
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol. , 09 January 2020
Sec. Autoimmune and Autoinflammatory Disorders
Volume 10 - 2019 | https://doi.org/10.3389/fimmu.2019.02980
 Yan-Mei Huang1,2†Xue-Zhi Hong1,3Jian Shen1,4Li-Jun Geng1,2Yan-Hong Pan1,2Wei Ling1,5Hai-Lu Zhao1,2,6*
Yan-Mei Huang1,2†Xue-Zhi Hong1,3Jian Shen1,4Li-Jun Geng1,2Yan-Hong Pan1,2Wei Ling1,5Hai-Lu Zhao1,2,6*Amyloid deposition is a histological hallmark of common human disorders including Alzheimer's disease (AD) and type 2 diabetes. Although some reports highlight that amyloid fibrils might activate the innate immunity system via pattern recognition receptors, here, we provide multiple lines of evidence for the protection by site-specific amyloid protein analogs and fibrils against autoimmune attacks: (1) strategies targeting clearance of the AD-related brain amyloid plaque induce high risk of deadly autoimmune destructions in subjects with cognitive dysfunction; (2) administration of amyloidogenic peptides with either full length or core hexapeptide structure consistently ameliorates signs of experimental autoimmune encephalomyelitis; (3) experimental autoimmune encephalomyelitis is exacerbated following genetic deletion of amyloid precursor proteins; (4) absence of islet amyloid coexists with T-cell-mediated insulitis in autoimmune diabetes and autoimmune polyendocrine syndrome; (5) use of islet amyloid polypeptide agonists rather than antagonists improves diabetes care; and (6) common suppressive signaling pathways by regulatory T cells are activated in both local and systemic amyloidosis. These findings indicate dual modulation activity mediated by amyloid protein monomers, oligomers, and fibrils to maintain immune homeostasis. The protection from autoimmune destruction by amyloid proteins offers a novel therapeutic approach to regenerative medicine for common degenerative diseases.
Amyloids refer to misfolding protein aggregates which convert from their soluble physiological monomers under certain endogenous or exogenous conditions (1, 2). The amyloid aggregates include soluble non-fibrillar intermediates, such as α-helix-rich oligomers and protofibrils, and insoluble deposits of β-sheet fibrils. Ever since amyloid was initially used to describe the specific microscopic abnormalities during autopsy examinations in 1639, the protein deposition has been recognized as a hallmark of a variety of disorders with disparate symptoms (3–6). At present, nearly 50 disorders are associated with the formation of extracellular amyloid fibrils or intracellular amyloid-like confirmations (7). Diseases associated with formation of extracellular amyloid deposits are designated as amyloidosis (8).
The innate immune mechanism induced by amyloid deposition plays a remarkable role in the pathogenesis of amyloidosis (9). In Alzheimer's disease (AD), microglia and astrocytes can recognize deposition of amyloid β (Aβ) via specialized pattern recognition receptors (PRRs), such as toll-like receptors (TLRs), nucleotide-binding oligomerization domain-like receptors (NLRs), complements, receptor for advanced glycation end product, and scavenger receptors (10). Ligation of CD36, TLR2, TLR4, and TLR6 leads to proinflammatory signal transduction (11, 12). While the proinflammatory cytokines derived from activated cells initially account for the phagocytosis of Aβ, the self-sustaining proinflammatory mediators elicit inducible isoform of nitric oxide (NO) synthase and local NO production to induce neuronal apoptosis, axonal and synaptic damage, and inhibition of mitochondrial respiration (11). Likewise, the interaction between Aβ deposition and chronic inflammation contributes to the malfunction and death of neurons and eventually leads to AD development and progression. Consistent with the roles of Aβ plaques in AD, type 2 diabetes (T2DM)-related islet amyloid polypeptide (IAPP) deposits (13, 14) in pancreatic islets also can activate the pyrin domain-containing 3 (NLRP3) inflammasome and generate mature proinflammatory cytokine interleukin (IL)-1β (15), resulting in β-cell dysfunction and insulin deficiency (16, 17).
However, all immunotherapies targeting the clearance of amyloid under the assumption of the amyloid pathogenesis have proven unsatisfactory (18–22). In this framework of these treatments, immunotherapy has been historically regarded as a promising approach. For instance, active anti-Aβ immunization AN-1792 in phase IIA trial provides evidence of amyloid amelioration, but fatal subacute aseptic meningoencephalitis occurred in vaccine recipients. The meningoencephalitis is autoimmune-related due to autoreactive CD4+T cells infiltrating after autopsy examination. More intriguingly, injection of Aβ peptide into experimental autoimmune encephalomyelitis (EAE) delays disease onset; conversely, EAE gets worse following genetic deletion of the amyloid precursor protein (APP) (23). Other amyloid-forming proteins consistently exhibit such therapeutic outcome with highly immune suppressive activity (24). Such improvement in EAE conferred by Aβ treatment, to some extent, indicates the potential protective role of amyloid in defending against autoimmunity.
Hence, we summarize amyloid researches pertaining to the roles of amyloids in immune regulation with particular emphasis on the protective role of autoimmunity.
The term amyloid was initially coined and popularized by Rudolph Virchow in 1854 to describe an abnormal change in the liver due to an iodine-staining reaction similar to that of starch. In fact, these “lardaceous” and “white stone” entities in other autopsy organs—consistent with the presence of amyloid—were described as early as in 1639 in autopsies by Nicolaus Fontanus. While there was no clear acknowledgment of the nature of amyloid between starch and cellulose until 1859, the absence of carbohydrate in a “mass” of amyloid and presence of highly proteinaceous species was established by Friedrich August Kekulé, who clarified that amyloid refers to a protein or a class of proteins (25).
Subsequent understanding of amyloid characteristics evolved with the development of more advanced techniques. The histochemical feature of binding Congo red with green birefringence was introduced by means of polarized light in the 1920s. In 1959, Cohen first identified characteristic elongated and unbranched fibrils, which differed from branching and thick collagen fibers under electron microscope (26). Furthermore, the biochemical feature of amyloid was identified in 1968 when Pras et al. solubilized and extracted insoluble amyloid fibrils (27). The two major types of breakdown subunits of amyloid fibrils were identified as amyloid light chain (AL) (28), derived from the light chain of an immunoglobulin and the other as amyloid A (AA) protein (29), which related to the primary and secondary form of the disease, respectively. Other common structures of amyloid fibrils include characteristic cross-β X-ray diffraction patterns, steric zippers of amyloid spine, and residues aligned in register (30–32).
Amyloid proteins mainly show three conformational structures: physiological monomers, pathological intermediates such α-helix-rich oligomers (e.g., dimers, trimers, dodecamers, and larger oligomers) and protofibrils, and eventually β-sheet amyloid fibrils. Intriguingly, amyloid fibrils with the common tinctorial properties and structural similarities are derived from non-homologous amyloid-forming proteins, which possess highly divergent sequence lists, secondary structure, and functions (Table 1). For example, some of the amyloidogenic proteins mainly display a high proportion of β-sheets; in other cases with α-helical and β-sheet mixture, even a rich α-helical structure is present. While during β-structure amyloid fibrils assembly, it has also been demonstrated that a similar kinetically intermediate state occurs—transient appearance of α-helical conformation (33–36). The phenomenon that physiologically disparate proteins from different sources convert to common structures of intermediates and fibrils remains an interesting mystery to explore. Furthermore, the terminal amyloid fibrils, beyond just the one core primary protein component, codeposit with common additional associated species, including metal ions, glycosaminoglycans, the serum amyloid P (SAP) component, apolipoprotein E, collagen, and many other minor components (2, 37, 38).
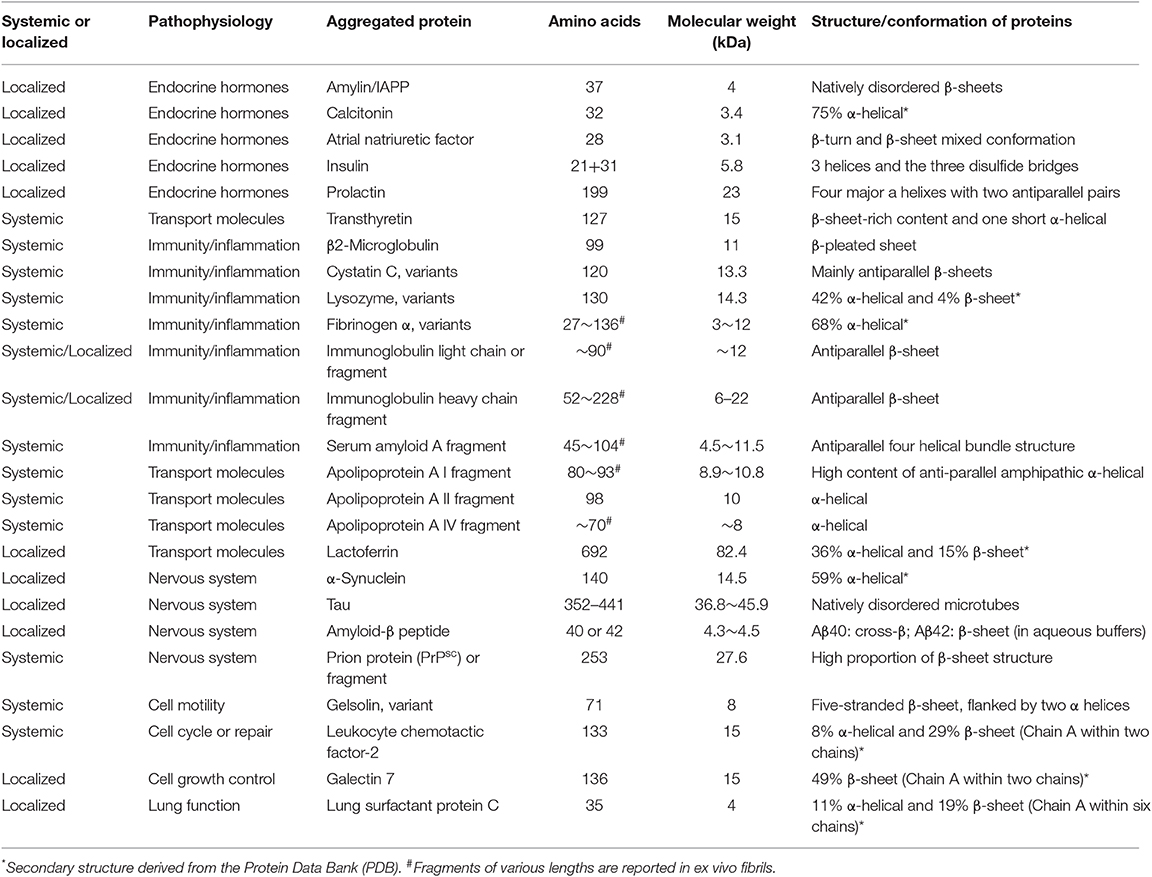
Table 1. Distinct structures of common amyloid proteins in clinical conditions.
