
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Immunol. , 17 January 2020
Sec. Vaccines and Molecular Therapeutics
Volume 10 - 2019 | https://doi.org/10.3389/fimmu.2019.03078
This article is part of the Research Topic Immunogenicity of Proteins Used as Therapeutics View all 27 articles
 David W. Scott*
David W. Scott* Kathleen P. Pratt
Kathleen P. PrattTherapeutic treatment of bleeds with FVIII can lead to an antibody response that effectively inhibits its function. Herein, we review the factors that contribute to this immunogenicity and possible ways to overcome it.
Self-non-self-discrimination is one of the basic tenets of the immune system. The general failure to respond immunologically to antigens in our bodies is learned during ontogeny, as aptly recognized by Ray Owen in the 1940's in seminal studies with dizygotic cattle twins which shared hematopoietic cells during fetal development (1). Thus, these siblings failed to react to red blood cell antigens or skin grafts of their unrelated twin because their immune systems had learned that they must be “self” during ontogeny. This phenomenon of “actively acquired (immunologic) tolerance” was experimentally verified by the Nobel Prize-winning experiments of Billingham, Brent and Medawar in mice (2). This process is specific because responses to unrelated antigens remains intact.
What happens in the case of a patient who fails to express a given human protein during ontogeny and is then subsequently exposed? The classic case is hemophilia A and B, where patients lack all or part of the factor VIII (FVIII) or factor IX procoagulant proteins, respectively, and therefore have never “acquired” tolerance to that protein as “self.” Prophylactic or on-demand treatment of bleeds with recombinant or plasma-derived FVIII can lead to an antibody response to this human (but “foreign”) protein that effectively neutralizes or inhibits its function in the coagulation pathway; these antibodies are called “inhibitors.”
We discuss here some recent approaches, focusing on several developed in our laboratories, to characterize anti-FVIII immune responses and to promote durable peripheral tolerance to exogenously administered FVIII.
Thus, while lack of tolerance (non-self) to FVIII explains its immunogenicity, there are other factors that potentially play a role. These are listed in Table 1 and are discussed below. Clearly, non-self-proteins tend to be recognized as foreign, as the process of c selection has not occurred. The developing immune system simply hasn't seen the T- and/or B-cell epitopes in the protein. Interestingly, FVIII is usually administered to patients intravenously (i.v.), a normally tolerogenic route to safely administer foreign antigens, yet it is highly immunogenic compared to many other therapeutic proteins, with approximately one in four patients developing a clinically significant inhibitor. Indeed, many foreign proteins that pass through the lymphatic or venous system into lymphoid organs are ignored, unless they provide additional signals or so-called adjuvanticity, often referred to as “danger” signals (3). This is because they lack properties that can stimulate the innate immune system, e.g., by interacting with Toll-like receptors (TLR) or through other innate immune processes either directly or indirectly. Efforts to demonstrate this property in FVIII have included culturing of human monocyte-derived dendritic cells (MoDCs) with FVIII, or thrombin-cleaved FVIII, or FVIII complexed with its carrier protein von Willebrand factor (VWF); interestingly, neither the maturation nor the T-cell stimulatory capacity of the MoDCs were affected (4). Skupsky et al. suggested that the biologic activity of FVIII in the clotting cascade, which leads to accelerated thrombin activation, provided an alternative mechanism of stimulating innate immune signaling (5). They found that treatment of mice with the anticoagulants warfarin or hirudin, which inactivate thrombin, reduced the immunogenicity of human FVIII in hemophilia A mice. In contrast, Meeks and co-workers, who engineered human FVIII proteins having several amino acid substitutions that neutralized its procoagulant activity, found that the immunogenicity of these non-active FVIII proteins was highly similar to that of active FVIII, thus leaving the role of its biological activity leading to immunologic “danger” as moot (6). If thrombin activation contributes to immunogenicity, then one might expect factor IX to also be unusually immunogenic when administered to hemophilia B patients. Inhibitor development in hemophilia B is actually rare, but this is likely due to the fact that most hemophilia B patients actually circulate a dysfunctional factor IX protein. They therefore could only respond to far fewer epitopes than patients with null mutations.
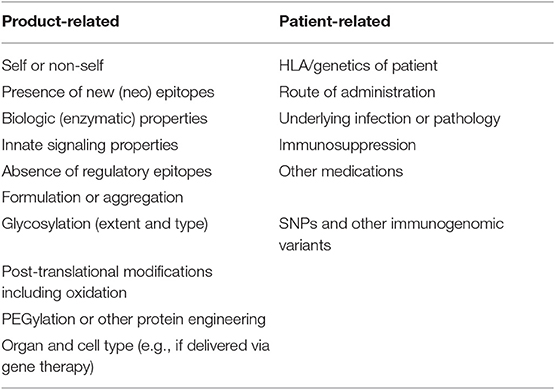
Table 1. Factors affecting the immunogenicity of human protein therapeutics.
Uptake and processing by antigen-presenting cells (APC) is the first step in the immune response to protein antigens. Proteolytic processing leads to presentation of peptides in major histocompatibility complexes on the APC surface, e.g., dendritic cells (DC). As noted above, peptides for which no thymic deletion has occurred may be immunogenic provided that they can be processed and presented on MHC Class I or Class II on mature DC, and that a T-cell receptor (TCR) on a circulating T cell recognizes and engages the resulting MHC-peptide complex. Interestingly, exposure to FVIII does not provoke a CD8+ immune response in hemophilia A patients or in murine FVIII−/− mice, whereas CD4+ T-cell help (7) is essential for the development of high-titer anti-FVIII antibodies (8). MHC Class II peptide presentation provides “signal one” to effector CD4 T cells in the peripheral repertoire. In contrast, it has been proposed that many proteins may contain promiscuous peptide sequences that preferentially activate T regulatory rather than CD4 effectors; these have been termed “Tregitopes” (9, 10). These peptide sequences are commonly found not only in immunoglobulins but in many infectious agents, which may enable them to modulate and reduce the immune response to those agents. The potential role of Tregitopes in modulating FVIII immunogenicity, however, has not yet been established.
