 Anu Kauppinen
Anu Kauppinen Kai Kaarniranta2,3
Kai Kaarniranta2,3- 1Faculty of Health Sciences, School of Pharmacy, University of Eastern Finland, Kuopio, Finland
- 2Department of Ophthalmology, Institute of Clinical Medicine, University of Eastern Finland, Kuopio, Finland
- 3Department of Ophthalmology, Kuopio University Hospital, Kuopio, Finland
- 4Department of Neurology, Institute of Clinical Medicine, University of Eastern Finland, Kuopio, Finland
Myeloid cells, such as granulocytes/neutrophils and macrophages, have responsibilities that include pathogen destruction, waste material degradation, or antigen presentation upon inflammation. During persistent stress, myeloid cells can remain partially differentiated and adopt immunosuppressive functions. Myeloid-derived suppressor cells (MDSCs) are primarily beneficial upon restoring homeostasis after inflammation. Because of their ability to suppress adaptive immunity, MDSCs can also ameliorate autoimmune diseases and semi-allogenic responses, e.g., in pregnancy or transplantation. However, immunosuppression is not always desirable. In certain conditions, such as cancer or chronically inflamed tissue, MDSCs prevent restorative immune responses and thereby aggravate disease progression. Age-related macular degeneration (AMD) is the most common disease in Western countries that severely threatens the central vision of aged people. The pathogenesis of this multifactorial disease is not fully elucidated, but inflammation is known to participate in both dry and wet AMD. In this paper, we provide an overview about the potential role of MDSCs in the pathogenesis of AMD.
Introduction
Hematopoietic stem cells (HSCs) produce lymphoid and myeloid blood cells. Lymphoid cells include T and B lymphocytes and natural killer (NK) cells, whereas myeloid cells include monocytes, macrophages, granulocytes, erythrocytes, megakaryocytes, and platelets (1). Despite their roles in the innate immune system, myeloid cells can also function as suppressors, although that task has more traditionally been associated with regulatory T cells. Myeloid-derived suppressor cells (MDSCs) remain immature and are a phenotypically and functionally heterogeneous cell population with immunosuppression as their common denominator. MDSCs are widely known for their capacity to suppress host T cell responses against tumor tissue, but they can also be generated upon other stressful conditions, from infections to autoimmunity and obesity, and suppress other cell types, including dendritic cells, NK cells, or macrophages (2–5). MDSCs are not uncommon in ocular diseases either, and they have been studied especially in experimental autoimmune uveitis, a murine model of posterior uveitis of autoimmune origin where retina-specific T cells promote local inflammation, leading to the breakdown of the blood—retinal barrier (BRB), as well as retinal granulomas, folding, and detachment (6, 7). Monocytic MDSCs have also been shown to protect retinal ganglion cells from glutamate-induced damage (8). Despite the potential of MDSCs to enter the retina, their role in other retinal diseases, such as age-related macular degeneration (AMD), has remained elusive.
Pathophysiology of AMD
AMD is the leading cause of severe vision loss among the elderly in developed countries (9). Prolonged life expectancies further amplify its prevalence and cause a vast economic and national health burden. AMD disturbs central vision due to the loss of photoreceptors in the macula, a photoreceptor-dense retinal area responsible for the fine visual acuity (Figure 1) (10). There are two forms of the disease; advanced retinal atrophy is known as dry AMD, and it slowly disturbs the central vision (11). In wet (also known as exudative or neovascular) AMD, which comprises ca. 10–15% of the cases, fragile blood vessels sprout from the choroid into the retina (11). Those neovessels rupture easily and cause edema and acute vision loss (12). It is commonly believed that the disease begins as a slowly progressing dry form that later in some people converts into wet AMD but mechanisms have remained elusive. In a recent study, Krogh-Nielsen et al. suggested that difference between the two disease forms could be associated with age and AMD pathology-related changes in the structure and the functionality of the Bruch's membrane. They showed a positive correlation in dry AMD patients between age and the expression of tissue inhibitor of metalloproteinase (TIMP)-1, a regulator of matrix metalloproteinases (MMPs) with anti-angiogenic properties (13). Conversely, plasma levels of TIMP-3/MMP-2 ratios were significantly lower in patients with wet AMD (13).
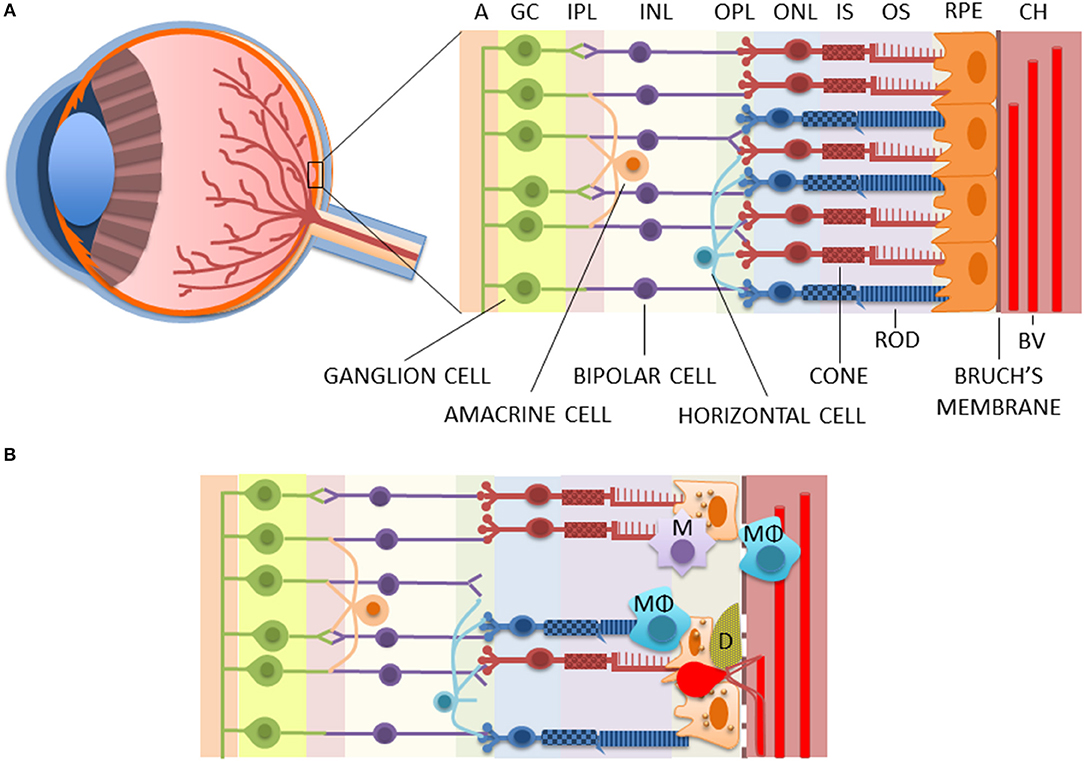
Figure 1. The structure of healthy (A) and diseased (B) retinae. AMD is associated with the death of RPE and photoreceptors, rupturing of the Bruch's membrane, the accumulation of macrophages and microglia in the choroidea and/or subretinal area, and the deposition of drusen between the RPE and the Bruch's membrane, as well as the accumulation of lipofuscin inside the RPE cells. Moreover, in wet AMD, fragile blood vessels sprout from the choroidea in a process called choroidal neovascularization (CNV) to the retina where they leak causing edema and rapid vision loss. A, axons of optic nerves; BV, blood vessel; CH, the choroidea; D, drusen; GC, ganglion cells; INL, inner nuclear layer; IPL, inner plexiform layer; IS, inner segments of photoreceptors; M, microglia; MΦ, macrophage; ONL, outer nuclear layer; OPL, outer plexiform layer; OS, outer segments of photoreceptors; RPE, retinal pigment epithelium cells.
As an age-related disease, changes resulting in clinical AMD accumulate over years, even decades. Normal aging contributes to retinal alterations, such as photoreceptor loss, Bruch's membrane thickening, choroid thinning, and formation of hard drusen in the retinal periphery; but in AMD, these become emphasized (11). In addition, AMD involves soft drusen formation at the macular area. Yellowish drusen are extracellular deposits of cellular debris, lipids, lipoproteins, amyloid deposits, and various proteins between the retinal pigment epithelium (RPE) and the Bruch's membrane (Figure 1) (11). They are typically among the first clinical signs upon diagnosis of AMD (14). Drusen-related proteins include various immune system-associated factors, such as complement components, and AMD-related soft drusen are also highly immunoreactive (11). Drusen material is known, for example, to activate inflammasome signaling in RPE cells and macrophage infiltration in the diseased retina (15–18).
The single-cell layer of retinal pigment epithelium (RPE) plays a significant role in the pathogenesis of AMD, and its degeneration is preceding the photoreceptor death in both dry and wet AMD (12, 19). In normal conditions, one major task of RPE cells is to phagocytize spent tips of photoreceptor outer segments (POS) and degrade them by autophagy (19). Efficient removal of waste material is critical, since ca. 10% of the photoreceptor layer volume becomes shed and degraded every day (20). However, aging deteriorates the functionality of intracellular degradation systems, which results in the accumulation of waste products in postmitotic and metabolically active RPE cells (19, 21). This results in increased oxidative stress and accumulation of non-degradable lipofuscin in lysosomes, which are responsible for waste removal by their enzymes (19). Lipofuscin inhibits autophagy, leading to the accumulation of aged mitochondria, which is one of the established characters of AMD (22, 23). In normal conditions, defective mitochondria become degraded by autophagy in a process called mitophagy; but upon autophagy blockade, they continue producing excessive amounts of reactive oxygen species (ROS). Dysfunctional autophagy promotes inflammation in RPE cells through inflammasome activation (24), and oxidative stress is the principal mechanism triggering the activation of NLRP3 receptor responsible for the inflammasome complex assembly (25). The vicious circle between dysfunctional autophagy, mitochondria, and inflammasome activation contributes to the development of pro-inflammatory retinal milieu that further promotes the deposition of lipofuscin and drusen material (19, 19). In addition to local pathology, systemic changes in the levels of certain factors, such as IL-6 or soluble TNF receptor II, have also been associated with AMD (26, 27).
