 Yuqian An1
Yuqian An1 Shichao Huang
Shichao Huang Gang Pei
Gang Pei
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Immunol. , 27 February 2020
Sec. Multiple Sclerosis and Neuroimmunology
Volume 11 - 2020 | https://doi.org/10.3389/fimmu.2020.00162
Neuroinflammation induced by overactivated glia cells is believed to be a major hallmark of Alzheimer's disease (AD) and a hopeful target against AD. A rhamnoside PL201 was previously reported to promote neurogenesis and ameliorate AD, and in this study, we revealed that PL201 also significantly reduced accumulation of the activated microglia and proinflammatory cytokines in APP/PS1 mice. In vitro, PL201 consistently suppressed the microglia induction of proinflammatory cytokines after stimulation with lipopolysaccharides and Aβ42. Further mechanistic studies demonstrated that PL201 considerably enhanced the expression level and the nuclear translocation of Nrf2, a key regulator of neuroinflammation. Moreover, PL201 effectively stimulated Nrf2 signaling cascade, including upregulation of HO-1 and downregulation of NF-κB pathway. Thus, our findings indicated the anti-neuroinflammatory effect by PL201 in vivo and suggested that PL201 or the like, with multiple functions such as neurogenesis, mitochondria maintenance, and anti-neuroinflammation, could be a promising candidate in AD treatment.
Alzheimer's disease (AD) is a chronic neurodegenerative disease and the most common cause of dementia (1). Mounting findings suggest a significant role of neuroinflammation in the pathogenesis of AD (2–4). Microglia, which are the major resident immune cells in the central nervous system (CNS), play a critical role in regulating neuroinflammation (5, 6). As a prominent feature of AD, microglia are recruited to amyloid beta (Aβ) plaques and being activated (7–9). Then, these activated microglia release chemokines, cytokines, and other inflammatory mediators such as IL-1β, IL-6, TNFα, MIP-1α, inducible nitric oxide synthase (iNOS), and cyclooxygenase-2 (COX-2), which will further aggravate neuronal damage and AD pathologies (2, 10–12).
Accumulating evidence suggest that nuclear factor erythroid 2-related factor 2 (Nrf2) is a transcription factor that critically involved in regulating the inflammatory response of microglia (13–15). Mechanistic studies reveal that, upon proper stimulation, Nrf2 translocates into the nucleus and activates its downstream targets including Heme oxygenase 1 (HO-1) (15–18). The upregulation of Nrf2 further inhibited NF-κB signaling to exert anti-inflammatory effects (19, 20). The initiation of this process is tightly regulated by Kelch-like ECH-associated protein 1 (Keap1)-mediated Nrf2 ubiquitination and degradation (21). Increasing lines of evidence reveal that Nrf2/HO-1 pathway is dysregulated in AD (22–27). For example, Ramsey et al. reported that the expression of nuclear Nrf2 was decreased in the hippocampi of AD patients (23). Similarly, Kanninen et al. found that overexpression of Nrf2 in the hippocampus reduces abnormal glia activation in a mouse model of AD (25). These findings indicated that proper Nrf2 signaling activators may serve as potential therapeutic interventions for AD.
AD is a multifactorial disease. Since their structural complexity and multitarget activity, natural products and their derivatives are recognized as an important source for developing AD drug leads (28–30). For instance, curcumin and its derivatives have been reported to ameliorate multiple AD pathologies including inhibiting Aβ production and tau phosphorylation, reducing neuroinflammation and oxidative stress, promoting neural stem cell proliferation and adult neurogenesis, and reversing cognitive deficits (31–33). Rhamnoside is a common kind of naturally occurring glycoside with antiaging effects. Moreover, it has also been revealed that rhamnoside may exert anti-AD effects by inhibiting production and aggregation of Aβ42 (34). We previously reported that a designed compound PL201, which was derived from rhamnoside, promoted neurogenesis and ameliorated cognitive impairment in the APP/PS1 mice (35). In this study, we found that PL201 significantly reduced microglia accumulation and proinflammatory cytokine production in the APP/PS1 mice. We also demonstrated that PL201 effectively suppressed LPS and Aβ induced proinflammatory cytokine expression in BV2 and human microglia cells. Furthermore, we elucidated that PL201 may exert its anti-inflammatory effect through Nrf2/HO-1/NF-κB signaling. In conclusion, PL201 may be served as a promising multitarget drug candidate for the treatment of AD.
All animal studies were carried out in accordance with the guidelines of the Institutional Animal Care and Use Committee (IACUC) of the Institute of Biochemistry and Cell Biology, Shanghai Institutes for Biological Sciences, Chinese Academy of Sciences. Both male and female mice were randomized in different experiment groups in this study. Nine-month-old APP/PS1 mice and wild-type (WT) littermates were used in this study. APP/PS1 transgenic mice expressing the mutated human app (Swedish mutations K595N M596L) and the human presenilin1 with a deletion of exon 9 were obtained from The Jackson Laboratory (stock no.004462) (36). All mice were bred and maintained in a temperature-controlled room and maintained on a 12-h light/dark cycle, with ad libitum access to food and water.
PL201 was synthesized as previously described (35). PL201 was dissolved in water. Nine-month-old APP/PS1 mice were orally administered with 30 mg/kg PL201 daily for 60 days. Control mice were treated with water as a vehicle. After drug treatment, mice of each group were euthanized, and the whole brains were removed. Brains were collected for measurement of proinflammatory and anti-inflammatory cytokines.
BV2 cells were cultured in DMEM (Invitrogen, Camarillo, CA); HMC3 cells were cultured in MEM (Invitrogen, Camarillo, CA). All medium was supplemented with 10% heat-inactivated fetal bovine serum (FBS), 100 U/ml penicillin, and 100 μg/ml streptomycin, and all cells were cultured at 37 °C in an atmosphere of 95% air and 5% CO2.
In all in vitro assays, BV2 and HMC3 cells were starved for 12 h in serum-free culture medium before PL201 treatment. To detect the expression of proinflammatory cytokines, cells were pretreated with PL201 at various concentrations (10–300 μM). After the pretreatment, 300 ng/ml LPS (Sigma, O55:B5) or 3 μM oligomer Aβ42 (GL Biochem, Shanghai) was added for further 24 h stimulation. To detect Nrf2′s downstream genes, cells were treated with PL201 at various concentrations (10–300 μM) for 6 h. To identify the nuclear translocation and expression of Nrf2, cells were treated with PL201 at various concentrations (10–300 μM) for 1 h. To assess the activation of NF-κB pathway, cells were pretreated with PL201 for 2 h followed by 1 h of LPS or oligomer Aβ42 stimulation.
Aβ42 peptide was prepared as previously described (37). In brief, 1 mg Aβ42 was dissolved in 1 ml cold hexafluoroisopropanol (Sigma, 52517), and the solution was aliquoted into Protein LoBind tubes (Eppendorf, 030108094); each tube containing 50 μg Aβ42 was dried overnight at RT. The residue was dissolved in 2.5 μl dimethyl sulfoxide (DMSO) followed by 117 μl cold phenol-free DMEM/F-12 medium (Thermo Fisher, 21041025) to obtain a 100-μM stock solution. After vortex for 60 s and incubated at 4 °C for 24 h, the solution was centrifuged at 16,000 × g for 10 min, and the supernatant containing Aβ42 oligomers was used for further experiments. The content of Aβ42 oligomers was confirmed by Western blot using anti-Aβ antibody (6E10, BioLegend, 803002; 1:1,000).
Mice cortex extract was obtained as reported previously (38, 39). In brief, cortex tissues were homogenized and sonicated 30 × 15 s with a 15-s interval between each sonication in Tris-buffered saline (TBS, pH 8.0) containing 0.1% Igepal. Samples were centrifuged at 16,000 × g at 4 °C for 30 min to remove any remaining insoluble debris in the solution. Samples were then detected by ELISA kits. The results were normalized to total protein.