Last, but not least, is the physical properties of the FVIII antigen that may influence immunogenicity, such as post-translational modifications or physical aggregation, especially when the antigen is stored or administered at high concentrations. This may be due to an intrinsic or extrinsic property of the antigen, e.g., caused by charge changes, or by physical perturbations resulting from heating or formulation (11, 12). Differences in glycosylation patterns, e.g., according to the type of cell expression system, and covalent modifications to extend protein half-life (PEGylation, fusions of FVIII with other proteins or domains, etc.), and B-domain removal all could affect the immunogenicity of FVIII. The recent, prospective SIPPET study showed a significantly higher inhibitor incidence in previously untreated patients receiving a recombinant FVIII product, compared to plasma-derived FVIII (13). The biological basis for this difference remains to be identified.
Beyond the above properties, one must consider additional factors that influence immunogenicity which may be manifested in the recipients of FVIII replacement therapy. While there is no clear linkage to the HLA of the patient, HLA does affect which peptides will bind to the MHC on DC. Indeed, HLA Class II-restricted epitopes in FVIII were identified years ago by peptide proliferation assays (14–19). Subsequent isolation of FVIII-specific T-cell clones by classical limiting dilution (20) or by using HLA Class II tetramers loaded with FVIII peptides (7, 21–24) provided unambiguous identification of specific high-avidity epitopes (25). At the level of the repertoire, one must consider the nature of the mutation in the FVIII gene (F8) that leads to delayed or absent pro-coagulant activity: patients with a major deletion or other mutation precluding expression of the FVIII protein should lack tolerance to all of the epitopes in FVIII. On the other hand, those with missense mutations, which generally are associated with mild or moderate severity hemophilia A due to a partially disabled but still full-length FVIII protein, have a lower risk of developing an inhibitor response following FVIII infusions. In addition to FVIII mutations, other genetic factors, as well as environmental differences including “danger” resulting from trauma or surgery, influence the risk of hemophilia A patients developing an inhibitor (26, 27). Meunier et al. recently determined the frequency of FVIII-specific CD4+ T cells in the periphery of non-hemophilic blood donors and found approximately equal numbers of memory and naïve cells (28). Earlier studies had documented both FVIII-reactive antibodies (29, 30) and FVIII-specific T cells (16) in healthy control subjects. These studies demonstrated that FVIII is an unusually immunogenic self-protein, as also indicated by the rare autoimmune antibody response to FVIII known as “acquired hemophilia A.”
Several studies have suggested that hemophilia A patients with Black African or Hispanic ancestry experience a higher incidence of inhibitors, compared to white patients (31–33). There are multiple naturally occurring, non-hemophilia causing variants of the F8 gene in the human population, including non-synonymous single nucleotide polymorphisms (ns-SNPs) that encode amino acid variants (34). Thus, it is conceivable that hemophilia A patients who express a dysfunctional FVIII protein, and are exposed to a therapeutic FVIII having a different amino acid sequence, could mount an immune response to the neo-epitope corresponding to this amino acid sequence (35). Although this is a plausible scenario, statistical analyses of inhibitor incidences in patients whose F8 sequence at these sites was known (33, 36–38), as well as tetramer-guided epitope mapping to detect CD4+ T cells specific for these “mismatched” sequence (36), indicated that immune responses to these potential neo-epitopes occur rarely, if at all, and are therefore unlikely to contribute significantly to the immunogenicity of therapeutic FVIII.
FVIII is usually administered intravenously (i.v.), whereupon it rapidly binds to von Willebrand factor, which may modify its immunogenicity (39–41). The i.v. route is usually tolerogenic when infusing aggregate-free proteins into mice (42). This has been interpreted to suggest that i.v.-administered proteins fail to activate DC and to be processed in an immunogenic manner. However, in contrast to soluble proteins like ovalbumin, which is not immunogenic without adjuvant, FVIII is highly immunogenic when administered i.v. to the majority of FVIII knockout (E16) mice (5, 43, 44). Indeed, administering FVIII mixed with OVA can lead to an anti-OVA response, consistent with the intrinsic adjuvanticity of FVIII (5).
Finally, one has to consider other extrinsic properties of the host aside from HLA or other genetic factors. That is, an underlying infection will create significant inflammation which can tilt the response from tolerance to immunity. This would be a potential concern if a hemophilia A patient has an indwelling cannula which gets infected. On the other hand, a number of medications, especially steroids, are immunosuppressive and can tilt the immune response non-specific toward tolerance (45). Interestingly, both murine model studies and statistical analyses of patient outcomes indicate that immunizations do not affect inhibitor risk (46, 47).
The immunogenicity of FVIII that results in formation of inhibitors is a major impediment for the prevention and treatment of bleeds. While bypassing agents, including the FVIII-mimetic antibody emicizumab (48), or recombinant factor VIIa (49, 50), or FEIBA (Factor Eight Inhibitor Bypassing Agent, which is essentially a plasma-derived pro-coagulant protein cocktail) can facilitate clotting, are critically important lifesaving agents (51), they do not overcome the need to induce tolerance to FVIII. In particular, FVIII remains an essential component of the clinical armamentarium to support surgery, and to restore hemostasis following trauma, whereas the bypassing agents may be less efficient and/or carry a risk of thrombosis if doses are not carefully monitored. The relative risk/benefit ratios of utilizing FVIII vs. recently introduced novel bypass agents to control bleeding in specific clinical scenarios will become more apparent with further research and clinical “real world” experience.
