
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Immunol. , 20 March 2020
Sec. Viral Immunology
Volume 11 - 2020 | https://doi.org/10.3389/fimmu.2020.00495
This article is part of the Research Topic Heterologous Immunity: Implications and Applications in Vaccines and Immunotherapies View all 14 articles
 Emma Connolly*
Emma Connolly* Tracy Hussell*
Tracy Hussell*That macrophages adapt to environmental cues is well-established. This adaptation has had several reiterations, first with innate imprinting and then with various combinations of trained, tolerant, paralyzed, or primed. Whatever the nomenclature, it represents a macrophage that is required to perform very different functions. First, alveolar macrophages are one of the sentinel cells that flag up damage and release mediators that attract other immune cells. Next, they mature to support T cell priming and survival. Finally they are critical in clearing inflammatory immune cells by phagocytosis and extracellular matrix turnover components by efferocytosis. At each functional stage they alter intrinsic components to guide their activity. Training therefore is akin to changing function. In this mini-review we focus on the lung and the specific role of type I interferons in altering macrophage activity. The proposed mechanisms of type I IFNs on lung-resident alveolar macrophages and their effect on host susceptibility to bacterial infection following influenza virus infection.
Bacteria entering the respiratory tract are generally tolerated well in healthy adults and their growth contained by the host commensal microbiome, antimicrobial peptides, phagocytic cells (predominantly macrophages), mucus entrapment, and ciliary clearance. Some bacteria associated with respiratory tract infections are part of the normal microbiome in health, such as Streptococcus pneumoniae, Haemophilus influenza, and S. aureus (1–3). However, severe consequences arise when the lung microenvironment is perturbed in some way. Perturbations can include underlying congenital abnormalities (e.g., primary ciliary dyskinesia), underlying chronic disease (e.g., asthma, chronic obstructive pulmonary disease, cystic fibrosis, idiopathic pulmonary fibrosis), the effect of the aging process, the premature lung and previous severe infections (4). In all cases, the outcome depends on the severity of the perturbation, the rate of bacterial growth, and whether the bacterium is contained in the airspaces or invades the lung tissue and systemic circulation.
Containment of bacteria relies on effective physical and chemical barriers, but also a timely immune response. Any delay in immunity allows the growth of bacteria to an over-whelming level. It is interesting to note that conditions associated with bacterial out-growth occur in situations where the lung has a heavy infiltration of the very cells (macrophages and neutrophils) required to clear the micro-organism, which suggests they are not functioning properly (5). The function and phenotype of any immune cell is influenced by the local microenvironment and the needs of the tissue at that time. We referred to this adaptation as “innate imprinting” in 2004 (6) that was superseded by the term “trained immunity” (7–10). However, the terminology continues to evolve and now trained immunity represents a “primed” state that is beneficial, whereas the more immune paralyzed state (as observed following viral infection of the lung) is referred to as a “tolerant” state. Trained/tolerant innate immunity is important in health, disease and disease resolution. The molecular mechanisms of trained immunity in health have been described extensively elsewhere (11). Here we will describe how alveolar macrophages are tolerised during and following inflammation with a specific emphasis on the role of type I interferons (type I IFNs).
Specificity and adaptation were once the hallmark of adaptive immunity alone. However, epidemiological studies as early as 1946 recognized that the Mycobacterium tuberculosis vaccine, BCG, also protected against childhood mortality caused by antigenically indistinct organisms, suggesting “adaptation” of cells of the innate, rather than adaptive, immune system (12, 13). Since then more recent studies have shown that innate immune cells can display adaptive characteristics (11). In terms of generating a specific response, it could be argued that pattern recognition receptors (PRRs), expressed by innate immune cells, confer specificity. PRRs are germline-encoded receptors and include the toll-like receptors (TLRs), RIG-I-like receptors (RLRs), NOD-like receptors (NLRs), and C-type lectins (14), among others. These receptors vary widely in the ligands that they bind to, allowing them to detect a substantial range of molecular patterns, known as pathogen- and damage-associated molecular pathogens (PAMPs and DAMPs, respectively) (15). This activates both divergent and convergent downstream signaling pathways enabling a tailored response to a specific pathogen (14). Furthermore, it is now recognized that innate immune cells, for example myeloid cells (7, 8, 16), NK cells (17, 18) and epithelial cells (19), can acquire “memory”, characterized as a heightened and quicker response upon re-exposure to a pathogen. Innate immune memory is well-defined in organisms that lack an adaptive immune system, including plants and invertebrates (20, 21). This is more controversial in vertebrates, partly due to the relatively short half-life of innate cells, which in the case of monocytes can be up to 1 day in the circulation (22). However, the presence of innate immune memory in monocytes has been observed for up to 3 months (13) and for macrophages 6 months or more (23). This innate immune memory or trained immunity likely serves as an evolutionary survival advantage with the innate immune system primed to combat a secondary pathogen encounter (11). However, training can lead to deleterious consequences if the outcome is a macrophage that is tolerant to stimulation. A slower macrophage response likely protects the host from further tissue damage, prioritizes a reparative state and prevents the development of autoimmunity. In the case of severe influenza virus infection, upon resolution macrophages are unable to respond quickly enough to curtail bacterial load leading to complications of secondary pneumonia (24).