The ELISA experiments were performed following manufacturer's instructions including the following: mouse TNFα (DAKEWE, 1217202), mouse IL-6 (DAKEWE, 1210602), mouse IL-1β (DAKEWE, 1210122), mouse MCP-1 (DAKEWE, 1217392), mouse IL-10 (DAKEWE, 1211002), and mouse MIP-1α (NEOBIOSCIENCE, EMC010a).
Total RNA was extracted from BV2 cells and HMC3 cells using TRIZOL Reagent according to the manufacturer's protocols. Equal amounts of RNA were reverse transcribed using a cDNA synthesis kit (Takara). qRT-PCR experiments were performed using synthetic primers and a 2 × HotStart SYBR Green qRT-PCR Master Mix kit (ExCell). Primers used in this study are listed in Table 1.

Table 1. Primers used for qRT-PCR.
Immunostaining was performed as previously described (28). In detail, mice were killed, and dissected brains were fixed in 4% paraformaldehyde (PFA) (Sigma) at 4 °C for 24 h. After fixation, tissues were washed in cold PBS for three times and then dehydrated in 30% sucrose overnight at 4 °C and embedded in optimal cutting temperature (OCT) (Thermo Fisher), followed by continuous serial cryostat sectioning at 30 μm. Sections were then washed with PBS (pH 7.4) and blocked with a blocking buffer (PBS supplemented with 0.5% Triton X-100 and 5% normal horse serum) for 1 h at room temperature. Afterwards, the sections were incubated with the following primary antibodies overnight at 4 °C: anti-Iba1 (Wako, 019-19741; 1:500), and anti-6E10 (BioLegend, 803002, 1:300). These sections were washed with PBS and incubated with Alexa fluorescence-conjugated secondary antibodies (Invitrogen, 1:1,000) for 1 h at room temperature followed by 10-min nuclei staining using 4′6-diamidino-2-phenylindole (DAPI Beyotime Biotechnology, 1:5,000). The images were acquired using a Zeiss confocal laser scanning microscope (Axio Scan.Z1). The instrument settings, including numeric gain, laser power, and magnification, were kept constant between animals to avoid potential technical artifacts.
Cultured BV2 cells and HMC3 cells were fixed in 4% PFA in PBS at room temperature for 15 min, and the immunostaining was carried out following the same procedure as brain sections. The following primary antibodies were used: anti-p65 (Santa Cruz, sc-372; 1:300) and anti-Nrf2 (Cell Signaling, 12721; 1:200). The images were captured using a Zeiss confocal laser scanning microscope (Zeiss 880 Airyscan). The instrument settings, including numeric gain, laser power, and magnification, were kept constant between animals to avoid potential technical artifacts.
As previously reported, ImageJ software was applied for quantitative analysis for Iba1 staining (39). First, red/green/blue (RGB) images were converted to 8-bit grayscale, and the threshold levels were adjusted to highlight positive signals. We quantified at least four sections per mouse. The Iba1 intensity from the cortex and hippocampus were qualified via Image J. Statistical analysis based on a blind review of data.
ImageJ software was applied for quantitative analysis for Nrf2 and p65 in cultured BV2 cells and HMC3 cells. The quantitative method was used following the same procedure as brain sections.
Western blotting were performed using the standard sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) method as previously reported (40). In brief, cultured BV2 and HMC3 cells were lysed using the protein lysis buffer (Sigma, R0278), and each sample containing 20 μg of total protein was electrophoresed with 8 or 10% SDS-PAGE. Then, these samples were transferred to nitrocellulose filter membranes (GE Healthcare Life science, A13711263). These membranes were blocked with 5% (w/v) skimmed milk for 1 h at room temperature. After that, the membrane was incubated with the following primary antibodies overnight at 4 °C: anti-iNOS (Cell Signaling, 13120S; 1:1,000), anti-COX-2 (Cell Signaling, 12282T; 1:1,000), anti-p65 (Santa Cruz, sc-372; 1:1,000), anti-p50/p105 (Beyotime Institute of Biotechnology, AF1246; 1:1,000), anti-Phospho-IκBα (Ser32) (Cell Signaling, 2859T; 1:1,000), anti-IκBα (Cell Signaling, 4814T; 1:1,000), anti-HO-1 (ABclonal, A1346; 1:1,000), anti-HO-2 (ABclonal, A2745; 1:1,000), anti-GCLM (ABclonal, A5314; 1:1,000), anti-NQO1 (Abclonal, A19586, 1:1,000), anti-Nrf2 (Cell Signaling, 12721; 1:1,000), anti-PCNA (Cell Signaling, 13110S, 1:1,000), and anti-Actin (Cell Signaling, 3700S, 1:1,000). After being washed with Tris-buffered saline with Tween 20 (TBST), these membranes were incubated with the secondary antibodies for 1 h at room temperature. The proteins of interest were visualized using an ECL Western blot detection kit (Bio-Rad). ImageJ software was applied to evaluate the densitometry. Actin or proliferating cell nuclear antigen (PCNA) were used as loading controls.
BV2 cells and HMC3 cells were cultured in six-well plates and grown for 24 h and then treated with various concentrations of PL201 for indicated time. Then, these cells were washed with PBS and lysed for 10 min in buffer A (10 mM Tris–HCl, pH 7.5, 10 mM NaCl, 3 mM MgCl2, and 1% NP-40) containing phosphatase inhibitor and protease inhibitor. After centrifugation at 3,000 × g for 5 min at 4 °C, the supernatant was collected to a new microfuge tube as the cytoplasmic fraction. The pellet was washed in buffer A for three times and resuspended in buffer B (20 mM HEPES, pH 7.9, 20% glycerol, 1.5 mM MgCl2, 0.2 mM EDTA, 1 mM dithiothreitol, and 1 mM phenylmethylsulfonyl fluoride). The suspension was then sonicated for 30 s, and centrifuged at 14,000 × g for 10 min at 4 °C to obtain the nuclear fraction.
Cell viability was detected by CellTiter-Glo Luminescent assay. One hundred microliters CellTiter-Glo Reagent was added to each well of 96-well plate and mixed for 2 min on an orbital shaker to lyse cells. The cell viability was judged according to the luminescence.
GraphPad Prism 7 (GraphPad Software, La Jolla, CA, United States) was used to draw graphs and perform data analysis. All values were represented as the mean ± SEM. One-way ANOVA followed by the Tukey multiple comparisons test were used to compare values from multiple groups. Two-tailed unpaired t-test was applied for comparisons between two groups. Values were considered to have significant difference when p < 0.05.
Accumulating evidence suggest that the pathogenesis of Alzheimer's disease is strongly associated with immunological alterations in the brain. In AD patients and mouse models, neuroinflammation has often been observed accompanied by excessive accumulation of microglia. To investigate whether PL201 treatment affected neuroinflammation in AD mice, we evaluated the microglial reactivity in the hippocampus and cortex by staining with ionized calcium binding adaptor molecule 1 (Iba1) antibody, which is a specific marker of microglia in the brain. We found that the microglia accumulating around the amyloid plagues significantly increased in the hippocampus and cortex of APP/PS1 mice when compared with the WT controls (Figures 1A,B). Moreover, reactive microgliosis was reduced in PL201-treated AD mice as well (Figures 1A–D). Since the activated microglia produce cytokines and chemokines, which contribute to AD pathology, we further detected the level of several cytokines and chemokines in brain samples from WT and AD mice. The results showed that, in AD mice, the levels of proinflammatory cytokines and chemokines including IL-6, TNFα, MIP-1α, and IL-1β were elevated, while the level of anti-inflammatory cytokine IL-10 was decreased (Figures 1E–I). After PL201 treatments, the levels of these proinflammatory cytokines were reduced in AD mice, but the levels of IL-10 remained unchanged. Collectively, these data demonstrated that PL201 treatment could suppress proinflammatory cytokine secretion and decrease neuroninflammation in AD mice. Emerging findings suggest that a significant increase in inflammatory response in AD is associated with Nrf2. We examined Nrf2 expression in mouse brains and found that PL201 had a tendency to increase Nrf2 expression in AD mice (Figures 1J,K).
