 Sebastien Lacroix-Desmazes
Sebastien Lacroix-Desmazes Jan Voorberg
Jan Voorberg David Lillicrap
David Lillicrap David W. Scott
David W. Scott Kathleen P. Pratt
Kathleen P. Pratt
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol. , 10 January 2020
Sec. Immunological Tolerance and Regulation
Volume 10 - 2019 | https://doi.org/10.3389/fimmu.2019.02991
This article is part of the Research Topic Tolerating Factor VIII: Novel Strategies to Prevent and Reverse Anti-FVIII Inhibitors View all 20 articles
Development of neutralizing antibodies against biotherapeutic agents administered to prevent or treat various clinical conditions is a longstanding and growing problem faced by patients, medical providers and pharmaceutical companies. The hemophilia A community has deep experience with attempting to manage such deleterious immune responses, as the lifesaving protein drug factor VIII (FVIII) has been in use for decades. Hemophilia A is a bleeding disorder caused by genetic mutations that result in absent or dysfunctional FVIII. Prophylactic treatment consists of regular intravenous FVIII infusions. Unfortunately, 1/4 to 1/3 of patients develop neutralizing anti-FVIII antibodies, referred to clinically as “inhibitors,” which result in a serious bleeding diathesis. Until recently, the only therapeutic option for these patients was “Immune Tolerance Induction,” consisting of intensive FVIII administration, which is extraordinarily expensive and fails in ~30% of cases. There has been tremendous recent progress in developing novel potential clinical alternatives for the treatment of hemophilia A, ranging from encouraging results of gene therapy trials, to use of other hemostatic agents (either promoting coagulation or slowing down anti-coagulant or fibrinolytic pathways) to “bypass” the need for FVIII or supplement FVIII replacement therapy. Although these approaches are promising, there is widespread agreement that preventing or reversing inhibitors remains a high priority. Risk profiles of novel therapies are still unknown or incomplete, and FVIII will likely continue to be considered the optimal hemostatic agent to support surgery and manage trauma, or to combine with other therapies. We describe here recent exciting studies, most still pre-clinical, that address FVIII immunogenicity and suggest novel interventions to prevent or reverse inhibitor development. Studies of FVIII uptake, processing and presentation on antigen-presenting cells, epitope mapping, and the roles of complement, heme, von Willebrand factor, glycans, and the microbiome in FVIII immunogenicity are elucidating mechanisms of primary and secondary immune responses and suggesting additional novel targets. Promising tolerogenic therapies include development of FVIII-Fc fusion proteins, nanoparticle-based therapies, oral tolerance, and engineering of regulatory or cytotoxic T cells to render them FVIII-specific. Importantly, these studies are highly applicable to other scenarios where establishing immune tolerance to a defined antigen is a clinical priority.
Factor VIII (FVIII) is an essential blood coagulation cofactor. Recombinant or plasma-derived FVIII is a lifesaving protein drug for hemophilia A (HA) patients, whose F8 gene mutations result in either a complete lack of endogenous FVIII or in a circulating dysfunctional FVIII. Unfortunately, immune responses to FVIII resulting in neutralizing anti-FVIII antibodies, or “inhibitors,” complicate or preclude effective FVIII replacement therapy in a substantial fraction of HA patients. Inhibitors typically develop early in the course of FVIII replacement therapy, with a peak incidence occurring within the first 10–15 exposure days (1, 2). Longer-term surveillance studies indicate, however, that a substantial fraction of inhibitors develop after age 5, and that incidences increase again after age 50 (3). Inhibitor development in non-HA individuals also occurs as a rare but serious autoimmune reaction that is typically diagnosed subsequent to unexplained bleeding (4), primarily in the elderly, or following trauma, surgery or childbirth. Both allo- and autoimmune FVIII-specific antibodies are class-switched, as is typical for CD4+ T-cell driven immune responses (5, 6).
This review focuses on mechanisms of factor VIII immunogenicity and novel approaches to promote immune tolerance to this important protein drug. Despite decades of clinical experience with both plasma-derived and recombinant (r)FVIII products, there is still much to be learned about risk factors for inhibitor development and mechanisms of the anti-FVIII immune response. It is hoped that improved mechanistic understanding will lead to identification of reliable prognostic biomarkers and, even more significantly, of novel targets to promote immune tolerance to FVIII. An ideal therapeutic intervention would tolerize the individual specifically to FVIII, thereby avoiding the potential side effects of general immunosuppression. We focus on recent advances, some of which are being tested in current clinical trials, and others that have the potential for future clinical translation, e.g., animal model studies and in vitro experiments utilizing donated human blood samples.
The armamentarium available to treat HA patients has expanded significantly over the past decade. It currently includes rFVIII products produced in mammalian cell culture systems and rFVIII proteins that have been engineered to create sequence-modified or fusion proteins, or covalently modified, e.g., by PEGylation to extend their half-life. In addition, non-FVIII therapies that either mimic FVIII cofactor activity, or that target specific pro-coagulant or anti-coagulant pathways by shifting hemostasis to a more pro-coagulant phenotype and thereby prevent hemophilic bleeds, are now available, in preclinical testing, and in clinical trials. Three recently introduced non-FVIII options to treat HA are the bispecific antibody emicizumab (Hemlibra) (7, 8), the anti-Tissue Factor Pathway Inhibitor (TFPI) monoclonal antibody concizumab (9) and an RNAi targeting antithrombin (Fitusiran). These products, and several others that are in various stages in the translational pipeline, are described in more detail below. They present patients with non-FVIII options; this is particularly important for those who have developed inhibitors that preclude effective prevention or treatment of bleeds with FVIII. Some also show promise as therapeutics for hemophilia B (lack of functional factor IX) and other bleeding disorders. Because of its earlier introduction and approval, there is more clinical experience with emicizumab, which a growing number of patients are choosing for prophylactic management of HA. All of these therapies offer significant benefits in terms of convenience, as they do not involve the frequent intravenous infusions required for FVIII prophylaxis. Importantly, however, they cannot induce tolerance to FVIII unless they are administered in formulations that include the FVIII antigen. Furthermore, there is still limited experience with their use, effectiveness, and risk profiles in settings of trauma and surgery, when FVIII supplementation may well be required to prevent or reverse breakthrough bleeds. Therefore, the induction and maintenance of immune tolerance to FVIII remains a vital issue for all HA patients, regardless of which therapeutic product they utilize for routine prophylaxis.
Animal model studies continue to be essential for understanding mechanisms of FVIII immunogenicity and peripheral tolerance, as well as for testing novel therapies to identify candidates for possible clinical translation (10). Most animal studies of anti-FVIII immune responses have utilized HA mice with a targeted disruption of the F8 gene, due to their lower cost and the greater availability of appropriate reagents and well-defined genetic strains, compared to larger animal models. However, large animal models have provided essential models of hemophilia A and B, especially for preclinical testing of various therapies. Gene therapy studies have relied for years on the use of HA dog colonies (11, 12). Interestingly, successful delivery of an F8 transgene has not only corrected the HA bleeding phenotype, in some cases for years, but it has also shown promise as a potential therapy to achieve peripheral tolerance to FVIII (13). Human F8 gene therapy trials have so far enrolled only adults with no prior FVIII inhibitor, so the possibility that gene therapy may have a tolerogenic effect on the naïve human immune response to FVIII remains untested.
Clinical Immune Tolerance Induction (ITI) therapy currently consists of intensive intravenous FVIII administration, which is challenging for patients/families, extraordinarily expensive, and fails in 25–30% of patients (14, 15). Unfortunately, attempts to “tolerize” HA mice via intensive FVIII infusions, analogous to clinical ITI protocols, have not yet been successful (16), although high-dose FVIII administration has been shown to suppress memory B cells in vitro (17). Therefore, further human studies are needed to identify biomarkers and potential new targets that could be manipulated to improve current clinical ITI success rates. Animal models allow studies of immune compartments in addition to the periphery, notably of the spleen and possibly the liver as major sites for the naïve response to intravenously administered FVIII. Recent advances in both animal and human studies of FVIII immunogenicity and tolerance are summarized below.
Inhibitors are, by definition, neutralizing anti-FVIII antibodies, with titers reported in “Bethesda units” as measured by a clotting assay (18, 19). More comprehensive immunoprofiling efforts have incorporated measurements of total anti-FVIII antibody titers (expressed as dilution factors) and antibody isotypes/subclasses using ELISAs (20–22), surface plasmon resonance (23), and fluorescent bead-based assays (21, 24). FVIII-specific antibodies isolated from HA patients with an inhibitor response are primarily of subclasses IgG1 and IgG4 (25, 26), although lower levels of FVIII-specific IgG2 and IgG3 have also been detected and quantified in patients' plasma (23). Analysis of samples from 371 HA subjects (21% inhibitor-positive) showed a correlation of anti-FVIII IgG1, IgG2, and IgG4 with inhibitor development (27), indicating involvement of both Th1 and Th2 CD4+ T cells (5), while a separate study of 101 HA subjects (24% inhibitor-positive) and 19 autoimmune subjects revealed that neutralizing antibodies had higher apparent affinities for FVIII compared to non-neutralizing antibodies (22). Anaphylaxis is not a feature of anti-FVIII allo- or autoimmune responses.
A seminal 1992 study of 500 plasma samples from healthy, non-hemophilic donors revealed that an appreciable fraction contained anti-FVIII antibodies that were detectable by Bethesda and/or ELISA assays (28). A subsequent study reported isolation of anti-FVIII antibodies from non-hemophilic plasma, epitope mapping via competition ELISA assays, and characterization of anti-idiotypic antibodies that could block the interactions between FVIII and the natural anti-FVIII antibodies (29). In a more recent study, non-neutralizing anti-FVIII antibodies were detected in ~20% of plasma samples from >600 healthy non-HA blood donors (21, 30), although their predominant recognition of the heavily glycosylated B domain of FVIII indicated the binding of many of these “natural” antibodies may not have been strictly specific for FVIII. Anti-FVIII antibodies have also been quantified using a sensitive Luminex-based assay (24), which detected low-titer FVIII-binding antibodies in the vast majority of ~400 HA subjects, most of whom did not have a current inhibitor detectable by a clotting (Bethesda) assay. Interestingly, this study also detected low-titer FVIII-specific antibodies in an appreciable fraction of healthy non-HA control plasma samples, although titers were significantly lower (31).
The uptake of blood coagulation FVIII by antigen presenting cells has been studied intensively in the last decade. Most of these studies have been performed in model systems like human monocyte-derived dendritic cells as well as mouse bone marrow-derived dendritic cells (32–34). In parallel, immuno-localization of FVIII in the spleen following its infusion into mice has generated very interesting findings on the topology of FVIII association with, and possibly uptake by, different populations of antigen presenting cells in specific niches within the splenic architecture (Figure 1). The afferent small vessels in the spleen are lined by fenestrated endothelial cells that allow for interaction of circulating antigens with antigen presenting cells underlying this layer of endothelial cells.
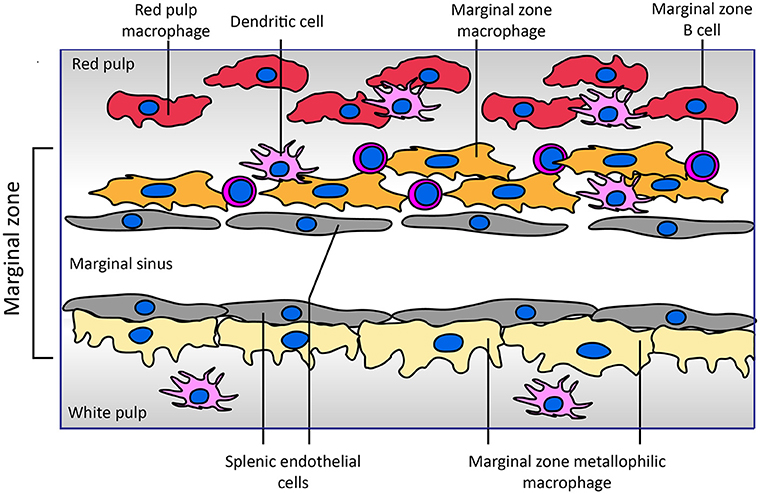
Figure 1. Architecture of the marginal zone of the mouse spleen. The image, adapted from Figure 4 of Mebius and Kraal (35), displays the cellular composition of the marginal zone of the spleen. Sampling of antigens from the circulating blood by different populations of antigen presenting cells surrounding the marginal sinus appears to be crucial for development of an adaptive immune response to FVIII. Both SIGNR1-positive marginal zone macrophages and CD169-positive marginal zone metallophilic macrophages have been implicated in the endocytosis of FVIII. Splenic endothelial cells lining the marginal sinus or red pulp sinus (not displayed in figure) expressing stabilin-2 may also contribute to FVIII internalization. Functional involvement of marginal zone B cells present within the marginal zone in the immune response has been inferred from depletion approaches. Different populations of dendritic cells reside in the spleen; the role of specific subsets of these cells in the development of FVIII inhibitors has not yet been explored. In contrast to that of the mouse spleen, the cellular architecture of the human spleen has not yet been fully elucidated (36, 37). In human spleen the border between the white and red pulp is formed by a perifollicular zone whose cellular composition has not yet been fully characterized.
A landmark study by Navarrete et al. provided compelling evidence for the association of FVIII with marginal zone metallophilic macrophages (38, 39). These results were confirmed in studies in which co-localization of infused FVIII with the marginal zone macrophages was observed based on its co-localization with macrophage marker proteins SIGNR1 and MARCO (35, 39). In addition, based on the co-localization of FVIII with SIGLEC-1, marginal zone macrophages located in the white pulp have also been implicated in processing of FVIII. The role of (metallophilic) marginal zone macrophages in in vivo uptake does not exclude that other populations of splenic cells also contribute to FVIII immunogenicity. Indeed, the depletion of splenic marginal zone B cells abrogated inhibitor development in a mouse model of HA (40). Marginal zone B cells are efficient scavengers for antigens that circulate in blood. Based on their localization in the spleen and their role in mounting an immune response, it is possible that FVIII is transported by them to different populations of antigen presenting cells following its retrieval from blood. However, recent Bruton's tyrosine kinase inhibition pre-clinical studies suggest no role for naïve B cells in development of a primary anti-FVIII immune response (41).
The architecture of the spleen promotes intimate contact between blood borne antigens and antigen capturing and presenting cells that are localized in the marginal zone (36, 37). Fenestrated splenic endothelial cells are well-positioned to filter blood borne antigens like FVIII and von Willebrand factor (VWF) from the circulating blood, thus promoting capture of antigens via scavenger receptors such as stabilin-2 (42). Marginal zone macrophages, marginal zone B cells and populations of marginal zone-located dendritic cells are also capable of capturing and/or processing blood borne antigens (36, 37). Transfer of antigens from marginal zone macrophages and endothelial cells to more dynamic marginal zone B cells and marginal zone-residing dendritic cells is most likely required for their transport to the T cell-enriched white pulp. Subsequent steps of FVIII transport are not yet well-defined, but antigen transfer to dendritic cells is required for primary immune responses, and FVIII presentation to naïve T cells and B cells is expected to occur primarily in the spleen (38). T follicular helper cells within germinal centers both select FVIII-specific B cells and drive affinity maturation and class-switching of their B-cell receptors, ultimately generating plasma cells that secrete high-affinity antibodies. Indeed, FVIII-deficient mice showed increased germinal center formation, proliferation of splenic T-follicular helper cells in vitro, and accumulation of T-follicular CD4+ T cells in the spleen following FVIII immunization (43).
