014-9620-7
89. Viby NE, Isidor MS, Buggeskov KB, Poulsen SS, Hansen JB, Kissow H. Glucagon-like peptide-1 (GLP-1) reduces mortality and improves lung function in a model of experimental obstructive lung disease in female mice. Endocrinology. (2013) 154:4503–11. doi: 10.1210/en.2013-1666
90. Choi W, Choe S, Lin J, Borchers MT, Kosmider B, Vassallo R, et al. Exendin-4 restores airway mucus homeostasis through the GLP1R-PKA-PPARγ-FOXA2-phosphatase signaling. Mucosal Immunol. (in press). doi: 10.1038/s41385-020-0262-1
91. Dailey MJ, Moran TH. Glucagon-like peptide 1 and appetite. Trends Endocrinol Metab. (2013) 24:85–91. doi: 10.1016/j.tem.2012.11.008
92. Nasr SZ, Drury D. Appetite stimulants use in cystic fibrosis. Pediatric Pulmonol. (2008) 43:209–19. doi: 10.1002/ppul.20766
Keywords: chronic lung diseases, mucus homeostasis, mucociliary clearance, FOXA2, EGFR, STAT6, MUC5AC, MUC5B
Citation: Choi W, Choe S and Lau GW (2020) Inactivation of FOXA2 by Respiratory Bacterial Pathogens and Dysregulation of Pulmonary Mucus Homeostasis. Front. Immunol. 11:515. doi: 10.3389/fimmu.2020.00515
Received: 18 November 2019; Accepted: 06 March 2020;
Published: 25 March 2020.
Edited by:
Markus M. Heimesaat, Charité—Universitätsmedizin Berlin, GermanyReviewed by:
Pieter Sicco Hiemstra, Leiden University Medical Center, NetherlandsBurton F. Dickey, University of Texas MD Anderson Cancer Center, United States
Copyright © 2020 Choi, Choe and Lau. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Gee W. Lau, Z2VlbGF1QGlsbGlub2lzLmVkdQ==