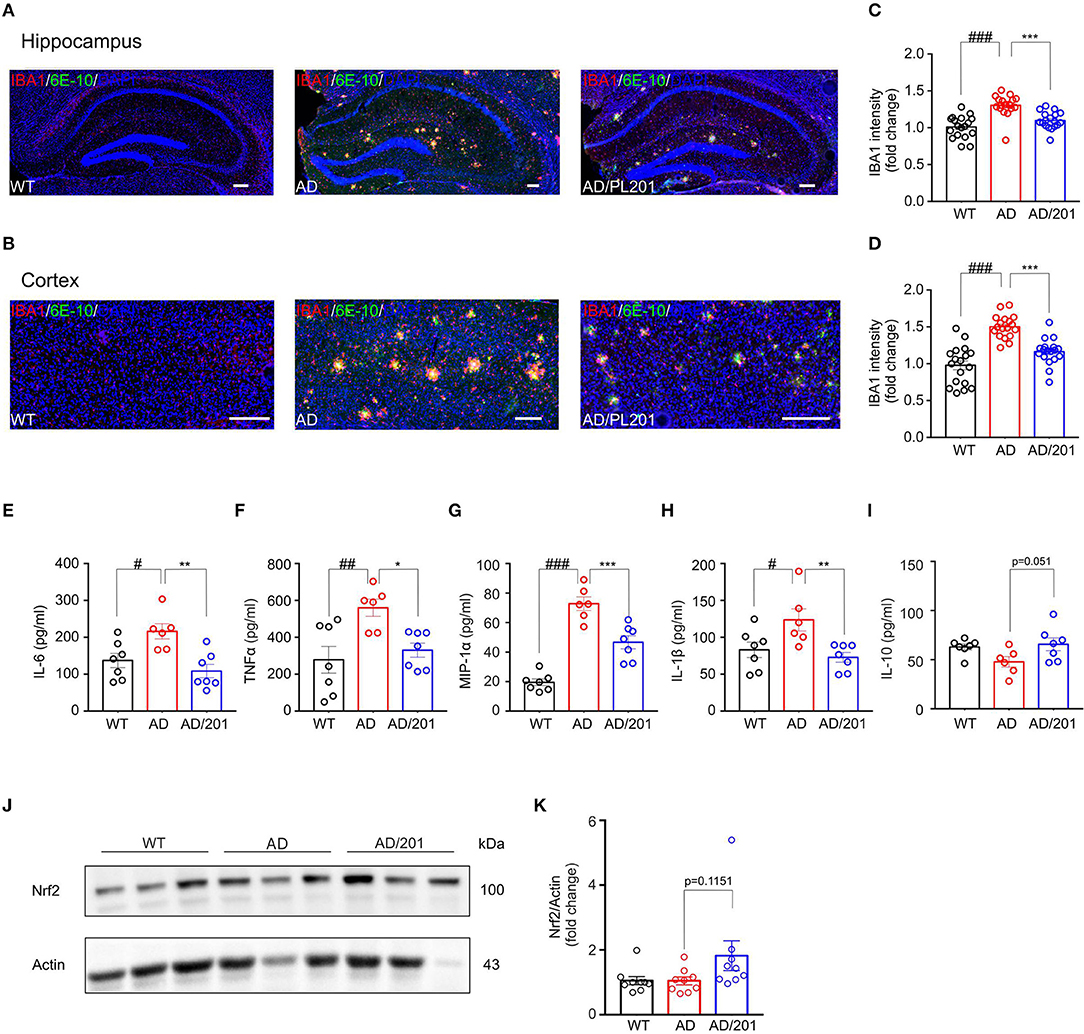
Figure 1. PL201 treatment decreases microglia reactivity and proinflammatory cytokine production in APP/PS1 mice. (A–D) Brain sections of WT, AD, and PL201 treated AD mice were immunostained with the microglial markers Iba1. (A,B) Representative images of Iba1 staining of hippocampus regions and cortex regions from WT, AD, and PL201 treated AD mice. Scale bar = 100 μm. (C,D) Quantification of Iba1 intensity from sections. n = 4 mice per group. (E–I) Indicated cytokine or chemokine levels in mouse brain were detected by ELISA. n = 7 (WT), 6 (AD), and 7 (AD/PL201) mice. (J,K) Nrf2 activation in mouse brains were detected by Western blot. Quantifications were expressed as mean ± SEM (compared with WT, #p < 0.05, ##p < 0.01, ###p < 0.005; compared with AD mice, *p < 0.05, **p < 0.01, ***p < 0.005).
Having established that PL201 decreases neuroinflammation in AD mice, we next tested the direct effects of PL201 on microglia cells in vitro. First, we examined the cytotoxicity of PL201. Twenty-Four hours treatment of PL201 (10–300 μM) exhibited no toxicity in BV2 cells (Figures 2A,B). Then, we investigated the effect of PL201 on LPS-induced cytokines and chemokines release. After pretreatment for 2 h with PL201, BV2 cells were exposed to LPS for 24 h (Figures 2C–O). We found that PL201 dose dependently inhibited the production of proinflammatory cytokines including IL-6, TNFα, and MCP-1, which were induced by 300 ng/ml LPS (Figures 2C–E). Similarly, PL201 effectively reduced the LPS-induced mRNA expression of IL-6, TNFα, MCP-1, IL-1β, MIP-1α, iNOS, and COX-2 (Figures 2F–L). PL201 also decreased protein level of iNOS and COX-2 in a concentration-dependent manner (Figures 2M–O). Besides, we tested the effect of PL201 on the expression profile of anti-inflammatory cytokines including IL-4, IL-10, and CD206; no statistically significant change was observed (Figures S1A–C). In AD, Aβ stimulates the microglia to aggravate neuroinflammation. Therefore, in the present study, we stimulated HMC3 human microglia cells with the soluble oligomer Aβ42, and then tested whether PL201 could exert similar anti-inflammatory effect. The results showed that PL201 also significantly reduced the levels of Aβ42-induced proinflammatory cytokines including IL-6, TNFα, MCP-1, IL-1β, iNOS, and COX-2 (Figures S2A–H).
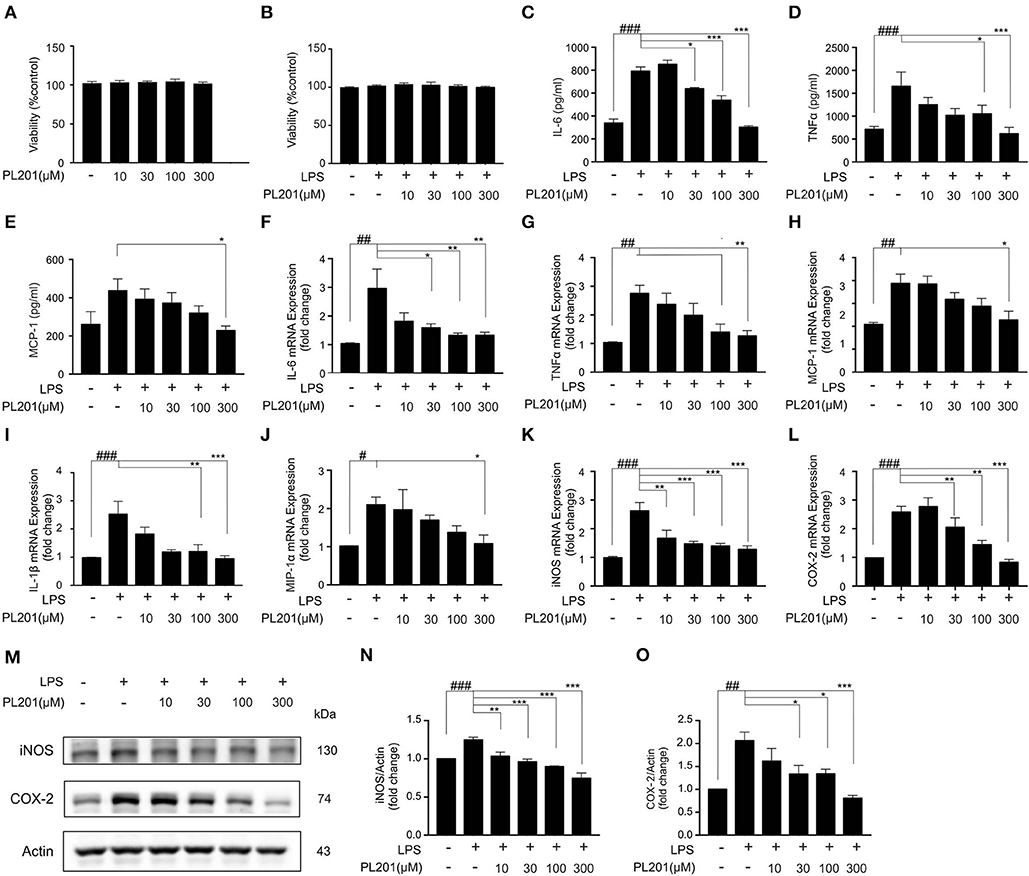
Figure 2. PL201 attenuates LPS-induced proinflammatory factor release in vitro. (A,B) PL201 has no cytotoxicity on BV2 cells. (C–O) BV2 cells were pretreated with PL201 for 2 h followed by LPS stimulation for further 24 h. (C–E) The levels of IL-6, TNFα, and MCP-1 in culture supernatant were measured by ELISA. (F–L) The mRNA expressions of IL-6, TNFα, MCP-1, IL-1β, MIP-1α, iNOS, and COX-2 were measured by qRT-PCR. (M–O) Representative Western blot analysis of iNOS and COX-2. Quantifications were expressed as mean ± SEM (compared with control, #p < 0.05, ##p < 0.01, ###p < 0.005; compared with LPS-stimulated condition, *p < 0.05, **p < 0.01, ***p < 0.005).
