 Melinda A. Biernacki
Melinda A. Biernacki Marie Bleakley
Marie Bleakley- 1Clinical Research Division, Fred Hutchinson Cancer Research Center, Seattle, WA, United States
- 2Department of Medicine, University of Washington, Seattle, WA, United States
- 3Department of Pediatrics, University of Washington, Seattle, WA, United States
T cell cancer neoantigens are created from peptides derived from cancer-specific aberrant proteins, such as mutated and fusion proteins, presented in complex with human leukocyte antigens on the cancer cell surface. Because expression of the aberrant target protein is exclusive to malignant cells, immunotherapy directed against neoantigens should avoid “on-target, off-tumor” toxicity. The efficacy of neoantigen vaccines in melanoma and glioblastoma and of adoptive transfer of neoantigen-specific T cells in epithelial tumors indicates that neoantigens are valid therapeutic targets. Improvements in sequencing technology and innovations in antigen discovery approaches have facilitated the identification of neoantigens. In comparison to many solid tumors, hematologic malignancies have few mutations and thus fewer potential neoantigens. Despite this, neoantigens have been identified in a wide variety of hematologic malignancies. These include mutated nucleophosmin1 and PML-RARA in acute myeloid leukemia, ETV6-RUNX1 fusions and other mutated proteins in acute lymphoblastic leukemia, BCR-ABL1 fusions in chronic myeloid leukemia, driver mutations in myeloproliferative neoplasms, immunoglobulins in lymphomas, and proteins derived from patient-specific mutations in chronic lymphoid leukemias. We will review advances in the field of neoantigen discovery, describe the spectrum of identified neoantigens in hematologic malignancies, and discuss the potential of these neoantigens for clinical translation.
Introduction
Neoantigens are composed of peptides derived from full-length aberrant cancer-specific proteins through a multi-step intracellular process that has been extensively reviewed (1–3) and presented in complex with human leukocyte antigen (HLA) molecules. This peptide-HLA complex is recognized by T cell receptors (TCRs). Non-viral neoantigens can be potentially be generated from any protein-coding mutations, fusion proteins, and cancer-specific splice isoforms (Figure 1), although not every aberrant protein will yield neoantigens. In the treatment of solid tumors, clinical successes have been seen with adoptive transfer of neoantigen-specific tumor-infiltrating lymphocytes (TIL) (4–7) and neoantigen vaccines (8–10), highlighting the importance of this class of antigens in effective anti-tumor immunity. T cell responses against neoantigens also appear to contribute to the efficacy of immune checkpoint blockade therapy (11, 12) and allogeneic hematopoietic cell transplantation (HCT) (13). Therapies targeting a neoantigen derived from an oncogenic driver in the founding clone could be curative, and tumor escape through loss of the target protein is unlikely when the neoantigen is from a protein critical for maintaining the malignant phenotype. As neoantigens are presented solely on malignant cells and not on healthy equivalents, the risk of “on-target, off-tumor” toxicity is minimized.
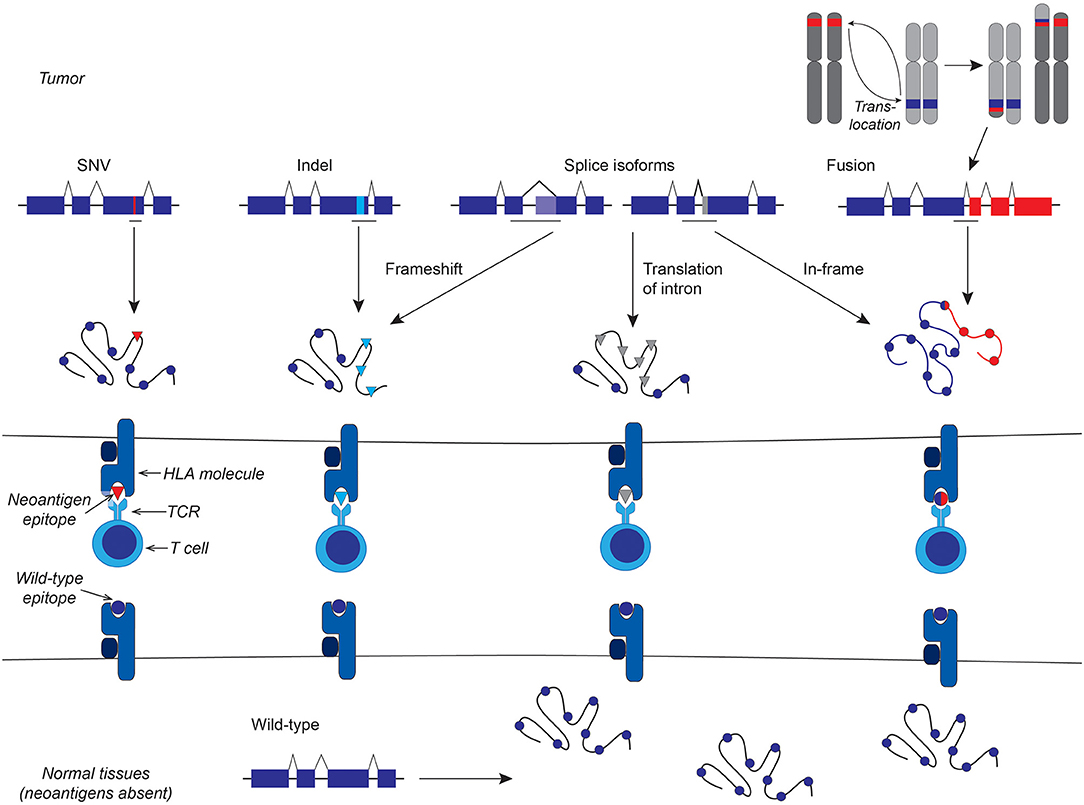
Figure 1. Schematic representation of different classes of non-viral neoantigens. From left to right: Protein-coding single nucleotide variants (SNV) lead to neoantigens that differ from the wild-type antigen by a single amino acid that alters HLA and/or TCR binding. Frameshift insertion-deletion (indels) mutations result in a novel amino acid sequence downstream of the indel. Cancer-specific splice isoforms can lead to frameshifts, if the splice is out of frame, or, like genomic fusions, juxtapose two usually separate amino acid sequences, or produce entirely novel amino acid sequence from introns or other portions of the genome that are not normally translated.
There are three major limitations to therapeutically targeting neoantigens in hematologic malignancies. First, most hematologic malignancies have relatively few protein-coding mutations and/or gene fusions, and thus fewer potential neoantigens than solid tumors, which may carry hundreds or even thousands of mutations in an individual patient (14). Second, therapies directed against any one neoantigen will apply only to the subset of patients who have both the mutation or fusion and restricting HLA allele, making neoantigens a less broadly applicable target than antigens from overexpressed wild-type proteins. However, in contrast to most solid tumors, many hematologic malignancies have recurrent mutations and/or fusions that are common within subgroups of patients and represent shared neoantigens. Finally, though targeting driver-derived neoantigens may prevent escape through loss of the target protein, other mechanism of escape from neoantigen-directed immunotherapy are possible, including downregulation or loss of HLA expression (15–24) or altered proteasomal processing of the epitope (25) by the malignant cell. However, the potential limitations of neoantigens as therapeutic targets are outweighed by their benefits: the high specificity for tumor and absent expression on normal cells; the ability to target intracellular as well as cell surface proteins; and, in some cases, the indispensable role of the aberrant protein in the malignant phenotype (26). Targeting a single high-quality neoantigen can be sufficient for disease control or even cure (4, 6, 7).
Neoantigen Discovery
Innovations in high-throughput genomic and transcriptomic sequencing techniques have greatly facilitated the identification of protein-coding mutations and fusions that produce potential neoantigens. However, there is still no reliable comprehensive in silico method for identifying immunogenic neoantigen epitopes from protein-coding mutations, splice variants, or other amino acid sequence-altering abnormalities. One challenge is determining which peptides will be presented on HLA molecules (27). In silico HLA-binding prediction algorithms [including but not limited to (28–36)] can predict binding of peptides to HLA molecules with reasonable accuracy and thereby identify candidate neoantigen epitopes. HLA-binding prediction algorithms are quite robust for prevalent HLA class I molecules, and active research by multiple groups has led to a greater understanding of and an improved ability to reliably predict peptide binding to uncommon class I molecules and most class II molecules (37–44). However, HLA-binding predictions do not specify whether the peptides are processed and presented on cell surfaces, although separate predictive tools for antigen processing have been developed (45–53). HLA-binding prediction from the parent amino acid sequence will additionally miss non-canonical epitopes (54), such as post-translationally modified or spliced peptides (55), and will also miss epitopes that are not predicted to bind HLA but actually do (56). An alternative approach is to directly determine the peptidome of malignant cells by immunoprecipitating HLA complexes and then to elute and identify peptides by tandem liquid chromatography–mass spectrometry (57–59). This unbiased approach can identify peptides as they are naturally presented on cells of interest but has significant technical hurdles (60–63). Modifications, such as the use of monoallelic cells (43, 60) should help to overcome some of these technical issues. Since predictive algorithms rely on datasets of peptides that are naturally processed and HLA-binding, improvements to direct identification of HLA ligands will in turn increase the reliability of predictive tools (64).
Determining which epitopes are immunogenic is also a challenge. Presentation of a peptide epitope on an HLA molecule is necessary but not sufficient for T cell recognition. Currently there are no reliable in silico tools to assess the immunogenicity of a neoantigen peptide, although this is an active area of research (65–70). There are three starting pools of cells in which one can assess the immunogenicity of a putative neoantigen: patient TIL or marrow-infiltrating lymphocytes (MIL), patient peripheral blood T cells, and healthy donor peripheral blood T cells after primary in vitro stimulation. The T cell repertoire of patients may be enriched for neoantigen-specific T cells (71, 72) due to antigen-induced expansion, but immunosuppressive tumor environments can render such T cells dysfunctional (73) or even absent (74). Finally, while neoantigen-specific T cells may exist in the repertoires of patients, unbiased methods to determine the cognate antigens of TCRs from their sequence are still in their infancy (23, 75). Stimulating healthy donor T cells with neoantigen-bearing antigen-presenting cells in vitro can be used to isolate reactive T cells without confounding T cell dysfunction (76, 77).
Shared vs. Personal Neoantigens
Shared or public neoantigens derive from aberrant proteins that are present in all or a sizeable subset of patients with a given disease. In contrast, private or personal neoantigens are those that result from mutations, fusions, or other abnormal amino acid sequences that occur rarely in a disease or are idiosyncratic to an individual's malignancy.
Whole genome and whole exome sequencing of hematologic malignancies (78–85) has revealed the spectrum of fusions and mutations (also referred to as the mutanome) of these diseases, including events that range from rare to highly recurrent. Many of these genetic abnormalities may give rise to neoantigens. Mutanomes provide a rich source of cancer-specific aberrant amino acid sequences that can be interrogated with HLA-binding prediction algorithms to identify candidate neoantigens (70). However, with the exception of one study in the myeloproliferative neoplasms (MPNs) (85), the mutanomes of hematologic malignancies have not yet been thoroughly explored as sources of neoantigens. Another source of both public and personal candidate neoantigens is the HLA peptidome, which is the comprehensive library of peptides eluted from HLA molecules isolated from malignant primary cells and/or cell lines and characterized by mass spectrometry. HLA peptidomes have been defined in acute myeloid leukemia (AML) (57), chronic lymphocytic leukemia (CLL) (59, 86), multiple myeloma (87), and chronic myeloid leukemia (CML) (58). However, mutation-derived candidate neoantigen epitopes have only been identified in more focused HLA peptidome studies [for example, in a subset of AML (74, 88)], reflecting both the heterogeneity of these diseases and the currently limited sensitivity of this approach.