Role of Myeloid Cells in AMD Pathology
Inflammation is a physiological first-line response to any factor endangering cellular homeostasis (28). Cytokines and chemokines produced in response to pattern-recognition receptor (PRR) activation-induced signaling cascades alert the immune system to restore homeostasis. Chemokines are messengers specialized in attracting leukocytes to the inflamed tissue. In normal conditions, the eye has immune privilege maintained by the BRB, which restricts the infiltration of blood-derived leukocytes, but does not entirely prevent it, especially during aging (29). Microglia are resident inflammatory cells at the retina, which normally locate in the inner layers of the neural retina near to retinal blood vessels (10). Microglia cells play an important role in maintaining homeostasis at the retina where they migrate back and forth to the subretinal space between RPE cells and photoreceptors. In AMD, prolonged existence of stress factors contributes to the tendency of microglia to accumulate in the subretinal space, which aggravates retinal degeneration (10, 30). Relocation of microglia to the subretinal space induces extravasation of myeloid cells through retinal vessels to replace the lack of microglia in the inner retina (31). C-C chemokine receptor type 2-positive (CCR2+) monocytes even differentiate into microglia-like cells after arrival (31).
AMD is associated with the rupture of BRB, allowing chemokines to recruit leukocytes also from the underlying choroid and systemic circulation to the retina (10). It has been shown both in dry AMD patients and the Cryba1 cKO mouse model with conditional knockout of the gene encoding βA3/A1-crystallin that early AMD is associated with infiltration of neutrophils to the choroid and the retina (32, 33). Infiltration of monocytes and their differentiation to macrophages upon retinal damage has been proven by various studies (34–37). Still, the fate of immune cells, especially microglia and monocyte/macrophages upon retinal damage is inadequately known (36). Despite observed leukocyte infiltration in the retina during the development of both AMD forms, it is possible that reduced oxygen consumption due to degeneration of photoreceptors alleviates the attraction of leukocytes in dry AMD. This view is supported by the fact that patients with advanced dry AMD lack significant macular edema or immune cell infiltration (38).
AMD-related leukocyte infiltration can be inflicted by impairment in receptors responding to chemokines that yield an increasing concentration gradient toward the inflamed tissue. C-X3-C Motif Receptor 1 (CX3CR1) and CCR2 are chemokine receptors implicated in drusen formation and the development of AMD (39). Interestingly, monocytes expressing both CX3CR1 and CCR2 receptors have been classified as inflammatory, whereas cells expressing only CX3CR1 have been termed anti-inflammatory (40). CX3CR1 and CCR2 ligands C-X3-C Motif Ligand 1 (CX3CL1 or fractalkine/human, neurotactin/mouse) and Monocyte Chemoattractant Protein 1 (MCP-1 or C-C Motif Chemokine Ligand 2, CCL2), respectively, recruit especially macrophages to inflamed tissue as well as microglia to and from the subretinal space (39, 41). CCL2 is also capable of attracting effector T cells, regulatory T (T reg) cells, and MDSCs (42, 43).
CX3CL1 is a transmembrane protein with integrin-like ability to bind monocytes and T cells, which can also be cleaved into a soluble form with chemotactic capacity (44). Several ocular tissues, including the RPE, constantly expresses CX3CL1 to control the redistribution and activity of CX3CR1-expressing microglia (40, 45). Dysfunctionality or loss of CX3CR1 results in the subretinal accumulation of microglia, which contributes to drusen-like lesions, retinal degeneration, and neovascularization (40). Also, prominent infiltration of inflammatory monocytes in the subretinal space has been associated with photoreceptor death through the P2X7R-dependent NLRP3 inflammasome activation and IL-1β production in Cx3cr1-deficient mice (37, 46, 47). Dysregulated microglia-mediated neurotoxicity upon CX3CR1 deficiency is also known in central nervous system (CNS)-related conditions, such as CNS response to systemic endotoxin-induced inflammation, Parkinson's disease, and amyotrophic lateral sclerosis (ALS) (48). On the other hand, subretinal accumulation of microglia and macrophages also increase during aging irrespective of CX3CR1 expression (49, 50). In a prospective case-control study, higher proportions of CX3CR1+ and CCR2+ non-classical monocytes were found from peripheral blood of patients suffering from wet AMD when compared to age-matched control subjects devoid of AMD (51). Together, the data suggest that mononuclear cells accumulated at the subretinal space contribute to the retinal degeneration and photoreceptor loss. The role of CX3CR1 in the cell infiltration remains elusive but its increased expression in the peripheral monocytes of wet AMD patients does not exclude disease-specific changes in chemokine receptors.
Adaptive Immunity in AMD Patients
In the case that acute inflammation cannot be resolved cannot be resolved, the adaptive immune system becomes activated (52). According to current knowledge, in AMD, this refers to T lymphocyte-dependent responses more than B lymphocytes. No differences in the levels of B cells between AMD patients and control subjects have been observed, but oxidative stress-induced neoepitopes and increased concentrations of retinal auto-antibodies in patients with either dry or wet AMD imply that B lymphocytes can be associated with the disease pathogenesis (53–55). In contrast to that, more findings point to the role of T lymphocytes in AMD. T cells can roughly be categorized as helper T (Th) cells, cytotoxic T (Tc) cells, γδ-T cells, and T reg cells (56).
Healthy RPE contributes to the formation of physical BRB and actively removes infiltrating T cells by killing them through Fas-mediated apoptosis or rendering them anergic (57, 58). There is evidence suggesting that in AMD, T lymphocytes can escape elimination or anergization or be functionally altered or impaired from being capable of responding to regulatory signals (52, 59). Also, disease-associated deviations in aging immune system have been reported. For example, increased levels of CD28−CD56+ T cells in AMD patients point toward an immunosenescent phenotype, as with similar association, found also in coronary artery disease, rheumatoid arthritis, and Behçet's uveitis (60, 61). Moreover, age-dependent reduction in the amounts of Th1 cells was observed in peripheral blood of healthy relatives but not in patients suffering either from dry or wet AMD (62). In a small study by Yu et al., higher levels of IFN-γ and IL-4 were measured from PHA-stimulated PBMC cultures of wet AMD patients in comparison to control subjects, suggesting reactivity of Th1 and Th2 cells in patients (63). The finding on Th1 cells is supported by Chen et al. who showed increased levels of IFN-γ and IL-17-expressing CD4+ T cells in the circulation of wet AMD patients when compared to control subjects (64). Th1 and Th17 cells isolated from patients also shifted monocytes toward a pro-inflammatory M1 macrophage phenotype that has been associated with retinal damage (64). In addition to data implying systemic activation of adaptive immunity, in an experimental model of AMD, cytotoxic CD8+ T cells directly facilitated RPE degeneration following the immunization of mice with carboxyethylpyrrole (CEP)-modified albumin in complete Freund's adjuvant (65). In another mouse model, laser-induced CNV resulted in the infiltration of IL-17-producing γδ-T cells into the eye, which subsequently promoted inflammation in RPE cells (56). Retinal infiltration of IL-17-producing γδ-T cells was also detected in mice deficient in anti-oxidant system-regulating nuclear erythroid 2-related factor 2 (Nrf2) exposed to a high-fat, cholesterol-rich diet (59). The findings of deleterious effects are contradictory to the role of γδ-T cells as intraepithelial lymphocyte (IEL)-like cells with protective functions upon inflammatory environment and RPE degeneration (66). The outcome is probably related to local conditions, since inflammasome-associated cytokine IL-1β and the alarmin protein high-mobility group box 1 (HMGB1) are capable of promoting IL-17 expression by γδ-T cells (67). Conversely, inhibition of IL-1β and HMGB1 or depletion of γδ-T cells prevented experimental CNV in laser-treated mice (67). Keeping in mind that inflammasome activity and autophagy are inversely dependent on each other, dysfunctional autophagy in aged RPE cells may promote infiltration of IL-17-producing γδ-T cells through inflammasome activation.