Since Nrf2 signaling plays an important role in modulating neuroinflammation, we next tested whether PL201 could activate Nrf2 signaling in both BV2 and HMC3 cells. The results revealed that PL201 activated Nrf2 nuclear translocation in a dose-dependent manner. Meanwhile, Nrf2 levels in the cytoplasm were correspondingly reduced after PL201 treatment (Figures 3A–D). To detect the specificity of Nrf2 antibody (41), we used tbhQ (15 μM) as a positive control and showed the whole extension of lanes in the Western blot. (Figures S3A,B). Similarly, using immunostaining, we observed that nuclear accumulation of Nrf2 was markedly increased after 1 h treatment with PL201 in BV2 (Figure 3E) and HMC3 cells (Figure 3F). We also determined gene level of Nrf2 after PL201 stimulation; the results indicated that PL201 effectively activated the mRNA expression of Nrf2, with a significant effect at the concentration of 300 μM in BV2 and HMC3 cells (Figures S3C,D). HO-1 is a major Nrf2 target gene; therefore, we tested the level of HO-1. The expression of the HO-1 significantly increased after PL201 treatment for 6 h in BV2 cells (Figures 3G–I). Similar result was also observed in HMC3 cells (Figures 3J–L). Moreover, the function of Nrf2 in the regulation of HO-1 induced by PL201 was studied using Nrf2 special inhibitor—brusatol. BV2 cells were pretreated with brusatol followed by PL201 for 6 h; brusatol completely abolished PL201-induced HO-1 expression (Figures S3E,F). Interestingly, we tested HO-2 in parallel to HO-1 and found that the protein level of HO-2 was not altered after PL201 treatment (Figures S4A–F). Besides HO-1, 6 h treatment of PL201 also dose-dependently increased the levels of GCLM and NQO1, which are both NRF2-target genes (Figures S5A–F). Taken together, these results indicated that PL201 treatment activates Nrf2 signaling pathway.
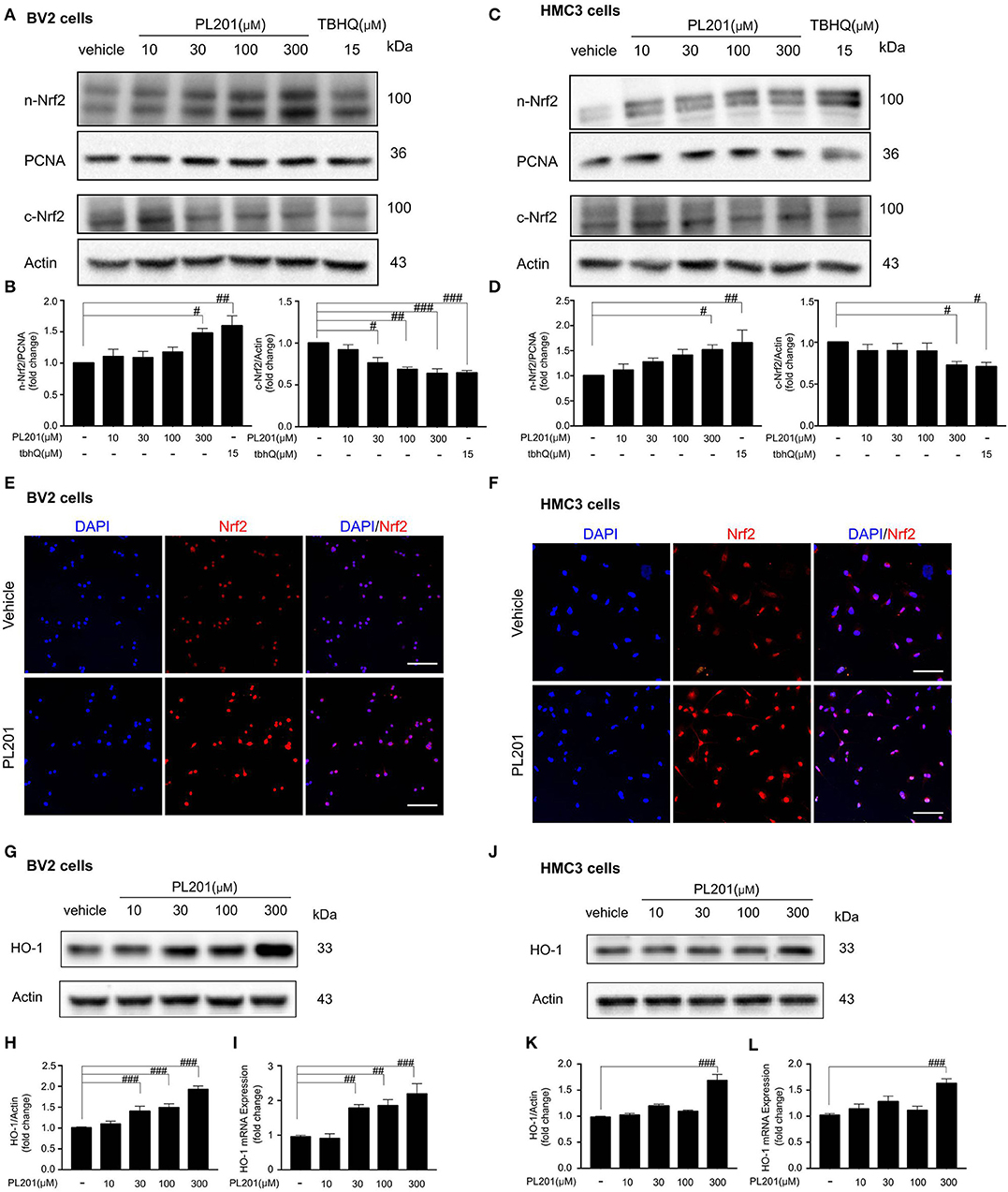
Figure 3. PL201 activates Nrf2 signaling. (A,B) The expression of Nrf2 in nuclear and cytoplasmic fractions from BV2 cells treated with various concentrations of PL201 for 1 h was measured via Western blot. tbhQ as a positive control. (C,D) The expression of Nrf2 in nuclear and cytoplasmic fractions from HMC3 cells treated with various concentrations of PL201 for 1 h was measured via Western blot. tbhQ as a positive control. (E) BV2 cells were pretreated with PL201 for 1 h; the nuclear translocation of Nrf2 was detected by immunostaing. Scale bar = 100 μm. (F) HMC3 cells were pretreated with PL201 for 1 h; the nuclear translocation of Nrf2 was detected by immunostaing. Scale bar = 100 μm. (G–I) BV2 cells were treated with PL201 for 6 h, representative Western blot analysis of HO-1 and mRNA expression of HO-1. (J–L) Representative Western blot analysis of HO-1 and mRNA expression of HO-1 in HMC3 cells were treated with PL201 for 6 h. Quantifications were expressed as mean ± SEM (compared with control, #p < 0.05, ##p < 0.01, ###p < 0.005).