Numerous methods to induce specific tolerance have been described for decades (52, 53). In terms of tolerance therapies to eradicate and prevent reoccurrence of inhibitors in hemophilia A patients, the standard clinical practice is intravenous repeated FVIII administration, which is called Immune Tolerance Induction (ITI). This protocol, first described by Brackmann and Gormsen in 1977 (54), is based on the high dose tolerance described by Mitchison in the 1960's (55) and essentially entails antigen overload, as well as maintaining higher trough levels of FVIII for continuous antigen exposure. This procedure is more often successful with patients having low tittered inhibitors but often fails in patients with higher titers. Moreover, it is expensive and challenging for patients and families, due to the need for frequent (often daily) infusions. Alternative methods to induce tolerance have primarily been tested in animal models, and most have not reached standard clinical practice. Below is a summary of several approaches in our labs, but it is not meant to be inclusive.
Following on the work of Weigle and colleagues (42, 56) with ultracentrifuged IgG as a model tolerogen, Borel utilized fusions or haptens and antigens on IgG carriers as tolerogens (57, 58), the latter being dependent on the presence of the IgG Fc fragment (9, 10). This would presumably crosslink the B-cell receptor with inhibitory Fc gamma receptors, an approach we will return to below. Based on the tolerogenicity of IgG fusions, we used retroviral transduction of FVIII domains with an IgG heavy chain in B cells as a tolerogenic protocol. This platform was successful in several autoimmune model systems as well (59–62); ironically, this approach was dependent on MHC class 2 presentation of peptides by B cells that led to the generation of regulatory T cells (Tregs) for both its induction and maintenance (63, 64).
Indeed, recent development of Fc fusions of clotting factors FVIII and FIX, designed for a longer half-life in vivo (65), have turned out to be tolerogenic in murine models and to induce Tregs (66, 67). This was initially supported by anecdotal cases reports of hemophilia A patients that suggest that FVIII-Fc is potentially tolerogenic (68–70); more highly powered clinical trials are in progress (NCT02234323, NCT03093480, and NCT03103542). Whether the tolerogenicity of Fc fusions is due to the regulatory epitopes in the constant region (9, 71) that turn on Tregs, and/or inhibitory Fc receptors (72, 73) is not clear.
While polyclonal human regulatory T cells (Tregs) have been proposed to treat autoimmune diseases and transplant rejection, and are already in clinical trials, the frequency of specific Tregs is very low. Moreover, the risk of non-specific immunosuppression and viral reactivation is real (74). Expansion of specific Tregs using peptide/APC and IL-2 has recently been achieved (75). Our labs have approached this issue by expressing specific receptors (or antigen) in expanded polyclonal Tregs or CD8 T cells, based on the seminal work by Eshhar (76, 77) and on clinical success of chimeric antigen receptor (CAR) T cells as reported by June and colleagues (78, 79). Since these studies have recently been published (80–83) and reviewed (52, 84), we will provide only a brief outline of these approaches to induce immune tolerance.
Starting with Tregs purified from healthy donors, our efforts to engineer specificity into polyclonal Tregs used retroviral transduction of specific T-cell receptors (TCR) (80) or CARs (scFv) (81), or even antigen (as B-cell Antibody Receptor = BAR) (83). In the first application, we cloned TCRs from FVIII reactive T-cell clones obtained from mild hemophilia patients (24). These clones recognized a peptide in the FVIII C2 domain restricted to HLA DRB1*01:01(21,22,24). The expanded TCR-transduced human Tregs suppressed proliferation and cytokine production by effector CD4 T cells even when the responders were in excess. Interestingly, the TCR-transduced Tregs also suppressed anti-FVIII B-cell responses in vitro and in vivo across a xenogeneic barrier (80)! Interestingly, although the engineered TCR recognizes a single peptide in the large FVIII protein, the antibody response to other major epitopes of FVIII was also suppressed. Thus, engineered FVIII-specific Tregs exhibit bystander suppression, an effect also seen with a TCR specific for a myelin peptide in a model of multiple sclerosis, an effect which appears to be due to uptake of IL-2 produced by effector T cells (85).
TCR-transduced Tregs are MHC class II restricted, thus limiting their eventual utility only to patients sharing the same HLA allele. Therefore, in the second approach, we collaborated with Anja Schmidt and Christoph Königs in Frankfurt, who provided a single chain Fv (scFv) that recognized the FVIII A2 domain. Like the CARs used in cancer therapy, these recognize conformational determinants and are not MHC restricted. Transduction of one of these scFv, called ANS8, into human Tregs also led to significant suppression of anti-FVIII responses in vitro and in vivo. Extensive dose response comparisons have not been performed as yet with these two types of engineered Tregs; the advantages (and disadvantages) of these specific Tregs are discussed elsewhere (Scott DW, Molecular Therapy submitted 2019).
Lastly, we hypothesized that it might be possible to directly target FVIII-specific B cells by expressing FVIII domains on the surface of Tregs. We refer to these as BAR Tregs, reflecting the fact that surface IgM/IgD on B cells react with these antigens. This was successfully achieved with both human BAR Tregs that expressed FVIII A2 and/or C2 domains and suppressed anti-FVIII responses in vitro and in vivo (83). The target of these BAR Tregs was proven to be the B cell, based on cell mixing experiments (83). Interestingly, in an allergy model, the target may also include sensitized mast cells, based on results of passive anaphylaxis experiments (86).
