 Darius Häusler1†
Darius Häusler1† Martin S. Weber
Martin S. Weber- 1Department of Neuropathology, Institute of Neuropathology, University Medical Center, Göttingen, Germany
- 2Department of Neurology, University Medical Center, Göttingen, Germany
The exact cause of multiple sclerosis (MS) is unknown; however, it is considered to be an inflammatory disease of the central nervous system (CNS) triggered by a combination of both environmental and genetic factors. Vitamin D deficiency is also discussed as a possible disease-promoting factor in MS, as low vitamin D status is associated with increased formation of CNS lesions, elevated number of relapses and accelerated disease progression. However, it remains unclear whether this association is causal and related and most importantly, whether vitamin D supplementation in MS is of direct therapeutic benefit. Recently, we could show that in a murine model of MS, administration of a moderate vitamin D dose was of clinical benefit, while excessive vitamin D supplementation had a negative effect on disease severity. Of note, disease exacerbation was associated with high-dose vitamin D caused secondary hypercalcemia. Mechanistically dissecting this outcome, we found that hypercalcemia independent of vitamin D similarly triggered activation of disease-perpetuating T cells. These findings caution that vitamin D should be supplemented in a controlled and moderate manner in patients with MS and concomitantly highlight calcium as a novel potential MS risk factor by itself. In this review, we will summarize the current evidence from animal and clinical studies aiming to assess whether vitamin D may be of benefit in patients with MS. Furthermore, we will discuss any possible secondary effects of vitamin D with a particular focus on the role of calcium on immune cells and in the pathogenesis of CNS demyelinating disease.
Introduction
A poor vitamin D status is implicated as a possible risk factor in many autoimmune diseases, including, systemic lupus erythematosus (SLE) (1), diabetes mellitus type I (2), rheumatoid arthritis (RA) (3), vasculitis (4) and multiple sclerosis (MS) (5). In patients with MS, low serum levels of vitamin D are associated with a higher relapse rate and an earlier development of persistent disability (6, 7). Based on this association and its assumed harmlessness, physicians frequently recommend high-dose supplementation with cholecalciferol the inactive form of vitamin D, although clear evidence for the efficacy of this interventional regimen is missing.
Beneficial Effects of Moderate Vitamin D Doses
In the most widely used animal model of MS, experimental autoimmune encephalomyelitis (EAE), intraperitoneal administration of the biologically active form of vitamin D (1,25(OH)2D3) showed beneficial results, both in a preventive (8, 9) and therapeutic (9–11) treatment regimen. This clinical benefit was associated with a reduced activation and accumulation of monocytes/macrophages and autoreactive T lymphocytes within the CNS. A moderate long-term oral supplementation of the inactive form of vitamin D (cholecalciferol), which represents the most commonly supplemented metabolite in humans confirmed these beneficial results (12, 13). The majority of biologic functions induced by vitamin D is mediated by its binding to the vitamin D (VDR) receptor, which triggers a chain of genomic events leading to the transcriptional control of vitamin D regulated genes (14–16). The VDR is expressed in the nucleus of almost all immune cells, including T cells (17), activated B cells (18), dendritic cells (19), monocytes and macrophages (17, 20), neuronal and glial cells (21–29) and the level of expression can be increased upon activation (17, 30–34). Growing evidence indicates that vitamin D exerts its beneficial effects by promoting the frequency and function of regulatory T cells (Tregs) (35, 36).
In MS patients, several smaller clinical trials of vitamin D supplementation have been conducted. A 96 weeks supplementation study of moderate vitamin D doses of 20,000 IU/week in 68 MS patients increased mean serum 25(OH)D3 levels from 55.6 nmol/L at baseline to 123.1 nmol/L. Treatment showed no clear effect on secondary outcomes such as disease activity with respect to relapse rate, functional tests, or fatigue severity (37). By contrast in 39 MS patients, moderate vitamin D supplementation (1,000 IU/day) for 6 months elevated mean serum 25(OH)D3 levels from 42.5 nmol/L at baseline to 70 nmol/L. Treatment was associated with increased serum level of transforming growth factor beta (TGF-ß) and diminished IL-2 mRNA in peripheral blood mononuclear cells (PBMCs) (38). Another study analyzed the effect of weekly supplementation with 20,000 IU vitamin D in 66 MS patients for 1 year (39). Mean serum 25(OH)D3 levels increased from 54 nmol/L at baseline to 110 nmol/L. Treated groups tended to have reduced disability accumulation, improved time tandem walk and significantly fewer T1 enhancing lesions. Golan et al. (40) compared moderately high (4,370 IU/ day) to low (800 IU/day) vitamin D supplementation in 45 patients on IFN-ß maintenance therapy. Mean serum 25(OH)D3 levels increased from 48 to 68 nmol/L and from 48.2 to 122.6 nmol/L in the low- and moderately high vitamin D group, respectively. Moderately high vitamin D supplementation was associated with a heterogeneous IL-17 response, while in comparison, IL-17 levels were significantly increased in the low dose group.
Recently, high dose vitamin D supplementation provided conflicting results in the treatment of MS. A 52 weeks safety study with escalating doses of vitamin D up to 40,000 IU/day reached a mean peak serum 25(OH)D3 level of 413 nmol/L and showed a trend toward reduced relapse rate (41). A 48 weeks study (SOLAR) of oral high-dose vitamin D (14,000 IU/day) as add-on therapy to IFN-ß, which has been the largest study to date showed a significant reduction in the number of new MRI lesions (42). However, the primary endpoint of the study defined as no evidence of disease activity (NEDA-3) was not reached. Another study (CHOLINE) compared 100,000 IU vitamin D supplementation as add-on therapy to IFN-ß every other week for 96 weeks (43). Mean serum 25(OH)D3 levels increased from 49.19 nmol/L at baseline to 156.92 nmol/L. Vitamin D supplementation was associated with annualized relapse rate reduction, less new hypointense T1-weighted lesions, a lower volume of hypointense T1-weighted lesions, and a lower progression of EDSS. Nevertheless, the primary endpoint defined as reducing the relapse rate in all included patients was not met. By contrast, the comparison of high-dose (6,000 IU/twice daily) to moderate dose (1,000 IU/once daily) vitamin D in 23 MS patients for 6 months failed to show a significant clinical benefit and rather revealed a worsening effect of the higher dose (44). Along the same lines, a meta-analysis concluded little additional benefit from using supra-physiological doses of vitamin D (45). Moreover, the authors found that in a proportion of analyzed high-dose vitamin D studies, there might be an adverse effect on annualized relapse rate (ARR), expanded disability status scale (EDSS) and gadolinium-enhancing lesions. For simplicity and comparability, Table 1 provides a comprehensive summary of the relevant studies.
