 Aviad Ben-Shmuel
Aviad Ben-Shmuel Guy Biber
Guy Biber Mira Barda-Saad
Mira Barda-Saad- Laboratory of Molecular and Applied Immunology, The Mina and Everard Goodman Faculty of Life Sciences, Bar-Ilan University, Ramat Gan, Israel
The emergence of immunotherapy for cancer treatment bears considerable clinical promise. Nevertheless, many patients remain unresponsive, acquire resistance, or suffer dose-limiting toxicities. Immune-editing of tumors assists their escape from the immune system, and the tumor microenvironment (TME) induces immune suppression through multiple mechanisms. Immunotherapy aims to bolster the activity of immune cells against cancer by targeting these suppressive immunomodulatory processes. Natural Killer (NK) cells are a heterogeneous subset of immune cells, which express a diverse array of activating and inhibitory germline-encoded receptors, and are thus capable of directly targeting and killing cancer cells without the need for MHC specificity. Furthermore, they play a critical role in triggering the adaptive immune response. Enhancing the function of NK cells in the context of cancer is therefore a promising avenue for immunotherapy. Different NK-based therapies have been evaluated in clinical trials, and some have demonstrated clinical benefits, especially in the context of hematological malignancies. Solid tumors remain much more difficult to treat, and the time point and means of intervention of current NK-based treatments still require optimization to achieve long term effects. Here, we review recently described mechanisms of cancer evasion from NK cell immune surveillance, and the therapeutic approaches that aim to potentiate NK function. Specific focus is placed on the use of specialized monoclonal antibodies against moieties on the cancer cell, or on both the tumor and the NK cell. In addition, we highlight newly identified mechanisms that inhibit NK cell activity in the TME, and describe how biochemical modifications of the TME can synergize with current treatments and increase susceptibility to NK cell activity.
Introduction
NK Cells and Cancer
In recent years, the field of immunotherapy has emerged as one of the most promising approaches for treating cancer (1). Though most immunotherapies have traditionally focused on T-cells, NK cell-based therapies are rapidly emerging in research and in the clinic (2, 3).
NK cells are cytotoxic innate lymphoid cells (ILCs), which can target and eliminate cancer cells through secretion of cytolytic granules, and trigger an immune response via secretion of immunomodulatory cytokines (4). In contrast to T- and B-cells, NK cells express a multitude of intrinsic germline-encoded activating and inhibiting membrane receptors, and therefore do not require antigen specificity (5, 6). Immune-editing of the tumor promotes evasion from the anti-tumor immune response; a common remodeling event is downregulation of β2-microglobulin, which leads to reduced MHC presentation. The selective pressure leading to low MHC presentation impairs T-cell anti-tumor activity (7). NK cell function is therefore partially complementary to T-cells, as they can target and lyse MHC-I deficient cells, in a process known as “missing-self recognition” (8, 9). In addition to missing-self recognition, activation of NK cells relies on the equilibrium between activating and inhibitory signals derived from surface receptors engaged with cognate ligands on target cells (10, 11). Central activating and co-activating NK cell receptors include the natural cytotoxicity receptors (NCRs) NKp46, NKp30, and NKp44, CD16, NKG2D, NKG2C, DNAX Accessory Molecule-1 (DNAM-1), and 2B4 (12, 13). In parallel, the important inhibitory receptors on NK cells engage with MHC-I ligands to down modulate the NK cell response, and these include the Killer-cell immunoglobulin-like receptors (KIRs), and the CD94/NKG2A heterodimer (12). NK cells express additional checkpoint inhibitory receptors, which play important roles in constraining their activity when engaged with cognate ligands, as will be discussed below.
Various approaches have been developed to bolster NK cell activity against cancer, some of which are being utilized in pre-clinical and clinical trials (3, 14). Significant hurdles still persist, however, for immunotherapeutic treatments in general, and for NK cells in particular. These include concerns regarding potential autoimmune cytotoxicity for therapies such as cytokine administration and immune checkpoint inhibitors, the magnitude of the patient response to treatment, and patient relapse due to innate or acquired resistance (15–18). Moreover, though some NK cell based treatments have shown promising results for hematological malignancies, NK cells generally have low capacity to infiltrate solid tumors, and so far, the efficacy against advanced cancers and solid tumors remains relatively low (19). Cytolytic immune cells such as NK cells and CD8+ T-cells are also suppressed through multiple pathways in the tumor microenvironment (TME) (20, 21).
Recent comprehensive reviews provide detailed descriptions and delineate the obstacles remaining in multiple NK-based treatments that have been developed and tested in pre-clinical and clinical trials. These reviews cover topics including cytokine therapy, hematopoietic stem cell transplantation (HSCT), adoptive cell transfer, and CAR-NK therapy (2, 3, 22–25). Here, we summarize recent pre-clinical and clinical evidence regarding NK cell expression of checkpoint molecules and some of the most recent NK-based immunotherapeutic strategies. These approaches include targeting NK surface receptors, NK cell ligands on tumors, and identification and modulation of pathways in the TME for sensitization to NK cell activity. We highlight newly developed technologies that may increase NK cell activation, infiltration, survival, and proliferation.
Triggering of Adaptive Immunity and Cancer Immune Surveillance by Subsets of NK Cells
Besides their potential for direct elimination of cancer cells, the crosstalk between NK cells and additional immune cells such as T-cells and dendritic cells (DCs) in the TME initiates potent anti-tumor effects. This interplay between NK cells and DCs can promote DC uptake of tumor antigens in secondary lymph nodes for presentation to and activation of T-cells, which facilitate additional anti-tumor responses (23). Recently, it was shown that NK cells recruit conventional type 1 DCs through secretion of the chemo attractants CCL5, XCL1, and XCL2, and the extent of this process correlates with cancer patient survival (26). Furthermore, production of the cytokine FLT3L by NK cells increased the frequency of conventional type 1 DCs in tumors, and frequencies of NK cells and conventional type 1 DCs correlate with responses to anti-PD-1 therapy (27). Moreover, in a recently established murine model of lung adenocarcinoma, expression of NK cell ligands on tumors and their activation facilitated the adaptive response of tumor-specific T-cells and reduction in tumor growth (28). These studies and others (29, 30) illustrate some of the indirect roles NK cells may play in mediating the immune response in the TME, warranting development of novel NK-based immunotherapies.
NK cells are not a uniform immune cell subset, but rather very heterogeneous, and can be segregated into subsets with distinct tissue residency, phenotypes, and functional activity. NK cells are traditionally classified into the CD56bright CD16dim subset and CD56dim CD16bright subset. CD56bright CD16dim cells are generally found in certain peripheral tissues (such as lymph nodes, gut, tonsils, uterus, and skin) and considered immature, with low KIR expression and high expression of CD94/NKG2A; these cells primarily secrete chemokines and cytokines such as IFNγ and TNFα (13, 31, 32). The CD56dim CD16bright subset is the major subset in peripheral blood and is considered terminally differentiated with high KIR expression; these cells exert their cytotoxic activity through secretion of perforin and granzyme (33). Over the last few years, additional NK cell subpopulations with distinct phenotypic expression profiles have been further identified and classified in different tissues (34) and in both solid and hematological malignancies (35). In addition to these findings, the characterization of ILCs over the past decade has demonstrated overlapping surface marker expression between NK cells and other ILC subsets in certain tissues (such as ILC1 and 3) (36). ILC1s, which have poor cytotoxic capacity but potent cytokine secretion share multiple common markers with NK cells, including NKp46 (37). Therefore, due to the similarity between ILCs and NK cell subtypes, their classification has considerable importance in the prism of immunotherapy for the specificity of treatment. Recently, it was shown that NK cells can be converted into ILC1s in the TME by a TGF-β dependent mechanism, and these converted ILC1s lose the capacity to control tumor growth and dissemination, and may promote tumorigenicity (38). The plasticity of NK cells in response to TGF-β is further exemplified by evidence of their conversion to decidual NK-like cells in the TME, which promote angiogenesis (39). The heterogeneity and plasticity of NK subsets adds an additional layer of complexity to treatment, yet it also opens doors toward fine tuning and enhancing the NK-based response; in the case of TGF-β mediated conversion, targeting TGF-β pathways (as discussed below) or identifying the intracellular pathways leading to NK cell conversion can offer new therapeutic opportunities. Further identification of NK cell/ILC markers and phenotypes, as well as molecular mechanisms regulating their effector function during tissue homeostasis or disease should greatly contribute to future therapeutic efficacy.
The Tumor Microenvironment Restrains NK Cell Activity
The extent of NK cell infiltration into solid tumors is an intensively debated subject, though there is consensus that such infiltration may contribute to improved clinical prognosis (40–42). Though some tumors, such as colorectal cancers, may almost completely exclude NK cell subsets (43), others such as breast, renal, lung, and head and neck squamous cell carcinoma (HNSCC) tumors are infiltrated by NK cells with possible clinical benefit (44–47). Poor NK cell homing and infiltration to the TME may be attributed to interference with NK chemotactic signaling and activation, and physical properties of the tumor bed such as vasculature density and ECM composition (40, 48–50).
Inside the tumor, immune cells are confronted with a suppressive milieu. The TME harbors an assortment of cell types that down modulate the immune response, including stromal cells, fibroblasts, regulatory T-cells (Tregs), and myeloid derived suppressor cells (MDSCs) (51, 52) which can induce specific changes in cytotoxic lymphocyte phenotype, metabolic program, transcriptional profile, and epigenetic profile (21, 53, 54). The TME induces inhibitory effects on immune cells through a variety of processes, specifically chronic immune cell activation and an inflammatory microenvironment, secretion of immunomodulatory cytokines and soluble factors, induction of hypoxia, and upregulation of inhibitory checkpoint ligands, which down-regulate immune cell activation when engaged with immune checkpoint receptors, such as Programmed cell death protein 1 (PD-1) and cytotoxic T-lymphocyte-associated protein 4 (CTLA-4) (51, 55). For example, MDSCs (which include myeloid progenitor cells and immature mononuclear cells) are recruited to the TME and can induce immune cell down-modulation through production of arginase, nitric oxide, TGF-β, and IL-10 (56). Tumor-derived macrophages are also recruited to the TME, and can suppress immune cells via secretion of TGF-β and IL-10. They can also recruit Tregs, which have immune-suppressive properties (57, 58). Tregs-enriched in the TME can inhibit CD8+ T-cells and NK cells directly or via secretion of TGF-β, and IL-10 (59, 60), inducing upregulation of inhibitory checkpoint receptors (61). Although IL-10 has immunosuppressive effects on NK cells, it has pleotropic effects on the regulation of tumor-promoting inflammation, and thus has been tested in clinical trials to augment CD8+ T-cell function against cancer (62). Therefore, it appears that multiple immunosuppressive pathways in the TME collaborate in the down-modulation of immune cell activity.
Exhausted T-cells demonstrate impaired effector functions including reduced cytotoxicity and cytokine secretion, impaired proliferation, reduced responses to cytokine stimulation, upregulation of inhibitory checkpoint receptors, and may ultimately undergo apoptosis (63–68). NK cells also undergo functional exhaustion when exposed to the conditions prevalent in the TME. These phenotypes in NK cells are induced by high expression of checkpoint ligands on tumors (such as Programmed death-ligand 1 (PD-L1) and HLA-E) (69, 70), inhibitory cytokines and inhibitory soluble factors (such as TGF-β and IL-10) (71–73), hypoxia (74, 75), exposure to tumor suppressor cells (i.e., Tregs, tumor associated macrophages, and MDSCs) (76–78), and sustained chronic activation (such as sustained activation through the activating NKG2D receptor) (79, 80).
Checkpoint Receptors Modulating NK Cell Activity
Exhausted NK cells share some phenotypes with exhausted CD8+ T-cells, namely downregulation of effector cytokines such as IFN-γ, impaired degranulation and cytotoxicity, downregulation of activating receptors such as NKG2D, upregulation of inhibitory receptors such as NKG2A, and transcriptional changes that promote exhaustion, i.e., downregulation of transcription factors including Eomesodermin and T-bet (20, 81–86). Recent studies further characterized inhibitory checkpoint receptors that promote NK cell dysfunction during malignancy. These studies show that NK cells, in addition to T-cells, can contribute to the efficacy of inhibitory checkpoint inhibition (ICI) through direct or indirect anti-tumor immunity. Therefore, understanding which checkpoint receptor is upregulated on specific NK subsets, and how such upregulation modulates the cell's capacity to generate an immune response, provides valuable information for treatment and combinatorial strategies. Table 1 lists some of the markers found on NK cells that may act as inhibitory checkpoint molecules.
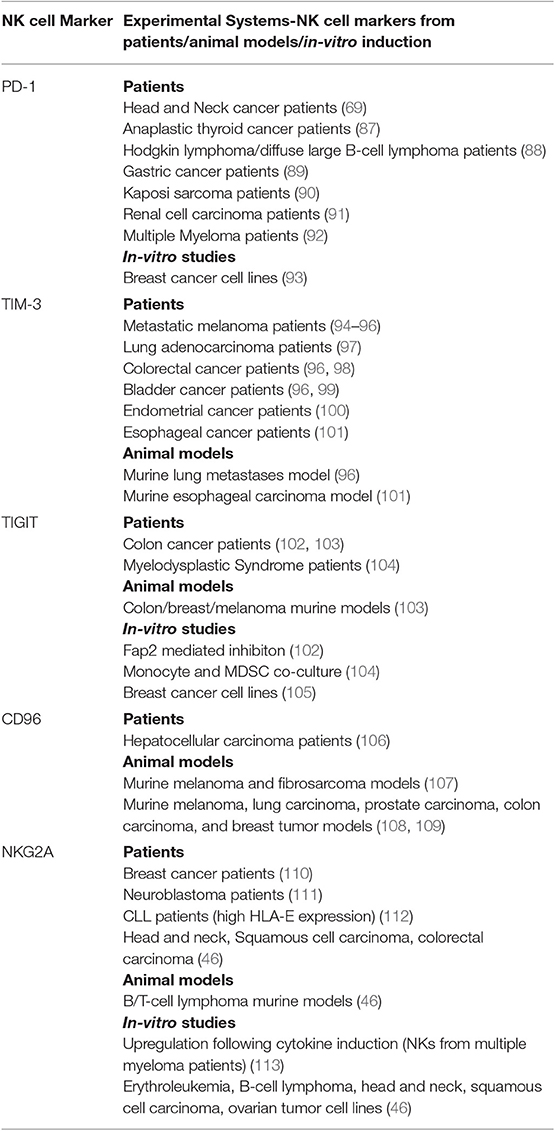
Table 1. NK cell inhibitory checkpoint receptors: Expression on NK cells and modulation of NK cell effector functions in different cancers-evidence from patients, animal models, and in-vitro studies.
PD-1
PD-1 is an inhibitory checkpoint molecule expressed by activated T-cells (114, 115), and was also shown to be expressed on NK cells (116, 117). It marks CD56dimNKG2A−KIR+CD57+ mature NK cells from Human Cytomegalovirus (HCMV) seropositive subjects (117), and may indicate an exhausted NK cell subset with memory-like features (118). PD-1 expression on NK cells is upregulated in several cancers, including head and neck cancer (69), thyroid cancer (87), Hodgkin lymphoma (HL) (88), digestive cancers (esophageal, liver, colorectal, gastric and biliary) (89), breast cancer (93), NK/T cell lymphomas (119), Kaposi sarcoma (90), renal cell carcinoma (91), and multiple myeloma (92). Such upregulated expression of PD-1 by NK cells in the TME is associated with the down-modulation of NK cell activity, manifested by reductions in cytotoxicity, cytokine secretion (e.g., IFN-γ, TNF-α, and GM-CSF), and proliferation (20). PD-1 blockade can unleash T-cells against PD-L1-expressing tumors; however, MHC-I loss on the tumor surface can impact the efficacy of treatment. Therefore, contribution of NK cells also appears important in PD-1 blockade, especially in the context of MHC-I loss on tumors. Indeed, PD-1/PD-L1 blockade in mice bearing PD-L1+ MHC-I− tumors demonstrated the importance of NK cells for the efficacy of these treatments (120). Interestingly, some PD-L1 negative tumors respond to anti-PD-L1 therapy, and a recent study demonstrated that this effect may be mediated by PD-L1+ NK cells. PD-L1+ NK cells treated with anti-PD-L1 showed enhanced activation and effector function, possibly identifying a novel biomarker of the NK PD-L1+ subset for immunotherapy (121).
TIM-3
Activation of T-cell immunoglobulin and mucin-domain containing-3 (TIM-3) by antibody cross-linking initially showed significant decrease of NK cell function (122), and its expression marks mature and exhausted NK cells (122). TIM-3+ NK cells isolated from peripheral blood of metastatic melanoma patients are functionally exhausted, and inhibitory antibodies against TIM-3 can reverse this NK cell dysfunction (94, 95). Higher expression of TIM-3+ NK cells is also apparent in lung adenocarcinoma with lymph node metastases at the progressive tumor stage, and is correlated with decreased patient survival (97). Here, as well, blocking TIM-3 with antibodies increased NK cell cytotoxicity and cytokine secretion. Additional recent studies identified TIM-3 expression as a marker of NK cell dysfunction and disease severity in colorectal cancer, esophageal cancer, endometrial cancer, and bladder cancer (96, 98–101). Interestingly, TIM-3 engagement was initially shown to increase the expression of IFN-γ by NK cells in response to galectin-9, the β-galactoside binding lectin (123). Since TIM-3 can bind additional ligands [such as phosphatidylserine and CEA-related cell adhesion molecule-1 (124, 125)], its activity, whether stimulatory or repressive, may be ligand dependent. Therefore, additional characterization of TIM-3 ligands and their downstream signaling mechanisms is needed for a better understanding of its targeting in immunotherapy. TIM-3 upregulation may also be associated with adaptive resistance to PD-1 blockade, emphasizing its importance as an alternative checkpoint, which should be considered in patients with anti-PD-1 resistant tumors (126). Therefore, anti-TIM-3 monoclonal antibodies such as TSR-022 (NCT02817633, NCT03680508) and Sym023 (NCT03311412) are under evaluation in clinical trials in combination with anti-PD-1 therapy for advanced solid tumors.
TIGIT
T cell immunoreceptor with Ig and ITIM domains (TIGIT) is a suppressive receptor in T-cells (127) that was recently shown to contribute to NK cell dysfunction in cancer. TIGIT is expressed on activated human NK cells, competing for the DNAM-1 ligands PVR/CD155 and PVRL2/CD112 (128) and thereby downregulating NK cell activation. Recent studies demonstrated that blockade of TIGIT significantly enhances anti-tumor NK cell activity against breast cancer and endometrial cancer (100, 105), and can bypass MDSC-mediated supression (104). Interestingly, NK cell inhibition through the TIGIT receptor in the tumor microenvironment may also contribute to carcinogenesis by binding to inhibitory ligands of the onco-bacteria, Fusobacterium nucleatum (FN) (102). The Fap2 protein expressed on FN directly binds TIGIT on NK cells in colon adenocarcinoma tumors, reducing NK cell activity and enabling tumor evasion. Though TIGIT clearly suppresses NK cell activity, the role it plays in maintaining NK cell tolerance in cancer was not fully understood until recently. Zhang et al. revealed that TIGIT expressing NK cells are a functionally exhausted subset in colon cancer and mouse models of melanoma and breast cancer (103). TIGIT+ NK cells demonstrated low cytotoxicity and cytokine production, a phenotype that was reversed in TIGIT-deficient mice or under TIGIT antibody blockade. Interestingly, blockade of TIGIT in NK cells alone appeared necessary for the most potent anti-tumor responses mediated by CD8+ T-cells, and generated a memory response against secondary tumor challenge. This requires further study, but raises the exciting possibility of memory responses against cancer mediated by NK cell activity. Since TIGIT blockade synergizes with anti-PD-1 treatment (129), it is also undergoing testing in combination in multiple clinical trials for solid tumors (NCT04047862, NCT03563716, NCT03628677).
CD96
T cell activation, increased late expression (tactile)/CD96 shares sequence similarity to TIGIT, and promotes NK cell adhesion by binding CD155 (130). More recently, it was also shown to down modulate NK cell activity (107). CD96-deficient mice are resistant to carcinogenesis and lung metastases (107), and blocking CD96 unleashes NK cell activity in mouse models of lung metastases, significantly suppressing metastatic growth (108, 109). Recent work by Sun et al. identified a subset of CD96+ NK cell infiltrates in hepatocellular carcinoma associated with a transient increase in disease-free survival in patients, and enhanced overall survival (106). This NK cell subset had low production of IFNγ and TNFα, as well as low perforin and granzyme B gene expression. Mechanistically, TGF-β1 appears to sustain CD96 expression on NK cells from hepatocellular carcinoma patients, further emphasizing its role in promoting NK cell exhaustion in the TME. These data potentially suggest a dual CD96/TGF-β1 targeting approach (106) which may also synergize with anti-TIGIT blockade, as they both share the CD155/CD112 ligands, and both may provide a synergistic effect with other immune checkpoint receptors such has PD-1 (103). It is interesting to speculate that CD96+, like TIGIT+ NK cells represent an exhausted subset, and the interplay between TIGIT/CD96 and DNAM-1 expression may provide a credible biomarker for identifying and characterizing exhausted NK cells in the TME.