An alternative approach utilized transduced cytotoxic CD8 T cells expressing the targeted FVIII domains (82), as was done by Ellebrecht et al. with desmoglein 3 for possible therapy of pemphigus vulgaris, a devastating skin disease (87). [They refer to their antigen-expressing CD8 T cells as chimeric autoantigen receptor T cells (CAAR)]. Whether they are called BAR or CAAR, the cytotoxic T cells are highly specific and do not display bystander effects. There are situations when such specificity and lack of bystander effect might be necessary to carefully target part of a large antigen, but Tregs might be preferred if one doesn't know the targeted domains, and in the case of large multi-domain protein antigens such as FVIII.
Finally, a nanoparticle approach has been developed that can provide an alternative to engineered cellular therapies for tolerance; nanoparticles have also been used for drug delivery and vaccine development (88). Such nanoparticles can contain drugs such as rapamycin and are delivered with the target antigen (either attached or concomitantly) and presumably are taken up by dendritic cells, which act as tolerogenic APC and induce Tregs (89, 90). The use of rapamycin-containing nanoparticles for tolerance was successfully used by our group for FVIII (91), and by others for modulating autoimmunity (89) or the immune response to therapeutic immunotoxins, which are highly immunogenic (92, 93).
Several other approaches, in addition to the above strategies, are being developed to promote tolerance to FVIII. These include hepatic gene therapy, oral tolerance, and trans-placental delivery of FVIII. These are discussed more comprehensively in a recent review (84).
In conclusion, while there are multiple factors that influence the immunogenicity of therapeutic proteins, novels approaches such as those described here have the potential to modulate such immunogenicity. Time will tell which of these approaches may become cost-effective clinical therapies in the future.
DS and KP wrote and edited this manuscript.
The research in the authors' laboratories was supported by grants from the NIH [RO1 HL126727 (DS), HL130448 (KP)], and the National Multiple Sclerosis Society.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
1. Owen RD. Immunogenetic consequences of vascular anastomoses between catle twins. Science. (1945) 102:400–1. doi: 10.1126/science.102.2651.400
2. Billingham RE, Brent L, Medawar PB. ‘Actively acquired tolerance' of foreign cells. 1953. Transplantation. (2003) 76:1409–12. doi: 10.1097/01.TP.0000102675.72061.88
3. Matzinger P. Tolerance, danger, and the extended family. Annu Rev Immunol. (1994) 12:991–1045. doi: 10.1146/annurev.iy.12.040194.005015
4. Pfistershammer K, Stockl J, Siekmann J, Turecek PL, Schwarz HP, Reipert BM. Recombinant factor VIII and factor VIII-von Willebrand factor complex do not present danger signals for human dendritic cells. Thromb Haemost. (2006) 96:309–16. doi: 10.1160/TH05-11-0729
5. Skupsky J, Zhang AH, Su Y, Scott DW. A role for thrombin in the initiation of the immune response to therapeutic factor VIII. Blood. (2009) 114:4741–8. doi: 10.1182/blood-2008-10-186452
6. Meeks SL, Cox CL, Healey JF, Parker ET, Doshi BS, Gangadharan B, et al. A major determinant of the immunogenicity of factor VIII in a murine model is independent of its procoagulant function. Blood. (2012) 120:2512–20. doi: 10.1182/blood-2012-02-412361
7. Ettinger RA, James EA, Kwok WW, Thompson AR, Pratt KP. Lineages of human T-cell clones, including T helper 17/T helper 1 cells, isolated at different stages of anti-factor VIII immune responses. Blood. (2009) 114:1423–8. doi: 10.1182/blood-2009-01-200725
8. Bray GL, Kroner BL, Arkin S, Aledort LW, Hilgartner MW, Eyster ME, et al. Loss of high-responder inhibitors in patients with severe hemophilia A and human immunodeficiency virus type 1 infection: a report from the Multi-Center Hemophilia Cohort Study. Am J Hematol. (1993) 42:375–9. doi: 10.1002/ajh.2830420408
9. De Groot AS, Moise L, Mcmurry JA, Wambre E, Van Overtvelt L, Moingeon P, et al. Activation of natural regulatory T cells by IgG Fc-derived peptide “Tregitopes”. Blood. (2008) 112:3303–11. doi: 10.1182/blood-2008-02-138073
10. Cousens LP, Najafian N, Mingozzi F, Elyaman W, Mazer B, Moise L, et al. In vitro and in vivo studies of IgG-derived Treg epitopes (Tregitopes): a promising new tool for tolerance induction and treatment of autoimmunity. J Clin Immunol. (2013) 33(Suppl. 1):S43–9. doi: 10.1007/s10875-012-9762-4
11. Schellekens H. Immunogenicity of therapeutic proteins. Nephrol Dial Transplant. (2003) 18:1257–9. doi: 10.1093/ndt/gfg164