Apart from the spleen, FVIII has also been shown to accumulate in macrophages in the liver (44). The precise population of antigen presenting cells in this organ have not yet been defined, but both liver sinusoidal endothelial cells and Kupffer cells have been shown to express endocytic receptors capable of internalizing FVIII and/or VWF (44). Both FVIII and VWF were shown to be endocytosed primarily by CD69+ Kupffer cells (44). More recently, liver sinusoidal endothelial cells have also been implicated in FVIII and VWF internalization (45). The liver is considered to provide a tolerogenic environment which supports the generation as well as proliferation of CD4+ T cells with a regulatory phenotype. LSEC are instrumental in the generation of regulatory CD4+ T cells, as they can efficiently endocytose and process blood borne antigens, thereby sequestering them in a relatively tolerogenic compartment, at least in the absence of significant inflammation (46). LSEC have been shown to express MHC class II, which can be further up-regulated upon inflammatory stimulation. Unlike dendritic cells, LSEC express limited amounts of co-stimulatory molecules and therefore are unable to direct the formation of classical CD4+ T helper cells (46). In apparent contrast with the tolerogenic role of LSEC, it was recently shown that stabilin-2 driven internalization of human FVIII/VWF complexes provides a crucial step in FVIII inhibitor development (45). Since stabilin-2 is expressed by both liver and splenic endothelial cells (42), uptake of FVIII-VWF complexes by these cells may also explain the modulating effects of stabilin-2 on FVIII immunogenicity.
Most of our knowledge of surface receptors implicated in FVIII endocytosis has been derived from in vitro studies. Different families of endocytic receptors have emerged during evolution to promote early processing of foreign antigens. Most of these receptors recognize distinct pattern-like structures which include glycans such as sialic acid (Siglec family of surface receptors), mannose or galactose structures on protein antigens (26, 47, 48). Additional classes of surface receptors recognizing more heterologous structures on protein antigens have also been implicated in FVIII endocytosis (32, 33, 47). The LDL-related low-density receptor 1 (LRP1) was identified as an endocytic receptor for FVIII (49, 50). The physiological importance of this receptor and other members of this receptor family is thought to be related primarily to FVIII clearance. Despite its abundance on antigen presenting cells, current evidence suggests that its role in FVIII presentation and immunogenicity is limited (51, 52). In contrast to LRP1, the mannose receptor has been firmly implicated in FVIII endocytosis by human dendritic cells (48). The mannose receptor is composed of a discrete series of repeated carbohydrate ligand-binding sites, one of which binds with high affinity to FVIII. This binding and endocytosis could be partially abrogated by mannan, and incubation of FVIII with dendritic cells in the presence of mannan completely inhibited proliferation of a FVIII-specific T-cell clone (48). Therefore, it was concluded that the mannose receptor is involved in immune recognition of FVIII by antigen presenting cells (48). Asn239 in the A1 domain of FVIII and Asn2118 in the C1 domain are attached to a glycan terminating in mannose, suggesting a mechanism for FVIII internalization by antigen presenting cells via the mannose receptor (53, 54). Complementary experiments employing murine bone marrow-derived dendritic cells, however, did not support a direct role for the mannose receptor in FVIII internalization (55). These findings suggest that, at least in mice, the mannose receptor may not be directly involved in FVIII immunogenicity; the extent to which FVIII immunogenicity in humans depends on mannose receptors remains to be established. Apart from the mannose-ending glycans linked to Asn239 and Asn2118, exposed surface loops in the C-terminal FVIII C1 and C2 domains containing positively-charged residues have been implicated in the uptake of FVIII by both human (monocyte-derived) and murine (bone marrow-derived) dendritic cells (52, 56, 57). Modification of residues in the C1 domain surface loop containing Arg2090, Lys2092, and Met2093 resulted in reduced FVIII inhibitor titers in FVIII-deficient mice (56). Interestingly, in one study the reduced immunogenicity was observed only in FVIII-VWF deficient mice, suggesting that the immunogenicity of this engineered FVIII variant was modulated by its binding to VWF (57). Likewise, modification of residues in the C2 domain surface loop containing Arg2215 and Arg2220 resulted in drastically reduced FVIII uptake by human dendritic cells (57). The roles of specific FVIII regions and receptors in binding, uptake by various cell types, antigenic processing and clearance of FVIII are the subject of ongoing research. An overview of our current understanding of FVIII uptake, processing and presentation on immunogenicity vs. tolerance is shown in Figure 2.
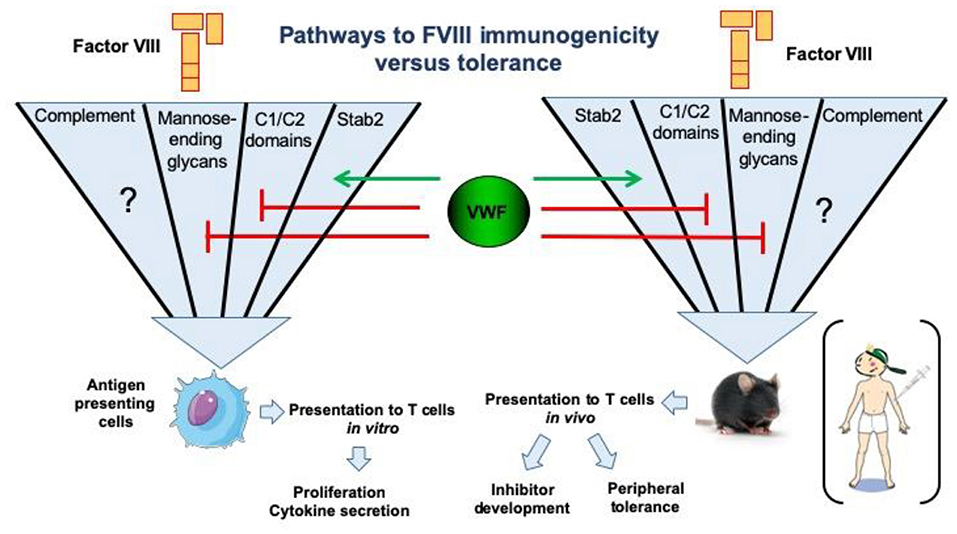
Figure 2. Pathways for endocytosis of FVIII by antigen-presenting cells and repercussions on FVIII immunogenicity or tolerance. The uptake of human FVIII has been studied in vitro using human antigen presenting cells, such as monocyte-derived dendritic cells. The major pathways identified to date include key positively charged amino-acids in the C1 and C2 domains of the FVIII molecule, as well as mannose-ending glycans at position N2118 in the C1 domain. Importantly, the later endocytic pathways are blocked in the presence of VWF. Conversely, activation of complement is able to restore the uptake of the FVIII C1 domain mutant Arg2090Ala, Lys2092Ala, and Met2093Ala. It is not known as yet whether complement activation overcomes the blocking effect of VWF in FVIII endocytosis. The stabilin-2 (Stab2)-dependent internalization pathway was demonstrated using Stab2-expressing HEK293 cells and also in vivo in mice. It is dependent on the presence of VWF. Interestingly, the neutralization of some endocytic pathways reduces the immunogenicity of FVIII in FVIII-deficient mice, particularly in the case of the Stab2- and complement C3b-dependent pathways (58).
Since FVIII circulates in complex with its physiological, multimeric carrier protein VWF, it is not surprising that its endocytosis and potential immunogenicity are both modulated by VWF. In vitro studies have firmly established that in the presence of VWF the amount of FVIII that is internalized by antigen presenting cells is greatly reduced (59, 60). Following this internalization, FVIII is processed into peptides by endo-lysosomal proteases for subsequent loading onto MHC class II and presentation to CD4+ T cells (61). In agreement with its role in decreasing FVIII internalization, it has also been shown that VWF modulates the efficiency of FVIII peptide presentation (60, 62). These effects of VWF have been suggested as an underlying mechanism for the apparent reduced immunogenicity of VWF-containing (plasma-derived) FVIII concentrates, compared to rFVIII products, that was reported in the recent SIPPET study (2).
Studies in which monocyte-derived dendritic cells were pulsed with FVIII, followed by peptide elution and identification by LC-mass spectrometry, have yielded valuable insights into the repertoires of FVIII-derived peptides that can be presented on various MHC class II, and thereby made available for potential recognition by CD4+ T cells (60–65). Together with complementary studies characterizing CD4+ T-cell responses, which are discussed in detail later in this review, this provides detailed information identifying naturally processed FVIII peptides. The immunodominant T-cell epitopes that elicit immune responses in patients with HA must necessarily be contained within the repertoire of peptides presented on the relevant HLA Class II. Depending on their micro-environment and immunological context, regulatory and other subsets of CD4+ T cells may also recognize FVIII-derived peptides in the context of MHC class II. Retrospective studies based on chart reviews of HLA-typed patient populations have not revealed any clearly immunodominant HLA Class II alleles associated with inhibitor development (66–69).
In view of the intimate relationship between FVIII and VWF, it is not surprising that internalization of FVIII by antigen presenting cells or phagocytic cells involved in clearance can be influenced by VWF. Based on large genetic studies, single nucleotide polymorphisms in several proteins have been associated with circulating levels of VWF and FVIII (70). Polymorphic sites within genes linked to biosynthetic pathways such as STX2 and STXBP5 most likely affect circulating VWF levels through modulation of biosynthetic pathways in platelets and endothelial cells (71, 72). Conversely, polymorphic sites within surface receptors are expected to modulate circulating FVIII and VWF levels through their effect on the clearance of FVIII, VWF or the FVIII-VWF complex. Following up on the observations of the Cohorts for Heart and Aging Research in Genomic Epidemiology (CHARGE) Consortium has yielded unexpected insights into the clearance and immunogenicity of FVIII, pointing toward a prominent role for liver sinusoidal endothelial cells. The C-type lectin CLEC4M is a candidate receptor for regulating FVIII and VWF levels. In vitro expression studies revealed that CLEC4M can bind to both FVIII and VWF, positioning it as a potential regulator of FVIII clearance and immunogenicity (73, 74). Interestingly, CLEC4M is expressed exclusively in sinusoidal endothelial cells; infusion studies in CLEC4M deficient animals did not reveal major differences in levels of FVIII and VWF (73, 74). Another candidate receptor that arose from the CHARGE study is stabilin-2 (57). Stabilin-2 is a hyaluronan-binding receptor that is expressed primarily by liver sinusoidal endothelial cells. Like many endocytic receptors, stabilin-2 is a highly modular protein that is composed of a series of repeated domains. In vitro expression studies have identified both VWF and FVIII as potential ligands of stabilin-2 (45). Interestingly, infusion of FVIII-VWF complexes into stabilin-2 deficient mice resulted in a reduced immune response when compared to infusion of highly purified recombinant FVIII (45). Infusion of hyaluronic acid also resulted in a reduced immune response to FVIII. Altogether, these findings suggest that stabilin-2 can regulate the immunogenicity of FVIII, and that liver sinusoidal endothelial cells not only serve as a major, possibly exclusive site of FVIII synthesis (75, 76) but are also implicated in FVIII catabolism and immunogenicity.
The involvement of CD4+ T cells in inhibitor development was first suggested by the clinical observation that HIV-positive HA patients, who tragically became infected through tainted blood products in the 1980s and were unfortunate enough to also have an inhibitor, showed a reduction in inhibitor titers as their T-cell counts declined (77). Qian et al. characterized FVIII-specific CD4+ T cells in a murine HA model (78) and demonstrated the critical role of T-cell dependent CD40-CD154 interactions in driving the antibody response (79). In a subsequent study, they identified an immunodominant CD4+ T-cell epitope recognized by these mice (80). The recent demonstration of expanded CD4+ T-follicular helper cells in spleens of FVIII-deficient mice with an inhibitor response has confirmed the expected essential role of this T-cell subset in providing B-cell help (43). Murine studies have also demonstrated an essential role for activated T cells in the memory B-cell response to FVIII, and the requirement for direct T-cell contact in order to re-stimulate these cells (81). Studies of patient blood samples have demonstrated CD4+ T-cell proliferation and cytokine secretion in response to FVIII protein and to synthetic peptides spanning the sequences of several FVIII domains (82–86). More recent investigations of the hemophilic immune response to FVIII have included isolation and characterization of human FVIII-specific T-cell clones and polyclonal lines (87–92), identifying immunodominant epitopes and phenotypes of minimally-expanded cells.
As mentioned earlier, autoimmune responses to self-FVIII can occur. Anti-FVIII antibodies isolated from acquired HA patient plasmas are class switched (23), indicating these inhibitors are flare-ups of a pre-existing but clinically insignificant autoimmune reaction. Interestingly, there is growing support for the notion that low-level T-cell auto-reactivity to endogenous FVIII may be a fairly common phenomenon in the healthy non-HA population. Several intriguing studies have indicated that many healthy individuals possess circulating CD4+ T cells that proliferate and secrete cytokines when stimulated with FVIII in vitro (93–95). Specificity of this T-cell response was further confirmed by a recent, elegant study in which FVIII-specific T-cell lines, which contained both naïve and memory subsets, were expanded from 16/16 non-HA blood donors (96). The calculated precursor frequency of FVIII-specific T cells was ~1.7 per million CD4+ T cells. These results indicate that thymic deletion of clones specific for the “self-protein” FVIII is incomplete, and that FVIII-specific memory T cells in non-HA individuals persist but do not expand [except in patients who develop neutralizing auto-antibodies to FVIII (4)]. A more recent study has identified FVIII epitopes recognized by CD4+ T cells from non-HA individuals using peptide ELISPOT assays and HLA Class II tetramers (97). Furthermore, peripheral blood mononuclear cells (PBMC) depleted of CD25+FoxP3+ cells showed enhanced proliferation compared to responses of non-depleted samples from non-HA subjects, suggesting that regulatory T cells (Tregs) play an important role in maintaining tolerance to endogenous FVIII under physiological conditions (93).
Thus, FVIII appears to be inherently more immunogenic than many other self-proteins (98, 99), and as-yet-undefined mechanisms maintain peripheral tolerance to self-FVIII in the vast majority of non-hemophilic individuals by preventing expansion of auto-reactive cells. Compare and contrast this situation with the development of anti-FVIII antibodies in severe HA, in which infused FVIII is a foreign protein: despite its inherent immunogenicity, ~¾ of these patients develop no neutralizing antibody (inhibitor) responses. Those who do develop an inhibitor may experience a transient neutralizing antibody response that either resolves spontaneously (100) or subsides following intensive FVIII infusions (ITI). Although low-titer anti-FVIII antibodies can often be detected in plasma/serum from these “tolerized” individuals, and FVIII-specific T-cell clones may still be isolated and expanded from their blood in vitro (88, 90), it is quite clear that peripheral tolerogenic mechanisms, which are still poorly defined, result in the desired clinical outcome of making FVIII replacement therapy possible by preventing or eliminating neutralizing FVIII-specific antibodies. Further studies of cellular responses to FVIII in HA patients with and without a high-titer inhibitor response, and in normal control blood donors, as well as animal model studies, are needed to clarify these mechanisms and potentially identify novel therapeutic targets.
Proliferation assays in which cells are stimulated with FVIII protein or peptides ex vivo (82–86, 95) have indicated that epitopes within multiple FVIII domains drive the anti-FVIII immune response. Analyses of FVIII-specific T-cell clones isolated using classic limiting dilution (87) or staining with peptide-loaded MHC Class II tetramers followed by cell sorting and expansion (88, 90–92) have unambiguously identified immunodominant T-cell epitopes in FVIII. Clones isolated from subjects with mild HA due to a missense mutation were, unsurprisingly, specific for epitopes corresponding to the wild-type FVIII sequence at the missense substitution site, as this is the only amino acid sequence in the infused FVIII that would be “foreign” to their immune system.