Myeloid-Derived Suppressor Cells
Acute inflammation induces myelopoiesis, strongly expanding neutrophils and monocytes, the latter of which differentiate into macrophages or dendritic cells in tissues depending on local conditions (68, 69). Those cells are active in restoring tissue homeostasis by phagocytosis, respiratory bursts, and promotion of further immune responses by secreting cytokines and activating adaptive immunity (69). Upon persistent stress, myeloid cells remain partially differentiated and adopt immunosuppressive functions (69). MDSCs were initially characterized in mice on their expression of Gr-1 in addition to the classical myeloid marker CD11b (70). Gr-1 is comprised of Ly6C and Ly6G that represent monocytic and granulocytic MDSCs, respectively (71). Due to the lack of homolog for Gr-1, human MDSCs are generally called mononuclear/monocytic (M-MDSC) and polymorphonuclear/granuclocytic (PMN-MDSC) cells (72). M-MDSCs are defined as CD11b+CD14+HLA-DR−/loCD15− and PMN-MDSCs as CD11b+CD14−CD15+ or CD11b+CD14−CD66b+ (71). Also, the myeloid marker CD33 can be used to define MDSCs; it is expressed by M-MDSCs, whereas PMN-MDSCs display DC33dim staining (71). Lin−HLA-DR−CD33+ are early stage MDSCs that can be found from human, but their equivalent in mice is not known yet (71). Physiologically, MDSCs are advantageous in semi-allogenic situations, such as pregnancy or transplantation, but they are better known for their disadvantageous immunosuppressive functions in chronic pathologies, for example, in chronic infections or cancer (72, 73) (Figure 2). MDSCs can regulate both innate and adaptive immune responses, e.g., by modulating macrophages, inhibiting NK or T cell responses or by inducing regulatory T cells (2, 5, 74–79). Their non-immunological functions include promotion of angiogenesis and metastasis (2, 80). Factors by which MDSCs execute their effects include arginase 1 (Arg-1), indoleamine dioxygenase (IDO), IL-10, inducible nitric oxide synthase (iNOS), nitric oxide (NO), heme oxygenase 1 (HO-1), carbon monoxide (CO), prostaglandin E2 (PGE2), ROS, and cysteine depletion (2, 73). The anergization of NK cells is dependent on the membrane-bound TGF-β1 of MDSCs, and intracellular HO-1 of MDSCs regulates T cell proliferation by CO production (73, 75). HO-1, as a stress-responsive enzyme with immunoregulatory and cytoprotective properties, is capable of protecting against oxidative stress, regulating cell proliferation, modulating inflammatory responses, and facilitating angiogenesis (81). HO-1 has been observed as a central mediator in MDSC-associated suppression upon transplantation (82). Along with the expression of regulatory cytokine IL-10, HMOX-1 encoding for HO-1 was reduced in monocytic MDSCs of secondary progressive multiple sclerosis (MS) patients when compared to relapsing–remitting MS patients or healthy subjects (83). HO-1 also participates in the tumor microenvironment to protect tumor cells from apoptosis, to improve their growth and potentially also metastasis (81, 84). Collectively, MDSCs are beneficial in quenching acute inflammation, autoimmune diseases, and responses against (semi-)allografts but become detrimental upon chronic inflammatory and neoplastic conditions.

Figure 2. Potential functions of MDSCs upon acute versus chronic conditions. Green and orange boxes refer to positive and negative effects, respectively. EAU, experimental autoimmune uveitis; M1, type 1 macrophage; MDSCs, myeloid-derived suppressor cells; Th1, type 1 helper T cell.
MDSCs in the Maintenance of Retinal Homeostasis
Besides the contribution to physical BRB, RPE cells have also other means to maintain homeostasis at the retina (38). Secretion of TGF-β or thrombospondin-1 (TSP-1) and the expression of programmed death-ligand 1 (PD-L1/CD247/B7-H1) on the plasma membrane alleviate T and B cell responses, and the production of cathepsin L inhibitor CTLA-2α promotes the induction of CD4+CD25+Foxp3+ T reg cells (85–90). RPE cells also produce soluble neuropeptides with immunomodulatory capacities. Kawanaka et al. showed that neuropeptides alpha-melanocyte stimulating factor (α-MSF) and neuropeptide Y (NPY) induced co-expression of Arg1 and NOS2 in resting macrophages, converting them into MDSC-resembling cells that showed significantly reduced secretion of pro-inflammatory cytokines upon exposure to lipopolysaccharide (LPS) (91). In contrast, cells expressing only Arg1 or NOS2 but not both were found from laser-wounded retina (91). Cells lacking co-expression were less efficient in inducing apoptosis in T cells and contributed to the development of pro-inflammatory milieu. As an additional mechanism, RPE cells have been shown capable of inducing MDSC differentiation from co-cultured bone marrow (BM) progenitor cells upon exposure to granulocyte–macrophage colony-stimulating factor (GM-CSF) and IL-4 (7). In the absence of RPE cells, BM cells were differentiated into dendritic cells in the presence of given cytokines, as expected (7). CD11b+Gr-1+ MDSCs shared similar surface markers with tumor MDSCs, they efficiently inhibited T cell proliferation and inflammatory cytokine production, and their systemic delivery to mice immunized with the interphotoreceptor retinoid-binding protein (IRBP)1−−20 peptide in complete Freund's adjuvant (CFA) prevented EAU-related retinal injury (7). The pro-inflammatory cytokine IL-6 was needed for the process since its blockade reduced the RPE cell-induced MDSC differentiation. Instead, TGF-β did not participate in the differentiation process (7). Both IL-6 and TGF-β can be very harmful due to their capacity to induce inflammation and fibrosis, respectively, but probably concentrations and locations influence their effects (86).
Potential of MDSCs in Provoking Angiogenesis in Diseased Retina
Chronic inflammation is known as a promoter for MDSC differentiation and functionality. MDSCs have also been experimentally induced using different cytokine cocktails (92). Bunt et al. showed in murine cells that IL-1β-induced inflammation activated MDSCs through the TLR4/CD14 pathway making MDSCs to regulate macrophages by increasing their IL-10 and reducing IL-12 productions (93). That may refer to a homeostatic process where M2-type macrophages producing immunosuppressive IL-10 prevent the expansion of M1 population, which by releasing pro-inflammatory cytokines, such as IL-12 and TNF-α, is associated with tissue destruction and T helper 1 (Th1) cell activation (65). On the other hand, anti-inflammatory but pro-angiogenic M2 macrophages have been associated with choroidal neovascularization and the development of wet AMD (94, 95).
Drusen material accumulating between the RPE and the Bruch's membrane diminish the oxygen supply from the choroid to the retina (96). RPE cells suffering from hypoxia can result in the release of vascular endothelial growth factor (VEGF) already prior to the production of inflammatory markers (97). Upon laser-induced vessel formation, peak in the VEGF production coincided with the arrival of macrophages, supporting the link between RPE-associated promotion of neovascularization (98). Macrophages sense the need for oxygen by retinal cells and further potentiate the VEGF-mediated neovessel formation (10). In cancer, inflammation-induced MDSCs inhibit immunosurveillance to allow persistence and proliferation of premalignant and malignant cells (99). The capacity of MDSCs to induce angiogenesis has not been shown only in cancer studies but also in the neovascularization of an ischemic hind-limb of mice (80, 100).
It is intriguing to hypothesize that cyclooxygenase-2 (COX-2) in choroidal neovascular membranes could maintain elevated MDSC levels such that neovessel growth is promoted. COX-2 is the predominant cyclooxygenase in human RPE cells, and inflammatory cytokines further promote its expression (101). COX-2 activity is capable of recruiting MDSCs through CCL2 or PGE2. COX-2 converts arachidonic acid to PGE2 that plays a major role in the physiologic induction of MDSCs (102). Direct effects of PGE2 through the E-prostanoid (EP) receptor provide an alternative pathway for Ccl-2- or Ccr-2-deficient mice to develop atrophic (dry) and neovascular (wet) pathologies in the absence of the chemokine or its receptor (103). In an in vitro study with mouse primary RPE cells, the major lipofuscin component bis-retinoid N-retinylidene-N-retinylethanolamine (A2E) reduced PGE2 levels and promoted RPE cells to induce Th1 cell differentiation in IL-1β-dependent way, which might thereby contribute to further retinal degeneration (104, 105).
COX-2 inhibition by acetylsalisylic acid (aspirin, ASA) prevented the CCL2-mediated accumulation of CD11b+Ly6GhiLy6Clo granulocytic MDSCs to the tumor microenviroment in mice with glioma (43). COX-2/CCL2 blockade also increased the expression of C-X-C Motif Chemokine 10 (CXCL10/Interferon γ-induced Protein 10/IP-10) that inhibits VEGF-mediated angiogenesis (43, 52). COX-2 is expressed by human choroidal neovascular membranes (106), and promotion of CXCL10 could result in its inhibition. CXCL10 is a ligand of C-X-C Motif Chemokine Receptor 3 (CXCR3 also known as GPR9 or CD183) that, along with C-C Chemokine Receptor Type 3 (CCR3), is associated with the development of wet AMD (52). Percentage of both CD4+ Th and CD8+ Tc cells expressing CXCR3 has been observed to be lower in the peripheral blood of patients with wet AMD in comparison to control subjects (62, 107), which may diminish the benefit of increased CXCL10 production following the COX-2 inhibition. Acetylsalisylic acid is a non-steroidal anti-inflammatory drug (NSAID) and COX-2 inhibitor that is commonly used at low doses for long periods due to its anti-thrombotic effects. A retrospective study on AREDS and AREDS2 data supports the inability of COX-2 inhibition to protect from neovascularization since the use of acetylsalisylic acid was not significantly associated with progression of either dry or wet AMD (108). Instead, a prospective double-blind randomized human study on the therapy of wet AMD with photodynamic therapy (PDT) supplemented with oral intake of the COX-2 inhibitor nabumetone resulted in the progression of macular atrophy (109). Collectively, the data on COX-2 inhibition suggest no beneficial effects on wet AMD but potential aggravation in the progression of dry AMD, thereby conflicting with the idea of COX-2-induced and MDSC-mediated neovascularization.
Complement and MDSCs
Due to facts concerning genetic predisposition and composition of drusen, it is evident that the complement pathway contributes to the pathogenesis of AMD. Excessive complement activation has also been observed in various studies to be harmful in pathologic retinal degenerative and angiogenic conditions (110).
Complement components produced by RPE cells contribute to the formation of sub-RPE deposits through complement-driven proteasome inhibition and release pro-inflammatory cytokines, such as IL-6 and IL-1β, that can amplify the response by further promoting expression of the C3a receptor (111, 112). Unstimulated RPE cells express significantly higher levels of complement regulator genes in comparison to complement component genes but under inflammatory conditions, activated macrophages induce complement factor B (CFB) and C3 expression in RPE cells (113). Laser photocoagulation has also shown to induce the deposition of C3 and membrane attack complex (MAC) in the neovascular complex (114). Furthermore, MAC formation is capable of releasing angiogenesis-related growth factors, such as β-fibroblast growth factor (β-FGF), VEGF, and platelet-derived growth factor (PDGF) (114). The role of MDSCs has not been studied in relation to ocular complement activation but there is evidence on the MDSC contribution from other disease models. Complement inhibition reduces tumor growth with accompanying decrease in the both C3 and MDSC levels (115). Hsieh et al. also showed that C3 plays a major role in the conversion of bone marrow progenitor cells into MDSC by hepatic stellate cells (116, 117). Those MDSCs efficiently inhibited T cell responses in vitro and in vivo, and alleviated experimental autoimmune myasthenia gravis also by reducing anti-actylcholine receptor IgG levels and decreasing complement activation at the endplates of neuromuscular junctions (116–118).