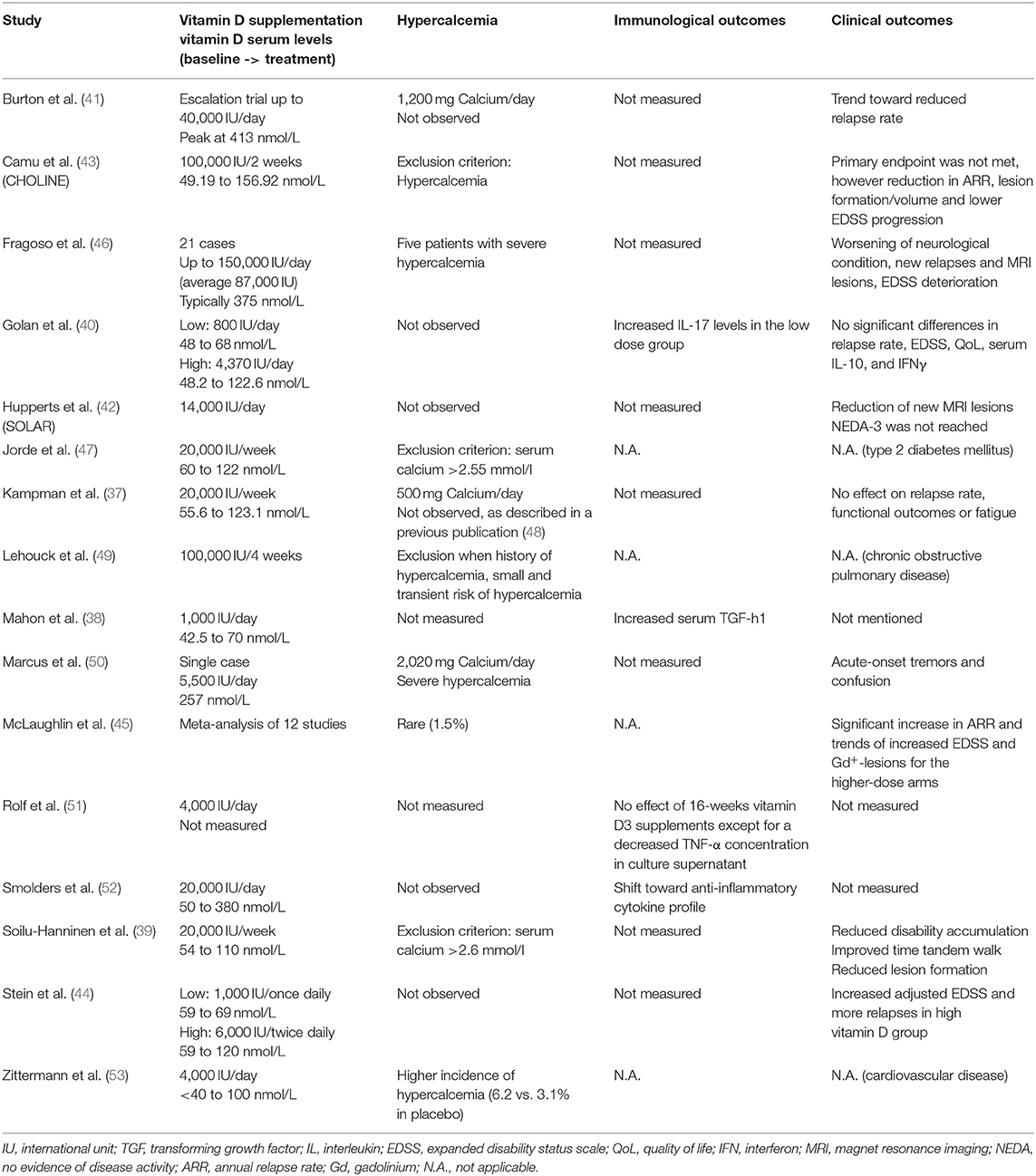
Table 1. Overview of relevant vitamin D supplementation studies.
These findings jointly corroborate that therapeutic correction of vitamin D deficiency by moderate vitamin D supplementation may exert an ameliorating effect in MS, but also indicate that supra-physiological doses fail to provide any additional therapeutic benefit.