Personal neoantigens can arise from truly patient-specific gene mutations and fusions. In addition, some recurrently affected single genes and gene fusions are highly heterogenous across individuals and would be expected to yield semi-personal rather than shared neoantigens. For example, fusions involving the mixed lineage leukemia (MLL)/ histone-lysine N-methyltransferase 2A (KMT2A) gene in acute lymphoblastic leukemia (ALL) and AML produce diverse amino acid sequences among patients because the fusions may occur at multiple breakpoints in the MLL/KMT2A genes and with multiple (>100) partner genes (89, 90). Recurrently mutated gene in hematologic malignancies that are likely to produce semi-personal rather than shared neoantigens include Wilms tumor 1 (WT1) in AML (91–94) and T cell ALL (95), Notch1 and FBXW7 in T cell ALL (96–98), and TP53 in multiple malignancies (99–101). In these examples, mutations occur at a variety of sites in the gene and involve multiple different nucleotide substitutions, insertions, and/or deletions, such that few, if any, of the resulting amino acid sequences and resulting potential neoantigens would be shared among patients even with the same disease.
At the other end of the spectrum are highly recurrent fusions and mutations, exemplified by the RUNX1-RUNX1T1 fusion (89) and exon 12 mutations in nucleophosmin1 (NPM1) (102, 103) in AML. Virtually all patients with such fusions or mutations will have identical aberrant amino acid sequences. Neoantigens created from these abnormalities are shared among patients who have the mutations or fusions and are potential therapeutic targets for these individuals as a group.
Whether optimal therapies should target shared neoantigens, personal neoantigens, or both is currently unknown. Some key features of neoantigen quality have been postulated [reviewed in (104)], including: clonality, dissimilarity to self-antigens, similarity to microbial antigens (105), high protein expression, binding to HLA, and low likelihood that genetic abnormality yielding the neoantigen will be lost (for example, driver mutations or genes involved in cell survival where loss would harm cancer fitness) through deletion or transcriptional repression. Neoantigens with high-quality features are likely to be suitable therapeutic targets whether they are personal or shared. One note of caution with personal neoantigens is that unless autologous tumor is available, there may be no way to validate that a given putative neoantigen is in fact presented on primary tumor, and thus no way to confidently predict therapeutic efficacy.
The feasibility of targeting personal neoantigens is currently under investigation. As the accessibility of whole genome and whole exome sequencing increases, defining an individual patient's mutanome is becoming increasingly practical, although the ability to reliably predict personal neoantigens remains imperfect (27). Personalized neoantigen vaccines based on patient mutanomes have shown efficacy in solid tumors (8–10), and as of December 2019, 14 clinical trials of personalized neoantigen vaccines were recruiting in the United States, although only one of these studies includes patients with a hematologic malignancy (NCT03631043, multiple myeloma). In addition, increasingly sophisticated T cell engineering technologies have made the production of personalized neoantigen-specific engineered T cell therapies more practical; currently three trials of such therapies for patients with advanced solid tumors (NCT03412877, NCT04102436, NCT03970382) are enrolling.
Although both personal and shared neoantigens have therapeutic promise, in this review we will focus primarily on shared neoantigens (summarized in Table 1), which make up the bulk of the data to date.
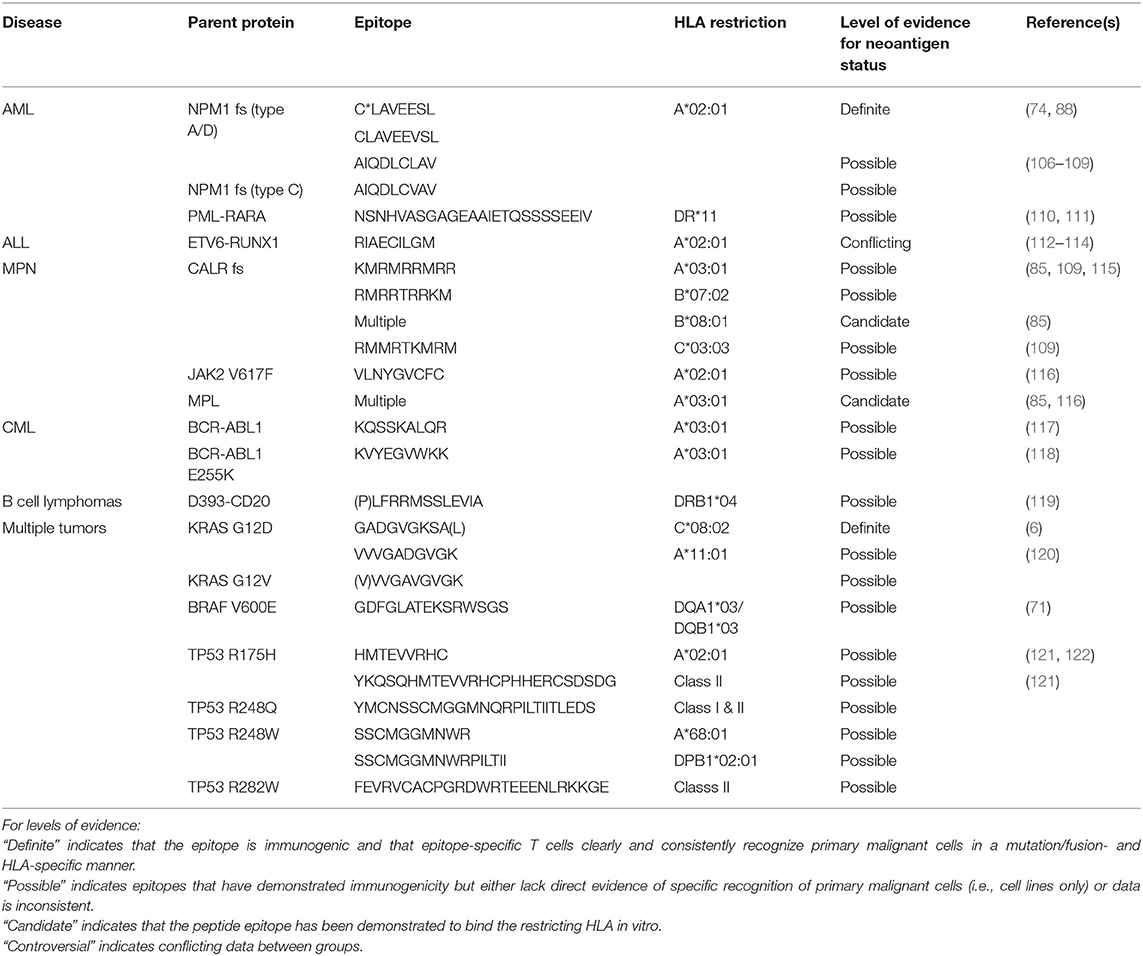
Table 1. Shared or potentially shared neoantigens relevant in hematologic malignancies (fs, frameshift).
Neoantigens in Specific Hematologic Malignancies
Acute Myeloid Leukemia
AML is the most common acute leukemia in adults, and mutations in nucleophosmin1 (NPM1) occur in 30–35% of adult patients (102, 103). The majority of NPM1 mutations are insertions of four nucleotides in exon 12, resulting in a frameshift that produces a novel C-terminal 11 amino acid sequence (123). NPM1 mutations are stable across the disease course and considered to be driver events, thus an optimal immunotherapy target. Eighty-five percent of patients with NPM1-mutated (NPM1mut) AML share the type A/D mutations that produce an identical abnormal amino acid sequence. Epitopes from the mutated region were independently identified as HLA ligands by two groups that used mass spectrometry to determine the amino acid sequences of peptides eluted off HLA molecules from primary leukemic blasts (74, 88) or AML cell lines (88).
Van der Lee et al. subsequently identified CD8+ T cell clones from healthy donors that were specific for the NPM1mut HLA-A*02:01-restricted epitopes CLAVEEVSL and C*LAVEEVSL (74). These clones specifically recognized HLA-A*02:01+ peptide-pulsed targets and NPM1mut AML blasts. One C*LAVEEVSL-specific TCR was sequenced and transferred into CD8+ T cells using a viral vector. T cells with transferred NPM1mut TCRs could lyse NPM1mut but not NPM1 wild-type AML in vitro and partially controlled leukemia in vivo in an NPM1mut OCI-AML3 cell-line-derived xenograft murine model. These results convincingly demonstrate that CLAVEEVSL and C*LAVEEVSL are naturally presented on HLA-A*02:01 on leukemic blasts, are immunogenic, and are thus bona fide AML neoantigens. Curiously, although the peptide was immunogenic, the authors were unable to identify naturally occurring epitope-specific T cell responses in HLA-A*02:01+ patients with NPM1mut AML. While a subsequent publication (123) suggested that NPM1mut -specific responses could be elicited ex vivo in patients, these studies were less stringently controlled.
Earlier studies identified candidate NPM1mut-derived epitopes predicted to bind HLA-A*02:01 (106–108), against which the authors elicited CD8+ T cells responses in patients and healthy donors after ex vivo stimulation. CD8+ T cells specific for two epitopes (AIQDLCLAV and AIQDLCVAV) identified in these publications lysed an NPM1mut AML sample (106), suggesting that these epitopes were naturally processed and presented. However, these peptides were not identified among HLA-A*02:01 ligands in either of two subsequent studies that directly examined peptide epitopes eluted from HLA-A*02:01 on NPM1mut primary blasts (74, 88) or cell lines (88), and identification of AIQDLCL/VAV-specific CD8+ T cells has not been reproduced by other groups.
In around 13% of AML cases (124, 125), a fusion of the retinoic acid receptor (RARA) gene on chromosome 17 and the promyelocytic leukemia (PML) gene on chromosome 15 occurs as a result of the chromosomal translocation, t(15; 17)(q24.1;q21.1), the classic translocation that produces the distinct entity of acute promyelocytic leukemia (APL). The resulting PML-RARA fusion protein not only serves as a driver of the leukemic phenotype but also represents a potential shared neoantigen at the fusion junction. Gambacorti-Passerini et al. investigated the immunogenicity of the PML-RARA fusion region by stimulating peripheral blood mononuclear cells (PBMC) from healthy volunteer donors with a 25mer peptide spanning the fusion (110, 126). CD4+ T cell clones from one donor proliferated specifically in response to exogenous PML-RARA peptide presented on HLA-DR*11 in autologous target cells. One T cell clone could lyse peptide-pulsed autologous target cells and recognize autologous target cells transduced to express the PML-RARA fusion protein. However, in a subsequent study, no PML-RARA-specific CD4+ T cell responses could be elicited from any of four HLA-DR*11+ individuals in remission after treatment for APL (111). Since neither study evaluated whether PML-RARA-specific T cells could recognize primary APL cells, the PML-RARA/HLA-DR*11 epitope would still currently be considered a possible, rather than definite, AML neoantigen pending confirmation that the epitope is naturally presented on APL cells.
Acute Lymphoblastic Leukemia
ALL is the most common childhood cancer. Like other hematologic and pediatric malignancies, there are few non-synonymous mutations (14, 127) and thus few potential neoantigens. However, in recent studies, Zamora et al. found surprisingly abundant neoantigen-specific CD8+ T cell responses in MIL from pediatric patients with ALL (112). To identify putative patient-specific neoantigens, cancer-specific mutations were identified from genomic sequencing of diagnostic biopsies and matched germline tissues from six patients. HLA typing was extrapolated from sample mRNA sequencing data, and the amino acid sequences of protein-coding mutations were interrogated using HLA-binding prediction algorithms. Mutation- or fusion-derived 15mer synthetic peptides were used to evaluate patient T cell specificity ex vivo. Functional CD8+ T cell responses against at least one neoantigen were detected in all patients and encompassed 31 of 36 putative neoantigens mostly originating from patient-specific single gene mutations.
The Zamora study also identified T cells responsive to several epitopes from the recurrent ETV6-RUNX1 fusion in five patients. The ETV6-RUNX1 fusion results from the t(12; 21)(p13.2;q22.1) chromosomal translocation and is the most common genetic event in childhood B-lineage ALL, occurring in 15–20% of patients (128–131). ETV6-RUNX1 epitopes eliciting T cell responses in this study were predicted to bind to HLA-A*02:01, HLA-A*11:01, and HLA-B*15:01, and ETV6-RUNX1-specific T cells were identified by positive staining with HLA-A*02:01 or HLA-A*11:01 peptide/HLA tetramers. In earlier studies, the same HLA-A*02:01 epitope (RIAECILGM) was identified as binding stably to HLA-A2 in in vitro competitive-binding assays by a group that also isolated CD8+ T cell lines specific for the epitope (113). The two RIAECILGM-specific lines that were isolated from healthy donors lysed fusion-expressing cell lines, and one T cell line from a patient with ETV6-RUNX1+ ALL lysed autologous leukemic blasts at low levels. However, a subsequent study disputed whether the ETV6-RUNX1 epitope is in fact naturally processed and presented, as it showed that the native RIAECILGM peptide had virtually no binding to HLA-A*02:01 in vitro, was not processed by cells transduced to express the ETV6-RUNX1 epitope, and was not cleaved at the relevant position by human proteasomes in vitro (114). Given the conflicting data, it remains unclear whether the RIAECILGM epitope is truly an ALL neoantigen.