There are many mechanisms associated with susceptibility to bacterial complications following lung viral infection. However, type I interferons (IFNs) stand out as particularly important as they directly impair, or lead to downstream consequences affecting, bacterial clearance (Figure 1) (25–30). All three types of interferons (Types I – III) play a major role in innate and adaptive immunity (14). Of the eight (-α, -β, -δ, -ε, -ζ, -κ, -τ, and –ω) type I IFNs, the -α, -β forms, which bind to the IFNAR receptor complex (IFNAR1 and IFNAR2), have received the most attention with regards to lung viral infection (32). Receptor binding recruits janus kinase 1 (JAK1) and tyrosine kinase 2 (TYK2) that leads to the phosphorylation of Signal Transducer and Activator of Transcription (STATs). Phosphorylation leads to homodimers and heterodimers; the precise combination dictating the final transcriptional outcome. STAT 1 and 2 heterodimers bind to IRF9 to form the ISG (Interferon Stimulated Gene) factor 3 complex−9 (33, 34). Type I IFNs also activate the p38-associated MAPK (mitogen-activated protein kinase pathway) (35). Type I IFNs have a myriad of functions in the lung where they are both crucial for the clearance of viral infection and resolution of inflammation. However, it is these diverse functions that are thought to contribute to host susceptibility to bacterial infections following viral infection.
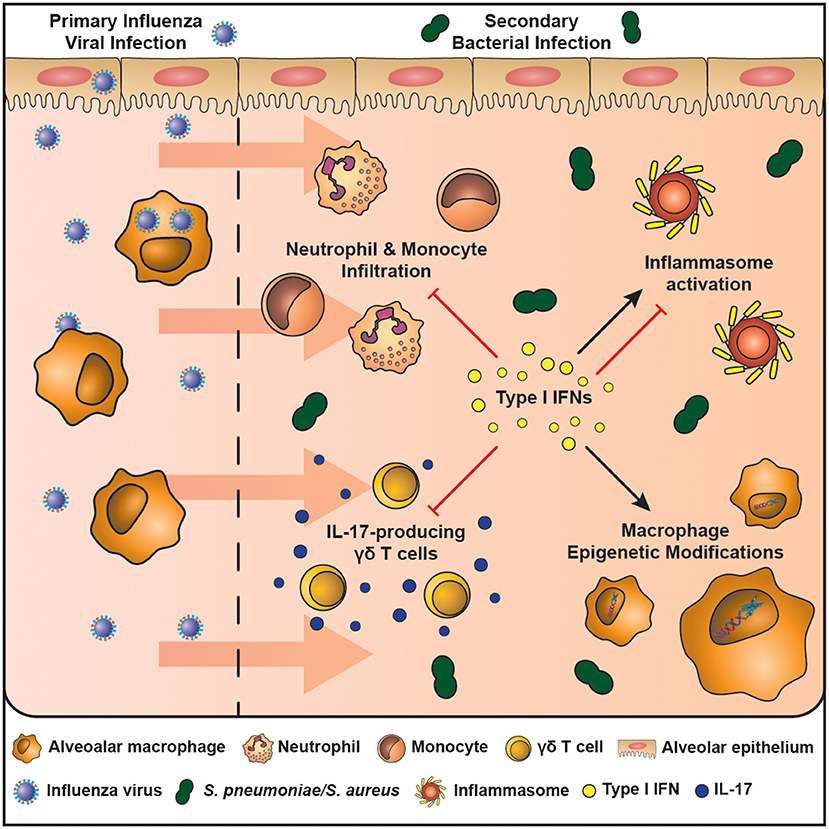
Figure 1. The mechanisms of enhanced host susceptibility to secondary bacterial infection by type I IFNs. Airway epithelial cells and alveolar macrophages are cells of the innate immune system that are at the first line of defense against infection in the airspaces. The influenza virus activates pattern recognition receptors expressed by airway epithelial cells and macrophages and leads to the production of type I IFNs, which are crucial in combating the infection. However, type I IFNs also induce an immunosuppressive state in the resolution phase of infection that enhances host susceptibility to secondary bacterial infection. These mechanisms include: (1) inhibition of IL-17-producing γδ T cells, (2) Induces macrophage epigenetic modifications, (3) Induces or inhibits inflammasome activation in a context-dependent manner, (4) Inhibits neutrophil and monocyte infiltration. These mechanisms result in a lung environment ill equipped to fight an increasing bacterial burden.
The contribution of type I IFNs to host susceptibly to bacterial infection is well-established (Table 1). In 2001, Biron's group defined a role for IFN α/β in viral-induced sensitization to bacterial products (36). Viral mimics, such as Poly I:C, also impair anti-bacterial immunity by induction of type I IFNs (37). Since then the field has expanded rapidly to show that type I IFNs decrease neutrophil chemoattractants (CXCL1/2) (25, 26), reduce IL-17 producing γδ T cells (27), and impair CCL2- mediated recruitment of macrophages following viral infection (28). Furthermore, depending on the context type I IFNs can promote or inhibit NLRP3 inflammasome activation, causing either an increase in IL-1β that limits γδ T cell activity with subsequent susceptibility to S. pneumoniae (27) or decreases IL-1β production enhancing susceptibility to S. aureus (29, 30), respectively. Additionally, type I IFN induced by viral infection alters cellular metabolism that may favor bacterial replication, uptake and adhesion (38). These mechanisms have been reviewed extensively elsewhere (39).
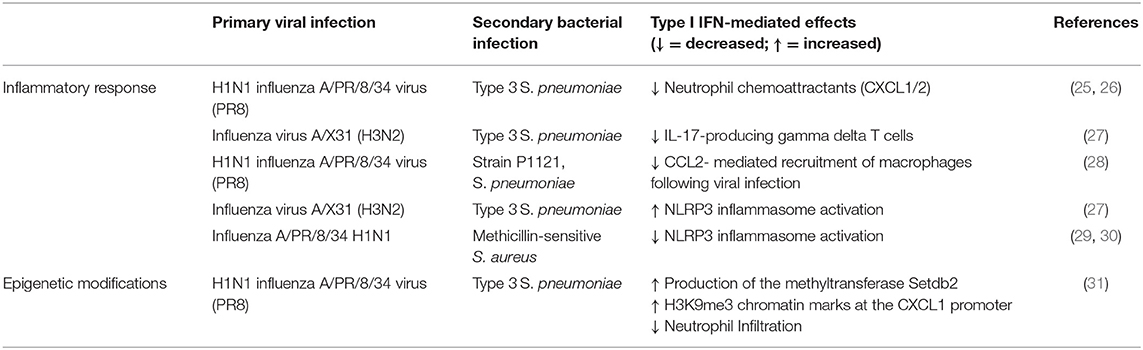
Table 1. The effects of viral—induced type I IFN on the inflammatory response to secondary bacterial infections.