In another recent study, blood from a severe HA subject with a major F8 gene deletion, who had failed ITI and had a persistent inhibitor, was analyzed by systematic epitope mapping using MHC Class II (HLA-DRA-DRB1*01:01 and HLA-DRA-DRB1*10:01) tetramers loaded with synthetic peptides spanning the FVIII A2, C1 and C2 domains (92). Given that FVIII is a large (~220 kDa) protein, one would expect to find a polyclonal T-cell response targeting multiple epitopes. Interestingly, and counter-intuitively, only one FVIII epitope produced tetramer staining above background levels. Furthermore, analysis of the T-cell receptor (TCR) repertoire of these FVIII-specific cells showed cells that stained most strongly for this tetramer (likely indicating high-avidity binding) had a very narrow, oligoclonal TCR repertoire. Together, these results are consistent with a role for clonal deletion and anergy, and perhaps regulatory T cells, as important components of the functional “peripheral tolerance” that most HA patients achieve, whereas clones that escape this elimination or down-regulation following exposure to infused FVIII (including high-intensity FVIII treatment as part of ITI therapy) can persist and continue to provide help to B cells leading to antibody secretion. It is worth mentioning that T-cell clones specific for this same HLA-DRA-DRB1*01:01-restricted epitope have also been identified in two mild HA subjects with the same allele (88–90). Together, these studies suggest that ITI deletes or anergizes the vast majority of FVIII-specific T-cell clones, and in cases where ITI fails, a polyclonal T-cell response has still been converted to a monoclonal or oligoclonal response. Further human studies are required to determine if this narrowing of the FVIII-specific T-cell repertoire is a general feature of ITI. If so, this may provide support for novel immune interventions, based on a limited number of HLA-restricted T-cell epitopes, to promote tolerance to the entire FVIII molecule in patients who have failed ITI (101).
It is important to note that the study described above, which utilized HLA Class II tetramers, identified T-cell clones with high-avidity binding to a FVIII epitope. More recent studies employing ELISPOT assays to detect Th1 or Th2 cytokine secretion in response to FVIII peptides have identified responses to a larger epitope repertoire in a series of severe HA subjects (102) (Figure 3). This may well reflect lower-avidity peptide binding by these responsive cells, compared to the tetramer-positive cells, which is nevertheless physiologically relevant. Indeed, the respective roles of high- and low-avidity TCR binding interactions are of interest in studies of multiple allo-immune responses including allograft rejection and vaccine efficacy (103–105). As mentioned earlier, both antibody phenotyping and analysis of secreted cytokines have confirmed the involvement of both Th1 and Th2 subsets of CD4+ T-effectors in inhibitor development. Serial samples from one mild HA subject identified transient FVIII-specific (tetramer-positive) Th17/Th1 T cells 3–5 months following his initial inhibitor diagnosis, and parallel analysis of his FVIII-stimulated CD4+ T cells depleted of CD25hi cells showed stronger tetramer staining, consistent with some suppression by CD25hi Tregs (88). The ability of FoxP3+ Tregs to suppress inhibitor development in mice was demonstrated by Miao and colleagues, who found that transgenic HA mice that overexpressed FoxP3, unlike HA mice with unmodified FoxP3 expression, did not develop inhibitors following exposure to FVIII via plasmid-based gene therapy. Furthermore, adoptive transfer of CD4+CD25+FoxP3+ cells from the FVIII-exposed transgenic mice to the non-transgenic mice protected the recipient mice from developing high-titer inhibitors, and these Tregs also suppressed proliferation of FVIII-stimulated CD4+ T-effectors in vitro (106).
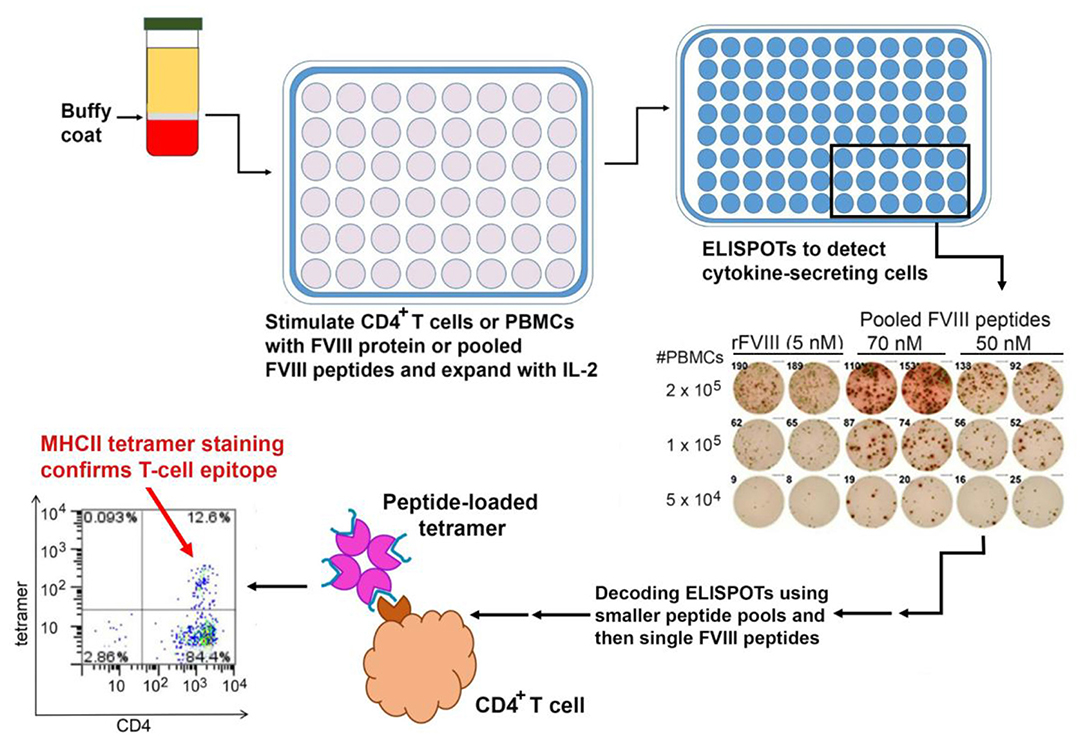
Figure 3. Mapping of HLA-restricted epitopes in FVIII recognized by CD4+ T cells from HA subjects. PBMCs are isolated from buffy coats of centrifuged blood and either used immediately or frozen. ELISPOT assays are carried out using either PBMCs or CD4+ T cells stimulated with FVIII protein or peptides, with unstimulated cells and tetanus-stimulated cells as negative and positive controls, respectively. Positive results from larger peptide pools are “decoded” by subsequent ELISPOT assays using smaller pools and then individual FVIII peptides as stimulants. Finally, ELISPOT results may be confirmed by staining CD4+ T cells using the appropriate peptide-loaded HLA Class II tetramers.
Further mechanistic studies in animal models and longitudinal studies of the anti-FVIII immune response in HA subjects are needed. The roles of FVIII-specific T-effector cells in patients with a persistent inhibitor also require further clarification; are these cells essential for maintenance of longstanding inhibitor responses, which are primarily driven by memory B cells?
The study of the immunogenicity of therapeutic FVIII relies on the use of different in vivo and in vitro experimental models. The in vitro analysis of the endocytosis of FVIII by purified APCs presents the advantage of a controlled study system; it however fails to encompass the diversity of the populations of APCs that may co-exist at a given time point and in a given microenvironment in vivo, and fails to account for the varying flow and shear stress conditions that pre-exist in the different body compartments where therapeutic FVIII may be encountered. More importantly, the use in cell culture of serum-free medium or medium containing heat-inactivated serum leaves aside numerous clotting factors and other plasma proteins. These include molecules that have developed a specific relationship with FVIII, in particular VWF, which was shown to reduce FVIII uptake (59, 107), as well as molecules that are essential for innate and adaptive immunity, such as circulating immunoglobulins or complement.
The complement system is an integral part of the innate and adaptive host defense (108, 109). Activation of the complement cascade can occur through at least three pathways: (1) the classical pathway is activated when C1q binds to immune complexes; (2) the lectin pathway is elicited by the binding of mannose-binding lectin to mannose residues on pathogens; and (3) the alternative pathway is spontaneously and continuously activated at a low rate (i.e., spontaneous C3 tick-over) (110, 111). The inappropriate activation of complement is pathogenic and has been associated with autoimmune reactions (112).
Recent studies have investigated whether complement activation plays a role in the onset of the anti-FVIII immune response. The administration of humanized cobra venom factor (hCVF) to mice was followed by an exhaustion of C3 from the circulation without generation of the down-stream pro-inflammatory anaphylatoxin C5a (113). The treatment of FVIII-deficient mice with hCVF prior to replacement therapy resulted in a 4-fold reduction in the levels of neutralizing anti-FVIII IgG, as compared to PBS-treated mice (114). To gain molecular insight into the implication of complement C3 in FVIII immunogenicity, the endocytosis of FVIII by APCs was studied in the presence of heat inactivated (i.e., de-complemented) or non-heat-inactivated human AB serum. Heat inactivation of serum resulted in a 2-fold decrease in FVIII uptake by both immature monocyte-derived dendritic cells (MODCs) and conventional blood DCs (114). Decreased FVIII internalization resulted in a proportional decreased activation of FVIII-specific T cells. Interestingly, elevated levels of FVIII uptake (and T-cell activation) were restored when MODCs were co-incubated with the reconstituted C3 activation complex or with the C3 activation fragment C3b alone. In agreement with this, although the specific endocytic receptor has not yet been identified, FVIII and C3b co-localized at the cell surface. Of note, an engineered FVIII protein with three amino acid changes in its C1 domain, which showed reduced immunogenicity, was described a couple of years ago (52, 57). While this mutant FVIII was not endocytosed by MODCs in vitro, its uptake was rescued in the presence of complement activation (114). It is tempting to propose temporary C3 depletion with hCVF as a therapeutic strategy to prevent the development of anti-FVIII antibodies during initial FVIII infusions of naïve HA patients (Figure 2).
Bleeding is typically associated with hemolysis that leads to the release of hemoglobin and free heme, and with the release at the site of injury of several pro-inflammatory mediators, including reactive oxygen species (ROS) (115). ROS have been demonstrated to alter the structure, function and immunogenicity of various proteins (116–118). The production of rFVIII under oxygen-free conditions preserves its pro-coagulant activity (119), possibly owing to its sensitivity to oxidation (120). Controlling oxidation in vivo in FVIII-deficient mice using N-acetyl-cysteine (NAC) was demonstrated to significantly reduce the intensity of the immune response to therapeutic FVIII (121). Conversely, ex vivo oxidation of FVIII prior to administration to mice resulted in increased immunogenicity as compared with non-oxidized FVIII. The immunogenicity of the oxidized FVIII was, however, not reduced when mice were treated with NAC, suggesting that NAC does not merely affect the immune response but may act directly by preventing FVIII oxidation. An earlier study also identified FVIII as a heme-binding protein (122). The binding of heme to FVIII resulted in a partial loss of pro-coagulant activity, which was at least in part consecutive to a reduced capacity of FVIII to interact with activated FIX. The effects of oxidation of heme-bound FVIII on FVIII functions and immunogenicity remain to be investigated.
Since the publication of the prospective randomized SIPPET study data, demonstrating a 1.87-fold increase in FVIII inhibitor incidence in previously untreated patients (PUPs) with the use of recombinant as opposed to plasma-derived FVIII, there has been a further search for factors that might provide a biological explanation for this difference (123). Furthermore, four independent cohort studies evaluating FVIII inhibitor incidence in PUPs have documented significant differences between a 2nd generation full-length rFVIII product and 3rd generation rFVIII concentrates, with the 2nd generation concentrate demonstrating a 1.6 to 2.8-fold increase in inhibitor incidence (124–128). Notably, the full-length 2nd generation product is expressed in baby hamster kidney (BHK) cells and the 3rd generation concentrates in Chinese hamster ovary (CHO) cells. While there are several possible explanations for the results of this series of epidemiological findings, a biologically plausible association relates to differences in the post-translational modifications, and specifically the glycosylation patterns found on various recombinant FVIII products produced in different cell types, and the plasma-derived protein derived from native human endothelial cells (54). A recent exploration of this glycosylation hypothesis has indeed provided supportive evidence for this proposal (129).
In this recent report, a range of methodologies have been used to document the glycan difference between rFVIII products and the subsequent effects on the FVIII immune response in vitro and in both “regular” (fully murine) F8-KO HA mice (F8 exon-16 deletion) and in humanized hemophilic mice expressing a mutant human F8 transgene product (with the hemophilia-causing mutation FVIII-Arg593Cys). Lectin binding and mass spectrometry analysis of the 2nd generation rFVIII and a full-length 3rd generation rFVIII concentrate showed a reduction in occupied N-linked glycosylation sites in the 2nd generation product and significant differences in the content of sialic acid and high mannose glycans. These structural differences were associated with increased immunogenicity in the mouse models of HA. In studies involving mice with the mutant human FVIII transgene (Arg593Cys), subcutaneous delivery of the BHK-expressed 2nd generation rFVIII resulted in a 94% incidence of FVIII inhibitors vs. 47% incidence following subcutaneous administration of the 3rd generation rFVIII. Anti-FVIII IgG titers were also significantly higher following exposure to the 2nd generation rFVIII.
In conclusion, these studies documented significant differences in the pattern of glycan occupancy and the types of glycans attached to rFVIII expressed in BHK vs. CHO cells. These differences were associated with variances in FVIII immunogenicity in mouse models of HA. These findings suggest that one of the factors influencing FVIII immunogenicity is the glycan profile, including both quantitative and qualitative details, at least in mice. They also suggest that there may be strategies involving glycan bioengineering that would be protective against FVIII immunogenicity. Preliminary results of an ongoing clinical trial testing a rFVIII product from a human cell line indicated that 17.6% of previously untreated patients developed a high-titer inhibitor (https://www.octapharma.com/news/corporate-news/2019/new-data-isth-2019/), suggesting that fine differences in glycosylation may not play a predominant role in FVIII immunogenicity in humans.
Over the past decade, there has been rapidly growing evidence that alterations of the gut microbiome play a key role in regulating both local and systemic immune responses (130–132). The mechanisms underlying this influence are still being investigated, but they include molecular mimicry from gut microbial antigens and immunomodulatory effects of microbial-derived metabolites (133).
Whether the gut microbiome contributes to the risk profile for FVIII immunogenicity has yet to be investigated in detail, but preliminary results in a mouse model system suggest that this may indeed be the case. Furthermore, the timing of initial FVIII exposure in severe HA children, in the first 1–2 years of life, not only represents the peak period for FVIII inhibitor generation but also represents the period when inter-individual microbiome differences are at their most extreme, and when exposure to immunologic challenges such as vaccinations are also initiated (134).
In a series of studies involving HA mice whose gut microbiome has been disrupted by oral administration of a broad-spectrum antibiotic, follow-up after repetitive FVIII challenge has been associated with significantly increased titers of anti-FVIII antibodies in the dysbiotic animals (135). Microbial analysis of the cecal contents in antibiotic-treated mice demonstrated significant reductions in Lactobacillus and Clostridia class immunomodulatory strains of bacteria. Detailed phenotyping of the antibiotic-treated and control mice at the time of initial FVIII exposure showed no differences in mesenteric lymph node and splenic regulatory T cell numbers, dendritic cell subsets and cytokine levels, but the cecal contents at this time demonstrated significantly reduced levels of the immune modulatory short chain fatty acids, acetate, propionate and butyrate. These initial observations provide a rationale for further evaluation of the gut microbiome as a contributing influence for FVIII immunogenicity. Whether interventions involving probiotic supplementation, specific immunomodulatory metabolite administration or microbiome- facilitated oral tolerance protocols can impact FVIII inhibitor development will require a considerable expansion of our current knowledge of this component of the body's immune system (136) (Figure 4).