Myeloproliferative Neoplasms
Philadelphia (Ph) chromosome-negative myeloproliferative neoplasms (MPNs) comprise a group of disorders, including essential thrombocytosis (ET), polycythemia vera (PV), and primary myelofibrosis (PMF). MPNs arise from an abnormal hematopoietic progenitor cell, in most cases consequent to the acquisition of one of three driver mutations in JAK2 (Janus kinase 2), CALR (calreticulin), or MPL (c-mpl proto-oncogene; thrombopoietin receptor), along with a variety of passenger mutations (132) that can all produce neoantigens. Recently, Schischlik et al. comprehensively evaluated potential neoantigens in 113 patients with MPNs (85). Using whole-transcriptome sequencing to define the MPN mutanome, they identified 13 fusions, 221 non-synonymous single nucleotide variants, 31 insertion or deletion mutations, and 20 frameshift-producing splicing abnormalities. HLA-binding predictions for the 12 most prevalent HLA-A, -B, and -C alleles in their patient cohort yielded 541 patient-specific peptides predicted to bind to at least one of the HLA alleles. Subsequent in vitro HLA binding studies of 35 peptides derived from aberrantly spliced proteins associated with SF3B1 mutations and from mutated CALR (CALRmut) and MPL validated binding of 23 peptides to HLA-A*03:01, -A*11:01, -B*07:02, and -B*08:01.
Although Schischlik et al. did not evaluate processing or immunogenicity of their putative neoantigens, others have identified T cell responses to CALRmut and JAK2 V617F. Cimen Bozkus et al. used in vitro stimulation to elicit T cell responses to CALRmut peptides that were primarily CD4+ T cells in patients with MPNs and both CD4+ and CD8+ in healthy donors. Inhibition of the PD-1 and CTLA-4 immune checkpoint molecules in vitro, and PD-1 in vivo (in a patient treated with pembrolizumab), enhanced T cell responses. An immunogenic HLA-C*03:03-restricted 10mer epitope was identified, and T cells specific for this epitope produced cytokine in response to antigen-presenting cells pulsed with a 15mer peptide, indicating that the epitope was processed from the longer peptide (109). While this finding is encouraging, data conclusively demonstrating that the CALRmut epitope is processed from the full-length protein and presented on HLA-C*03:03 on primary MPN cells is currently lacking. Another group described cytokine production, primarily by CD4+ T cells, in response to ex vivo stimulation of peripheral blood mononuclear cells from patients with MPNs with long (31mer) CALRmut peptides (133). CD8+ T cells specific for CALRmut peptides presented on HLA-A*03:01 and -B*07:02 were identified by another group, but the low avidity of the T cells prevented them from assessing whether the epitopes were naturally processed and presented on CALRmut cells (115). Additionally, a 9mer peptide spanning the JAK2 V617F mutation (VLNYGVCFC) was identified as a ligand of HLA-A*02:01 by HLA-binding prediction; epitope-specific CD8+ T cells lysed target cells either pulsed with the mutant peptide or naturally expressing JAK2 V617F, but also recognized targets pulsed with wild-type JAK2 peptide with lower efficiency (116). While this is a promising possible neoantigen with broad applicability for patients with MPNs, especially PV, further study is needed to definitively show that VLNYGVCFC is presented on primary malignant cells.
Philadelphia Chromosome-Positive Malignancies
The BCR-ABL1 fusion derives from translocation t(9; 22)(q34;q11), also called the Ph chromosome, which is highly recurrent in chronic myeloid leukemia (CML) and Ph-positive ALL (Ph+-ALL). Most patients have one of two fusions resulting from different breakpoints, namely p210BCR-ABL1 and p190BCR-ABL1. p210BCR-ABL1 is found in both CML and Ph+-ALL, while p190BCR-ABL1 is primarily associated with Ph+-ALL (134). As an oncogenic driver, the BCR-ABL1 fusion is essential to the malignant phenotype and an ideal therapeutic target. Small molecule tyrosine kinase inhibitors (TKIs) are now key components of therapy for Ph+ malignancies, but resistance does occur. Because the fusion is highly recurrent and disease-specific, it is a potential source for shared neoantigens. BCR-ABL1 was first described as a neoantigen in 1992 (135), and additional BCR-ABL1 epitopes were subsequently investigated by multiple groups [reviewed in (136)]. However, evidence for the natural CML presentation of BCR-ABL1 fusion peptides is conflicting: one group eluted an immunogenic fusion peptide from HLA-A*03:01 in primary CML (117), but a recent comprehensive evaluation of the HLA-ligandome in CML found no BCR-ABL1 epitopes presented on class I or class II molecules (58). Because the biology and specific fusions differ in the two diseases, the CML and Ph+-ALL peptidomes may differ. Interestingly, adoptive transfer of ex vivo-expanded p190BCR-ABL1-specific CD8+ T cells showed encouraging anti-leukemic activity in three patients with Ph+-ALL (137). Specific BCR-ABL1 mutations that confer TKI resistance might also serve as neoantigens; one group identified donor-derived CD8+ T cell responses to an HLA-A*03:01-restricted epitope from BCR-ABL1 E255K in a patient with the mutation who had achieved remission after HCT (118). While the BCR-ABL1 E255K-specific T cell clones could recognize minigene-transduced target cells, recognition of primary CML was not tested and thus it remains unclear whether the epitope represents a bona fide CML neoantigen.
Lymphomas and Chronic Lymphocytic Leukemia
In B cell malignancies, such as lymphomas and myelomas, neoplastic B cell-produced clonal immunoglobulin (Ig) was first described as a tumor-specific antigen in 1972 (138). Ig idiotypes have been extensively investigated as neoantigens with varying degrees of success, including in clinical trials (139). More recently, Khodadoust et al. recovered peptides representing somatic mutations in Ig heavy and light chain genes from the peptidomes of both class I and class II molecules in 17 primary mantle cell lymphomas (MCL) and two MCL cell lines, and detected circulating functional CD4+ T cells specific for one Ig neoantigen that could kill autologous lymphoma (140). Subsequent studies by this group identified primarily class II-restricted Ig-derived neoantigens in other B cell malignancies, including follicular lymphoma, diffuse large B cell lymphoma, and chronic lymphocytic leukemia (CLL) (141). A cytoplasmic variant of CD20 (D393-CD20), produced by alternative splicing of the CD20 transcript, is detectable in malignant primary B cells and B cell lines, but not normal resting B cells (142). CD4+ T cell responses to an epitope of D393-CD20 could be elicited from both healthy donors and patients with B cell lymphomas after in vitro peptide stimulation and blocked with anti-HLA-DR monoclonal antibody, but the exact HLA restriction could not be determined (119). MYD88 is recurrently mutated in a variety of B cell malignancies and has been proposed as a potential neoantigen (143). Separately, in a small cohort of CLL patients evaluated after HCT, CD8+ responses to neoantigens created from patient-specific non-Ig somatic mutations were identified; one well-studied patient-derived T cell clone could lyse autologous primary CLL cells, indicating that the epitope the clone recognized was a true personal neoantigen (13).
Neoantigens With General Applicability in Hematologic Malignancies
While some genetic abnormalities are specific to or even defining of particular cancer types, others, especially mutations in oncogenes or tumor suppressor genes, can be found in numerous cancers with a wide variety of cellular origins, including hematopoietic tissues. For example, somatic mutations affecting members of the Ras-MAPK pathway are among the most common in human cancers and are found across diverse cancer types (144–147). Similarly, TP53 is the most commonly mutated gene in human cancer, with TP53 mutations estimated to occur in ~25% of all cancers (99). Neoantigens derived from these mutations may thus be shared not just among patients with a single disease but across patients with many different cancers, including hematologic cancers (Table 2).
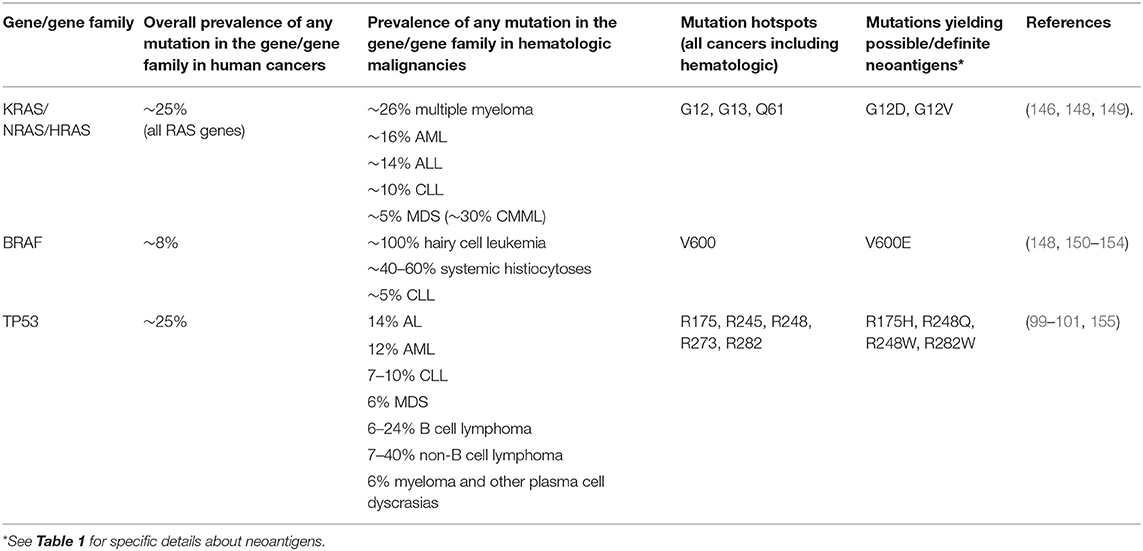
Table 2. Recurrently mutated genes in cancers, including hematologic malignancies, for which possible or definite public neoantigens have been identified.
Mutations in KRAS or NRAS are found in ~5–26% of hematologic malignancies (146, 148) (Table 2). The most recurrent oncogenic mutations that occur in the RAS genes (NRAS, KRAS, HRAS) across cancers occur at codons 12, 13, and 61. As such, neoantigens derived from these recurrent mutations in RAS genes are attractive therapeutic targets with applicability in multiple diseases, including blood cancers. Moreover, the amino acid sequences of RAS family members are highly similar, such that identical epitopes may be derived from different proteins. Although there are no publications specifically investigating RAS-derived neoantigens in hematologic malignancies, findings from studies in solid tumors have potential applicability. For example, Tran et al. studied tumor-infiltrating lymphocytes (TIL) from a patient with metastatic KRAS G12D-mutated colorectal cancer and identified CD8+ T cell clones specific for a KRAS G12D epitope presented on HLA-C*08:02 (6). KRAS G12D specific T cells expanded in the patient's peripheral blood after re-infusion of TIL, were persistently detectable ~9 months after TIL infusion, and mediated at least a transient regression of metastatic lung lesions. Subsequently, Cafri et al. performed in vitro stimulation of memory T cells isolated from two patients with KRAS-mutated solid tumors (one with endometrial cancer, one with rectal cancer) and identified CD8+ T cells specific for an HLA-A*11:01-restricted epitope from KRAS G12V and CD4+ T cells specific for an HLA-DRB1*08:01 restricted epitope from KRAS G12D (156). Earlier studies also identified murine TCRs with specificity for HLA-A*11:01-restricted epitopes from KRAS G12D and G12V in HLA-A*11:01+ transgenic mice immunized with KRAS peptides (120). Retroviral transfer of the KRAS-specific TCRs into human T cells conferred KRAS neoantigen-specific anti-tumor activity in vitro and in vivo. These findings have been translated into clinical trials of transgenic TCR T cell immunotherapy for HLA-A*11:01+ patients with certain KRAS G12D- or G12V-mutated solid tumors (NCT03190941 and NCT03745326). Although this clinical trial is directed toward patients with solid tumors, such therapies also have applicability to those with hematologic malignancies; for example, alterations at codon G12 of NRAS occur in a subset of patients with AML and produce identical amino acid sequences to the equivalent KRAS mutations, and so should yield the same epitope that could be targeted with KRAS G12D or G12V-directed T cells.