12. Buttel IC, Chamberlain P, Chowers Y, Ehmann F, Greinacher A, Jefferis R, et al. Taking immunogenicity assessment of therapeutic proteins to the next level. Biologicals. (2011) 39:100–9. doi: 10.1016/j.biologicals.2011.01.006
13. Peyvandi F, Mannucci PM, Garagiola I, El-Beshlawy A, Elalfy M, Ramanan V, et al. A randomized trial of factor VIII and neutralizing antibodies in hemophilia A. N Engl J Med. (2016) 374:2054–64. doi: 10.1056/NEJMoa1516437
14. Pratt KP, Qian J, Ellaban E, Okita DK, Diethelm-Okita BM, Conti-Fine B, et al. Immunodominant T-cell epitopes in the factor VIII C2 domain are located within an inhibitory antibody binding site. Thromb Haemost. (2004) 92:522–8. doi: 10.1160/TH03-12-0755
15. Reding MT, Okita DK, Diethelm-Okita BM, Anderson TA, Conti-Fine BM. Epitope repertoire of human CD4(+) T cells on the A3 domain of coagulation factor VIII. J Thromb Haemost. (2004) 2:1385–94. doi: 10.1111/j.1538-7836.2004.00850.x
16. Reding MT, Wu H, Krampf M, Okita DK, Diethelm-Okita BM, Key NS, et al. CD4+ T cell response to factor VIII in hemophilia A, acquired hemophilia, and healthy subjects. Thromb Haemost. (1999) 82:509–15. doi: 10.1055/s-0037-1615873
17. Reding MT, Wu H, Krampf M, Okita DK, Diethelm-Okita BM, Christie BA, et al. Sensitization of CD4+ T cells to coagulation factor VIII: response in congenital and acquired hemophilia patients and in healthy subjects. Thromb Haemost. (2000) 84:643–52. doi: 10.1055/s-0037-1614081
18. Reding MT, Okita DK, Diethelm-Okita BM, Anderson TA, Conti-Fine BM. Human CD4+ T-cell epitope repertoire on the C2 domain of coagulation factor VIII. J Thromb Haemost. (2003) 1:1777–84. doi: 10.1046/j.1538-7836.2003.00251.x
19. Hu GL, Okita DK, Conti-Fine BM. T cell recognition of the A2 domain of coagulation factor VIII in hemophilia patients and healthy subjects. J Thromb Haemost. (2004) 2:1908–17. doi: 10.1111/j.1538-7836.2004.00918.x
20. Jacquemin M, Vantomme V, Buhot C, Lavend'homme R, Burny W, Demotte N, et al. CD4+ T-cell clones specific for wild-type factor VIII: a molecular mechanism responsible for a higher incidence of inhibitor formation in mild/moderate hemophilia A. Blood. (2003) 101:1351–8. doi: 10.1182/blood-2002-05-1369
21. James EA, Kwok WW, Ettinger RA, Thompson AR, Pratt KP. T-cell responses over time in a mild hemophilia A inhibitor subject: epitope identification and transient immunogenicity of the corresponding self-peptide. J Thromb Haemost. (2007) 5:2399–407. doi: 10.1111/j.1538-7836.2007.02762.x
22. Ettinger RA, James EA, Kwok WW, Thompson AR, Pratt KP. HLA-DR-restricted T-cell responses to factor VIII epitopes in a mild haemophilia A family with missense substitution A2201P. Haemophilia. (2010) 16:44–55. doi: 10.1111/j.1365-2516.2008.01905.x
23. James EA Van Haren SD, Ettinger RA, Fijnvandraat K, Liberman JA, Kwok WW, et al. T-cell responses in two unrelated hemophilia A inhibitor subjects include an epitope at the factor VIII R593C missense site. J Thromb Haemost. (2011) 9:689–99. doi: 10.1111/j.1538-7836.2011.04202.x
24. Ettinger RA, Paz P, James EA, Gunasekera D, Aswad F, Thompson AR, et al. T cells from hemophilia A subjects recognize the same HLA-restricted FVIII epitope with a narrow TCR repertoire. Blood. (2016) 128:2043–54. doi: 10.1182/blood-2015-11-682468
25. Pratt KP, Thompson AR. B-cell and T-cell epitopes in anti-factor VIII immune responses. Clin Rev Allergy Immunol. (2009) 37:80–95. doi: 10.1007/s12016-009-8120-7
26. Astermark J, Oldenburg J, Escobar M, White GC II, Berntorp E, Malmö International Brother Study Study Group. The Malmö International Brother Study (MIBS). Genetic defects and inhibitor development in siblings with severe hemophilia A. Haematologica. (2005) 90:924–31.
27. Astermark J, Donfield SM, Gomperts ED, Schwarz J, Menius ED, Pavlova A, et al. The polygenic nature of inhibitors in hemophilia A: results from the Hemophilia Inhibitor Genetics Study (HIGS) Combined Cohort. Blood. (2013) 121:1446–54. doi: 10.1182/blood-2012-06-434803
28. Meunier S, Menier C, Marcon E, Lacroix-Desmazes S, Maillere B. CD4 T cells specific for factor VIII are present at high frequency in healthy donors and comprise naive and memory cells. Blood Adv. (2017) 1:1842–7. doi: 10.1182/bloodadvances.2017008706
29. Lacroix-Desmazes S, Misra N, Bayry J, Mohanty D, Kaveri SV, Kazatchkine MD. Autoantibodies to factor VIII. Autoimmun Rev. (2002) 1:105–10. doi: 10.1016/S1568-9972(01)00017-9
30. Whelan SF, Hofbauer CJ, Horling FM, Allacher P, Wolfsegger MJ, Oldenburg J, et al. Distinct characteristics of antibody responses against factor VIII in healthy individuals and in different cohorts of hemophilia A patients. Blood. (2013) 121:1039–48. doi: 10.1182/blood-2012-07-444877
31. Aledort LM, Dimichele DM. Inhibitors occur more frequently in African-American and Latino haemophiliacs. Haemophilia. (1998) 4:68. doi: 10.1046/j.1365-2516.1998.0146c.x