The immune suppressive outcome of enhanced type I IFNs is exemplified by strategies to inhibit its action. Inhibition of IFN receptor I- and III-associated TYK2 restores anti-bacterial immunity in a human ex vivo lung co-infection model (40). An absence of STAT2 that is downstream of IFN-αR 1/2 makes influenza infection more severe, but prevents the development of secondary bacterial pneumonia (41). Furthermore, blocking Toll-like receptor 4 (TLR4) after influenza virus infection decreases bacterial growth by reducing IFNβ (26). Type I IFN induction may also contribute to the risk of bacterial infection following the administration of anesthetics prior to surgery. Infectious risk due to the immune modulatory effects of anesthetics delays surgical procedures in patients suspected of a respiratory infection. However, not all anesthetics cause this problem (42) and halothane actually reduces bacterial burden in influenza infected mice by decreasing type I IFN in the mouse lung (43). These observations suggest that type I IFN-induced tolerance following severe lung viral infection, although beneficial in limiting excess tissue damage and restoring tissue to homeostasis, results in a macrophage unable to deal with a growing bacterial burden.
Type I IFNs are directly involved in important processes necessary to restore the lung to health. A reduction of inflammatory responses during apoptotic cell clearance is critical to prevent autoimmunity to self-antigens. Type I IFN receptor signaling induces suppressor of cytokine signaling (SOCS) 1 and 3 activation during efferocytosis of apoptotic cells by the receptor tyrosine kinase AXL (44). The combination of AXL and IFNAR1 signaling causes reduced macrophage responses and subsequent bacterial complications (45, 46). Furthermore, macrophages are also “tolerised” during the uptake of extracellular matrix turnover by-products; again an important function to restore homeostasis (47). The glycosaminoglycan, hyaluronan for example, is a prevalent extracellular matrix component in the lung (48), but it suppresses alveolar macrophage activity and is maintained at a higher level following resolution of a severe viral infection (49). Similarly, versican, a chondroitin sulfate proteoglycan, is expressed at low levels in the healthy lungs, but upregulated by TLR agonists LPS and Poly I:C and requires TLR, TRIF and type I IFN signaling. In turn versican up-regulates IL-10 and IFNβ, leading to an immune suppressive state (50). Therefore, repairing the damaged lung and restoring the steady state, impairs inflammation and involves type I IFNs. This raises the possibility that trained immunity in macrophages simply represents a change in function from inflammation to homeostatic maintenance.
The longevity of alterations in lung immunity following severe viral infection is surprising considering the relatively short life of innate immune cells. However, alveolar macrophages in particular, turnover relatively slowly in health (51). Of particular relevance to the altered reactivity of alveolar macrophages, is their re-wiring by epigenetic changes (52). Epigenetic changes are mediated by (micro) miRNAs, DNA methylation, and histone modifications, amongst others and regulate chromatin accessibility (53). Chromatin accessibility determines which genes are visible and therefore impacts on cellular signaling and gene expression.
Monocyte/Macrophage adaptation is accompanied by fundamental epigenetic changes (54, 55) and is often associated with alterations in cellular metabolism (56, 57). Trained monocytes, producing excess TNFα and IL-6 protect RAG-/- mice (lacking functional T and B lymphocytes) against reinfection with Candida albicans due to stable histone trimethylation at H3K4 (8). Candida binding to Dectin-1 causes stable changes in histone trimethylation at H3K4 and increases the immune responsiveness of monocytes (8). Similarly, chromatin modifications by BCG vaccination provide protection to unrelated infections (13). Tolerance induction in macrophages cultured with LPS results in methylation at H3K9me2 and H3K9me3 and protects against S. aureus infection (58). Looking beyond pro-inflammatory processes, it is clear that in tolerised macrophages not all genes are repressed in all circumstances. For example, LPS-stimulation of murine macrophages in vitro represses pro-inflammatory genes, but enhances genes encoding anti-microbial effector proteins (16). However, this is often not the case in vivo, where reduced anti-bacterial immunity and macrophage effector function are observed following viral infection. This discrepancy, represents an opportunity since it suggests that some stimuli lead to a different macrophage outcome. A recent study of influenza infection followed by a S. pnemoniae strain lacking the major virulence factor pneumolysin, shows that not all macrophages are affected equally and that long term epigenetic changes differ between recruited and resident macrophages (59). Understanding how to achieve a bactericidal vs. an anti-inflammatory macrophage outcome could provide strategies to combat post-viral bacterial pneumonia.