Mutations in BRAF, another component of the Ras-MAPK pathway, are present in about 8% of all human cancers (150). While the majority of BRAF-mutated malignancies are solid tumors, BRAF mutations do occur in a subset of hematologic malignancies. The BRAF V600E mutation is highly prevalent in hairy cell leukemia (151–153) and systemic histiocytoses (Erdheim-Chester disease and Langerhans cell histiocytosis) (154) and have also been identified in CLL (148) (Table 2). BRAF-derived neoantigens, particularly those originating from the V600E mutation, thus have applicability in a subset of hematologic malignancies. By examining peripheral blood lymphocytes from a patient with BRAF V600E+ melanoma who had a clinical response after TIL therapy, Veatch et al. identified a CD4+ T cell clone specific for an HLA-DQB1*03-restricted epitope of BRAF V600E (71). Lentiviral transfer of the BRAF V600E-specific TCR to donor CD4+ conferred recognition of BRAF V600E-expressing target cells. An earlier study also detected CD4+ T cell responses to HLA class II-restricted epitopes from BRAF V600E in patients with BRAF V600E melanoma, although not in the context of clinical response after immunotherapy (157).
TP53 mutations occur in malignancies of all origins (99), including all types of hematologic malignancies (100, 101, 158) (Table 2). While TP53 mutations can be quite heterogeneous, there are mutation hotspots at R175, R245, R248, R273, and R282 that are shared across multiple kinds of cancers, including hematologic cancers (99, 101). Neoantigens created from TP53 mutations thus have broad potential applicability in blood cancers as well as solid tumors. Malekzadeh et al. isolated T cells specific for HLA class I- and class II-restricted epitopes from five different recurrent TP53 mutations from TIL generated from patients with a variety of epithelial tumors (colorectal, ovarian, and pancreatic) (121). Both this publication and a subsequent report from the same group (122) identified an HLA-A*02:01-restricted epitope from TP53 R175H that appears to be naturally presented on a number of tumor cell lines. CD4+ and CD8+ responses to epitopes from patient-specific TP53 mutations have also been identified (122, 159, 160). Because the codon distribution of TP53 mutations is not specific to the tissue origin of a cancer, therapy targeting the TP53 R175H epitope, for example, should be equally applicable in a TP53 R175H+ hematologic cancer as in a TP53 R175H+ solid tumor, assuming the epitope is processed and presented appropriately. Similarly, specific mutations that are identical in many different malignancies, like the ones described in this section, are sources for neoantigens that are shared across cancers.
Therapeutic Applications of Neoantigens
While neoantigens are attractive targets for therapy because of their high specificity for malignant cells, there are challenges in translating neoantigen-directed immunotherapies to the clinic and the best approach to neoantigen-directed therapy is currently unknown. One strategy is to adoptively transfer neoantigen-specific T cells. T cells can be isolated from patient peripheral blood, TIL, or MIL (in hematologic malignancies), then expanded ex vivo non-specifically or against a defined antigen and re-infused (4, 6, 7, 161). Alternatively, T cells can be engineered to express a transgenic neoantigen-specific TCR (TCR-T), allowing infusion of a rapidly generated product with defined specificity and composition. Preclinical studies have shown that transfer of neoantigen-specific TCR-Ts is feasible (71, 74, 122), and two clinical trials of autologous TCR-T targeting HLA-A*11:01-restricted epitopes derived from point mutations in KRAS are enrolling (NCT03190941 and NCT03745326). Although TCR-T targeting epitopes from wild-type WT1 have shown safety (162–164) and efficacy (165), no clinical trials of neoantigen-specific TCR-T immunotherapy for hematologic malignancies have opened to date. TCR constructs can be modified to include other features to improve TCR-T safety and function: a CD8 co-stimulatory receptor enables CD4+ T cells to function with a class I-restricted TCR and provide targeted help to neoantigen-specific CD8+ T cells (166–168), a safety switch (167, 169) enables rapid removal of transgenic TCR-T cells in the event of toxicity, and other elements have been advanced [reviewed in (170)]. Lastly, vaccines do not require adoptive cell transfer, have shown clinical efficacy in solid tumors (8–10), and are particularly attractive for targeting highly immunogenic but less prevalent neoantigens.
Many factors influence which immunotherapy strategy is optimal for a given antigen. For neoantigens, the relatively low prevalence of each neoantigen among individuals with a given hematologic malignancy is a significant consideration, as immunotherapy for one neoantigen will apply only to a subset of patients. For example, the NPM1mut epitope described above (74) is only presented by the ~15% of AML patients with NPM1mut (30–35%) and HLA-A*02:01 (~50% in the U.S.A.) and this represents one of the most broadly applicable recurrent neoantigens in hematologic malignancies. Producing neoantigen-directed TCR-T therapies using currently standard viral transfer methods is probably not cost-effective for less common neoantigens given their narrow applicability, but vaccines could be. Moreover, the growing use of non-viral techniques for TCR gene transfer, such as transposon-based technologies (159, 171, 172), nanoparticles (173), and RNA electroporation (174) should facilitate the development of TCR-T immunotherapy for all neoantigens, as illustrated by a recently opened clinical trial of gene-edited TCR-T immunotherapy for personal neoantigens (NCT03970382). The use of “universal donor cells” that have been engineered to be HLA-negative and express natural killer (NK) cell inhibitory molecules (175), in combination with silencing or editing the endogenous TCR (163), could also facilitate neoantigen-directed TCR-T immunotherapy.
Another consideration is the natural immunologic landscape of a particular malignancy. Ex vivo expansion and vaccination rely largely on the presence of existing anti-tumor responses that can be boosted in vivo or ex vivo and would be challenging in an immunosuppressive environment. Because hematologic malignancies have multiple mechanisms for blocking effective naturally occurring anti-leukemic immune responses (11, 176–183), TCR-T immunotherapy may be preferable for these diseases. For example, transgenic TCRs can used to modify selected virus-specific memory T cells for therapeutic transfer (165). Immune checkpoint blockade has been used alone in MPNs (109); combining them with neoantigen-specific immunotherapies could potentiate their effect in these and other hematological malignancies.
Discussion
Much progress has been made in the field of neoantigens generally and in hematologic malignancies specifically. A number of promising bona fide and potential shared neoantigens have been identified for hematologic malignancies, most of which are derived from well-established mutations and fusions. However, growing access to comprehensive sequencing technologies has greatly enhanced the ability to define disease- and patient-specific mutanomes, which are valuable sources of potential neoantigens. Combined with improvements in T cell antigen discovery approaches, sequencing advances will facilitate the discovery of additional shared and personal neoantigens derived from known as well as new genetic abnormalities, expanding the repertoire of potential targets and moving the field forward. While there are a number of challenges in translating neoantigen-directed immunotherapies to the clinic, the rapid evolution of neoantigen discovery methods and the immunotherapy field is making barriers to clinical translation surmountable. Experience gained from T cell immunotherapy and vaccine studies in solid tumors and from cell therapy engineering for non-neoantigen targets will provide a critical foundation for building potent neoantigen-directed immunotherapies that are viable treatment strategies for hematologic malignancies.
Author Contributions
MAB and MB reviewed the literature and wrote and edited the manuscript.
Funding
This work was supported by Hyundai Hope on Wheels (TE 6705) and by a Stand Up To Cancer Innovative Research Grant, Grant Number SU2C-AACR-IRG 14-17. Stand Up To Cancer is a division of the Entertainment Industry Foundation. Research grants are administered by the American Association for Cancer Research, the scientific partner of SU2C. MAB received additional support from a National Cancer Institute Paul Calabresi Career Development Award for Clinical Oncology (5 K12 CA076930-18) and from Alex's Lemonade Stand-Foundation for Childhood Cancer.
Conflict of Interest
MB has received compensation from Miltenyi Biotec for presentations at conferences and corporate symposia pertaining to research unrelated to that presented in the current manuscript. MB is a Founder and Scientific Advisory Board member of HighPassBio and Scientific Advisory Board member of Orca Bio.
The remaining author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank Drs. Deborah Banker and Michelle Brault for the editorial assistance.