32. Lochan A, Macaulay S, Chen WC, Mahlangu JN, Krause A. Genetic factors influencing inhibitor development in a cohort of South African haemophilia A patients. Haemophilia. (2014) 20:687–92. doi: 10.1111/hae.12436
33. Miller CH, Benson J, Ellingsen D, Driggers J, Payne A, Kelly FM, et al. F8 and F9 mutations in US haemophilia patients: correlation with history of inhibitor and race/ethnicity. Haemophilia. (2012) 18:375–82. doi: 10.1111/j.1365-2516.2011.02700.x
34. Viel KR, Machiah DK, Warren DM, Khachidze M, Buil A, Fernstrom K, et al. A sequence variation scan of the coagulation factor VIII (FVIII) structural gene and associations with plasma FVIII activity levels. Blood. (2007) 109:3713–24. doi: 10.1182/blood-2006-06-026104
35. Viel KR, Ameri A, Abshire TC, Iyer RV, Watts RG, Lutcher C, et al. Inhibitors of factor VIII in black patients with hemophilia. N Engl J Med. (2009) 360:1618–27. doi: 10.1056/NEJMoa075760
36. Gunasekera D, Ettinger RA, Nakaya Fletcher S, James EA, Liu M, Barrett JC, et al. Factor VIII gene variants and inhibitor risk in African American hemophilia A patients. Blood. (2015) 126:895–904. doi: 10.1182/blood-2014-09-599365
37. Schwarz J, Astermark J, Menius ED, Carrington M, Donfield SM, Gomperts ED, et al. F8 haplotype and inhibitor risk: results from the Hemophilia Inhibitor Genetics Study (HIGS) combined cohort. Haemophilia. (2013) 19:113–8. doi: 10.1111/hae.12004
38. Astermark J, Voorberg J, Lenk H, Dimichele D, Shapiro A, Tjonnfjord G, et al. Impact of inhibitor epitope profile on the neutralizing effect against plasma-derived and recombinant factor VIII concentrates in vitro. Haemophilia. (2003) 9:567–72. doi: 10.1046/j.1365-2516.2003.00802.x
39. Repesse Y, Costa C, Palla R, Moshai EF, Borel-Derlon A, D'oiron R, et al. Role of factor VIII-binding capacity of endogenous von Willebrand factor in the development of factor VIII inhibitors in patients with severe hemophilia A. Haematologica. (2019) 104:e369–72. doi: 10.3324/haematol.2018.212001
40. Lai J, Hough C, Tarrant J, Lillicrap D. Biological considerations of plasma-derived and recombinant factor VIII immunogenicity. Blood. (2017) 129:3147–54. doi: 10.1182/blood-2016-11-750885
41. Pipe SW, Montgomery RR, Pratt KP, Lenting PJ, Lillicrap D. Life in the shadow of a dominant partner: the FVIII-VWF association and its clinical implications for hemophilia A. Blood. (2016) 128:2007–16. doi: 10.1182/blood-2016-04-713289
42. Chiller JM, Habicht GS, Weigle WO. Cellular sites of immunologic unresponsiveness. Proc Natl Acad Sci U S A. (1970) 65:551–6. doi: 10.1073/pnas.65.3.551
43. Qian J, Saenko E, Scott D. Co-stimulation blockade, hemophilic inhibitors and tolerance. Thromb Haemost. (2001) 86:1343–4. doi: 10.1055/s-0037-1616732
44. Zhang AH, Skupsky J, Scott DW. Effect of B-cell depletion using anti-CD20 therapy on inhibitory antibody formation to human FVIII in hemophilia A mice. Blood. (2011) 117:2223–6. doi: 10.1182/blood-2010-06-293324
45. Georgescu MT, Moorehead PC, Van Velzen AS, Nesbitt K, Reipert BM, Steinitz KN, et al. Dexamethasone promotes durable factor VIII-specific tolerance in hemophilia A mice via thymic mechanisms. Haematologica. (2018) 103:1403–13. doi: 10.3324/haematol.2018.189852
46. Lai JD, Moorehead PC, Sponagle K, Steinitz KN, Reipert BM, Hough C, et al. Concurrent influenza vaccination reduces anti-FVIII antibody responses in murine hemophilia A. Blood. (2016) 127:3439–49. doi: 10.1182/blood-2015-11-679282
47. Platokouki H, Fischer K, Gouw SC, Rafowicz A, Carcao M, Kenet G, et al. Vaccinations are not associated with inhibitor development in boys with severe haemophilia A. Haemophilia. (2018) 24:283–90. doi: 10.1111/hae.13387
48. Shima M, Hanabusa H, Taki M, Matsushita T, Sato T, Fukutake K, et al. Factor VIII-Mimetic Function of Humanized Bispecific Antibody in Hemophilia A. N Engl J Med. (2016) 374:2044–53. doi: 10.1056/NEJMoa1511769
49. Meeks SL, Leissinger CA. The evolution of factor VIIa in the treatment of bleeding in haemophilia with inhibitors. Haemophilia. (2019) 25:911–8. doi: 10.1111/hae.13845
50. Jankowski W, Mcgill J, Lagasse HaD, Surov S, Bembridge G, Bunce C, et al. Mitigation of T-cell dependent immunogenicity by reengineering factor VIIa analogue. Blood Adv. (2019) 3:2668–78. doi: 10.1182/bloodadvances.2019000338
51. Shapiro AD, Mitchell IS, Nasr S. The future of bypassing agents for hemophilia with inhibitors in the era of novel agents. J Thromb Haemost. (2018) 16:2362–74. doi: 10.1111/jth.14296
52. Scott DW. From IgG fusion proteins to engineered-specific human regulatory T cells: a life of tolerance. Front Immunol. (2017) 8:1576. doi: 10.3389/fimmu.2017.01576