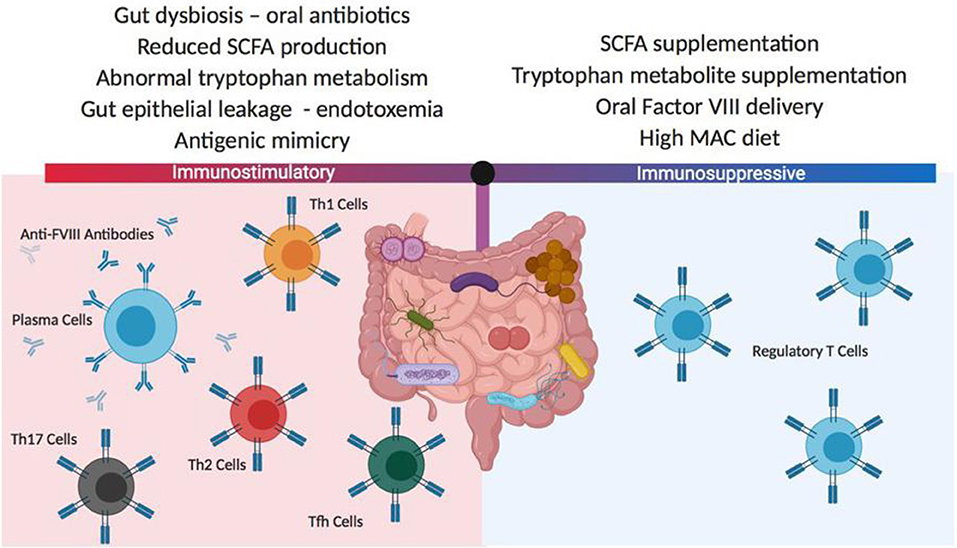
Figure 4. Potential gut microbiota influences on FVIII immunogenicity. The composition of the gut microbiome can have significant positive or negative effects on FVIII immunogenicity. These differences in the microbiome composition can derive from changes in diet, medications and coincident disease states. In most instances, the mechanisms responsible for changes to FVIII immune reactivity are not well-characterized but it is thought that antigenic mimicry and gut inflammation may play roles in initiating and boosting FVIII immunogenicity, while the delivery or generation of gut-derived immunomodulatory metabolites can mediate tolerogenic immune responses. SCFA, short chain fatty acids; MAC, microbiome accessible carbohydrate.
It has long been appreciated that monomeric, heterologous immunoglobulin G (IgG), as well as model antigens coupled to Ig heavy chains (IgG H), are tolerogenic (137–140). The IgG heavy chain Fc region binds to Fc receptors, thereby inhibiting B-cell receptor signaling (141) to facilitate antigen uptake and tolerance. Indeed, early studies showed that coupling of haptens to IgG Fc led to tolerance, but coupling to F(ab')2 did not (142). The recent fusion of FVIII with a human IgG1 Fc has created a therapeutic FVIII with extended half-life due to recycling of the protein via the neonatal Fc receptor (FcRn) (143). Initial pre-clinical and clinical studies (144–147) have suggested that FVIII-Fc may also lower the incidence of inhibitor development and increase ITI success rates (148). A recent study based on retrospective chart review indicated that patients receiving FVIII-Fc for ITI tolerized faster than with standard ITI protocols utilizing non-Fc fusion products, and several patients receiving “rescue ITI,” i.e., who had failed an earlier ITI regimen, became tolerized during ITI with FVIII-Fc and were able to resume standard replacement therapy (149). Clinical trials to test the safety and efficacy of FVIII-Fc in previously untreated HA patients (NCT02234323), and in patients undergoing ITI (NCT03093480 and NCT03103542), are currently under way. In addition, a FVIII-Fc fused to the VWF D'-D3 fragment and to an XTEN polypeptide to further extend its half-life, and which can be delivered subcutaneously, is in phase 1 testing (NCT03205163).
In humans, maternal IgG are transferred to the fetus through the placenta during the third trimester of pregnancy. This transfer of IgG is not passive but involves binding of the Fc fragment of the IgG to the FcRn expressed by the syncytiotrophoblast (150). The binding between FcRn and IgG occurs after uptake of IgG into the acidic endosome and prevents routing of the internalized IgG to the lysosomes and degradation, thereby favoring transcytosis to the fetal circulation instead. The same occurs in mice, albeit in a different time frame, with maternal IgG being transferred to the fetus from day 15 of pregnancy onwards. Such a phenomenon was exploited in mice, wherein an Fc fusion version of β-glucuronidase injected into the pregnant animals was detected in the fetus (151). Incidentally, the third trimester of pregnancy in the human, and days 14–20 of pregnancy in the mouse, witness the development of the fetal immune system and establishment of tolerance to self (152). Administration to pregnant FVIII-deficient mice of Fc-fused A2 and C2 domains of FVIII is followed by the FcRn-dependent transfer of these molecules to the fetal compartment, followed by their transport by SIRPα+ dendritic cells to the thymus (153). The introduction of FVIII-A2-Fc and FVIII-C2-Fc during fetal life induced FVIII-specific regulatory T cells that were detected after birth, and that protected against alloimmunization to therapeutic FVIII later in life (154). Interestingly, preliminary data from the Lillicrap group showed that injection of high dose therapeutic FVIII-Fc (Eloctate) to pregnant mice allows detection of FVIII activity in the fetuses, which is not the case when recombinant FVIII alone is injected (155). Together, these data provide proof of concept for the antenatal induction of active and long-lasting immune tolerance to therapeutic FVIII, if FVIII is administered at the appropriate gestational stage. A similar approach was tested, with success, in animal models of Type I diabetes (156). In principle, this could be extended to prevent alloimmune responses to therapeutic agents used to treat other monogenic diseases too, such as hemophilia B (157) or Pompe disease (158).
Fc-FVIII fusions may be employed in novel cellular-based therapies designed to induce specific tolerance to the Fc-conjugated protein. Based on studies that demonstrated that B-cell presentation of antigens could be tolerogenic (159, 160), it was demonstrated that retroviral transduction of the FVIII A2 and C2 domains inserted in-frame at the N-terminus of isologous IgG H chains into B cells blocked and even reversed inhibitor formation (161). This system required MHC class II expression on the B cells, and led to the generation of regulatory T cells (Tregs) in multiple models of adverse immune responses (uveitis, EAE, diabetes, arthritis) in addition to HA (161–165).
Polyclonal Tregs can be suppressive both in vitro and in vivo. However, because they contain multiple TCR from the entire repertoire, a potential drawback to their clinical application is that they could be non-specifically immunosuppressive. Indeed, there are anecdotal examples of viral re-activation in some clinical trials (166) and concerns about lowering immune barriers to cancer. Efforts to expand antigen-specific Tregs from polyclonal precursors are challenging; however, this has been achieved recently for HA mouse models (167, 168). The use of antigen-specific Tregs has also been significantly refined in an alternative approach using retroviral transduction to express a single TCR, derived from a HA subject and recognizing a well-defined HLA-restricted T-cell epitope in FVIII (89), on human Tregs. When this TCR was expressed in sorted human Tregs (CD25+, CD127lo, FoxP3+, Helios+), these engineered Tregs suppressed proliferation and cytokine secretion by FVIII-specific CD4+ T-effector clones. An important advance that made this tolerogenic approach possible was the development of methods to expand human Tregs ex vivo (169). Moreover, when the TCR-transduced Tregs were added to spleen cells from FVIII-immunized mice, antibody formation to FVIII was significantly inhibited, thereby demonstrating that bystander suppression to multiple epitopes in other domains of FVIII was occurring. This suppression was also demonstrated in vivo, despite the xenogeneic barrier and rejection of the human Tregs within 1–2 weeks. A fully murine system needs to be developed to test the durability of this tolerogenic effect.
These studies provided proof-of-principle for the utility of engineered Tregs, but the HLA restriction of TCRs would require development of patient-specific Treg lines, a formidable barrier. In order to develop antigen-specific Tregs that are not TCR/HLA-restricted, Tregs were next engineered to express a single chain Fv (isolated from a phage display library) recognizing the FVIII A2 domain (170). Tregs expressing this chimeric antigen receptor (CAR) were similarly effective as the TCR-engineered Tregs at suppressing FVIII antibody and inhibitor response in vitro and in vivo. Mechanistic studies suggested that contact between Tregs and T-effectors enhanced suppressive function driven by IL-2 (171), but the targets of the TCR- and scFv CAR Tregs might be different. The former act on antigen-presenting cells (expressing peptide:MHC) while the latter would be activated by conformational epitopes of the properly folded FVIII protein.
As a further approach, FVIII domains have now been expressed on the surfaces of both Tregs and CD8+ cytotoxic cells, in order to directly target B cells. These engineered cells are referred to as B-cell Antigen Receptor, or “BAR” T cells, since the expressed domains would be recognized by FVIII-specific B-cell receptors (BCR). The Treg and CD8+ BARs suppressed and killed, respectively, FVIII-specific B cells, thereby blocking anti-FVIII antibody production (172, 173). Thus, these various engineered FVIII-specific Tregs (Figure 5) are demonstrably functional, with different targets and advantages. Ongoing studies are now testing their tolerogenic properties in the presence of high-titer FVIII inhibitors.
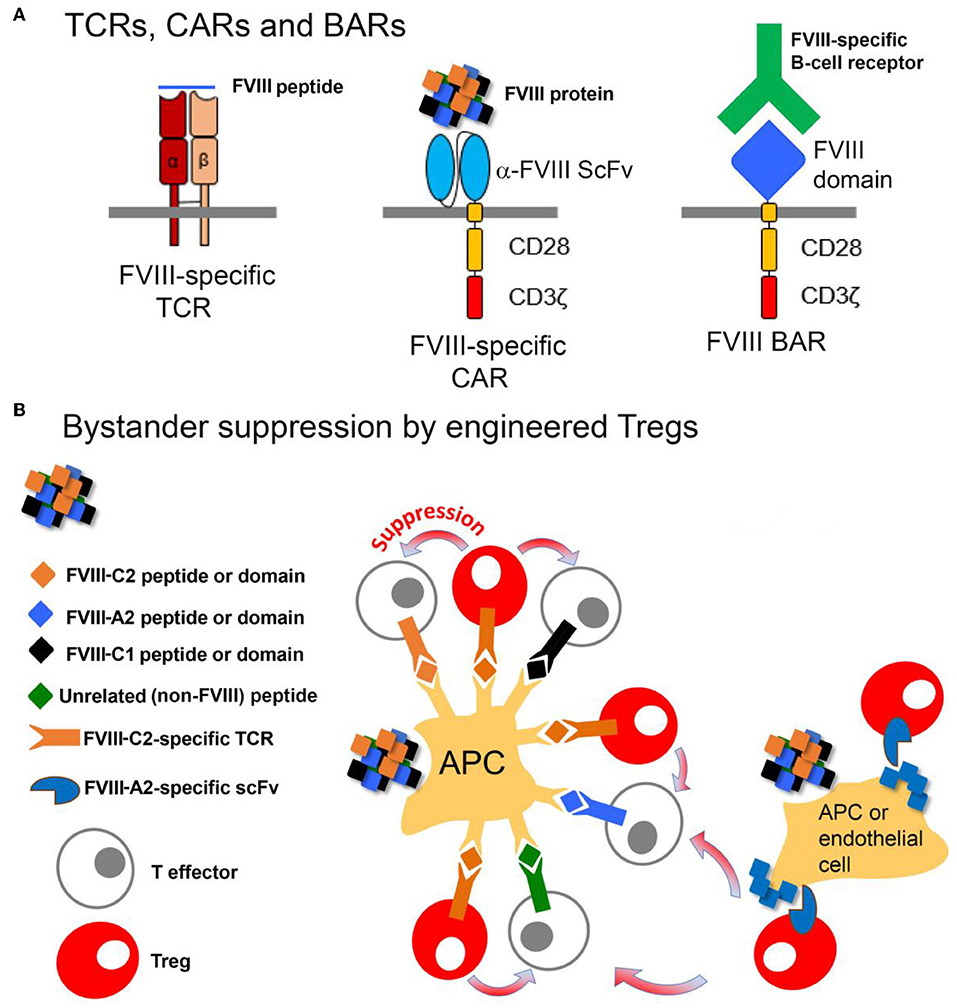
Figure 5. Design and function of Tregs engineered to express FVIII-specific TCRs, CARs or BARs. (A) The FVIII-TCR Tregs will recognize FVIII peptides with specific HLA-restricted T-cell epitopes. The FVIII-specific scFv Tregs will recognize FVIII domains, with no HLA restriction. The FVIII-BAR T cells will recognize B cells expressing FVIII-specific BCRs. FVIII-BAR Tregs are expected to down-regulate or prevent FVIII-specific B-cell activation, while FVIII-BAR CD8+ T cells should specifically kill only the FVIII-specific B cells. (B) Cartoon showing possible mechanisms by which engineered FVIII-specific Tregs may exert bystander suppression, i.e., create a tolerogenic environment that will suppress nearby T-effectors having specificity for multiple epitopes in FVIII.
Delivery of antigens via the mucosal route has been known to be tolerogenic for decades, and this route of antigen exposure may lead to the induction of Tregs as well. One challenge for oral tolerance is that it requires large amounts of protein, the cost of which would be prohibitive with FVIII. Daniell and Herzog have overcome this challenge by designing a system in which encapsulated FVIII fused with cholera toxin subunit B (which enables transfer across the gut epithelium but is itself nontoxic) is expressed in lettuce leaves (174–176). The lettuce is then processed into powder in a GMP facility. When this FVIII-containing powder was fed to hemophilic mice and dogs, tolerance was induced in both prophylactic and therapeutic experiments (176). This approach appears to hold tremendous promise as a non-invasive method to promote tolerance to FVIII, even in advance of initial FVIII infusions (and hence before inhibitors could develop). Furthermore, the application of oral tolerance protocols in the context of immune modulatory gut microbial environments may provide additional benefits in the induction of tolerogenic responses to FVIII.
In the last few years, nanoparticles (NP) that had been designed for drug delivery were also found to be efficient vehicles for tolerance induction. These self-assembling, biodegradable poly(lactide-co-glycolide) tolerogenic NPs (tNP) that contain the immune modulator, rapamycin, with or without protein or peptide antigens are capable of inducing durable antigen-specific tolerance that controls adaptive immune responses and can withstand multiple immunogenic challenges with antigen. Thus, Maldonado et al. utilized tNPs containing rapamycin given together with repeated doses of FVIII (177). This protocol blocked FVIII inhibitor production even with repeated FVIII challenge for over 200 days and led to Treg development. Moreover, this protocol was successful in previously immunized mice, albeit requiring additional (multiple) treatments with tNPs. Further commercial development of such tNPs (SEL-212) is proceeding to promote tolerance to uricase, an immunogenic agent used for gout therapy (https://www.selectabio.com/immtor/gouttherapy/phase2results/). There is considerable interest in testing nanoparticle + rapamycin therapy in other immunogenic therapies, and rapid advances in this field are expected over the next several years.
Over the past decade, a range of innovative non-factor replacement approaches for the treatment of HA have been under development, and several of these therapies are now in the clinic (178). The influence of these treatments on the FVIII immune response and FVIII tolerance will be variable, with some approaches (e.g., rebalancing hemostasis strategies) having a less obvious potential impact on FVIII inhibitor development, while for other novel therapies there will be clear, either direct or indirect, consequences for the FVIII immune response. The most clearly influential of these treatments to date are the humanized bispecific antibody, emicizumab (“Hemlibra”), and FVIII gene therapy.