Distinct native proteins in humans serve different biological functions in vivo, such as endocrine hormones, transport molecules, immunity response, and normal cell function control. Usually, protein aggregation has unique biophysical characteristics with dramatical impact on cell activity. The smaller species of amyloids such as oligomers and protofibrils are considered to be the most toxic form in the process of membrane perforation and cell degeneration.
Soluble Aβ oligomers have been shown to induce memory deficits and cognitive impairment in transgenic mice (39, 40). In addition, these purified soluble assemblies from brains of impaired Tg2576 mice disrupt memory when administered to young rats. The reduction in oligomer levels, on the other hand, corresponded to improved memory in these mice (41). Tg2576 mice with amyloid plaques did not show memory function and behavioral deficits during episodes of markedly reduced levels of Aβ oligomers. Moreover, these soluble oligomers were also toxic to cultured neuronal cells by inducing membrane depolarization (42–44). Similarly, IAPP oligomers induce insulin-producing β cell mass loss and apoptosis in vitro cell cultures and in transgenic mice (45, 46), consistent with the findings of oligomer-specific immunoreactivity and β cell depletion in islet cells of patients with T2DM (47). Such cell toxicity exhibited by oligomeric species have also been seen in other amyloid-related disorders including Parkinson's disease (α-synuclein), spongiform encephalopathies [prion protein (PrP)], Huntington's disease, and spinocerebellar ataxias (polyQ proteins) (43).
The toxicity of oligomers is not specific, and they interact with many targets, including membrane disruption interaction, mitochondrial dysfunction, oxidative stress, and reactive oxygen species production, suggesting that toxicity is associated with the formation process rather than a specific oligomeric species. It is generally assumed that toxicities of oligomers of different proteins are mediated by a common sequence-independent conformation, implying a common mechanism of pathogenesis of all the amyloidoses (48–50).
Instead of acting as an etiological agent, amyloid fibrils have three major disparate roles as described for differential amyloid deposition according to growing evidence (Table 2). First, the formation of amyloid fibrils does not necessarily denote causality with diseases for the following reasons: (1) weak correlation between Aβ deposits and cognitive status (18, 53, 54); (2) lack of correlation between loss neural function within the regions responsible for memory and the extent of Aβ deposits in that brain region (55–58); (3) oxidative stress precedes fibrillar depositions of Aβ (59–61); (4) amyloid fibrils are the product of the innate immune response (62–64); (5) Aβ plaques were identified in cognitively normal elderly people (65–67); (6) animals with Aβ deposition do not develop the clinical signs of the cognitive impairment (68); and (7) treatments targeting on Aβ plaques have been unsuccessful. Second, biophysical or functional amyloids have been described broadly from bacteria to humans (95–97), such as curli biogenesis (69–72), silkmoth chorion generation (73), melanin and other hormones synthesis (74, 75), epigenetic control of polyamines (76, 77), and other biological functions (78–81). Furthermore, to a far greater extent than anyone suspected, amyloid wields potential for protective roles (98), including neuroprotective action (82–85), defending against oxidative damage (86–89), prion (90, 91), and metal-induced toxicity (92–94), and protecting against microbial infection and autoimmune destruction.
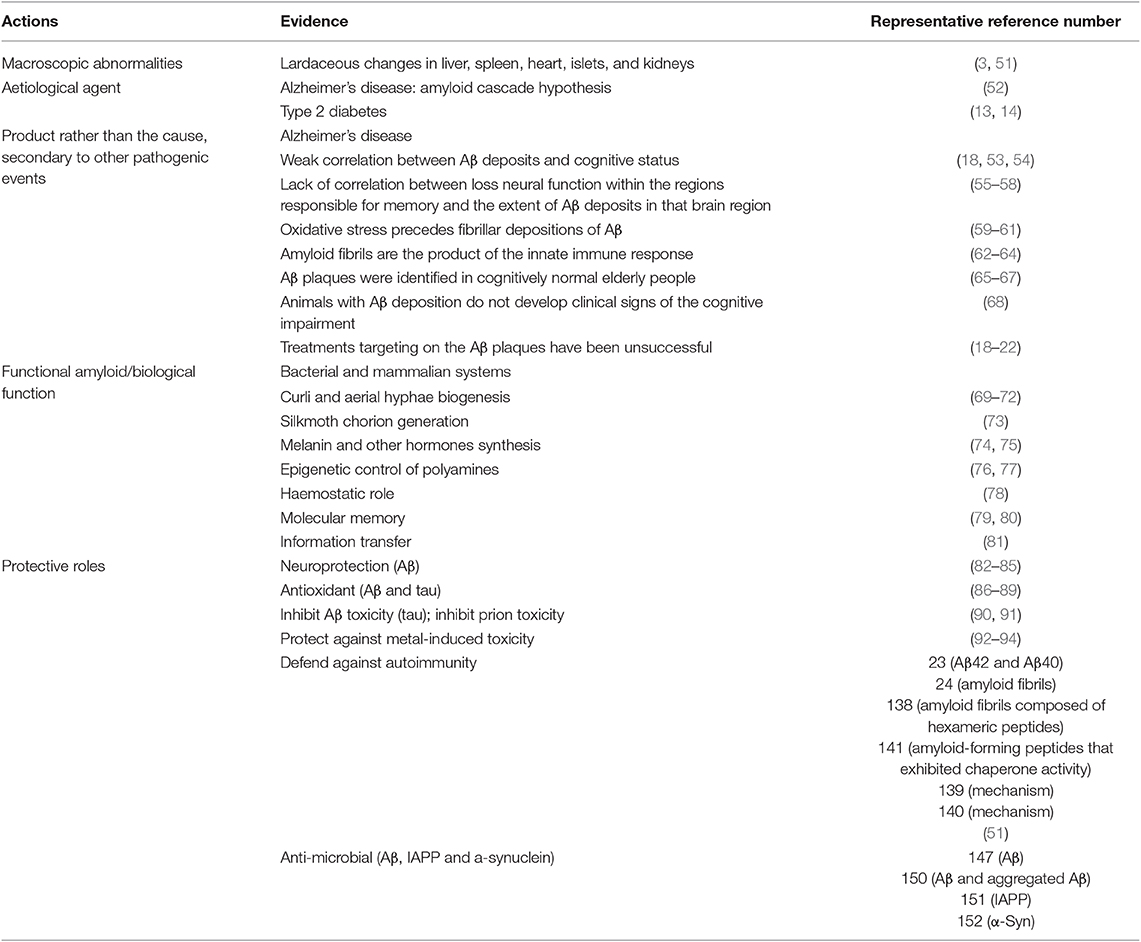
Table 2. Specific actions of amyloid protein and deposition denoted in research evidence.
Different amyloid-forming proteins are associated with different diseases. According to the International Society of Amyloidosis, there are 36 known extracellular amyloid fibril proteins associated with amyloidoses in humans, 2 of which are iatrogenic in nature and 9 of which have also been identified in animals (8).
The amyloidoses are classified as systemic or localized forms based on location and extent of amyloid protein buildup. Three common conditions associated with systemic amyloidosis are primary amyloidosis (also called AL), familial (hereditary) amyloidosis, and secondary amyloidosis (AA amyloidosis) such as tuberculosis or rheumatoid arthritis. Secondary amyloidosis, characterized by the deposition of serum amyloid A (SAA), occurs as a complication of an existing chronic infection or chronic inflammatory disease. Infections and inflammation stimulate human liver to produce high levels of SAA. With ongoing inflammation, a small portion of the SAA protein, called AA protein, might separate from SAA and deposit in tissues as AA amyloid. Although the mechanism of partial breakdown of SAA to AA is not well-understood, tuberculosis with chronic states of inflammation usually induces renal complications of SAA-induced amyloidosis. Besides, β2-microglobulin amyloidosis affects people who undergo long-term hemodialysis or continuous ambulatory peritoneal dialysis. T2DM and AD represent two common clinical conditions of localized amyloidosis. Another study broadly groups amyloidoses into neurodegenerative conditions, non-neuropathic localized amyloidosis, and non-neuropathic systemic amyloidosis (2). The chemical characterization of the precursor protein is also recommended as an approved classification criterion (8).