LAG-3
The inhibitory function of Lymphocyte-activation gene 3 (LAG-3) on NK cells is less well-characterized. LAG-3 is expressed on activated NK cells, binding MHC II, and the C-type lectin receptor, LSECtin, which is upregulated on various types of tumors (131–133). Early reports based on LAG-3 deficient mice showed that NK cells from these mice were defective in their ability to lyse certain tumor targets, suggesting a role for this receptor in facilitating NK cell killing (134). However, MHC-I mismatched targets were successfully lysed by NK cells from these LAG-3 deficient mice. Moreover, blocking LAG-3 with mAbs had no effect on NK cell lysis of various target cells (135). This may suggest a role for LAG-3 in the NK cell maturation and receptor repertoire, affecting the cell's capacity to distinguish between MHC-I matched healthy and transformed cells, but not in directly inhibiting NK cell killing efficacy. By contrast, a recent report showed that chronic stimulation of adaptive NK cells from HCMV seropositive donors via NKG2C and IL-15, NKp30, or NKG2D increased PD-1 and LAG-3 surface expression, downregulating NK activity during subsequent interactions with tumor targets (136). These experiments provide primary evidence of LAG-3 as a potential exhaustion marker on NK cells following chronic stimulation. It would be interesting to speculate that these NK cells could have their cytotoxic capacity restored by blocking LAG-3 on their surface. This would provide further insight regarding its function on the NK cell surface. Accordingly, blocking LAG-3 in a murine lung metastases model restored the capacity of NK cells to clear metastasis (137). In line with these findings, a role for LAG-3 was also recently demonstrated in NK cells with a deficiency in Wiskott-Aldrich syndrome protein (WASp). WASp is a critical actin nucleation promoting factor in hematopoietic cells, and along with the WASp interacting protein (WIP), plays a critical role in NK cell activation (138–142). WASp-deficient NK cells display deficient effector functions against tumor targets, and were shown to express increased exhaustion markers, including LAG-3 (143). Thus, it is appears that LAG-3 expression correlates with low overall NK cell activation status and effector function, and NK cells could contribute to the effects of anti-LAG-3 mAb therapy. Several anti-LAG-3 mAbs are being tested in multiple clinical trials for advanced solid tumors, either alone, or in combination with anti-PD-1 (3) (e.g., NCT03489369, NCT04080804, NCT03250832, NCT01968109).
KIR/NKG2A
HLA-E, a ligand for CD94/NKG2A is upregulated in certain cancers (144), and tumors can inhibit NK cells through expression of HLA molecules (145). Targeting the KIR and CD94/NKG2A inhibitory NK cell receptors to generate missing-self recognition is therefore another primary strategy to unleash NK cell activity (84, 110–113, 146, 147). Drugs introduced to block these inhibitory molecules, such as antibodies against KIRs- Lirilumab, which targets KIR2DL1-3 and KIR2DS1/2, and IPH4102, which targets KIR3DL2, are currently being tested in various clinical trials (https://clinicaltrials.gov) (146, 148, 149). In pre-clinical models, anti-KIR antibodies showed potent effects on NK cell activity (145, 150, 151). Lirilumab is effective in AML and multiple myeloma (MM) patients with low toxicity and minimal adverse events (152, 153). In the study by Vey et al., however, Lirilumab treatment did not significantly improve leukemia-free survival in AML (154), and no clinical efficacy was observed in a phase II trial of smoldering MM (155). Interestingly, Carlsten et al. suggest that one possible mechanism impeding the efficacy of this treatment is the downregulation of KIR2D on the NK cell surface following infusion of anti-KIR antibodies. Since NK cell licensing requires engagement of KIR receptors with cognate MHC-I ligands (156, 157), administration of anti-KIR mAbs may counterintuitively lead to down-modulation of NK cell activity (158). One possible solution involves lower or alternating dosing regimens (30). Furthermore, anti-KIR mAbs can synergize well with other mAb therapies such as Rituximab and anti-PD-1 (151, 159). Additional evaluation of Lirilumab for clinical use is warranted, and it is currently being tested as a combination treatment for various malignancies, including bladder cancer (NCT03532451) and advanced and metastatic tumors (NCT03203876, NCT01714739, NCT03347123).
The inhibitory NKG2A receptor was also shown to be a beneficial target for NK-based therapy. NKG2A targeting may be especially significant given the higher HLA-E expression on certain cancerous tissues relative to other classical HLAs (85, 144, 160). Recent work from the Vivier group has shown the potent effect of anti-NKG2A antibody (Monalizumab) on NK cell and CD8+ cell effector functions. Monalizumab increases NK and CD8+ antibody-dependent cellular cytotoxicity (ADCC) and synergizes with anti-PD-1/PD-L1 blockade and anti-epidermal growth factor receptor (EGFR) (Cetuximab) treatment (46). Interim results from clinical studies of Monalizumab + Cetuximab/ Durvalumab (anti-PD-L1) appear well-tolerated, with encouraging efficacy profiles (NCT02643550, NCT02671435) (46, 161).
Targeting Tumors for Reinvigoration of NK Cell Activity
Toxicities and poor patient response to immunotherapies, especially against advanced/solid tumors, remain challenging (15, 162). Non-specific augmentation of immune cell activity can generate immune-related adverse events, and advanced tumors have powerful immunosuppressive properties and are difficult to infiltrate (163, 164). For example, cytokine therapy, such as administration of IL-2, 12, 15, and 21 can promote the proliferation, survival, and activation of NK cells in-vitro and in-vivo. However, their use in clinical trials has shown dose-dependent toxicities such as hypotension, thrombocytopenia, neutropenia, and an increase of Alanine transaminase/Aspartate transaminase (ALT/AST) levels. NK cells, in addition to other immune cell subsets such as T-cells, may also be involved in some of these toxic side effects (165–168). In addition, though therapies targeting inhibitory checkpoint receptors expressed on NK cells, such as anti-PD-1 and the PD-1 ligand, PD-L1, increase NK cell activity (121, 169, 170), they may inadvertently cause various dose-dependent immune-related adverse events, such as pneumonitis, colitis, hepatotoxicity, endocrinopathies, and neurological and cardiac toxicities (171).
Harnessing modalities that can target ligands expressed predominantly on tumors and not on healthy tissues, rather than unleashing immune cell activity in a systemic fashion (e.g., systemic administration of cytokines and checkpoint inhibitors), have the potential to overcome some of these obstacles (Figure 1). Such strategies may increase the activation and infiltration of NK cell subsets in-situ by promoting localized anti-tumor immune responses.
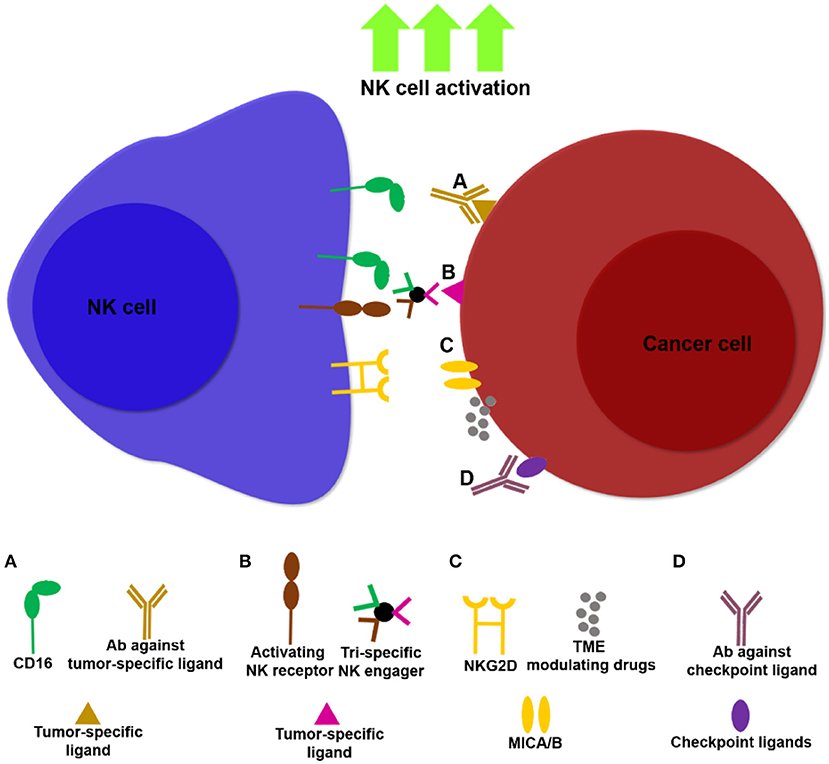
Figure 1. Examples of NK cell immunotherapies targeting NK cells and tumors. Multiple approaches are being developed to unleash NK cell activity against tumors, thereby increasing potency and specificity of NK cell based treatments. (A) Antibodies that bind tumor ligands (e.g., anti-EGFR, anti-CD20) induce NK cell ADCC through ligation to CD16. (B) NK cell engagers can target multiple activating NK cell receptors in addition to CD16 (172), together with tumor ligands such as CD20, thereby facilitating NK cell activation and ADCC. (C) Drugs that modulate the TME, such as histone deactetylase inhibitors, induce upregulation of the NKG2D ligands, MICA/B, promoting NK cell infiltration and cancer cell lysis. (D) Masking tumor checkpoint ligands, such as PD-L1 and PCNA can unleash NK cell activity in the TME.
Antibodies to Direct NK Cell Activity Toward Cancer Cells
Antibodies for NK Cell Mediated ADCC
Monoclonal antibodies (mAbs) which target tumor-specific ligands have been used for treatment of hematological and solid malignancies for the last two decades (173). mAbs can have several mechanisms of action, including initiation of ADCC, mediated by engagement of the CD16 (FcγRIII) activating receptor on the NK cell with the Fc portion of the antibody that is directed toward the tumor ligand (174). The relative contribution of NK cells to the success of mAb therapies is not completely clear, but evidence of their role has increased in recent years (175). Several recent studies demonstrated enhanced efficacy of NK cell mediated ADCC using novel tumor-specific antibodies (Table 2).

Table 2. Antibody-based treatments reported to augment NK cell activity.
Clinically utilized mAbs- recent evidence of NK cell contribution and optimization of treatment
Anti-EGFR: cetuximab. Cetuximab, is an anti-EGFR mAb used clinically for cancer treatment. Cetuximab can activate NK cell ADCC in-vitro and in-vivo, and initiate an adaptive immune response through activation of NK cell:DC crosstalk (180–182). Ex-vivo stimulation of NK cell ADCC with Cetuximab may also be predictive of clinical responses in patients (183). The efficacy of mAb treatment, including that of Cetuximab, is influenced by FCγR polymorphism (184). The homozygous genotype of valine at position 158 on the FcγRIIIa receptor (158 V/V genotypes), as opposed to the 158 V/F or 158 F/F FcγRIIIa genotypes is often, but not always, associated with improved clinical outcomes (185–189). Moreover, FCγ receptor affinity is impacted by patterns of antibody glycosylation (190) and by particular IgG variants. For example, though IgG1 and IgG3 appear to bind all Fcgamma receptors, IgG2 and IgG4 demonstrate varying affinity for specific Fcgamma receptors and their variants (191). In order to overcome these obstacles, much effort has been placed in IgG engineering to enhance the efficacy of ADCC. Glyco-engineering of antibodies, such as removal of fucose from the Fc region to generate non-fucosylated antibodies can significantly enhance interaction between antibodies and FcγRIIIa (192). Furthermore, screening for amino acid point mutations on antibody Fc regions which enhance ADCC has yielded mAbs which generate superior ADCC relative to their WT counterparts (190). Cytokine administration together with Cetuximab increases efficacy of NK cell activation and function (180, 193), and therefore Cetuximab was recently utilized in clinical trials for unresectable primary or recurrent HNSCC in conjunction with IL-12 administration (194). Combination treatment was generally well-tolerated. About half of the patients in this study demonstrated an average prolonged progression-free survival of 6.5 months, although neither complete nor partial responses to therapy were observed. It is important to note that patients in the study were already heavily pretreated and many underwent chemotherapy, radiotherapy, and surgical regimens without therapeutic effect. Moreover, peripheral blood mononuclear cells (PBMCs) from patients enrolled in the second phase of this trial, who were treated with IL-12, or patients with progression-free survival over 100 days showed an increase in ADCC against tumor cells coated with Cetuximab. It is possible, as suggested by the authors in this study, that Cetuximab may exhibit an off target effect by increasing the frequency of CTLA-4+ Tregs in the TME, thereby suppressing NK cell function (194). Accordingly, treatments incorporating anti-CTLA-4 mAbs or transient Treg depletion could potentially increase efficacy (195). Cetuximab in conjunction with toll-like receptor agonists, PD-1 blockade, and CD137 agonists, may also enhance efficacy, in addition to cytokine administration (69, 181, 182, 196–200). Moreover, combination treatment with super agonist cytokines (discussed below) may substantially potentiate Cetuximab activity, as recently described for the IL-15 super agonist ALT-803 (201). Interestingly, treatment with Nimotuzumab, a different anti-EGFR mAb, also promoted cross-talk between NK cells and DCs, leading to EGFR-specific CD8+ T-cell priming, and unlike Cetuximab, did not increase the abundance of Tregs (202). Nevertheless, both Cetuximab and Nimotuzumab may upregulate the expression of TIM-3 on the NK cell surface (202), potentially leading to exhaustion. Thus, the anti-EGFR pathway may require additional investigation in future treatments and could possibly be bypassed through a combination treatment of Cetuximab/Nimotuzumab with anti-TIM-3 antibodies.
Anti-HER2: Trastuzumab. Trastuzumab is a clinically used anti-HER2 mAb commonly used for treating HER2+ tumors. NK cells can mediate ADCC against Trastuzumab coated target cells and recruit T-cells through chemokine secretion. Trastuzumab treatment is also associated with high numbers of tumor infiltrating NK cells (203–205), and higher NK cell function and expansion may also correlate with response to Trastuzumab treatment (206, 207). A recent phase I clinical study used adoptive NK cell therapy together with Trastuzumab administration, showing good tolerability, and induction of immune activity (208). Nevertheless, several patients still display intrinsic and acquired resistance to Trastuzumab therapy (209). Interestingly, a recent study demonstrated that the number of circulating CD57+ NK cells from primary breast cancer patients receiving Trastuzumab therapy correlates with resistance to therapy (210). These NK cells showed reduced CXCR3 expression and proliferation, suggesting a phenotype exhibiting poor homing to and survival in the tumor niche. CD57+ circulating NK cells may therefore provide a novel prognostic biomarker for Trastuzumab treatment, and it would be interesting to consider reinvigorating this NK subset to increase the efficacy of Trastuzumab administration; it is possible that approaches involving upregulation of chemokine receptors (211) (such as CXCR3) or utilization of antibody-based homing proteins designed to increase NK cell homing along chemokine gradients (212) may restore their redirection to tumors, however a broader understanding of the molecular mechanisms and surface receptors governing the responsivity of the subset during Trastuzumab treatment are required. Enhancing the activation and proliferation of NK cells may thus benefit anti-HER2 targeting approaches. Indeed, aside from cytokine administration (IL-2/15/12) and glyco-optimization, additional approaches, which include anti-PD-1/TIGIT blockade, and CD137/Toll-like receptor agonists have been shown to potentially augment Trastuzumab efficacy in combinatorial studies (213). Another effective strategy to improve NK cell ADCC is incorporation of Trastuzumab in the bispecific antibody format (discussed below). Bispecific Trastuzumab antibody [(HER2)2xCD16] significantly enhanced ADCC compared to Trastuzumab treatment (214).
Anti CD20: Rituximab/Obinutuzumab. Rituximab and Obinutuzumab are clinically utilized mAbs targeting CD-20, which is expressed on the surface of mature B-cells and on the surface of most malignant B-cells (215). Evidence for the involvement of ADCC in the mechanism of Rituximab stems from the correlation between FcγR polymorphism and clinical responses in some hematologic malignancies, in addition to lack of therapeutic effects in FcγR deficient mice (216–218). Therefore, in order to improve effector function mediated by mAbs targeting CD-20, Obinutuzumab was glyco-modified to reduce Fc fucosylation (219). Indeed, Obinutuzumab demonstrated superior ADCC in xenograft models and in-vitro efficacy assays compared to Rituximab (218). Furthermore, the glyco-engineered Obinutuzumab appears to be less affected by the inhibitory effects of KIR/HLA ligation compared to Rituximab (220). Several lines of evidence, from in-vitro and animal studies show that NK cells are involved in the mechanism of action of Rituximab/ Obinutuzumab (221–224), yet their exact role in clinical settings are not completely understood. A recent clinical study suggested that lower peripheral NK cell counts in follicular lymphoma and lower peripheral and tumor infiltrated NK cell numbers in diffuse large B-cell lymphoma associate with transient progression-free survival in response to therapy (225). NK cell count could therefore potentially serve as a prognostic biomarker for efficacy of anti-CD20 treatment. As additional means to augment NK-mediated anti-CD20 therapy (apart from glyco-optimization), combination with cytokines such as IL-15/21 was shown to increase the efficacy of anti-CD20 treatment (226–228). Lenalidomide, an immunomodulatory drug commonly used for multiple myeloma also enhances the effect of anti-CD20 administration on NK cell effector function, in part by promoting nanoscale reorganization of the cytoskeleton at the immunological synapse (229–231). Moreover, a recent study revealed a CD56hi CD16− PD-1hi NK population in HL patients. It is tempting therefore to speculate that anti-CD20 treatment might synergize well with PD-1/PD-L1 blockade in HL (88), and that incorporation of modulatory drugs such as lenalidomide may even enhance these combinations further.
mAbs in clinical trials- targeting tumor ligands for optimization of NK cell activity
Though it is clear that NK cells mediate some of the effects of clinically utilized immunotherapeutic mAbs, additional clinical studies are required to understand the full extent of their contribution. In addition to the caveats of mAb therapy mentioned above, it has become clear that reduction of ligand expression (internalization and shedding) and ligand specificity are critical parameters, which are starting to be addressed in recent pre-clinical and clinical studies (232).
One of the major challenges of current treatments for acute lymphoblastic leukemia (ALL), which target CD19 (such as CAR T-cell therapy), is the loss of CD19 surface expression (233). Therefore, identification of additional antigens that retain their expression on drug-resistant and relapsed ALL cells, may provide additional avenues for treatment (234). A recent study addressed this need through an optimized anti- B-cell activating factor receptor, BAFF-R antibody (VAY736), based on BAFF-R overexpression on B-ALL cells (234). Drug resistant and relapsed ALL cells maintained expression of BAFF-R, and VAY736 treatment decreased disease burden and increased NK cell activity. Nevertheless, advanced stages of the disease showed resistance to therapy, possibly due in part to NK cell exhaustion. Thus, patients with advanced stages of ALL may benefit from additional combinatorial treatments. Indeed, resistance to VAY736 therapy could be overcome with combination treatment using anti TGF-β antibody in this study. VAY736 is currently in phase I trials for CLL (NCT03400176), and it will be interesting to determine if it may synergize well with other ICIs that could restore NK cell functionality in patients.
In acute myeloid leukemia (AML), Krupka et al. demonstrated high CD157 expression on a majority of primary AML cells in samples from both newly diagnosed and relapsed patients. Since the antigen is not rapidly internalized, it provides a stable and attractive target for potential antibody-based therapy for AML. Utilizing a non-fucosylated anti-CD157 antibody (MEN1112) can induce potent anti-leukemic activity mediated through NK cells ex-vivo (235). Though MEN1112 showed promising effects mediated through NK cells against AML cell lines and primary AML cells, it had varying effects on NK cells derived from AML patients, emphasizing that NK cells from AML patients are functionally impaired. A recently described subset of ILC1-like cells in AML patients, with NK cell-like cytotoxic capabilities is functionally impaired by high levels of TGF-β in patients' serum and high HLA-E expression on leukemic blasts (236). Therefore, MEN1112 treatment may benefit from specific timing of administration during disease progression, and may also benefit from additional combinatorial approaches such as ICI (NKG2A blockade)/TGFβ blockade to negate NK cell exhaustion and to maximize efficiency. A phase I study for relapsed/refractory AML patients using MEN1112 (NCT02353143) is expected to shed light on its tolerability and potential future use in single/combination therapy.