We next investigated whether the anti-inflammatory effects of PL201 were mediated by Nrf2 signaling. To test this, we pretreated BV2 cells with an Nrf2-specific inhibitor brusatol before adding PL201 and found that brusatol attenuated the inhibitory effect of PL201 on the release of proinflammatory cytokines in LPS-treated BV2 cells (Figures 4A–E). We next blocked HO-1, which is a critical downstream mediator of Nrf2, by its inhibitor SNPP. BV2 cells were pretreated in the absence or presence of SNPP or PL201, followed by LPS stimulation for 24 h. Similar to that of Nrf2 inhibition, SNPP also attenuated the inhibitory effects of PL201 on proinflammatory cytokines such as IL-6, TNFα, and IL-1β (Figures 4F–J). We investigated the effect of PL201, brusatol, and Snpp on cytokines expression alone. They did not have effect on the expression of proinflammatory cytokines (Figures S6A–C). In summary, these data suggested that Nrf2 signaling is required for the anti-inflammatory effects of PL201.
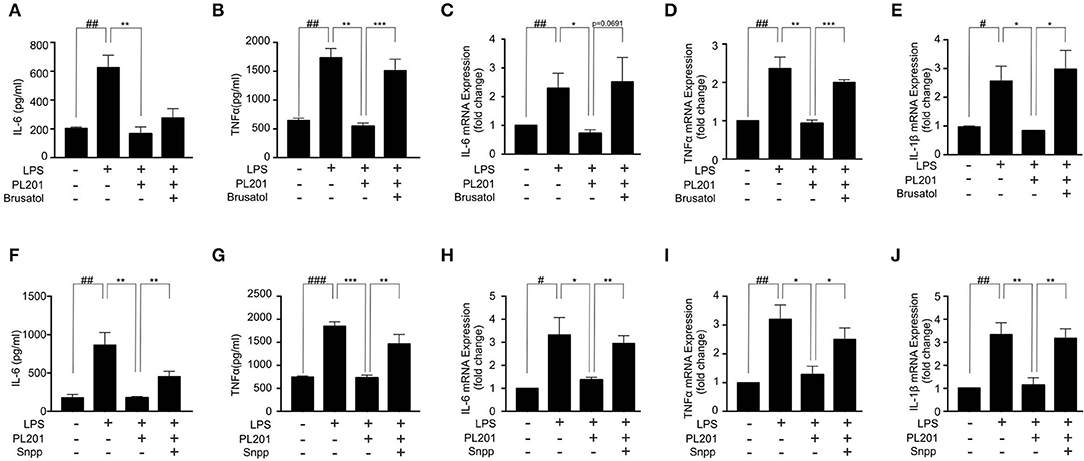
Figure 4. Nrf2 signaling is required for the anti-inflammatory effects of PL201. (A–E) BV2 cells were treated with PL201 in the presence or absence of brusatol (30 nM) and then exposed to LPS for 24 h. (A,B) The levels of IL-6 and TNFα in culture supernatant were measured by ELISA. (C–E) The mRNA expression of IL-6, TNFα, and IL-1β were measured by qRT-PCR. (F–J) BV2 cells were treated with PL201 in the presence or absence of Snpp (10 μM) and then exposed to LPS for 24 h. (F,G) The levels of IL-6 and TNFα in culture supernatant were measured by ELISA. (H–J) The mRNA expression of IL-6, TNFα, and IL-1β were measured by qRT-PCR. Quantifications were expressed as mean ± SEM (compared with control, #p < 0.05, ##p < 0.01, ###p < 0.005; compared with LPS-stimulated condition, *p < 0.05, **p < 0.01, ***p < 0.005).
Emerging evidence suggest that the cross-talk between Nrf2 and NF-κB signaling is important for the regulation of inflammation. To explore the effect of PL201 on NF-κB pathway, BV2 cells were pretreated with PL201 for 2 h followed by LPS stimulation for another 1 h. After that, nuclear protein was extracted as previously reported. The nuclear accumulation of p50 and p65 markedly increased after LPS treatment, which is reduced by PL201 treatment (Figures 5A–C). Using immunofluorescence microscopy, we also observed that PL201 significantly inhibited the nuclear translocation of the NF-κB p65 subunit in LPS or oligomer Aβ42-activated microglia cells (Figures 5D,E). IκB-α is a negative regulator of NF-κB. Previous reports showed that elevating the expression of Nrf2 inhibited NF-κB activity. We hypothesized that PL201 might suppress NF-κB activity by inducing Nrf2. To test this, we pretreated BV2 cells in the absence or presence of brusatol or PL201, followed by LPS stimulation for 1 h, and found that brusatol reversed the inhibitory effect of PL201 on IκB-α phosphorylation, while the basal protein levels of Nrf2 and IκB-α have no changes after brusatol stimulation (Figures 5F–H). These results demonstrated that Nrf2 mediates PL201-dependent downregulation of NF-κB activity.
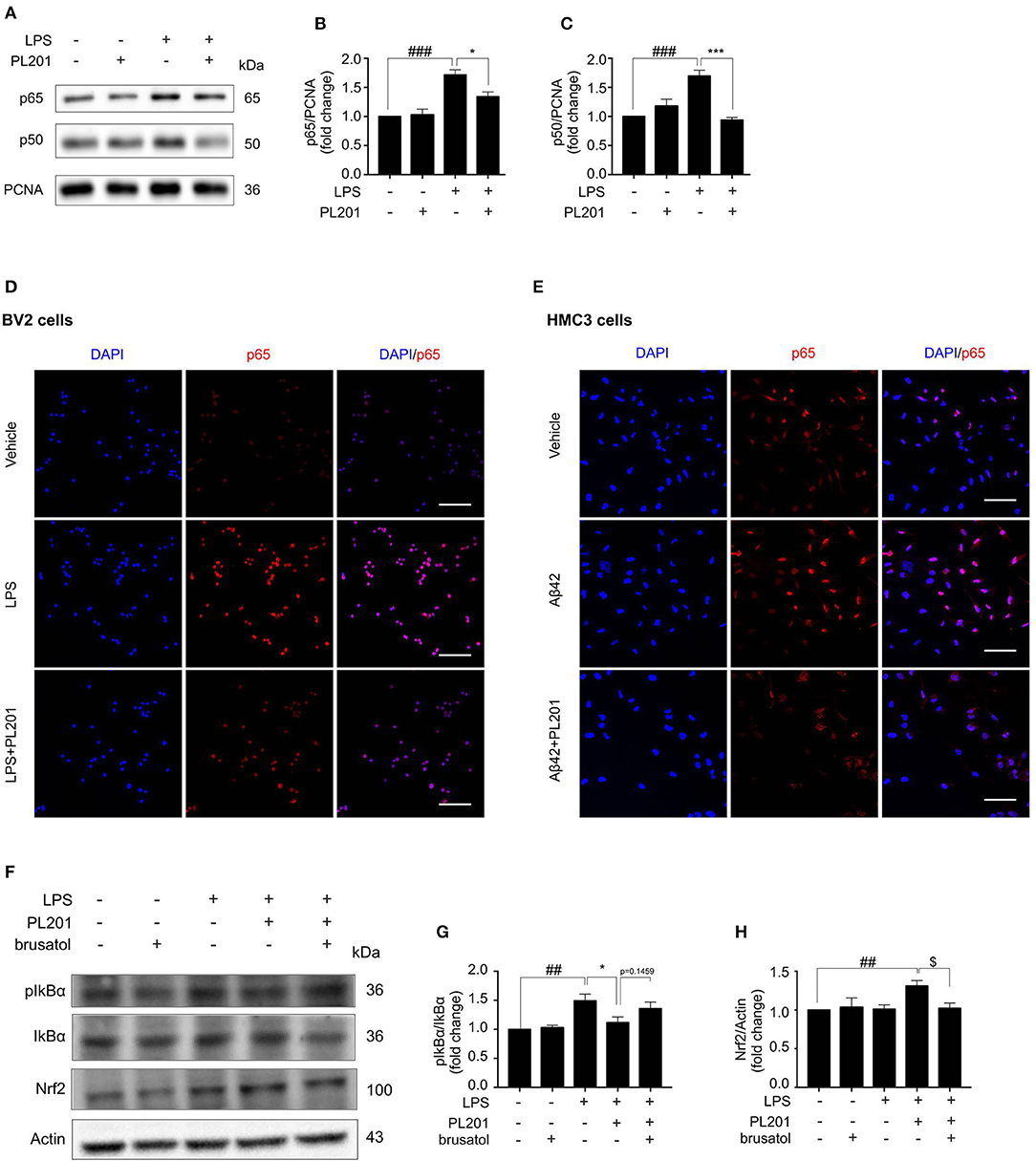
Figure 5. PL201 suppresses the NF-κB pathway. (A–D, F–H) BV2 cells were pretreated with PL201 for 2 h followed by LPS for further 1 h. (A–C) The intranuclear protein expression of p50 and p65 was measured via Western blot. (D) The nuclear translocation of p65 in BV2 cells was detected by immunostaining. Scale bar = 100 μm. (E) HMC3 cells were pretreated with PL201 for 2 h followed by Aβ42 for further 1 h. The nuclear translocation of p65 in HMC3 cells was detected by immunostaining. Scale bar = 100 μm. (F–H) BV2 cells were treated with PL201 in the presence or absence of brusatol (30 nM) and then exposed to LPS for 1 h; the protein levels of phosphorylated IkBα and total IkBα were analyzed by Western blot, the expression of Nrf2 was measured by Western blot. Quantifications were expressed as mean ± SEM (compared with vehicle, #p < 0.05, ##p < 0.01, ###p < 0.005; compared with LPS-stimulated condition, *p < 0.05, **p < 0.01, ***p < 0.005; compared with PL201 under brusatol treatment, $p < 0.05, $$p < 0.01, $$$p < 0.005).
In AD, microglia accumulate and secrete proinflammatory cytokines, which accelerate neuronal injury (42, 43). Our results demonstrated that PL201 treatment significantly reduced the levels of microglia activation and proinflammatory cytokines in the brains of APP/PS1 mice. Furthermore, we found that PL201 inhibited proinflammatory cytokine production in microglia cells after LPS or Aβ stimulation, which indicated that PL201 may affect microglia directly. In summary, these data demonstrated that PL201 may suppress the inflammatory response mediated by microglia both in vivo and in vitro.