Secondary Hypercalcemia as a risk of Vitamin D High-dose Supplementation
Vitamin D and its metabolites regulate the secretion of hormones and have complex functions in calcium and phosphorus homeostasis, as for instance regulation of intestinal calcium and phosphate absorption, calcium mobilization from bones and reabsorption of calcium in the kidneys (54). Hypercalcemia may occur in humans supplemented with high doses of vitamin D (55, 56), especially when combined with calcium intake (57–59). In addition, hypercalcemia was also reported in the treatment of chronic obstructive pulmonary disease (49) and type 2 diabetes mellitus (47) upon high-dose vitamin D. In cardiovascular disease (CVD) the incidence of hypercalcemia tended to be higher upon moderately high vitamin D supplementation (53). Most of the vitamin D high-dose trials in MS described no hypercalcemia (42, 52) and few cases of hypercalciuria (51), with no significant difference to controls. However, in individual MS patients hypercalcemia was detected in conjunction with high-dose vitamin D supplementation (50) not reaching the well-accepted critical serum 25(OH)D3 level of > 375 nmol/L (60) and, most importantly, this was associated with increased relapse rate and MRI activity (46). These data indicate that hypercalcemia may also occur at lower 25(OH)D3 level and might individually develop depending on calcium intake. Moreover, it has been shown that already serum 25(OH)D3 level > 125 nmol/L are related to an increased morbidity and mortality risk, proposing to define a variable upper intake level of vitamin D depending on baseline 25(OH)D3 level and body weight as vitamin D is a fat-soluble vitamin (61). In a recent study (13), we fed mice with three different vitamin D (cholecalciferol) concentrations reflective of MS patients who are vitamin D-deficient, moderately supplemented and treated in MS high-dose supplementation trials (41, 52) resulting in mean serum 25(OH)D3 levels of 23, 96, and 282 nmol/L, respectively (13, 62). We found that moderate vitamin D supplementation ameliorated the course of EAE when compared to vitamin D-deprived mice. By contrast, the group of mice receiving high doses of vitamin D, in which serum 25(OH)D3 levels exceeded 200 nmol/L developed severely aggravated EAE, with a fulminant CNS infiltration by activated myeloid cells, Th1 and Th17 cells. Mechanistically dissecting these opposite clinical outcomes, we found that the benefit of moderate vitamin D levels related to a direct anti-inflammatory effect of vitamin D and its metabolites on T cells. In contrast, disease worsening in the group of mice continuously treated with high-dose vitamin D referred to secondary hypercalcemia, which was associated with promoted development of encephalitogenic T cells. Raising murine serum calcium level directly by calcium gluconate injections resulted in increased expression of activation markers on CD4+ and CD8+ T cells, confirming that this effect occurs in vivo independent of vitamin D. Directly exposing murine or human T cells to equivalent calcium concentrations enhanced its influx, caused an increased susceptibility to activation, an upregulation of pro-inflammatory gene products as well as an enhanced capacity of activated T cells to transmigrate across a blood-brain-barrier model. Most of the previous EAE studies used the biologically active form (1,25(OH)2D3) in a short-term treatment regimen (8–11). Contrary to our observations Cantorna et al. described a clinical benefit upon high-dose 1,25(OH)2D3 treatment with concomitantly occurring hypercalcemia (63). However, in this model hypercalcemia was also accompanied by a significant loss of body weight, which might influence a proper development of EAE. Interestingly, a decrease in calcium diet was associated with a reduction in EAE incidence.
Taken together, these findings highlight that an elevation of available extracellular calcium may directly activate T cells and promote their pro-inflammatory maturation and function. This points toward calcium as a novel risk factor in inflammatory CNS-demyelinating disease. Understanding calcium signaling in T lymphocytes may therefore help fathom the efficacy of various available therapies targeting the immune system and additionally open up novel therapeutic pathways.
T cell Receptor Signaling and Calcium Mobilization
The main mechanism of raising calcium (Ca2+) levels in T cells is store-operated calcium entry (SOCE) through calcium release-activated calcium (CRAC) channels. The importance of efficient calcium entry in lymphocyte function is highlighted by the fact that several severe combined immunodeficiency (SCID) have been described as a result of defects in SOCE and CRAC which severely impairs overall lymphocyte function (64, 65). CRAC channels are activated after initial T cell receptor (TCR) engagement. TCR activation leads to the phosphorylation of immunoreceptor tyrosine-based activation motifs (ITAM) by Src kinases. This leads to the recruitment of the Syk family kinase zeta-activated protein 70 (ZAP70) and the subsequent phosphorylation of the linker for activation of T cells (LAT). LAT recruits the scaffold protein SH2-domain containing leucocyte protein of 76kDa (SLP-76) which, after a phosphorylation by ZAP70 interacts with the Interleukin-2 inducible tyrosine kinase (ITK). This leads to the activation of phospholipase C gamma 1 (PLC-γ1) which creates the second messengers diacylglycerol (DAG) and inositol triphosphate (IP3) by cleaving phosphatidylinositol triphosphate (PIP2). While DAG activates kinases such as protein kinase C (PKC) and Ras guanyl nucleotide-releasing protein (RasGRP), IP3 stimulates a calcium release from the endoplasmic reticulum (ER) into the cytosol (66–68). This transient elevation of cytoplasmic calcium from the ER is known as “store depletion” and results in the activation of the calcium-sensitive CRAC channels in the plasma membrane which triggers a greater influx into the cytosol from the extracellular space. The responsible CRAC channel in lymphocytes is CRAC modulator 1 (CRACM1, also called ORAI1) which is stimulated by the stromal interaction molecule 1 (STIM1). STIM1, which is mainly located in the ER, senses the store depletion and oligomerizes to form distinct “punctae” at the ER-plasma membrane junctions which leads to the activation of CRACM1 (69, 70). In addition, several other channels, such as members of the transient receptor potential (TRP) family, P2X receptors or voltage-gated calcium-channels (Cav), may contribute to the elevation of calcium (71). The resulting sustained calcium elevation activates several calcium-sensitive signaling proteins such as the phosphatase calcineurin and its target nuclear factor of activated T cells (NFAT), nuclear factor κB (NFκB) or the Ca+2 calmodulin-dependent kinase (CaMK) (Figure 1).
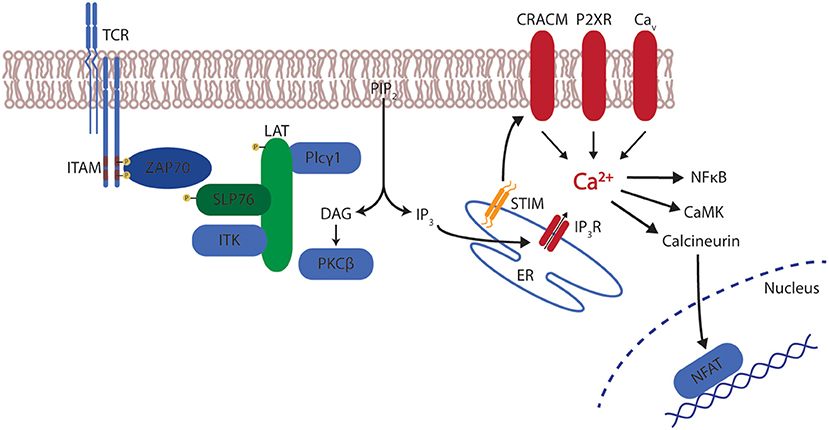
Figure 1. T cell receptor signaling and calcium. After stimulation of the T cell receptor (TCR), the signal is relayed via immunoreceptor tyrosine-based activation motifs (ITAM) leading to the recruitment of zeta-activated protein 70 (ZAP70) and the phosphorylation of linker for activation of T cells (LAT). LAT recruits SH2-domain containing leucocyte protein of 76 kDa (SLP-76) which activates Interleukin-2 inducible tyrosine kinase (ITK). Subsequently, phospholipase C gamma 1 (PLC-γ1) creates diacylglycerol (DAG) and inositol triphosphate (IP3) by cleaving phosphatidylinositol triphosphate (PIP2). DAG activates protein kinase C (PKC), IP3 stimulates a calcium release from the endoplasmic reticulum (ER) via the IP3 receptor (IP3R). This calcium release induces the interaction of stromal interaction molecule 1 (STIM1) with CRAC modulator 1 (CRACM1). In addition membrane-bound channels from the transient receptor potential (TRP) family, P2X receptors or voltage-gated calcium-channels (Cav) contribute to calcium mobilization. The resulting calcium elevation activates several signaling proteins such as calcineurin and its target nuclear factor of activated T cells (NFAT), nuclear factor κB (NFκB) or the Ca+2 calmodulin-dependent kinase (CaMK).