References
1. Cresswell P. Antigen processing and presentation. Immunol Rev. (2005) 207:5–7. doi: 10.1111/j.0105-2896.2005.00320.x
2. Vyas JM, Van der Veen AG, Ploegh HL. The known unknowns of antigen processing and presentation. Nat Rev Immunol. (2008) 8:607–18. doi: 10.1038/nri2368
3. Blum JS, Wearsch PA, Cresswell P. Pathways of antigen processing. Annu Rev Immunol. (2013) 31:443–73. doi: 10.1146/annurev-immunol-032712-095910
4. Tran E, Turcotte S, Gros A, Robbins PF, Lu YC, Dudley ME, et al. Cancer immunotherapy based on mutation-specific CD4+ T cells in a patient with epithelial cancer. Science. (2014) 344:641–5. doi: 10.1126/science.1251102
5. Robbins PF, Lu YC, El-Gamil M, Li YF, Gross C, Gartner J, et al. Mining exomic sequencing data to identify mutated antigens recognized by adoptively transferred tumor-reactive T cells. Nat Med. (2013) 19:747–52. doi: 10.1038/nm.3161
6. Tran E, Robbins PF, Lu YC, Prickett TD, Gartner JJ, Jia L, et al. T-Cell Transfer Therapy Targeting Mutant KRAS in Cancer. N Engl J Med. (2016) 375:2255–62. doi: 10.1056/NEJMoa1609279
7. Zacharakis N, Chinnasamy H, Black M, Xu H, Lu YC, Zheng Z, et al. Immune recognition of somatic mutations leading to complete durable regression in metastatic breast cancer. Nat Med. (2018) 24:724–30. doi: 10.1038/s41591-018-0040-8
8. Ott PA, Hu Z, Keskin DB, Shukla SA, Sun J, Bozym DJ, et al. An immunogenic personal neoantigen vaccine for patients with melanoma. Nature. (2017) 547:217–21. doi: 10.1038/nature22991
9. Sahin U, Derhovanessian E, Miller M, Kloke BP, Simon P, Lower M, et al. Personalized RNA mutanome vaccines mobilize poly-specific therapeutic immunity against cancer. Nature. (2017) 547:222–6. doi: 10.1038/nature23003
10. Keskin DB, Anandappa AJ, Sun J, Tirosh I, Mathewson ND, Li S, et al. Neoantigen vaccine generates intratumoral T cell responses in phase Ib glioblastoma trial. Nature. (2019) 565:234–9. doi: 10.1038/s41586-018-0792-9
11. Yarchoan M, Johnson BA III, Lutz ER, Laheru DA, Jaffee EM. Targeting neoantigens to augment antitumour immunity. Nat Rev Cancer. (2017) 17:209–22. doi: 10.1038/nrc.2016.154
12. Yang W, Lee KW, Srivastava RM, Kuo F, Krishna C, Chowell D, et al. Immunogenic neoantigens derived from gene fusions stimulate T cell responses. Nat Med. (2019) 25:767–75. doi: 10.1038/s41591-019-0434-2
13. Rajasagi M, Shukla SA, Fritsch EF, Keskin DB, DeLuca D, Carmona E, et al. Systematic identification of personal tumor-specific neoantigens in chronic lymphocytic leukemia. Blood. (2014) 124:453–62. doi: 10.1182/blood-2014-04-567933
14. Lawrence MS, Stojanov P, Polak P, Kryukov GV, Cibulskis K, Sivachenko A, et al. Mutational heterogeneity in cancer and the search for new cancer-associated genes. Nature. (2013) 499:214–8. doi: 10.1038/nature12213
15. Koopman LA, Corver WE, van der Slik AR, Giphart MJ, Fleuren GJ. Multiple genetic alterations cause frequent and heterogeneous human histocompatibility leukocyte antigen class I loss in cervical cancer. J Exp Med. (2000) 191:961–76. doi: 10.1084/jem.191.6.961
16. Campoli M, Chang CC, Ferrone S. HLA class I antigen loss, tumor immune escape and immune selection. Vaccine. (2002) 20 (Suppl. 4):A40–5. doi: 10.1016/S0264-410X(02)00386-9
17. Garrido F, Ruiz-Cabello F, Aptsiauri N. Rejection versus escape: the tumor MHC dilemma. Cancer Immunol Immunother. (2017) 66:259–71. doi: 10.1007/s00262-016-1947-x
18. Shukla SA, Rooney MS, Rajasagi M, Tiao G, Dixon PM, Lawrence MS, et al. Comprehensive analysis of cancer-associated somatic mutations in class I HLA genes. Nat Biotechnol. (2015) 33:1152–8. doi: 10.1038/nbt.3344
19. Benitez R, Godelaine D, Lopez-Nevot MA, Brasseur F, Jimenez P, Marchand M, et al. Mutations of the β2-microglobulin gene result in a lack of HLA class I molecules on melanoma cells of two patients immunized with MAGE peptides. Tissue Antigens. (1998) 52:520–9. doi: 10.1111/j.1399-0039.1998.tb03082.x
20. Sade-Feldman M, Jiao YJ, Chen JH, Rooney MS, Barzily-Rokni M, Eliane JP, et al. Resistance to checkpoint blockade therapy through inactivation of antigen presentation. Nat Commun. (2017) 8:1136. doi: 10.1038/s41467-017-01062-w
21. Gettinger S, Choi J, Hastings K, Truini A, Datar I, Sowell R, et al. Impaired HLA class I antigen processing and presentation as a mechanism of acquired resistance to immune checkpoint inhibitors in lung cancer. Cancer Discov. (2017) 7:1420–35. doi: 10.1158/2159-8290.CD-17-0593
22. McGranahan N, Rosenthal R, Hiley CT, Rowan AJ, Watkins TBK, Wilson GA, et al. Allele-specific HLA loss and immune escape in lung cancer evolution. Cell. (2017) 171:1259–71.e11. doi: 10.1016/j.cell.2017.10.001
23. Paulson KG, Voillet V, McAfee MS, Hunter DS, Wagener FD, Perdicchio M, et al. Acquired cancer resistance to combination immunotherapy from transcriptional loss of class I HLA. Nat Commun. (2018) 9:3868. doi: 10.1038/s41467-018-06300-3
24. Christopher MJ, Petti AA, Rettig MP, Miller CA, Chendamarai E, Duncavage EJ, et al. Immune escape of relapsed AML cells after allogeneic transplantation. N Engl J Med. (2018) 379:2330–41. doi: 10.1056/NEJMoa1808777
25. Ebstein F, Keller M, Paschen A, Walden P, Seeger M, Burger E, et al. Exposure to Melan-A/MART-126–35 tumor epitope specific CD8(+)T cells reveals immune escape by affecting the ubiquitin-proteasome system (UPS). Sci Rep. (2016) 6:25208. doi: 10.1038/srep25208
26. Yamamoto TN, Kishton RJ, Restifo NP. Developing neoantigen-targeted T cell-based treatments for solid tumors. Nat Med. (2019) 25:1488–99. doi: 10.1038/s41591-019-0596-y
28. Bocchia M, Wentworth PA, Southwood S, Sidney J, McGraw K, Scheinberg DA, et al. Specific binding of leukemia oncogene fusion protein peptides to HLA class I molecules. Blood. (1995) 85:2680–4. doi: 10.1182/blood.V85.10.2680.bloodjournal85102680
29. Sturniolo T, Bono E, Ding J, Raddrizzani L, Tuereci O, Sahin U, et al. Generation of tissue-specific and promiscuous HLA ligand databases using DNA microarrays and virtual HLA class II matrices. Nat Biotechnol. (1999) 17:555–61. doi: 10.1038/9858
30. Singh H, Raghava GP. ProPred: prediction of HLA-DR binding sites. Bioinformatics. (2001) 17:1236–7. doi: 10.1093/bioinformatics/17.12.1236
31. Peters B, Sette A. Generating quantitative models describing the sequence specificity of biological processes with the stabilized matrix method. BMC Bioinformatics. (2005) 6:132. doi: 10.1186/1471-2105-6-132
32. Guan P, Hattotuwagama CK, Doytchinova IA, Flower DR. MHCPred 2.0: an updated quantitative T-cell epitope prediction server. Appl Bioinformatics. (2006) 5:55–61. doi: 10.2165/00822942-200605010-00008
33. Sidney J, Assarsson E, Moore C, Ngo S, Pinilla C, Sette A, et al. Quantitative peptide binding motifs for 19 human and mouse MHC class I molecules derived using positional scanning combinatorial peptide libraries. Immunome Res. (2008) 4:2. doi: 10.1186/1745-7580-4-2
34. Wang P, Sidney J, Dow C, Mothe B, Sette A, Peters B. A systematic assessment of MHC class II peptide binding predictions and evaluation of a consensus approach. PLoS Comput Biol. (2008) 4:e1000048. doi: 10.1371/journal.pcbi.1000048
35. Hoof I, Peters B, Sidney J, Pedersen LE, Sette A, Lund O, et al. NetMHCpan, a method for MHC class I binding prediction beyond humans. Immunogenetics. (2009) 61:1–13. doi: 10.1007/s00251-008-0341-z
36. Kim Y, Ponomarenko J, Zhu Z, Tamang D, Wang P, Greenbaum J, et al. Immune epitope database analysis resource. Nucleic Acids Res. (2012) 40:W525–30. doi: 10.1093/nar/gks438
37. Jurtz V, Paul S, Andreatta M, Marcatili P, Peters B, Nielsen M. NetMHCpan-4.0: Improved peptide-MHC class I interaction predictions integrating eluted ligand and peptide binding affinity data. J Immunol. (2017) 199:3360–8. doi: 10.4049/jimmunol.1700893
38. Jensen KK, Andreatta M, Marcatili P, Buus S, Greenbaum JA, Yan Z, et al. Improved methods for predicting peptide binding affinity to MHC class II molecules. Immunology. (2018) 154:394–406. doi: 10.1111/imm.12889
39. Barra C, Alvarez B, Paul S, Sette A, Peters B, Andreatta M, et al. Footprints of antigen processing boost MHC class II natural ligand predictions. Genome Med. (2018) 10:84. doi: 10.1186/s13073-018-0594-6
40. Bulik-Sullivan B, Busby J, Palmer CD, Davis MJ, Murphy T, Clark A, et al. Deep learning using tumor HLA peptide mass spectrometry datasets improves neoantigen identification. Nat Biotechnol. (2018) 37, 55–63. doi: 10.1038/nbt.4313
41. Chen B, Khodadoust MS, Olsson N, Wagar LE, Fast E, Liu CL, et al. Predicting HLA class II antigen presentation through integrated deep learning. Nat Biotechnol. (2019) 37, 1332–43. doi: 10.1038/s41587-019-0280-2
42. Racle J, Michaux J, Rockinger GA, Arnaud M, Bobisse S, Chong C, et al. Robust prediction of HLA class II epitopes by deep motif deconvolution of immunopeptidomes. Nat Biotechnol. (2019) 37, 1283–6. doi: 10.1038/s41587-019-0289-6
43. Abelin JG, Harjanto D, Malloy M, Suri P, Colson T, Goulding SP, et al. Defining HLA-II ligand processing and binding rules with mass spectrometry enhances cancer epitope prediction. Immunity. (2019) 51:766–79.e17. doi: 10.1016/j.immuni.2019.08.012
44. Garde C, Ramarathinam SH, Jappe EC, Nielsen M, Kringelum JV, Trolle T, et al. Improved peptide-MHC class II interaction prediction through integration of eluted ligand and peptide affinity data. Immunogenetics. (2019) 71:445–54. doi: 10.1007/s00251-019-01122-z
45. Besser H, Louzoun Y. Cross-modality deep learning-based prediction of TAP binding and naturally processed peptide. Immunogenetics. (2018) 70:419–28. doi: 10.1007/s00251-018-1054-6
46. Tenzer S, Peters B, Bulik S, Schoor O, Lemmel C, Schatz MM, et al. Modeling the MHC class I pathway by combining predictions of proteasomal cleavage, TAP transport and MHC class I binding. Cell Mol Life Sci. (2005) 62:1025–37. doi: 10.1007/s00018-005-4528-2
47. Calis JJ, Reinink P, Keller C, Kloetzel PM, Kesmir C. Role of peptide processing predictions in T cell epitope identification: contribution of different prediction programs. Immunogenetics. (2015) 67:85–93. doi: 10.1007/s00251-014-0815-0
48. Diez-Rivero CM, Lafuente EM, Reche PA. Computational analysis and modeling of cleavage by the immunoproteasome and the constitutive proteasome. BMC Bioinformatics. (2010) 11:479. doi: 10.1186/1471-2105-11-479
49. Paul S, Karosiene E, Dhanda SK, Jurtz V, Edwards L, Nielsen M, et al. Determination of a predictive cleavage motif for eluted major histocompatibility complex class II ligands. Front Immunol. (2018) 9:1795. doi: 10.3389/fimmu.2018.01795
50. Zhang GL, Petrovsky N, Kwoh CK, August JT, Brusic V. PRED(TAP): a system for prediction of peptide binding to the human transporter associated with antigen processing. Immunome Res. (2006) 2:3. doi: 10.1186/1745-7580-2-3
51. Boehm KM, Bhinder B, Raja VJ, Dephoure N, Elemento O. Predicting peptide presentation by major histocompatibility complex class I: an improved machine learning approach to the immunopeptidome. BMC Bioinformatics. (2019) 20:7. doi: 10.1186/s12859-018-2561-z
52. Larsen MV, Lundegaard C, Lamberth K, Buus S, Lund O, Nielsen M. Large-scale validation of methods for cytotoxic T-lymphocyte epitope prediction. BMC Bioinformatics. (2007) 8:424. doi: 10.1186/1471-2105-8-424
53. Nielsen M, Lundegaard C, Lund O, Kesmir C. The role of the proteasome in generating cytotoxic T-cell epitopes: insights obtained from improved predictions of proteasomal cleavage. Immunogenetics. (2005) 57:33–41. doi: 10.1007/s00251-005-0781-7
54. Liepe J, Ovaa H, Mishto M. Why do proteases mess up with antigen presentation by re-shuffling antigen sequences? Curr Opin Immunol. (2018) 52:81–6. doi: 10.1016/j.coi.2018.04.016
55. Liepe J, Marino F, Sidney J, Jeko A, Bunting DE, Sette A, et al. A large fraction of HLA class I ligands are proteasome-generated spliced peptides. Science. (2016) 354:354–8. doi: 10.1126/science.aaf4384
56. Fritsch EF, Rajasagi M, Ott PA, Brusic V, Hacohen N, Wu CJ. HLA-binding properties of tumor neoepitopes in humans. Cancer Immunol Res. (2014) 2:522–9. doi: 10.1158/2326-6066.CIR-13-0227
57. Berlin C, Kowalewski DJ, Schuster H, Mirza N, Walz S, Handel M, et al. Mapping the HLA ligandome landscape of acute myeloid leukemia: a targeted approach toward peptide-based immunotherapy. Leukemia. (2015) 29:647–59. doi: 10.1038/leu.2014.233
58. Bilich T, Nelde A, Bichmann L, Roerden M, Salih HR, Kowalewski DJ, et al. The HLA ligandome landscape of chronic myeloid leukemia delineates novel T-cell epitopes for immunotherapy. Blood. (2019) 133:550–65. doi: 10.1182/blood-2018-07-866830