Some hypotheses have been put forward for the pathogenesis of amyloidoses, in which normally soluble proteins aggregate into regular, insoluble amyloid fibrils. Any methods that enhance the sources of amyloidogenic peptide and/or decrease clearance of amyloid-forming proteins contribute to amyloid formation and lead to amyloidoses eventually (99). Genetic variability of amyloidogenic protein as a negative factor leads to a vulnerable process of amyloid assembly. For instance, amyloid transthyretin amyloidosis is caused by mutations in the TTR gene (100, 101). Cleavage of the precursor protein contributes to an overproduction of the amyloidogenic protein and enhances the propensity for encouraging amyloid fibrils formation. One typical instance is that Alzheimer-related Aβ, which is cleaved by β-secretase and γ-secretase (102). It is also theoretically recognized that the “nucleated growth” mechanism is one of the characteristics of amyloid fibril formation. Once lag phase “nuclei” form, mature fibrils grow rapidly by association with various oligomers or monomers (2). In addition, intrinsic properties—referring to amino acid sequences and conformation states—and external conditions—such as pH, temperature and pressure—both serve as important factors to the susceptibility of a given polypeptide chain to convert into amyloid fibrils (103).
Extracellular deposits of Aβ protein forming senile plaques represents pathognomonic for AD. The triggering of immune reactions leads to inflammatory processes in the brain (Figure 1). The most influential model has been the “amyloid cascade hypothesis” proposed by Hardy nearly 30 years ago (52). The innate immune system, as the first line of defense against pathogen-associated molecular patterns or damage-associated molecular patterns, is triggered in response to sensing emerging Aβ plaques via the PRRs expressed in microglia and astrocytes. Activated microglia play an indispensable role in surveying the brain milieu by producing the proinflammatory cytokines IL-1, IL-6, and tumor necrosis factor alpha (TNF-α), and chemokines IL-8, macrophage inflammatory protein-1, monocyte chemoattractant peptide-1, the growth factor macrophage-colony stimulating factor and complement cascade proteins (104, 105). Reactive astrocytes also can secrete proinflammatory mediators such as monocyte chemoattractant peptide-1 (MCP-1), regulated on activation, normal T cell expressed and secreted (RANTES), TNF-α, and IL-1 in response to stimulation with Aβ42 (106). A wide array of PRRs in glial cells and neurons are implicated in Aβ hypothesis cascade, such as TLRs, NLRs, receptor for advanced glycation end products, complements, and scavenger receptors (11, 12, 107).
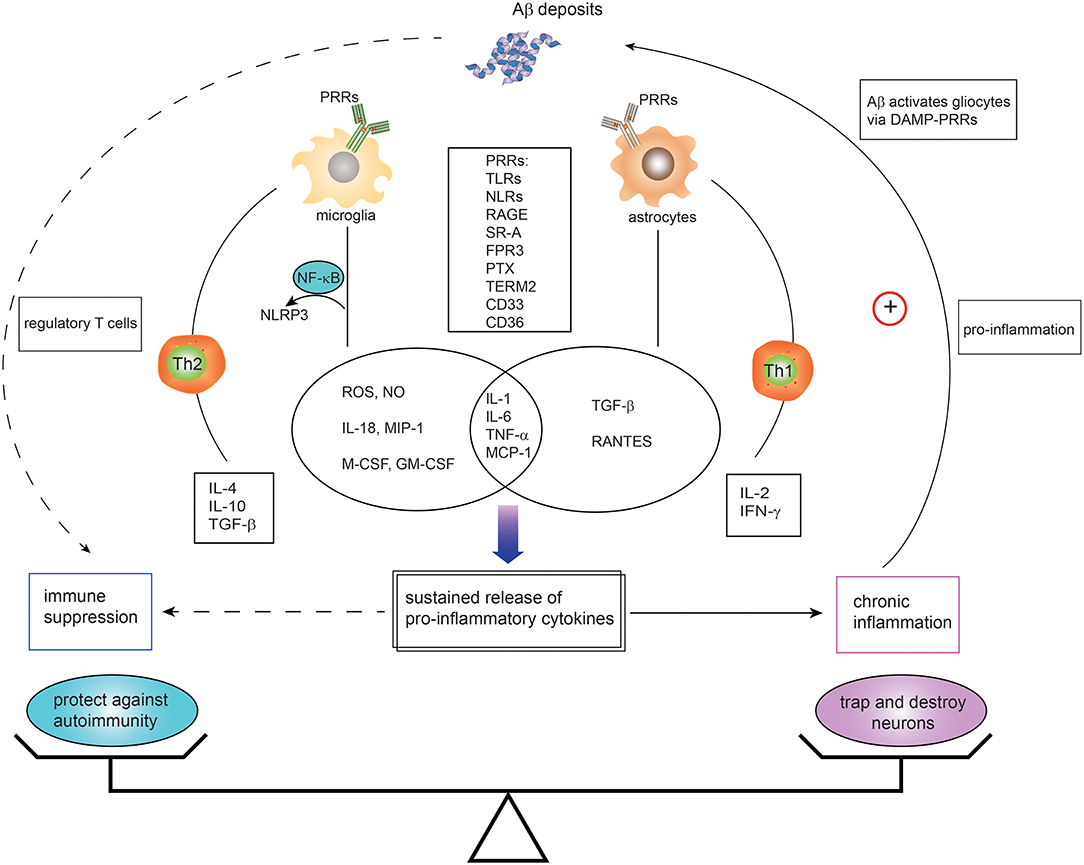
Figure 1. Dual immunomodulation of brain amyloid β deposits in the context of Alzheimer's disease. Microglia and astrocytes, expressing innate immune receptors—pattern recognition receptors (PRRs)—can be activated in response to amyloid-β (Aβ) via DAMPs-PRRs ligation. Such PRRs include toll-like receptors (TLRs), nucleotide-binding oligomerization domain-like receptors (NLRs), receptor for advanced glycation end products (RAGE), scavenger receptors (SRs), N-formyl peptide receptors (FPRs), pentraxin (PTX), triggering receptor expressed by myeloid cells 2 (TREM2), CD36, and CD33. The activation of microglia and astrocytes leads to secretion of proinflammatory cytokines and chemokines. Activated microglia with TLRs agonist or cytokines also activate NALP3 inflammasomes via nuclear factor kappa B (NF-κB) mediated signaling, resulting in production of proinflammatory mediators. While the proinflammatory cytokines derived from activated cells initially account for the phagocytosis of Aβ deposition, the self-sustaining proinflammatory mediators and chronic inflammation contribute to the malfunction and death of neurons and eventually leads to AD development. Likewise, the chronic neuroinflammation in turn hastens cycle reinforcing Aβ deposition. In addition, Th1 cells and Th2-polarized cells are activated with implication in proinflammatory and anti-inflammatory regulation. However, direct and indirect evidence is emerging that Aβ has immune suppressive activity and protects against autoimmune disorders. It is likely that the states and activities of brain Aβ might be orchestrated by a whole variety of different factors. TNF, tumor necrosis factor; TGF, transforming growth factor; IFN, interferon; DAMPs, damage-associated molecular patterns; TREM2, triggering receptor expressed by myeloid cells 2; IL, interleukin. G-CSF, granulocyte colony-stimulating factor; GM-CSF, granulocyte macrophage colony-stimulating factor; IFN-γ, interferon-gamma; M-CSF, macrophage colony stimulating factor; NLRP3, NOD-like receptor, type 3; NO, nitric oxide; ROS, reactive oxygen species; SRs, scavenger receptors; TLRs, Toll-like receptors; MIP-1, macrophage inflammatory protein 1; MCP-1, monocyte chemoattractant protein-1; RANTES, regulated on activation, normal T cell expressed and secreted.
In addition, insoluble fibrillar Aβ can activate the NALP3 inflammasome dependent on lysosomal damage in mouse brain phagocytic cells (108, 109), which results in the release of IL-1β and cathepsin B followed by caspase-1 activation and secretion of several proinflammatory and chemotactic mediators (110). Plasmacytoid dendritic cells as another innate immune cell population are potently activated by nucleic-acid-containing amyloid fibrils (111).
The activated signaling pathways in the early stage of AD play a protective role by phagocytosis and degradation of amyloid aggregates (9, 112). However, along with continued production of the amyloid clumps, chronic stimulation of the innate immune system leads to persistent inflammation. Such chronic inflammation results in serious disadvantages, such as immune-competent cells deprivation, more APP synthesis, and cellular dysfunction or death (113, 114). This is the dichotomous role of amyloid-induced inflammation. Therefore, since amyloid plaque usually present with chronic inflammation, a plethora of studies have generally connected amyloid with the pathogenesis process of AD (115–117).
Consistent with the role of Aβ in AD, T2DM-related IAPP deposits in islets of the pancreas can activate the NLRP3 inflammasome and generate mature proinflammatory cytokine IL-1β in transgenic mice model (15). The proposed mechanism underlying the release of proinflammatory cytokine is attributed to intraislet macrophages with increased expression of cell surface Ly6C and CD11c as induced by IAPP aggregates (118). In macrophage-prime cultured medium, IL-1β secretion is TLR2 dependent or NLRP3 dependent based on distinct IAPP aggregation states (119). Islet β cells are also a potential source of proinflammatory cytokines, which are associated with β-cell dysfunction in patients with T2DM (16, 17).
Indeed, other amyloidogenic peptides, including SAA (120, 121), show common activation pathways that trigger an innate immune response via binding multiple cell surface PRRs comprised of RIG-I-like receptor, NLR and TLR.
The common immunosuppressive regulation signaling pathways include stimulation of cytokines production and several TLRs, as well as lipopolysaccharide-induced tolerance of innate immunity system. The overexpressed pleiotropic cytokines IL-10, IL-6, and IL-1 (122–126) and increased CD33 expression (127, 128) within affected cerebral cortical regions of AD have anti-inflammatory and immunosuppressive properties. Variant triggering receptors expressed on myeloid cells 2 binding tyrosine kinase-binding protein expressed by microglia cells downregulate inflammatory signaling in response to TLR ligation (129, 130). Hickman et al. also reported that the inflammatory cascade initiator of receptor CD36 in microglia for Aβ binding displayed a major decrease in the AD transgenic mice (131). In addition, suppressors of cytokine signaling highly expressed in the astrocytes and microglia of AD patients are indicative of cytokine-JAK-STAT pathway inhibition, as well as adaptive and innate immune response downregulation (132, 133).