Nrf2 has been reported to regulate the inflammatory response of microglia, and involved in the pathogenesis of AD (18, 44). Thus, we evaluated whether the effect of PL201 was mediated by Nrf2. The results indicated that PL201 could activate Nrf2 signaling by promoting Nrf2 metastasis into the nuclei of microglia cells. PL201 also elevated the mRNA expression of Nrf2 in a dose-dependent manner. Meanwhile, the expression of Nrf2 target genes concluding HO-1, NQO1, and GCLM could be induced by PL201. Interestingly, the anti-neuroinflammatory effect of PL201 can be blocked by Nrf2 inhibitor. PL201 could inhibit NF-κB signal dependent on Nrf2 activation. Collectively, we proposed that PL201 is a Nrf2 signaling activator. Emerging evidence indicated that Nrf2 may play an important role in tau pathology (27, 45, 46). Maybe PL201 could affect tau phosphorylation. Nrf2 also acts as a regulator of autophagy and takes an important role in regulating AD-related protein (47). Future work will be needed to detect if pharmacological induction of Nrf2 may be a valid strategy to facilitate degradation of toxic proteins such as Aβ or tau in the brain.
A large number of anti-inflammatory drugs, including non-steroidal anti-inflammatory drug (NSAID), peroxisome proliferator-activated receptor gamma (PPAR-γ) activators, and TNFα inhibitors, have been assessed in clinical trials for AD, but the results are not yet conclusive (2, 4). Since the complicated etiology of AD, developing multitarget drugs might be a promising strategy to treat this disease. We previously reported that PL201 treatment improved mitochondrial function of neural stem cells and promoted neurogenesis in AD mice. In this study, we further demonstrated that PL201 stimulates Nrf2 signaling in microglia and significantly suppresses neuroinflammation. Taken together, our results indicated that PL201 may serve as a potential multitarget treatment for AD.
In summary, we found that PL201 significantly reduced neuroinflammation in microglia via Nrf2/HO-1/NF-κB signaling pathway. Our findings indicated that PL201 might serve as a promising therapeutic agent for AD.
All datasets generated for this study are included in the article/Supplementary Material.
The animal study was reviewed and approved by the Institutional Animal Care and Use Committee (IACUC) of the Institute of Biochemistry and Cell Biology, Shanghai Institutes for Biological Sciences, Chinese Academy of Sciences.
YA designed this research and performed the cellular experiments and wrote the manuscript. YA and HZ conducted the animal experiments. GP and SH revised the manuscript. All authors read and approved the final manuscript.
This work was supported by the National Key Research and Development Program of China (2018YFA0108000 and 2018YFA0108003), the Strategic Priority Research Program of the Chinese Academy of Sciences (XDA16010309), the National Key Research and Development Program of China Stem Cell and Translational Research (2016YFA0101200 and 2016YFA0101202), and the National Science Foundation for Young Scientists of China (81901094).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2020.00162/full#supplementary-material
Figure S1. PL201 has no effects on the expression of anti-neuroinflammatory cytokines. BV2 cells were pretreated with PL201 for 2 h followed by LPS stimulation for further 24 h. mRNA expressions of IL-4 (A), IL-10 (B), and CD206 (C) were measured by qRT-PCR. Quantifications were expressed as mean ± SEM.
Figure S2. PL201 attenuates Aβ -induced pro-inflammatory factor release in vitro. (A,B) PL201 has no cytotoxicity on HMC3 cells. (C–H) HMC3 cells were pretreated with PL201 for 2 h followed by Aβ42 stimulation for further 24 h. The mRNA expressions of IL-6, TNFα, MCP-1, IL-1β, iNOS, and COX-2 were measured by qRT-PCR. Quantifications were expressed as mean ± SEM (compared with control: #p < 0.05, ##p < 0.01, ###p < 0.005; compared with Aβ-stimulated condition: *p < 0.05, **p < 0.01, ***p < 0.005).
Figure S3. PL201 elevates Nrf2 expression. (A,B) BV2 cells and HMC3 cells were treated with PL201 for 1 h, Nrf2 expression was detected by western blot. (C,D) BV2 cells and HMC3 cells were treated with PL201 for 1 h, the mRNA expression of Nrf2 was measured by qRT-PCR. (E,F) BV2 cells were treated with PL201 in the presence or absence of brusatol (30 nM) for 6 h, the protein level of HO-1 was analyzed by western blot. Quantifications were expressed as mean ± SEM (compared with control: #p < 0.05, ##p < 0.01, ###p < 0.005).
Figure S4. PL201 has no effects on the expression of HO-2 protein. BV2 cells and HMC3 cells were pretreated with PL201 for 6 h. (A–C) Representative western blot analysis of HO-2 and mRNA expression of HO-2 in BV2 cells. (D–F) Representative western blot analysis of HO-2 and mRNA expression of HO-2 in HMC3 cells. Quantifications were expressed as mean ± SEM.