NK cells are largely excluded from the TME in HL, and peripheral blood and tumor infiltrating NK cells from HL patients are severely impaired (237). Therefore, enhancing their activity in HL may greatly benefit treatment. A recent study describes targeting CD123 (IL-3Rα), expressed on most HL cells with an anti-CD123 antibody (CSL362) (238) to induce NK cell ADCC as a potential therapy (239). CSL362 incorporates the S239D and I332E substitution in the Fc region to improve binding affinity (240). This study demonstrated for the first time the feasibility of targeting HL through NK cells using antibody therapy. CSL362 increased primary NK cell mediated-killing of HL targets, though it stimulated high-affinity Fcγ-receptor NK-92 cells (haNK cells) to a greater degree than primary NK cells isolated from donors (239). Ernst et al. suggest that as haNK cells are less impacted by CD16 polymorphism, it is possible that they display higher cytotoxicity compared to primary NK cells. Thus, it currently appears that this treatment may be most effective in combination with adoptive NK cell treatment. It would be informative to analyze Fc polymorphism of the cohorts in a clinical setting to determine if indeed this is the major mechanism explaining discrepancy between autologous NK and haNK cells. Furthermore, analysis of NK cell subsets from HL patients may reveal additional avenues of treatment that may synergize with CSL632. CSL362 is being evaluated in a phase II clinical study for patients with myelodysplastic syndromes (NCT03011034).
mAbs in pre-clinical development–targeting tumor antigens for augmentation of NK cell activities
Previous clinical trials for AML showed limited efficacy when antigens such as such as CD123 (241) were targeted for treatment. Moreover, CD33 was also targeted with mAb therapy for AML, yet its limited clinical efficacy may have been caused by antigenic shift (242). Therefore, antibody engineering and targeting of different antigens may enhance NK cell activity and efficacy of treatment. Koerner et al. previously reported an optimized anti-CD133 antibody, 293C3-SDIE for AML, which was also recently tested by Schmied et al. for colorectal cancer (176, 243). 293C3-SDIE was engineered with Fc portion containing the S239D and I332E substitutions to enhance binding affinity for ADCC. In the AML study, 293C3-SDIE significantly decreased AML burden in xenograft mouse models, and was superior in anti-leukemic activity in vitro compared to WT 293C3. Interestingly, there appears to be large variability in the anti-leukemic effects of 293C3-SDIE, possibly due to differing expression patterns of CD133 between patients. This may also be in part due to evidence of HLA dimorphism and NK cell ligand expression impacting AML therapy (244, 245). Furthermore, as described above, autologous patient NK cells may have been functionally impaired, relative to allogeneic NK cells (176). Additional examination of NK cell receptor repertoire from healthy individuals and patients may be informative, to possibly screen for specific checkpoints that could synergize with 293C3-SDIE. Though the colorectal study utilizing 293C3-SDIE was conducted in-vitro, the preclinical data demonstrate it has a potent effect on NK cell activity against colorectal cell lines (243). Though additional study is required to test the effect of 293C3-SDIE against physiological solid tumors, this treatment may have important significance for NK cells against the CD133 tumor antigen, which is implicated in multiple cancer types (246). It would be interesting to follow effects of this antibody in future clinical settings, to validate efficacy and the lack of off-target toxicity against healthy cells expressing CD133, such as healthy hematopoietic progenitor cells and others (246, 247).
An anti-IL-7 receptor (IL-7Rα) antibody, B12, has also recently been described to induce potent NK cell-mediated ADCC against T-ALL (177). Studies previously showed that IL-7 is an important factor in the progression of T-ALL, enhancing cell expansion and survival (248), making it an attractive target for antibody therapy. In addition to stimulating NK cell ADCC, B12 appears to be rapidly internalized and trafficked into lysosomes of leukemic cells, demonstrating its additional therapeutic potential, such as for intracellular drug delivery. B12 administration into mice was not sufficient to halt the progression of the disease but did prolong mouse survival. Thus, in addition to NK cell stimulation, the treatment could be beneficial against disease in combination with additional drugs such as dexamethasone, which the authors reported to have an additive effect with B12 (177). It is also possible that blocking pathways that were reported to interfere with T-ALL progression using small molecule inhibitors, such as ones targeting JAK/STAT signaling pathways, may also synergize well with anti IL-7Rα mAb treatments (249). Moreover, a recent report showed that NK cells from T-ALL patients have reduced expression of activating receptors and reduced terminal differentiation, which may be bypassed by pre-activation of autologous NK cells with IL-12, IL-15, and IL-18 (250). Therefore, combining B12 with pre-treated NK cells may potentially show synergistic effects.
An approach that may synergize well with additional antibody-based treatments aims at maintaining tumor antigens on the cancer cell surface in order to abrogate their shedding or internalization. A recent study aimed at increasing NK cell activity against cancer cells utilized antibodies against the proteolytic shedding domains of MICA and MICB (the tumor ligands for NKG2D), which are often shed by the TME to inhibit NK cell function (178). This strategy was shown to prevent MICA/B shedding and to increase their expression on the tumor surfaces, ultimately inducing potent NK cell activity and inhibiting tumor growth (178). This approach may be applicable to several cancer types that upregulate MICA/B antigens and may have an additive effect with additional treatments. It is interesting to suggest that additional ligands [e.g., PVR and NKp30 ligands (232)] that are shed by cancer cells may also be targeted in a similar fashion to increase ADCC. Moreover, studies focused on CD16-mediated-killing demonstrate that CD16 undergoes significant proteolytic downregulation following activation (251). CD16 shedding is regulated through metalloproteases such as ADAM17 (252, 253). This mechanism may serve to facilitate NK cell detachment from targets and thereby increase their mobility (254). Inhibition of CD16 shedding through ADAM17 blockade may increase NK cell activation in the TME, thereby potentiating antibody treatments targeting NK cells (251, 255, 256). Sustaining this CD16 mediated activity may be especially important, as the Cerwenka group showed that NK cell engagement through CD16 might prime NK cells against cancers by providing them with memory-like effects (257).
Additional antibodies potentially involving NK cell activation and ADCC as mechanism of action were reviewed by Wang et al. (175). More recent reports of NK cell activating antibodies with varying combinatorial treatments are listed in Table 2, including anti-SLAMF7, Elotuzumab, for multiple myeloma (258); anti-cathepsin-D, F1, for Triple Negative Breast Cancer (179); anti-GD2, Dinutuximab and hu14.18K322A, for neuroblastoma (259, 260); anti-PD-L1, Avelumab, for Triple Negative Breast Cancer (261); anti-CD38, Daratumumab, for multiple myeloma (262, 263); anti- GPC3, Codrituzumab, for hepatocellular carcinoma (264); and anti-CD33, BI 836858, for AML (265).
Engagers of NK Cells to Unleash NK Cell Activity
BiKEs and TriKEs are multi-specific antibodies composed of a single-chain variable fragment from the heavy and light variable chains of an antibody that are joined by a short peptide linker and connected to the single-chain variable fragment of an additional antibody (BiKE) or two antibodies (TriKE) of interest. Usually, one of the components incorporates an anti-CD16 moiety to induce NK cell-mediated ADCC (266, 267). Thus, BiKEs and TriKEs can overcome the limitations described above when utilizing standard antibodies to induce ADCC. For example, the affinity of CD16 for the IgG Fc region of therapeutic antibodies of different isotypes can be increased by incorporating anti-CD16 into BiKEs and TriKEs (268). Additional advantages include superior bio-distribution, low immunogenicity, and relatively rapid construction (266).
Utilization of BiKEs and TriKEs to boost NK cell activity as a therapeutic approach was demonstrated in multiple malignancies, among which are Non-HLs, leukemia, metastatic breast cancers, EGFR expressing tumors, and carcinomas (266). The Vivier group recently showed the potency of a multifunctional NK cell engager composed of two antibody domains targeting, on the one hand, the activating NK cell receptor, NKp46, and on the other hand, specific antigens such as CD19, CD20, and EGFR (172), with an additional optimized Fc fragment for the binding of CD16. The multifunctional NK cell engager was injected into mice at different concentrations after tumor inoculation, and demonstrated enhanced NK cell infiltration into tumors and promoted tumor clearance in in-vivo models. Importantly, this study demonstrated how NK cell engagers could surpass the efficacy of current antibodies in clinical use, such as Rituximab, Obinutuzumab, and Cetuximab in in-vitro and in-vivo models. An additional important aspect of this technology is that in addition to promoting NK cell infiltration, it leverages the expression of NKp46 in the tumor microenvironment of multiple solid tumors, overcoming the issue of low CD16 expression on tumor-infiltrating lymphocytes (269). Moreover, harnessing the activating potential of multiple stimulatory receptors (CD16 and NKp46) on the NK cell surface can synergize to overcome inhibition, and fully potentiate NK cell activity (270). Thus, it would be interesting to follow this technology in clinical settings, and observe how different potential modifications such as alternative targeting moieties may be optimal for treatment of certain solid/hematological tumors.
One potential modification for improving efficacy of NK cell engagers is the inclusion of IL-15 moieties to induce NK cell proliferation and activation. TriKE 161519 was recently described by Felices et al. (271) for treatment of chronic lymphocytic leukemia (CLL); this antibody consists of anti-CD19 and anti-CD16 fragments, with an additional IL-15 moiety. Inclusion of the IL-15 moiety may be beneficial in CLL patients, who display low numbers and survival of mature and functional NK cells (272). TriKE 161519 induced superior NK cell activity against lymphoma cell lines and primary CLL patient cells compared to Rituximab, and, importantly, promoted NK cell survival and proliferation. The IL-15 moiety on this TriKE may also reduce off-target toxicities caused by general IL-15 administration by activating NK cells selectively through CD16. Similarly, previously described TriKE constructs (TriKE 161533, and TriKE 1615133) (273), which incorporate an IL-15 linker and contain anti-CD16 and anti-CD33 fragments (161533 TriKE), or anti-CD16 and anti CD133 fragments (1615133 TriKE), were described for activating NK cells against neoplastic mast cells, myelodysplastic syndrome cells, and cancer stem cells (273–275), as well as providing NK cells with sustained survival and proliferation signals. The plasticity of NK cell engagers enables them to simultaneously target additional moieties, as demonstrated by Schmohl et al., who constructed a TetraKE that binds CD16 on NK cells, EpCAM on carcinoma cells, and CD133 on cancer stem cells, while containing an IL-15 linker (1615EpCAM133) (276). This construct improved NK cell-mediated ADCC, NK cell survival, and NK cell proliferation without excessive IFN-γ secretion. Furthermore, the simultaneous targeting of the small cancer stem cell niche (with the anti-CD133 moiety), which is resistant to most conventional therapies, could potentially be a powerful addition to other mAb-based approaches. Current NK cell engagers appear to enhance NK cell therapy in a safe manner by targeting tumor antigens, sustaining NK cell proliferation and survival, surpassing some clinically utilized mAbs. Clinical trials testing TriKE 161533 will be starting imminently (NCT03214666), and it will be interesting to observe the development and clinical progress of the additional constructs.
The challenges facing therapy involving NK cell engagers include the complexity of the design process, screening, production yield, and selection of the proper tumor antigen (277). Often, tumor antigens are expressed on otherwise healthy tissue, adding an obstacle to possible on-target/off-tumor toxicity, requiring proper screening prior to development (17). Variance in CD16 affinity for Fc IgG, CD16 polymorphism, and varying expression of CD16 on tumor-infiltrating lymphocytes also remain a challenge, perhaps to be overcome by Fc glyco-optimization and targeting alternative activating receptors on the NK cell surface, such as NKp46. Furthermore, the proteolytic cleavage of CD16 during NK cell activation is also an issue that may be overcome by using metalloprotease inhibitors in combination with current treatments (253, 267).
Sensitizing Tumors for NK Cell Killing
Additional strategies may synergize with current mAb therapy/ICI therapy by reducing the NK cell activation threshold. This could be achieved by masking tumor checkpoint ligands [i.e., anti-PD-L1, Avelumab, which can also initiate NK cell ADCC (69, 120, 169, 261, 278, 279), or the TIGIT ligands CD112/CD155 (105, 280, 281)], activating NK cells in-situ with cytokine variants, and modulating tumors via bio-chemical modifications or inhibitors, making them more susceptible to NK cell-mediated lysis.
Cytokines for NK Cell Activation in the TME
As mentioned above, systemic administration of cytokines such as IL-2/IL-12/ IL-15 is associated with adverse toxicities. Moreover, though IL-2 stimulates NK cell proliferation and activation, it concurrently enhances Treg expansion through the high affinity IL-2 receptor subunit-α (IL-2Rα) expressed on Tregs; these cells downregulate NK cell activity through secretion of TGF-β (76). In order to circumvent Treg expansion, much effort has been directed at generation of cytokine variants. Prominent examples includes the IL-2 “superkine,” with increases binding for IL-2Rβ expressed on resting T effector and NK cells, which can preferentially activate them without Treg expansion, and PEGylated IL-2, which preferentially activates IL-2Rβ (282, 283). Furthermore, IL-15 can substantially increase NK cell expansion and activation without stimulating Treg development; however it exhibits a short half-life in-vivo and thus requires frequent dosing for efficacy (284). Therefore, an IL-15 super agonist (ALT-803) was generated with a longer in-vivo half-life, and it is being tested in 25 clinical trials at the recruiting and active stages (www.clinicaltrials.gov) (285–287). Nevertheless, delivery of immune-stimulatory cytokines directly to the TME for local enhancement of NK cell survival and proliferation may be preferable to avoid systemic adverse immune events and to promote immune cell activation in-situ. Hutmacher et al. recently reported utilization of the fusion protein F8-IL2, consisting of IL-2 fused to the F8 antibody, specific to the alternatively-spliced EDA domain of fibronectin, which is expressed in multiple solid tumors and lymphomas (288), to increase NK cell activity in murine models. F8 can therefore direct IL-2 activation of NK cells to the TME. F8-IL2 was most effective when used in combination with immune checkpoint blockade (anti-CTLA-4) to increase NK cell infiltration and to unleash NK cells in the TME, delaying tumor growth in mouse models. Besides minimizing potential autoimmune toxicities, this approach can also increase the time window of cytokine stimulation overcoming the relatively short half-life of IL-2 (289). Conversely, inhibitory immunomodulatory cytokines such as TGF-β may be targeted in situ, specifically in the TME, to reduce their suppressive effects on NK cells. Once again, specific targeting of TGF-β in the TME is important due to potential toxicities associated with anti-TGF-β therapies administered systemically (290). Knudson et al. reported an anti-PD-L1/TGF-β fusion protein (M7824), which depletes TGF-β signaling in the TME. This treatment promoted an activated NK cell phenotype and tumor clearance in murine models (291). Thus, this treatment appears to have a dual effect, both blocking PD-L1 and sequestering TGF-β to overcome tumor resistance to immune cell function. In initial clinical trials, M7824 appeared to be well-tolerated in patients with pre-treated advanced solid tumors (NCT02517398) with early signs of efficacy (292), and is involved in multiple clinical trials as part of different combination therapies (www.clinicaltrials.gov).
Novel NK Cell Inhibitory Checkpoints/Tumor Antigens for Blockade
Novel NK cell checkpoint ligands expressed on cancer cells could be desirable targets for new immune checkpoint blockers with favorable toxicity profiles compared to other established immune checkpoints (171). Recently, it was demonstrated that the NKp44 isoform1 receptor, a splice variant of the activating NKp44 receptor, can bind membrane proliferating cell nuclear antigen (PCNA) upregulated on cancer cells, inhibiting NK cell activation (293, 294). This is a potentially attractive target due to its different expression profile in healthy proliferating cells vs. cancer cells. In healthy cells, PCNA appears to be expressed in the nucleus and is involved in DNA replication and repair (295), whereas in cancer cells, it is overexpressed on the cell surface (296). By constructing a monoclonal antibody against PCNA (14-25-9), the Porgador group was able to demonstrate that blocking this tumor antigen unleashes NK cell activity in-vitro and in-vivo, and promotes tumor clearance in murine xenograft models of autologous HNSCC patient NK cells (294). It is interesting to suggest that splice variants of additional activating NK receptors may restrain NK cell activity when interacting with atypical ligands, and their expression on tissue resident/tumor infiltrating NK cell subsets should be characterized. This could potentially provide multiple new avenues for mAb-mediated blockade and combinations with established mAb therapy. Thura et al. have also recently taken advantage of high cell surface expression of the oncogenic phosphatase of regenerating liver 3 (PRL3) on hepatocellular carcinoma cells for targeted therapy. Overexpression of PRL3 is associated with heightened tumorigenicity and metastatic spread and is thus an attractive immunotherapeutic target (297). Blocking PRL3 with a humanized antibody (PRL3-zumab) enriched NK cells in the tumor niche and promoted tumor clearance in mouse models (298). Since this study also demonstrated elevated expression of PRL3 on multiple patient-derived tumor samples with little expression on the matched healthy tissue, this therapeutic approach may be beneficial for additional malignancies, and may provide enhanced specificity for immunotherapy. PRL3-zumab is involved in two clinical trials for advanced solid tumors (NCT03191682, NCT04118114).
Turning “Cold” Tumors “Hot” for NK Cell Immune Surveillance With Synthetic Targeting
Identification of tumor-specific antigens for immunotherapy remains a difficult task due to the heterogeneity of the tumor and tumor infiltrating immunological niche, antigen internalization and shedding, antigen mutations, and differential expression profiles between patients (232, 299). Furthermore, many antigens on tumors may also be expressed on healthy tissue. Therefore, several recent studies suggested modulating tumors through synthetic biochemical modifications in order to enhance NK cell infiltration and function. Two recent studies demonstrate that tumors can be coated with antibodies and antibody Fc fragments, exposing them to NK cell-mediated ADCC. These studies take advantage of specific biochemical characteristics of the TME, such as lower pH levels and high sialic acid expression, to directly coat tumors with antibody fragments (300, 301). Subsequently, NK cells can recognize these Fc IgG fragments and become activated in-situ to promote tumor clearance in in-vivo models. The antibody coating treatments also appear to be non-toxic and to additionally promote adaptive immune responses against tumors. It would be interesting to consider whether additional ligands for NK cell receptors [such as NCRs to deliver synergistic signals (302)] can be coated together with Fc IgG fragments to provide additive effects and to ensure the safety and efficacy of these approaches in pre-clinical and clinical studies. Furthermore, combinations of treatments to stimulate NK cell expansion and activation (such as IL-15 administration) and checkpoint blockade may potentially enhance the efficacy of these approaches.
A similar recent approach includes induced expression of certain genes in tumors that may synergize with other immunotherapies. Meraz et al. demonstrated “forced” expression in tumors of tumor suppressor candidate 2 (TUSC2/ FUS1) by an engineered expression plasmid delivery system, in a mouse model of lung cancer. TUSC2 is reduced in a significant number of lung cancers, and this reduced expression is associated with worse prognosis (303). Therefore, counteracting this down-regulated state may be an effective strategy to sensitize tumors for immunotherapy. Indeed, forced induction of TUSC2 expression in lung cancer cells increased NK cell infiltration and stimulated NK cell activation. Interestingly, TUSC2 modulated the TME to facilitate homing and altered the infiltrating immune populations through an increase of IL-15 and of the chemo-attractants Ccl3/4. This is especially important due to low NK cell infiltration and proliferation in solid tumors, and may open avenues for targeting tumor suppressor genes to increase IL-15 in the tumor bed, since its expression by the pro-inflammatory milieu in tumors was recently shown to play a critical role in NK cell infiltration and activation (304). TUSC2 treatment promoted the survival of mice in the lung metastasis model, and synergized with conventional anti-PD1/CTLA-4 therapy (305). An active trial is currently underway with TUSC2 in combination with the EGFR inhibitor Erlotinib for stage IV lung cancer (NCT01455389). It would be interesting to evaluate TUSC2 administration in future clinical settings in combination with immune checkpoint blockade.
Upregulation of NK Cell Ligands
Additional therapeutic approaches harness chemical compounds that induce the expression of NK cell-activating ligands on tumors, or that inhibit tumor pathways promoting NK cell immunosuppression (306). Such strategies may increase tumor susceptibly to immune surveillance, and synergize with current therapies such as mAb treatment and checkpoint blockade, since downregulation of activating ligands promotes tumor escape (29). For example, recently reported approaches include upregulation of the activating NKG2D NK cell ligands on colon cancer cells, multiple myeloma cells, and hepatocellular carcinoma cells, through administration of spironolactone (SPIR) (307), upregulation of Liver X receptor (308), and inhibition of Enhancer of zeste homolog 2 (EZH2) (309). Previous studies also reported sensitizing tumors to NK cell activity by targeting alternate signaling pathways in tumors, including inhibition of BRAF (V600E) (310), STAT3 inhibition (311), and modulation of cancer cell autophagy (312, 313).