To explore the amyloid deposition mechanism that underlies multiple sclerosis-like brain inflammation known as EAE, in 2012, Steinman et al. injected synthetic Aβ40 or Aβ42 peptides peripherally into four different models of EAE (23). Unexpectedly, Aβ could neither cross the blood–brain barrier nor stimulate immune cells to attack neurons, whereas it relieved symptoms associated with EAE. To further explain the mechanism underlying Aβ-mediated suppression of EAE, the researchers measured more than 20 cytokines and chemokines in the blood of Aβ-treated mice and found overall dampening of proinflammatory signaling molecules, most notably IL-6, IL-2, transforming growth factor-β and IL-17, as well as key components of the well-known EAE-pathogenetic Th1 and Th17 pathways. In the periphery, treated mice exhibited an increased subset of lymphocytes effector T cells (CD4+CD62L+) that produced higher amounts of interferon-gamma (IFN-γ), which acted to suppress the immune system in EAE. Flow cytometry of immune cell subsets (T cells: CD4+ T cells, CD8+ T cells, CD4+CD62L+IFNγ + T cells, B cells: B1 and B2 subsets, and myeloid cells: neutrophils, mast cells, large peripheral macrophages, and small peripheral macrophages) revealed significant changes in the distribution and subtle expansion/depletion of immune subsets in the peritoneal cavity. Furthermore, in both mouse and human in vitro experiments, Aβ directly suppressed thymidine incorporation of activated EAE-specific CD4+ T cells. These in vitro experiments demonstrate that activated mouse and human CD4+ T cells are direct targets of Aβ immunosuppression. Thus, the collection of findings means that Aβ-induced amelioration of EAE may be related to production of immune suppressive cytokines, modulation of lymph node and myeloid compartments, and reduction in serum levels of proinflammatory signaling molecules.
Moreover, exacerbated clinical EAE occurs in mice with the genetic deletion of the AβPP (23). “Loss of function” experiments consolidate the observation with gene deletion of other amyloid-forming related molecules of PrP (134), tau (135), SAP (136), and molecular chaperone αB crystallin (Cryab). Subsequently, a core hexapeptide structure in amyloid-forming molecules was identified from a variety of notorious amyloid proteins including PrP, tau, Aβ, amylin, SAA, insulin, and molecular chaperone Cryab. Congruent with EAE protection, when these hexapeptides (Table 3) were given to engineered EAE mice with early clinical symptoms, they showed remarkable and consistent autoimmune therapeutic results (24). In addition, we have found that human IAPP could also induce CD4+CD25+Foxp3+ regulatory T cells and reduce the risk of autoimmune diabetes (51, 137). These findings collectively suggest localized autoimmune protection conferred by some kinds of amyloid forming proteins. More recent studies propose the benefits arising from several pathways of immune suppression, such as activating B-1a lymphocytes and F4/80+ macrophages, inducing a set of immune-suppressive cell-surface proteins (BTLA, IRF4, and Siglec G) and IL-10 (138). Two independent pathways are stimulated by amyloid fibrils, reduction in IL-6, TNF-α, and IFN-γ levels, and induction of type 1 IFN by plasmacytoid dendritic cells. These pathways act in concert to be immunosuppressive in Th1 indications (139, 140). Molecular chaperone activity and the capacity of binding proinflammatory proteins at the elevated temperatures within inflammatory foci are also correlated with the therapeutic activity of amyloid fibrils (24, 141). In addition, amyloid hexapeptide treatments improve the clinical outcomes of C57BL/6J (B6), but not B10 mice, indicating that genetic background might influence therapeutic efficacy (142).
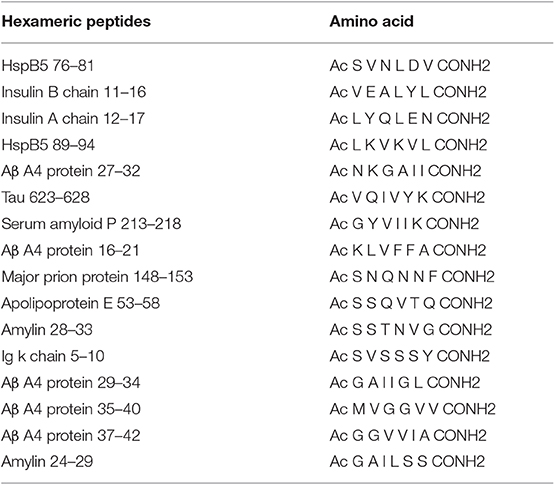
Table 3. Hexapeptide-containing amyloid fibrils with therapeutic potentials for experimental autoimmune encephalomyelitis.
Indeed, as early as 2007, Steinman et al. found Cryab can attenuate inflammation in several models of inflammation, including ongoing EAE, downregulate antigen-specific Th1 and Th17 responses, and impact key inflammatory pathways such as nuclear factor kappa B and p38MAPK (143, 144). As these results conflict with van Noort's findings of multiple sclerosis in 1995 (145), the hint of amyloid-forming peptides' therapeutic potential has not yet come into consideration. It was Tanaka et al. who reported that the protein worked as a chaperone when it formed amyloid fibrils (146) that the unexpected idea that the molecule might be functioning in an amyloid state first dawned on various research fields.
In vitro, synthetic Aβ exhibits potent antimicrobial activity toward microbial pathogens, fungi (147), and viruses (148, 149). The biological relevance of this protection by in vitro Aβ activities was validated in transgenic mice overexpressing human Aβ, demonstrating defense against meningitis. Intriguingly, Aβ aggregates entrap and imprison these pathogens (150). Particularly germane to Aβ's role in anti-inflammation, several other amyloidogenic peptides including IAPP (151), α-synuclein (152), serum amyloid A, microcin E492, temporins, and protegrin-1 also show antimicrobial properties (153).
In addition, Moir et al. propose that Aβ belongs to a family of antimicrobial proteins (AMPs) based on characteristics that Aβ shares with an AMP called LL-37 (147). Intriguingly, LL-37 plays the dual role of rousing inflammation in some scenarios and squelching it in others (154, 155). In this regard, the discovery implies that Aβ deposition appears to exert action in immune regulation. On the other hand, biophysical analysis showed an extensive β-sheet structure present in various species of bacterial fimbriae, suggesting that amyloids share common structural components of the extracellular matrix in bacterial biofilms (71, 156, 157). The established biofilms are stable communities and function as antimicrobials agents generally resistant to harsh denaturing. In addition, biofilms from both commensal and pathogenic microbiota recognized by TLR2 resulted in the production of an immunomodulatory cytokine of intestinal homeostasis that ameliorate inflammation in a mouse model of colitis (155, 158, 159). The amyloid fibrils with structural and functional similarity to the protective shell of bacterial biofilm implies that amyloid fibril structure exhibits beneficial behaviors in defending against autoimmune attacks.
The CD4+CD25+Foxp3+ regulatory T cells (Tregs) play a critical role in modulating the balance between inflammation and immune tolerance to prevent autoimmune diseases (160). In peripheral lymphocytes, the frequency and suppressive activity of Tregs are increased in patients with AD as compared to non-demented controls (161). Additional evidence reinforces the significant augmentation of the strongest suppressive subset of PD1-Tregs subpopulation in patients with mild cognitive impairment; however, this augmentation is lost in patients with severe AD (162). Previous review has revealed that Tregs non-specifically inhibit CD4+ T cell proliferation both in vitro and in EAE (163). It is worth noting, according to the Dorothee group, that Tregs critically control the magnitude of CD4+ T cell targeting to amyloid-β in response to amyloid-β vaccination in both physiological and pathological settings (164). Consistently, transient depletion of Tregs in mouse models of AD accelerates the onset of cognitive deficits; conversely, amplification of Tregs improves cognitive functions (165). Therefore, increased frequency and suppressive activities of Tregs within AD patients suggest a protective role in autoimmune disorders (166, 167).
In contrast to intensive exploration of innate immunity in amyloid diseases, there has been comparatively limited research focusing on the impact of the adaptive immune system, in which self-reactive T cells or autoantibodies underlie autoimmune diseases.
In the 1970s, studies demonstrated that common amyloid-inducing agents were polyclonal B-cell activators in vitro as well as in vivo (168, 169). The role of adaptive immunity on AD pathogenesis remains ambiguous. Samuel et al. revealed dramatically increased Aβ plaque load in the “Rag-5xfAD” mice model, which lacked an adaptive immune response. Furthermore, genetic depletion of immunoglobulin G producing B cells exacerbated amyloid deposition, and this effect would be conversely ameliorated when again given immunoglobulin G through direct injection or bone marrow transplantation (170). Another study showed contradictory results that established PSAPP mice model of AD lacking functional B and T cells revealed reduced brain Aβ-peptide levels (171). The favorable and negative roles of the adaptive immune system, at least, suggest an impact of the adaptive immunity on cerebral β-amyloid pathology.
However, in view of the evidence from clinical trials applying Aβ-immunized vaccine showing the occurrence of specific CD4+ T cells targeting Aβ, we pay close attention to the changes of CD4+T cells in the brain, cerebrospinal fluid (CSF), and perivascular blood.