Active pro-inflammatory complement components C3a and C5a (anaphylatoxins) are included in drusen material and efficiently promote choroidal neovascularization in mice by stimulating VEGF production in RPE and choroid cells (98). Conversely, normalized anaphylatoxin levels promoted recovery from CNV lesions and prevented fibrotic scar formation (119). A study on the ARPE-19 cell line originating from spontaneously transformed human RPE cells emphasized the role of C5a over C3a in the induction of VEGF production, but Nozaki et al. also found C3a to stimulate VEGF production in primary human RPE cells in addition to D407 RPE cell line (120, 121). Both C3a and C5a were found from the drusen, the proximity of RPE cells, and the Bruch's membrane of an AMD patient, whereas neither of them were present in the eye of a control subject without AMD (98). A study on C3aR−/− and C5aR−/− mice showed that complement components also play a role in the recruitment of neutrophils and macrophages that were observed to peak one and three days after the insult, respectively (98). Neutrophils further pave the way to other leukocytes by expressing MMP-9 that disrupts the integrity of BRB (122). In dry AMD, photoreceptor outer segments exposed to human serum following the BRB breakdown resulted in complement activation, C5a-mediated attraction of peripheral blood monocytes, and diminished survival of the RPE and neural retina (123). Complement activation in subretinal macrophages can potentially play a role in the pathogenesis of AMD (124). Also systemic complement activation in AMD patients has been shown (125, 126).
Substitution of histidine to tyrosine (Y402H) is one of the best-known polymorphic complement factor H (CFH) variants associated with AMD. By diminishing the functionality of the regulatory CFH, it results in overactive alternative complement pathway (10). Incomplete CFH activity has been shown to enhance C3 deposition and C5a release in non-small lung cancer cell lines (127–130). In addition to direct receptor binding, C5a can stimulate MDSCs also indirectly through IL-6, IL-1β, or VEGF (131). The role of C5a in cancer promotion is well known, and it is also considered as a key player in poor cancer prognosis on subjects suffering from an autoimmune disease with complement activation by immune complexes (the Arthus reaction) (132). According to a recent discovery, CFH(Y402H) also efficiently binds CD11b, preventing CD47-mediated elimination of mononuclear cells, such as macrophages and microglia, attracted to the subretinal space between the RPE and photoreceptor outer segments to restore homeostasis (133, 134). This may extend the presence of mononuclear cells at the retina beyond the need for homeostasis restoration, which predisposes to adverse effects, such as photoreceptor degeneration and choroidal neovascularization (41, 135, 136). CD11b is also expressed by both monocytic and granulocytic MDSCs (71) predisposing the retina to their prolonged impact in susceptible persons. Overall, complement and CFH(Y402H) are efficient in activating MDSC and may thereby prevent the removal of drusen material by immune cells.
MDSCs Enhance Fibrosis
Choroidal neovascularization does not only cause rapid vision loss due to bleeding and swelling at the retina, but it also results in subretinal fibrosis that can develop despite the anti-VEGF therapy that prevents the neovessel formation (137). Conversely, epithelial-mesenchymal transition (EMT) can also cause resistance to anti-VEGF drugs, as suggested by studies on pancreatic cancer cells (138). Subretinal fibrosis is associated with the end stage pathogenesis of wet AMD (137). TGF-β signaling in myeloid cells promotes CCL2-dependent MDSC recruitment to the tumor microenvironment, and granulocytic MDSC-derived TGF-β promotes EMT upon metastasis, which is the advanced form of malignancy (139, 140). TGF-β is a multifunctional growth factor physiologically needed for the development and tissue repair, but excessive concentrations are associated with inflammation and tissue fibrosis, with detrimental consequences in the eye (141, 142).
Concluding Remarks
MDSCs have been found beneficial in various CNS disorders and autoimmune diseases (Figure 2). Both monocytic and granulocytic MDSCs have been shown to alleviate EAE (143–145). Monocytic MDSCs participated in resolving damage following experimental spinal cord injury in mice, alleviated inflammation after stroke in mice, and were found in increased numbers in the peripheral blood of ischemic stroke patients (146, 147). MDSCs induced by hepatic stellate cells efficiently inhibited T cell responses in vitro and in vivo and attenuated experimental autoimmune myasthenia gravis also by reducing anti-actylcholine receptor IgG levels and decreasing complement activation at the endplates of neuromuscular junctions (116–118). MDSCs were observed beneficial also especially at the beginning of autoimmune uveoretinitis (6, 148–150). Adoptive transfer of RPE-induced MDSCs reduced EAU severity when detected 21 days after immunization when the acute phase peaks (7). However, upon combined deletion of CCL2 and CX3CR1, deficiency in macrophages and MDSCs during chronic EAU (90 days post-immunization) contributed to reduced retinal damage and angiogenesis (150). The data supports the overall idea of inflammation where the timing matters. In the acute phase, MDSCs probably participate in the restoration of homeostasis, but upon chronic inflammation, their capacity to promote atrophy and angiogenesis may overcome the benefits.
Author Contributions
AK contributed to the planning of the content, writing of the manuscript, and preparing the figures. AS and KK contributed to the planning of the content and the critical evaluation of the text.
Funding
This study was financially supported by the grants from the Academy of Finland (AK297267, AK307341, KK296840, and AK328443), the Kuopio University Hospital VTR grant (KK5503743), the Emil Aaltonen Foundation, the Sigrid Juselius Foundation, the Päivikki and Sakari Sohlberg Foundation, the Finnish Cultural Foundation, and the Finnish Eye Foundation.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Kondo M. Lymphoid and myeloid lineage commitment in multipotent hematopoietic progenitors. Immunol Rev. (2010) 238:37–46. doi: 10.1111/j.1600-065X.2010.00963.x
2. Gabrilovich DI, Nagaraj S. Myeloid-derived suppressor cells as regulators of the immune system. Nat Rev Immunol. (2009) 9:162–74. doi: 10.1038/nri2506
3. Kwak Y, Kim HE, Park SG. Insights into myeloid-derived suppressor cells in inflammatory diseases. Arch Immunol Ther Exp. (2015) 63:269–85. doi: 10.1007/s00005-015-0342-1
4. Ostrand-Rosenberg S, Beury DW, Parker KH, Horn LA. Survival of the fittest: how myeloid-derived suppressor cells survive in the inhospitable tumor microenvironment. Cancer Immunol Immunother. (2019) 69:215–21. doi: 10.1007/s00262-019-02388-8
5. Veglia F, Perego M, Gabrilovich D. Myeloid-derived suppressor cells coming of age. Nat Immunol. (2018) 19:108–19. doi: 10.1038/s41590-017-0022-x
6. Kerr EC, Raveney BJ, Copland DA, Dick AD, Nicholson LB. Analysis of retinal cellular infiltrate in experimental autoimmune uveoretinitis reveals multiple regulatory cell populations. J Autoimmun. (2008) 31:354–61. doi: 10.1016/j.jaut.2008.08.006
7. Tu Z, Li Y, Smith D, Doller C, Sugita S, Chan CC, et al. Myeloid suppressor cells induced by retinal pigment epithelial cells inhibit autoreactive T-cell responses that lead to experimental autoimmune uveitis. Invest Ophthalmol Vis Sci. (2012) 53:959–66. doi: 10.1167/iovs.11-8377
8. London A, Itskovich E, Benhar I, Kalchenko V, Mack M, Jung S, Schwartz M. Neuroprotection and progenitor cell renewal in the injured adult murine retina requires healing monocyte-derived macrophages. J Exp Med. (2011) 208:23–39. doi: 10.1084/jem.20101202
9. Wong WL, Su X, Li X, Cheung CM, Klein R, Cheng CY, Wong TY. Global prevalence of age-related macular degeneration disease burden projection for 2020 2040: a systematic review meta-analysis. Lancet Glob Health. (2014) 2:e106–16. doi: 10.1016/S2214-109X(13)70145-1
10. Ambati J, Atkinson JP, Gelfand BD. Immunology of age-related macular degeneration. Nat Rev Immunol. (2013) 13:438–51. doi: 10.1038/nri3459
11. Ardeljan D, Chan CC. Aging is not a disease: Distinguishing age-related macular degeneration from aging. Prog Retin Eye Res. (2013) 37:68–9. doi: 10.1016/j.preteyeres.2013.07.003
12. Ambati J, Fowler BJ. Mechanisms of age-related macular degeneration. Neuron. (2012) 75:26–39. doi: 10.1016/j.neuron.2012.06.018
13. Krogh Nielsen M, Subhi Y, Rue Molbech C, Nilsson LL, Nissen MH, Sorensen TL. Imbalances in tissue inhibitors of metalloproteinases differentiate choroidal neovascularization from geographic atrophy. Acta Ophthalmol. (2019) 97:84–90. doi: 10.1111/aos.13894
14. Lim LS, Mitchell P, Seddon JM, Holz FG, Wong TY. Age-related macular degeneration. Lancet. (2012) 379:1728–38. doi: 10.1016/S0140-6736(12)60282-7
15. Doyle SL, Campbell M, Ozaki E, Salomon RG, Mori A, Kenna PF, et al. NLRP3 has a protective role in age-related macular degeneration through the induction of IL-18 by drusen components. Nat Med. (2012) 18:791–8. doi: 10.1038/nm.2717
16. Gao J, Cui JZ, To E, Cao S, Matsubara JA. Evidence for the activation of pyroptotic and apoptotic pathways in RPE cells associated with NLRP3 inflammasome in the rodent eye. J Neuroinflammation. (2018) 15:15. doi: 10.1186/s12974-018-1062-3