Type I IFN modification of the epigenetic landscape is mostly via their regulation of interferon-stimulated genes (ISGs) (60, 61). ISGs encode a wide range of proteins that restrict viral infection and spread, including inhibition of viral transcription, translation and replication, the degradation of viral nucleic acids and the alteration of cellular lipid metabolism (62, 63). Approximately 2,000 human and mouse ISGs have been identified and cataloged in the Interferome database (64). All classes of IFNs have overlapping ISGs (65, 66) and so it remains unclear how ISGs are regulated in order to produce a unique and tailored response to a given pathogen. Epigenetic modifications are proposed as one mechanism by which ISG transcription can be context specific (65). The ISGs induced may depend on the cell type, the exposure of the cell to other stimuli, such as PAMPs or DAMPs, or the strength and duration of the interferon stimulus. All these variables may affect the chromatin landscape and provide another level of ISG regulation to different environmental cues. Evidence shows that enhanced transcription of ISGs upon re-stimulation is not due to increased expression of the required transcription factors or IFN signaling molecules, but rather as a result of altered chromatin marks at ISG promoters, thereby priming or repressing certain ISGs. Of the 1,000 s of ISGs known, only half are reported to become primed or display “memory” upon restimulation (61). Other inflammatory factors present in the microenvironment also affect the profile of ISGs available. For example, in response to LPS, type I IFNs prevent the silencing of inflammatory genes driven by prior TNF exposure of macrophages. This is mediated by an altered chromatin state, with increased recruitment of H4ac and H3K4me3 histone marks that are generally associated with transcriptional activity, and increased chromatin accessibility at tolerised genes (60). In addition to driving alterations in the epigenome, type I IFNs can also be regulated by epigenetic modifications. For example, miR146a (67), and miR26a (68) promote type I IFNs and reduce influenza infection in experimental models, whereas miR29a reduces IFNAR1 and has the opposite effect (69).
An important aspect of viral-induced macrophage tolerance to consider is whether it can be overcome or reversed in order to unleash the full inflammatory potential of macrophages and promote anti-bacterial responses. One possibility could be via manipulation of epigenetic changes. For example, histone modifications are reversible and therefore can be altered. The methyltransferase Setdb2 is an ISG that regulates the production of the neutrophil chemoattractant CXCL1. Deletion of Setdb2 decreases H3K9me3 chromatin marks, releases the CXCL1 promoter from inhibition, enhances airway neutrophil infiltration and reduces susceptibility to secondary S. pneumoniae (31). Furthermore, β-glucan can overcome the tolerised phenotype of macrophages following LPS exposure (70) and monocytes from experimental endotoxemia in healthy volunteers. This suggests that it is possible to improve the antibacterial function of macrophages. A tolerance phenotype is also observed in other cells. Airway epithelial cells, for example, are also refractory to TLR agonists following stimulation that can be restored by histone deacetylase inhibitors (71). Although not specifically identified to our knowledge, it would be interesting to determine whether these inhibitors could potentially reverse macrophage tolerance and reduce susceptibility to secondary bacterial infections.
The post-viral lung effects of type I IFNs span multiple bacterial species, including Streptococcus pneumoniae (25), Pseudomonas aeruginosa (72), Staphylococcus aureus (73) [including multi-drug resistant forms (41)] and Escherichia coli (74). Furthermore, the preceding viral infection need not be in the lung. For example, systemic Lymphocytic choriomeningitis Virus (LCMV) infection causes apoptosis of granulocytes in the bone marrow leading to reduced recruitment of neutrophils to the airways during Listeria monocytogenes or S. aureus infection (75). Therefore, manipulation of type I IFNs may represent a therapeutic option once bacterial complications arise following severe viral lung infection. Targeting of type I IFN responses is currently used in the treatment of several inflammatory and autoimmune diseases. For instance, IFNβ is an effective therapy for multiple sclerosis patients and IFNα has been approved for the treatment of hepatitis B and C (76). In contrast, the blockade of the type I IFN receptor with anti-IFNAR, has been an attractive therapeutic for autoimmune diseases including systemic lupus erythematosus (SLE) and rheumatoid arthritis, as these diseases are characterized by a profound IFN gene signature (77, 78). However, difficulties in developing effective therapies that target the type I IFN system relies upon selecting the specific type I IFN to administer or block, and the timing of drug delivery, which can lead to opposing outcomes. This is observed by the pro-inflammatory and immunosuppressive mechanisms that type I IFNs can generate in the tumor microenvironment. Although IFNα immunotherapy has proven effective in the treatment of hematological malignancies (79, 80), type I IFN treatment of solid tumors has shown less potential (81). Conversely, type I IFN inhibition can promote an anti-tumor responses by unleashing the inflammatory potential of exhausted T cells and removing the requirement for combinatorial immune checkpoint inhibitor immunotherapies (82). Further understanding of the roles of individual interferons in different inflammatory contexts and the divergent downstream signaling pathways they trigger is still required to generate effective treatment options. Currently, research is lacking for targeting type I IFNs to treat secondary bacterial pneumonia. However, studies suggest that targeting the epigenome of ISGs may be a more successful avenue of investigation. This would more likely limit potential negative side effects that may arise from removing type I IFNs themselves.
Type I IFNs clearly play a central role in bacterial super infections following lung damage, particularly that caused by pulmonary viral infection. Here we have focused on the effect of, predominantly, influenza infection on macrophages. However, similar processes may exist following infection with other respiratory viruses, such as respiratory syncytial virus. Collectively, the evidence suggests that overcoming type I IFN driven immune suppression may be beneficial for viral-induced bacterial super infection. Anti-IFNAR (e.g., Sifalimumab) is already used in the treatment of SLE (83) and could be repurposed for post-viral lung conditions. However, any strategy would need to be carefully timed and type I IFN administration during influenza infection may enhance viral immunopathogenesis. Bacterial infections mostly arise when the bulk of viral titer has been eliminated. Sometimes there is a sufficient and visible window between viral infection and bacterial outgrowth that would allow timed treatment to be administered. However, ultimately the problem is dependent in the first place on the severity of the viral infection. Studies to date show that any strategy that reduces the impact of lung viral infection reduces the chances of developing subsequent bacterial complications. Vaccination would therefore still seem the best policy; as long as any attenuated forms do not induce excess type I IFNs themselves. Finally, we should remember that macrophages attune to the needs of the tissue. Their trained/tolerant/primed state is therefore not abnormal, but rather represents a macrophage that has to first inflame to recruit immune cells, then change to professionally instruct them and finally clear up the mess afterwards.
EC and TH drafted the manuscript, made substantial contributions to the conception and design of the work, approved the submitted version of the manuscript, and agreed to be accountable for all aspects of the work.