In several studies of the brains of patients with AD, CD4+ T cells have on occasion been found in the brain. In these cases, these CD4+ T cells have mainly been situated in the hippocampus and cortical regions of the brain and have only rarely appeared to colocalize with parenchymal Aβ deposits (172, 173). Similarly, the increased T cell in leptomeningeal and cortical vessels parallel with Aβ deposits indicate little evidence of intraparenchymal Aβ plaque driving T-cell infiltration into the AD brain (174). One study described a majority of T cells as existing in the parenchyma of AD patients; however, these T cells were memory cells expressing CD45RO+ and with the absence of proliferating nuclear proteins (i.e., Ki67 and PCNA) (172). A recent investigation further examined the activation state of T cells in transgenic mice models of AD-like cerebral amyloidosis and revealed hypo-responsiveness of the adaptive T cells response to Aβ, including the weak activation and proliferation, reduced expression of the effector cytokine IFN-γ, and downregulation of antigen presentation (175), which was in line with the concept of Aβ-induced immune hyporesponsiveness as early as in 2001 (176). Those findings, taken together, illustrate that the appearance of amyloid accompanied with regional declined/anergized disease-producing T-cell activation may imply a suppressive role of Aβ on localized autoimmune response.
In contrast to pathological brain-related T cells, T cells retained in the blood–CSF barrier of the choroid plexus usually mediate physiological immune surveillance. As such, in the research on T cells in CSF and T cells in peripheral circulating lymphocytes, there exist conflicting results.
One study pointed out that in circulating blood, there were no appreciable differences in the percentage of CD4+T cells between AD patients in different stages of dementia and the health control (177), whereas another study revealed that CD4+T lymphocytes exhibited a significant decline in the CSF as Aβ deposits in brain increased (178). However, several researches have illustrated that in both CSF and peripheral blood of mild AD patients, the proportion of activated CD4+ T cells was enhanced in comparison with age-matched, elderly controls (179, 180). The human leukocyte antigen DR isotype independent Aβ-reactive CD4+ T cells were also increased, while the magnitude of T-cell response showed great variable in different stages of dementia (181). With enhanced functioning of effector T cells, AD progression slows down (182). Whereas, as the severity of AD progressed, the number of CD4+T cells showed significant reduction in comparison with mild to moderate symptoms and controls (183). The interaction that exists between perivascular T subsets and Aβ pathological autoreactive T cells in the brain remains perplexing (184); however, the findings above may supply the beneficial role of Aβ and concomitant increased surveillance of circulating T cells.
Since the notion of amyloid causality has become a dogma within the research field, the passion for removing amyloid fibrils has gone unabated.
Dating back to 2001, AN-1792 reached clinical trials as the first active immunotherapy targeting β-amyloid clearance. The preclinical results from the vaccine were promising. However, the phase IIA trial was halted completely in 2002 due to subacute aseptic meningoencephalitis that developed in 6% (18 of 298) vaccine recipients (185, 186). Neuropathological examinations of brain tissues showed clearance of amyloid plaques while notably accompanied with elevated levels of CD4+ T cells in perivascular distribution. Depletion of brain Aβ with T-cell activation was also noted in another post-mortem examination of a patient who participated in the phase I study (187). These autoreactive CD4+ T cells that infiltrate the brain can induce autoimmune reactions and are thought to be responsible for the presence of encephalitis (188). A potential mechanism that underlies post-vaccination meningoencephalitis currently remains debated. Adjuvant (QS-21)-related TH1 lymphocytes activation and presence of T-cell recognition epitopes (within amino acids 15–42 of Aβ) (186) in the Aβ peptide have been proposed to initiate the T responses that triggered the autoimmune meningoencephalitis in some AN1792-vaccinated patients (189).
We previously summarized frequent occurrence of another autoimmune response against cerebral amyloid angiopathy—amyloid-related imaging abnormities (ARIA)—in all the clinical trials using amyloid-centric agents (21). Alarmingly, 20 times higher risk of ARIA occurred in patients with reduced brain amyloid burden than those without amyloid plaque clearance. Moreover, ARIA often colocalized at sites with the highest level of amyloid reduction.
We propose that the side effect of active anti-Aβ immunization rarely derives from vaccine adjuvant or any other reasons. The presence of autoimmune-related side effects in the absence of Aβ deposition raises the concern of amyloid plaque's potential role in retarding or inhibiting immune responses of an organism against its own healthy tissue. The overlap of the appearance of inflammatory signs with the time of microglial activation after the Aβ immunization provides an explanation that Aβ clearance from the brain are concomitant with periods of inflammation (185). Thus, the removal of amyloid plaques may prevent it from serving its natural protective role (22) and lead to an autoimmunity impairment of brain function.
SAP component is confirmed as a minor constituent binding to specific determinants shared by all types of amyloid fibrils (190). The concentration of SAP has been shown to be positively associated with amyloid deposits both in vivo and in vitro (191).
In many autoimmune disorders, improper clearance of nuclear debris released by apoptotic and necrotic cells is a potential source of autoantigens (192–195). Studies in mice with targeted deletion of the SAP gene spontaneously developed significant levels of antinuclear autoantibodies and autoimmune disease (191, 196). In addition, supplement of SAP into mice with systemic lupus erythematosus could effectively alleviate the autoimmune disease (197). The underlying mechanism indicates that SAP participates in handling of non-immunogenic apoptosis through reacting with nuclear autoantigens and inhibiting the formation of pathogenic autoantibodies targeted in systemic autoimmunity (198, 199).
Therefore, amyloid binding with SAP may potentially imply an important physiological role with regards to defense against autoimmune disorders.
Amylin, or IAPP, a 37-amino-acid peptide with amyloidogenic properties, is synthesized and cosecreted with insulin from pancreatic β cells and plays a critical role in modulating peripheral glucose balance. The deposition of islet amyloid is paralleled by progressive β-cell dysfunction found in 40–90% patients with type 2 diabetes (200, 201).
Type 1 diabetes (T1DM) is a form of autoimmune diabetes in which not enough amylin is produced. Then, the absence of islet amyloid deposition in patients with T1DM results in a greater risk of many autoimmune disorders, both organ and non-organ specific: thyroid disease, Addison's disease, coeliac disease, rheumatic disease, among others (202, 203). Similar latent autoimmune diabetes in adults (LADA) occurs with damaged β cells in the pancreas with inadequate amylin production. Various circulating autoantibodies including gliadin, endomysial, and thyroid antibodies are reported more frequently in patients with LADA than in T2DM patients (204).
Common histocompatibility antigens shared in both diseases that may explain autoimmune diseases often appear in clusters. Owing to islet amyloid formation not found in all T2DM patients and a limited number of studies, we cannot directly find more evidence that patients with IAPP deposition defend against localized or systematic autoimmune diseases. However, the coexistence of autoantibodies or autoimmune diseases present in T1DM and LADA broaden the view that lack of specific amyloid protein fibrils has some bearing on the presence of these autoimmune disorders.
The prevailing idea casting insoluble amyloid fibrils as strictly harmful has dominated the scientific literature for many centuries. However, based on the pathogenetic activation of proinflammatory states, anti-inflammatory strategies do not obtain any clinically satisfactory results. Moreover, autoimmune diseases that occur accompany with clearance of amyloid. Such results, along with the presence of toxic oligomers in pathogenesis of amyloidosis, represent a paradigm shift in our understanding of amyloid. An unconventional, new view is emerging in which some amyloid-forming proteins have a potential for doing good—far more so than anyone suspected; furthermore, their benefit is not just limited to antioxidant defense or an antibacterial function but has also been shown to exhibit therapeutic effects on autoimmune disorders, according to the latest evidence. The protective function from localized and systematic autoimmune disorders should be distinguished as exemplified in Aβ and IAPP. Indeed, amyloid with robust ordered β-sheet conformation resembles an armor, which to some extent traps and constrains cell dysfunctions while possessing a common protective role in different sites such as defensive biofilm in bacteria, antimicrobial peptide of IAPP and Aβ, and in our study, defending the body against immune attacks.
In summary, these arguments suggest that immune regulation mediated by amyloid plays a critical role in maintaining homeostasis between stimulating inflammation and defending against autoimmune responses. The appearance of amyloid represents a self-protective physiological phenomenon whereby the body engages in an elaborate orchestration to protect itself against a harmful disorder. Such insight into amyloid and autoimmunity may offer a novel therapeutic approach to regenerative medicine for neurodegenerative diseases, diabetes, and arthritis.
Y-MH and H-LZ contributed to the conception and design of the study. Y-MH wrote the first draft of the manuscript. X-ZH, JS, L-JG, Y-HP, and WL performed the study research. Y-MH, X-ZH, and H-LZ revised the manuscript for the final version. Y-MH is the guarantor of this work and takes responsibility for the integrity of the data and the accuracy of the data analysis. All authors read and approved the submitted version.
This work was supported by the National Natural Science Foundation of China (81471054) and Innovation Project of Guangxi Graduate Education (YCSZ2015213 and JGY2015128). The sponsor of the study had no role in the study design, data collection, data analysis, data interpretation, or the writing of the manuscript.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We are grateful to Dr. Yu-Zhong Ouyang for his help in drawing the graph and Dr. Petr Novak for his editing and proofreading.