An approach that may activate NK cell activity in the TME and is being investigated in the clinic is inhibition of histone deacetylases (HDACs) (314). Through epigenetic alteration of their genomes, cancer cells may acquire advantages that facilitate their immune escape. HDAC inhibition may increase antigen presentation on the surface of cancer cells, making them more susceptible to immune cell surveillance, thereby potentially enhancing the efficacy of additional immunotherapies (315). Hicks et al. recently studied the use of HDAC inhibitors in conjunction with standard anti PD-1/PD-L1 therapy (316). HDAC inhibitors induced upregulation of the NKG2D stress ligands MICA/B on different carcinoma cell lines, increased the activated phenotype of patient-derived NK cells, and sensitized tumors to NK cell killing. Likewise, Stone et al. demonstrated use of DNA methyltransferase and histone deacetylase inhibitors on ovarian cancer cells, showing that this treatment increases NK cell infiltration and activation in the TME, as well as extending the life of animal models (317). Therefore, screening and identification of tumors with low NK activating ligand expression (such as MICA/B) in patients may favor utilization of HDAC inhibitors to increase their susceptibility to NK cell immune surveillance. Ongoing clinical trials involving HDAC inhibitors with anti-PD-1/PD-L1 therapy (NCT02708680, NCT02915523, and NCT02900560) may shed additional light on the efficacy and safety of these treatments.
Friedman et al. recently utilized an inhibitor for the WEE1 kinase (AZD1775) on head and neck cancer cells, which reversed G2/M cell cycle checkpoint activation of the cancer cells, resulting in increased DNA damage and susceptibility to NK cell-mediated killing (318). Interestingly, DNA cell cycle checkpoint in this study did not impact surface expression of MHC-I, PD-L1, or NKG2D ligands (RAE, H60, MULT-1) on cancer cells, despite increasing their susceptibility to NK cells. These data may suggest a global sensitization mediated through AZD1775 that could bypass the need for anti-PD-1 or anti-NKG2D checkpoint blockade, and it is interesting to consider utilizing it universally for multiple NK-based approaches on tumors with low PD-L1/NKG2D ligand expression. Stimulation of NK cells in the TME via administration of the Heparan sulfate (HS) mimetic Pixatimod (PG545) has also recently been reported. This treatment inhibits the tumor Heparanase enzyme, which promotes extracellular matrix degradation and tumor invasion/metastases. By activating DC production of IL-12, PG545 enhanced the anti-tumor function of NK cells in murine lymphoma models. Therefore, this treatment may synergize well with other immunotherapeutic treatments to stimulate NK cell activity in the TME due to the pleotropic effects of IL-12, and it is interesting to consider its utilization together with mAb therapies to maintain infiltrating NK cell activity. Pixatimod can also be utilized in combination with anti-PD-1 treatment to elicit an even greater anti-tumor effect (319, 320). Furthermore, inhibition of another common enzyme in the TME, arginase 1 (Arg1), using the small molecule inhibitor CB-1158 (clinical trials in combination with checkpoint inhibitors for advanced solid tumors- NCT02903914), was also shown to activate NK cells, promote their infiltration into the TME, and synergize with checkpoint inhibition to promote tumor clearance. This pathway is mediated through inhibition of MDSCs in the TME, which express Arg1 and lead to a decrease of L-arginine, which is required for NK cell proliferation and function (321). It is possible that enzymes such as Arg1 and IDO-1 (322) confer some resistance of tumors to current treatments such as checkpoint blockade, by depleting essential metabolites such as arginine and tryptophan in the TME. Therefore, identification and targeting of novel inhibitory pathways in the TME is warranted, and may significantly bolster the efficacy of current treatments if administered in combination. It is also tempting to consider simultaneous targeting of multiple suppressive pathways in addition to clinically utilized treatments, though first testing for efficacy and specificity of treatment.
Modulating Tumor Metabolites
Finally, modulation of metabolites in the TME appears to be an attractive strategy to increase anti-tumor NK cell activity. Cancer cells metabolize glucose more rapidly compared to normal cells, producing large amounts of lactate, in a process known as the Warburg effect (323). This metabolic shift has important ramifications on the TME, and thus, on tumor-infiltrating immune cells (324). One metabolite that is present at higher concentrations in the TME is adenosine. Studies show the inhibitory effect of adenosine accumulation in the TME on immune cell function in general, and NK cell activity, primarily through engagement with the adenosine receptor A2AR (325, 326). Adenosine signaling in the TME can reduce NK cell proliferation and activation, as well as its effector function (327–329). Blocking adenosine-catalyzing enzymes and purinergic signaling in the TME (such as antibodies against CD73, CD39, and CD38, which mediate the production of extracellular adenosine, and blockade of A2AR) is therefore being investigated in multiple clinical trials in combination with immune checkpoint blockade (330) and may synergistically enhance tumor-resident NK cell activity (330–338).
Perspective
It is evident that NK cells elicit potent anti-tumor responses by complimenting T-cell recognition, and by initiating cross-talk with the tumor-resident immune-cell milieu. As recently demonstrated, they are an attractive addition to T-cell therapy due to their ability to trigger potent immune responses in the TME, promoting adaptive anti-tumor activity. Harnessing NK cells for immunotherapy is also highly attractive due to their innate ability to recognize transformed cells. Evidence is mounting demonstrating the potential for activation of NK cells in-vivo, and though we are beginning to understand the mechanisms that suppress NK cell immune surveillance in cancer, more research is required to clarify the circuits that ultimately dictate their capacity to elicit anti-tumor responses. Current in-vivo approaches mostly seek to increase the activation and expansion of the overall NK population. It would be interesting to consider, given our increasing understanding of NK cell subpopulations and NK cell tissue/intratumor residency, the harnessing of specific NK effector populations in-vivo that may generate the most robust anti-tumor responses, while avoiding tolerant subsets or regulatory ILCs, which can inadvertently hamper therapy. Furthermore, current approaches such as CAR-T therapy, ICI therapy, and small molecule inhibition are subject to adaptive and acquired resistance, resulting from the complex immune-cancer landscape and immune-editing (15). Alternatively, it may be possible that adopting a “bottom-up” approach, that is, first enhancing the innate immune landscape (such as NK cells and ILCs) over different time regimens, secondly, targeting the suppressive TME milieu to negate exhaustion, and finally, enhancing adaptive immune responses, could sustain greater anti-tumor effects. Though this is an intriguing concept, additional studies and safety measures must be implemented to evaluate its efficacy. It is likely that a combinatorial approach, involving more than one facet of therapy, will be most effective, and could hold great promise in clinical settings (339).
Author Contributions
AB-S, GB, and MB-S wrote the manuscript.
Funding
This research was funded by the Israel Science Foundation grant no. 747/13 and from the Chief Scientist Office of the Ministry of Health grant no. 3-10151, and a Taubenblatt Family Foundation Bio-Medicine excellence grant.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Farkona S, Diamandis EP, Blasutig IM. Cancer immunotherapy: the beginning of the end of cancer? BMC Med. (2016) 14:73. doi: 10.1186/s12916-016-0623-5
2. Hu W, Wang G, Huang D, Sui M, Xu Y. Cancer immunotherapy based on natural killer cells: current progress and new opportunities. Front Immunol. (2019) 10:1205. doi: 10.3389/fimmu.2019.01205
3. Sanchez-Correa B, Lopez-Sejas N, Duran E, Labella F, Alonso C, Solana R, et al. Modulation of NK cells with checkpoint inhibitors in the context of cancer immunotherapy. Cancer Immunol Immunother. (2019) 68:861–70. doi: 10.1007/s00262-019-02336-6
4. Vivier E, Tomasello E, Baratin M, Walzer T, Ugolini S. Functions of natural killer cells. Nat Immunol. (2008) 9:503–10. doi: 10.1038/ni1582
5. Lanier LL. Natural killer cell receptor signaling. Curr Opin Immunol. (2003) 15:308–14. doi: 10.1016/S0952-7915(03)00039-6
6. Lanier LL. NK cell receptors. Annu Rev Immunol. (1998) 16:359–93. doi: 10.1146/annurev.immunol.16.1.359
7. Sade-Feldman M, Jiao YJ, Chen JH, Rooney MS, Barzily-Rokni M, Eliane J-P, et al. Resistance to checkpoint blockade therapy through inactivation of antigen presentation. Nat Commun. (2017) 8:1136. doi: 10.1038/s41467-017-01062-w
8. Kärre K, Ljunggren HG, Piontek G, Kiessling R. Selective rejection of H−2-deficient lymphoma variants suggests alternative immune defence strategy. Nature. (1986) 319:675–8. doi: 10.1038/319675a0
9. Ljunggren HG, Kärre K. In search of the ‘missing self’: MHC molecules and NK cell recognition. Immunol Today. (1990) 11:237–44. doi: 10.1016/0167-5699(90)90097-S
10. Lanier LL. NK cell recognition. Annu Rev Immunol. (2005) 23:225–74. doi: 10.1146/annurev.immunol.23.021704.115526
11. Long EO, Kim HS, Liu D, Peterson ME, Rajagopalan S. Controlling natural killer cell responses: integration of signals for activation and inhibition. Annu Rev Immunol. (2013) 31:227–58. doi: 10.1146/annurev-immunol-020711-075005
12. Moretta L, Moretta A. Unravelling natural killer cell function: triggering and inhibitory human NK receptors. EMBO J. (2004) 23:255–9. doi: 10.1038/sj.emboj.7600019
13. Vivier E, Raulet DH, Moretta A, Caligiuri MA, Zitvogel L, Lanier LL, et al. Innate or adaptive immunity? The example of natural killer cells. Science. (2011) 331:44–9. doi: 10.1126/science.1198687
14. Suen WC-W, Lee WY-W, Leung K-T, Pan X-H, Li G. Natural killer cell-based cancer immunotherapy: a review on 10 years completed clinical trials. Cancer Invest. (2018) 36:431–57. doi: 10.1080/07357907.2018.1515315
15. Sharma P, Hu-Lieskovan S, Wargo JA, Ribas A. Primary, adaptive, and acquired resistance to cancer immunotherapy. Cell. (2017) 168:707–23. doi: 10.1016/j.cell.2017.01.017
16. Byun DJ, Wolchok JD, Rosenberg LM, Girotra M. Cancer immunotherapy — immune checkpoint blockade and associated endocrinopathies. Nat Rev Endocrinol. (2017) 13:195–207. doi: 10.1038/nrendo.2016.205
17. Kroschinsky F, Stölzel F, von Bonin S, Beutel G, Kochanek M, Kiehl M, et al. New drugs, new toxicities: severe side effects of modern targeted and immunotherapy of cancer and their management. Crit Care. (2017) 21:89. doi: 10.1186/s13054-017-1678-1
18. Prasad V, Kaestner V. Nivolumab and pembrolizumab: monoclonal antibodies against programmed cell death-1 (PD-1) that are interchangeable. Semin Oncol. (2017) 44:132–5. doi: 10.1053/j.seminoncol.2017.06.007
19. Nayyar G, Chu Y, Cairo MS. Overcoming resistance to natural killer cell based immunotherapies for solid tumors. Front Oncol. (2019) 9:51. doi: 10.3389/fonc.2019.00051
22. Lupo KB, Matosevic S. Natural killer cells as allogeneic effectors in adoptive cancer immunotherapy. Cancers. (2019) 11:769. doi: 10.3390/cancers11060769
23. Souza-Fonseca-Guimaraes F, Cursons J, Huntington ND. The emergence of natural killer cells as a major target in cancer immunotherapy. Trends Immunol. (2019) 40:142–58. doi: 10.1016/j.it.2018.12.003
24. Kim N, Kim HS. Targeting checkpoint receptors and molecules for therapeutic modulation of natural killer cells. Front Immunol. (2018) 9:2041. doi: 10.3389/fimmu.2018.02041
25. Daher M, Rezvani K. Next generation natural killer cells for cancer immunotherapy: the promise of genetic engineering. Curr Opin Immunol. (2018) 51:146–53. doi: 10.1016/j.coi.2018.03.013
26. Böttcher JP, Bonavita E, Chakravarty P, Blees H, Cabeza-Cabrerizo M, Sammicheli S, et al. NK cells stimulate recruitment of cDC1 into the tumor microenvironment promoting cancer immune control. Cell. (2018) 172:1022–37.e14. doi: 10.1016/j.cell.2018.01.004
27. Barry KC, Hsu J, Broz ML, Cueto FJ, Binnewies M, Combes AJ, et al. A natural killer–dendritic cell axis defines checkpoint therapy–responsive tumor microenvironments. Nat Med. (2018) 24:1178–91. doi: 10.1038/s41591-018-0085-8
28. Schmidt L, Eskiocak B, Kohn R, Dang C, Joshi NS, DuPage M, et al. Enhanced adaptive immune responses in lung adenocarcinoma through natural killer cell stimulation. Proc Natl Acad Sci USA. (2019) 116:17460–9. doi: 10.1073/pnas.1904253116
29. Malmberg K-J, Carlsten M, Björklund A, Sohlberg E, Bryceson YT, Ljunggren H-G. Natural killer cell-mediated immunosurveillance of human cancer. Semin Immunol. (2017) 31:20–9. doi: 10.1016/j.smim.2017.08.002
30. Chiossone L, Dumas P-Y, Vienne M, Vivier E. Natural killer cells and other innate lymphoid cells in cancer. Nat Rev Immunol. (2018) 18:671–88. doi: 10.1038/s41577-018-0061-z
31. Caligiuri MA. Human natural killer cells. Blood. (2008) 112:461–9. doi: 10.1182/blood-2007-09-077438
32. Björkström NK, Ljunggren H-G, Michaëlsson J. Emerging insights into natural killer cells in human peripheral tissues. Nat Rev Immunol. (2016) 16:310–20. doi: 10.1038/nri.2016.34
33. Cichocki F, Schlums H, Theorell J, Tesi B, Miller JS, Ljunggren H-G, et al. Diversification and functional specialization of human NK cell subsets. Curr Topics Microbiol Immunol. (2015) 395:63–93. doi: 10.1007/82_2015_487
34. Freud AG, Mundy-Bosse BL, Yu J, Caligiuri MA. The broad spectrum of human natural killer cell diversity. Immunity. (2017) 47:820–33. doi: 10.1016/j.immuni.2017.10.008
35. Stabile H, Fionda C, Gismondi A, Santoni A. Role of distinct natural killer cell subsets in anticancer response. Front Immunol. (2017) 8:293. doi: 10.3389/fimmu.2017.00293
36. Vivier E, Artis D, Colonna M, Diefenbach A, Di Santo JP, Eberl G, et al. Innate lymphoid cells: 10 years on. Cell. (2018) 174:1054–66. doi: 10.1016/j.cell.2018.07.017
37. Colonna M. Innate lymphoid cells: diversity, plasticity, and unique functions in immunity. Immunity. (2018) 48:1104–17. doi: 10.1016/j.immuni.2018.05.013
38. Gao Y, Souza-Fonseca-Guimaraes F, Bald T, Ng SS, Young A, Ngiow SF, et al. Tumor immunoevasion by the conversion of effector NK cells into type 1 innate lymphoid cells. Nat Immunol. (2017) 18:1004–15. doi: 10.1038/ni.3800
39. Bruno A, Focaccetti C, Pagani A, Imperatori AS, Spagnoletti M, Rotolo N, et al. The proangiogenic phenotype of natural killer cells in patients with non-small cell lung cancer. Neoplasia. (2013) 15:133–42. doi: 10.1593/neo.121758
40. Vitale M, Cantoni C, Pietra G, Mingari MC, Moretta L. Effect of tumor cells and tumor microenvironment on NK-cell function. Eur J Immunol. (2014) 44:1582–92. doi: 10.1002/eji.201344272
41. Cooper MA, Fehniger TA, Caligiuri MA. The biology of human natural killer-cell subsets. Trends Immunol. (2001) 22:633–40. doi: 10.1016/S1471-4906(01)02060-9
42. Habif G, Crinier A, André P, Vivier E, Narni-Mancinelli E. Targeting natural killer cells in solid tumors. Cell Mol Immunol. (2019) 16:415–22. doi: 10.1038/s41423-019-0224-2
43. Halama N, Braun M, Kahlert C, Spille A, Quack C, Rahbari N, et al. Natural killer cells are scarce in colorectal carcinoma tissue despite high levels of chemokines and cytokines. Clin Cancer Res. (2011) 17:678–89. doi: 10.1158/1078-0432.CCR-10-2173
44. Muntasell A, Rojo F, Servitja S, Rubio-Perez C, Cabo M, Tamborero D, et al. NK cell infiltrates and HLA class I expression in primary HER2 + breast cancer predict and uncouple pathological response and disease-free survival. Clin Cancer Res. (2019) 25:1535–45. doi: 10.1158/1078-0432.CCR-18-2365
45. Eckl J, Buchner A, Prinz PU, Riesenberg R, Siegert SI, Kammerer R, et al. Transcript signature predicts tissue NK cell content and defines renal cell carcinoma subgroups independent of TNM staging. J Mol Med. (2012) 90:55–66. doi: 10.1007/s00109-011-0806-7
46. André P, Denis C, Soulas C, Bourbon-Caillet C, Lopez J, Arnoux T, et al. Anti-NKG2A mAb is a checkpoint inhibitor that promotes anti-tumor immunity by unleashing both T and NK cells. Cell. (2018) 175:1731–43.e13. doi: 10.1016/j.cell.2018.10.014
47. Lavin Y, Kobayashi S, Leader A, Amir ED, Elefant N, Bigenwald C, et al. Innate immune landscape in early lung adenocarcinoma by paired single-cell analyses. Cell. (2017) 169:750–65.e17. doi: 10.1016/j.cell.2017.04.014
48. Castriconi R, Dondero A, Bellora F, Moretta L, Castellano A, Locatelli F, et al. Neuroblastoma-derived TGF-β1 modulates the chemokine receptor repertoire of human resting NK cells. J Immunol. (2013) 190:5321–8. doi: 10.4049/jimmunol.1202693
49. Ponzetta A, Benigni G, Antonangeli F, Sciume G, Sanseviero E, Zingoni A, et al. Multiple myeloma impairs bone marrow localization of effector natural killer cells by altering the chemokine microenvironment. Cancer Res. (2015) 75:4766–77. doi: 10.1158/0008-5472.CAN-15-1320
50. Yang Q, Goding S, Hagenaars M, Carlos T, Albertsson P, Kuppen P, et al. Morphological appearance, content of extracellular matrix and vascular density of lung metastases predicts permissiveness to infiltration by adoptively transferred natural killer and T cells. Cancer Immunol Immunother. (2006) 55:699–707. doi: 10.1007/s00262-005-0043-4
51. Whiteside TL. The tumor microenvironment and its role in promoting tumor growth. Oncogene. (2008) 27:5904–12. doi: 10.1038/onc.2008.271
52. Weber R, Fleming V, Hu X, Nagibin V, Groth C, Altevogt P, et al. Myeloid-derived suppressor cells hinder the anti-cancer activity of immune checkpoint inhibitors. Front Immunol. (2018) 9:1310. doi: 10.3389/fimmu.2018.01310
53. Wherry EJ, Kurachi M. Molecular and cellular insights into T cell exhaustion. Nat Rev Immunol. (2015) 15:486–99. doi: 10.1038/nri3862
54. McLane LM, Abdel-Hakeem MS, Wherry EJ. CD8 T cell exhaustion during chronic viral infection and cancer. Annu Rev Immunol. (2019) 37:457–95. doi: 10.1146/annurev-immunol-041015-055318
55. Jiang Y, Li Y, Zhu B. T-cell exhaustion in the tumor microenvironment. Cell Death Dis. (2015) 6:e1792. doi: 10.1038/cddis.2015.162
56. Marvel D, Gabrilovich DI. Myeloid-derived suppressor cells in the tumor microenvironment: expect the unexpected. J Clin Invest. (2015) 125:3356–64. doi: 10.1172/JCI80005
57. Quatromoni JG, Eruslanov E. Tumor-associated macrophages: function, phenotype, and link to prognosis in human lung cancer. Am J Transl Res. (2012) 4:376–89.