Modulation of Calcium Signaling in T Lymphocytes
Calcium homeostasis can to a large part be regulated by mitochondria. Mitochondria can sense changes in the calcium micro domains on proximal ER membranes, leading to an active transport of calcium across the inner mitochondrial membrane by the mitochondrial calcium-uniporter (MCU). They can thereby function as calcium buffers, taking up between 25 and 50% of calcium released from the ER, depending on the specific cell type and CRAC channels (72). This buffering leads to a decreased calcium-mediated inactivation of the IP3-receptor (IP3R) and thereby a greater overall calcium mobilization. Additionally, since this takes place adjacent to the ER, mitochondrial buffering of calcium diminishes the available calcium that can be transported back into the ER by the sarco/endoplasmic reticulum calcium ATPase (SERCA) and thereby prolongs the cytoplasmic elevation of calcium (73).
Furthermore, calcium entry can be regulated be the availability of reactive oxygen species (ROS). It has been shown that an increase in ROS, such as hydrogen peroxide (H2O2), can lead to a dysregulation of the interaction between STIM1 and CRACM1. This is mediated either by a ROS-mediated alteration of cysteine 56 on STIM1, leading to a S-glutathionylation with subsequent oligomerization and translocation toward the plasma membrane or by a CRAC-independent activation of IP3R (74–76). Regardless of the mechanism, this points toward direct effects of ROS levels on the mobilization of calcium.
Calcium as a Potential Therapeutic Target in CNS Autoimmunity
Calcium not only plays a role in the activation of T cells but also their differentiation and regulation. Therapeutic blocking of CRACM1 impaired the ability of T cells to in vitro differentiate into the Th17 subset and reduced the severity of EAE (77). Additionally, selective interference with calcium mobilization via the blocking of CRAC channels or by administration of CRACM1-specific antibodies have shown an inhibition of cytokine production and proliferation of T cells in vitro (78, 79). In MS patients, calcium-modulation has mainly played a role in symptomatic therapy. Baclofen, a specific agonist to GABA B receptors supresses' neuronal activity by indirectly inhibiting postsynaptic potentials with an increase in intracellular calcium levels. Additionally, baclofen acts directly on voltage-gated calcium channels and blocks the influx of calcium, a mechanism that has also been described for pregabalin, an agent prescribed for the relief of neuropathic pain (80, 81). Consequently, this inhibition of voltage-gated calcium-channels diminishes the amount of available neurotransmitters in the synaptic cleft. When tested in an animal model of MS, pregabalin reduced the calcium-mediated neuronal cytotoxicity and neuronal damage (82). A similar attenuation of EAE was observed when mice were treated with the calcium antagonist nimodipine. Additional to the direct and calcium-mediated clinical and histological effects, nimodipine induced microglial apoptosis in a calcium-independent manner, resulting in lowered levels of nitric oxide (NO) and an increase in oligodendrocytes promoting remyelination (83). Another calcium-targeting drug is dantrolene, an anti-spastic agent that inhibits the calcium-release from the ER by blocking ryanodine receptors (84). Blocking voltage-gated calcium-channels in EAE by administration of bepridil or nitrendipine significantly ameliorated EAE with reduced spinal cord inflammation and axonal pathology (85) (Figure 2).
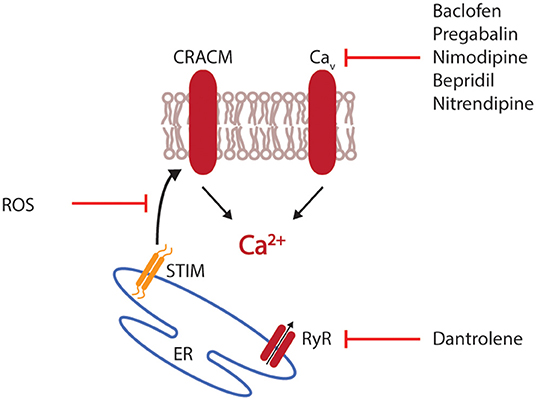
Figure 2. Interference with calcium mobilization. Therapeutic intervention with calcium mobilization can target the initial release of calcium from the endoplasmic reticulum (ER), for example by interfering with the ryanodine receptor (RyR). Alternatively, the interaction of stromal interaction molecule 1 (STIM1) with CRAC modulator 1 (CRACM1) can be disturbed by reactive oxygen species (ROS). Most available drugs target voltage-gated calcium channels (Cav).
Taken together, these animal studies suggest that calcium targeting approaches are not only relevant for the symptomatic treatment of patients with spastic motor function or neuropathic pain but may also reduce neuronal damage and promote remyelination.
In conclusion, physiologically normal levels of vitamin D are safe and most likely beneficial for patients with autoimmune diseases (86). The therapeutic aim of supplementation should be to reach and hold these levels, preferably with moderate supplementation. However, there is also a need for monitoring patients, especially in the initial phase of treatment to prevent a supra-physiological elevation of vitamin D. In addition, patients should be informed about potential risks of combining vitamin D therapy with increased calcium intake, and calcium levels should be monitored as well.