1. Ramirez-Alvarado M, Merkel JS, Regan L. A systematic exploration of the influence of the protein stability on amyloid fibril formation in vitro. Proc Natl Acad Sci USA. (2000) 97:8979–84. doi: 10.1073/pnas.150091797
2. Chiti F, Dobson CM. Protein misfolding, functional amyloid, and human disease. Annu Rev Biochem. (2006) 75:333–66. doi: 10.1146/annurev.biochem.75.101304.123901
3. Hoppener JW, Ahren B, Lips CJ. Islet amyloid and type 2 diabetes mellitus. N Engl J Med. (2000) 343:411–9. doi: 10.1056/NEJM200008103430607
4. Hardy J, Selkoe DJ. The amyloid hypothesis of Alzheimer's disease: progress and problems on the road to therapeutics. Science. (2002) 297:353–6. doi: 10.1126/science.1072994
5. Merlini G, Westermark P. The systemic amyloidoses: clearer understanding of the molecular mechanisms offers hope for more effective therapies. J Intern Med. (2004) 255:159–78. doi: 10.1046/j.1365-2796.2003.01262.x
7. Knowles TP, Vendruscolo M, Dobson CM. The amyloid state and its association with protein misfolding diseases. Nat Rev Mol Cell Biol. (2014) 15:384–96. doi: 10.1038/nrm3810
8. Benson MD, Buxbaum JN, Eisenberg DS, Merlini G, Saraiva MJM, Sekijima Y, et al. Amyloid nomenclature 2018: recommendations by the International Society of Amyloidosis (ISA) nomenclature committee. Amyloid. (2018) 25:215–9. doi: 10.1080/13506129.2018.1549825
9. Guillot-Sestier MV, Doty KR, Town T. Innate immunity fights Alzheimer's disease. Trends Neurosci. (2015) 38:674–81. doi: 10.1016/j.tins.2015.08.008
10. Salminen A, Ojala J, Kauppinen A, Kaarniranta K, Suuronen T. Inflammation in Alzheimer's disease: amyloid-beta oligomers trigger innate immunity defence via pattern recognition receptors. Prog Neurobiol. (2009) 87:181–94. doi: 10.1016/j.pneurobio.2009.01.001
11. Heneka MT, Kummer MP, Latz E. Innate immune activation in neurodegenerative disease. Nat Rev Immunol. (2014) 14:463–77. doi: 10.1038/nri3705
12. Heneka MT, Golenbock DT, Latz E. Innate immunity in Alzheimer's disease. Nat Immunol. (2015) 16:229–36. doi: 10.1038/ni.3102
13. Cooper GJ, Willis AC, Clark A, Turner RC, Sim RB, Reid KB. Purification and characterization of a peptide from amyloid-rich pancreases of type 2 diabetic patients. Proc Natl Acad Sci USA. (1987) 84:8628–32. doi: 10.1073/pnas.84.23.8628
14. Westermark P, Wernstedt C, O'Brien TD, Hayden DW, Johnson KH. Islet amyloid in type 2 human diabetes mellitus and adult diabetic cats contains a novel putative polypeptide hormone. Am J Pathol. (1987) 127:414–7.
15. Masters SL, Dunne A, Subramanian SL, Hull RL, Tannahill GM, Sharp FA, et al. Activation of the NLRP3 inflammasome by islet amyloid polypeptide provides a mechanism for enhanced IL-1beta in type 2 diabetes. Nat Immunol. (2010) 11:897–904. doi: 10.1038/ni.1935
16. Maedler K, Sergeev P, Ris F, Oberholzer J, Joller-Jemelka HI, Spinas GA, et al. Glucose-induced beta cell production of IL-1beta contributes to glucotoxicity in human pancreatic islets. J Clin Invest. (2002) 110:851–60. doi: 10.1172/JCI200215318
17. Zhou R, Tardivel A, Thorens B, Choi I, Tschopp J. Thioredoxin-interacting protein links oxidative stress to inflammasome activation. Nat Immunol. (2010) 11:136–40. doi: 10.1038/ni.1831
18. Holmes C, Boche D, Wilkinson D, Yadegarfar G, Hopkins V, Bayer A, et al. Long-term effects of Abeta42 immunisation in Alzheimer's disease: follow-up of a randomised, placebo-controlled phase I trial. Lancet. (2008) 372:216–23. doi: 10.1016/S0140-6736(08)61075-2
19. Herrup K. The case for rejecting the amyloid cascade hypothesis. Nat Neurosci. (2015) 18:794–9. doi: 10.1038/nn.4017
20. Sevigny J, Chiao P, Bussiere T, Weinreb PH, Williams L, Maier M, et al. The antibody aducanumab reduces Aβ plaques in Alzheimer's disease. Nature. (2016) 537:50–6. doi: 10.1038/nature19323
21. Huang YM, Shen J, Zhao HL. Major clinical trials failed the amyloid hypothesis of Alzheimer's disease. J Am Geriatr Soc. (2019) 67:841–4. doi: 10.1111/jgs.15830
22. Zhao HL, Huang YM. Reply to: why amyloid is still a target for Alzheimer's disease. J Am Geriatr Soc. (2019) 67:850. doi: 10.1111/jgs.15827
23. Grant JL, Ghosn EE, Axtell RC, Herges K, Kuipers HF, Woodling NS, et al. Reversal of paralysis and reduced inflammation from peripheral administration of beta-amyloid in TH1 and TH17 versions of experimental autoimmune encephalomyelitis. Sci Transl Med. (2012) 4:145ra105. doi: 10.1126/scitranslmed.3004145
24. Kurnellas MP, Adams CM, Sobel RA, Steinman L, Rothbard JB. Amyloid fibrils composed of hexameric peptides attenuate neuroinflammation. Sci Transl Med. (2013) 5:179ra142. doi: 10.1126/scitranslmed.3005681
25. Cohen AS. General introduction and a brief history of the amyloid fibril. In Marrink J, Van Rijswijk MH, editos. Amyloidosis. Dordrecht: Nijhoff (1986). p. 3–19. doi: 10.1007/978-94-009-4309-4_1
26. Cohen AS, Calkins E. Electron microscopic observations on a fibrous component in amyloid of diverse origins. Nature. (1959) 183:1202–3. doi: 10.1038/1831202a0
27. Pras M, Schubert M, Zucker-Franklin D, Rimon A, Franklin EC. The characterization of soluble amyloid prepared in water. J Clin Invest. (1968) 47:924–33. doi: 10.1172/JCI105784
28. Glenner GG, Terry W, Harada M, Isersky C, Page D. Amyloid fibril proteins: proof of homology with immunoglobulin light chains by sequence analyses. Science. (1971) 172:1150–1. doi: 10.1126/science.172.3988.1150
30. Toyama BH, Weissman JS. Amyloid structure: conformational diversity and consequences. Annu Rev Biochem. (2011) 80:557–85. doi: 10.1146/annurev-biochem-090908-120656
31. Eisenberg D, Jucker M. The amyloid state of proteins in human diseases. Cell. (2012) 148:1188–203. doi: 10.1016/j.cell.2012.02.022
32. Wu H, Fuxreiter M. The structure and dynamics of higher-order assemblies: amyloids, signalosomes, and granules. Cell. (2016) 165:1055–66. doi: 10.1016/j.cell.2016.05.004
33. Liu G, Prabhakar A, Aucoin D, Simon M, Sparks S, Robbins KJ, et al. Mechanistic studies of peptide self-assembly: transient alpha-helices to stable beta-sheets. J Am Chem Soc. (2010) 132:18223–32. doi: 10.1021/ja1069882
34. Hauser CA, Deng R, Mishra A, Loo Y, Khoe U, Zhuang F, et al. Natural tri- to hexapeptides self-assemble in water to amyloid beta-type fiber aggregates by unexpected alpha-helical intermediate structures. Proc Natl Acad Sci USA. (2011) 108:1361–6. doi: 10.1073/pnas.1014796108
35. Ghosh D, Singh PK, Sahay S, Jha NN, Jacob RS, Sen S, et al. Structure based aggregation studies reveal the presence of helix-rich intermediate during α-Synuclein aggregation. Sci Rep. (2015) 5:9228. doi: 10.1038/srep09228
36. Zhang XX, Pan YH, Huang YM, Zhao HL. Neuroendocrine hormone amylin in diabetes. World J Diabetes. (2016) 7:189–97. doi: 10.4239/wjd.v7.i9.189
37. Hirschfield GM, Hawkins PN. Amyloidosis: new strategies for treatment. Int J Biochem Cell Biol. (2003) 35:1608–13. doi: 10.1016/S1357-2725(03)00169-9
38. Alexandrescu AT. Amyloid accomplices and enforcers. Protein Sci. (2005) 14:1–12. doi: 10.1110/ps.04887005
39. Lesne S, Koh MT, Kotilinek L, Kayed R, Glabe CG, Yang A, et al. A specific amyloid-beta protein assembly in the brain impairs memory. Nature. (2006) 440:352–7. doi: 10.1038/nature04533
40. Gandy S, Simon AJ, Steele JW, Lublin AL, Lah JJ, Walker LC, et al. Days to criterion as an indicator of toxicity associated with human Alzheimer amyloid-beta oligomers. Ann Neurol. (2010) 68:220–30. doi: 10.1002/ana.22052
41. Lesne S, Kotilinek L, Ashe KH. Plaque-bearing mice with reduced levels of oligomeric amyloid-beta assemblies have intact memory function. Neuroscience. (2008) 151:745–9. doi: 10.1016/j.neuroscience.2007.10.054
42. Lambert MP, Barlow AK, Chromy BA, Edwards C, Freed R, Liosatos M, et al. Diffusible, nonfibrillar ligands derived from Abeta1-42 are potent central nervous system neurotoxins. Proc Natl Acad Sci USA. (1998) 95:6448–53. doi: 10.1073/pnas.95.11.6448
43. Glabe CG. Common mechanisms of amyloid oligomer pathogenesis in degenerative disease. Neurobiol Aging. (2006) 27:570–5. doi: 10.1016/j.neurobiolaging.2005.04.017