This work was supported in part by grants from the Wellcome Trust (202865/Z/16/Z).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We thank Joshua Casulli for assistance with Figure 1.
1. Dickson RP, Huffnagle GB. The lung microbiome: new principles for respiratory bacteriology in health and disease. PLoS Pathog. (2015) 11:e1004923. doi: 10.1371/journal.ppat.1004923
2. Dethlefsen L, McFall-Ngai M, Relman DA. An ecological and evolutionary perspective on human-microbe mutualism and disease. Nature. (2007) 449:811–8. doi: 10.1038/nature06245
3. Wang H, Anthony D, Selemidis S, Vlahos R, Bozinovski S. Resolving viral-induced secondary bacterial infection in COPD: a concise review. Front Immunol. (2018) 9:2345. doi: 10.3389/fimmu.2018.02345
4. Rynda-Apple A, Robinson KM, Alcorn JF. Influenza and bacterial superinfection: illuminating the immunologic mechanisms of disease. Infect Immun. (2015) 83:3764–70. doi: 10.1128/IAI.00298-15
5. Warr GA, Jakab GJ. Pulmonary inflammatory responses during viral pneumonia and secondary bacterial infection. Inflammation. (1983) 7:93–104. doi: 10.1007/BF00917815
6. Williams AE, Edwards L, Humphreys IR, Snelgrove R, Rae A, Rappuoli R, et al. Innate imprinting by the modified heat-labile toxin of Escherichia coli (LTK63) provides generic protection against lung infectious disease. J Immunol. (2004) 173:7435–43. doi: 10.4049/jimmunol.173.12.7435
7. Saeed S, Quintin J, Kerstens HH, Rao NA, Aghajanirefah A, Matarese F, et al. Epigenetic programming of monocyte-to-macrophage differentiation and trained innate immunity. Science. (2014) 345:1251086. doi: 10.1126/science.1251086
8. Quintin J, Saeed S, Martens JHA, Giamarellos-Bourboulis EJ, Ifrim DC, et al. Candida albicans infection affords protection against reinfection via functional reprogramming of monocytes. Cell Host Microbe. (2012) 12:223–32. doi: 10.1016/j.chom.2012.06.006
9. Cheng SC, Quintin J, Cramer RA, Shepardson KM, Saeed S, Kumar V, et al. mTOR- and HIF-1alpha-mediated aerobic glycolysis as metabolic basis for trained immunity. Science. (2014) 345:1250684. doi: 10.1126/science.1250684
10. Bordon Y. Macrophages: innate memory training. Nat Rev Immunol. (2014) 14:713. doi: 10.1038/nri3759
11. Netea MG, Joosten LA, Latz E, Mills KH, Natoli G, Stunnenberg HG, et al. Trained immunity: a program of innate immune memory in health and disease. Science. (2016) 352:aaf1098. doi: 10.1126/science.aaf1098
12. Levine MI, Sackett MF. Results of BCG immunization in New York City. Am Rev Tuberculosis. (1946) 53:517–32.
13. Kleinnijenhuis J, Quintin J, Preijers F, Joosten LA, Ifrim DC, Saeed S, et al. Bacille Calmette-Guerin induces NOD2-dependent nonspecific protection from reinfection via epigenetic reprogramming of monocytes. Proc Natl Acad Sci USA. (2012) 109:17537–42. doi: 10.1073/pnas.1202870109
14. McNab F, Mayer-Barber K, Sher A, Wack A, O'Garra A. Type I interferons in infectious disease. Nat Rev Immunol. (2015) 15:87–103. doi: 10.1038/nri3787
15. Medzhitov R. Toll-like receptors and innate immunity. Nat Rev Immunol. (2001) 1:135–45. doi: 10.1038/35100529
16. Foster SL, Hargreaves DC, Medzhitov R. Gene-specific control of inflammation by TLR-induced chromatin modifications. Nature. (2007) 447:972–8. doi: 10.1038/nature05836
17. Lee J, Zhang T, Hwang I, Kim A, Nitschke L, Kim M, et al. Epigenetic modification and antibody-dependent expansion of memory-like NK cells in human cytomegalovirus-infected individuals. Immunity. (2015) 42:431–42. doi: 10.1016/j.immuni.2015.02.013
18. Schlums H, Cichocki F, Tesi B, Theorell J, Beziat V, Holmes TD, et al. Cytomegalovirus infection drives adaptive epigenetic diversification of NK cells with altered signaling and effector function. Immunity. (2015) 42:443–56. doi: 10.1016/j.immuni.2015.02.008
19. Hamada A, Torre C, Drancourt M, Ghigo E. Trained immunity carried by non-immune cells. Front Microbiol. (2018) 9:3225. doi: 10.3389/fmicb.2018.03225
20. Durrant WE, Dong X. Systemic acquired resistance. Ann Rev Phytopathol. (2004) 42:185–209. doi: 10.1146/annurev.phyto.42.040803.140421
21. Kurtz J. Specific memory within innate immune systems. Trends Immunol. (2005) 26:186–92. doi: 10.1016/j.it.2005.02.001
22. Yona S, Kim KW, Wolf Y, Mildner A, Varol D, Breker M, et al. Fate mapping reveals origins and dynamics of monocytes and tissue macrophages under homeostasis. Immunity. (2013) 38:79–91. doi: 10.1016/j.immuni.2013.05.008
23. Didierlaurent A, Goulding J, Patel S, Snelgrove R, Low L, Bebien M, et al. Sustained desensitization to bacterial Toll-like receptor ligands after resolution of respiratory influenza infection. J Exp Med. (2008) 205:323–9. doi: 10.1084/jem.20070891
24. Hussell T, Bell TJ. Alveolar macrophages: plasticity in a tissue-specific context. Nat Rev Immunol. (2014) 14:81–93. doi: 10.1038/nri3600