58. Curiel TJ, Coukos G, Zou L, Alvarez X, Cheng P, Mottram P, et al. Specific recruitment of regulatory T cells in ovarian carcinoma fosters immune privilege and predicts reduced survival. Nat Med. (2004) 10:942–9. doi: 10.1038/nm1093
59. Jarnicki AG, Lysaght J, Todryk S, Mills KHG. Suppression of antitumor immunity by IL-10 and TGF-beta-producing T cells infiltrating the growing tumor: influence of tumor environment on the induction of CD4+ and CD8+ regulatory T cells. J Immunol. (2006) 177:896–904. doi: 10.4049/jimmunol.177.2.896
60. Pitt JM, Marabelle A, Eggermont A, Soria J-C, Kroemer G, Zitvogel L. Targeting the tumor microenvironment: removing obstruction to anticancer immune responses and immunotherapy. Ann Oncol. (2016) 27:1482–92. doi: 10.1093/annonc/mdw168
61. Bauer CA, Kim EY, Marangoni F, Carrizosa E, Claudio NM, Mempel TR. Dynamic Treg interactions with intratumoral APCs promote local CTL dysfunction. J Clin Invest. (2014) 124:2425–40. doi: 10.1172/JCI66375
62. Autio K, Oft M. Pegylated interleukin-10: clinical development of an immunoregulatory cytokine for use in cancer therapeutics. Curr Oncol Rep. (2019) 21:19. doi: 10.1007/s11912-019-0760-z
63. Pauken KE, Wherry EJ. Overcoming T cell exhaustion in infection and cancer. Trends Immunol. (2015) 36:265–76. doi: 10.1016/j.it.2015.02.008
64. Wherry EJ, Blattman JN, Murali-Krishna K, van der Most R, Ahmed R. Viral persistence alters CD8 T-cell immunodominance and tissue distribution and results in distinct stages of functional impairment. J Virol. (2003) 77:4911–27. doi: 10.1128/JVI.77.8.4911-4927.2003
65. Baitsch L, Baumgaertner P, Devêvre E, Raghav SK, Legat A, Barba L, et al. Exhaustion of tumor-specific CD8+ T cells in metastases from melanoma patients. J Clin Invest. (2011) 121:2350–60. doi: 10.1172/JCI46102
66. Fourcade J, Sun Z, Benallaoua M, Guillaume P, Luescher IF, Sander C, et al. Upregulation of Tim-3 and PD-1 expression is associated with tumor antigen-specific CD8+ T cell dysfunction in melanoma patients. J Exp Med. (2010) 207:2175–86. doi: 10.1084/jem.20100637
67. Gros A, Robbins PF, Yao X, Li YF, Turcotte S, Tran E, et al. PD-1 identifies the patient-specific CD8+ tumor-reactive repertoire infiltrating human tumors. J Clin Invest. (2014) 124:2246–59. doi: 10.1172/JCI73639
68. Mumprecht S, Schürch C, Schwaller J, Solenthaler M, Ochsenbein AF. Programmed death 1 signaling on chronic myeloid leukemia-specific T cells results in T-cell exhaustion and disease progression. Blood. (2009) 114:1528–36. doi: 10.1182/blood-2008-09-179697
69. Concha-Benavente F, Kansy BA, Moskovitz J, Moy JD, Chandran UR, Ferris RL. PD-L1 mediates dysfunction in activated PD-1+ NK cells in head and neck cancer patients. Cancer Immunol Res. (2018) 6:1548–60. doi: 10.1158/2326-6066.CIR-18-0062
70. Richards JO, Chang X, Blaser BW, Caligiuri MA, Zheng P, Liu Y. Tumor growth impedes natural-killer-cell maturation in the bone marrow. Blood. (2006) 108:246–52. doi: 10.1182/blood-2005-11-4535
71. Baltz KM, Krusch M, Baessler T, Schmiedel BJ, Bringmann A, Brossart P, et al. Neutralization of tumor-derived soluble glucocorticoid-induced TNFR-related protein ligand increases NK cell anti-tumor reactivity. Blood. (2008) 112:3735–43. doi: 10.1182/blood-2008-03-143016
72. Castriconi R, Cantoni C, Della Chiesa M, Vitale M, Marcenaro E, Conte R, et al. Transforming growth factor beta 1 inhibits expression of NKp30 and NKG2D receptors: consequences for the NK-mediated killing of dendritic cells. Proc Natl Acad Sci USA. (2003) 100:4120–5. doi: 10.1073/pnas.0730640100
73. Vacca P, Munari E, Tumino N, Moretta F, Pietra G, Vitale M, et al. Human natural killer cells and other innate lymphoid cells in cancer: friends or foes? Immunol Lett. (2018) 201:14–9. doi: 10.1016/j.imlet.2018.11.004
74. Balsamo M, Manzini C, Pietra G, Raggi F, Blengio F, Mingari MC, et al. Hypoxia downregulates the expression of activating receptors involved in NK-cell-mediated target cell killing without affecting ADCC. Eur J Immunol. (2013) 43:2756–64. doi: 10.1002/eji.201343448
75. Parodi M, Raggi F, Cangelosi D, Manzini C, Balsamo M, Blengio F, et al. Hypoxia modifies the transcriptome of human NK cells, modulates their immunoregulatory profile, and influences NK cell subset migration. Front Immunol. (2018) 9:2358. doi: 10.3389/fimmu.2018.02358
76. Ghiringhelli F, Ménard C, Terme M, Flament C, Taieb J, Chaput N, et al. CD4+CD25+ regulatory T cells inhibit natural killer cell functions in a transforming growth factor–β-dependent manner. J Exp Med. (2005) 202:1075–85. doi: 10.1084/jem.20051511
77. Li H, Han Y, Guo Q, Zhang M, Cao X. Cancer-expanded myeloid-derived suppressor cells induce anergy of NK cells through membrane-bound TGF-beta 1. J Immunol. (2009) 182:240–9. doi: 10.4049/jimmunol.182.1.240
78. Peng L, Zhang J, Teng Y, Zhao Y, Wang T, Mao F, et al. Tumor-associated monocytes/macrophages impair NK-cell function via TGFβ1 in human gastric cancer. Cancer Immunol. Res. (2017) 5:248–56. doi: 10.1158/2326-6066.CIR-16-0152
79. Gasser S, Orsulic S, Brown EJ, Raulet DH. The DNA damage pathway regulates innate immune system ligands of the NKG2D receptor. Nature. (2005) 436:1186–90. doi: 10.1038/nature03884
80. Alvarez M, Simonetta F, Baker J, Pierini A, Wenokur AS, Morrison AR, et al. Regulation of murine NK cell exhaustion through the activation of the DNA damage repair pathway. JCI Insight. (2019) 5:127729. doi: 10.1172/jci.insight.127729
81. Gill S, Vasey AE, De Souza A, Baker J, Smith AT, Kohrt HE, et al. Rapid development of exhaustion and down-regulation of eomesodermin limit the antitumor activity of adoptively transferred murine natural killer cells. Blood. (2012) 119:5758–68. doi: 10.1182/blood-2012-03-415364
82. Paul S, Kulkarni N, Shilpi Lal G. Intratumoral natural killer cells show reduced effector and cytolytic properties and control the differentiation of effector Th1 cells. Oncoimmunology. (2016) 5:e1235106. doi: 10.1080/2162402X.2016.1235106
83. Peng Y-P, Zhu Y, Zhang J-J, Xu Z-K, Qian Z-Y, Dai C-C, et al. Comprehensive analysis of the percentage of surface receptors and cytotoxic granules positive natural killer cells in patients with pancreatic cancer, gastric cancer, and colorectal cancer. J Transl Med. (2013) 11:262. doi: 10.1186/1479-5876-11-262
84. Sun C, Xu J, Huang Q, Huang M, Wen H, Zhang C, et al. High NKG2A expression contributes to NK cell exhaustion and predicts a poor prognosis of patients with liver cancer. Oncoimmunology. (2017) 6:e1264562. doi: 10.1080/2162402X.2016.1264562
85. Platonova S, Cherfils-Vicini J, Damotte D, Crozet L, Vieillard V, Validire P, et al. Profound coordinated alterations of intratumoral NK cell phenotype and function in lung carcinoma. Cancer Res. (2011) 71:5412–22. doi: 10.1158/0008-5472.CAN-10-4179
86. Mamessier E, Sylvain A, Thibult M-L, Houvenaeghel G, Jacquemier J, Castellano R, et al. Human breast cancer cells enhance self tolerance by promoting evasion from NK cell antitumor immunity. J Clin Invest. (2011) 121:3609–22. doi: 10.1172/JCI45816
87. Yin M, Di G, Bian M. Dysfunction of natural killer cells mediated by PD-1 and Tim-3 pathway in anaplastic thyroid cancer. Int Immunopharmacol. (2018) 64:333–9. doi: 10.1016/j.intimp.2018.09.016
88. Vari F, Arpon D, Keane C, Hertzberg MS, Talaulikar D, Jain S, et al. Immune evasion via PD-1/PD-L1 on NK cells and monocyte/macrophages is more prominent in Hodgkin lymphoma than DLBCL. Blood. (2018) 131:1809–19. doi: 10.1182/blood-2017-07-796342
89. Liu Y, Cheng Y, Xu Y, Wang Z, Du X, Li C, et al. Increased expression of programmed cell death protein 1 on NK cells inhibits NK-cell-mediated anti-tumor function and indicates poor prognosis in digestive cancers. Oncogene. (2017) 36:6143–53. doi: 10.1038/onc.2017.209
90. Beldi-Ferchiou A, Lambert M, Dogniaux S, Vély F, Vivier E, Olive D, et al. PD-1 mediates functional exhaustion of activated NK cells in patients with Kaposi sarcoma. Oncotarget. (2016) 7:72961–77. doi: 10.18632/oncotarget.12150
91. MacFarlane AW, Jillab M, Plimack ER, Hudes GR, Uzzo RG, Litwin S, et al. PD-1 Expression on peripheral blood cells increases with stage in renal cell carcinoma patients and is rapidly reduced after surgical tumor resection. Cancer Immunol Res. (2014) 2:320–31. doi: 10.1158/2326-6066.CIR-13-0133
92. Benson DM, Bakan CE, Mishra A, Hofmeister CC, Efebera Y, Becknell B, et al. The PD-1/PD-L1 axis modulates the natural killer cell versus multiple myeloma effect: a therapeutic target for CT-011, a novel monoclonal anti-PD-1 antibody. Blood. (2010) 116:2286–94. doi: 10.1182/blood-2010-02-271874
93. Park IH, Yang HN, Lee KJ, Kim T-S, Lee ES, Jung S-Y, et al. Tumor-derived IL-18 induces PD-1 expression on immunosuppressive NK cells in triple-negative breast cancer. Oncotarget. (2017) 8:32722–30. doi: 10.18632/oncotarget.16281
94. da Silva IP, Gallois A, Jimenez-Baranda S, Khan S, Anderson AC, Kuchroo VK, et al. Reversal of NK-cell exhaustion in advanced melanoma by Tim-3 blockade. Cancer Immunol Res. (2014) 2:410–22. doi: 10.1158/2326-6066.CIR-13-0171
95. Tallerico R, Cristiani CM, Staaf E, Garofalo C, Sottile R, Capone M, et al. IL-15, TIM-3 and NK cells subsets predict responsiveness to anti-CTLA-4 treatment in melanoma patients. Oncoimmunology. (2017) 6:e1261242. doi: 10.1080/2162402X.2016.1261242
96. Seo H, Jeon I, Kim B-S, Park M, Bae E-A, Song B, et al. IL-21-mediated reversal of NK cell exhaustion facilitates anti-tumour immunity in MHC class I-deficient tumours. Nat Commun. (2017) 8:15776. doi: 10.1038/ncomms15776
97. Xu L, Huang Y, Tan L, Yu W, Chen D, Lu C, et al. Increased Tim-3 expression in peripheral NK cells predicts a poorer prognosis and Tim-3 blockade improves NK cell-mediated cytotoxicity in human lung adenocarcinoma. Int Immunopharmacol. (2015) 29:635–41. doi: 10.1016/j.intimp.2015.09.017
98. Wang Y, Sun J, Gao W, Song B, Shao Q, Zhao L, et al. Preoperative Tim-3 expression on peripheral NK cells is correlated with pathologic TNM staging in colorectal cancer. Mol Med Rep. (2017) 15:3810–8. doi: 10.3892/mmr.2017.6482
99. Zhang W, Feng H, Chen Q, Lu X, Ge J. The functional potency of natural killer cells in response to IL-2/IL-15/IL-21 stimulation is limited by a concurrent upregulation of Tim-3 in bladder cancer. Exp Cell Res. (2018) 372:92–8. doi: 10.1016/j.yexcr.2018.09.013
100. Degos C, Heinemann M, Barrou J, Boucherit N, Lambaudie E, Savina A, et al. Endometrial tumor microenvironment alters human NK cell recruitment, and resident NK cell phenotype and function. Front Immunol. (2019) 10:877. doi: 10.3389/fimmu.2019.00877
101. Zheng Y, Li Y, Lian J, Yang H, Li F, Zhao S, et al. TNF-α-induced Tim-3 expression marks the dysfunction of infiltrating natural killer cells in human esophageal cancer. J Transl Med. (2019) 17:165. doi: 10.1186/s12967-019-1917-0
102. Gur C, Ibrahim Y, Isaacson B, Yamin R, Abed J, Gamliel M, et al. Binding of the Fap2 protein of fusobacterium nucleatum to human inhibitory receptor TIGIT protects tumors from immune cell attack. Immunity. (2015) 42:344–55. doi: 10.1016/j.immuni.2015.01.010
103. Zhang Q, Bi J, Zheng X, Chen Y, Wang H, Wu W, et al. Blockade of the checkpoint receptor TIGIT prevents NK cell exhaustion and elicits potent anti-tumor immunity. Nat Immunol. (2018) 19:723–32. doi: 10.1038/s41590-018-0132-0
104. Sarhan D, Cichocki F, Zhang B, Yingst A, Spellman SR, Cooley S, et al. Adaptive NK cells with low TIGIT expression are inherently resistant to myeloid-derived suppressor cells. Cancer Res. (2016) 76:5696–706. doi: 10.1158/0008-5472.CAN-16-0839
105. Xu F, Sunderland A, Zhou Y, Schulick RD, Edil BH, Zhu Y. Blockade of CD112R and TIGIT signaling sensitizes human natural killer cell functions. Cancer Immunol Immunother. (2017) 66:1367–75. doi: 10.1007/s00262-017-2031-x
106. Sun H, Huang Q, Huang M, Wen H, Lin R, Zheng M, et al. Human CD96 correlates to natural killer cell exhaustion and predicts the prognosis of human hepatocellular carcinoma. Hepatology. (2019) 70:168–83. doi: 10.1002/hep.30347
107. Chan CJ, Martinet L, Gilfillan S, Souza-Fonseca-Guimaraes F, Chow MT, Town L, et al. The receptors CD96 and CD226 oppose each other in the regulation of natural killer cell functions. Nat Immunol. (2014) 15:431–8. doi: 10.1038/ni.2850
108. Blake SJ, Stannard K, Liu J, Allen S, Yong MCR, Mittal D, et al. Suppression of Metastases using a new lymphocyte checkpoint target for cancer immunotherapy. Cancer Discov. (2016) 6:446–59. doi: 10.1158/2159-8290.CD-15-0944
109. Roman Aguilera A, Lutzky VP, Mittal D, Li X-Y, Stannard K, Takeda K, et al. CD96 targeted antibodies need not block CD96-CD155 interactions to promote NK cell anti-metastatic activity. Oncoimmunology. (2018) 7:e1424677. doi: 10.1080/2162402X.2018.1424677
110. Frazao A, Messaoudene M, Nunez N, Dulphy N, Roussin F, Sedlik C, et al. CD16 + NKG2A high natural killer cells infiltrate breast cancer–draining lymph nodes. Cancer Immunol Res. (2019) 7:208–18. doi: 10.1158/2326-6066.CIR-18-0085
111. Modak S, Le Luduec J-B., Cheung IY, Goldman DA, Ostrovnaya I, Doubrovina E, et al. Adoptive immunotherapy with haploidentical natural killer cells and Anti-GD2 monoclonal antibody m3F8 for resistant neuroblastoma: Results of a phase I study. Oncoimmunology. (2018) 7:e1461305. doi: 10.1080/2162402X.2018.1461305
112. McWilliams EM, Mele JM, Cheney C, Timmerman EA, Fiazuddin F, Strattan EJ, et al. Therapeutic CD94/NKG2A blockade improves natural killer cell dysfunction in chronic lymphocytic leukemia. Oncoimmunology. (2016) 5:e1226720. doi: 10.1080/2162402X.2016.1226720
113. Tognarelli S, Wirsching S, von Metzler I, Rais B, Jacobs B, Serve H, et al. Enhancing the activation and releasing the brakes: a double hit strategy to improve NK cell cytotoxicity against multiple myeloma. Front Immunol. (2018) 9:2743. doi: 10.3389/fimmu.2018.02743
114. Ishida Y, Agata Y, Shibahara K, Honjo T. Induced expression of PD-1, a novel member of the immunoglobulin gene superfamily, upon programmed cell death. EMBO J. (1992) 11:3887–95. doi: 10.1002/j.1460-2075.1992.tb05481.x
115. Blank C, Mackensen A. Contribution of the PD-L1/PD-1 pathway to T-cell exhaustion: an update on implications for chronic infections and tumor evasion. Cancer Immunol Immunother. (2007) 56:739–45. doi: 10.1007/s00262-006-0272-1
116. Mariotti FR, Petrini S, Ingegnere T, Tumino N, Besi F, Scordamaglia F, et al. PD-1 in human NK cells: evidence of cytoplasmic mRNA and protein expression. Oncoimmunology. (2019) 8:1557030. doi: 10.1080/2162402X.2018.1557030
117. Pesce S, Greppi M, Tabellini G, Rampinelli F, Parolini S, Olive D, et al. Identification of a subset of human natural killer cells expressing high levels of programmed death 1: A phenotypic and functional characterization. J Allergy Clin Immunol. (2017) 139:335–6.e3. doi: 10.1016/j.jaci.2016.04.025
118. Della Chiesa M, Pesce S, Muccio L, Carlomagno S, Sivori S, Moretta A, et al. Features of Memory-Like and PD-1+ Human NK Cell Subsets. Front Immunol. (2016) 7:351. doi: 10.3389/fimmu.2016.00351
119. Nagato T, Ohkuri T, Ohara K, Hirata Y, Kishibe K, Komabayashi Y, et al. Programmed death-ligand 1 and its soluble form are highly expressed in nasal natural killer/T-cell lymphoma: a potential rationale for immunotherapy. Cancer Immunol Immunother. (2017) 66:877–90. doi: 10.1007/s00262-017-1987-x
120. Hsu J, Hodgins JJ, Marathe M, Nicolai CJ, Bourgeois-Daigneault M-C, Trevino TN, et al. Contribution of NK cells to immunotherapy mediated by PD-1/PD-L1 blockade. J Clin Investig. (2018) 128:4654–68. doi: 10.1172/JCI99317
121. Dong W, Wu X, Ma S, Wang Y, Nalin AP, Zhu Z, et al. The mechanism of anti-PD-L1 antibody efficacy against PD-L1 negative tumors identifies NK cells expressing PD-L1 as a cytolytic effector. Cancer Discov. (2019) 9:1422–37. doi: 10.1158/2159-8290.CD-18-1259
122. Ndhlovu LC, Lopez-Verges S, Barbour JD, Jones RB, Jha AR, Long BR, et al. Tim-3 marks human natural killer cell maturation and suppresses cell-mediated cytotoxicity. Blood. (2012) 119:3734–43. doi: 10.1182/blood-2011-11-392951