44. Kayed R, Lasagna-Reeves CA. Molecular mechanisms of amyloid oligomers toxicity. J Alzheimers Dis. (2013) 33(Suppl. 1):S67–78. doi: 10.3233/JAD-2012-129001
45. Lin CY, Gurlo T, Kayed R, Butler AE, Haataja L, Glabe CG, et al. Toxic human islet amyloid polypeptide (h-IAPP) oligomers are intracellular, and vaccination to induce anti-toxic oligomer antibodies does not prevent h-IAPP-induced beta-cell apoptosis in h-IAPP transgenic mice. Diabetes. (2007) 56:1324–32. doi: 10.2337/db06-1579
46. Kim J, Cheon H, Jeong YT, Quan W, Kim KH, Cho JM, et al. Amyloidogenic peptide oligomer accumulation in autophagy-deficient beta cells induces diabetes. J Clin Invest. (2014) 124:3311–24. doi: 10.1172/JCI69625
47. Park JY, No HS, Ahn YR, Oh SH, Kim YS, Kim SY, et al. Pathologic changes and glucose homeostasis according to expression of human islet amyloid polypeptide in type 2 diabetic patients. J Histochem Cytochem. (2010) 58:731–40. doi: 10.1369/jhc.2010.955906
48. Kayed R, Head E, Thompson JL, McIntire TM, Milton SC, Cotman CW, et al. Common structure of soluble amyloid oligomers implies common mechanism of pathogenesis. Science. (2003) 300:486–9. doi: 10.1126/science.1079469
49. Kayed R, Glabe CG. Conformation-dependent anti-amyloid oligomer antibodies. Methods Enzymol. (2006) 413:326–44. doi: 10.1016/S0076-6879(06)13017-7
50. Campioni S, Mannini B, Zampagni M, Pensalfini A, Parrini C, Evangelisti E, et al. A causative link between the structure of aberrant protein oligomers and their toxicity. Nat Chem Biol. (2010) 6:140–7. doi: 10.1038/nchembio.283
51. Ling W, Huang YM, Qiao YC, Zhang XX, Zhao HL. Human amylin: from pathology to physiology and pharmacology. Curr Protein Pept Sci. (2019) 20:944–57. doi: 10.2174/1389203720666190328111833
52. Hardy JA, Higgins GA. Alzheimer's disease: the amyloid cascade hypothesis. Science. (1992) 256:184–5. doi: 10.1126/science.1566067
53. Terry RD, Masliah E, Salmon DP, Butters N, DeTeresa R, Hill R, et al. Physical basis of cognitive alterations in Alzheimer's disease: synapse loss is the major correlate of cognitive impairment. Ann Neurol. (1991) 30:572–80. doi: 10.1002/ana.410300410
54. Dickson DW, Crystal HA, Mattiace LA, Masur DM, Blau AD, Davies P, et al. Identification of normal and pathological aging in prospectively studied nondemented elderly humans. Neurobiol Aging. (1992) 13:179–89. doi: 10.1016/0197-4580(92)90027-U
55. Arriagada PV, Growdon JH, Hedley-Whyte ET, Hyman BT. Neurofibrillary tangles but not senile plaques parallel duration and severity of Alzheimer's disease. Neurology. (1992) 42:631–9. doi: 10.1212/WNL.42.3.631
56. Bierer LM, Hof PR, Purohit DP, Carlin L, Schmeidler J, Davis KL, et al. Neocortical neurofibrillary tangles correlate with dementia severity in Alzheimer's disease. Arch Neurol. (1995) 52:81–8. doi: 10.1001/archneur.1995.00540250089017
57. Wujek JR, Dority MD, Frederickson RC, Brunden KR. Deposits of A beta fibrils are not toxic to cortical and hippocampal neurons in vitro. Neurobiol Aging. (1996) 17:107–13. doi: 10.1016/0197-4580(95)02020-9
58. Giannakopoulos P, Herrmann FR, Bussiere T, Bouras C, Kovari E, Perl DP, et al. Tangle and neuron numbers, but not amyloid load, predict cognitive status in Alzheimer's disease. Neurology. (2003) 60:1495–500. doi: 10.1212/01.WNL.0000063311.58879.01
59. Nunomura A, Perry G, Aliev G, Hirai K, Takeda A, Balraj EK, et al. Oxidative damage is the earliest event in Alzheimer disease. J Neuropathol Exp Neurol. (2001) 60:759–67. doi: 10.1093/jnen/60.8.759
60. Pratico D, Uryu K, Leight S, Trojanoswki JQ, Lee VM. Increased lipid peroxidation precedes amyloid plaque formation in an animal model of Alzheimer amyloidosis. J Neurosci. (2001) 21:4183–7. doi: 10.1523/JNEUROSCI.21-12-04183.2001
61. Drake J, Link CD, Butterfield DA. Oxidative stress precedes fibrillar deposition of Alzheimer's disease amyloid beta-peptide (1-42) in a transgenic Caenorhabditis elegans model. Neurobiol Aging. (2003) 24:415–20. doi: 10.1016/S0197-4580(02)00225-7
62. Kim C, Lv G, Lee JS, Jung BC, Masuda-Suzukake M, Hong CS, et al. Exposure to bacterial endotoxin generates a distinct strain of alpha-synuclein fibril. Sci Rep. (2016) 6:30891. doi: 10.1038/srep30891
63. Pulze L, Bassani B, Gini E, D'Antona P, Grimaldi A, Luini A, et al. NET amyloidogenic backbone in human activated neutrophils. Clin Exp Immunol. (2016) 183:469–79. doi: 10.1111/cei.12730
64. Franchi N, Ballarin L, Peronato A, Cima F, Grimaldi A, Girardello R, et al. Functional amyloidogenesis in immunocytes from the colonial ascidian Botryllus schlosseri: evolutionary perspective. Dev Comp Immunol. (2019) 90:108–20. doi: 10.1016/j.dci.2018.09.010
65. Mintun MA, Larossa GN, Sheline YI, Dence CS, Lee SY, Mach RH, et al. [11C]PIB in a nondemented population: potential antecedent marker of Alzheimer disease. Neurology. (2006) 67:446–52. doi: 10.1212/01.wnl.0000228230.26044.a4
66. Aizenstein HJ, Nebes RD, Saxton JA, Price JC, Mathis CA, Tsopelas ND, et al. Frequent amyloid deposition without significant cognitive impairment among the elderly. Arch Neurol. (2008) 65:1509–17. doi: 10.1001/archneur.65.11.1509
67. Villemagne VL, Pike KE, Chetelat G, Ellis KA, Mulligan RS, Bourgeat P, et al. Longitudinal assessment of Aβ and cognition in aging and Alzheimer disease. Ann Neurol. (2011) 69:181–92. doi: 10.1002/ana.22248
68. Jucker M. The benefits and limitations of animal models for translational research in neurodegenerative diseases. Nat Med. (2010) 16:1210–4. doi: 10.1038/nm.2224
69. Chapman MR, Robinson LS, Pinkner JS, Roth R, Heuser J, Hammar M, et al. Role of Escherichia coli curli operons in directing amyloid fiber formation. Science. (2002) 295:851–5. doi: 10.1126/science.1067484
70. Claessen D, Rink R, de Jong W, Siebring J, de Vreugd P, Boersma FG, et al. A novel class of secreted hydrophobic proteins is involved in aerial hyphae formation in Streptomyces coelicolor by forming amyloid-like fibrils. Genes Dev. (2003) 17:1714–26. doi: 10.1101/gad.264303
71. Gebbink MF, Claessen D, Bouma B, Dijkhuizen L, Wosten HA. Amyloids–a functional coat for microorganisms. Nat Rev Microbiol. (2005) 3:333–41. doi: 10.1038/nrmicro1127
72. Barnhart MM, Chapman MR. Curli biogenesis and function. Annu Rev Microbiol. (2006) 60:131–47. doi: 10.1146/annurev.micro.60.080805.142106
73. Iconomidou VA, Vriend G, Hamodrakas SJ. Amyloids protect the silkmoth oocyte and embryo. FEBS Lett. (2000) 479:141–5. doi: 10.1016/S0014-5793(00)01888-3
74. Fowler DM, Koulov AV, Alory-Jost C, Marks MS, Balch WE, Kelly JW. Functional amyloid formation within mammalian tissue. PLoS Biol. (2006) 4:e6. doi: 10.1371/journal.pbio.0040006
75. Maji SK, Perrin MH, Sawaya MR, Jessberger S, Vadodaria K, Rissman RA, et al. Functional amyloids as natural storage of peptide hormones in pituitary secretory granules. Science. (2009) 325:328–32. doi: 10.1126/science.1173155
76. Wickner RB, Edskes HK, Shewmaker F, Nakayashiki T. Prions of fungi: inherited structures and biological roles. Nat Rev Microbiol. (2007) 5:611–8. doi: 10.1038/nrmicro1708
77. Namy O, Galopier A, Martini C, Matsufuji S, Fabret C, Rousset JP. Epigenetic control of polyamines by the prion [PSI+]. Nat Cell Biol. (2008) 10:1069–75. doi: 10.1038/ncb1766
78. Kranenburg O, Bouma B, Kroon-Batenburg LM, Reijerkerk A, Wu YP, Voest EE, et al. Tissue-type plasminogen activator is a multiligand cross-beta structure receptor. Curr Biol. (2002) 12:1833–9. doi: 10.1016/S0960-9822(02)01224-1
79. Si K, Lindquist S, Kandel ER. A neuronal isoform of the aplysia CPEB has prion-like properties. Cell. (2003) 115:879–91. doi: 10.1016/S0092-8674(03)01020-1
80. Shorter J, Lindquist S. Prions as adaptive conduits of memory and inheritance. Nat Rev Genet. (2005) 6:435–50. doi: 10.1038/nrg1616
81. Crist CG, Nakamura Y. Cross-talk between RNA and prions. J Biochem. (2006) 140:167–73. doi: 10.1093/jb/mvj163