17. Liu RT, Gao J, Cao S, Sandhu N, Cui JZ, Chou CL, et al. Inflammatory mediators induced by amyloid-beta in the retina and RPE in vivo: implications for inflammasome activation in age-related macular degeneration. Invest Ophthalmol Vis Sci. (2013) 54:2225–37. doi: 10.1167/iovs.12-10849
18. Zhao T, Gao J, Van J, To E, Wang A, Cao S, et al. Age-related increases in amyloid beta and membrane attack complex: evidence of inflammasome activation in the rodent eye. J Neuroinflammation. (2015) 12:121. doi: 10.1186/s12974-015-0337-1
19. Kaarniranta K, Sinha D, Blasiak J, Kauppinen A, Vereb Z, Salminen A, et al. Autophagy and heterophagy dysregulation leads to retinal pigment epithelium dysfunction and development of age-related macular degeneration. Autophagy. (2013) 9:973–84. doi: 10.4161/auto.24546
20. Kevany BM, Palczewski K. Phagocytosis of retinal rod and cone photoreceptors. Physiology. (2010) 25:8–15. doi: 10.1152/physiol.00038.2009
21. Kaarniranta K, Salminen A, Haapasalo A, Soininen H, Hiltunen M. Age-related macular degeneration (AMD): Alzheimer's disease in the eye? J Alzheimers Dis. (2011) 24:615–31. doi: 10.3233/JAD-2011-101908
22. Ao J, Wood JP, Chidlow G, Gillies MC, Casson RJ. Retinal pigment epithelium in the pathogenesis of age-related macular degeneration and photobiomodulation as a potential therapy? Clin Exp Ophthalmol. (2018) 46:670–86. doi: 10.1111/ceo.13121
23. Green DR, Galluzzi L, Kroemer G. Mitochondria and the autophagy-inflammation-cell death axis in organismal aging. Science. (2011) 333:1109–12. doi: 10.1126/science.1201940
24. Piippo N, Korkmaz A, Hytti M, Kinnunen K, Salminen A, Atalay M, et al. Decline in cellular clearance systems induces inflammasome signaling in human ARPE-19 cells. Biochim Biophys Acta. (2014) 1843:3038–46. doi: 10.1016/j.bbamcr.2014.09.015
25. Piippo N, Korhonen E, Hytti M, Kinnunen K, Kaarniranta K, Kauppinen A. Oxidative stress is the principal contributor to inflammasome activation in retinal pigment epithelium cells with defunct proteasomes and autophagy. Cell Physiol Biochem. (2018) 49:359–67. doi: 10.1159/000492886
26. Faber C, Jehs T, Juel HB, Singh A, Falk MK, Sorensen TL, Nissen MH. Early exudative age-related macular degeneration is associated with increased plasma levels of soluble TNF receptor II. Acta Ophthalmol. (2015) 93:242–7. doi: 10.1111/aos.12581
27. Krogh Nielsen M, Subhi Y, Molbech CR, Falk MK, Nissen MH, Sorensen TL. Systemic Levels of Interleukin-6 Correlate With Progression Rate of Geographic Atrophy Secondary to Age-Related Macular Degeneration. Invest Ophthalmol Vis Sci. (2019) 60:202–8. doi: 10.1167/iovs.18-25878
28. Kauppinen A, Paterno JJ, Blasiak J, Salminen A, Kaarniranta K. Inflammation and its role in age-related macular degeneration. Cell Mol Life Sci. (2016) 73:1765–86. doi: 10.1007/s00018-016-2147-8
29. Forrester JV, Xu H. Good news-bad news: the Yin and Yang of immune privilege in the eye. Front Immunol. (2012) 3:338. doi: 10.3389/fimmu.2012.00338
30. Gupta N, Brown KE, Milam AH. Activated microglia in human retinitis pigmentosa, late-onset retinal degeneration, and age-related macular degeneration. Exp Eye Res. (2003) 76:463–71. doi: 10.1016/S0014-4835(02)00332-9
31. Ma W, Zhang Y, Gao C, Fariss RN, Tam J, Wong WT. Monocyte infiltration and proliferation reestablish myeloid cell homeostasis in the mouse retina following retinal pigment epithelial cell injury. Sci Rep. (2017) 7:1538–43. doi: 10.1038/s41598-017-08702-7
32. Ghosh S, Padmanabhan A, Vaidya T, Watson AM, Bhutto IA, Hose S, et al. Neutrophils homing into the retina trigger pathology in early age-related macular degeneration. Commun Biol. (2019) 2:348. doi: 10.1038/s42003-019-0588-y
33. Ghosh S, Shang P, Yazdankhah M, Bhutto I, Hose S, Montezuma SR, et al. Activating the AKT2-nuclear factor-kappaB-lipocalin-2 axis elicits an inflammatory response in age-related macular degeneration. J Pathol. (2017) 241:583–8. doi: 10.1002/path.4870
34. Caicedo A, Espinosa-Heidmann DG, Pina Y, Hernandez EP, Cousins SW. Blood-derived macrophages infiltrate the retina and activate Muller glial cells under experimental choroidal neovascularization. Exp Eye Res. (2005) 81:38–47. doi: 10.1016/j.exer.2005.01.013
35. Chen M, Lechner J, Zhao J, Toth L, Hogg R, Silvestri G, et al. STAT3 activation in circulating monocytes contributes to neovascular age-related macular degeneration. Curr Mol Med. (2016) 16:412–23. doi: 10.2174/1566524016666160324130031
36. Saban DR. New concepts in macrophage ontogeny in the adult neural retina. Cell Immunol. (2018) 330:79–85. doi: 10.1016/j.cellimm.2018.04.008
37. Sennlaub F, Auvynet C, Calippe B, Lavalette S, Poupel L, Hu SJ, et al. CCR2 (+) monocytes infiltrate atrophic lesions in age-related macular disease and mediate photoreceptor degeneration in experimental subretinal inflammation in Cx3cr1 deficient mice. EMBO Mol Med. (2013) 5:1775–93. doi: 10.1002/emmm.201302692
38. Chen M, Luo C, Zhao J, Devarajan G, Xu H. Immune regulation in the aging retina. Prog Retin Eye Res. (2019) 69:159–72. doi: 10.1016/j.preteyeres.2018.10.003
39. Falk MK, Singh A, Faber C, Nissen MH, Hviid T, Sorensen TL. CX3CL1/CX3CR1 CCL2/CCR2 chemokine/chemokine receptor complex in patients with AMD. PLoS ONE. (2014) 9:e112473. doi: 10.1371/journal.pone.0112473
40. Combadiere C, Feumi C, Raoul W, Keller N, Rodero M, Pezard A, et al. CX3CR1-dependent subretinal microglia cell accumulation is associated with cardinal features of age-related macular degeneration. J Clin Invest. (2007) 117:2920–8. doi: 10.1172/JCI31692
41. Raoul W, Auvynet C, Camelo S, Guillonneau X, Feumi C, Combadiere C, Sennlaub F. CCL2/CCR2 and CX3CL1/CX3CR1 chemokine axes and their possible involvement in age-related macular degeneration. J Neuroinflammation. (2010) 7:87. doi: 10.1186/1742-2094-7-87
42. Deshmane SL, Kremlev S, Amini S, Sawaya BE. Monocyte chemoattractant protein-1 (MCP-1): an overview. J Interferon Cytokine Res. (2009) 29:313–26. doi: 10.1089/jir.2008.0027
43. Fujita M, Kohanbash G, Fellows-Mayle W, Hamilton RL, Komohara Y, Decker SA, et al. COX-2 blockade suppresses gliomagenesis by inhibiting myeloid-derived suppressor cells. Cancer Res. (2011) 71:2664–74. doi: 10.1158/0008-5472.CAN-10-3055
44. Bazan JF, Bacon KB, Hardiman G, Wang W, Soo K, Rossi D, et al. A new class of membrane-bound chemokine with a CX3C motif. Nature. (1997) 385:640–4. doi: 10.1038/385640a0
45. Silverman MD, Zamora DO, Pan Y, Texeira PV, Baek SH, Planck SR, Rosenbaum JT. Constitutive and inflammatory mediator-regulated fractalkine expression in human ocular tissues and cultured cells. Invest Ophthalmol Vis Sci. (2003) 44:1608–15. doi: 10.1167/iovs.02-0233
46. Eandi CM, Charles Messance H, Augustin S, Dominguez E, Lavalette S, Forster V, et al. Subretinal mononuclear phagocytes induce cone segment loss via IL-1beta. Elife. (2016) 5:16490. doi: 10.7554/eLife.16490
47. Hu SJ, Calippe B, Lavalette S, Roubeix C, Montassar F, Housset M, et al. Upregulation of P2RX7 in Cx3cr1-deficient mononuclear phagocytes leads to increased interleukin-1beta secretion and photoreceptor neurodegeneration. J Neurosci. (2015) 35:6987–96. doi: 10.1523/JNEUROSCI.3955-14.2015
48. Cardona AE, Pioro EP, Sasse ME, Kostenko V, Cardona SM, Dijkstra IM, et al. Control of microglial neurotoxicity by the fractalkine receptor. Nat Neurosci. (2006) 9:917–24. doi: 10.1038/nn1715
49. Chinnery HR, McLenachan S, Humphries T, Kezic JM, Chen X, Ruitenberg MJ, McMenamin PG. Accumulation of murine subretinal macrophages: effects of age, pigmentation and CX3CR1. Neurobiol Aging. (2012) 33:1769–76. doi: 10.1016/j.neurobiolaging.2011.03.010
50. Xu H, Chen M, Manivannan A, Lois N, Forrester JV. Age-dependent accumulation of lipofuscin in perivascular and subretinal microglia in experimental mice. Aging Cell. (2008) 7:58–68. doi: 10.1111/j.1474-9726.2007.00351.x
51. Subhi Y, Krogh Nielsen M, Molbech CR, Sorensen TL. Altered proportion of CCR2 (+) and CX3CR1 (+) circulating monocytes in neovascular age-related macular degeneration and polypoidal choroidal vasculopathy. Clin Exp Ophthalmol. (2018) 46:661–9. doi: 10.1111/ceo.13152
52. Niazi S, Krogh Nielsen M, Sorensen TL, Subhi Y. Neutrophil-to-lymphocyte ratio in age-related macular degeneration: a systematic review and meta-analysis. Acta Ophthalmol. (2019) 97:558–66. doi: 10.1111/aos.14072
53. Adamus G, Chew EY, Ferris FL, Klein ML. Prevalence of anti-retinal autoantibodies in different stages of Age-related macular degeneration. BMC Ophthalmol. (2014) 14:154. doi: 10.1186/1471-2415-14-154
54. Hector SM, Sorensen TL. Circulating monocytes and B-lymphocytes in neovascular age-related macular degeneration. Clin Ophthalmol. (2017) 11:179–84. doi: 10.2147/OPTH.S121332
55. Joseph K, Kulik L, Coughlin B, Kunchithapautham K, Bandyopadhyay M, Thiel S, et al. Oxidative stress sensitizes retinal pigmented epithelial (RPE) cells to complement-mediated injury in a natural antibody-, lectin pathway-, and phospholipid epitope-dependent manner. J Biol Chem. (2013) 288:12753–65. doi: 10.1074/jbc.M112.421891
56. Coughlin B, Schnabolk G, Joseph K, Raikwar H, Kunchithapautham K, Johnson K, et al. Connecting the innate and adaptive immune responses in mouse choroidal neovascularization via the anaphylatoxin C5a and gammadeltaT-cells. Sci Rep. (2016) 6:23794. doi: 10.1038/srep23794
57. Gregerson DS, Heuss ND, Lew KL, McPherson SW, Ferrington DA. Interaction of retinal pigmented epithelial cells and CD4 T cells leads to T-cell anergy. Invest Ophthalmol Vis Sci. (2007) 48:4654–63. doi: 10.1167/iovs.07-0286
58. Jorgensen A, Wiencke AK, la Cour M, Kaestel CG, Madsen HO, Hamann S, et al. Human retinal pigment epithelial cell-induced apoptosis in activated T cells. Invest Ophthalmol Vis Sci. (1998) 39:1590–9.