59. Nelde A, Kowalewski DJ, Backert L, Schuster H, Werner JO, Klein R, et al. HLA ligandome analysis of primary chronic lymphocytic leukemia (CLL) cells under lenalidomide treatment confirms the suitability of lenalidomide for combination with T-cell-based immunotherapy. Oncoimmunology. (2018) 7:e1316438. doi: 10.1080/2162402X.2017.1316438
60. Abelin JG, Keskin DB, Sarkizova S, Hartigan CR, Zhang W, Sidney J, et al. Mass Spectrometry profiling of HLA-associated peptidomes in mono-allelic cells enables more accurate epitope prediction. Immunity. (2017) 46:315–26. doi: 10.1016/j.immuni.2017.02.007
61. Hawkins OE, Vangundy RS, Eckerd AM, Bardet W, Buchli R, Weidanz JA, et al. Identification of breast cancer peptide epitopes presented by HLA-A*0201. J Proteome Res. (2008) 7:1445–57. doi: 10.1021/pr700761w
62. Trolle T, McMurtrey CP, Sidney J, Bardet W, Osborn SC, Kaever T, et al. The length distribution of class I-restricted T cell epitopes is determined by both peptide supply and MHC allele-specific binding preference. J Immunol. (2016) 196:1480–7. doi: 10.4049/jimmunol.1501721
63. Bassani-Sternberg M, Pletscher-Frankild S, Jensen LJ, Mann M. Mass spectrometry of human leukocyte antigen class I peptidomes reveals strong effects of protein abundance and turnover on antigen presentation. Mol Cell Proteomics. (2015) 14:658–73. doi: 10.1074/mcp.M114.042812
64. Gfeller D, Bassani-Sternberg M. Predicting antigen presentation-what could we learn from a million peptides? Front Immunol. (2018) 9:1716. doi: 10.3389/fimmu.2018.01716
65. Luksza M, Riaz N, Makarov V, Balachandran VP, Hellmann MD, Solovyov A, et al. A neoantigen fitness model predicts tumour response to checkpoint blockade immunotherapy. Nature. (2017) 551:517–20. doi: 10.1038/nature24473
66. Smith CC, Chai S, Washington AR, Lee SJ, Landoni E, Field K, et al. Machine-learning prediction of tumor antigen immunogenicity in the selection of therapeutic epitopes. Cancer Immunol Res. (2019) 7:1591–604. doi: 10.1158/2326-6066.CIR-19-0155
67. Riley TP, Keller GLJ, Smith AR, Davancaze LM, Arbuiso AG, Devlin JR, et al. Structure based prediction of neoantigen immunogenicity. Front Immunol. (2019) 10:2047. doi: 10.3389/fimmu.2019.02047
68. Richman LP, Vonderheide RH, Rech AJ. Neoantigen dissimilarity to the self-proteome predicts immunogenicity and response to immune checkpoint blockade. Cell Syst. (2019) 9:375–82.e4. doi: 10.1016/j.cels.2019.08.009
69. Kosaloglu-Yalcin Z, Lanka M, Frentzen A, Logandha Ramamoorthy Premlal A, Sidney J, Vaughan K, et al. Predicting T cell recognition of MHC class I restricted neoepitopes. Oncoimmunology. (2018) 7:e1492508. doi: 10.1080/2162402X.2018.1492508
70. Yadav M, Jhunjhunwala S, Phung QT, Lupardus P, Tanguay J, Bumbaca S, et al. Predicting immunogenic tumour mutations by combining mass spectrometry and exome sequencing. Nature. (2014) 515:572–6. doi: 10.1038/nature14001
71. Veatch JR, Lee SM, Fitzgibbon M, Chow IT, Jesernig B, Schmitt T, et al. Tumor-infiltrating BRAFV600E-specific CD4+ T cells correlated with complete clinical response in melanoma. J Clin Invest. (2018) 128:1563–8. doi: 10.1172/JCI98689
72. Gros A, Parkhurst MR, Tran E, Pasetto A, Robbins PF, Ilyas S, et al. Prospective identification of neoantigen-specific lymphocytes in the peripheral blood of melanoma patients. Nat Med. (2016) 22:433–8. doi: 10.1038/nm.4051
73. Sakuishi K, Apetoh L, Sullivan JM, Blazar BR, Kuchroo VK, Anderson AC. Targeting Tim-3 and PD-1 pathways to reverse T cell exhaustion and restore anti-tumor immunity. J Exp Med. (2010) 207:2187–94. doi: 10.1084/jem.20100643
74. van der Lee DI, Reijmers RM, Honders MW, Hagedoorn RS, de Jong RC, Kester MG, et al. Mutated nucleophosmin 1 as immunotherapy target in acute myeloid leukemia. J Clin Invest. (2019) 129:774–85. doi: 10.1172/JCI97482
75. Kula T, Dezfulian MH, Wang CI, Abdelfattah NS, Hartman ZC, Wucherpfennig KW, et al. T-Scan: a genome-wide method for the systematic discovery of T cell epitopes. Cell. (2019) 178:1016–28.e13. doi: 10.1016/j.cell.2019.07.009
76. Bleakley M, Otterud BE, Richardt JL, Mollerup AD, Hudecek M, Nishida T, et al. Leukemia-associated minor histocompatibility antigen discovery using T-cell clones isolated by in vitro stimulation of naive CD8+ T cells. Blood. (2010) 115:4923–33. doi: 10.1182/blood-2009-12-260539
77. Stronen E, Toebes M, Kelderman S, van Buuren MM, Yang W, van Rooij N, et al. Targeting of cancer neoantigens with donor-derived T cell receptor repertoires. Science. (2016) 352:1337–41. doi: 10.1126/science.aaf2288
78. Ding LW, Sun QY, Tan KT, Chien W, Mayakonda A, Yeoh AEJ, et al. Mutational landscape of pediatric acute lymphoblastic leukemia. Cancer Res. (2017) 77:390–400. doi: 10.1158/0008-5472.CAN-16-1303
79. Iacobucci I, Mullighan CG. Genetic basis of acute lymphoblastic leukemia. J Clin Oncol. (2017) 35:975–83. doi: 10.1200/JCO.2016.70.7836
80. Bolouri H, Farrar JE, Triche T Jr, Ries RE, Lim EL, Alonzo TA, et al. The molecular landscape of pediatric acute myeloid leukemia reveals recurrent structural alterations and age-specific mutational interactions. Nat Med. (2018) 24:103–12. doi: 10.1038/nm.4439
81. Cancer Genome Atlas Research N. Genomic and epigenomic landscapes of adult de novo acute myeloid leukemia. N Engl J Med. (2013) 368:2059–74. doi: 10.1056/NEJMoa1301689
82. Landau DA, Tausch E, Taylor-Weiner AN, Stewart C, Reiter JG, Bahlo J, et al. Mutations driving CLL and their evolution in progression and relapse. Nature. (2015) 526:525–30. doi: 10.1038/nature15395
83. Puente XS, Bea S, Valdes-Mas R, Villamor N, Gutierrez-Abril J, Martin-Subero JI, et al. Non-coding recurrent mutations in chronic lymphocytic leukaemia. Nature. (2015) 526:519–24. doi: 10.1038/nature14666
84. Bose P, Nazha A, Komrokji RS, Patel KP, Pierce SA, Al-Ali N, et al. Mutational landscape of myelodysplastic/myeloproliferative neoplasm-unclassifiable. Blood. (2018) 132:2100–3. doi: 10.1182/blood-2018-05-848473
85. Schischlik F, Jager R, Rosebrock F, Hug E, Schuster M, Holly R, et al. Mutational landscape of the transcriptome offers putative targets for immunotherapy of myeloproliferative neoplasms. Blood. (2019) 134:199–210. doi: 10.1182/blood.2019000519
86. Kowalewski DJ, Schuster H, Backert L, Berlin C, Kahn S, Kanz L, et al. HLA ligandome analysis identifies the underlying specificities of spontaneous antileukemia immune responses in chronic lymphocytic leukemia (CLL). Proc Natl Acad Sci USA. (2015) 112:E166–75. doi: 10.1073/pnas.1416389112
87. Walz S, Stickel JS, Kowalewski DJ, Schuster H, Weisel K, Backert L, et al. The antigenic landscape of multiple myeloma: mass spectrometry (re)defines targets for T-cell-based immunotherapy. Blood. (2015) 126:1203–13. doi: 10.1182/blood-2015-04-640532
88. Narayan R, Olsson N, Wagar LE, Medeiros BC, Meyer E, Czerwinski D, et al. Acute myeloid leukemia immunopeptidome reveals HLA presentation of mutated nucleophosmin. PLoS ONE. (2019) 14:e0219547. doi: 10.1371/journal.pone.0219547
89. van Dongen JJ, Macintyre EA, Gabert JA, Delabesse E, Rossi V, Saglio G, et al. Standardized RT-PCR analysis of fusion gene transcripts from chromosome aberrations in acute leukemia for detection of minimal residual disease. Report of the BIOMED-1 Concerted Action: investigation of minimal residual disease in acute leukemia. Leukemia. (1999) 13:1901–28. doi: 10.1038/sj.leu.2401592
90. Meyer C, Burmeister T, Groger D, Tsaur G, Fechina L, Renneville A, et al. The MLL recombinome of acute leukemias in 2017. Leukemia. (2018) 32:273–84. doi: 10.1038/leu.2017.213
91. King-Underwood L, Renshaw J, Pritchard-Jones K. Mutations in the Wilms' tumor gene WT1 in leukemias. Blood. (1996) 87:2171–9. doi: 10.1182/blood.V87.6.2171.bloodjournal8762171
92. Pronier E, Bowman RL, Ahn J, Glass J, Kandoth C, Merlinsky TR, et al. Genetic and epigenetic evolution as a contributor to WT1-mutant leukemogenesis. Blood. (2018) 132:1265–78. doi: 10.1182/blood-2018-03-837468
93. Niktoreh N, Walter C, Zimmermann M, von Neuhoff C, von Neuhoff N, Rasche M, et al. Mutated WT1, FLT3-ITD, and NUP98-NSD1 fusion in various combinations define a poor prognostic group in pediatric acute myeloid leukemia. J Oncol. (2019) 2019:1609128. doi: 10.1155/2019/1609128
94. Potter N, Miraki-Moud F, Ermini L, Titley I, Vijayaraghavan G, Papaemmanuil E, et al. Single cell analysis of clonal architecture in acute myeloid leukaemia. Leukemia. (2019) 33:1113–23. doi: 10.1038/s41375-018-0319-2
95. Tosello V, Mansour MR, Barnes K, Paganin M, Sulis ML, Jenkinson S, et al. WT1 mutations in T-ALL. Blood. (2009) 114:1038–45. doi: 10.1182/blood-2008-12-192039
96. Weng AP, Ferrando AA, Lee W, Morris JPt, Silverman LB, Sanchez-Irizarry C, et al. Activating mutations of NOTCH1 in human T cell acute lymphoblastic leukemia. Science. (2004) 306:269–71. doi: 10.1126/science.1102160
97. Grabher C, von Boehmer H, Look AT. Notch 1 activation in the molecular pathogenesis of T-cell acute lymphoblastic leukaemia. Nat Rev Cancer. (2006) 6:347–59. doi: 10.1038/nrc1880
98. Girardi T, Vicente C, Cools J, De Keersmaecker K. The genetics and molecular biology of T-ALL. Blood. (2017) 129:1113–23. doi: 10.1182/blood-2016-10-706465
99. Bykov VJN, Eriksson SE, Bianchi J, Wiman KG. Targeting mutant p53 for efficient cancer therapy. Nat Rev Cancer. (2018) 18:89–102. doi: 10.1038/nrc.2017.109
100. Stengel A, Kern W, Haferlach T, Meggendorfer M, Fasan A, Haferlach C. The impact of TP53 mutations and TP53 deletions on survival varies between AML, ALL, MDS and CLL: an analysis of 3307 cases. Leukemia. (2017) 31:705–11. doi: 10.1038/leu.2016.263
101. Xu-Monette ZY, Medeiros LJ, Li Y, Orlowski RZ, Andreeff M, Bueso-Ramos CE, et al. Dysfunction of the TP53 tumor suppressor gene in lymphoid malignancies. Blood. (2012) 119:3668–83. doi: 10.1182/blood-2011-11-366062
102. Falini B, Martelli MP, Bolli N, Sportoletti P, Liso A, Tiacci E, et al. Acute myeloid leukemia with mutated nucleophosmin (NPM1): is it a distinct entity? Blood. (2011) 117:1109–20. doi: 10.1182/blood-2010-08-299990
103. Falini B, Mecucci C, Tiacci E, Alcalay M, Rosati R, Pasqualucci L, et al. Cytoplasmic nucleophosmin in acute myelogenous leukemia with a normal karyotype. N Engl J Med. (2005) 352:254–66. doi: 10.1056/NEJMoa041974
104. McGranahan N, Swanton C. Neoantigen quality, not quantity. Sci Transl Med. (2019) 11:eaax7918. doi: 10.1126/scitranslmed.aax7918
105. Balachandran VP, Luksza M, Zhao JN, Makarov V, Moral JA, Remark R, et al. Identification of unique neoantigen qualities in long-term survivors of pancreatic cancer. Nature. (2017) 551:512–6. doi: 10.1038/nature24462
106. Greiner J, Schneider V, Schmitt M, Gotz M, Dohner K, Wiesneth M, et al. Immune responses against the mutated region of cytoplasmatic NPM1 might contribute to the favorable clinical outcome of AML patients with NPM1 mutations (NPM1mut). Blood. (2013) 122:1087–8. doi: 10.1182/blood-2013-04-496844
107. Hofmann S, Gotz M, Schneider V, Guillaume P, Bunjes D, Dohner H, et al. Donor lymphocyte infusion induces polyspecific CD8+ T-cell responses with concurrent molecular remission in acute myeloid leukemia with NPM1 mutation. J Clin Oncol. (2013) 31:e44–7. doi: 10.1200/JCO.2011.41.1116
108. Schneider V, Zhang L, Bullinger L, Rojewski M, Hofmann S, Wiesneth M, et al. Leukemic stem cells of acute myeloid leukemia patients carrying NPM1 mutation are candidates for targeted immunotherapy. Leukemia. (2014) 28:1759–62. doi: 10.1038/leu.2014.116
109. Cimen Bozkus C, Roudko V, Finnigan JP, Mascarenhas J, Hoffman R, Iancu-Rubin C, et al. Immune checkpoint blockade enhances shared neoantigen-induced T-cell immunity directed against mutated calreticulin in myeloproliferative neoplasms. Cancer Discov. (2019) 9:1192–207. doi: 10.1158/2159-8290.CD-18-1356
110. Gambacorti-Passerini C, Grignani F, Arienti F, Pandolfi PP, Pelicci PG, Parmiani G. Human CD4 lymphocytes specifically recognize a peptide representing the fusion region of the hybrid protein pml/RAR alpha present in acute promyelocytic leukemia cells. Blood. (1993) 81:1369–75. doi: 10.1182/blood.V81.5.1369.1369
111. Dermime S, Bertazzoli C, Marchesi E, Ravagnani F, Blaser K, Corneo GM, et al. Lack of T-cell-mediated recognition of the fusion region of the pml/RAR-alpha hybrid protein by lymphocytes of acute promyelocytic leukemia patients. Clin Cancer Res. (1996) 2:593–600.