Figure S5. PL201 activates the expression of GCLM and NQO1. BV2 cells were pretreated with PL201 for 6 h. (A–C) Representative western blot analysis of GCLM and mRNA expression of GCLM. (D–F) Representative western blot analysis of NQO1 and mRNA expression of NQO1. Quantifications were expressed as mean ± SEM (compared with vehicle: #p < 0.05, ##p < 0.01, ###p < 0.005).
Figure S6. The effect of PL201, brusatol and Snpp on cytokines expression alone. (A–C) BV2 cells were treated with PL201, brusatol and Snpp alone stimulation for 24 h. The expressions of IL-6, TNF-α, and IL-1β were detected by qRT-PCR.
AD, Alzheimer's disease; Aβ42, amyloid beta42; CNS, central nervous system; DMEM, Dulbecco's minimal essential medium; MEM, minimal essential medium; DMEM/F12, Dulbecco's modified Eagle's medium nutrient mixture F-12; ELISA, enzyme-linked immunosorbent assay; FBS, fetal bovine serum; HFIP, hexafluoroisopropanol; DMSO, dimethyl sulfoxide; LPS, lipopolysaccharides; IL-1β, interleukin-1 beta; IL-6, interleukin-6; TNFα, tumor necrosis factor alpha; MCP-1, monocyte chemoattractant protein; iNOS, inducible nitric oxide synthase; MIP-1α, macrophage inflammatory protein 1alpha; COX-2, cyclooxygenase-2; IL-4, interleukin 4; IL-10, interleukin 10; CD206, cluster of differentiation 206; Iba1, ionized calcium binding adaptor molecule; GFAP, glial fibrillary acidic protein; Nrf2, nuclear factor erythroid 2-related factor; HO-1, heme oxygenase-1; HO-2, heme oxygenase-2; NQO1, NAD(P)H dehydrogenase quinone1; GCLM, glutamate-cysteine ligase modifier; SNPP, Tin protoporphyrin IX; PBS, phosphate-buffered saline; TBS, Tris-buffered saline; DAPI, 4′6-diamidino-2-phenylindole; NF-κB, nuclear factor-kappa B; qRT-PCR, quantitative real-time polymerase chain reaction; RT, room temperature; SDS-PAGE, sodium dodecyl sulfate polyacrylamide gel electrophoresis; IκBα, nuclear factor of kappa light polypeptide gene enhancer in B-cell inhibitor, alpha.
1. Waterloo HW, LaFerla FM. Alzheimer's disease. N Engl J Med. (2010) 362:329–44. doi: 10.1056/NEJMra0909142
2. Heneka MT, Carson MJ, El Khoury J, Landreth GE, Brosseron F, Feinstein DL, et al. Neuroinflammation in Alzheimer's disease. Lancet Neurol. (2015) 14:388–405. doi: 10.1016/S1474-4422(15)70016-5
3. Heneka MT, O'Banion MK, Terwel D, Kummer MP. Neuroinflammatory processes in Alzheimer's disease. J Neural Transm. (2010) 117:919–47. doi: 10.1007/s00702-010-0438-z
4. Calsolaro V, Edison P. Neuroinflammation in Alzheimer's disease: Current evidence and future directions. Alzheimers Dement. (2016) 12:719–732. doi: 10.1016/j.jalz.2016.02.010
5. Streit WJ, Mrak RE, Griffin WS. Microglia and neuroinflammation: a pathological perspective. J Neuroinflammation. (2004) 1:14. doi: 10.1186/1742-2094-1-14
6. Perry VH, Teeling J. Microglia and macrophages of the central nervous system: the contribution of microglia priming and systemic inflammation to chronic neurodegeneration. Semin Immunopathol. (2013) 35:601–12. doi: 10.1007/s00281-013-0382-8
7. Cameron B, Landreth GE. Inflammation, microglia, and Alzheimer's disease. Neurobiol Dis. (2010) 37:503–9. doi: 10.1016/j.nbd.2009.10.006
8. Hansen DV, Hanson JE, Sheng M. Microglia in Alzheimer's disease. J Cell Biol. (2018) 217:459–72. doi: 10.1083/jcb.201709069
9. Heneka MT, Golenbock DT, Latz E. Innate immunity in Alzheimer's disease. Nat Immunol. (2015) 16:229–236. doi: 10.1038/ni.3102
10. Sarlus H, Heneka MT. Microglia in Alzheimer's disease. J Clin Invest. (2017) 127:3240–9. doi: 10.1172/JCI90606
11. Patel NS, Paris D, Mathura V, Quadros AN, Crawford FC, Mullan MJ. Inflammatory cytokine levels correlate with amyloid load in transgenic mouse models of Alzheimer's disease. J Neuroinflammation. (2005) 2:9. doi: 10.1186/1742-2094-2-9
12. Mrak RE, Sheng JG, Griffin WS. Glial cytokines in Alzheimer's disease: review and pathogenic implications. Hum Pathol. (1995) 26:816–23. doi: 10.1016/0046-8177(95)90001-2
13. Wang Y, Zhao H, Lin C, Ren J, Zhang S. Forsythiaside a exhibits anti-inflammatory effects in LPS-stimulated BV2 microglia cells through activation of Nrf2/HO-1 signaling pathway. Neurochem Res. (2016) 41:659–65. doi: 10.1007/s11064-015-1731-x
14. Park SY, Kim YH, Park G. Cucurbitacins attenuate microglial activation and protect from neuroinflammatory injury through Nrf2/ARE activation and STAT/NF-kappaB inhibition. Neurosci Lett. (2015) 609:129–36. doi: 10.1016/j.neulet.2015.10.022
15. Lee DS, Jeong GS. Butein provides neuroprotective and anti-neuroinflammatory effects through Nrf2/ARE-dependent haem oxygenase 1 expression by activating the PI3K/Akt pathway. Br J Pharmacol. (2016) 173:2894–909. doi: 10.1111/bph.13569
16. Kensler TW, Wakabayashi N, Biswal S. Cell survival responses to environmental stresses via the Keap1-Nrf2-ARE pathway. Annu Rev Pharmacol Toxicol. (2007) 47:89–116. doi: 10.1146/annurev.pharmtox.46.120604.141046
17. Quinti L, Naidu SD, Träger U, Chen X, Kegel-Gleason K, Llères D, et al. KEAP1-modifying small molecule reveals muted NRF2 signaling responses in neural stem cells from Huntington's disease patients. Proc Natl Acad Sci USA. (2017) 114:E4676–85. doi: 10.1073/pnas.1614943114
18. Ahmed SM, Luo L, Namani A, Wang XJ, Tang X. Nrf2 signaling pathway: pivotal roles in inflammation. Biochim Biophys Acta Mol Basis Dis. (2017) 1863:585–97. doi: 10.1016/j.bbadis.2016.11.005
19. Bellezza I, Tucci A, Galli F, Grottelli S, Mierla AL, Pilolli F, Minelli A. Inhibition of NF-kappaB nuclear translocation via HO-1 activation underlies alpha-tocopheryl succinate toxicity. J Nutr Biochem. (2012) 23:1583–91. doi: 10.1016/j.jnutbio.2011.10.012
20. Thimmulappa RK, Lee H, Rangasamy T, Reddy SP, Yamamoto M, Kensler TW, et al. Nrf2 is a critical regulator of the innate immune response and survival during experimental sepsis. J Clin Invest. (2006) 116:984–95. doi: 10.1172/JCI25790
21. Taguchi K, Motohashi H, Yamamoto M. Molecular mechanisms of the Keap1-Nrf2 pathway in stress response and cancer evolution. Genes Cells. (2011) 16:123–40. doi: 10.1111/j.1365-2443.2010.01473.x
22. Buendia I, Michalska P, Navarro E, Gameiro I, Egea J, Leon R. Nrf2-ARE pathway: an emerging target against oxidative stress and neuroinflammation in neurodegenerative diseases. Pharmacol Ther. (2016) 157:84–104. doi: 10.1016/j.pharmthera.2015.11.003