Emicizumab has demonstrated partial FVIII mimetic properties in a variety of in vitro tests (179) and has been shown in phase 3 studies of HA patients both with (7) and without (8) FVIII inhibitors using prophylactic emicizumab treatment to very significantly reduce annualized bleed rates. The bispecific antibody does not induce or exacerbate anti-FVIII antibody responses, and anti-emicizumab antibodies have been detected in <5% of treated patients to date. A major question that is currently unresolved is whether FVIII inhibitor patients being successfully treated with emicizumab prophylaxis should undergo ITI in an attempt to eliminate their neutralizing anti-FVIII antibody response. Initial clinical studies are now underway aimed at addressing this question, but basic immunologic principles would suggest that the efficacy of ITI should not be decreased in emicizumab users. On the contrary, their reduced inflammatory status accompanying restoration of hemostasis may even improve ITI success rates. Induction of tolerance to FVIII would enable the preferential use of FVIII to treat episodes of breakthrough bleeding in these patients and would avoid the less predictable outcomes obtained with bypass product treatment. Importantly, administration of FEIBA as a bypass agent when emicizumab is “on board” may be contra-indicated due to a possibly increased thrombotic risk when these therapeutics are combined (180, 181); ongoing and future monitoring of patients treated with FEIBA while still on emicizumab will generate sufficient data to properly evaluate this potential risk. The only other currently approved bypass agent is recombinant factor (F)VIIa, which is expensive and has a short half-life. However, recent studies have indicated that concomitant treatment with emicizumab + rFVIIa does not change the safety profile of rFVIIa. The relative effectiveness of FEIBA vs. rFVIIa in treating breakthrough bleeds for patients on emicizumab has not yet been established, due to the limited amount of time this bispecific antibody has been on the market. Anecdotally, some inhibitor patients seem to respond better to one bypass agent than to another during serious bleeds, so removing FEIBA from the available armamentarium could prove problematic in some cases. Therefore, a lack of tolerance to FVIII could constitute an additional clinical risk factor even for patients successfully receiving emicizumab prophylaxis, by narrowing the options to staunch potentially dangerous bleeding following accidents, trauma or surgery. We therefore suggest that ITI continues to be an entirely appropriate therapy for inhibitor patients, regardless of whether they are being successfully treated with emicizumab, as ongoing tolerance to FVIII provides a clear clinical benefit by increasing the available options to treat or prevent bleeds.
The other scenario that may require consideration concerns the use of emicizumab in infants (previously untreated or treated with any FVIII concentrate), where the practical convenience of infrequent sub-cutaneous administration will provide a significant advantage for care givers and patients. In this situation, FVIII might only be administered at the time of breakthrough bleeding, and thus there may be a potential for increasing the inhibitor risk through FVIII delivery only at times of “high immunologic danger” due to associated bleeding and inflammation. This concern could be mitigated by early low dose FVIII prophylaxis to induce peripheral tolerance to FVIII. Furthermore, it is highly likely that regular, intermittent re-exposures to FVIII will be required to maintain this tolerance in infants, children and adults who choose emicizumab (or other non-FVIII therapies) for prophylactic prevention of bleeding. The necessity of antigen persistence for immunologic unresponsiveness has been demonstrated in many scenarios and is an accepted principle of immunology (182). Further research will be required to determine the optimal FVIII doses and maximum intervals between these doses to maintain peripheral tolerance. This would preserve the option of future FVIII replacement therapy for HA patients who choose alternative, non-FVIII therapies.
After 25 years of pre-clinical development, FVIII gene therapy is now being successfully applied in late stage clinical trials (183). There is a strong likelihood that the first licensed FVIII gene therapy product will be available within the next 12 months. All clinical trials to date have involved adeno-associated viral vector (AAV) liver-directed gene transfer of a B-domain-deleted FVIII transgene construct. All enrolled patients (until now) have had no prior history of FVIII inhibitor development, and no FVIII inhibitors have been documented in their follow up post-vector administration (out to a maximum of 3 years). In previous pre-clinical animal studies, transient anti-FVIII immune responses have been seen in a few animals, but all have been eliminated with persistent expression of the FVIII transgene. Furthermore, in studies of HA dogs with pre-existing FVIII inhibitory antibodies, AAV-mediated delivery of a canine FVIII transgene has been successful in mediating tolerance to FVIII, and the dogs eventually demonstrated persistent, therapeutically relevant levels of FVIII expression (184). Based on these results, it is reasonable to propose that in patients with FVIII inhibitors where routine ITI has failed, a trial of liver-directed FVIII gene therapy might prove effective. Development of a formal clinical trial protocol for evaluation of this intervention would be essential. Theoretical advantages that a gene therapy strategy for ITI might have include the persistent as opposed to intermittent exposure to the FVIII antigen, the relatively stable concentration of circulating FVIII, and the fact that FVIII production is from the liver, a well-documented location for supporting tolerogenic immune responses (185). FVIII gene therapy has not been approved for pediatric patients, so inhibitor risk for previously untreated severe HA patients is unknown. Further animal model studies will allow improved estimates of this and other potential safety issues that must be addressed adequately before offering this experimental therapy to infants and children.
Another creative approach to re-balance hemostasis in the absence of functional FVIII is to inhibit specific anticoagulation pathways by targeting activated protein C, which is generated in vivo by the thrombin/thrombomodulin complex on the surface of endothelial cells. Activated protein C is a serine protease that cleaves and thereby inactivates the cofactors FVIIIa and factor Va. Its natural inhibitor is the serpin (serine protease inhibitor) protein C inhibitor, which despite its name is a promiscuous inhibitor of multiple serine proteases. James Huntington and colleagues recently engineered a novel serpin that is highly selective for activated protein C, and that corrected bleeding in HA mice (186, 187). Further testing to evaluate the safety of this approach is needed, but at present it offers the intriguing possibility of achieving hemostasis by administering this relatively long-lived protein therapeutic, even in the presence of neutralizing anti-FVIII antibodies. As with other non-FVIII therapies, no effect on FVIII immunogenicity would be expected, although co-administration with FVIII to either induce or maintain tolerance would allow patients to resume FVIII replacement therapy either prophylactically, or as needed to treat traumatic or breakthrough bleeds.
In this review we have presented an overview of recent insights into the immune response to FVIII and novel approaches to prevent, circumvent or reverse the development of neutralizing anti-FVIII antibodies. It is an exciting time to be working in this field, with some new therapies already in clinical trials and others showing promising results in animal models. Several practical challenges remain, many of them inherent to studies of rare clinical disorders such as HA. For example, given that the anti-FVIII immune response is most likely to develop during initial infusions of infants and toddlers, there is limited availability of the required blood volumes for mechanistic studies of inhibitor development in humans. Yet, these studies are vital to understand the basis of different clinical outcomes, especially given the many differences between the diverse human population vs. other species, notably inbred mice (188). The number of cells available from genetically well-characterized mice, and even large animal models, can also be a limiting factor for studies, e.g., when attempting to characterize splenic marginal zone cells, or vascular and sinusoidal endothelial cells. The emergence of increasingly sophisticated techniques to analyze small samples, e.g., by flow cytometry-based immunophenotyping (189, 190), mass cytometry (189, 191), TCRαβ repertoire profiling (192, 193), and improved “-omics” methodologies combined with bioinformatics (194, 195) holds tremendous promise for furthering research into antigen-specific immune responses, including the basis for FVIII immunogenicity and maintenance of peripheral tolerance to FVIII.
Increasing coordination between hemophilia care providers, funding agencies and, of course, the hemophilia community itself is enabling initiatives from large-scale registries and associated data sets (196) to the establishment of sample and data repositories (197). Studies utilizing these resources will not only further our understanding of FVIII immunogenicity and tolerance, but they are also likely to provide insights into immunogenicity of various other biotherapeutics, which comprise a growing proportion of biotechnology and pharmaceutical company portfolios.
The introduction of cryoprecipitate, over 50 years ago, to treat HA patients was followed by development of additional plasma-derived and recombinant FVIII concentrates with improved safety profiles, and more recent advances have included modest half-life extension through various modifications of the FVIII protein. Non-FVIII therapies are now beginning to transform the lives of inhibitor (and some non-inhibitor) patients, allowing them to re-balance hemostasis to avoid most breakthrough bleeds, even in the presence of neutralizing anti-FVIII antibodies. Possible longer-term risks associated with these alternative therapies are unknown, and there is still little experience with their use in settings of trauma or surgery. Also, many patients and families who have evaluated recently available novel therapies choose to begin or remain on FVIII therapy. Therefore, maintenance of tolerance to FVIII remains a high priority. It is hoped that further development of tolerogenic approaches such as those described in this review will lead to new therapies allowing HA patients to “tolerate,” and fully benefit from, FVIII replacement therapy.
All authors contributed equally to strategy discussions and to writing of this article.
Study-related expenses, including publication charges and travel support for face-to-face meetings in conjunction with international scientific meetings and symposia, were funded under an investigator-initiated educational grant to KP from Grifols, Inc. Grifols was not involved in the study design, collection, analysis, interpretation of data, the writing of this article or the decision to submit it for publication. Additional funding from NIH HL 130448 (to KP).
The opinions or assertions contained herein are the private ones of the authors and are not to be construed as official or reflecting the views of the Department of Defense or the Uniformed Services University of the Health Sciences (USUHS).
KP, DS, and SL-D are inventors on FVIII-related patents. JV is an inventor on FVIII-related patents and has received research funding from Novo Nordisk and has acted as an advisor for Biotest.
The remaining author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
1. Gouw SC, Van Den Berg HM, Fischer K, Auerswald G, Carcao M, Chalmers E, et al. Intensity of factor VIII treatment and inhibitor development in children with severe hemophilia A: the RODIN study. Blood. (2013) 121:4046–55. doi: 10.1182/blood-2012-09-457036
2. Peyvandi F, Cannavo A, Garagiola I, Palla R, Mannucci PM, Rosendaal FR, et al. Timing and severity of inhibitor development in recombinant versus plasma-derived factor VIII concentrates: a SIPPET analysis. J Thromb Haemost. (2018) 16:39–43. doi: 10.1111/jth.13888
3. Hay CR, Palmer B, Chalmers E, Liesner R, Maclean R, Rangarajan S, et al. Incidence of factor VIII inhibitors throughout life in severe hemophilia A in the United Kingdom. Blood. (2011) 117:6367–70. doi: 10.1182/blood-2010-09-308668
4. Janbain M, Leissinger CA, Kruse-Jarres R. Acquired hemophilia A: emerging treatment options. J Blood Med. (2015) 6:143–50. doi: 10.2147/JBM.S77332
5. Reding MT, Lei S, Lei H, Green D, Gill J, Conti-Fine BM. Distribution of Th1- and Th2-induced anti-factor VIII IgG subclasses in congenital and acquired hemophilia patients. Thromb Haemost. (2002) 88:568–75. doi: 10.1055/s-0037-1613257
6. Van Den Brink EN, Turenhout EA, Davies J, Bovenschen N, Fijnvandraat K, Ouwehand WH, et al. Human antibodies with specificity for the C2 domain of factor VIII are derived from VH1 germline genes. Blood. (2000) 95:558–63. doi: 10.1182/blood.V95.2.558
7. Oldenburg J, Mahlangu JN, Kim B, Schmitt C, Callaghan MU, Young G, et al. Emicizumab prophylaxis in hemophilia A with Inhibitors. N Engl J Med. (2017) 377:809–18. doi: 10.1056/NEJMoa1703068
8. Mahlangu J, Oldenburg J, Paz-Priel I, Negrier C, Niggli M, Mancuso ME, et al. Emicizumab prophylaxis in patients who have hemophilia A without inhibitors. N Engl J Med. (2018) 379:811–22. doi: 10.1056/NEJMoa1803550
9. Shapiro AD, Angchaisuksiri P, Astermark J, Benson G, Castaman G, Chowdary P, et al. Subcutaneous concizumab prophylaxis in hemophilia A and hemophilia A/B with inhibitors: Phase 2 trial results. Blood. (2019) 134:1973–82. doi: 10.1182/blood.2019001542
10. Lozier JN, Nichols TC. Animal models of hemophilia and related bleeding disorders. Semin Hematol. (2013) 50:175–84. doi: 10.1053/j.seminhematol.2013.03.023
12. Nichols TC, Hough C, Agerso H, Ezban M, Lillicrap D. Canine models of inherited bleeding disorders in the development of coagulation assays, novel protein replacement and gene therapies. J Thromb Haemost. (2016) 14:894–905. doi: 10.1111/jth.13301
13. Arruda VR, Samelson-Jones BJ. Gene therapy for immune tolerance induction in hemophilia with inhibitors. J Thromb Haemost. (2016) 14:1121–34. doi: 10.1111/jth.13331
14. Hay CR, Dimichele DM, International Immune Tolerance S. The principal results of the International Immune Tolerance Study: a randomized dose comparison. Blood. (2012) 119:1335–44. doi: 10.1182/blood-2011-08-369132