112. Zamora AE, Crawford JC, Allen EK, Guo XJ, Bakke J, Carter RA, et al. Pediatric patients with acute lymphoblastic leukemia generate abundant and functional neoantigen-specific CD8+ T cell responses. Sci Transl Med. (2019) 11:eaat8549. doi: 10.1126/scitranslmed.aat8549
113. Yotnda P, Garcia F, Peuchmaur M, Grandchamp B, Duval M, Lemonnier F, et al. Cytotoxic T cell response against the chimeric ETV6-AML1 protein in childhood acute lymphoblastic leukemia. J Clin Invest. (1998) 102:455–62. doi: 10.1172/JCI3126
114. Popovic J, Li LP, Kloetzel PM, Leisegang M, Uckert W, Blankenstein T. The only proposed T-cell epitope derived from the TEL-AML1 translocation is not naturally processed. Blood. (2011) 118:946–54. doi: 10.1182/blood-2010-12-325035
115. Tubb VM, Schrikkema DS, Croft NP, Purcell AW, Linnemann C, Freriks MR, et al. Isolation of T cell receptors targeting recurrent neoantigens in hematological malignancies. J Immunother Cancer. (2018) 6:70. doi: 10.1186/s40425-018-0386-y
116. Holmstrom MO, Hjortso MD, Ahmad SM, Met O, Martinenaite E, Riley C, et al. The JAK2V617F mutation is a target for specific T cells in the JAK2V617F-positive myeloproliferative neoplasms. Leukemia. (2017) 31:495–8. doi: 10.1038/leu.2016.290
117. Clark RE, Dodi IA, Hill SC, Lill JR, Aubert G, Macintyre AR, et al. Direct evidence that leukemic cells present HLA-associated immunogenic peptides derived from the BCR-ABL b3a2 fusion protein. Blood. (2001) 98:2887–93. doi: 10.1182/blood.V98.10.2887
118. Cai A, Keskin DB, DeLuca DS, Alonso A, Zhang W, Zhang GL, et al. Mutated BCR-ABL generates immunogenic T-cell epitopes in CML patients. Clin Cancer Res. (2012) 18:5761–72. doi: 10.1158/1078-0432.CCR-12-1182
119. Vauchy C, Gamonet C, Ferrand C, Daguindau E, Galaine J, Beziaud L, et al. CD20 alternative splicing isoform generates immunogenic CD4 helper T epitopes. Int J Cancer. (2015) 137:116–26. doi: 10.1002/ijc.29366
120. Wang QJ, Yu Z, Griffith K, Hanada K, Restifo NP, Yang JC. Identification of T-cell receptors targeting KRAS-mutated human tumors. Cancer Immunol Res. (2016) 4:204–14. doi: 10.1158/2326-6066.CIR-15-0188
121. Malekzadeh P, Pasetto A, Robbins PF, Parkhurst MR, Paria BC, Jia L, et al. Neoantigen screening identifies broad TP53 mutant immunogenicity in patients with epithelial cancers. J Clin Invest. (2019) 129:1109–14. doi: 10.1172/JCI123791
122. Lo W, Parkhurst M, Robbins PF, Tran E, Lu YC, Jia L, et al. Immunologic recognition of a shared p53 mutated neoantigen in a patient with metastatic colorectal cancer. Cancer Immunol Res. (2019) 7:534–43. doi: 10.1158/2326-6066.CIR-18-0686
123. Forghieri F, Riva G, Lagreca I, Barozzi P, Vallerini D, Morselli M, et al. Characterization and dynamics of specific T cells against nucleophosmin-1 (NPM1)-mutated peptides in patients with NPM1-mutated acute myeloid leukemia. Oncotarget. (2019) 10:869–82. doi: 10.18632/oncotarget.26617
124. Grimwade D, Hills RK, Moorman AV, Walker H, Chatters S, Goldstone AH, et al. Refinement of cytogenetic classification in acute myeloid leukemia: determination of prognostic significance of rare recurring chromosomal abnormalities among 5876 younger adult patients treated in the United Kingdom Medical Research Council trials. Blood. (2010) 116:354–65. doi: 10.1182/blood-2009-11-254441
125. Grimwade D, Ivey A, Huntly BJ. Molecular landscape of acute myeloid leukemia in younger adults and its clinical relevance. Blood. (2016) 127:29–41. doi: 10.1182/blood-2015-07-604496
126. Papaemmanuil E, Gerstung M, Malcovati L, Tauro S, Gundem G, Van Loo P, et al. Clinical and biological implications of driver mutations in myelodysplastic syndromes. Blood. (2013) 122:3616–27; quiz 99. doi: 10.1182/blood-2013-08-518886
127. Chalmers ZR, Connelly CF, Fabrizio D, Gay L, Ali SM, Ennis R, et al. Analysis of 100,000 human cancer genomes reveals the landscape of tumor mutational burden. Genome Med. (2017) 9:34. doi: 10.1186/s13073-017-0424-2
128. Shurtleff SA, Buijs A, Behm FG, Rubnitz JE, Raimondi SC, Hancock ML, et al. TEL/AML1 fusion resulting from a cryptic t(12;21) is the most common genetic lesion in pediatric ALL and defines a subgroup of patients with an excellent prognosis. Leukemia. (1995) 9:1985–9.
129. Romana SP, Mauchauffe M, Le Coniat M, Chumakov I, Le Paslier D, Berger R, et al. The t(12;21) of acute lymphoblastic leukemia results in a tel-AML1 gene fusion. Blood. (1995) 85:3662–70. doi: 10.1182/blood.V85.12.3662.bloodjournal85123662
130. Romana SP, Poirel H, Leconiat M, Flexor MA, Mauchauffe M, Jonveaux P, et al. High frequency of t(12;21) in childhood B-lineage acute lymphoblastic leukemia. Blood. (1995) 86:4263–9. doi: 10.1182/blood.V86.11.4263.bloodjournal86114263
131. Rubnitz JE, Downing JR, Pui CH, Shurtleff SA, Raimondi SC, Evans WE, et al. TEL gene rearrangement in acute lymphoblastic leukemia: a new genetic marker with prognostic significance. J Clin Oncol. (1997) 15:1150–7. doi: 10.1200/JCO.1997.15.3.1150
132. Vainchenker W, Kralovics R. Genetic basis and molecular pathophysiology of classical myeloproliferative neoplasms. Blood. (2017) 129:667–79. doi: 10.1182/blood-2016-10-695940
133. Holmstrom MO, Riley CH, Svane IM, Hasselbalch HC, Andersen MH. The CALR exon 9 mutations are shared neoantigens in patients with CALR mutant chronic myeloproliferative neoplasms. Leukemia. (2016) 30:2413–6. doi: 10.1038/leu.2016.233
134. Voncken JW, Kaartinen V, Pattengale PK, Germeraad WT, Groffen J, Heisterkamp N. BCR/ABL P210 and P190 cause distinct leukemia in transgenic mice. Blood. (1995) 86:4603–11. doi: 10.1182/blood.V86.12.4603.bloodjournal86124603
135. Chen W, Peace DJ, Rovira DK, You SG, Cheever MA. T-cell immunity to the joining region of p210BCR-ABL protein. Proc Natl Acad Sci USA. (1992) 89:1468–72. doi: 10.1073/pnas.89.4.1468
136. Riley CL, Mathieu MG, Clark RE, McArdle SE, Rees RC. Tumour antigen-targeted immunotherapy for chronic myeloid leukaemia: is it still viable? Cancer Immunol Immunother. (2009) 58:1489–99. doi: 10.1007/s00262-009-0675-x
137. Comoli P, Basso S, Riva G, Barozzi P, Guido I, Gurrado A, et al. BCR-ABL-specific T-cell therapy in Ph+ ALL patients on tyrosine-kinase inhibitors. Blood. (2017) 129:582–6. doi: 10.1182/blood-2016-07-731091
138. Lynch RG, Graff RJ, Sirisinha S, Simms ES, Eisen HN. Myeloma proteins as tumor-specific transplantation antigens. Proc Natl Acad Sci USA. (1972) 69:1540–4. doi: 10.1073/pnas.69.6.1540
139. Bendandi M. Idiotype vaccines for lymphoma: proof-of-principles and clinical trial failures. Nat Rev Cancer. (2009) 9:675–81. doi: 10.1038/nrc2717
140. Khodadoust MS, Olsson N, Wagar LE, Haabeth OA, Chen B, Swaminathan K, et al. Antigen presentation profiling reveals recognition of lymphoma immunoglobulin neoantigens. Nature. (2017) 543:723–7. doi: 10.1038/nature21433
141. Khodadoust MS, Olsson N, Chen B, Sworder B, Shree T, Liu CL, et al. B-cell lymphomas present immunoglobulin neoantigens. Blood. (2019) 133:878–81. doi: 10.1182/blood-2018-06-845156
142. Henry C, Deschamps M, Rohrlich PS, Pallandre JR, Remy-Martin JP, Callanan M, et al. Identification of an alternative CD20 transcript variant in B-cell malignancies coding for a novel protein associated to rituximab resistance. Blood. (2010) 115:2420–9. doi: 10.1182/blood-2009-06-229112
143. Nelde A, Walz JS, Kowalewski DJ, Schuster H, Wolz OO, Peper JK, et al. HLA class I-restricted MYD88 L265P-derived peptides as specific targets for lymphoma immunotherapy. Oncoimmunology. (2017) 6:e1219825. doi: 10.1080/2162402X.2016.1219825
144. Malumbres M, Barbacid M. RAS oncogenes: the first 30 years. Nat Rev Cancer. (2003) 3:459–65. doi: 10.1038/nrc1097
145. Schubbert S, Shannon K, Bollag G. Hyperactive Ras in developmental disorders and cancer. Nat Rev Cancer. (2007) 7:295–308. doi: 10.1038/nrc2109
146. Ward AF, Braun BS, Shannon KM. Targeting oncogenic Ras signaling in hematologic malignancies. Blood. (2012) 120:3397–406. doi: 10.1182/blood-2012-05-378596
147. Prior IA, Lewis PD, Mattos C. A comprehensive survey of Ras mutations in cancer. Cancer Res. (2012) 72:2457–67. doi: 10.1158/0008-5472.CAN-11-2612
148. Vendramini E, Bomben R, Pozzo F, Benedetti D, Bittolo T, Rossi FM, et al. KRAS, NRAS, and BRAF mutations are highly enriched in trisomy 12 chronic lymphocytic leukemia and are associated with shorter treatment-free survival. Leukemia. (2019) 33:2111–5. doi: 10.1038/s41375-019-0444-6