53. Scott DW, Alés-Martínez JE, Chace JH, Locascio NJ, Silver L, Warner GL. Models of B-cell unresponsiveness. Cold Spring Harb Symp Quant Biol. (1989) 54(Pt 2):899–905. doi: 10.1101/SQB.1989.054.01.105
54. Brackmann HH, White GC II, Berntorp E, Andersen T, Escuriola-Ettingshausen C. Immune tolerance induction: What have we learned over time? Haemophilia. (2018) 24(Suppl. 3):3–14. doi: 10.1111/hae.13445
55. Mitchison NA. Induction of immunological paralysis in two zones of dosage. Proc R Soc Lond B Biol Sci. (1964) 161:275–92. doi: 10.1098/rspb.1964.0093
56. Chiller JM, Habicht GS, Weigle WO. Kinetic differences in unresponsiveness of thymus and bone marrow cells. Science. (1971) 171:813–5. doi: 10.1126/science.171.3973.813
57. Borel Y. Isologous IgG-induced immunologic tolerance to haptens: a model of self versus non-self recognition. Transplant Rev. (1976) 31:3–22. doi: 10.1111/j.1600-065X.1976.tb01450.x
58. Borel Y. Induction of immunological tolerance by a hapten (DNP) bound to a non-immunogenic protein carrier. Nat New Biol. (1971) 230:180–2. doi: 10.1038/newbio230180a0
59. Zambidis ET, Kurup A, Scott DW. Genetically transferred central and peripheral immune tolerance via retroviral-mediated expression of immunogenic epitopes in hematopoietic progenitors or peripheral B lymphocytes. Mol Med. (1997) 3:212–24. doi: 10.1007/BF03401674
60. El-Amine M, Melo M, Kang Y, Nguyen H, Qian J, Scott DW. Mechanisms of tolerance induction by a gene-transferred peptide-IgG fusion protein expressed in B lineage cells. J Immunol. (2000) 165:5631–6. doi: 10.4049/jimmunol.165.10.5631
61. Melo ME, El-Amine M, Tonnetti L, Fleischman L, Scott DW. Gene therapeutic approaches to induction and maintenance of tolerance. Int Rev Immunol. (2001) 20:627–45. doi: 10.3109/08830180109045582
62. Agarwal RK, Kang Y, Zambidis E, Scott DW, Chan CC, Caspi RR. Retroviral gene therapy with an immunoglobulin-antigen fusion construct protects from experimental autoimmune uveitis. J Clin Invest. (2000) 106:245–52. doi: 10.1172/JCI9168
63. Soukhareva N, Jiang Y, Scott DW. Treatment of diabetes in NOD mice by gene transfer of Ig-fusion proteins into B cells: role of T regulatory cells. Cell Immunol. (2006) 240:41–6. doi: 10.1016/j.cellimm.2006.06.004
64. Litzinger MT, Su Y, Lei TC, Soukhareva N, Scott DW. Mechanisms of gene therapy for tolerance: B7 signaling is required for peptide-IgG gene-transferred tolerance induction. J Immunol. (2005) 175:780–7. doi: 10.4049/jimmunol.175.2.780
65. Baker K, Qiao SW, Kuo TT, Aveson VG, Platzer B, Andersen JT, et al. Neonatal Fc receptor for IgG (FcRn) regulates cross-presentation of IgG immune complexes by CD8-CD11b+ dendritic cells. Proc Natl Acad Sci U S A. (2011) 108:9927–32. doi: 10.1073/pnas.1019037108
66. Blumberg RS, Lillicrap D, Ig GFITG. Tolerogenic properties of the Fc portion of IgG and its relevance to the treatment and management of hemophilia. Blood. (2018) 131:2205–14. doi: 10.1182/blood-2017-12-822908
67. Krishnamoorthy S, Liu T, Drager D, Patarroyo-White S, Chhabra ES, Peters R, et al. Recombinant factor VIII Fc (rFVIIIFc) fusion protein reduces immunogenicity and induces tolerance in hemophilia A mice. Cell Immunol. (2016) 301:30–9. doi: 10.1016/j.cellimm.2015.12.008
68. Malec LM, Journeycake J, Ragni MV. Extended half-life factor VIII for immune tolerance induction in haemophilia. Haemophilia. (2016) 22:e552–e4. doi: 10.1111/hae.13064
69. Groomes CL, Gianferante DM, Crouch GD, Parekh DS, Scott DW, Lieuw K. Reduction of Factor VIII Inhibitor Titers During Immune Tolerance Induction With Recombinant Factor VIII-Fc Fusion Protein. Pediatr Blood Cancer. (2016) 63:922–4. doi: 10.1002/pbc.25874
70. Carcao M, Shapiro A, Staber JM, Hwang N, Druzgal C, Lieuw K, et al. Recombinant factor VIII Fc fusion protein for immune tolerance induction in patients with severe haemophilia A with inhibitors-A retrospective analysis. Haemophilia. (2018) 24:245–52. doi: 10.1111/hae.13413
71. De Groot AS, Scott DW. Immunogenicity of protein therapeutics. Trends Immunol. (2007) 28:482–90. doi: 10.1016/j.it.2007.07.011
73. Klaus GG, Bijsterbosch MK, O'garra A, Harnett MM, Rigley KP. Receptor signalling and crosstalk in B lymphocytes. Immunol Rev. (1987) 99:19–38. doi: 10.1111/j.1600-065X.1987.tb01170.x
74. Brunstein CG, Blazar BR, Miller JS, Cao Q, Hippen KL, Mckenna DH, et al. Adoptive transfer of umbilical cord blood-derived regulatory T cells and early viral reactivation. Biol Blood Marrow Transplant. (2013) 19:1271–3. doi: 10.1016/j.bbmt.2013.06.004
75. Smith BM, Lyle MJ, Chen AC, Miao CH. Antigen-specific in vitro expansion of factor VIII-specific regulatory T cells induces tolerance in hemophilia A mice. J Thromb Haemost. (2019). doi: 10.1111/jth.14659. [Epub ahead of print].