59. Zhao Z, Xu P, Jie Z, Zuo Y, Yu B, Soong L, et al. gammadelta T cells as a major source of IL-17 production during age-dependent RPE degeneration. Invest Ophthalmol Vis Sci. (2014) 55:6580–9. doi: 10.1167/iovs.14-15166
60. Faber C, Singh A, Kruger Falk M, Juel HB, Sorensen TL, Nissen MH. Age-related macular degeneration is associated with increased proportion of CD56 (+) T cells in peripheral blood. Ophthalmology. (2013) 120:2310–6. doi: 10.1016/j.ophtha.2013.04.014
61. Subhi Y, Nielsen MK, Molbech CR, Oishi A, Singh A, Nissen MH, Sorensen TL. T-cell differentiation and CD56+ levels in polypoidal choroidal vasculopathy and neovascular age-related macular degeneration. Aging. (2017) 9:2436–52. doi: 10.18632/aging.101329
62. Singh A, Subhi Y, Krogh Nielsen M, Falk MK, Matzen SMH, Sellebjerg F, Sorensen TL. Systemic frequencies of T helper 1 and T helper 17 cells in patients with age-related macular degeneration: a case-control study. Sci Rep. (2017) 7:605. doi: 10.1038/s41598-017-00741-4
63. Yu Y, Ren XR, Wen F, Chen H, Su SB. T-helper-associated cytokines expression by peripheral blood mononuclear cells in patients with polypoidal choroidal vasculopathy and age-related macular degeneration. BMC Ophthalmol. (2016) 16:80. doi: 10.1186/s12886-016-0251-z
64. Chen J, Wang W, Li Q. Increased Th1/Th17 responses contribute to low-grade inflammation in age-related macular degeneration. Cell Physiol Biochem. (2017) 44:357–67. doi: 10.1159/000484907
65. Cruz-Guilloty F, Saeed AM, Duffort S, Cano M, Ebrahimi KB, Ballmick A, et al. T cells and macrophages responding to oxidative damage cooperate in pathogenesis of a mouse model of age-related macular degeneration. PLoS ONE. (2014) 9:e88201. doi: 10.1371/journal.pone.0088201
66. Zhao Z, Liang Y, Liu Y, Xu P, Flamme-Wiese MJ, Sun D, et al. Choroidal gammadelta T cells in protection against retinal pigment epithelium and retinal injury. FASEB J. (2017) 31:4903–4916. doi: 10.1096/fj.201700533R
67. Hasegawa E, Sonoda KH, Shichita T, Morita R, Sekiya T, Kimura A, et al. IL-23-independent induction of IL-17 from gammadeltaT cells and innate lymphoid cells promotes experimental intraocular neovascularization. J Immunol. (2013) 190:1778–87. doi: 10.4049/jimmunol.1202495
68. Consonni FM, Porta C, Marino A, Pandolfo C, Mola S, Bleve A, Sica A. Myeloid-derived suppressor cells: ductile targets in disease. Front Immunol. (2019) 10:949. doi: 10.3389/fimmu.2019.00949
69. Gabrilovich DI. Myeloid-derived suppressor cells. Cancer Immunol Res. (2017) 5:3–8. doi: 10.1158/2326-6066.CIR-16-0297
70. Talmadge JE, Gabrilovich DI. History of myeloid-derived suppressor cells. Nat Rev Cancer. (2013) 13:739–52. doi: 10.1038/nrc3581
71. Bronte V, Brandau S, Chen SH, Colombo MP, Frey AB, Greten TF, et al. Recommendations for myeloid-derived suppressor cell nomenclature and characterization standards. Nat Commun. (2016) 7:12150. doi: 10.1038/ncomms12150
72. Pawelec G, Verschoor CP, Ostrand-Rosenberg S. Myeloid-derived suppressor cells: not only in tumor immunity. Front Immunol. (2019) 10:1099. doi: 10.3389/fimmu.2019.01099
73. Zhao Y, Wu T, Shao S, Shi B, Zhao Y. Phenotype, development, and biological function of myeloid-derived suppressor cells. Oncoimmunology. (2016) 5:e1004983. doi: 10.1080/2162402X.2015.1004983
74. Huang B, Pan PY, Li Q, Sato AI, Levy DE, Bromberg J, et al. Gr-1+CD115+ immature myeloid suppressor cells mediate the development of tumor-induced T regulatory cells and T-cell anergy in tumor-bearing host. Cancer Res. (2006) 66:1123–31. doi: 10.1158/0008-5472.CAN-05-1299
75. Li H, Han Y, Guo Q, Zhang M, Cao X. Cancer-expanded myeloid-derived suppressor cells induce anergy of NK cells through membrane-bound TGF-beta 1. J Immunol. (2009) 182:240–9. doi: 10.4049/jimmunol.182.1.240
76. Pan PY, Ma G, Weber KJ, Ozao-Choy J, Wang G, Yin B, et al. Immune stimulatory receptor CD40 is required for T-cell suppression and T regulatory cell activation mediated by myeloid-derived suppressor cells in cancer. Cancer Res. (2010) 70:99–108. doi: 10.1158/0008-5472.CAN-09-1882
77. Serafini P, Mgebroff S, Noonan K, Borrello I. Myeloid-derived suppressor cells promote cross-tolerance in B-cell lymphoma by expanding regulatory T cells. Cancer Res. (2008) 68:5439–49. doi: 10.1158/0008-5472.CAN-07-6621
78. Sinha P, Clements VK, Bunt SK, Albelda SM, Ostrand-Rosenberg S. Cross-talk between myeloid-derived suppressor cells and macrophages subverts tumor immunity toward a type 2 response. J Immunol. (2007) 179:977–83. doi: 10.4049/jimmunol.179.2.977
79. Suzuki E, Kapoor V, Jassar AS, Kaiser LR, Albelda SM. Gemcitabine selectively eliminates splenic Gr-1+/CD11b+ myeloid suppressor cells in tumor-bearing animals and enhances antitumor immune activity. Clin Cancer Res. (2005) 11:6713–21. doi: 10.1158/1078-0432.CCR-05-0883
80. Kim JA, March K, Chae HD, Johnstone B, Park SJ, Cook T, et al. Muscle-derived Gr1 (dim) CD11b (+) cells enhance neovascularization in an ischemic hind limb mouse model. Blood. (2010) 116:1623–6. doi: 10.1182/blood-2009-08-237040
81. Was H, Dulak J, Jozkowicz A. Heme oxygenase-1 in tumor biology and therapy. Curr Drug Targets. (2010) 11:1551–70. doi: 10.2174/1389450111009011551
82. De Wilde V, Van Rompaey N, Hill M, Lebrun JF, Lemaître P, Lhommé F, et al. Endotoxin-induced myeloid-derived suppressor cells inhibit alloimmune responses via heme oxygenase-1. Am J Trans. (2009) 9:2034–47. doi: 10.1111/j.1600-6143.2009.02757.x
83. Iacobaeus E, Douagi I, Jitschin R, Marcusson-Ståhl M, Andrén AT, Gavin C, et al. Phenotypic and functional alterations of myeloid-derived suppressor cells during the disease course of multiple sclerosis. Immunol Cell Biol. (2018) 96:820–30. doi: 10.1111/imcb.12042
84. Sunthamala N, Pientong C, Ohno T, Zhang C, Bhingare A, Kondo Y, et al. HPV16 E2 protein promotes innate immunity by modulating immunosuppressive status. Biochem Biophys Res Commun. (2014) 446:977–82. doi: 10.1016/j.bbrc.2014.03.042
85. Futagami Y, Sugita S, Vega J, Ishida K, Takase H, Maruyama K, et al. Role of thrombospondin-1 in T cell response to ocular pigment epithelial cells. J Immunol. (2007) 178:6994–7005. doi: 10.4049/jimmunol.178.11.6994
86. Holtkamp GM, Kijlstra A, Peek R de Vos AF. Retinal pigment epithelium-immune system interactions: cytokine production and cytokine-induced changes. Prog Retin Eye Res. (2001) 20:29–48. doi: 10.1016/S1350-9462(00)00017-3
87. Ke Y, Sun D, Jiang G, Kaplan HJ, Shao H. PD-L1 (hi) retinal pigment epithelium (RPE) cells elicited by inflammatory cytokines induce regulatory activity in uveitogenic T cells. J Leukoc Biol. (2010) 88:1241–9. doi: 10.1189/jlb.0610332
88. Mochizuki M, Sugita S, Kamoi K. Immunological homeostasis of the eye. Prog Retin Eye Res. (2013) 33:10–27. doi: 10.1016/j.preteyeres.2012.10.002
89. Sugita S, Horie S, Nakamura O, Futagami Y, Takase H, Keino H, et al. Retinal pigment epithelium-derived CTLA-2alpha induces TGFbeta-producing T regulatory cells. J Immunol. (2008) 181:7525–36. doi: 10.4049/jimmunol.181.11.7525
90. Sugita S, Horie S, Yamada Y, Mochizuki M. Inhibition of B-cell activation by retinal pigment epithelium. Invest Ophthalmol Vis Sci. (2010) 51:5783–8. doi: 10.1167/iovs.09-5098
91. Kawanaka N, Taylor AW. Localized retinal neuropeptide regulation of macrophage and microglial cell functionality. J Neuroimmunol. (2011) 232:17–25. doi: 10.1016/j.jneuroim.2010.09.025
92. Condamine T, Gabrilovich DI. Molecular mechanisms regulating myeloid-derived suppressor cell differentiation and function. Trends Immunol. (2011) 32:19–25. doi: 10.1016/j.it.2010.10.002