123. Gleason MK, Lenvik TR, McCullar V, Felices M, O'Brien MS, Cooley SA, et al. Tim-3 is an inducible human natural killer cell receptor that enhances interferon gamma production in response to galectin-9. Blood. (2012) 119:3064–72. doi: 10.1182/blood-2011-06-360321
124. Nakayama M, Akiba H, Takeda K, Kojima Y, Hashiguchi M, Azuma M, et al. Tim-3 mediates phagocytosis of apoptotic cells and cross-presentation. Blood. (2009) 113:3821–30. doi: 10.1182/blood-2008-10-185884
125. Huang Y-H, Zhu C, Kondo Y, Anderson AC, Gandhi A, Russell A, et al. CEACAM1 regulates TIM-3-mediated tolerance and exhaustion. Nature. (2015) 517:386–90. doi: 10.1038/nature13848
126. Koyama S, Akbay EA, Li YY, Herter-Sprie GS, Buczkowski KA, Richards WG, et al. Adaptive resistance to therapeutic PD-1 blockade is associated with upregulation of alternative immune checkpoints. Nat Commun. (2016) 7:10501. doi: 10.1038/ncomms10501
127. Yu X, Harden K, C Gonzalez L, Francesco M, Chiang E, Irving B, et al. The surface protein TIGIT suppresses T cell activation by promoting the generation of mature immunoregulatory dendritic cells. Nat Immunol. (2009) 10:48–57. doi: 10.1038/ni.1674
128. Stanietsky N, Simic H, Arapovic J, Toporik A, Levy O, Novik A, et al. The interaction of TIGIT with PVR and PVRL2 inhibits human NK cell cytotoxicity. Proc Natl Acad Sci USA. (2009) 106:17858–63. doi: 10.1073/pnas.0903474106
129. Chauvin J-M, Pagliano O, Fourcade J, Sun Z, Wang H, Sander C, et al. TIGIT and PD-1 impair tumor antigen–specific CD8+ T cells in melanoma patients. J Clin Invest. (2015) 125:2046–58. doi: 10.1172/JCI80445
130. Fuchs A, Cella M, Giurisato E, Shaw AS, Colonna M. Cutting edge: CD96 (Tactile) promotes NK cell-target cell adhesion by interacting with the poliovirus receptor (CD155). J Immunol. (2004) 172:3994–8. doi: 10.4049/jimmunol.172.7.3994
131. Xu F, Liu J, Liu D, Liu B, Wang M, Hu Z, et al. LSECtin expressed on melanoma cells promotes tumor progression by inhibiting antitumor T-cell responses. Cancer Res. (2014) 74:3418–28. doi: 10.1158/0008-5472.CAN-13-2690
132. Triebel F, Jitsukawa S, Baixeras E, Roman-Roman S, Genevee C, Viegas-Pequignot E, et al. LAG-3, a novel lymphocyte activation gene closely related to CD4. J Exp Med. (1990) 171:1393–405. doi: 10.1084/jem.171.5.1393
133. Baixeras E. Characterization of the lymphocyte activation gene 3-encoded protein. A new ligand for human leukocyte antigen class II antigens. J Exp Med. (1992) 176:327–37. doi: 10.1084/jem.176.2.327
134. Miyazaki T, Dierich A, Benoist C, Mathis D. Independent modes of natural killing distinguished in mice lacking Lag3. Science. (1996) 272:405–8. doi: 10.1126/science.272.5260.405
135. Huard B, Tournier M, Triebel F. LAG-3 does not define a specific mode of natural killing in human. Immunol Lett. (1998) 61:109–12. doi: 10.1016/S0165-2478(97)00170-3
136. Merino AM, Zhang B, Dougherty PR, Luo X, Wang J, Blazar BR, et al. Chronic stimulation drives human NK cell dysfunction and epigenetic reprograming. J Clin Invest. (2019) 130: 3770–85. doi: 10.1172/JCI125916
137. Ohs I, Ducimetière L, Marinho J, Kulig P, Becher B, Tugues S. Restoration of natural killer cell antimetastatic activity by IL12 and checkpoint blockade. Cancer Res. (2017) 77:7059–71. doi: 10.1158/0008-5472.CAN-17-1032
138. Fried S, Matalon O, Noy E, Barda-Saad M. WIP: more than a WASp-interacting protein. J Leukoc Biol. (2014) 96:713–27. doi: 10.1189/jlb.2RU0314-162R
139. Orange JS, Ramesh N, Remold-O'Donnell E, Sasahara Y, Koopman L, Byrne M, et al. Wiskott-Aldrich syndrome protein is required for NK cell cytotoxicity and colocalizes with actin to NK cell-activating immunologic synapses. Proc Natl Acad Sci USA. (2002) 99:11351–6. doi: 10.1073/pnas.162376099
140. Krzewski K, Chen X, Strominger JL. WIP is essential for lytic granule polarization and NK cell cytotoxicity. Proc Natl Acad Sci USA. (2008) 105:2568–73. doi: 10.1073/pnas.0711593105
141. Fried S, Reicher B, Pauker MH, Eliyahu S, Matalon O, Noy E, et al. Triple-color FRET analysis reveals conformational changes in the WIP-WASp actin-regulating complex. Sci. Signal. (2014) 7:ra60. doi: 10.1126/scisignal.2005198
142. Haba NY, Gross R, Novacek J, Shaked H, Zidek L, Barda-Saad M, et al. NMR determines transient structure and dynamics in the disordered C-terminal domain of WASp interacting protein. Biophys J. (2013) 105:481–93. doi: 10.1016/j.bpj.2013.05.046
143. Kritikou JS, Dahlberg CIM, Baptista MAP, Wagner AK, Banerjee PP, Gwalani LA, et al. IL-2 in the tumor microenvironment is necessary for Wiskott-Aldrich syndrome protein deficient NK cells to respond to tumors in vivo. Sci Rep. (2016) 6:30636. doi: 10.1038/srep30636
144. Monaco EL, Tremante E, Cerboni C, Melucci E, Sibilio L, Zingoni A, et al. Human leukocyte antigen E contributes to protect tumor cells from lysis by natural killer cells. Neoplasia. (2011) 13:822-IN14. doi: 10.1593/neo.101684
145. Romagné F, André P, Spee P, Zahn S, Anfossi N, Gauthier L, et al. Preclinical characterization of 1-7F9, a novel human anti-KIR receptor therapeutic antibody that augments natural killer-mediated killing of tumor cells. Blood. (2009) 114:2667–77. doi: 10.1182/blood-2009-02-206532
146. Pende D, Falco M, Vitale M, Cantoni C, Vitale C, Munari E, et al. Killer Ig-Like receptors (KIRs): their role in NK cell modulation and developments leading to their clinical exploitation. Front Immunol. (2019) 10:1179. doi: 10.3389/fimmu.2019.01179
147. Zaghi E, Calvi M, Marcenaro E, Mavilio D, Di Vito C. Targeting NKG2A to elucidate natural killer cell ontogenesis and to develop novel immune-therapeutic strategies in cancer therapy. J Leukoc Biol. (2019) 105:1243–51. doi: 10.1002/JLB.MR0718-300R
148. Benson DM, Cohen AD, Jagannath S, Munshi NC, Spitzer G, Hofmeister CC, et al. A phase I trial of the anti-KIR antibody IPH2101 and lenalidomide in patients with relapsed/refractory multiple myeloma. Clin Cancer Res. (2015) 21:4055–61. doi: 10.1158/1078-0432.CCR-15-0304
149. Van Der Weyden C, Bagot M, Neeson P, Darcy PK, Prince HM. IPH4102, a monoclonal antibody directed against the immune receptor molecule KIR3DL2, for the treatment of cutaneous T-cell lymphoma. Expert Opin Investig Drugs. (2018) 27:691–7. doi: 10.1080/13543784.2018.1498081
150. Benson DM, Bakan CE, Zhang S, Collins SM, Liang J, Srivastava S, et al. IPH2101, a novel anti-inhibitory KIR antibody, and lenalidomide combine to enhance the natural killer cell versus multiple myeloma effect. Blood. (2011) 118:6387–91. doi: 10.1182/blood-2011-06-360255
151. Kohrt HE, Thielens A, Marabelle A, Sagiv-Barfi I, Sola C, Chanuc F, et al. Anti-KIR antibody enhancement of anti-lymphoma activity of natural killer cells as monotherapy and in combination with anti-CD20 antibodies. Blood. (2014) 123:678–86. doi: 10.1182/blood-2013-08-519199
152. Benson DM, Hofmeister CC, Padmanabhan S, Suvannasankha A, Jagannath S, Abonour R, et al. A phase 1 trial of the anti-KIR antibody IPH2101 in patients with relapsed/refractory multiple myeloma. Blood. (2012) 120:4324–33. doi: 10.1182/blood-2012-06-438028
153. Vey N, Bourhis J-H, Boissel N, Bordessoule D, Prebet T, Charbonnier A, et al. A phase 1 trial of the anti-inhibitory KIR mAb IPH2101 for AML in complete remission. Blood. (2012) 120:4317–23. doi: 10.1182/blood-2012-06-437558
154. Vey N, Dumas P-Y, Recher C, Gastaud L, Lioure B, Bulabois C-E, et al. Blood. (2017) 130:889. doi: 10.1182/blood.V130.Suppl_1.889.889
155. Korde N, Carlsten M, Lee M-J, Minter A, Tan E, Kwok M, et al. A phase II trial of pan-KIR2D blockade with IPH2101 in smoldering multiple myeloma. Haematologica. (2014) (2014) 99:e81–3. doi: 10.3324/haematol.2013.103085
156. Yokoyama WM, Kim S. Licensing of natural killer cells by self-major histocompatibility complex class I. Immunol Rev. (2006) 214:143–54. doi: 10.1111/j.1600-065X.2006.00458.x
157. Anfossi N, André P, Guia S, Falk CS, Roetynck S, Stewart CA, et al. Human NK cell education by inhibitory receptors for MHC class I. Immunity. (2006) 25:331–42. doi: 10.1016/j.immuni.2006.06.013
158. Carlsten M, Korde N, Kotecha R, Reger R, Bor S, Kazandjian D, et al. Checkpoint inhibition of KIR2D with the monoclonal antibody IPH2101 induces contraction and hyporesponsiveness of NK cells in patients with myeloma. Clin Cancer Res. (2016) 22:5211–22. doi: 10.1158/1078-0432.CCR-16-1108
159. He Y, Liu S, Mattei J, Bunn Jr P, Zhou C, Chan D. The combination of anti-KIR monoclonal antibodies with anti-PD-1/PD-L1 monoclonal antibodies could be a critical breakthrough in overcoming tumor immune escape in NSCLC. Drug Des Dev Ther. (2018) 12:981–6. doi: 10.2147/DDDT.S163304
160. Levy EM, Bianchini M, Von Euw EM, Barrio MM, Bravo AI, Furman D, et al. Human leukocyte antigen-E protein is overexpressed in primary human colorectal cancer. Int J Oncol. (2008) 32:633–41. doi: 10.3892/ijo.32.3.633
161. Segal NH, Naidoo J, Curigliano G, Patel S, Sahebjam S, Papadopoulos KP, et al. First-in-human dose escalation of monalizumab plus durvalumab, with expansion in patients with metastatic microsatellite-stable colorectal cancer. J Clin Oncol. (2018) 36:3540. doi: 10.1200/JCO.2018.36.15_suppl.3540
162. Weber JS, Yang JC, Atkins MB, Disis ML. Toxicities of immunotherapy for the practitioner. J Clin Oncol. (2015) 33:2092–9. doi: 10.1200/JCO.2014.60.0379
163. Gajewski TF, Woo S-R, Zha Y, Spaapen R, Zheng Y, Corrales L, et al. Cancer immunotherapy strategies based on overcoming barriers within the tumor microenvironment. Curr Opin Immunol. (2013) 25:268–76. doi: 10.1016/j.coi.2013.02.009
164. Vasievich EA, Huang L. The suppressive tumor microenvironment: a challenge in cancer immunotherapy. Mol Pharm. (2011) 8:635–41. doi: 10.1021/mp1004228
165. Conlon KC, Lugli E, Welles HC, Rosenberg SA, Fojo AT, Morris JC, et al. Redistribution, hyperproliferation, activation of natural killer cells and CD8 T cells, and cytokine production during first-in-human clinical trial of recombinant human interleukin-15 in patients with cancer. J Clin Oncol. (2015) 33:74–82. doi: 10.1200/JCO.2014.57.3329
166. Grünwald V, Desar IME, Haanen J, Fiedler W, Mouritzen U, Olsen MWB, et al. A phase I study of recombinant human interleukin-21 (rIL-21) in combination with sunitinib in patients with metastatic renal cell carcinoma (RCC). Acta Oncol. (2011) 50:121–6. doi: 10.3109/0284186X.2010.509104
167. Leonard JP, Sherman ML, Fisher GL, Buchanan LJ, Larsen G, Atkins MB, et al. Effects of single-dose interleukin-12 exposure on interleukin-12-associated toxicity and interferon-gamma production. Blood. (1997) 90:2541–8.
168. Guo Y, Luan L, Rabacal W, Bohannon JK, Fensterheim BA, Hernandez A, et al. IL-15 Superagonist-mediated immunotoxicity: role of NK cells and IFN-γ. J Immunol. (2015) 195:2353–64. doi: 10.4049/jimmunol.1500300
169. Oyer JL, Gitto SB, Altomare DA, Copik AJ. PD-L1 blockade enhances anti-tumor efficacy of NK cells. Oncoimmunology. (2018) 7:e1509819. doi: 10.1080/2162402X.2018.1509819
170. Makowska A, Braunschweig T, Denecke B, Shen L, Baloche V, Busson P, et al. Interferon β and anti-PD-1/PD-L1 checkpoint blockade cooperate in NK cell-mediated killing of nasopharyngeal carcinoma cells. Transl Oncol. (2019) 12:1237–56. doi: 10.1016/j.tranon.2019.04.017
171. Wang DY, Johnson DB, Davis EJ. Toxicities associated with PD-1/PD-L1 blockade. Cancer J. (2018) 24:36–40. doi: 10.1097/PPO.0000000000000296
172. Gauthier L, Morel A, Anceriz N, Rossi B, Blanchard-Alvarez A, Grondin G, et al. Multifunctional natural killer cell engagers targeting NKp46 trigger protective tumor immunity. Cell. (2019) 177:1701–13.e16. doi: 10.1016/j.cell.2019.04.041
173. Boyiadzis M, Foon KA. Approved monoclonal antibodies for cancer therapy. Expert Opin Biol Ther. (2008) 8:1151–8. doi: 10.1517/14712598.8.8.1151
174. Scott AM, Wolchok JD, Old LJ. Antibody therapy of cancer. Nat Rev Cancer. (2012) 12:278–87. doi: 10.1038/nrc3236
175. Wang W, Erbe AK, Hank JA, Morris ZS, Sondel PM. NK cell-mediated antibody-dependent cellular cytotoxicity in cancer immunotherapy. Front Immunol. (2015) 6:368. doi: 10.3389/fimmu.2015.00368
176. Koerner SP, André MC, Leibold JS, Kousis PC, Kübler A, Pal M, et al. An Fc-optimized CD133 antibody for induction of NK cell reactivity against myeloid leukemia. Leukemia. (2017) 31:459–69. doi: 10.1038/leu.2016.194
177. Akkapeddi P, Fragoso R, Hixon JA, Ramalho AS, Oliveira ML, Carvalho T, et al. A fully human anti-IL-7Rα antibody promotes antitumor activity against T-cell acute lymphoblastic leukemia. Leukemia. (2019) 33:2155–68. doi: 10.1038/s41375-019-0434-8
178. Ferrari de Andrade L, Tay RE, Pan D, Luoma AM, Ito Y, Badrinath S, et al. Antibody-mediated inhibition of MICA and MICB shedding promotes NK cell–driven tumor immunity. Science. (2018) 359:1537–42. doi: 10.1126/science.aao0505
179. Ashraf Y, Mansouri H, Laurent-Matha V, Alcaraz LB, Roger P, Guiu S, et al. Immunotherapy of triple-negative breast cancer with cathepsin D-targeting antibodies. J Immunother Cancer. (2019) 7:29. doi: 10.1186/s40425-019-0498-z
180. Roda JM, Joshi T, Butchar JP, McAlees JW, Lehman A, Tridandapani S, et al. The activation of natural killer cell effector functions by cetuximab-coated, epidermal growth factor receptor positive tumor cells is enhanced by cytokines. Clin Cancer Res. (2007) 13:6419–28. doi: 10.1158/1078-0432.CCR-07-0865
181. McMichael EL, Jaime-Ramirez AC, Guenterberg KD, Luedke E, Atwal LS, Campbell AR, et al. IL-21 Enhances N2atural killer cell response to cetuximab-coated pancreatic tumor cells. Clin Cancer Res. (2017) 23:489–502. doi: 10.1158/1078-0432.CCR-16-0004
182. Srivastava RM, Lee SC, Andrade Filho PA, Lord CA, Jie H-B, Davidson HC, et al. Cetuximab-activated natural killer and dendritic cells collaborate to trigger tumor antigen-specific T-cell immunity in head and neck cancer patients. Clin Cancer Res. (2013) 19:1858–72. doi: 10.1158/1078-0432.CCR-12-2426
183. Taylor RJ, Saloura V, Jain A, Goloubeva O, Wong S, Kronsberg S, et al. Ex vivo antibody-dependent cellular cytotoxicity inducibility predicts efficacy of cetuximab. Cancer Immunol Res. (2015) 3:567–74. doi: 10.1158/2326-6066.CIR-14-0188
184. López-Albaitero A, Lee SC, Morgan S, Grandis JR, Gooding WE, Ferrone S, et al. Role of polymorphic Fc gamma receptor IIIa and EGFR expression level in cetuximab mediated, NK cell dependent in vitro cytotoxicity of head and neck squamous cell carcinoma cells. Cancer Immunol Immunother. (2009) 58:1853–62. doi: 10.1007/s00262-009-0697-4
185. Musolino A, Naldi N, Bortesi B, Pezzuolo D, Capelletti M, Missale G, et al. Immunoglobulin G fragment C receptor polymorphisms and clinical efficacy of trastuzumab-based therapy in patients with HER-2/neu-positive metastatic breast cancer. J Clin Oncol. (2008) 26:1789–96. doi: 10.1200/JCO.2007.14.8957
186. Mellor JD, Brown MP, Irving HR, Zalcberg JR, Dobrovic A. A critical review of the role of Fc gamma receptor polymorphisms in the response to monoclonal antibodies in cancer. J Hematol Oncol. (2013) 6:1. doi: 10.1186/1756-8722-6-1
187. Trotta AM, Ottaiano A, Romano C, Nasti G, Nappi A, De Divitiis C, et al. Prospective evaluation of cetuximab-mediated antibody-dependent cell cytotoxicity in metastatic colorectal cancer patients predicts treatment efficacy. Cancer Immunol Res. (2016) 4:366–74. doi: 10.1158/2326-6066.CIR-15-0184
188. Liu G, Tu D, Lewis M, Cheng D, Sullivan LA, Chen Z, et al. Fc-receptor polymorphisms, cetuximab therapy, and survival in the NCIC CTG CO.17 trial of colorectal cancer. Clin Cancer Res. (2016) 22:2435–44. doi: 10.1158/1078-0432.CCR-15-0414
189. Kaifu T, Nakamura A. Polymorphisms of immunoglobulin receptors and the effects on clinical outcome in cancer immunotherapy and other immune diseases: a general review. Int Immunol. (2017) 29:319–25. doi: 10.1093/intimm/dxx041