25. Shahangian A, Chow EK, Tian X, Kang JR, Ghaffari A, Liu SY, et al. Type I IFNs mediate development of postinfluenza bacterial pneumonia in mice. J Clin Investig. (2009) 119:1910–20. doi: 10.1172/JCI35412
26. Shirey KA, Perkins DJ, Lai W, Zhang W, Fernando LR, Gusovsky F, et al. Influenza trains the host for enhanced susceptibility to secondary bacterial infection. mBio. (2019) 10:e00810–9. doi: 10.1128/mBio.00810-19
27. Li W, Moltedo B, Moran TM. Type I interferon induction during influenza virus infection increases susceptibility to secondary Streptococcus pneumoniae infection by negative regulation of gammadelta T cells. J Virol. (2012) 86:12304–12. doi: 10.1128/JVI.01269-12
28. Nakamura S, Davis KM, Weiser JN. Synergistic stimulation of type I interferons during influenza virus coinfection promotes Streptococcus pneumoniae colonization in mice. J Clin Investig. (2011) 121:3657–65. doi: 10.1172/JCI57762
29. Robinson KM, Choi SM, McHugh KJ, Mandalapu S, Enelow RI, Kolls JK, et al. Influenza A exacerbates Staphylococcus aureus pneumonia by attenuating IL-1beta production in mice. J Immunol. (2013) 191:5153–9. doi: 10.4049/jimmunol.1301237
30. Shepardson KM, Larson K, Morton RV, Prigge JR, Schmidt EE, Huber VC, et al. Differential type I interferon signaling is a master regulator of susceptibility to postinfluenza bacterial superinfection. mBio. (2016) 7:e00506–16. doi: 10.1128/mBio.00506-16
31. Schliehe C, Flynn EK, Vilagos B, Richson U, Swaminanthan S, Bosnjak B, et al. The methyltransferase Setdb2 mediates virus-induced susceptibility to bacterial superinfection. Nat Immunol. (2015) 16:67–74. doi: 10.1038/ni.3046
32. Wang BX, Fish EN. Global virus outbreaks: Interferons as 1st responders. Semin Immunol. (2019) 43:101300. doi: 10.1016/j.smim.2019.101300
33. Fu XY, Kessler DS, Veals SA, Levy DE, Darnell JE Jr. ISGF3, the transcriptional activator induced by interferon alpha, consists of multiple interacting polypeptide chains. Proc Natl Acad Sci USA. (1990) 87:8555–9. doi: 10.1073/pnas.87.21.8555
34. Qureshi SA, Salditt-Georgieff M, Darnell JE Jr. Tyrosine-phosphorylated Stat1 and Stat2 plus a 48-kDa protein all contact DNA in forming interferon-stimulated-gene factor 3. Proc Natl Acad Sci USA. (1995) 92:3829–33. doi: 10.1073/pnas.92.9.3829
35. Uddin S, Majchrzak B, Woodson J, Arunkumar P, Alsayed Y, Pine R, et al. Activation of the p38 mitogen-activated protein kinase by type I interferons. J Biol Chem. (1999) 274:30127–31. doi: 10.1074/jbc.274.42.30127
36. Doughty L, Nguyen K, Durbin J, Biron C. A role for IFN-alpha beta in virus infection-induced sensitization to endotoxin. J Immunol. (2001) 166:2658–64. doi: 10.4049/jimmunol.166.4.2658
37. Tian X, Xu F, Lung WY, Meyerson C, Ghaffari AA, Cheng G, et al. Poly I:C enhances susceptibility to secondary pulmonary infections by gram-positive bacteria. PLoS ONE. (2012) 7:e41879. doi: 10.1371/journal.pone.0041879
38. Eisenreich W, Rudel T, Heesemann J, Goebel W. How viral and intracellular bacterial pathogens reprogram the metabolism of host cells to allow their intracellular replication. Front Cell Infect Microbiol. (2019) 9:42. doi: 10.3389/fcimb.2019.00042
39. Mehta D, Petes C, Gee K, Basta S. The role of virus infection in deregulating the cytokine response to secondary bacterial infection. J Interf Cytokine Res. (2015) 35:925–34. doi: 10.1089/jir.2015.0072
40. Berg J, Zscheppang K, Fatykhova D, Tonnies M, Bauer TT, Schneider P, et al. Tyk2 as a target for immune regulation in human viral/bacterial pneumonia. Eur Respir J. (2017) 50:1601953. doi: 10.1183/13993003.01953-2016
41. Gopal R, Lee B, McHugh KJ, Rich HE, Ramanan K, Mandalapu S, et al. STAT2 signaling regulates macrophage phenotype during influenza and bacterial super-infection. Front Immunol. (2018) 9:2151. doi: 10.3389/fimmu.2018.02151
42. Sanders RD, Godlee A, Fujimori T, Goulding J, Xin G, Salek-Ardakani S, et al. Benzodiazepine augmented gamma-amino-butyric acid signaling increases mortality from pneumonia in mice. Crit Care Med. (2013) 41:1627–36. doi: 10.1097/CCM.0b013e31827c0c8d
43. MacDonald BA, Chakravarthy KV, Davidson BA, Mullan BA, Alluri R, Hakansson AP, et al. Halothane modulates the type i interferon response to influenza and minimizes the risk of secondary bacterial pneumonia through maintenance of neutrophil recruitment in an animal model. Anesthesiology. (2015) 123:590–602. doi: 10.1097/ALN.0000000000000766
44. Delgado-Ortega M, Marc D, Dupont J, Trapp S, Berri M, Meurens F. SOCS proteins in infectious diseases of mammals. Vet Immunol Immunopathol. (2013) 151:1–19. doi: 10.1016/j.vetimm.2012.11.008