93. Bunt SK, Clements VK, Hanson EM, Sinha P, Ostrand-Rosenberg S. Inflammation enhances myeloid-derived suppressor cell cross-talk by signaling through Toll-like receptor 4. J Leukoc Biol. (2009) 85:996–1004. doi: 10.1189/jlb.0708446
94. Cao X, Shen D, Patel MM, Tuo J, Johnson TM, Olsen TW, Chan CC. Macrophage polarization in the maculae of age-related macular degeneration: a pilot study. Pathol Int. (2011) 61:528–35. doi: 10.1111/j.1440-1827.2011.02695.x
95. Yang Y, Liu F, Tang M, Yuan M, Hu A, Zhan Z, et al. Macrophage polarization in experimental and clinical choroidal neovascularization. Sci Rep. (2016) 6:30933. doi: 10.1038/srep30933
96. McHugh KJ, Li D, Wang JC, Kwark L, Loo J, Macha V, et al. Computational modeling of retinal hypoxia and photoreceptor degeneration in patients with age-related macular degeneration. PLoS ONE. (2019) 14:e0216215. doi: 10.1371/journal.pone.0216215
97. Arjamaa O, Aaltonen V, Piippo N, Csont T, Petrovski G, Kaarniranta K, Kauppinen A. Hypoxia and inflammation in the release of VEGF and interleukins from human retinal pigment epithelial cells. Graefes Arch Clin Exp Ophthalmol. (2017) 255:1757–62. doi: 10.1111/j.1755-3768.2016.0421
98. Nozaki M, Raisler BJ, Sakurai E, Sarma JV, Barnum SR, Lambris JD, et al. Drusen complement components C3a and C5a promote choroidal neovascularization. Proc Natl Acad Sci U S A. (2006) 103:2328–33. doi: 10.1073/pnas.0408835103
99. Sinha P, Clements VK, Fulton AM, Ostrand-Rosenberg S. Prostaglandin E2 promotes tumor progression by inducing myeloid-derived suppressor cells. Cancer Res. (2007) 67:4507–13. doi: 10.1158/0008-5472.CAN-06-4174
100. Yang L, DeBusk LM, Fukuda K, Fingleton B, Green-Jarvis B, Shyr Y, et al. Expansion of myeloid immune suppressor Gr+CD11b+ cells in tumor-bearing host directly promotes tumor angiogenesis. Cancer Cell. (2004) 6:409–21. doi: 10.1016/j.ccr.2004.08.031
101. Chin MS, Nagineni CN, Hooper LC, Detrick B, Hooks JJ. Cyclooxygenase-2 gene expression and regulation in human retinal pigment epithelial cells. Invest Ophthalmol Vis Sci. (2001) 42:2338–46.
102. Obermajer N, Muthuswamy R, Lesnock J, Edwards RP, Kalinski P. Positive feedback between PGE2 and COX2 redirects the differentiation of human dendritic cells toward stable myeloid-derived suppressor cells. Blood. (2011) 118:5498–505. doi: 10.1182/blood-2011-07-365825
103. Ambati J, Anand A, Fernandez S, Sakurai E, Lynn BC, Kuziel WA, et al. An animal model of age-related macular degeneration in senescent Ccl-2- or Ccr-2-deficient mice. Nat Med. (2003) 9:1390–7. doi: 10.1038/nm950
104. Ekkens MJ, Shedlock DJ, Jung E, Troy A, Pearce EL, Shen H, Pearce EJ. Th1 and Th2 cells help CD8 T-cell responses. Infect Immun. (2007) 75:2291–6. doi: 10.1128/IAI.01328-06
105. Shi Q, Wang Q, Li J, Zhou X, Fan H, Wang F, et al. A2E suppresses regulatory function of RPE cells in Th1 cell differentiation via production of IL-1beta and inhibition of PGE2. Invest Ophthalmol Vis Sci. (2015) 56:7728–38. doi: 10.1167/iovs.15-17677
106. Maloney SC, Fernandes BF, Castiglione E, Antecka E, Martins C, Marshall JC, et al. Expression of cyclooxygenase-2 in choroidal neovascular membranes from age-related macular degeneration patients. Retina. (2009) 29:176–80. doi: 10.1097/IAE.0b013e3181884fa6
107. Falk MK, Singh A, Faber C, Nissen MH, Hviid T, Sorensen TL. Dysregulation of CXCR3 expression on peripheral blood leukocytes in patients with neovascular age-related macular degeneration. Invest Ophthalmol Vis Sci. (2014) 55:4050–6. doi: 10.1167/iovs.14-14107
108. Keenan TD, Wiley HE, Agron E, Aronow ME, Christen WG, Clemons TE, et al. The association of aspirin use with age-related macular degeneration progression in the age-related eye disease studies: age-related eye disease study 2 report no. 20. Ophthalmology. (2019) 126:1647–56. doi: 10.1016/j.ophtha.2019.06.023
109. Sin M, Chrapek O, Karhanova M, Pracharova Z, Langova K, Rehak J. Progression of macular atrophy after PDT combined with the COX-2 inhibitor Nabumetone in the treatment of neovascular ARMD. Biomed Pap Med Fac Univ Palacky Olomouc Czech Repub. (2014) 158:138–43. doi: 10.5507/bp.2012.066
110. Xu H, Chen M. Targeting the complement system for the management of retinal inflammatory and degenerative diseases. Eur J Pharmacol. (2016) 787:94–104. doi: 10.1016/j.ejphar.2016.03.001
111. Fernandez-Godino R, Garland DL, Pierce EA. A local complement response by RPE causes early-stage macular degeneration. Hum Mol Genet. (2015) 24:5555–69. doi: 10.1093/hmg/ddv287
112. Fernandez-Godino R, Pierce EA. C3a triggers formation of sub-retinal pigment epithelium deposits via the ubiquitin proteasome pathway. Sci Rep. (2018) 8:9679. doi: 10.1038/s41598-018-28143-0
113. Luo C, Zhao J, Madden A, Chen M, Xu H. Complement expression in retinal pigment epithelial cells is modulated by activated macrophages. Exp Eye Res. (2013) 112:93–101. doi: 10.1016/j.exer.2013.04.016
114. Bora PS, Sohn JH, Cruz JM, Jha P, Nishihori H, Wang Y, et al. Role of complement and complement membrane attack complex in laser-induced choroidal neovascularization. J Immunol. (2005) 174:491–7. doi: 10.4049/jimmunol.174.1.491
115. Downs-Canner S, Magge D, Ravindranathan R, O'Malley ME, Francis L, Liu Z, et al. Complement inhibition: a novel form of immunotherapy for colon cancer. Ann Surg Oncol. (2016) 23:655–62. doi: 10.1245/s10434-015-4778-7
116. Chou HS, Hsieh CC, Yang HR, Wang L, Arakawa Y, Brown K, et al. Hepatic stellate cells regulate immune response by way of induction of myeloid suppressor cells in mice. Hepatology. (2011) 53:1007–19. doi: 10.1002/hep.24162
117. Hsieh CC, Chou HS, Yang HR, Lin F, Bhatt S, Qin J, et al. The role of complement component 3 (C3) in differentiation of myeloid-derived suppressor cells. Blood. (2013) 121:1760–8. doi: 10.1182/blood-2012-06-440214
118. Li Y, Tu Z, Qian S, Fung JJ, Markowitz SD, Kusner LL, et al. Myeloid-derived suppressor cells as a potential therapy for experimental autoimmune myasthenia gravis. J Immunol. (2014) 193:2127–34. doi: 10.4049/jimmunol.1400857
119. Parsons N, Annamalai B, Obert E, Schnabolk G, Tomlinson S, Rohrer B. Inhibition of the alternative complement pathway accelerates repair processes in the murine model of choroidal neovascularization. Mol Immunol. (2019) 108:8–12. doi: 10.1016/j.molimm.2019.02.001
120. Cortright DN, Meade R, Waters SM, Chenard BL, Krause JE. C5a, but not C3a, increases VEGF secretion in ARPE-19 human retinal pigment epithelial cells. Curr Eye Res. (2009) 34:57–61. doi: 10.1080/02713680802546658
121. Lee D, Son HG, Jung Y, Lee SV. The role of dietary carbohydrates in organismal aging. Cell Mol Life Sci. (2017) 74:1793–803. doi: 10.1007/s00018-016-2432-6
122. Zhou J, He S, Zhang N, Spee C, Zhou P, Ryan SJ, et al. Neutrophils compromise retinal pigment epithelial barrier integrity. J Biomed Biotechnol. (2010) 2010:289360. doi: 10.1155/2010/289360
123. Katschke KJ Jr, Xi H, Cox C, Truong T, Malato Y, Lee WP, et al. Classical and alternative complement activation on photoreceptor outer segments drives monocyte-dependent retinal atrophy. Sci Rep. (2018) 8:7348. doi: 10.1038/s41598-018-30162-w
124. Jiao H, Rutar M, Fernando N, Yednock T, Sankaranarayanan S, Aggio-Bruce R, et al. Subretinal macrophages produce classical complement activator C1q leading to the progression of focal retinal degeneration. Mol Neurodegener. (2018) 13:45. doi: 10.1186/s13024-018-0278-0
125. Lynch AM, Mandava N, Patnaik JL, Frazer-Abel AA, Wagner BD, Palestine AG, et al. Systemic activation of the complement system in patients with advanced age-related macular degeneration. Eur J Ophthalmol. (2019) 1120672119857896. doi: 10.1177/1120672119857896. [Epub ahead of print].