190. Wang X, Mathieu M, Brezski RJ. IgG Fc engineering to modulate antibody effector functions. Protein Cell. (2018) 9:63–73. doi: 10.1007/s13238-017-0473-8
191. Bruhns P, Iannascoli B, England P, Mancardi DA, Fernandez N, Jorieux S, et al. Specificity and affinity of human Fcgamma receptors and their polymorphic variants for human IgG subclasses. Blood. (2009) 113:3716–25. doi: 10.1182/blood-2008-09-179754
192. Ferrara C, Grau S, Jager C, Sondermann P, Brunker P, Waldhauer I, et al. Unique carbohydrate-carbohydrate interactions are required for high affinity binding between Fc RIII and antibodies lacking core fucose. Proc Natl Acad Sci USA. (2011) 108:12669–74. doi: 10.1073/pnas.1108455108
193. Kondadasula SV, Roda JM, Parihar R, Yu J, Lehman A, Caligiuri MA, et al. Colocalization of the IL-12 receptor and FcgammaRIIIa to natural killer cell lipid rafts leads to activation of ERK and enhanced production of interferon-gamma. Blood. (2008) 111:4173–83. doi: 10.1182/blood-2007-01-068908
194. McMichael EL, Benner B, Atwal LS, Courtney NB, Mo X, Davis ME, et al. A Phase I/II trial of cetuximab in combination with interleukin-12 administered to patients with unresectable primary or recurrent head and neck squamous cell carcinoma. Clin Cancer Res. (2019) 25:4955–65. doi: 10.1158/1078-0432.CCR-18-2108
195. Jie H-B, Schuler PJ, Lee SC, Srivastava RM, Argiris A, Ferrone S, et al. CTLA-4 + regulatory T cells increased in cetuximab-treated head and neck cancer patients suppress NK cell cytotoxicity and correlate with poor prognosis. Cancer Res. (2015) 75:2200–10. doi: 10.1158/0008-5472.CAN-14-2788
196. Roberti MP, Barrio MM, Bravo AI, Rocca YS, Arriaga JM, Bianchini M, et al. IL-15 and IL-2 increase Cetuximab-mediated cellular cytotoxicity against triple negative breast cancer cell lines expressing EGFR. Breast Cancer Res Treat. (2011) 130:465–75. doi: 10.1007/s10549-011-1360-2
197. Steele N, Anthony A, Saunders M, Esmarck B, Ehrnrooth E, Kristjansen PEG, et al. A phase 1 trial of recombinant human IL-21 in combination with cetuximab in patients with metastatic colorectal cancer. Br J Cancer. (2012) 106:793–8. doi: 10.1038/bjc.2011.599
198. Luedke E, Jaime-Ramirez AC, Bhave N, Roda J, Choudhary MM, Kumar B, et al. Cetuximab therapy in head and neck cancer: Immune modulation with interleukin-12 and other natural killer cell–activating cytokines. Surgery. (2012) 152:431–40. doi: 10.1016/j.surg.2012.05.035
199. Ming Lim C, Stephenson R, Salazar AM, Ferris RL. TLR3 agonists improve the immunostimulatory potential of cetuximab against EGFR + head and neck cancer cells. Oncoimmunology. (2013) 2:e24677. doi: 10.4161/onci.24677
200. Srivastava RM, Trivedi S, Concha-Benavente F, Gibson SP, Reeder C, Ferrone S, et al. CD137 stimulation enhances cetuximab-induced natural killer: dendritic cell priming of antitumor T-cell immunity in patients with head and neck cancer. Clin Cancer Res. (2017) 23:707–16. doi: 10.1158/1078-0432.CCR-16-0879
201. Pinette A, McMichael E, Courtney NB, Duggan M, Benner BN, Choueiry F, et al. An IL-15-based superagonist ALT-803 enhances the NK cell response to cetuximab-treated squamous cell carcinoma of the head and neck. Cancer Immunol Immunother. (2019) 68:1379–89. doi: 10.1007/s00262-019-02372-2
202. Mazorra Z, Lavastida A, Concha-Benavente F, Valdés A, Srivastava RM, García-Bates TM, et al. Nimotuzumab induces NK cell activation, cytotoxicity, dendritic cell maturation and expansion of EGFR-specific T cells in head and neck cancer patients. Front Pharmacol. (2017) 8:382. doi: 10.3389/fphar.2017.00382
203. Roda JM, Parihar R, Magro C, Nuovo GJ, Tridandapani S, Carson WE. Natural killer cells produce T cell–recruiting chemokines in response to antibody-coated tumor cells. Cancer Res. (2006) 66:517–26. doi: 10.1158/0008-5472.CAN-05-2429
204. Arnould L, Gelly M, Penault-Llorca F, Benoit L, Bonnetain F, Migeon C, et al. Trastuzumab-based treatment of HER2-positive breast cancer: an antibody-dependent cellular cytotoxicity mechanism? Br J Cancer. (2006) 94:259–67. doi: 10.1038/sj.bjc.6602930
205. Roda JM, Parihar R, Lehman A, Mani A, Tridandapani S, Carson WE. Interleukin-21 enhances NK cell activation in response to antibody-coated targets. J Immunol. (2006) 177:120–9. doi: 10.4049/jimmunol.177.1.120
206. Beano A, Signorino E, Evangelista A, Brusa D, Mistrangelo M, Polimeni M, et al. Correlation between NK function and response to trastuzumab in metastatic breast cancer patients. J Transl Med. (2008) 6:25. doi: 10.1186/1479-5876-6-25
207. Mani A, Roda J, Young D, Caligiuri MA, Fleming GF, Kaufman P, et al. A phase II trial of trastuzumab in combination with low-dose interleukin-2 (IL-2) in patients (PTS) with metastatic breast cancer (MBC) who have previously failed trastuzumab. Breast Cancer Res Treat. (2009) 117:83–9. doi: 10.1007/s10549-008-0251-7
208. Ishikawa T, Okayama T, Sakamoto N, Ideno M, Oka K, Enoki T, et al. Phase I clinical trial of adoptive transfer of expanded natural killer cells in combination with I g G 1 antibody in patients with gastric or colorectal cancer. Int J Cancer. (2018) 142:2599–609. doi: 10.1002/ijc.31285
209. Rexer BN, Arteaga CL. Intrinsic and acquired resistance to HER2-targeted therapies in HER2 gene-amplified breast cancer: mechanisms and clinical implications. Crit Rev Oncog. (2012) 17:1–16. doi: 10.1615/CritRevOncog.v17.i1.20
210. Muntasell A, Servitja S, Cabo M, Bermejo B, Pérez-Buira S, Rojo F, et al. High numbers of circulating CD57 + NK cells associate with resistance to HER2-specific therapeutic antibodies in HER2 + primary breast cancer. Cancer Immunol Res. (2019) 7:1280–92. doi: 10.1158/2326-6066.CIR-18-0896
211. Levy E, Reger R, Segerberg F, Lambert M, Leijonhufvud C, Baumer Y, et al. Enhanced bone marrow homing of natural killer cells following mRNA transfection with gain-of-function variant CXCR4R334X. Front Immunol. (2019) 10:1262. doi: 10.3389/fimmu.2019.01262
212. Lee J, Kang TH, Yoo W, Choi H, Jo S, Kong K, et al. An antibody designed to improve adoptive NK-cell therapy inhibits pancreatic cancer progression in a murine model. Cancer Immunol Res. (2018) 7:219–29. doi: 10.1158/2326-6066.CIR-18-0317
213. Muntasell A, Cabo M, Servitja S, Tusquets I, Martínez-García M, Rovira A, et al. Interplay between natural killer cells and anti-HER2 antibodies: perspectives for breast cancer immunotherapy. Front Immunol. (2017) 8:1544. doi: 10.3389/fimmu.2017.01544
214. Oberg HH, Kellner C, Gonnermann D, Sebens S, Bauerschlag D, Gramatzki M, et al. Tribody [(HER2)2xCD16] is more effective than trastuzumab in enhancing γδ T cell and natural killer cell cytotoxicity against HER2-expressing cancer cells. Front Immunol. (2018) 9:814. doi: 10.3389/fimmu.2018.00814
215. Motta G, Cea M, Moran E, Carbone F, Augusti V, Patrone F, et al. Monoclonal antibodies for non-Hodgkin's lymphoma: state of the art and perspectives. Clin Dev Immunol. (2010) 2010:428253. doi: 10.1155/2010/428253
216. Dall'Ozzo S, Tartas S, Paintaud G, Cartron G, Colombat P, Bardos P, et al. Rituximab-dependent cytotoxicity by natural killer cells. Cancer Res. (2004) 64:4664–9. doi: 10.1158/0008-5472.CAN-03-2862
217. Clynes RA, Towers TL, Presta LG, Ravetch JV. Inhibitory Fc receptors modulate in vivo cytoxicity against tumor targets. Nat Med. (2000) 6:443–6. doi: 10.1038/74704
218. Gonzalez-Rodriguez AP, Villa-Álvarez M, Sordo-Bahamonde C, Lorenzo-Herrero S, Gonzalez S. NK cells in the treatment of hematological malignancies. J Clin Med. (2019) 8:1557. doi: 10.3390/jcm8101557
219. Mössner E, Brünker P, Moser S, Püntener U, Schmidt C, Herter S, et al. Increasing the efficacy of CD20 antibody therapy through the engineering of a new type II anti-CD20 antibody with enhanced direct and immune effector cell–mediated B-cell cytotoxicity. Blood. (2010) 115:4393–402. doi: 10.1182/blood-2009-06-225979
220. Terszowski G, Klein C, Stern M. KIR/HLA interactions negatively affect rituximab- but not GA101 (Obinutuzumab)-induced antibody-dependent cellular cytotoxicity. J Immunol. (2014) 192:5618–24. doi: 10.4049/jimmunol.1400288
221. Bologna L, Gotti E, Manganini M, Rambaldi A, Intermesoli T, Introna M, et al. Mechanism of action of type II, glycoengineered, anti-CD20 MONOCLONAL ANTIBODY GA101 in B-Chronic lymphocytic leukemia whole blood assays in comparison with rituximab and alemtuzumab. J Immunol. (2011) 186:3762–9. doi: 10.4049/jimmunol.1000303
222. Merkt W, Lorenz H-M, Watzl C. Rituximab induces phenotypical and functional changes of NK cells in a non-malignant experimental setting. Arthritis Res Ther. (2016) 18:206. doi: 10.1186/s13075-016-1101-3
223. Rudnicka D, Oszmiana A, Finch DK, Strickland I, Schofield DJ, Lowe DC, et al. Rituximab causes a polarization of B cells that augments its therapeutic function in NK-cell–mediated antibody-dependent cellular cytotoxicity. Blood. (2013) 121:4694–702. doi: 10.1182/blood-2013-02-482570
224. Veuillen C, Aurran-Schleinitz T, Castellano R, Rey J, Mallet F, Orlanducci F, et al. Primary B-CLL resistance to NK cell cytotoxicity can be overcome in vitro and in vivo by priming NK cells and monoclonal antibody therapy. J Clin Immunol. (2012) 32:632–46. doi: 10.1007/s10875-011-9624-5
225. Klanova M, Oestergaard MZ, Trněný M, Hiddemann W, Marcus R, Sehn LH, et al. Prognostic impact of natural killer cell count in follicular lymphoma and diffuse large B-cell lymphoma patients treated with immunochemotherapy. Clin Cancer Res. (2019) 25:4634–43. doi: 10.1158/1078-0432.CCR-18-3270
226. Zhang M, Wen B, Anton OM, Yao Z, Dubois S, Ju W, et al. IL-15 enhanced antibody-dependent cellular cytotoxicity mediated by NK cells and macrophages. Proc Natl Acad Sci USA. (2018) 115:E10915–24. doi: 10.1073/pnas.1811615115
227. Bachanova V, Sarhan D, DeFor TE, Cooley S, Panoskaltsis-Mortari A, Blazar BR, et al. Haploidentical natural killer cells induce remissions in non-Hodgkin lymphoma patients with low levels of immune-suppressor cells. Cancer Immunol Immunother. (2018) 67:483–94. doi: 10.1007/s00262-017-2100-1
228. Rosario M, Liu B, Kong L, Collins LI, Schneider SE, Chen X, et al. The IL-15-based ALT-803 complex enhances Fc RIIIa-triggered NK cell responses and in vivo clearance of B Cell lymphomas. Clin Cancer Res. (2016) 22:596–608. doi: 10.1158/1078-0432.CCR-15-1419
229. Vo D-N, Alexia C, Allende-Vega N, Morschhauser F, Houot R, Menard C, et al. NK cell activation and recovery of NK cell subsets in lymphoma patients after obinutuzumab and lenalidomide treatment. Oncoimmunology. (2018) 7:e1409322. doi: 10.1080/2162402X.2017.1409322
230. Wu L, Adams M, Carter T, Chen R, Muller G, Stirling D, et al. Lenalidomide enhances natural killer cell and monocyte-mediated antibody-dependent cellular cytotoxicity of rituximab-treated CD20+ tumor cells. Clin Cancer Res. (2008) 14:4650–7. doi: 10.1158/1078-0432.CCR-07-4405
231. Lagrue K, Carisey A, Morgan DJ, Chopra R, Davis DM. Lenalidomide augments actin remodeling and lowers NK-cell activation thresholds. Blood. (2015) 126:50–60. doi: 10.1182/blood-2015-01-625004
232. Molfetta R, Zingoni A, Santoni A, Paolini R. Post-translational mechanisms regulating NK cell activating receptors and their ligands in cancer: potential targets for therapeutic intervention. Front Immunol. (2019) 10:2557. doi: 10.3389/fimmu.2019.02557
233. Mejstríková E, Hrusak O, Borowitz MJ, Whitlock JA, Brethon B, Trippett TM, et al. CD19-negative relapse of pediatric B-cell precursor acute lymphoblastic leukemia following blinatumomab treatment. Blood Cancer J. (2017) 7:659. doi: 10.1038/s41408-017-0023-x
234. Vicioso Y, Gram H, Beck R, Asthana A, Zhang K, Wong DP, et al. Combination therapy for treating advanced drug-resistant acute lymphoblastic leukemia. Cancer Immunol Res. (2019) 7:1106–1119. doi: 10.1158/2326-6066.CIR-19-0058
235. Krupka C, Lichtenegger FS, Köhnke T, Bögeholz J, Bücklein V, Roiss M, et al. Targeting CD157 in AML using a novel, Fc-engineered antibody construct. Oncotarget. (2017) 8:35707–17. doi: 10.18632/oncotarget.16060
236. Salomé B, Gomez-Cadena A, Loyon R, Suffiotti M, Salvestrini V, Wyss T, et al. CD56 as a marker of an ILC1-like population with NK cell properties that is functionally impaired in AML. Blood Adv. (2019) 3:3674–87. doi: 10.1182/bloodadvances.2018030478
237. Chiu J, Ernst DM, Keating A. Acquired natural killer cell dysfunction in the tumor microenvironment of classic hodgkin lymphoma. Front Immunol. (2018) 9:267. doi: 10.3389/fimmu.2018.00267
238. Busfield SJ, Biondo M, Wong M, Ramshaw HS, Lee EM, Ghosh S, et al. Targeting of acute myeloid leukemia in vitro and in vivo with an anti-CD123 mAb engineered for optimal ADCC. Leukemia. (2014) 28:2213–21. doi: 10.1038/leu.2014.128
239. Ernst D, Williams BA, Wang X-H, Yoon N, Kim K-P, Chiu J, et al. Humanized anti-CD123 antibody facilitates NK cell antibody-dependent cell-mediated cytotoxicity (ADCC) of Hodgkin lymphoma targets via ARF6/PLD-1. Blood Cancer J. (2019) 9:6. doi: 10.1038/s41408-018-0168-2
240. Lazar GA, Dang W, Karki S, Vafa O, Peng JS, Hyun L, et al. Engineered antibody Fc variants with enhanced effector function. Proc Natl Acad Sci USA. (2006) 103:4005–10. doi: 10.1073/pnas.0508123103
241. Roberts AW, He S, Ritchie D, Hertzberg MS, Kerridge I, Durrant ST, et al. A phase I study of anti-CD123 monoclonal antibody (mAb) CSL360 targeting leukemia stem cells (LSC) in AML. J Clin Oncol. (2010) 28:e13012. doi: 10.1200/jco.2010.28.15_suppl.e13012
242. Feldman EJ, Brandwein J, Stone R, Kalaycio M, Moore J, O'Connor J, et al. Phase III randomized multicenter study of a humanized anti-CD33 monoclonal antibody, lintuzumab, in combination with chemotherapy, versus chemotherapy alone in patients with refractory or first-relapsed acute myeloid leukemia. J Clin Oncol. (2005) 23:4110–6. doi: 10.1200/JCO.2005.09.133
243. Schmied BJ, Riegg F, Zekri L, Grosse-Hovest L, Bühring H-J, Jung G, et al. An Fc-optimized CD133 antibody for induction of natural killer cell reactivity against colorectal cancer. Cancers. (2019) 11:789. doi: 10.3390/cancers11060789
244. Hallner A, Bernson E, Hussein BA, Ewald Sander F, Brune M, Aurelius J, et al. The HLA-B −21 dimorphism impacts on NK cell education and clinical outcome of immunotherapy in acute myeloid leukemia. Blood. (2019) 133:1479–88. doi: 10.1182/blood-2018-09-874990
245. Mastaglio S, Wong E, Perera T, Ripley J, Blombery P, Smyth MJ, et al. Natural killer receptor ligand expression on acute myeloid leukemia impacts survival and relapse after chemotherapy. Blood Adv. (2018) 2:335–46. doi: 10.1182/bloodadvances.2017015230
246. Glumac PM, LeBeau AM. The role of CD133 in cancer: a concise review. Clin Transl Med. (2018) 7:18. doi: 10.1186/s40169-018-0198-1
247. Yin AH, Miraglia S, Zanjani ED, Almeida-Porada G, Ogawa M, Leary AG, et al. AC133, a novel marker for human hematopoietic stem and progenitor cells. Blood. (1997) 90:5002–12. doi: 10.1182/blood.V90.12.5002
248. Oliveira ML, Akkapeddi P, Ribeiro D, Melão A, Barata JT. IL-7R-mediated signaling in T-cell acute lymphoblastic leukemia: an update. Adv Biol Regul. (2019) 71:88–96. doi: 10.1016/j.jbior.2018.09.012
249. Shouse G, Nikolaenko L. Targeting the JAK/STAT pathway in T cell lymphoproliferative disorders. Curr Hematol Malig Rep. (2019) 14:570–6. doi: 10.1007/s11899-019-00545-5
250. Boieri M, Ulvmoen A, Sudworth A, Lendrem C, Collin M, Dickinson AM, et al. IL-12, IL-15, and IL-18 pre-activated NK cells target resistant T cell acute lymphoblastic leukemia and delay leukemia development in vivo. Oncoimmunology. (2017) 6:e1274478. doi: 10.1080/2162402X.2016.1274478
251. Wu J, Mishra HK, Walcheck B. Role of ADAM17 as a regulatory checkpoint of CD16A in NK cells and as a potential target for cancer immunotherapy. J Leukoc Biol. (2019) 105:1297–303. doi: 10.1002/JLB.2MR1218-501R
252. Harrison D, Phillips JH, Lanier LL. Involvement of a metalloprotease in spontaneous and phorbol ester-induced release of natural killer cell-associated Fc gamma RIII (CD16-II). J Immunol. (1991) 147:3459–65.