45. Fujimori T, Grabiec AM, Kaur M, Bell TJ, Fujino N, Cook PC, et al. The Axl receptor tyrosine kinase is a discriminator of macrophage function in the inflamed lung. Mucosal Immunol. (2015) 8:1021–30. doi: 10.1038/mi.2014.129
46. Grabiec AM, Denny N, Doherty JA, Happonen KE, Hankinson J, Connolly E, et al. Diminished airway macrophage expression of the Axl receptor tyrosine kinase is associated with defective efferocytosis in asthma. J Allergy Clin Immunol. (2017) 140:1144–6.e4. doi: 10.1016/j.jaci.2017.03.024
47. Grabiec AM, Hussell T. The role of airway macrophages in apoptotic cell clearance following acute and chronic lung inflammation. Semin Immunopathol. (2016) 38:409–23. doi: 10.1007/s00281-016-0555-3
48. Tighe RM, Garantziotis S. Hyaluronan interactions with innate immunity in lung biology. Matrix Biol. (2019) 79:84–99. doi: 10.1016/j.matbio.2018.01.027
49. Bell TJ, Brand OJ, Morgan DJ, Salek-Ardakani S, Jagger C, Fujimori T, et al. Defective lung function following influenza virus is due to prolonged, reversible hyaluronan synthesis. Matrix Biol. (2019) 80:14–28. doi: 10.1016/j.matbio.2018.06.006
50. Chang MY, Kang I, Gale M Jr, Manicone AM, Kinsella MG, Braun KR, et al. Versican is produced by Trif- and type I interferon-dependent signaling in macrophages and contributes to fine control of innate immunity in lungs. Am J Physiol. (2017) 313:L1069–86. doi: 10.1152/ajplung.00353.2017
51. Maus UA, Janzen S, Wall G, Srivastava M, Blackwell TS, Christman JW, et al. Resident alveolar macrophages are replaced by recruited monocytes in response to endotoxin-induced lung inflammation. Am J Respir Cell Mol Biol. (2006) 35:227–35. doi: 10.1165/rcmb.2005-0241OC
52. Zhang Q, Cao X. Epigenetic regulation of the innate immune response to infection. Nat Rev Immunol. (2019) 19:417–32. doi: 10.1038/s41577-019-0151-6
53. Sun P, Zhang SJ, Maksim S, Yao YF, Liu HM, Du J. Epigenetic modification in macrophages: a promising target for tumor and inflammation-associated disease therapy. Curr Top Med Chem. (2019) 19:1350–62. doi: 10.2174/1568026619666190619143706
54. Kaufmann E, Sanz J, Dunn JL, Khan N, Mendonca LE, Pacis A, et al. BCG Educates hematopoietic stem cells to generate protective innate immunity against tuberculosis. Cell. (2018) 172:176–90.e19. doi: 10.1016/j.cell.2017.12.031
55. Mitroulis I, Ruppova K, Wang B, Chen LS, Grzybek M, Grinenko T, et al. Modulation of Myelopoiesis progenitors is an integral component of trained immunity. Cell. (2018) 172:147–61.e12. doi: 10.1016/j.cell.2017.11.034
56. Penkov S, Mitroulis I, Hajishengallis G, Chavakis T. Immunometabolic crosstalk: an ancestral principle of trained immunity? Trends Immunol. (2019) 40:1–11. doi: 10.1016/j.it.2018.11.002
57. Russell DG, Huang L, VanderVen BC. Immunometabolism at the interface between macrophages and pathogens. Nat Rev Immunol. (2019) 19:291–304. doi: 10.1038/s41577-019-0124-9
58. Yoshida K, Maekawa T, Zhu Y, Renard-Guillet C, Chatton B, Inoue K, et al. The transcription factor ATF7 mediates lipopolysaccharide-induced epigenetic changes in macrophages involved in innate immunological memory. Nat Immunol. (2015) 16:1034–43. doi: 10.1038/ni.3257
59. Aegerter H, Kulikauskaite J, Crotta S, Patel H, Kelly G, Hessel EM, et al. Influenza-induced monocyte-derived alveolar macrophages confer prolonged antibacterial protection. Nat Immunol. (2020) 21:145–57. doi: 10.1038/s41590-019-0568-x
60. Park SH, Kang K, Giannopoulou E, Qiao Y, Kang K, Kim G, et al. Type I interferons and the cytokine TNF cooperatively reprogram the macrophage epigenome to promote inflammatory activation. Nat Immunol. (2017) 18:1104–16. doi: 10.1038/ni.3818
61. Kamada R, Yang W, Zhang Y, Patel MC, Yang Y, Ouda R, et al. Interferon stimulation creates chromatin marks and establishes transcriptional memory. Proc Natl Acad Sci USA. (2018) 115:E9162–71. doi: 10.1073/pnas.1720930115
62. MacMicking JD. Interferon-inducible effector mechanisms in cell-autonomous immunity. Nat Rev Immunol. (2012) 12:367–82. doi: 10.1038/nri3210
63. Saka HA, Valdivia R. Emerging roles for lipid droplets in immunity and host-pathogen interactions. Ann Rev Cell Dev Biol. (2012) 28:411–37. doi: 10.1146/annurev-cellbio-092910-153958
64. Hertzog P, Forster S, Samarajiwa S. Systems biology of interferon responses. J Interf Cytokine Res. (2011) 31:5–11. doi: 10.1089/jir.2010.0126
65. Mesev EV, LeDesma RA, Ploss A. Decoding type I and III interferon signalling during viral infection. Nat Microbiol. (2019) 4:914–24. doi: 10.1038/s41564-019-0421-x
66. Lazear HM, Schoggins JW, Diamond MS. Shared and distinct functions of type I and type III interferons. Immunity. (2019) 50:907–23. doi: 10.1016/j.immuni.2019.03.025
67. Zhang F, Sun X, Zhu Y, Qin W. Downregulation of miR-146a inhibits influenza A virus replication by enhancing the type I interferon response in vitro and in vivo. Biomed Pharmacother. (2019) 111:740–50. doi: 10.1016/j.biopha.2018.12.103