126. Scholl HP, Charbel Issa P, Walier M, Janzer S, Pollok-Kopp B, Borncke F, et al. Systemic complement activation in age-related macular degeneration. PLoS ONE. (2008) 3:e2593. doi: 10.1371/journal.pone.0002593
127. Ajona D, Hsu YF, Corrales L, Montuenga LM, Pio R. Down-regulation of human complement factor H sensitizes non-small cell lung cancer cells to complement attack and reduces in vivo tumor growth. J Immunol. (2007) 178:5991–8. doi: 10.4049/jimmunol.178.9.5991
128. Corrales L, Ajona D, Rafail S, Lasarte JJ, Riezu-Boj JI, Lambris JD, et al. Anaphylatoxin C5a creates a favorable microenvironment for lung cancer progression. J Immunol. (2012) 189:4674–4683. doi: 10.4049/jimmunol.1201654
129. Markiewski MM, DeAngelis RA, Benencia F, Ricklin-Lichtsteiner SK, Koutoulaki A, Gerard C, et al. Modulation of the antitumor immune response by complement. Nat Immunol. (2008) 9:1225–1235. doi: 10.1038/ni.1655
130. Nitta H, Murakami Y, Wada Y, Eto M, Baba H, Imamura T. Cancer cells release anaphylatoxin C5a from C5 by serine protease to enhance invasiveness. Oncol Rep. (2014) 32:1715–9. doi: 10.3892/or.2014.3341
131. Darling VR, Hauke RJ, Tarantolo S, Agrawal DK. Immunological effects and therapeutic role of C5a in cancer. Expert Rev Clin Immunol. (2015) 11:255–63. doi: 10.1586/1744666X.2015.983081
132. Yoneda M, Imamura R, Nitta H, Taniguchi K, Saito F, Kikuchi K, et al. Enhancement of cancer invasion and growth via the C5a-C5a receptor system: Implications for cancer promotion by autoimmune diseases and association with cervical cancer invasion. Oncol Lett. (2019) 17:913–920. doi: 10.3892/ol.2018.9715
133. Calippe B, Augustin S, Beguier F, Charles-Messance H, Poupel L, Conart JB, et al. Complement factor H inhibits CD47-mediated resolution of inflammation. Immunity. (2017) 46:261–72. doi: 10.1016/j.immuni.2017.01.006
134. Toomey CB, Kelly U, Saban DR, Bowes Rickman C. Regulation of age-related macular degeneration-like pathology by complement factor H. Proc Natl Acad Sci USA. (2015) 112:E3040–9. doi: 10.1073/pnas.1424391112
135. Ma W, Zhao L, Fontainhas AM, Fariss RN, Wong WT. Microglia in the mouse retina alter the structure function of retinal pigmented epithelial cells: a potential cellular interaction relevant to AMD. PLoS ONE. (2009) 4:e7945. doi: 10.1371/journal.pone.0007945
136. Raoul W, Keller N, Rodero M, Behar-Cohen F, Sennlaub F, Combadiere C. Role of the chemokine receptor CX3CR1 in the mobilization of phagocytic retinal microglial cells. J Neuroimmunol. (2008) 198:56–61. doi: 10.1016/j.jneuroim.2008.04.014
137. Ishikawa K, Sreekumar PG, Spee C, Nazari H, Zhu D, Kannan R, Hinton DR. alphaB-crystallin regulates subretinal fibrosis by modulation of epithelial-mesenchymal transition. Am J Pathol. (2016) 186:859–73. doi: 10.1016/j.ajpath.2015.11.014
138. Carbone C, Moccia T, Zhu C, Paradiso G, Budillon A, Chiao PJ, et al. Anti-VEGF treatment-resistant pancreatic cancers secrete proinflammatory factors that contribute to malignant progression by inducing an EMT cell phenotype. Clin Cancer Res. (2011) 17:5822–32. doi: 10.1158/1078-0432.CCR-11-1185
139. Condamine T, Ramachandran I, Youn JI, Gabrilovich DI. Regulation of tumor metastasis by myeloid-derived suppressor cells. Annu Rev Med. (2015) 66:97–110. doi: 10.1146/annurev-med-051013-052304
140. Fan Q, Gu D, Liu H, Yang L, Zhang X, Yoder MC, et al. Defective TGF-beta signaling in bone marrow-derived cells prevents hedgehog-induced skin tumors. Cancer Res. (2014) 74:471–83. doi: 10.1158/0008-5472.CAN-13-2134-T
141. Dvashi Z, Goldberg M, Adir O, Shapira M, Pollack A. TGF-beta1 induced transdifferentiation of rpe cells is mediated by TAK1. PLoS ONE. (2015) 10:e0122229. doi: 10.1371/journal.pone.0122229
142. Saika S. TGFbeta pathobiology in the eye. Lab Invest. (2006) 86:106–15. doi: 10.1038/labinvest.3700375
143. Ioannou M, Alissafi T, Lazaridis I, Deraos G, Matsoukas J, Gravanis A, et al. Crucial role of granulocytic myeloid-derived suppressor cells in the regulation of central nervous system autoimmune disease. J Immunol. (2012) 188:1136–46. doi: 10.4049/jimmunol.1101816
144. Sendo S, Saegusa J, Morinobu A. Myeloid-derived suppressor cells in non-neoplastic inflamed organs. Inflamm Regen. (2018) 38:19. doi: 10.1186/s41232-018-0076-7
145. Zhu B, Kennedy JK, Wang Y, Sandoval-Garcia C, Cao L, Xiao S, et al. Plasticity of Ly-6C (hi) myeloid cells in T cell regulation. J Immunol. (2011) 187:2418–32. doi: 10.4049/jimmunol.1100403
146. Liesz A, Dalpke A, Mracsko E, Antoine DJ, Roth S, Zhou W, et al. DAMP signaling is a key pathway inducing immune modulation after brain injury. J Neurosci. (2015) 35:583–98. doi: 10.1523/JNEUROSCI.2439-14.2015
147. Saiwai H, Kumamaru H, Ohkawa Y, Kubota K, Kobayakawa K, Yamada H, et al. Ly6C+ Ly6G- Myeloid-derived suppressor cells play a critical role in the resolution of acute inflammation and the subsequent tissue repair process after spinal cord injury. J Neurochem. (2013) 125:74–88. doi: 10.1111/jnc.12135
148. Jeong HJ, Lee HJ, Ko JH, Cho BJ, Park SY, Park JW, et al. Myeloid-derived suppressor cells mediate inflammation resolution in humans and mice with autoimmune uveoretinitis. J Immunol. (2018) 200:1306–15. doi: 10.4049/jimmunol.1700617
149. Lee HJ, Ko JH, Jeong HJ, Ko AY, Kim MK, Wee WR, et al. Mesenchymal stem/stromal cells protect against autoimmunity via CCL2-dependent recruitment of myeloid-derived suppressor cells. J Immunol. (2015) 194:3634–45. doi: 10.4049/jimmunol.1402139
Keywords: myeloid-derived suppressor cell, age-related macular degeneration, inflammation, innate immunity, adaptive immunity
Citation: Kauppinen A, Kaarniranta K and Salminen A (2020) Potential Role of Myeloid-Derived Suppressor Cells (MDSCs) in Age-Related Macular Degeneration (AMD). Front. Immunol. 11:384. doi: 10.3389/fimmu.2020.00384
Received: 19 November 2019; Accepted: 18 February 2020;
Published: 20 March 2020.
Edited by:
Darren James Lee, University of Oklahoma Health Sciences Center, United StatesReviewed by:
Alain Le Moine, Université Libre de Bruxelles, BelgiumKevin K. Fuller, University of Oklahoma Health Sciences Center, United States
Copyright © 2020 Kauppinen, Kaarniranta and Salminen. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Anu Kauppinen, YW51LmthdXBwaW5lbkB1ZWYuZmk=