15. Schep SJ, Schutgens REG, Fischer K, Boes ML. Review of immune tolerance induction in hemophilia A. Blood Rev. (2018) 32:326–38. doi: 10.1016/j.blre.2018.02.003
16. Zhang AH, Skupsky J, Scott DW. Effect of B-cell depletion using anti-CD20 therapy on inhibitory antibody formation to human FVIII in hemophilia A mice. Blood. (2011) 117:2223–6. doi: 10.1182/blood-2010-06-293324
17. Hausl C, Ahmad RU, Sasgary M, Doering CB, Lollar P, Richter G, et al. High-dose factor VIII inhibits factor VIII-specific memory B cells in hemophilia A with factor VIII inhibitors. Blood. (2005) 106:3415–22. doi: 10.1182/blood-2005-03-1182
18. Kasper CK, Aledort L, Aronson D, Counts R, Edson JR, Van Eys J, et al. Proceedings: A more uniform measurement of factor VIII inhibitors. Thromb Diath Haemorrh. (1975) 34:612. doi: 10.1055/s-0039-1689482
19. Verbruggen B, Novakova I, Wessels H, Boezeman J, Van Den Berg M, Mauser-Bunschoten E. The Nijmegen modification of the Bethesda assay for factor VIII:C inhibitors: improved specificity and reliability. Thromb Haemost. (1995) 73:247–51. doi: 10.1055/s-0038-1653759
20. Lavigne-Lissalde G, Rothschild C, Pouplard C, Lapalud P, Gruel Y, Schved JF, et al. Characteristics, mechanisms of action, and epitope mapping of anti-factor VIII antibodies. Clin Rev Allergy Immunol. (2009) 37:67–79. doi: 10.1007/s12016-009-8119-0
21. Whelan SF, Hofbauer CJ, Horling FM, Allacher P, Wolfsegger MJ, Oldenburg J, et al. Distinct characteristics of antibody responses against factor VIII in healthy individuals and in different cohorts of hemophilia A patients. Blood. (2013) 121:1039–48. doi: 10.1182/blood-2012-07-444877
22. Hofbauer CJ, Whelan SF, Hirschler M, Allacher P, Horling FM, Lawo JP, et al. Affinity of FVIII-specific antibodies reveals major differences between neutralizing and nonneutralizing antibodies in humans. Blood. (2015) 125:1180–8. doi: 10.1182/blood-2014-09-598268
23. Lewis KB, Hughes RJ, Epstein MS, Josephson NC, Kempton CL, Kessler CM, et al. Phenotypes of allo- and autoimmune antibody responses to FVIII characterized by surface plasmon resonance. PLoS ONE. (2013) 8:e61120. doi: 10.1371/journal.pone.0061120
24. Krudysz-Amblo J, Parhami-Seren B, Butenas S, Brummel-Ziedins KE, Gomperts ED, Rivard GE, et al. Quantitation of anti-factor VIII antibodies in human plasma. Blood. (2009) 113:2587–94. doi: 10.1182/blood-2008-08-174987
25. Reipert BM. Risky business of inhibitors: HLA haplotypes, gene polymorphisms, and immune responses. Hematol Am Soc Hematol Educ Program. (2014) 2014:372–8. doi: 10.1182/asheducation.V2014.1.372.3884771
26. Wroblewska A, Reipert BM, Pratt KP, Voorberg J. Dangerous liaisons: how the immune system deals with factor VIII. J Thromb Haemost. (2013) 11:47–55. doi: 10.1111/jth.12065
27. Boylan B, Rice AS, Dunn AL, Tarantino MD, Brettler DB, Barrett JC, et al. Characterization of the anti-factor VIII immunoglobulin profile in patients with hemophilia A by use of a fluorescence-based immunoassay. J Thromb Haemost. (2015) 13:47–53. doi: 10.1111/jth.12768
28. Algiman M, Dietrich G, Nydegger UE, Boieldieu D, Sultan Y, Kazatchkine MD. Natural antibodies to factor VIII. (anti-hemophilic factor) in healthy individuals. Proc Natl Acad Sci U S A. (1992) 89:3795–9. doi: 10.1073/pnas.89.9.3795
29. Gilles JG, Saint-Remy JM. Healthy subjects produce both anti-factor VIII and specific anti-idiotypic antibodies. J Clin Invest. (1994) 94:1496–505. doi: 10.1172/JCI117489
30. Lacroix-Desmazes S, Misra N, Bayry J, Mohanty D, Kaveri SV, Kazatchkine MD. Autoantibodies to factor VIII. Autoimmun Rev. (2002) 1:105–10. doi: 10.1016/S1568-9972(01)00017-9
31. Pratt KP, Gunasekera D, Vir P, Peters R, Tan S, Pierce G, et al. Neutralizing and non-neutralizing anti-FVIII antibodies in black and white hemophilia a subjects: a natural history profile. Blood. (2019) 134(S1):1131. doi: 10.1182/blood-2019-124743
32. Lacroix-Desmazes S, Navarrete AM, Andre S, Bayry J, Kaveri SV, Dasgupta S. Dynamics of factor VIII interactions determine its immunologic fate in hemophilia A. Blood. (2008) 112:240–9. doi: 10.1182/blood-2008-02-124941
33. Varthaman A, Lacroix-Desmazes S. Pathogenic immune response to therapeutic factor VIII: exacerbated response or failed induction of tolerance? Haematologica. (2019) 104:236–44. doi: 10.3324/haematol.2018.206383
34. Hartholt RB, Van Velzen AS, Peyron I, Ten Brinke A, Fijnvandraat K, Voorberg J. To serve and protect: the modulatory role of von Willebrand factor on factor VIII immunogenicity. Blood Rev. (2017) 31:339–47. doi: 10.1016/j.blre.2017.07.001
35. Mebius RE, Kraal G. Structure and function of the spleen. Nat Rev Immunol. (2005) 5:606–16. doi: 10.1038/nri1669
36. Eisenbarth SC. Dendritic cell subsets in T cell programming: location dictates function. Nat Rev Immunol. (2019) 19:89–103. doi: 10.1038/s41577-018-0088-1
37. Lewis SM, Williams A, Eisenbarth SC. Structure and function of the immune system in the spleen. Sci Immunol. (2019) 4:eaau6085. doi: 10.1126/sciimmunol.aau6085
38. Navarrete A, Dasgupta S, Delignat S, Caligiuri G, Christophe OD, Bayry J, et al. Splenic marginal zone antigen-presenting cells are critical for the primary allo-immune response to therapeutic factor VIII in hemophilia A. J Thromb Haemost. (2009) 7:1816–23. doi: 10.1111/j.1538-7836.2009.03571.x
39. Lai JD, Cartier D, Hartholt RB, Swystun LL, Van Velzen AS, Den Haan JMM, et al. Early cellular interactions and immune transcriptome profiles in human factor VIII-exposed hemophilia A mice. J Thromb Haemost. (2018) 16:533–45. doi: 10.1111/jth.13936
40. Zerra PE, Cox C, Baldwin WH, Patel SR, Arthur CM, Lollar P, et al. Marginal zone B cells are critical to factor VIII inhibitor formation in mice with hemophilia A. Blood. (2017) 130:2559–68. doi: 10.1182/blood-2017-05-782912
41. Delignat S, Russick J, Gangadharan B, Rayes J, Ing M, Voorberg J, et al. Prevention of the anti-factor VIII memory B-cell response by inhibition of Bruton tyrosine kinase in experimental hemophilia A. Haematologica. (2019) 104:1046–54. doi: 10.3324/haematol.2018.200279
42. Harris EN, Weigel JA, Weigel PH. The human hyaluronan receptor for endocytosis. (HARE/Stabilin-2) is a systemic clearance receptor for heparin. J Biol Chem. (2008) 283:17341–50. doi: 10.1074/jbc.M710360200
43. Jing W, Chen J, Cai Y, Chen Y, Schroeder JA, Johnson BD, et al. Induction of activated T follicular helper cells is critical for anti-FVIII inhibitor development in hemophilia A mice. Blood Adv. (2019) 3:3099–110. doi: 10.1182/bloodadvances.2019000650
44. Van Schooten CJ, Shahbazi S, Groot E, Oortwijn BD, Van Den Berg HM, Denis CV, et al. Macrophages contribute to the cellular uptake of von Willebrand factor and factor VIII in vivo. Blood. (2008) 112:1704–12. doi: 10.1182/blood-2008-01-133181
45. Swystun LL, Lai JD, Notley C, Georgescu I, Paine AS, Mewburn J, et al. The endothelial cell receptor stabilin-2 regulates VWF-FVIII complex half-life and immunogenicity. J Clin Invest. (2018) 128:4057–73. doi: 10.1172/JCI96400
46. Knolle PA, Wohlleber D. Immunological functions of liver sinusoidal endothelial cells. Cell Mol Immunol. (2016) 13:347–53. doi: 10.1038/cmi.2016.5
47. Van Haren SD, Wroblewska A, Fischer K, Voorberg J, Herczenik E. Requirements for immune recognition and processing of factor VIII by antigen-presenting cells. Blood Rev. (2012) 26:43–9. doi: 10.1016/j.blre.2011.10.001
48. Dasgupta S, Navarrete AM, Bayry J, Delignat S, Wootla B, Andre S, et al. A role for exposed mannosylations in presentation of human therapeutic self-proteins to CD4+ T lymphocytes. Proc Natl Acad Sci USA. (2007) 104:8965–70. doi: 10.1073/pnas.0702120104
49. Lenting PJ, Neels JG, Van Den Berg BM, Clijsters PP, Meijerman DW, Pannekoek H, et al. The light chain of factor VIII comprises a binding site for low density lipoprotein receptor-related protein. J Biol Chem. (1999) 274:23734–9. doi: 10.1074/jbc.274.34.23734
50. Saenko EL, Yakhyaev AV, Mikhailenko I, Strickland DK, Sarafanov AG. Role of the low density lipoprotein-related protein receptor in mediation of factor VIII catabolism. J Biol Chem. (1999) 274:37685–92. doi: 10.1074/jbc.274.53.37685
51. Dasgupta S, Navarrete AM, Andre S, Wootla B, Delignat S, Repesse Y, et al. Factor VIII bypasses CD91/LRP for endocytosis by dendritic cells leading to T-cell activation. Haematologica. (2008) 93:83–9. doi: 10.3324/haematol.11535
52. Herczenik E, Van Haren SD, Wroblewska A, Kaijen P, Van Den Biggelaar M, Meijer AB, et al. Uptake of blood coagulation factor VIII by dendritic cells is mediated via its C1 domain. J Allergy Clin Immunol. (2012) 129:501–9. doi: 10.1016/j.jaci.2011.08.029
53. Canis K, Anzengruber J, Garenaux E, Feichtinger M, Benamara K, Scheiflinger F, et al. In-depth comparison of N-glycosylation of human plasma-derived factor VIII and different recombinant products: from structure to clinical implications. J Thromb Haemost. (2018) 16:1592–603. doi: 10.1111/jth.14204
54. Kannicht C, Ramstrom M, Kohla G, Tiemeyer M, Casademunt E, Walter O, et al. Characterisation of the post-translational modifications of a novel, human cell line-derived recombinant human factor VIII. Thromb Res. (2013) 131:78–88. doi: 10.1016/j.thromres.2012.09.011
55. Delignat S, Repesse Y, Navarrete AM, Meslier Y, Gupta N, Christophe OD, et al. Immunoprotective effect of von Willebrand factor towards therapeutic factor VIII in experimental haemophilia A. Haemophilia. (2012) 18:248–54. doi: 10.1111/j.1365-2516.2011.02679.x
56. Wroblewska A, Van Haren SD, Herczenik E, Kaijen P, Ruminska A, Jin SY, et al. Modification of an exposed loop in the C1 domain reduces immune responses to factor VIII in hemophilia A mice. Blood. (2012) 119:5294–300. doi: 10.1182/blood-2011-11-391680
57. Gangadharan B, Ing M, Delignat S, Peyron I, Teyssandier M, Kaveri SV, et al. The C1 and C2 domains of blood coagulation factor VIII mediate its endocytosis by dendritic cells. Haematologica. (2017) 102:271–81. doi: 10.3324/haematol.2016.148502
58. Rayes J, Ing M, Delignat S, Peyron I, Gilardin L, Vogel CW, et al. Complement C3 is a novel modulator of the anti-factor VIII immune response. Haematologica. (2018) 103:351–60. doi: 10.3324/haematol.2017.165720
59. Dasgupta S, Repesse Y, Bayry J, Navarrete AM, Wootla B, Delignat S, et al. VWF protects FVIII from endocytosis by dendritic cells and subsequent presentation to immune effectors. Blood. (2007) 109:610–2. doi: 10.1182/blood-2006-05-022756
60. Sorvillo N, Hartholt RB, Bloem E, Sedek M, Ten Brinke A, Van Der Zwaan C, et al. von Willebrand factor binds to the surface of dendritic cells and modulates peptide presentation of factor VIII. Haematologica. (2016) 101:309–18. doi: 10.3324/haematol.2015.137067
61. Hartholt RB, Peyron I, Voorberg J. Hunting down factor VIII in the immunopeptidome. Cell Immunol. (2016) 301:59–64. doi: 10.1016/j.cellimm.2015.11.001
62. Jankowski W, Park Y, Mcgill J, Maraskovsky E, Hofmann M, Diego VP, et al. Peptides identified on monocyte-derived dendritic cells: a marker for clinical immunogenicity to FVIII products. Blood Adv. (2019) 3:1429–40. doi: 10.1182/bloodadvances.2018030452
63. Van Haren SD, Herczenik E, Ten Brinke A, Mertens K, Voorberg J, Meijer AB. HLA-DR-presented peptide repertoires derived from human monocyte-derived dendritic cells pulsed with blood coagulation factor VIII. Mol Cell Proteomics. (2011) 10:M110 002246. doi: 10.1074/mcp.M110.002246
64. Van Haren SD, Wroblewska A, Herczenik E, Kaijen PH, Ruminska A, Ten Brinke A, et al. Limited promiscuity of HLA-DRB1 presented peptides derived of blood coagulation factor VIII. PLoS ONE. (2013) 8:e80239. doi: 10.1371/journal.pone.0080239
65. Peyron I, Hartholt RB, Pedro-Cos L, Van Alphen F, Brinke AT, Lardy N, et al. Comparative profiling of HLA-DR and HLA-DQ associated factor VIII peptides presented by monocyte-derived dendritic cells. Haematologica. (2018) 103:172–8. doi: 10.3324/haematol.2017.175083
66. Hay CR, Ollier W, Pepper L, Cumming A, Keeney S, Goodeve AC, et al. HLA class II profile: a weak determinant of factor VIII inhibitor development in severe haemophilia A. UKHCDO Inhibitor Working Party. Thromb Haemost. (1997) 77:234–7. doi: 10.1055/s-0038-1655944
67. Oldenburg J, Picard JK, Schwaab R, Brackmann HH, Tuddenham EG, Simpson E. HLA genotype of patients with severe haemophilia A due to intron 22 inversion with and without inhibitors of factor VIII. Thromb Haemost. (1997) 77:238–42. doi: 10.1055/s-0038-1655945
68. Pavlova A, Delev D, Lacroix-Desmazes S, Schwaab R, Mende M, Fimmers R, et al. Impact of polymorphisms of the major histocompatibility complex class II, interleukin-10, tumor necrosis factor-alpha and cytotoxic T-lymphocyte antigen-4 genes on inhibitor development in severe hemophilia A. J Thromb Haemost. (2009) 7:2006–15. doi: 10.1111/j.1538-7836.2009.03636.x
69. Gunasekera D, Ettinger RA, Nakaya Fletcher S, James EA, Liu M, Barrett JC, et al. Factor VIII gene variants and inhibitor risk in African American hemophilia A patients. Blood. (2015) 126:895–904. doi: 10.1182/blood-2014-09-599365
70. Smith NL, Chen MH, Dehghan A, Strachan DP, Basu S, Soranzo N, et al. Novel associations of multiple genetic loci with plasma levels of factor VII, factor VIII, and von Willebrand factor: the CHARGE. (Cohorts for Heart and Aging Research in Genome Epidemiology) Consortium. Circulation. (2010) 121:1382–92. doi: 10.1161/CIRCULATIONAHA.109.869156
71. Schillemans M, Karampini E, Kat M, Bierings R. Exocytosis of Weibel-Palade bodies: how to unpack a vascular emergency kit. J Thromb Haemost. (2019) 17:6–18. doi: 10.1111/jth.14322
72. Zhu Q, Yamakuchi M, Ture S, De La Luz Garcia-Hernandez M, Ko KA, Modjeski KL, et al. Syntaxin-binding protein STXBP5 inhibits endothelial exocytosis and promotes platelet secretion. J Clin Invest. (2014) 124:4503–16. doi: 10.1172/JCI71245
73. Rydz N, Swystun LL, Notley C, Paterson AD, Riches JJ, Sponagle K, et al. The C-type lectin receptor CLEC4M binds, internalizes, and clears von Willebrand factor and contributes to the variation in plasma von Willebrand factor levels. Blood. (2013) 121:5228–37. doi: 10.1182/blood-2012-10-457507
74. Swystun LL, Notley C, Georgescu I, Lai JD, Nesbitt K, James PD, et al. The endothelial lectin clearance receptor CLEC4M binds and internalizes factor VIII in a VWF-dependent and independent manner. J Thromb Haemost. (2019) 17:681–94. doi: 10.1111/jth.14404
75. Fahs SA, Hille MT, Shi Q, Weiler H, Montgomery RR. A conditional knockout mouse model reveals endothelial cells as the principal and possibly exclusive source of plasma factor VIII. Blood. (2014) 123:3706–13. doi: 10.1182/blood-2014-02-555151
76. Everett LA, Cleuren AC, Khoriaty RN, Ginsburg D. Murine coagulation factor VIII is synthesized in endothelial cells. Blood. (2014) 123:3697–705. doi: 10.1182/blood-2014-02-554501
77. Bray GL, Kroner BL, Arkin S, Aledort LW, Hilgartner MW, Eyster ME, et al. Loss of high-responder inhibitors in patients with severe hemophilia A and human immunodeficiency virus type 1 infection: a report from the Multi-Center Hemophilia Cohort Study. Am J Hematol. (1993) 42:375–9. doi: 10.1002/ajh.2830420408