76. Eshhar Z. From the mouse cage to human therapy: a personal perspective of the emergence of T-bodies/chimeric antigen receptor T cells. Hum Gene Ther. (2014) 25:773–8. doi: 10.1089/hum.2014.2532
77. Eshhar Z, Waks T, Bendavid A, Schindler DG. Functional expression of chimeric receptor genes in human T cells. J Immunol Methods. (2001) 248:67–76. doi: 10.1016/S0022-1759(00)00343-4
78. Maus MV, Grupp SA, Porter DL, June CH. Antibody-modified T cells: CARs take the front seat for hematologic malignancies. Blood. (2014) 123:2625–35. doi: 10.1182/blood-2013-11-492231
79. Grupp SA, Kalos M, Barrett D, Aplenc R, Porter DL, Rheingold SR, et al. Chimeric antigen receptor-modified T cells for acute lymphoid leukemia. N Engl J Med. (2013) 368:1509–18. doi: 10.1056/NEJMoa1215134
80. Kim YC, Zhang AH, Su Y, Rieder SA, Rossi RJ, Ettinger RA, et al. Engineered antigen-specific human regulatory T cells: immunosuppression of FVIII-specific T- and B-cell responses. Blood. (2015) 125:1107–15. doi: 10.1182/blood-2014-04-566786
81. Yoon J, Schmidt A, Zhang AH, Königs C, Kim YC, Scott DW. FVIII-specific human chimeric antigen receptor T-regulatory cells suppress T- and B-cell responses to FVIII. Blood. (2017) 129:238–45. doi: 10.1182/blood-2016-07-727834
82. Parvathaneni K, Scott DW. Engineered FVIII-expressing cytotoxic T cells target and kill FVIII-specific B cells in vitro and in vivo. Blood Adv. (2018) 2:2332–40. doi: 10.1182/bloodadvances.2018018556
83. Zhang A-H, Parvathaneni K, Yoon J, Kim Y, Scott D. Targeting antigen-specific B cells using BAR-transduced cytotoxic and regulatory T cells. J Immunol. (2016) 196(Suppl. 1):70. doi: 10.1182/blood.V128.22.329.329
84. Lacroix-Desmazes S, Voorberg J, Lillicrap D, Scott DW, Pratt KP. Tolerating factor VIII: recent progress. Front. Immunol. (2020) 10:2991. doi: 10.3389/fimmu.2019.02991
85. Kim YC, Zhang AH, Yoon J, Culp WE, Lees JR, Wucherpfennig KW, et al. Engineered MBP-specific human Tregs ameliorate MOG-induced EAE through IL-2-triggered inhibition of effector T cells. J Autoimmun. (2018) 92:77–86. doi: 10.1016/j.jaut.2018.05.003
86. Abdeladhim M, Zhang AH, Kropp LE, Lindrose AR, Venkatesha SH, Mitre E, et al. Engineered ovalbumin-expressing regulatory T cells protect against anaphylaxis in ovalbumin-sensitized mice. Clin Immunol. (2019) 207:49–54. doi: 10.1016/j.clim.2019.07.009
87. Ellebrecht CT, Bhoj VG, Nace A, Choi EJ, Mao X, Cho MJ, et al. Reengineering chimeric antigen receptor T cells for targeted therapy of autoimmune disease. Science. (2016) 353:179–84. doi: 10.1126/science.aaf6756
88. Ilyinskii PO, Roy CJ, O'neil CP, Browning EA, Pittet LA, Altreuter DH, et al. Adjuvant-carrying synthetic vaccine particles augment the immune response to encapsulated antigen and exhibit strong local immune activation without inducing systemic cytokine release. Vaccine. (2014) 32:2882–95. doi: 10.1016/j.vaccine.2014.02.027
89. Maldonado RA, Lamothe RA, Ferrari JD, Zhang AH, Rossi RJ, Kolte PN, et al. Polymeric synthetic nanoparticles for the induction of antigen-specific immunological tolerance. Proc Natl Acad Sci U S A. (2015) 112:E156–65. doi: 10.1073/pnas.1408686111
90. Getts DR, Shea LD, Miller SD, King NJ. Harnessing nanoparticles for immune modulation. Trends Immunol. (2015) 36:419–27. doi: 10.1016/j.it.2015.05.007
91. Zhang AH, Rossi RJ, Yoon J, Wang H, Scott DW. Tolerogenic nanoparticles to induce immunologic tolerance: Prevention and reversal of FVIII inhibitor formation. Cell Immunol. (2016) 301:74–81. doi: 10.1016/j.cellimm.2015.11.004
92. Mazor R, King EM, Onda M, Cuburu N, Addissie S, Crown D, et al. Tolerogenic nanoparticles restore the antitumor activity of recombinant immunotoxins by mitigating immunogenicity. Proc Natl Acad Sci USA. (2018) 115:E733–42. doi: 10.1073/pnas.1717063115
Keywords: factor VIII, tolerance, hemophilia, inhibitors, regulatory T (Treg) cells
Citation: Scott DW and Pratt KP (2020) Factor VIII: Perspectives on Immunogenicity and Tolerogenic Strategies. Front. Immunol. 10:3078. doi: 10.3389/fimmu.2019.03078
Received: 06 November 2019; Accepted: 17 December 2019;
Published: 17 January 2020.
Edited by:
Zuben E. Sauna, United States Food and Drug Administration, United StatesReviewed by:
Pete Lollar, Emory University, United StatesCopyright © 2020 Scott and Pratt. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: David W. Scott, c2NvdHRkNDNAZ21haWwuY29t
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.