Author Contributions
DH and ST prepared the figures and wrote the manuscript. DH, ST, and MW participated in reviewing and editing the manuscript.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Kamen DL, Cooper GS, Bouali H, Shaftman SR, Hollis BW, Gilkeson GS. Vitamin D deficiency in systemic lupus erythematosus. Autoimmun Rev. (2006) 5:114–7. doi: 10.1016/j.autrev.2005.05.009
2. Hypponen E, Laara E, Reunanen A, Jarvelin MR, Virtanen SM. Intake of vitamin D and risk of type 1 diabetes: a birth-cohort study. Lancet. (2001) 358:1500–3. doi: 10.1016/S0140-6736(01)06580-1
3. Cutolo M, Otsa K, Paolino S, Yprus M, Veldi T, Seriolo B. Vitamin D involvement in rheumatoid arthritis and systemic lupus erythaematosus. Ann Rheum Dis. (2009) 68:446–7. doi: 10.1136/ard.2008.093476
4. Gatenby PA, Lucas RM, Engelsen O, Ponsonby AL, Clements M. Antineutrophil cytoplasmic antibody-associated vasculitides: could geographic patterns be explained by ambient ultraviolet radiation? Arthritis Rheum. (2009) 61:1417–24. doi: 10.1002/art.24790
5. Ascherio A, Munger KL, Simon KC. Vitamin D and multiple sclerosis. Lancet Neurol. (2010) 9:599–612. doi: 10.1016/S1474-4422(10)70086-7
6. Munger KL, Levin LI, Hollis BW, Howard NS, Ascherio A. Serum 25-hydroxyvitamin D levels and risk of multiple sclerosis. JAMA. (2006) 296:2832–8. doi: 10.1001/jama.296.23.2832
7. Smolders J, Menheere P, Kessels A, Damoiseaux J, Hupperts R. Association of vitamin D metabolite levels with relapse rate and disability in multiple sclerosis. Mult Scler. (2008) 14:1220–4. doi: 10.1177/1352458508094399
8. Lemire JM, Archer DC. 1,25-dihydroxyvitamin D3 prevents the in vivo induction of murine experimental autoimmune encephalomyelitis. J Clin Invest. (1991) 87:1103–7. doi: 10.1172/JCI115072
9. Cantorna MT, Hayes CE, Deluca HF. 1,25-Dihydroxyvitamin D3 reversibly blocks the progression of relapsing encephalomyelitis, a model of multiple sclerosis. Proc Natl Acad Sci USA. (1996) 93:7861–4. doi: 10.1073/pnas.93.15.7861
10. Nataf S, Garcion E, Darcy F, Chabannes D, Muller JY, Brachet P. 1,25 Dihydroxyvitamin D3 exerts regional effects in the central nervous system during experimental allergic encephalomyelitis. J Neuropathol Exp Neurol. (1996) 55:904–14. doi: 10.1097/00005072-199608000-00006
11. Nashold FE, Miller DJ, Hayes CE. 1,25-dihydroxyvitamin D3 treatment decreases macrophage accumulation in the CNS of mice with experimental autoimmune encephalomyelitis. J Neuroimmunol. (2000) 103:171–9. doi: 10.1016/S0165-5728(99)00247-7
12. Spach KM, Hayes CE. Vitamin D3 confers protection from autoimmune encephalomyelitis only in female mice. J Immunol. (2005) 175:4119–26. doi: 10.4049/jimmunol.175.6.4119
13. Häusler D, Torke S, Peelen E, Bertsch T, Djukic M, Nau R, et al. High dose vitamin D exacerbates central nervous system autoimmunity by raising T-cell excitatory calcium. Brain. (2019) 142:2737–55. doi: 10.1093/brain/awz190
14. Haussler MR, Haussler CA, Jurutka PW, Thompson PD, Hsieh JC, Remus LS, et al. The vitamin D hormone and its nuclear receptor: molecular actions and disease states. J Endocrinol. (1997) 154(Suppl.):S57–73.
15. Haussler MR, Whitfield GK, Haussler CA, Hsieh JC, Thompson PD, Selznick SH, et al. The nuclear vitamin D receptor: biological and molecular regulatory properties revealed. J Bone Miner Res. (1998) 13:325–49. doi: 10.1359/jbmr.1998.13.3.325