78. Qian J, Collins M, Sharpe AH, Hoyer LW. Prevention and treatment of factor VIII inhibitors in murine hemophilia A. Blood. (2000) 95:1324–9. doi: 10.1182/blood.V95.4.1324.004k25_1324_1329
79. Qian J, Burkly LC, Smith EP, Ferrant JL, Hoyer LW, Scott DW, et al. Role of CD154 in the secondary immune response: the reduction of pre-existing splenic germinal centers and anti-factor VIII inhibitor titer. Eur J Immunol. (2000) 30:2548–54. doi: 10.1002/1521-4141(200009)30:9<2548::AID-IMMU2548>3.0.CO;2-H
80. Pratt KP, Qian J, Ellaban E, Okita DK, Diethelm-Okita BM, Conti-Fine B, et al. Immunodominant T-cell epitopes in the factor VIII C2 domain are located within an inhibitory antibody binding site. Thromb Haemost. (2004) 92:522–8. doi: 10.1160/TH03-12-0755
81. Hausl C, Ahmad RU, Schwarz HP, Muchitsch EM, Turecek PL, Dorner F, et al. Preventing restimulation of memory B cells in hemophilia A: a potential new strategy for the treatment of antibody-dependent immune disorders. Blood. (2004) 104:115–22. doi: 10.1182/blood-2003-07-2456
82. Reding MT, Wu H, Krampf M, Okita DK, Diethelm-Okita BM, Key NS, et al. CD4+ T cell response to factor VIII in hemophilia A, acquired hemophilia, and healthy subjects. Thromb Haemost. (1999) 82:509–15. doi: 10.1055/s-0037-1615873
83. Reding MT, Wu H, Krampf M, Okita DK, Diethelm-Okita BM, Christie BA, et al. Sensitization of CD4+ T cells to coagulation factor VIII: response in congenital and acquired hemophilia patients and in healthy subjects. Thromb Haemost. (2000) 84:643–52. doi: 10.1055/s-0037-1614081
84. Reding MT, Okita DK, Diethelm-Okita BM, Anderson TA, Conti-Fine BM. Human CD4+ T-cell epitope repertoire on the C2 domain of coagulation factor VIII. J Thromb Haemost. (2003) 1:1777–84. doi: 10.1046/j.1538-7836.2003.00251.x
85. Reding MT, Okita DK, Diethelm-Okita BM, Anderson TA, Conti-Fine BM. Epitope repertoire of human CD4+ T cells on the A3 domain of coagulation factor VIII. J Thromb Haemost. (2004) 2:1385–94. doi: 10.1111/j.1538-7836.2004.00850.x
86. Hu GL, Okita DK, Conti-Fine BM. T cell recognition of the A2 domain of coagulation factor VIII in hemophilia patients and healthy subjects. J Thromb Haemost. (2004) 2:1908–17. doi: 10.1111/j.1538-7836.2004.00918.x
87. Jacquemin M, Vantomme V, Buhot C, Lavend'homme R, Burny W, Demotte N, et al. CD4+ T-cell clones specific for wild-type factor VIII: a molecular mechanism responsible for a higher incidence of inhibitor formation in mild/moderate hemophilia A. Blood. (2003) 101:1351–8. doi: 10.1182/blood-2002-05-1369
88. James EA, Kwok WW, Ettinger RA, Thompson AR, Pratt KP. T-cell responses over time in a mild hemophilia A inhibitor subject: epitope identification and transient immunogenicity of the corresponding self-peptide. J Thromb Haemost. (2007) 5:2399–407. doi: 10.1111/j.1538-7836.2007.02762.x
89. Ettinger RA, James EA, Kwok WW, Thompson AR, Pratt KP. Lineages of human T-cell clones, including T helper 17/T helper 1 cells, isolated at different stages of anti-factor VIII immune responses. Blood. (2009) 114:1423–8. doi: 10.1182/blood-2009-01-200725
90. Ettinger RA, James EA, Kwok WW, Thompson AR, Pratt KP. HLA-DR-restricted T-cell responses to factor VIII epitopes in a mild haemophilia A family with missense substitution A2201P. Haemophilia. (2010) 16:44–55. doi: 10.1111/j.1365-2516.2008.01905.x
91. James EA, Van Haren SD, Ettinger RA, Fijnvandraat K, Liberman JA, Kwok WW, et al. T-cell responses in two unrelated hemophilia A inhibitor subjects include an epitope at the factor VIII R593C missense site. J Thromb Haemost. (2011) 9:689–99. doi: 10.1111/j.1538-7836.2011.04202.x
92. Ettinger RA, Paz P, James EA, Gunasekera D, Aswad F, Thompson AR, et al. T cells from hemophilia A subjects recognize the same HLA-restricted FVIII epitope with a narrow TCR repertoire. Blood. (2016) 128:2043–54. doi: 10.1182/blood-2015-11-682468
93. Kamate C, Lenting PJ, Van Den Berg HM, Mutis T. Depletion of CD4+/CD25high regulatory T cells may enhance or uncover factor VIII-specific T-cell responses in healthy individuals. J Thromb Haemost. (2007) 5:611–3. doi: 10.1111/j.1538-7836.2007.02336.x
94. Hu GL, Okita DK, Diethelm-Okita BM, Conti-Fine BM. Recognition of coagulation factor VIII by CD4+ T cells of healthy humans. J Thromb Haemost. (2003) 1:2159–66. doi: 10.1046/j.1538-7836.2003.00366.x
95. Hu G, Guo D, Key NS, Conti-Fine BM. Cytokine production by CD4+ T cells specific for coagulation factor VIII in healthy subjects and haemophilia A patients. Thromb Haemost. (2007) 97:788–94. doi: 10.1160/TH06-09-0519
96. Meunier S, Menier C, Marcon E, Lacroix-Desmazes S, Maillere B. CD4 T cells specific for factor VIII are present at high frequency in healthy donors and comprise naive and memory cells. Blood Adv. (2017) 1:1842–7. doi: 10.1182/bloodadvances.2017008706
97. Karim AF, Vir P, Gunasekera D, Stering AI, Lieuw K, Pratt KP. Elispots and ex vivo expansion of FVIII-specific T-cell clones confirm specificity of CD4 + T-cell responses to self-FVIII in healthy non-hemophilic blood donors. Blood. (2019) 134(S1). doi: 10.1182/blood-2019-124803
98. Skupsky J, Zhang AH, Su Y, Scott DW. A role for thrombin in the initiation of the immune response to therapeutic factor VIII. Blood. (2009) 114:4741–8. doi: 10.1182/blood-2008-10-186452
99. Meeks SL, Cox CL, Healey JF, Parker ET, Doshi BS, Gangadharan B, et al. A major determinant of the immunogenicity of factor VIII in a murine model is independent of its procoagulant function. Blood. (2012) 120:2512–20. doi: 10.1182/blood-2012-02-412361
100. Tagariello G, Iorio A, Matino D, Belvini D, Salviato R, Sartori R, et al. High rate of spontaneous inhibitor clearance during the long term observation study of a single cohort of 524 haemophilia A patients not undergoing immunotolerance. J Hematol Oncol. (2013) 6:63. doi: 10.1186/1756-8722-6-63
101. Lacroix-Desmazes S, Maillere B. Monoepitopic anti-FVIII T-cell response. Blood. (2016) 128:1999–2001. doi: 10.1182/blood-2016-08-732123
102. Gunasekera D, Karim AF, Ragni MV, Pratt KP. Hemophilia A subjects with an intron-22 inversion mutation show robust T-cell responses to epitopes in both the FVIII heavy and light chain. Spec Compil Res Pract Thromb Haemost. (2018) 93. doi: 10.1002/rth2.12125
103. Martinez RJ, Evavold BD. Lower affinity T cells are critical components and active participants of the immune response. Front Immunol. (2015) 6:468. doi: 10.3389/fimmu.2015.00468
104. Billeskov R, Wang Y, Solaymani-Mohammadi S, Frey B, Kulkarni S, Andersen P, et al. Low antigen dose in adjuvant-based vaccination selectively induces CD4 T cells with enhanced functional avidity and protective efficacy. J Immunol. (2017) 198:3494–506. doi: 10.4049/jimmunol.1600965
105. Miller ML, Mcintosh CM, Williams JB, Wang Y, Hollinger MK, Isaad NJ, et al. Distinct graft-specific TCR avidity profiles during acute rejection and tolerance. Cell Rep. (2018) 24:2112–26. doi: 10.1016/j.celrep.2018.07.067
106. Miao CH, Harmeling BR, Ziegler SF, Yen BC, Torgerson T, Chen L, et al. CD4+FOXP3+ regulatory T cells confer long-term regulation of factor VIII-specific immune responses in plasmid-mediated gene therapy-treated hemophilia mice. Blood. (2009) 114:4034–44. doi: 10.1182/blood-2009-06-228155
107. Bayry J, Lacroix-Desmazes S, Dasgupta S, Kazatchkine MD, Kaveri SV. Efficacy of regulatory T-cell immunotherapy: are inflammatory cytokines key determinants? Nat Rev Immunol. (2008) 8:86. doi: 10.1038/nri2138-c1
108. Merle NS, Church SE, Fremeaux-Bacchi V, Roumenina LT. Complement system part i - molecular mechanisms of activation and regulation. Front Immunol. (2015) 6:262. doi: 10.3389/fimmu.2015.00262
109. Merle NS, Noe R, Halbwachs-Mecarelli L, Fremeaux-Bacchi V, Roumenina LT. Complement system part ii: role in immunity. Front Immunol. (2015) 6:257. doi: 10.3389/fimmu.2015.00257
110. Bexborn F, Andersson PO, Chen H, Nilsson B, Ekdahl KN. The tick-over theory revisited: formation and regulation of the soluble alternative complement C3 convertase. (C3(H2O)Bb). Mol Immunol. (2008) 45:2370–9. doi: 10.1016/j.molimm.2007.11.003
111. Zewde N, Gorham RD Jr, Dorado A, Morikis D. Quantitative modeling of the alternative pathway of the complement system. PLoS ONE. (2016) 11:e0152337. doi: 10.1371/journal.pone.0152337
112. Ballanti E, Perricone C, Greco E, Ballanti M, Di Muzio G, Chimenti MS, et al. Complement and autoimmunity. Immunol Res. (2013) 56:477–91. doi: 10.1007/s12026-013-8422-y
113. Fritzinger DC, Hew BE, Thorne M, Pangburn MK, Janssen BJ, Gros P, et al. Functional characterization of human C3/cobra venom factor hybrid proteins for therapeutic complement depletion. Dev Comp Immunol. (2009) 33:105–16. doi: 10.1016/j.dci.2008.07.006
114. Delignat S, Rayes J, Russick J, Kaveri SV, Lacroix-Desmazes S, Consortium A. Inhibitor formation in congenital hemophilia A: an immunological perspective. Semin Thromb Hemost. (2018) 44:517–30. doi: 10.1055/s-0038-1657777
115. Alom-Ruiz SP, Anilkumar N, Shah AM. Reactive oxygen species and endothelial activation. Antioxid Redox Signal. (2008) 10:1089–100. doi: 10.1089/ars.2007.2007
116. Hermeling S, Crommelin DJ, Schellekens H, Jiskoot W. Structure-immunogenicity relationships of therapeutic proteins. Pharm Res. (2004) 21:897–903. doi: 10.1023/B:PHAM.0000029275.41323.a6
117. Weiskopf D, Schwanninger A, Weinberger B, Almanzar G, Parson W, Buus S, et al. Oxidative stress can alter the antigenicity of immunodominant peptides. J Leukoc Biol. (2010) 87:165–72. doi: 10.1189/jlb.0209065
118. Trujillo JA, Croft NP, Dudek NL, Channappanavar R, Theodossis A, Webb AI, et al. The cellular redox environment alters antigen presentation. J Biol Chem. (2014) 289:27979–91. doi: 10.1074/jbc.M114.573402
119. Osterberg T, Fatouros A, Neidhardt E, Warne N, Mikaelsson M. B-domain deleted recombinant factor VIII formulation and stability. Semin Hematol. (2001) 38:40–3. doi: 10.1016/S0037-1963(01)90107-6
120. Stief TW, Kurz J, Doss MO, Fareed J. Singlet oxygen inactivates fibrinogen, factor V, factor VIII, factor X, and platelet aggregation of human blood. Thromb Res. (2000) 97:473–80. doi: 10.1016/S0049-3848(99)00211-X
121. Peyron I, Dimitrov JD, Delignat S, Gangadharan B, Srivastava A, Kaveri SV, et al. Oxidation of factor VIII increases its immunogenicity in mice with severe hemophilia A. Cell Immunol. (2018) 325:64–8. doi: 10.1016/j.cellimm.2018.01.008
122. Repesse Y, Dimitrov JD, Peyron I, Farrokhi Moshai E, Kiger L, Dasgupta S, et al. Heme binds to factor VIII and inhibits its interaction with activated factor IX. J Thromb Haemost. (2012) 10:1062–71. doi: 10.1111/j.1538-7836.2012.04724.x
123. Peyvandi F, Mannucci PM, Garagiola I, El-Beshlawy A, Elalfy M, Ramanan V, et al. A randomized trial of factor VIII and neutralizing antibodies in hemophilia, A. N Engl J Med. (2016) 374:2054–64. doi: 10.1056/NEJMoa1516437
124. Collins PW, Palmer BP, Chalmers EA, Hart DP, Liesner R, Rangarajan S, et al. Factor VIII brand and the incidence of factor VIII inhibitors in previously untreated UK children with severe hemophilia A, 2000-2011. Blood. (2014) 124:3389–97. doi: 10.1182/blood-2014-07-580498
125. Gouw SC, Van Der Bom JG, Ljung R, Escuriola C, Cid AR, Claeyssens-Donadel S, et al. Factor VIII products and inhibitor development in severe hemophilia A. N Engl J Med. (2013) 368:231–9. doi: 10.1056/NEJMoa1208024
126. Calvez T, Chambost H, Claeyssens-Donadel S, D'oiron R, Goulet V, Guillet B, et al. Recombinant factor VIII products and inhibitor development in previously untreated boys with severe hemophilia A. Blood. (2014) 124:3398–408. doi: 10.1182/blood-2014-07-586347
127. Calvez T, Chambost H, D'oiron R, Dalibard V, Demiguel V, Doncarli A, et al. Analyses of the FranceCoag cohort support differences in immunogenicity among one plasma-derived and two recombinant factor VIII brands in boys with severe hemophilia A. Haematologica. (2018) 103:179–89. doi: 10.3324/haematol.2017.174706
128. Volkers P, Hanschmann KM, Calvez T, Chambost H, Collins PW, Demiguel V, et al. Recombinant factor VIII products and inhibitor development in previously untreated patients with severe haemophilia A: combined analysis of three studies. Haemophilia. (2019) 25:398–407. doi: 10.1111/hae.13747
129. Lai JD, Swystun LL, Cartier D, Nesbitt K, Zhang C, Hough C, et al. N-linked glycosylation modulates the immunogenicity of recombinant human factor VIII in hemophilia A mice. Haematologica. (2018) 103:1925–36. doi: 10.3324/haematol.2018.188219
130. Thaiss CA, Zmora N, Levy M, Elinav E. The microbiome and innate immunity. Nature. (2016) 535:65–74. doi: 10.1038/nature18847
131. Honda K, Littman DR. The microbiota in adaptive immune homeostasis and disease. Nature. (2016) 535:75–84. doi: 10.1038/nature18848
132. Levy M, Kolodziejczyk AA, Thaiss CA, Elinav E. Dysbiosis and the immune system. Nat Rev Immunol. (2017) 17:219–32. doi: 10.1038/nri.2017.7
133. Haase S, Haghikia A, Wilck N, Muller DN, Linker RA. Impacts of microbiome metabolites on immune regulation and autoimmunity. Immunology. (2018) 154:230–8. doi: 10.1111/imm.12933
134. Milani C, Duranti S, Bottacini F, Casey E, Turroni F, Mahony J, et al. The first microbial colonizers of the human gut: composition, activities, and health implications of the infant gut microbiota. Microbiol Mol Biol Rev. (2017) 81: e00036–17. doi: 10.1128/MMBR.00036-17
135. Cormier MC, Tarrant J, Nesbitt K, Dwyer C, Hough C, Lillicrap D. Dysbiotic gut microbiota diminishes short-chain fatty acid production and increases factor VIII immunogenicity in haemophilia A mice. Res Pract Thromb Haemost. (2019) 3:111. doi: 10.1002/rth2.12227
136. Wambre E, Jeong D. Oral tolerance development and maintenance. Immunol Allergy Clin North Am. (2018) 38:27–37. doi: 10.1016/j.iac.2017.09.003
137. Habicht GS, Chiller JM, Weigle WO. Termination of aquired and natural immunological tolerance with specific complexes. J Exp Med. (1975) 142:312–20. doi: 10.1084/jem.142.2.312
138. Borel Y. Induction of immunological tolerance by a hapten. (DNP) bound to a non-immunogenic protein carrier. Nat New Biol. (1971) 230:180–2. doi: 10.1038/newbio230180a0
139. Sanfilippo F, Cohn ML, Scott DW. Effect of carrier-specific tolerance on the generation of helper cell function. Eur J Immunol. (1976) 6:742–6. doi: 10.1002/eji.1830061016
140. Zambidis ET, Scott DW. Epitope-specific tolerance induction with an engineered immunoglobulin. Proc Natl Acad Sci USA. (1996) 93:5019–24. doi: 10.1073/pnas.93.10.5019
141. Bijsterbosch MK, Klaus GG. Crosslinking of surface immunoglobulin and Fc receptors on B lymphocytes inhibits stimulation of inositol phospholipid breakdown via the antigen receptors. J Exp Med. (1985) 162:1825–36. doi: 10.1084/jem.162.6.1825
142. Waldschmidt TJ, Borel Y, Vitetta ES. The use of haptenated immunoglobulins to induce B cell tolerance in vitro. The roles of hapten density and the Fc portion of the immunoglobulin carrier. J Immunol. (1983) 131:2204–9.