149. Hobbs GA, Der CJ, Rossman KL. RAS isoforms and mutations in cancer at a glance. J Cell Sci. (2016) 129:1287–92. doi: 10.1242/jcs.182873
150. Holderfield M, Deuker MM, McCormick F, McMahon M. Targeting RAF kinases for cancer therapy: BRAF-mutated melanoma and beyond. Nat Rev Cancer. (2014) 14:455–67. doi: 10.1038/nrc3760
151. Tiacci E, Trifonov V, Schiavoni G, Holmes A, Kern W, Martelli MP, et al. BRAF mutations in hairy-cell leukemia. N Engl J Med. (2011) 364:2305–15. doi: 10.1056/NEJMoa1014209
152. Arcaini L, Zibellini S, Boveri E, Riboni R, Rattotti S, Varettoni M, et al. The BRAF V600E mutation in hairy cell leukemia and other mature B-cell neoplasms. Blood. (2012) 119:188–91. doi: 10.1182/blood-2011-08-368209
153. Chung SS, Kim E, Park JH, Chung YR, Lito P, Teruya-Feldstein J, et al. Hematopoietic stem cell origin of BRAFV600E mutations in hairy cell leukemia. Sci Transl Med. (2014) 6:238ra71. doi: 10.1126/scitranslmed.3008004
154. Berres ML, Lim KP, Peters T, Price J, Takizawa H, Salmon H, et al. BRAF-V600E expression in precursor versus differentiated dendritic cells defines clinically distinct LCH risk groups. J Exp Med. (2014) 211:669–83. doi: 10.1084/jem.20130977
155. Bouaoun L, Sonkin D, Ardin M, Hollstein M, Byrnes G, Zavadil J, et al. TP53 Variations in human cancers: new lessons from the IARC TP53 database and genomics data. Hum Mutat. (2016) 37:865–76. doi: 10.1002/humu.23035
156. Cafri G, Yossef R, Pasetto A, Deniger DC, Lu YC, Parkhurst M, et al. Memory T cells targeting oncogenic mutations detected in peripheral blood of epithelial cancer patients. Nat Commun. (2019) 10:449. doi: 10.1038/s41467-019-08304-z
157. Sharkey MS, Lizee G, Gonzales MI, Patel S, Topalian SL. CD4+ T-cell recognition of mutated B-RAF in melanoma patients harboring the V599E mutation. Cancer Res. (2004) 64:1595–9. doi: 10.1158/0008-5472.CAN-03-3231
158. Kandoth C, McLellan MD, Vandin F, Ye K, Niu B, Lu C, et al. Mutational landscape and significance across 12 major cancer types. Nature. (2013) 502:333–9. doi: 10.1038/nature12634
159. Deniger DC, Pasetto A, Tran E, Parkhurst MR, Cohen CJ, Robbins PF, et al. Stable, nonviral expression of mutated tumor neoantigen-specific t-cell receptors using the sleeping beauty transposon/transposase system. Mol Ther. (2016) 24:1078–89. doi: 10.1038/mt.2016.51
160. Yossef R, Tran E, Deniger DC, Gros A, Pasetto A, Parkhurst MR, et al. Enhanced detection of neoantigen-reactive T cells targeting unique and shared oncogenes for personalized cancer immunotherapy. JCI Insight. (2018) 3:e122467. doi: 10.1172/jci.insight.122467
161. Noonan KA, Huff CA, Davis J, Lemas MV, Fiorino S, Bitzan J, et al. Adoptive transfer of activated marrow-infiltrating lymphocytes induces measurable antitumor immunity in the bone marrow in multiple myeloma. Sci Transl Med. (2015) 7:288ra78. doi: 10.1126/scitranslmed.aaa7014
162. Chapuis AG, Ragnarsson GB, Nguyen HN, Chaney CN, Pufnock JS, Schmitt TM, et al. Transferred WT1-reactive CD8+ T cells can mediate antileukemic activity and persist in post-transplant patients. Sci Transl Med. (2013) 5:174ra27. doi: 10.1126/scitranslmed.3004916
163. Ochi T, Fujiwara H, Okamoto S, An J, Nagai K, Shirakata T, et al. Novel adoptive T-cell immunotherapy using a WT1-specific TCR vector encoding silencers for endogenous TCRs shows marked antileukemia reactivity and safety. Blood. (2011) 118:1495–503. doi: 10.1182/blood-2011-02-337089
164. Ohminami H, Yasukawa M, Fujita S. HLA class I-restricted lysis of leukemia cells by a CD8+ cytotoxic T-lymphocyte clone specific for WT1 peptide. Blood. (2000) 95:286–93. doi: 10.1182/blood.V95.1.286
165. Chapuis AG, Egan DN, Bar M, Schmitt TM, McAfee MS, Paulson KG, et al. T cell receptor gene therapy targeting WT1 prevents acute myeloid leukemia relapse post-transplant. Nat Med. (2019) 25:1064–72. doi: 10.1038/s41591-019-0472-9
166. Hung K, Hayashi R, Lafond-Walker A, Lowenstein C, Pardoll D, Levitsky H. The central role of CD4+ T cells in the antitumor immune response. J Exp Med. (1998) 188:2357–68. doi: 10.1084/jem.188.12.2357
167. Dossa RG, Cunningham T, Sommermeyer D, Medina-Rodriguez I, Biernacki MA, Foster K, et al. Development of T-cell immunotherapy for hematopoietic stem cell transplantation recipients at risk of leukemia relapse. Blood. (2018) 131:108–20. doi: 10.1182/blood-2017-07-791608
168. Sommermeyer D, Hudecek M, Kosasih PL, Gogishvili T, Maloney DG, Turtle CJ, et al. Chimeric antigen receptor-modified T cells derived from defined CD8+ and CD4+ subsets confer superior antitumor reactivity in vivo. Leukemia. (2016) 30:492–500. doi: 10.1038/leu.2015.247
169. Di Stasi A, Tey SK, Dotti G, Fujita Y, Kennedy-Nasser A, Martinez C, et al. Inducible apoptosis as a safety switch for adoptive cell therapy. N Engl J Med. (2011) 365:1673–83. doi: 10.1056/NEJMoa1106152
170. Biernacki MA, Brault M, Bleakley M. T-cell receptor-based immunotherapy for hematologic malignancies. Cancer J. (2019) 25:179–90. doi: 10.1097/PPO.0000000000000378
171. Kebriaei P, Singh H, Huls MH, Figliola MJ, Bassett R, Olivares S, et al. Phase I trials using Sleeping Beauty to generate CD19-specific CAR T cells. J Clin Invest. (2016) 126:3363–76. doi: 10.1172/JCI86721
172. Bishop DC, Xu N, Tse B, O'Brien TA, Gottlieb DJ, Dolnikov A, et al. PiggyBac-engineered T cells expressing CD19-specific CARs that Lack IgG1 Fc spacers have potent activity against B-ALL xenografts. Mol Ther. (2018) 26:1883–95. doi: 10.1016/j.ymthe.2018.05.007
173. Wang J, Ma Z, Carr SA, Mertins P, Zhang H, Zhang Z, et al. Proteome profiling outperforms transcriptome profiling for coexpression based gene function prediction. Mol Cell Proteomics. (2017) 16:121–34. doi: 10.1074/mcp.M116.060301
174. Campillo-Davo D, Fujiki F, Van den Bergh JMJ, De Reu H, Smits E, Goossens H, et al. Efficient and non-genotoxic RNA-based engineering of human T cells using tumor-specific T cell receptors with minimal TCR mispairing. Front Immunol. (2018) 9:2503. doi: 10.3389/fimmu.2018.02503
175. Torikai H, Reik A, Soldner F, Warren EH, Yuen C, Zhou Y, et al. Toward eliminating HLA class I expression to generate universal cells from allogeneic donors. Blood. (2013) 122:1341–9. doi: 10.1182/blood-2013-03-478255
176. Lemos H, Huang L, Prendergast GC, Mellor AL. Immune control by amino acid catabolism during tumorigenesis and therapy. Nat Rev Cancer. (2019) 19:162–75. doi: 10.1038/s41568-019-0106-z
177. van der Burg SH, Arens R, Ossendorp F, van Hall T, Melief CJ. Vaccines for established cancer: overcoming the challenges posed by immune evasion. Nat Rev Cancer. (2016) 16:219–33. doi: 10.1038/nrc.2016.16
178. Fukuno K, Hara T, Tsurumi H, Shibata Y, Mabuchi R, Nakamura N, et al. Expression of indoleamine 2,3-dioxygenase in leukemic cells indicates an unfavorable prognosis in acute myeloid leukemia patients with intermediate-risk cytogenetics. Leuk Lymphoma. (2015) 56:1398–405. doi: 10.3109/10428194.2014.953150
179. Mussai F, De Santo C, Abu-Dayyeh I, Booth S, Quek L, McEwen-Smith RM, et al. Acute myeloid leukemia creates an arginase-dependent immunosuppressive microenvironment. Blood. (2013) 122:749–58. doi: 10.1182/blood-2013-01-480129
180. Zhang L, Gajewski TF, Kline J. PD-1/PD-L1 interactions inhibit antitumor immune responses in a murine acute myeloid leukemia model. Blood. (2009) 114:1545–52. doi: 10.1182/blood-2009-03-206672
181. Buggins AG, Milojkovic D, Arno MJ, Lea NC, Mufti GJ, Thomas NS, et al. Microenvironment produced by acute myeloid leukemia cells prevents T cell activation and proliferation by inhibition of NF-kappaB, c-Myc, and pRb pathways. J Immunol. (2001) 167:6021–30. doi: 10.4049/jimmunol.167.10.6021
182. Bruck O, Blom S, Dufva O, Turkki R, Chheda H, Ribeiro A, et al. Immune cell contexture in the bone marrow tumor microenvironment impacts therapy response in CML. Leukemia. (2018) 32:1643–56. doi: 10.1038/s41375-018-0175-0
Keywords: neoantigen, hematologic malignancies, human leukocyte antigen, T cell receptor, immunotherapy, mutations, fusion proteins
Citation: Biernacki MA and Bleakley M (2020) Neoantigens in Hematologic Malignancies. Front. Immunol. 11:121. doi: 10.3389/fimmu.2020.00121
Received: 23 October 2019; Accepted: 16 January 2020;
Published: 14 February 2020.
Edited by:
Robbert Spaapen, AMC-Sanquin Landsteiner Laboratory, NetherlandsCopyright © 2020 Biernacki and Bleakley. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Melinda A. Biernacki, bWJpZXJuYWMmI3gwMDA0MDtmcmVkaHV0Y2gub3Jn