68. Gao S, Li J, Song L, Wu J, Huang W. Influenza A virus-induced downregulation of miR-26a contributes to reduced IFNalpha/beta production. Virol Sinica. (2017) 32:261–70. doi: 10.1007/s12250-017-4004-9
69. Zhang Y, Yang L, Wang H, Zhang G, Sun X. Respiratory syncytial virus non-structural protein 1 facilitates virus replication through miR-29a-mediated inhibition of interferon-alpha receptor. Biochem Biophys Res Commun. (2016) 478:1436–41. doi: 10.1016/j.bbrc.2016.08.142
70. Novakovic B, Habibi E, Wang SY, Arts RJW, Davar R, Megchelenbrink W, et al. β-Glucan reverses the epigenetic state of lps-induced immunological tolerance. Cell. (2016) 167:1354–68.e14. doi: 10.1016/j.cell.2016.09.034
71. Neagos J, Standiford TJ, Newstead MW, Zeng X, Huang SK, Ballinger MN. Epigenetic regulation of tolerance to Toll-like receptor ligands in alveolar epithelial cells. Am J Respir Cell Mol Biol. (2015) 53:872–81. doi: 10.1165/rcmb.2015-0057OC
72. Lee B, Robinson KM, McHugh KJ, Scheller EV, Mandalapu S, Chen C, et al. Influenza-induced type I interferon enhances susceptibility to gram-negative and gram-positive bacterial pneumonia in mice. Am J Physiol. (2015) 309:L158–67. doi: 10.1152/ajplung.00338.2014
73. Kudva A, Scheller EV, Robinson KM, Crowe CR, Choi SM, Slight SR, et al. Influenza A inhibits Th17-mediated host defense against bacterial pneumonia in mice. J Immunol. (2011) 186:1666–74. doi: 10.4049/jimmunol.1002194
74. Kim YG, Park JH, Reimer T, Baker DP, Kawai T, Kumar H, et al. Viral infection augments Nod1/2 signaling to potentiate lethality associated with secondary bacterial infections. Cell Host Microbe. (2011) 9:496–507. doi: 10.1016/j.chom.2011.05.006
75. Navarini AA, Lang KS, Verschoor A, Recher M, Zinkernagel AS, Nizet V, et al. Innate immune-induced depletion of bone marrow neutrophils aggravates systemic bacterial infections. Proc Natl Acad Sci USA. (2009) 106:7107–12. doi: 10.1073/pnas.0901162106
76. Reder AT, Feng X. How type I interferons work in multiple sclerosis and other diseases: some unexpected mechanisms. J Interf Cytok Res. (2014) 34:589–99. doi: 10.1089/jir.2013.0158
77. Hall JC, Rosen A. Type I interferons: crucial participants in disease amplification in autoimmunity. Nat Rev Rheumatol. (2010) 6:40–9. doi: 10.1038/nrrheum.2009.237
78. Furie R, Toder K, Zapantis E. Lessons learned from the clinical trials of novel biologics and small molecules in lupus nephritis. Semin Nephrol. (2015) 35:509–20. doi: 10.1016/j.semnephrol.2015.08.012
79. Guilhot F, Chastang C, Michallet M, Guerci A, Harousseau JL, Maloisel F, et al. Interferon alfa-2b combined with cytarabine versus interferon alone in chronic myelogenous leukemia. French chronic myeloid leukemia study group. N Engl J Med. (1997) 337:223–9. doi: 10.1056/NEJM199707243370402
80. Snell LM, McGaha TL, Brooks DG. Type I interferon in chronic virus infection and cancer. Trends Immunol. (2017) 38:542–57. doi: 10.1016/j.it.2017.05.005
81. Alberts DS, Hannigan EV, Liu PY, Jiang C, Wilczynski S, Copeland L, et al. Randomized trial of adjuvant intraperitoneal alpha-interferon in stage III ovarian cancer patients who have no evidence of disease after primary surgery and chemotherapy: an intergroup study. Gynecol Oncol. (2006) 100:133–8. doi: 10.1016/j.ygyno.2005.07.117
82. Benci JL, Xu B, Qiu Y, Wu TJ, Dada H, Twyman-Saint Victor C, et al. Tumor interferon signaling regulates a multigenic resistance program to immune checkpoint blockade. Cell. (2016) 167:1540–54.e12. doi: 10.1016/j.cell.2016.11.022
83. Khamashta M, Merrill JT, Werth VP, Furie R, Kalunian K, Illei GG, et al. Sifalimumab, an anti-interferon-alpha monoclonal antibody, in moderate to severe systemic lupus erythematosus: a randomised, double-blind, placebo-controlled study. Ann Rheumat Dis. (2016) 75:1909–16. doi: 10.1136/annrheumdis-2015-208562
Keywords: type I IFN, trained immunity, alveolar macrophage, lung viral infection, secondary bacterial pneumonia, epigenome, tolerance
Citation: Connolly E and Hussell T (2020) The Impact of Type 1 Interferons on Alveolar Macrophage Tolerance and Implications for Host Susceptibility to Secondary Bacterial Pneumonia. Front. Immunol. 11:495. doi: 10.3389/fimmu.2020.00495
Received: 10 January 2020; Accepted: 04 March 2020;
Published: 20 March 2020.
Edited by:
Babita Agrawal, University of Alberta, CanadaReviewed by:
Jianzhong Zhu, Yangzhou University, ChinaCopyright © 2020 Connolly and Hussell. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Emma Connolly, ZW1tYS5jb25ub2xseUBtYW5jaGVzdGVyLmFjLnVr; Tracy Hussell, dHJhY3kuaHVzc2VsbEBtYW5jaGVzdGVyLmFjLnVr
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.