143. Rath T, Kuo TT, Baker K, Qiao SW, Kobayashi K, Yoshida M, et al. The immunologic functions of the neonatal Fc receptor for IgG. J Clin Immunol. (2013) 33 (Suppl. 1):S9–17. doi: 10.1007/s10875-012-9768-y
144. Krishnamoorthy S, Liu T, Drager D, Patarroyo-White S, Chhabra ES, Peters R, et al. Recombinant factor VIII Fc. (rFVIIIFc) fusion protein reduces immunogenicity and induces tolerance in hemophilia A mice. Cell Immunol. (2016) 301:30–9. doi: 10.1016/j.cellimm.2015.12.008
145. Groomes CL, Gianferante DM, Crouch GD, Parekh DS, Scott DW, Lieuw K. Reduction of factor VIII inhibitor titers during immune tolerance induction with recombinant factor VIII-Fc fusion protein. Pediatr Blood Cancer. (2016) 63:922–4. doi: 10.1002/pbc.25874
146. Malec LM, Journeycake J, Ragni MV. Extended half-life factor VIII for immune tolerance induction in haemophilia. Haemophilia. (2016) 22:e552–4. doi: 10.1111/hae.13064
147. Santagostino E, Young G, Carcao M, Mannucci PM, Halimeh S, Austin S. A contemporary look at FVIII inhibitor development: still a great influence on the evolution of hemophilia therapies. Expert Rev Hematol. (2018) 11:87–97. doi: 10.1080/17474086.2018.1419862
148. Blumberg RS, Lillicrap D, Ig GFITG. Tolerogenic properties of the Fc portion of IgG and its relevance to the treatment and management of hemophilia. Blood. (2018) 131:2205–14. doi: 10.1182/blood-2017-12-822908
149. Carcao M, Shapiro A, Staber JM, Hwang N, Druzgal C, Lieuw K, et al. Recombinant factor VIII Fc fusion protein for immune tolerance induction in patients with severe haemophilia A with inhibitors-A retrospective analysis. Haemophilia. (2018) 24:245–52. doi: 10.1111/hae.13413
150. Simister NE, Story CM, Chen HL, Hunt JS. An IgG-transporting Fc receptor expressed in the syncytiotrophoblast of human placenta. Eur J Immunol. (1996) 26:1527–31. doi: 10.1002/eji.1830260718
151. Grubb JH, Vogler C, Tan Y, Shah GN, Macrae AF, Sly WS. Infused Fc-tagged beta-glucuronidase crosses the placenta and produces clearance of storage in utero in mucopolysaccharidosis VII mice. Proc Natl Acad Sci USA. (2008) 105:8375–80. doi: 10.1073/pnas.0803715105
152. Mold JE, Michaelsson J, Burt TD, Muench MO, Beckerman KP, Busch MP, et al. Maternal alloantigens promote the development of tolerogenic fetal regulatory T cells in utero. Science. (2008) 322:1562–5. doi: 10.1126/science.1164511
153. Gupta N, Culina S, Meslier Y, Dimitrov J, Arnoult C, Delignat S, et al. Regulation of immune responses to protein therapeutics by transplacental induction of T cell tolerance. Sci Transl Med. (2015) 7:275ra21. doi: 10.1126/scitranslmed.aaa1957
154. Andre S, Meslier Y, Dimitrov JD, Repesse Y, Kaveri SV, Lacroix-Desmazes S, et al. A cellular viewpoint of anti-FVIII immune response in hemophilia A. Clin Rev Allergy Immunol. (2009) 37:105–13. doi: 10.1007/s12016-009-8117-2
155. Georgescu MT, Liu AW, Sponagle K, Hebert K, Drager D, Patarroyo-White S, et al. Factor VIII tolerance induction in haemophilia A mice via transplacental transfer of recombinant factor VIII Fc. J Thromb Haemost. (2015) 13(S2):68–9. doi: 10.1111/jth.12993
156. Culina S, Gupta N, Boisgard R, Afonso G, Gagnerault MC, Dimitrov J, et al. Materno-Fetal transfer of preproinsulin through the neonatal Fc receptor prevents autoimmune Diabetes. Diabetes. (2015) 64:3532–42. doi: 10.2337/db15-0024
157. Mannucci PM, Tuddenham EG. The hemophilias–from royal genes to gene therapy. N Engl J Med. (2001) 344:1773–9. doi: 10.1056/NEJM200106073442307
158. Mendelsohn NJ, Messinger YH, Rosenberg AS, Kishnani PS. Elimination of antibodies to recombinant enzyme in Pompe's disease. N Engl J Med. (2009) 360:194–5. doi: 10.1056/NEJMc0806809
160. Matzinger P. Tolerance, danger, and the extended family. Annu Rev Immunol. (1994) 12:991–1045. doi: 10.1146/annurev.iy.12.040194.005015
161. Lei TC, Su Y, Scott DW. Tolerance induction via a B-cell delivered gene therapy-based protocol: optimization and role of the Ig scaffold. Cell Immunol. (2005) 235:12–20. doi: 10.1016/j.cellimm.2005.06.007
162. Zambidis ET, Kurup A, Scott DW. Genetically transferred central and peripheral immune tolerance via retroviral-mediated expression of immunogenic epitopes in hematopoietic progenitors or peripheral B lymphocytes. Mol Med. (1997) 3:212–24. doi: 10.1007/BF03401674
163. Litzinger MT, Su Y, Lei TC, Soukhareva N, Scott DW. Mechanisms of gene therapy for tolerance: B7 signaling is required for peptide-IgG gene-transferred tolerance induction. J Immunol. (2005) 175:780–7. doi: 10.4049/jimmunol.175.2.780
164. Liang W, Karabekian Z, Mattapallil M, Xu Q, Viley AM, Caspi R, et al. B-cell delivered gene transfer of human S-Ag-Ig fusion protein protects from experimental autoimmune uveitis. Clin Immunol. (2006) 118:35–41. doi: 10.1016/j.clim.2005.08.007
165. Soukhareva N, Jiang Y, Scott DW. Treatment of diabetes in NOD mice by gene transfer of Ig-fusion proteins into B cells: role of T regulatory cells. Cell Immunol. (2006) 240:41–6. doi: 10.1016/j.cellimm.2006.06.004
166. Brunstein CG, Blazar BR, Miller JS, Cao Q, Hippen KL, Mckenna DH, et al. Adoptive transfer of umbilical cord blood-derived regulatory T cells and early viral reactivation. Biol Blood Marrow Transplant. (2013) 19:1271–3. doi: 10.1016/j.bbmt.2013.06.004
167. Smith BM, Lyle MJ, Chen AC, Miao CH. Antigen-specific in vitro expansion of factor VIII-specific regulatory T cells induces tolerance in hemophilia A mice. J Thromb Haemost. (2019). doi: 10.1111/jth.14659. [Epub ahead of print].
168. Herzog RW, Kuteyeva V, Saboungi R, Terhorst C, Biswas M. Reprogrammed CD4+ T cells that express FoxP3+ control inhibitory antibody formation in hemophilia A mice. Front Immunol. (2019) 10:274. doi: 10.3389/fimmu.2019.00274
169. Kim YC, Zhang AH, Su Y, Rieder SA, Rossi RJ, Ettinger RA, et al. Engineered antigen-specific human regulatory T cells: immunosuppression of FVIII-specific T- and B-cell responses. Blood. (2015) 125:1107–15. doi: 10.1182/blood-2014-04-566786
170. Yoon J, Schmidt A, Zhang AH, Konigs C, Kim YC, Scott DW. FVIII-specific human chimeric antigen receptor T-regulatory cells suppress T- and B-cell responses to FVIII. Blood. (2017) 129:238–45. doi: 10.1182/blood-2016-07-727834
171. Kim YC, Zhang AH, Yoon J, Culp WE, Lees JR, Wucherpfennig KW, et al. Engineered MBP-specific human Tregs ameliorate MOG-induced EAE through IL-2-triggered inhibition of effector T cells. J Autoimmun. (2018) 92:77–86. doi: 10.1016/j.jaut.2018.05.003
172. Zhang AH, Yoon J, Kim YC, Scott DW. Targeting antigen-specific B cells using antigen-expressing transduced regulatory T cells. J Immunol. (2018) 201:1434–41. doi: 10.4049/jimmunol.1701800
173. Parvathaneni K, Scott DW. Engineered FVIII-expressing cytotoxic T cells target and kill FVIII-specific B cells in vitro and in vivo. Blood Adv. (2018) 2:2332–40. doi: 10.1182/bloodadvances.2018018556
174. Kwon KC, Sherman A, Chang WJ, Kamesh A, Biswas M, Herzog RW, et al. Expression and assembly of largest foreign protein in chloroplasts: oral delivery of human FVIII made in lettuce chloroplasts robustly suppresses inhibitor formation in haemophilia A mice. Plant Biotechnol J. (2018) 16:1148–60. doi: 10.1111/pbi.12859
175. Herzog RW, Nichols TC, Su J, Zhang B, Sherman A, Merricks EP, et al. Oral tolerance induction in hemophilia B dogs fed with transplastomic lettuce. Mol Ther. (2017) 25:512–22. doi: 10.1016/j.ymthe.2016.11.009
176. Sherman A, Su J, Lin S, Wang X, Herzog RW, Daniell H. Suppression of inhibitor formation against FVIII in a murine model of hemophilia A by oral delivery of antigens bioencapsulated in plant cells. Blood. (2014) 124:1659–68. doi: 10.1182/blood-2013-10-528737
177. Maldonado RA, Lamothe RA, Ferrari JD, Zhang AH, Rossi RJ, Kolte PN, et al. Polymeric synthetic nanoparticles for the induction of antigen-specific immunological tolerance. Proc Natl Acad Sci USA. (2015) 112:E156–65. doi: 10.1073/pnas.1408686111
178. Weyand AC, Pipe SW. New therapies for hemophilia. Blood. (2019) 133:389–98. doi: 10.1182/blood-2018-08-872291
179. Shima M, Hanabusa H, Taki M, Matsushita T, Sato T, Fukutake K, et al. Factor VIII-mimetic function of humanized bispecific antibody in hemophilia, A. N Engl J Med. (2016) 374:2044–53. doi: 10.1056/NEJMoa1511769
180. Lenting PJ, Denis CV, Christophe OD. Emicizumab, a bispecific antibody recognizing coagulation factors IX and X: how does it actually compare to factor VIII? Blood. (2017) 130:2463–8. doi: 10.1182/blood-2017-08-801662
181. Makris M, Iorio A, Lenting PJ. Emicizumab and thrombosis: the story so far. J Thromb Haemost. (2019) 17:1269–72. doi: 10.1111/jth.14556
182. Chiller JM, Habicht GS, Weigle WO. Kinetic differences in unresponsiveness of thymus and bone marrow cells. Science. (1971) 171:813–5. doi: 10.1126/science.171.3973.813
183. Dunbar CE, High KA, Joung JK, Kohn DB, Ozawa K, Sadelain M. Gene therapy comes of age. Science. (2018) 359:eaan4672. doi: 10.1126/science.aan4672
184. Finn JD, Ozelo MC, Sabatino DE, Franck HW, Merricks EP, Crudele JM, et al. Eradication of neutralizing antibodies to factor VIII in canine hemophilia A after liver gene therapy. Blood. (2010) 116:5842–8. doi: 10.1182/blood-2010-06-288001
185. Loduca PA, Hoffman BE, Herzog RW. Hepatic gene transfer as a means of tolerance induction to transgene products. Curr Gene Ther. (2009) 9:104–14. doi: 10.2174/156652309787909490
186. Polderdijk SG, Adams TE, Ivanciu L, Camire RM, Baglin TP, Huntington JA. Design and characterization of an APC-specific serpin for the treatment of hemophilia. Blood. (2017) 129:105–13. doi: 10.1182/blood-2016-05-718635
187. Polderdijk SGI, Baglin TP, Huntington JA. Targeting activated protein C to treat hemophilia. Curr Opin Hematol. (2017) 24:446–52. doi: 10.1097/MOH.0000000000000364
188. Wagar LE, Difazio RM, Davis MM. Advanced model systems and tools for basic and translational human immunology. Genome Med. (2018) 10:73. doi: 10.1186/s13073-018-0584-8
189. Nicholas KJ, Greenplate AR, Flaherty DK, Matlock BK, Juan JS, Smith RM, et al. Multiparameter analysis of stimulated human peripheral blood mononuclear cells: a comparison of mass and fluorescence cytometry. Cytometry A. (2016) 89:271–80. doi: 10.1002/cyto.a.22799
190. Finak G, Langweiler M, Jaimes M, Malek M, Taghiyar J, Korin Y, et al. Standardizing flow cytometry immunophenotyping analysis from the human immunophenotyping consortium. Sci Rep. (2016) 6:20686. doi: 10.1038/srep20686
191. Simoni Y, Chng MHY, Li S, Fehlings M, Newell EW. Mass cytometry: a powerful tool for dissecting the immune landscape. Curr Opin Immunol. (2018) 51:187–96. doi: 10.1016/j.coi.2018.03.023
192. Dash P, Wang GC, Thomas PG. Single-cell analysis of T-cell receptor αβ repertoire. Methods Mol Biol. (2015) 1343:181–97. doi: 10.1007/978-1-4939-2963-4_15
193. Wong GK, Heather JM, Barmettler S, Cobbold M. Immune dysregulation in immunodeficiency disorders: the role of T-cell receptor sequencing. J Autoimmun. (2017) 80:1–9. doi: 10.1016/j.jaut.2017.04.002
194. Chaussabel D, Baldwin N. Democratizing systems immunology with modular transcriptional repertoire analyses. Nat Rev Immunol. (2014) 14:271–80. doi: 10.1038/nri3642
195. Stubbington MJT, Rozenblatt-Rosen O, Regev A, Teichmann SA. Single-cell transcriptomics to explore the immune system in health and disease. Science. (2017) 358:58–63. doi: 10.1126/science.aan6828
196. Fischer K, Ljung R, Platokouki H, Liesner R, Claeyssens S, Smink E, et al. Prospective observational cohort studies for studying rare diseases: the European PedNet Haemophilia Registry. Haemophilia. (2014) 20:e280–6. doi: 10.1111/hae.12448
Keywords: factor VIII, protein immunogenicity, hemophilia A, peripheral tolerance, immune tolerance induction, antigen presentation, T-cell engineering
Citation: Lacroix-Desmazes S, Voorberg J, Lillicrap D, Scott DW and Pratt KP (2020) Tolerating Factor VIII: Recent Progress. Front. Immunol. 10:2991. doi: 10.3389/fimmu.2019.02991
Received: 02 November 2019; Accepted: 05 December 2019;
Published: 10 January 2020.
Edited by:
Luis Graca, University of Lisbon, PortugalReviewed by:
Raymond John Steptoe, University of Queensland, AustraliaCopyright © 2020 Lacroix-Desmazes, Voorberg, Lillicrap, Scott and Pratt. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Kathleen P. Pratt, a2F0aGxlZW4ucHJhdHRAdXN1aHMuZWR1
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.