16. Darwish H, Deluca HF. Vitamin D-regulated gene expression. Crit Rev Eukaryot Gene Expr. (1993) 3:89–116.
17. Veldman CM, Cantorna MT, Deluca HF. Expression of 1,25-dihydroxyvitamin D(3) receptor in the immune system. Arch Biochem Biophys. (2000) 374:334–8. doi: 10.1006/abbi.1999.1605
18. Chen S, Sims GP, Chen XX, Gu YY, Chen S, Lipsky PE. Modulatory effects of 1,25-dihydroxyvitamin D3 on human B cell differentiation. J Immunol. (2007) 179:1634–47. doi: 10.4049/jimmunol.179.3.1634
19. Hewison M. Vitamin D and immune function: autocrine, paracrine or endocrine? Scand J Clin Lab Invest Suppl. (2012) 243:92–102. doi: 10.3109/00365513.2012.682862
20. Bhalla AK, Amento EP, Clemens TL, Holick MF, Krane SM. Specific high-affinity receptors for 1,25-dihydroxyvitamin D3 in human peripheral blood mononuclear cells: presence in monocytes and induction in T lymphocytes following activation. J Clin Endocrinol Metab. (1983) 57:1308–10. doi: 10.1210/jcem-57-6-1308
21. Clemens TL, Garrett KP, Zhou XY, Pike JW, Haussler MR, Dempster DW. Immunocytochemical localization of the 1,25-dihydroxyvitamin D3 receptor in target cells. Endocrinology. (1988) 122:1224–30. doi: 10.1210/endo-122-4-1224
22. Sutherland MK, Somerville MJ, Yoong LK, Bergeron C, Haussler MR, Mclachlan DR. Reduction of vitamin D hormone receptor mRNA levels in Alzheimer as compared to Huntington hippocampus: correlation with calbindin-28k mRNA levels. Brain Res Mol Brain Res. (1992) 13:239–50. doi: 10.1016/0169-328X(92)90032-7
23. Neveu I, Naveilhan P, Jehan F, Baudet C, Wion D, De Luca HF, et al. 1,25-dihydroxyvitamin D3 regulates the synthesis of nerve growth factor in primary cultures of glial cells. Brain Res Mol Brain Res. (1994) 24:70–6. doi: 10.1016/0169-328X(94)90119-8
24. Johnson JA, Grande JP, Windebank AJ, Kumar R. 1,25-Dihydroxyvitamin D(3) receptors in developing dorsal root ganglia of fetal rats. Brain Res Dev Brain Res. (1996) 92:120–4. doi: 10.1016/0165-3806(95)00204-9
25. Cornet A, Baudet C, Neveu I, Baron-Van Evercooren A, Brachet P, Naveilhan P. 1,25-Dihydroxyvitamin D3 regulates the expression of VDR and NGF gene in Schwann cells in vitro. J Neurosci Res. (1998) 53:742–6. doi: 10.1002/(SICI)1097-4547(19980915)53:6<742::AID-JNR11>3.0.CO;2-#
26. Veenstra TD, Prufer K, Koenigsberger C, Brimijoin SW, Grande JP, Kumar R. 1,25-Dihydroxyvitamin D3 receptors in the central nervous system of the rat embryo. Brain Res. (1998) 804:193–205. doi: 10.1016/S0006-8993(98)00565-4
27. Prufer K, Veenstra TD, Jirikowski GF, Kumar R. Distribution of 1,25-dihydroxyvitamin D3 receptor immunoreactivity in the rat brain and spinal cord. J Chem Neuroanat. (1999) 16:135–45. doi: 10.1016/S0891-0618(99)00002-2
28. Baas D, Prufer K, Ittel ME, Kuchler-Bopp S, Labourdette G, Sarlieve LL, et al. Rat oligodendrocytes express the vitamin D(3) receptor and respond to 1,25-dihydroxyvitamin D(3). Glia. (2000) 31:59–68. doi: 10.1002/(SICI)1098-1136(200007)31:1<59::AID-GLIA60>3.0.CO;2-Y
29. Langub MC, Herman JP, Malluche HH, Koszewski NJ. Evidence of functional vitamin D receptors in rat hippocampus. Neuroscience. (2001) 104:49–56. doi: 10.1016/S0306-4522(01)00049-5
30. Provvedini DM, Tsoukas CD, Deftos LJ, Manolagas SC. 1,25-dihydroxyvitamin D3 receptors in human leukocytes. Science. (1983) 221:1181–3. doi: 10.1126/science.6310748
31. Provvedini DM, Tsoukas CD, Deftos LJ, Manolagas SC. 1 alpha,25-Dihydroxyvitamin D3-binding macromolecules in human B lymphocytes: effects on immunoglobulin production. J Immunol. (1986) 136:2734–40.
32. Nashold FE, Hoag KA, Goverman J, Hayes CE. Rag-1-dependent cells are necessary for 1,25-dihydroxyvitamin D(3) prevention of experimental autoimmune encephalomyelitis. J Neuroimmunol. (2001) 119:16–29. doi: 10.1016/S0165-5728(01)00360-5
33. Chang SH, Chung Y, Dong C. Vitamin D suppresses Th17 cytokine production by inducing C/EBP homologous protein (CHOP) expression. J Biol Chem. (2010) 285:38751–5. doi: 10.1074/jbc.C110.185777
34. Palmer MT, Lee YK, Maynard CL, Oliver JR, Bikle DD, Jetten AM, et al. Lineage-specific effects of 1,25-dihydroxyvitamin D(3) on the development of effector CD4 T cells. J Biol Chem. (2011) 286:997–1004. doi: 10.1074/jbc.M110.163790
35. Barrat FJ, Cua DJ, Boonstra A, Richards DF, Crain C, Savelkoul HF, et al. In vitro generation of interleukin 10-producing regulatory CD4(+) T cells is induced by immunosuppressive drugs and inhibited by T helper type 1 (Th1)- and Th2-inducing cytokines. J Exp Med. (2002) 195:603–16. doi: 10.1084/jem.20011629
36. Correale J, Ysrraelit MC, Gaitan MI. Immunomodulatory effects of vitamin D in multiple sclerosis. Brain. (2009) 132:1146–60. doi: 10.1093/brain/awp033
37. Kampman MT, Steffensen LH, Mellgren SI, Jorgensen L. Effect of vitamin D3 supplementation on relapses, disease progression, and measures of function in persons with multiple sclerosis: exploratory outcomes from a double-blind randomised controlled trial. Mult Scler. (2012) 18:1144–51. doi: 10.1177/1352458511434607
38. Mahon BD, Gordon SA, Cruz J, Cosman F, Cantorna MT. Cytokine profile in patients with multiple sclerosis following vitamin D supplementation. J Neuroimmunol. (2003) 134:128–32. doi: 10.1016/S0165-5728(02)00396-X