253. Romee R, Foley B, Lenvik T, Wang Y, Zhang B, Ankarlo D, et al. NK cell CD16 surface expression and function is regulated by a disintegrin and metalloprotease-17 (ADAM17). Blood. (2013) 121:3599–608. doi: 10.1182/blood-2012-04-425397
254. Srpan K, Ambrose A, Karampatzakis A, Saeed M, Cartwright ANR, Guldevall K, et al. Shedding of CD16 disassembles the NK cell immune synapse and boosts serial engagement of target cells. J Cell Biol. (2018) 217:3267–83. doi: 10.1083/jcb.201712085
255. Liu Q, Sun Y, Rihn S, Nolting A, Tsoukas PN, Jost S, et al. Matrix metalloprotease inhibitors restore impaired NK cell-mediated antibody-dependent cellular cytotoxicity in human immunodeficiency virus type 1 infection. J Virol. (2009) 83:8705–12. doi: 10.1128/JVI.02666-08
256. Hatjiharissi E, Xu L, Santos DD, Hunter ZR, Ciccarelli BT, Verselis S, et al. Increased natural killer cell expression of CD16, augmented binding and ADCC activity to rituximab among individuals expressing the Fc RIIIa-158 V/V and V/F polymorphism. Blood. (2007) 110:2561–2564. doi: 10.1182/blood-2007-01-070656
257. Pahl JHW, Koch J, Götz J-J, Arnold A, Reusch U, Gantke T, et al. CD16A activation of NK cells promotes NK cell proliferation and memory-like cytotoxicity against cancer cells. Cancer Immunol Res. (2018) 6:517–27. doi: 10.1158/2326-6066.CIR-17-0550
258. Pazina T, James AM, MacFarlane AW, Bezman NA, Henning KA, Bee C, et al. The anti-SLAMF7 antibody elotuzumab mediates NK cell activation through both CD16-dependent and –independent mechanisms. Oncoimmunology. (2017) 6:e1339853. doi: 10.1080/2162402X.2017.1339853
259. Barry WE, Jackson JR, Asuelime GE, Wu H-W, Sun J, Wan Z, et al. Activated natural killer cells in combination with anti-GD2 antibody dinutuximab improve survival of mice after surgical resection of primary neuroblastoma. Clin Cancer Res. (2019) 25:325–33. doi: 10.1158/1078-0432.CCR-18-1317
260. Federico SM, McCarville MB, Shulkin BL, Sondel PM, Hank JA, Hutson P, et al. A pilot trial of humanized Anti-GD2 monoclonal antibody (hu14.18K322A) with chemotherapy and natural killer cells in children with recurrent/refractory neuroblastoma. Clin Cancer Res. (2017) 23:6441–9. doi: 10.1158/1078-0432.CCR-17-0379
261. Juliá EP, Amante A, Pampena MB, Mordoh J, Levy EM. Avelumab, an IgG1 anti-PD-L1 immune checkpoint inhibitor, triggers NK cell-mediated cytotoxicity and cytokine production against triple negative breast cancer cells. Front Immunol. (2018) 9:2140. doi: 10.3389/fimmu.2018.02140
262. Mahaweni NM, Bos GMJ, Mitsiades CS, Tilanus MGJ, Wieten L. Daratumumab augments alloreactive natural killer cell cytotoxicity towards CD38+ multiple myeloma cell lines in a biochemical context mimicking tumour microenvironment conditions. Cancer Immunol Immunother. (2018) 67:861–72. doi: 10.1007/s00262-018-2140-1
263. Casneuf T, Xu XS, Adams HC, Axel AE, Chiu C, Khan I, et al. Effects of daratumumab on natural killer cells and impact on clinical outcomes in relapsed or refractory multiple myeloma. Blood Adv. (2017) 1:2105–14. doi: 10.1182/bloodadvances.2017006866
264. Chen G, Chen Y-C, Reis B, Belousov A, Jukofsky L, Rossin C, et al. Combining expression of GPC3 in tumors and CD16 on NK cells from peripheral blood to identify patients responding to codrituzumab. Oncotarget. (2018) 9:10436–44. doi: 10.18632/oncotarget.23830
265. Vasu S, He S, Cheney C, Gopalakrishnan B, Mani R, Lozanski G, et al. Decitabine enhances anti-CD33 monoclonal antibody BI 836858-mediated natural killer ADCC against AML blasts. Blood. (2016) 127:2879–89. doi: 10.1182/blood-2015-11-680546
266. Davis ZB, Vallera DA, Miller JS, Felices M. Natural killer cells unleashed: checkpoint receptor blockade and BiKE/TriKE utilization in NK-mediated anti-tumor immunotherapy. Semin Immunol. (2017) 31:64–75. doi: 10.1016/j.smim.2017.07.011
267. Felices M, Lenvik TR, Davis ZB, Miller JS, Vallera DA. Generation of BiKEs and TriKEs to improve NK cell-mediated targeting of tumor cells. Methods Mol Biol. (2016) 1441:333–46. doi: 10.1007/978-1-4939-3684-7_28
268. Moore GL, Bautista C, Pong E, Nguyen D-HT, Jacinto J, Eivazi A, et al. A novel bispecific antibody format enables simultaneous bivalent and monovalent co-engagement of distinct target antigens. MAbs. (2011) 3:546–57. doi: 10.4161/mabs.3.6.18123
269. Levi I, Amsalem H, Nissan A, Darash-Yahana M, Peretz T, Mandelboim O, et al. Characterization of tumor infiltrating natural killer cell subset. Oncotarget. (2015) 6:13835–43. doi: 10.18632/oncotarget.3453
270. Kim HS, Das A, Gross CC, Bryceson YT, Long EO. Synergistic signals for natural cytotoxicity are required to overcome inhibition by c-Cbl ubiquitin ligase. Immunity. (2010) 32:175–86. doi: 10.1016/j.immuni.2010.02.004
271. Felices M, Kodal B, Hinderlie P, Kaminski MF, Cooley S, Weisdorf DJ, et al. Novel CD19-targeted TriKE restores NK cell function and proliferative capacity in CLL. Blood Adv. (2019) 3:897–907. doi: 10.1182/bloodadvances.2018029371
272. MacFarlane AW, Jillab M, Smith MR, Alpaugh RK, Cole ME, Litwin S, et al. NK cell dysfunction in chronic lymphocytic leukemia is associated with loss of the mature cells expressing inhibitory killer cell Ig-like receptors. Oncoimmunology. (2017) 6:e1330235. doi: 10.1080/2162402X.2017.1330235
273. Schmohl JU, Felices M, Oh F, Lenvik AJ, Lebeau AM, Panyam J, et al. Engineering of anti-CD133 trispecific molecule capable of inducing NK expansion and driving antibody-dependent cell-mediated cytotoxicity. Cancer Res Treat. (2017) 49:1140–52. doi: 10.4143/crt.2016.491
274. Sarhan D, Brandt L, Felices M, Guldevall K, Lenvik T, Hinderlie P, et al. 161533 TriKE stimulates NK-cell function to overcome myeloid-derived suppressor cells in MDS. Blood Adv. (2018) 2:1459–69. doi: 10.1182/bloodadvances.2017012369
275. Don Yun H, Felices M, Vallera DA, Hinderlie P, Cooley S, Arock M, et al. Trispecific killer engager CD16xIL15xCD33 potently induces NK cell activation and cytotoxicity against neoplastic mast cells. Blood Adv. (2018) 2:1580–4. doi: 10.1182/bloodadvances.2018018176
276. Schmohl JU, Felices M, Todhunter D, Taras E, Miller JS, Vallera DA. Tetraspecific scFv construct provides NK cell mediated ADCC and self-sustaining stimuli via insertion of IL-15 as a cross-linker. Oncotarget. (2016) 7:73830–44. doi: 10.18632/oncotarget.12073
277. Runcie K, Budman DR, John V, Seetharamu N. Bi-specific and tri-specific antibodies- the next big thing in solid tumor therapeutics. Mol Med. (2018) 24:50. doi: 10.1186/s10020-018-0051-4
278. Lian S, Xie R, Ye Y, Xie X, Li S, Lu Y, et al. Simultaneous blocking of CD47 and PD-L1 increases innate and adaptive cancer immune responses and cytokine release. EBioMedicine. (2019) 42:281–95. doi: 10.1016/j.ebiom.2019.03.018
279. Chen J, Chen Y, Feng F, Chen C, Zeng H, Wen S, et al. Programmed cell death protein-1/programmed death-ligand 1 blockade enhances the antitumor efficacy of adoptive cell therapy against non-small cell lung cancer. J Thorac Dis. (2018) 10:6711–21. doi: 10.21037/jtd.2018.10.111
280. Zhou X-M, Li W-Q, Wu Y-H, Han L, Cao X-G, Yang X-M, et al. Intrinsic expression of immune checkpoint molecule TIGIT could help tumor growth in vivo by suppressing the function of NK and CD8+ T cells. Front Immunol. (2018) 9:2821. doi: 10.3389/fimmu.2018.02821
281. Li X-Y, Das I, Lepletier A, Addala V, Bald T, Stannard K, et al. CD155 loss enhances tumor suppression via combined host and tumor-intrinsic mechanisms. J Clin Invest. (2018) 128:2613–25. doi: 10.1172/JCI98769
282. Levin AM, Bates DL, Ring AM, Krieg C, Lin JT, Su L, et al. Exploiting a natural conformational switch to engineer an interleukin-2 ‘superkine'. Nature. (2012) 484:529–33. doi: 10.1038/nature10975
283. Charych DH, Hoch U, Langowski JL, Lee SR, Addepalli MK, Kirk PB, et al. NKTR-214, an engineered cytokine with biased IL2 receptor binding, increased tumor exposure, and marked efficacy in mouse tumor models. Clin Cancer Res. (2016) 22:680–90. doi: 10.1158/1078-0432.CCR-15-1631
284. Berger C, Berger M, Hackman RC, Gough M, Elliott C, Jensen MC, et al. Safety and immunologic effects of IL-15 administration in nonhuman primates. Blood. (2009) 114:2417–26. doi: 10.1182/blood-2008-12-189266
285. Han K, Zhu X, Liu B, Jeng E, Kong L, Yovandich JL, et al. IL-15:IL-15 receptor alpha superagonist complex: high-level co-expression in recombinant mammalian cells, purification and characterization. Cytokine. (2011) 56:804–10. doi: 10.1016/j.cyto.2011.09.028
286. Romee R, Cooley S, Berrien-Elliott MM, Westervelt P, Verneris MR, Wagner JE, et al. First-in-human phase 1 clinical study of the IL-15 superagonist complex ALT-803 to treat relapse after transplantation. Blood. (2018) 131:2515–27. doi: 10.1182/blood-2017-12-823757
287. Wong HC, Jeng EK, Rhode PR. The IL-15-based superagonist ALT-803 promotes the antigen-independent conversion of memory CD8 + T cells into innate-like effector cells with antitumor activity. Oncoimmunology. (2013) 2:e26442. doi: 10.4161/onci.26442
288. Hutmacher C, Gonzalo Núñez N, Liuzzi AR, Becher B, Neri D. Targeted delivery of IL2 to the tumor stroma potentiates the action of immune checkpoint inhibitors by preferential activation of NK and CD8 + T cells. Cancer Immunol Res. (2019) 7:572–83. doi: 10.1158/2326-6066.CIR-18-0566
289. Tomala J, Chmelova H, Mrkvan T, Rihova B, Kovar M. in vivo expansion of activated naive CD8 + T cells and NK cells driven by complexes of IL-2 and anti-IL-2 monoclonal antibody as novel approach of cancer immunotherapy. J Immunol. (2009) 183:4904–12. doi: 10.4049/jimmunol.0900284
290. Akhurst RJ. Targeting TGF-β signaling for therapeutic gain. Cold Spring Harb Perspect Biol. (2017) 9:a022301. doi: 10.1101/cshperspect.a022301
291. Knudson KM, Hicks KC, Luo X, Chen J-Q, Schlom J, Gameiro SR. M7824, a novel bifunctional anti-PD-L1/TGFβ Trap fusion protein, promotes anti-tumor efficacy as monotherapy and in combination with vaccine. Oncoimmunology. (2018) 7:e1426519. doi: 10.1080/2162402X.2018.1426519
292. Strauss J, Heery CR, Schlom J, Madan RA, Cao L, Kang Z, et al. Phase I trial of M7824 (MSB0011359C), a bifunctional fusion protein targeting PD-L1 and TGFβ, in advanced solid tumors. Clin Cancer Res. (2018) 24:1287–95. doi: 10.1158/1078-0432.CCR-17-2653
293. Shemesh A, Kundu K, Peleg R, Yossef R, Kaplanov I, Ghosh S, et al. NKp44-derived peptide binds proliferating cell nuclear antigen and mediates tumor cell death. Front Immunol. (2018) 9:1114. doi: 10.3389/fimmu.2018.01114
294. Kundu K, Ghosh S, Sarkar R, Edri A, Brusilovsky M, Gershoni-Yahalom O, et al. Inhibition of the NKp44-PCNA immune checkpoint using a mAb to PCNA. Cancer Immunol Res. (2019) 7:1120–34. doi: 10.1158/2326-6066.CIR-19-0023
295. Boehm EM, Gildenberg MS, Washington MT. The many roles of PCNA in eukaryotic DNA replication. Enzym. (2016) 39:231–54. doi: 10.1016/bs.enz.2016.03.003
296. Horton NC, Mathew SO, Mathew PA. Novel interaction between proliferating cell nuclear antigen and HLA I on the surface of tumor cells inhibits NK cell function through NKp44. PLoS ONE. (2013) 8:e59552. doi: 10.1371/journal.pone.0059552
297. Bessette DC, Qiu D, Pallen CJ. PRL PTPs: mediators and markers of cancer progression. Cancer Metastasis Rev. (2008) 27:231–52. doi: 10.1007/s10555-008-9121-3
298. Thura M, Al-Aidaroos AQ, Gupta A, Chee CE, Lee SC, Hui KM, et al. PRL3-zumab as an immunotherapy to inhibit tumors expressing PRL3 oncoprotein. Nat Commun. (2019) 10:2484. doi: 10.1038/s41467-019-10127-x
299. Shembrey C, Huntington ND, Hollande F. Impact of tumor and immunological heterogeneity on the anti-cancer immune response. Cancers. (2019) 11:1217. doi: 10.3390/cancers11091217
300. Zheng C, Wang Q, Wang Y, Zhao X, Gao K, Liu Q, et al. In situ modification of the tumor cell surface with immunomodulating nanoparticles for effective suppression of tumor growth in mice. Adv Mater. (2019) 2019:1902542. doi: 10.1002/adma.201902542
301. Ji T, Lang J, Ning B, Qi F, Wang H, Zhang Y, et al. Enhanced natural killer cell immunotherapy by rationally assembling fc fragments of antibodies onto tumor membranes. Adv Mater. (2018) 31:1804395. doi: 10.1002/adma.201804395
302. Bryceson YT, March ME, Ljunggren H-G, Long EO. Synergy among receptors on resting NK cells for the activation of natural cytotoxicity and cytokine secretion. Blood. (2006) 107:159–66. doi: 10.1182/blood-2005-04-1351
303. Prudkin L, Behrens C, Liu DD, Zhou X, Ozburn NC, Bekele BN, et al. Loss and reduction of Fus1 protein expression is a frequent phenomenon in the pathogenesis of lung cancer. Clin Cancer Res. (2008) 14:41–7. doi: 10.1158/1078-0432.CCR-07-1252
304. Santana Carrero RM, Beceren-Braun F, Rivas SC, Hegde SM, Gangadharan A, Plote D, et al. IL-15 is a component of the inflammatory milieu in the tumor microenvironment promoting antitumor responses. Proc Natl Acad Sci USA. (2019) 116:599–608. doi: 10.1073/pnas.1814642116
305. Meraz IM, Majidi M, Cao X, Lin H, Li L, Wang J, et al. TUSC2 immunogene therapy synergizes with anti–PD-1 through enhanced proliferation and infiltration of natural killer cells in syngeneic Kras -mutant mouse lung cancer models. Cancer Immunol Res. (2018) 6:163–77. doi: 10.1158/2326-6066.CIR-17-0273
306. Raulet DH, Marcus A, Coscoy L. Dysregulated cellular functions and cell stress pathways provide critical cues for activating and targeting natural killer cells to transformed and infected cells. Immunol Rev. (2017) 280:93–101. doi: 10.1111/imr.12600
307. Leung W-H, Vong QP, Lin W, Janke L, Chen T, Leung W. Modulation of NKG2D ligand expression and metastasis in tumors by spironolactone via RXRγ activation. J Exp Med. (2013) 210:2675–92. doi: 10.1084/jem.20122292
308. Bilotta MT, Abruzzese MP, Molfetta R, Scarno G, Fionda C, Zingoni A, et al. Activation of liver X receptor up-regulates the expression of the NKG2D ligands MICA and MICB in multiple myeloma through different molecular mechanisms. FASEB J. (2019) 33:9489–504. doi: 10.1096/fj.201900319R
309. Bugide S, Green MR, Wajapeyee N. Inhibition of enhancer of zeste homolog 2 (EZH2) induces natural killer cell-mediated eradication of hepatocellular carcinoma cells. Proc Natl Acad Sci USA. (2018) 115:E3509–18. doi: 10.1073/pnas.1802691115
310. Ferrari de Andrade L, Ngiow SF, Stannard K, Rusakiewicz S, Kalimutho M, Khanna KK, et al. Natural killer cells are essential for the ability of BRAF inhibitors to control BRAF V600E -mutant metastatic melanoma. Cancer Res. (2014) 74:7298–308. doi: 10.1158/0008-5472.CAN-14-1339
311. Sun X, Sui Q, Zhang C, Tian Z, Zhang J. Targeting blockage of STAT3 in hepatocellular carcinoma cells augments NK cell functions via reverse hepatocellular carcinoma-induced immune suppression. Mol Cancer Ther. (2013) 12:2885–96. doi: 10.1158/1535-7163.MCT-12-1087
312. Noman MZ, Paggetti J, Moussay E, Berchem G, Janji B. Driving natural killer cells toward the melanoma tumor battlefield: Autophagy as a valuable therapeutic target. Oncoimmunology. (2018) 7:e1452583. doi: 10.1080/2162402X.2018.1452583
313. Yao C, Ni Z, Gong C, Zhu X, Wang L, Xu Z, et al. Rocaglamide enhances NK cell-mediated killing of non-small cell lung cancer cells by inhibiting autophagy. Autophagy. (2018) 14:1831–44. doi: 10.1080/15548627.2018.1489946
314. Booth L, Roberts JL, Poklepovic A, Kirkwood J, Dent P. HDAC inhibitors enhance the immunotherapy response of melanoma cells. Oncotarget. (2017) 8:83155–70. doi: 10.18632/oncotarget.17950
315. Park J, Thomas S, Munster PN. Epigenetic modulation with histone deacetylase inhibitors in combination with immunotherapy. Epigenomics. (2015) 7:641–52. doi: 10.2217/epi.15.16
316. Hicks KC, Fantini M, Donahue RN, Schwab A, Knudson KM, Tritsch SR, et al. Epigenetic priming of both tumor and NK cells augments antibody-dependent cellular cytotoxicity elicited by the anti-PD-L1 antibody avelumab against multiple carcinoma cell types. Oncoimmunology. (2018) 7:e1466018. doi: 10.1080/2162402X.2018.1466018
317. Stone ML, Chiappinelli KB, Li H, Murphy LM, Travers ME, Topper MJ, et al. Epigenetic therapy activates type I interferon signaling in murine ovarian cancer to reduce immunosuppression and tumor burden. Proc Natl Acad Sci USA. (2017) 114:E10981–90. doi: 10.1073/pnas.1712514114
318. Friedman J, Morisada M, Sun L, Moore EC, Padget M, Hodge JW, et al. Inhibition of WEE1 kinase and cell cycle checkpoint activation sensitizes head and neck cancers to natural killer cell therapies. J Immunother Cancer. (2018) 6:59. doi: 10.1186/s40425-018-0374-2
319. Brennan TV, Lin L, Brandstadter JD, Rendell VR, Dredge K, Huang X, et al. Heparan sulfate mimetic PG545-mediated antilymphoma effects require TLR9-dependent NK cell activation. J Clin Invest. (2015) 126:207–19. doi: 10.1172/JCI76566
320. Hammond E, Haynes NM, Cullinane C, Brennan TV, Bampton D, Handley P, et al. Immunomodulatory activities of pixatimod: emerging nonclinical and clinical data, and its potential utility in combination with PD-1 inhibitors. J Immunother Cancer. (2018) 6:54. doi: 10.1186/s40425-018-0363-5
321. Steggerda SM, Bennett MK, Chen J, Emberley E, Huang T, Janes JR, et al. Inhibition of arginase by CB-1158 blocks myeloid cell-mediated immune suppression in the tumor microenvironment. J Immunother Cancer. (2017) 5:101. doi: 10.1186/s40425-017-0308-4
322. Hornyák L, Dobos N, Koncz G, Karányi Z, Páll D, Szabó Z, et al. The role of indoleamine-2,3-dioxygenase in cancer development, diagnostics, and therapy. Front Immunol. (2018) 9:151. doi: 10.3389/fimmu.2018.00151
323. Liberti MV, Locasale JW. The warburg effect: how does it benefit cancer cells? Trends Biochem Sci. (2016) 41:211–8. doi: 10.1016/j.tibs.2015.12.001
324. O'Sullivan D, Sanin DE, Pearce EJ, Pearce EL. Metabolic interventions in the immune response to cancer. Nat Rev Immunol. (2019) 19:324–35. doi: 10.1038/s41577-019-0140-9
325. Chambers AM, Lupo KB, Matosevic S. Tumor microenvironment-induced immunometabolic reprogramming of natural killer cells. Front Immunol. (2018) 9:2517. doi: 10.3389/fimmu.2018.02517
326. Ohta A, Gorelik E, Prasad SJ, Ronchese F, Lukashev D, Wong MKK, et al. A2A adenosine receptor protects tumors from antitumor T cells. Proc Natl Acad Sci USA. (2006) 103:13132–7. doi: 10.1073/pnas.0605251103
327. Miller JS, Cervenka T, Lund J, Okazaki IJ, Moss J. Purine metabolites suppress proliferation of human NK cells through a lineage-specific purine receptor. J Immunol. (1999) 162:7376–82.
328. Raskovalova T, Huang X, Sitkovsky M, Zacharia LC, Jackson EK, Gorelik E. G s protein-coupled adenosine receptor signaling and lytic function of activated NK cells. J Immunol. (2005) 175:4383–91. doi: 10.4049/jimmunol.175.7.4383
329. Raskovalova T, Lokshin A, Huang X, Jackson EK, Gorelik E. Adenosine-mediated inhibition of cytotoxic activity and cytokine production by IL-2/NKp46-activated NK cells: involvement of protein kinase A isozyme I (PKA I). Immunol Res. (2006) 36:91–100. doi: 10.1385/IR:36:1:91
330. Sek K, Mølck C, Stewart G, Kats L, Darcy P, Beavis P. Targeting adenosine receptor signaling in cancer immunotherapy. Int J Mol Sci. (2018) 19:3837. doi: 10.3390/ijms19123837
331. Chambers AM, Wang J, Lupo KB, Yu H, Atallah Lanman NM, Matosevic S. Adenosinergic signaling alters natural killer cell functional responses. Front Immunol. (2018) 9:2533. doi: 10.3389/fimmu.2018.02533
332. Wang J, Matosevic S. Adenosinergic signaling as a target for natural killer cell immunotherapy. J Mol Med. (2018) 96:903–13. doi: 10.1007/s00109-018-1679-9
333. Young A, Ngiow SF, Gao Y, Patch A-M, Barkauskas DS, Messaoudene M, et al. A2AR adenosine signaling suppresses natural killer cell maturation in the tumor microenvironment. Cancer Res. (2018) 78:1003–16. doi: 10.1158/0008-5472.CAN-17-2826
334. Young A, Mittal D, Stannard K, Yong M, Teng MW, Allard B, et al. Co-blockade of immune checkpoints and adenosine A 2A receptor suppresses metastasis. Oncoimmunology. (2014) 3:e958952. doi: 10.4161/21624011.2014.958952
335. Bastid J, Regairaz A, Bonnefoy N, Dejou C, Giustiniani J, Laheurte C, et al. Inhibition of CD39 enzymatic function at the surface of tumor cells alleviates their immunosuppressive activity. Cancer Immunol Res. (2015) 3:254–65. doi: 10.1158/2326-6066.CIR-14-0018
336. Cekic C, Day Y-J, Sag D, Linden J. Myeloid expression of adenosine A2A receptor suppresses T and NK cell responses in the solid tumor microenvironment. Cancer Res. (2014) 74:7250–9. doi: 10.1158/0008-5472.CAN-13-3583
337. Häusler SF, Del Barrio IM, Diessner J, Stein RG, Strohschein J, Hönig A, et al. Anti-CD39 and anti-CD73 antibodies A1 and 7G2 improve targeted therapy in ovarian cancer by blocking adenosine-dependent immune evasion. Am J Transl Res. (2014) 6:129–39.
338. Beavis PA, Divisekera U, Paget C, Chow MT, John LB, Devaud C, et al. Blockade of A 2A receptors potently suppresses the metastasis of CD73 + tumors. Proc Natl Acad Sci USA. (2013) 110:14711–6. doi: 10.1073/pnas.1308209110
Keywords: natural killer cells, inhibitory checkpoints, immunotherapy, tumor microenvironment, tumor ligands
Citation: Ben-Shmuel A, Biber G and Barda-Saad M (2020) Unleashing Natural Killer Cells in the Tumor Microenvironment–The Next Generation of Immunotherapy? Front. Immunol. 11:275. doi: 10.3389/fimmu.2020.00275
Received: 13 August 2019; Accepted: 03 February 2020;
Published: 21 February 2020.
Edited by:
Carsten Watzl, Leibniz Research Centre for Working Environment and Human Factors (IfADo), GermanyReviewed by:
Michael R. Verneris, University of Colorado Denver, United StatesJörg Wischhusen, Julius Maximilian University of Würzburg, Germany
Copyright © 2020 Ben-Shmuel, Biber and Barda-Saad. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Mira Barda-Saad, bWlyYS5iYXJkYS1zYWFkJiN4MDAwNDA7Yml1LmFjLmls