23. Ramsey CP, Glass CA, Montgomery MB, Lindl KA, Ritson GP, Chia LA, et al. Expression of Nrf2 in neurodegenerative diseases. J Neuropathol Exp Neurol. (2007) 66:75–85. doi: 10.1097/nen.0b013e31802d6da9
24. Loboda A, Damulewicz M, Pyza E, Jozkowicz A, Dulak J. Role of Nrf2/HO-1 system in development, oxidative stress response and diseases: an evolutionarily conserved mechanism. Cell Mol Life Sci. (2016) 73:3221–47. doi: 10.1007/s00018-016-2223-0
25. Kanninen K, Heikkinen R, Malm T, Rolova T, Kuhmonen S, Leinonen H, et al. Intrahippocampal injection of a lentiviral vector expressing Nrf2 improves spatial learning in a mouse model of Alzheimer's disease. Proc Natl Acad Sci USA. (2009) 106:16505–10. doi: 10.1073/pnas.0908397106
26. Rojo AI, Pajares M, García-Yagüe AJ, Buendia I, Van Leuven F, Yamamoto M, et al. Deficiency in the transcription factor NRF2 worsens inflammatory parameters in a mouse model with combined tauopathy and amyloidopathy. Redox Biol. (2018) 18:173–80. doi: 10.1016/j.redox.2018.07.006
27. Rojo AI, Pajares M, Rada P, Nuñez A, Nevado-Holgado AJ, Killik R, et al. NRF2 deficiency replicates transcriptomic changes in Alzheimer's patients and worsens APP and TAU pathology. Redox Biol. (2017) 13:444–51. doi: 10.1016/j.redox.2017.07.006
28. Hou Y, Wang Y, Zhao J, Li X, Cui J, Ding J, et al. Smart Soup, a traditional Chinese medicine formula, ameliorates amyloid pathology and related cognitive deficits. PLoS ONE. (2014) 9:e111215. doi: 10.1371/journal.pone.0111215
29. Mao J, Huang S, Liu S, Feng X-L, Yu M, Liu J, et al. A herbal medicine for Alzheimer's disease and its active constituents promote neural progenitor proliferation. Aging Cell. (2015) 14:784–96. doi: 10.1111/acel.12356
30. Yang W, Huang S, Liu S, Feng XL, Yu M, Liu J, et al. Naringin Dihydrochalcone Ameliorates Cognitive Deficits and Neuropathology in APP/PS1 Transgenic Mice. Front Aging Neurosci. (2018) 10:169. doi: 10.3389/fnagi.2018.00169
31. Zheng K, Dai X, Wu X, Wei Z, Fang W, Zhu Y, et al. Curcumin ameliorates memory decline via inhibiting BACE1 expression and beta-amyloid pathology in 5xFAD transgenic mice. Mol Neurobiol. (2017) 54:1967–77. doi: 10.1007/s12035-016-9802-9
32. Yang J, Zhang X, Yuan P, Yang J, Xu Y, Grutzendler J, et al. Oxalate-curcumin-based probe for micro- and macroimaging of reactive oxygen species in Alzheimer's disease. Proc Natl Acad Sci USA. (2017) 114:12384–9. doi: 10.1073/pnas.1706248114
33. Ullah F, Liang A, Rangel A, Gyengesi E, Niedermayer G, Münch G, et al. High bioavailability curcumin: an anti-inflammatory and neurosupportive bioactive nutrient for neurodegenerative diseases characterized by chronic neuroinflammation. Arch Toxicol. (2017) 91:1623–34. doi: 10.1007/s00204-017-1939-4
34. Zhou L, Liao W, Chen X, Yue H, Li S, Ding K. An arabinogalactan from fruits of Lycium barbarum L. inhibits production and aggregation of Abeta42. Carbohydr Polym. (2018) 195:643–51. doi: 10.1016/j.carbpol.2018.05.022
35. Huang S, Cao X, Zhou Y, Shi F, Xin S, He S, et al. An analog derived from phenylpropanoids ameliorates Alzheimer's disease-like pathology and protects mitochondrial function. Neurobiol Aging. (2019) 80:187–95. doi: 10.1016/j.neurobiolaging.2019.05.002
36. Jankowsky JL, Fadale DJ, Anderson J, Xu GM, Gonzales V, Jenkins NA, et al. Mutant presenilins specifically elevate the levels of the 42 residue beta-amyloid peptide in vivo: evidence for augmentation of a 42-specific gamma secretase. Hum Mol Genet. (2004) 13:159–70. doi: 10.1093/hmg/ddh019
37. Cai Q, Li Y, Pei G. Polysaccharides from Ganoderma lucidum attenuate microglia-mediated neuroinflammation and modulate microglial phagocytosis and behavioural response. J Neuroinflammation. (2017) 14:63. doi: 10.1186/s12974-017-0839-0
38. Ou Z, Kong X, Sun X, He X, Zhang L, Gong Z, et al. Metformin treatment prevents amyloid plaque deposition and memory impairment in APP/PS1 mice. Brain Behav Immun. (2018) 69:351–63. doi: 10.1016/j.bbi.2017.12.009
39. Fang EF, Hou Y, Palikaras K, Adriaanse BA, Kerr JS, Yang B, et al. Mitophagy inhibits amyloid-beta and tau pathology and reverses cognitive deficits in models of Alzheimer's disease. Nat Neurosci. (2019) 22:401–12. doi: 10.1038/s41593-018-0332-9
40. Wang X, Wang C, Pei G. alpha-secretase ADAM10 physically interacts with beta-secretase BACE1 in neurons and regulates CHL1 proteolysis. J Mol Cell Biol. (2018) 10:411–22. doi: 10.1093/jmcb/mjy001
41. Kemmerer ZA, Ader NR, Mulroy SS, Eggler AL. Comparison of human Nrf2 antibodies: a tale of two proteins. Toxicol Lett. (2015) 238:83–9. doi: 10.1016/j.toxlet.2015.07.004
42. Norden DM, Godbout JP. Review: microglia of the aged brain: primed to be activated and resistant to regulation. Neuropathol Appl Neurobiol. (2013) 39:19–34. doi: 10.1111/j.1365-2990.2012.01306.x
43. Solito E, Sastre M. Microglia function in Alzheimer's disease. Front Pharmacol. (2012) 3:14. doi: 10.3389/fphar.2012.00014
44. Kobayashi EH, Suzuki T, Funayama R, Nagashima T, Hayashi M, Sekine H, et al. Nrf2 suppresses macrophage inflammatory response by blocking proinflammatory cytokine transcription. Nat Commun. (2016) 7:11624. doi: 10.1038/ncomms11624
45. Jo C, Gundemir S, Pritchard S, Jin YN, Rahman I, Johnson GV. Nrf2 reduces levels of phosphorylated tau protein by inducing autophagy adaptor protein NDP52. Nat Commun. (2014) 5:3496. doi: 10.1038/ncomms4496
46. Tang M, Ji C, Pallo S, Rahman I, Johnson GV W. Nrf2 mediates the expression of BAG3 and autophagy cargo adaptor proteins and tau clearance in an age-dependent manner. Neurobiol Aging. (2018) 63:128–39. doi: 10.1016/j.neurobiolaging.2017.12.001
Keywords: Alzheimer's disease, neuroinflammation, microglia, rhamnoside analog, nuclear factor erythroid 2-related factor 2, heme oxygenase 1, nuclear factor kappa B
Citation: An Y, Zhang H, Huang S and Pei G (2020) PL201, a Reported Rhamnoside Against Alzheimer's Disease Pathology, Alleviates Neuroinflammation and Stimulates Nrf2 Signaling. Front. Immunol. 11:162. doi: 10.3389/fimmu.2020.00162
Received: 01 October 2019; Accepted: 21 January 2020;
Published: 27 February 2020.
Edited by:
Jorge Matias-Guiu, Complutense University of Madrid, SpainReviewed by:
Feng Zhang, Zunyi Medical University, ChinaCopyright © 2020 An, Zhang, Huang and Pei. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Shichao Huang, aHVhbmdzaGljaGFvQHNpYmNiLmFjLmNu; Gang Pei, Z3BlaUBzaWJzLmFjLmNu
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.