39. Soilu-Hanninen M, Aivo J, Lindstrom BM, Elovaara I, Sumelahti ML, Farkkila M, et al. A randomised, double blind, placebo controlled trial with vitamin D3 as an add on treatment to interferon beta-1b in patients with multiple sclerosis. J Neurol Neurosurg Psychiatry. (2012) 83:565–71. doi: 10.1136/jnnp-2011-301876
40. Golan D, Halhal B, Glass-Marmor L, Staun-Ram E, Rozenberg O, Lavi I, et al. Vitamin D supplementation for patients with multiple sclerosis treated with interferon-beta: a randomized controlled trial assessing the effect on flu-like symptoms and immunomodulatory properties. BMC Neurol. (2013) 13:60. doi: 10.1186/1471-2377-13-60
41. Burton JM, Kimball S, Vieth R, Bar-Or A, Dosch HM, Cheung R, et al. A phase I/II dose-escalation trial of vitamin D3 and calcium in multiple sclerosis. Neurology. (2010) 74:1852–9. doi: 10.1212/WNL.0b013e3181e1cec2
42. Hupperts R, Smolders J, Vieth R, Holmoy T, Marhardt K, Schluep M, et al. Randomized trial of daily high-dose vitamin D3 in patients with RRMS receiving subcutaneous interferon beta-1a. Neurology. (2019) 93:e1906–16. doi: 10.1212/WNL.0000000000008445
43. Camu W, Lehert P, Pierrot-Deseilligny C, Hautecoeur P, Besserve A, Jean Deleglise AS, et al. Cholecalciferol in relapsing-remitting MS: a randomized clinical trial (CHOLINE). Neurol Neuroimmunol Neuroinflamm. (2019) 6:e597–603. doi: 10.1212/NXI.0000000000000597
44. Stein MS, Liu Y, Gray OM, Baker JE, Kolbe SC, Ditchfield MR, et al. A randomized trial of high-dose vitamin D2 in relapsing-remitting multiple sclerosis. Neurology. (2011) 77:1611–8. doi: 10.1212/WNL.0b013e3182343274
45. Mclaughlin L, Clarke L, Khalilidehkordi E, Butzkueven H, Taylor B, Broadley SA. Vitamin D for the treatment of multiple sclerosis: a meta-analysis. J Neurol. (2018) 265:2893–905. doi: 10.1007/s00415-018-9074-6
46. Fragoso YD, Adoni T, Damasceno A, De Albuquerque Damasceno CA, Ferreira ML, Finkelzstejn A, et al. Unfavorable outcomes during treatment of multiple sclerosis with high doses of vitamin D. J Neurol Sci. (2014) 346:341–2. doi: 10.1016/j.jns.2014.08.019
47. Jorde R, Sollid ST, Svartberg J, Schirmer H, Joakimsen RM, Njolstad I, et al. Vitamin D 20,000 IU per week for 5 years does not prevent progression from prediabetes to diabetes. J Clin Endocrinol Metab. (2016) 101:1647–55. doi: 10.1210/jc.2015-4013
48. Steffensen LH, Jørgensen L, Straume B, Mellgren SI, Kampman MT. Can vitamin D3 supplementation prevent bone loss in persons with MS? A placebo-controlled trial. J Neurol. (2011) 258:1624–31. doi: 10.1007/s00415-011-5980-6
49. Lehouck A, Mathieu C, Carremans C, Baeke F, Verhaegen J, Van Eldere J, et al. High doses of vitamin D to reduce exacerbations in chronic obstructive pulmonary disease: a randomized trial. Ann Intern Med. (2012) 156:105–14. doi: 10.7326/0003-4819-156-2-201201170-00004
50. Marcus JF, Shalev SM, Harris CA, Goodin DS, Josephson SA. Severe hypercalcemia following vitamin D supplementation in a patient with multiple sclerosis: a note of caution. Arch Neurol. (2012) 69:129–32. doi: 10.1001/archneurol.2011.1199
51. Rolf L, Smolders J, Van Den Ouweland J, Hupperts R, Damoiseaux J. Correlation of different cellular assays to analyze T cell-related cytokine profiles in vitamin D3-supplemented patients with multiple sclerosis. Mol Immunol. (2019) 105:198–204. doi: 10.1016/j.molimm.2018.12.001
52. Smolders J, Peelen E, Thewissen M, Cohen Tervaert JW, Menheere P, Hupperts R, et al. Safety and T cell modulating effects of high dose vitamin D3 supplementation in multiple sclerosis. PLoS ONE. (2010) 5:e15235. doi: 10.1371/journal.pone.0015235
53. Zittermann A, Ernst JB, Prokop S, Fuchs U, Dreier J, Kuhn J, et al. Effect of vitamin D on all-cause mortality in heart failure (EVITA): a 3-year randomized clinical trial with 4,000 IU vitamin D daily. Eur Heart J. (2017) 38:2279–86. doi: 10.1093/eurheartj/ehx235
54. Barragan M, Good M, Kolls JK. Regulation of dendritic cell function by vitamin D. Nutrients. (2015) 7:8127–51. doi: 10.3390/nu7095383
55. Malihi Z, Wu Z, Stewart AW, Lawes CM, Scragg R. Hypercalcemia, hypercalciuria, and kidney stones in long-term studies of vitamin D supplementation: a systematic review and meta-analysis. Am J Clin Nutr. (2016) 104:1039–51. doi: 10.3945/ajcn.116.134981
56. Malihi Z, Wu Z, Lawes CMM, Scragg R. Adverse events from large dose vitamin D supplementation taken for one year or longer. J Steroid Biochem Mol Biol. (2019) 188:29–37. doi: 10.1016/j.jsbmb.2018.12.002
57. Jansen TL, Janssen M, De Jong AJ. Severe hypercalcaemia syndrome with daily low-dose vitamin D supplementation. Br J Rheumatol. (1997) 36:712–3. doi: 10.1093/rheumatology/36.6.712
58. Avenell A, Mak JC, O'connell D. Vitamin D and vitamin D analogues for preventing fractures in post-menopausal women and older men. Cochrane Database Syst Rev. (2014) 4:CD000227. doi: 10.1002/14651858.CD000227.pub4
59. Bjelakovic G, Gluud LL, Nikolova D, Whitfield K, Wetterslev J, Simonetti RG, et al. Vitamin D supplementation for prevention of mortality in adults. Cochrane Database Syst Rev. (2014) 1:CD007470. doi: 10.1002/14651858.CD007470.pub3
60. Jones G. Pharmacokinetics of vitamin D toxicity. Am J Clin Nutr. (2008) 88:582S−6S. doi: 10.1093/ajcn/88.2.582S
61. Zittermann A, Prokop S, Gummert JF, Borgermann J. Safety issues of vitamin D supplementation. Anticancer Agents Med Chem. (2013) 13:4–10. doi: 10.2174/187152013804487290
62. Häusler D, Torke S, Peelen E, Bertsch T, Weber M, Heilmann M, et al. Reply: neither human nor mouse is hypercalcaemic with 250 nmol/l 25-hydroxyvitamin D. Brain. (2019) 143:e10. doi: 10.1093/brain/awz389
63. Cantorna MT, Humpal-Winter J, Deluca HF. Dietary calcium is a major factor in 1,25-dihydroxycholecalciferol suppression of experimental autoimmune encephalomyelitis in mice. J Nutr. (1999) 129:1966–71. doi: 10.1093/jn/129.11.1966
64. Feske S, Gwack Y, Prakriya M, Srikanth S, Puppel SH, Tanasa B, et al. A mutation in Orai1 causes immune deficiency by abrogating CRAC channel function. Nature. (2006) 441:179–85. doi: 10.1038/nature04702
65. Feske S, Picard C, Fischer A. Immunodeficiency due to mutations in ORAI1 and STIM1. Clin Immunol. (2010) 135:169–82. doi: 10.1016/j.clim.2010.01.011
66. Courtney AH, Lo WL, Weiss A. TCR signaling: mechanisms of initiation and propagation. Trends Biochem Sci. (2018) 43:108–23. doi: 10.1016/j.tibs.2017.11.008
67. Gaud G, Lesourne R, Love PE. Regulatory mechanisms in T cell receptor signaling. Nat Rev Immunol. (2018) 18:485–97. doi: 10.1038/s41577-018-0020-8
68. Muro R, Takayanagi H, Nitta T. T cell receptor signaling for gammadeltaT cell development. Inflamm Regen. (2019) 39:6. doi: 10.1186/s41232-019-0095-z
69. Vig M, Kinet JP. Calcium signaling in immune cells. Nat Immunol. (2009) 10:21–7. doi: 10.1038/ni.f.220
70. Oh-Hora M, Rao A. Calcium signaling in lymphocytes. Curr Opin Immunol. (2008) 20:250–8. doi: 10.1016/j.coi.2008.04.004
71. Toldi G. The regulation of calcium homeostasis in T lymphocytes. Front Immunol. (2013) 4:432. doi: 10.3389/fimmu.2013.00432
72. Pacher P, Csordas P, Schneider T, Hajnoczky G. Quantification of calcium signal transmission from sarco-endoplasmic reticulum to the mitochondria. J Physiol. (2000) 529:553–64. doi: 10.1111/j.1469-7793.2000.00553.x
73. Fracchia KM, Pai CY, Walsh CM. Modulation of T cell metabolism and function through calcium signaling. Front Immunol. (2013) 4:324. doi: 10.3389/fimmu.2013.00324
74. Bogeski I, Kummerow C, Al-Ansary D, Schwarz EC, Koehler R, Kozai D, et al. Differential redox regulation of ORAI ion channels: a mechanism to tune cellular calcium signaling. Sci Signal. (2010) 3:ra24. doi: 10.1126/scisignal.2000672
75. Grupe M, Myers G, Penner R, Fleig A. Activation of store-operated I(CRAC) by hydrogen peroxide. Cell Calcium. (2010) 48:1–9. doi: 10.1016/j.ceca.2010.05.005
76. Hawkins BJ, Irrinki KM, Mallilankaraman K, Lien YC, Wang Y, Bhanumathy CD, et al. S-glutathionylation activates STIM1 and alters mitochondrial homeostasis. J Cell Biol. (2010) 190:391–405. doi: 10.1083/jcb.201004152
77. Kim KD, Srikanth S, Tan YV, Yee MK, Jew M, Damoiseaux R, et al. Calcium signaling via Orai1 is essential for induction of the nuclear orphan receptor pathway to drive Th17 differentiation. J Immunol. (2014) 192:110–22. doi: 10.4049/jimmunol.1302586
78. Chen G, Panicker S, Lau KY, Apparsundaram S, Patel VA, Chen SL, et al. Characterization of a novel CRAC inhibitor that potently blocks human T cell activation and effector functions. Mol Immunol. (2013) 54:355–67. doi: 10.1016/j.molimm.2012.12.011
79. Cox JH, Hussell S, Sondergaard H, Roepstorff K, Bui JV, Deer JR, et al. Antibody-mediated targeting of the Orai1 calcium channel inhibits T cell function. PLoS ONE. (2013) 8:e82944. doi: 10.1371/journal.pone.0082944
80. Takeuchi H. Sensitivities of Achatina giant neurones to putative amino acid neurotransmitters. Comp Biochem Physiol C. (1992) 103:1–12. doi: 10.1016/0742-8413(92)90219-W
81. Vetulani J. Drug addiction. Part III. Pharmacotherapy of addiction. Pol J Pharmacol. (2001) 53:415–34.
82. Hundehege P, Fernandez-Orth J, Romer P, Ruck T, Muntefering T, Eichler S, et al. Targeting voltage-dependent calcium channels with pregabalin exerts a direct neuroprotective effect in an animal model of multiple sclerosis. Neurosignals. (2018) 26:77–93. doi: 10.1159/000495425
83. Schampel A, Volovitch O, Koeniger T, Scholz CJ, Jorg S, Linker RA, et al. Nimodipine fosters remyelination in a mouse model of multiple sclerosis and induces microglia-specific apoptosis. Proc Natl Acad Sci USA. (2017) 114:E3295–304. doi: 10.1073/pnas.1620052114
84. Davidoff RA. Antispasticity drugs: mechanisms of action. Ann Neurol. (1985) 17:107–16. doi: 10.1002/ana.410170202
85. Brand-Schieber E, Werner P. Calcium channel blockers ameliorate disease in a mouse model of multiple sclerosis. Exp Neurol. (2004) 189:5–9. doi: 10.1016/j.expneurol.2004.05.023
Keywords: T cells, calcium, hypercalemia, vitamin D, multiple sclerosis, neuroimmunology
Citation: Häusler D, Torke S and Weber MS (2020) High-Dose Vitamin D-Mediated Hypercalcemia as a Potential Risk Factor in Central Nervous System Demyelinating Disease. Front. Immunol. 11:301. doi: 10.3389/fimmu.2020.00301
Received: 17 December 2019; Accepted: 06 February 2020;
Published: 25 February 2020.
Edited by:
Joost Smolders, Erasmus Medical Center, NetherlandsReviewed by:
Armin Zittermann, Heart and Diabetes Center North Rhine-Westphalia, GermanyTrygve Holmøy, Akershus University Hospital, Norway
Copyright © 2020 Häusler, Torke and Weber. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Martin S. Weber, bWFydGluLndlYmVyJiN4MDAwNDA7bWVkLnVuaS1nb2V0dGluZ2VuLmRl
†These authors have contributed equally to this work