82. Whitson JS, Selkoe DJ, Cotman CW. Amyloid beta protein enhances the survival of hippocampal neurons in vitro. Science. (1989) 243:1488–90. doi: 10.1126/science.2928783
83. Whitson JS, Glabe CG, Shintani E, Abcar A, Cotman CW. Beta-amyloid protein promotes neuritic branching in hippocampal cultures. Neurosci Lett. (1990) 110:319–24. doi: 10.1016/0304-3940(90)90867-9
84. Koo EH, Park L, Selkoe DJ. Amyloid beta-protein as a substrate interacts with extracellular matrix to promote neurite outgrowth. Proc Natl Acad Sci USA. (1993) 90:4748–52. doi: 10.1073/pnas.90.10.4748
85. Luo Y, Sunderland T, Roth GS, Wolozin B. Physiological levels of beta-amyloid peptide promote PC12 cell proliferation. Neurosci Lett. (1996) 217:125–8. doi: 10.1016/0304-3940(96)13087-1
86. Cuajungco MP, Goldstein LE, Nunomura A, Smith MA, Lim JT, Atwood CS, et al. Evidence that the beta-amyloid plaques of Alzheimer's disease represent the redox-silencing and entombment of abeta by zinc. J Biol Chem. (2000) 275:19439–42. doi: 10.1074/jbc.C000165200
87. Kontush A. Alzheimer's amyloid-beta as a preventive antioxidant for brain lipoproteins. Cell Mol Neurobiol. (2001) 21:299–315. doi: 10.1023/A:1012629603390
88. Kontush A, Berndt C, Weber W, Akopyan V, Arlt S, Schippling S, et al. Amyloid-beta is an antioxidant for lipoproteins in cerebrospinal fluid and plasma. Free Radic Biol Med. (2001) 30:119–28. doi: 10.1016/S0891-5849(00)00458-5
89. Smith MA, Casadesus G, Joseph JA, Perry G. Amyloid-beta and tau serve antioxidant functions in the aging and Alzheimer brain. Free Radic Biol Med. (2002) 33:1194–9. doi: 10.1016/S0891-5849(02)01021-3
90. Douglas PM, Treusch S, Ren HY, Halfmann R, Duennwald ML, Lindquist S, et al. Chaperone-dependent amyloid assembly protects cells from prion toxicity. Proc Natl Acad Sci USA. (2008) 105:7206–11. doi: 10.1073/pnas.0802593105
91. Ittner A, Chua SW, Bertz J, Volkerling A, van der Hoven J, Gladbach A, et al. Site-specific phosphorylation of tau inhibits amyloid-beta toxicity in Alzheimer's mice. Science. (2016) 354:904–8. doi: 10.1126/science.aah6205
92. Zou K, Gong JS, Yanagisawa K, Michikawa M. A novel function of monomeric amyloid beta-protein serving as an antioxidant molecule against metal-induced oxidative damage. J Neurosci. (2002) 22:4833–41. doi: 10.1523/JNEUROSCI.22-12-04833.2002
93. Bishop GM, Robinson SR. Human Abeta1-42 reduces iron-induced toxicity in rat cerebral cortex. J Neurosci Res. (2003) 73:316–23. doi: 10.1002/jnr.10661
94. Rao KS, Hegde ML, Anitha S, Musicco M, Zucca FA, Turro NJ, et al. Amyloid beta and neuromelanin–toxic or protective molecules? The cellular context makes the difference. Prog Neurobiol. (2006) 78:364–73. doi: 10.1016/j.pneurobio.2006.03.004
95. Kelly JW, Balch WE. Amyloid as a natural product. J Cell Biol. (2003) 161:461–2. doi: 10.1083/jcb.200304074
96. Fowler DM, Koulov AV, Balch WE, Kelly JW. Functional amyloid–from bacteria to humans. Trends Biochem Sci. (2007) 32:217–24. doi: 10.1016/j.tibs.2007.03.003
97. Maury CP. The emerging concept of functional amyloid. J Intern Med. (2009) 265:329–34. doi: 10.1111/j.1365-2796.2008.02068.x
98. Castellani RJ, Lee HG, Siedlak SL, Nunomura A, Hayashi T, Nakamura M, et al. Reexamining Alzheimer's disease: evidence for a protective role for amyloid-beta protein precursor and amyloid-beta. J Alzheimers Dis. (2009) 18:447–52. doi: 10.3233/JAD-2009-1151
99. Lundmark K, Westermark GT, Olsen A, Westermark P. Protein fibrils in nature can enhance amyloid protein A amyloidosis in mice: cross-seeding as a disease mechanism. Proc Natl Acad Sci USA. (2005) 102:6098–102. doi: 10.1073/pnas.0501814102
100. Singleton A, Myers A, Hardy J. The law of mass action applied to neurodegenerative disease: a hypothesis concerning the etiology and pathogenesis of complex diseases. Hum Mol Genet. (2004) 13 Spec No 1:R123–126. doi: 10.1093/hmg/ddh093
101. Ueda M, Ageyama N, Nakamura S, Nakamura M, Chambers JK, Misumi Y, et al. Aged vervet monkeys developing transthyretin amyloidosis with the human disease-causing Ile122 allele: a valid pathological model of the human disease. Lab Invest. (2012) 92:474–84. doi: 10.1038/labinvest.2011.195
102. De Strooper B. Proteases and proteolysis in Alzheimer disease: a multifactorial view on the disease process. Physiol Rev. (2010) 90:465–94. doi: 10.1152/physrev.00023.2009
103. Marcon G, Plakoutsi G, Canale C, Relini A, Taddei N, Dobson CM, et al. Amyloid formation from HypF-N under conditions in which the protein is initially in its native state. J Mol Biol. (2005) 347:323–35. doi: 10.1016/j.jmb.2005.01.034
104. Meda L, Cassatella MA, Szendrei GI, Otvos L Jr, Baron P, Villalba M, et al. Activation of microglial cells by beta-amyloid protein and interferon-gamma. Nature. (1995) 374:647–50. doi: 10.1038/374647a0
105. Patel NS, Paris D, Mathura V, Quadros AN, Crawford FC, Mullan MJ. Inflammatory cytokine levels correlate with amyloid load in transgenic mouse models of Alzheimer's disease. J Neuroinflammation. (2005) 2:9. doi: 10.1186/1742-2094-2-9
106. Smits HA, Rijsmus A, van Loon JH, Wat JW, Verhoef J, Boven LA, et al. Amyloid-beta-induced chemokine production in primary human macrophages and astrocytes. J Neuroimmunol. (2002) 127:160–8. doi: 10.1016/S0165-5728(02)00112-1
107. Michaud JP, Halle M, Lampron A, Theriault P, Prefontaine P, Filali M, et al. Toll-like receptor 4 stimulation with the detoxified ligand monophosphoryl lipid A improves Alzheimer's disease-related pathology. Proc Natl Acad Sci USA. (2013) 110:1941–6. doi: 10.1073/pnas.1215165110
108. Martinon F, Mayor A, Tschopp J. The inflammasomes: guardians of the body. Annu Rev Immunol. (2009) 27:229–65. doi: 10.1146/annurev.immunol.021908.132715
109. Heneka MT, Kummer MP, Stutz A, Delekate A, Schwartz S, Vieira-Saecker A, et al. NLRP3 is activated in Alzheimer's disease and contributes to pathology in APP/PS1 mice. Nature. (2013) 493:674–8. doi: 10.1038/nature11729
110. Halle A, Hornung V, Petzold GC, Stewart CR, Monks BG, Reinheckel T, et al. The NALP3 inflammasome is involved in the innate immune response to amyloid-beta. Nat Immunol. (2008) 9:857–65. doi: 10.1038/ni.1636
111. Di Domizio J, Dorta-Estremera S, Gagea M, Ganguly D, Meller S, Li P, et al. Nucleic acid-containing amyloid fibrils potently induce type I interferon and stimulate systemic autoimmunity. Proc Natl Acad Sci USA. (2012) 109:14550–5. doi: 10.1073/pnas.1206923109
112. Meyer-Luehmann M, Spires-Jones TL, Prada C, Garcia-Alloza M, de Calignon A, Rozkalne A, et al. Rapid appearance and local toxicity of amyloid-beta plaques in a mouse model of Alzheimer's disease. Nature. (2008) 451:720–4. doi: 10.1038/nature06616
113. Thomas T, Thomas G, McLendon C, Sutton T, Mullan M. β-Amyloid-mediated vasoactivity and vascular endothelial damage. Nature. (1996) 380:168–71. doi: 10.1038/380168a0
114. Blasko I, Stampfer-Kountchev M, Robatscher P, Veerhuis R, Eikelenboom P, Grubeck-Loebenstein B. How chronic inflammation can affect the brain and support the development of Alzheimer's disease in old age: the role of microglia and astrocytes. Aging Cell. (2004) 3:169–76. doi: 10.1111/j.1474-9728.2004.00101.x
115. Hsiao K, Chapman P, Nilsen S, Eckman C, Harigaya Y, Younkin S, et al. Correlative memory deficits, Aβ elevation, and amyloid plaques in transgenic mice. Science. (1996) 274:99–102. doi: 10.1126/science.274.5284.99
116. Yan SD, Chen X, Fu J, Chen M, Zhu H, Roher A, et al. RAGE and amyloid-beta peptide neurotoxicity in Alzheimer's disease. Nature. (1996) 382:685–91. doi: 10.1038/382685a0
117. Walsh DM, Klyubin I, Fadeeva JV, Cullen WK, Anwyl R, Wolfe MS, et al. Naturally secreted oligomers of amyloid beta protein potently inhibit hippocampal long-term potentiation in vivo. Nature. (2002) 416:535–9. doi: 10.1038/416535a
118. Westwell-Roper CY, Ehses JA, Verchere CB. Resident macrophages mediate islet amyloid polypeptide-induced islet IL-1beta production and beta-cell dysfunction. Diabetes. (2014) 63:1698–711. doi: 10.2337/db13-0863
119. Westwell-Roper C, Denroche HC, Ehses JA, Verchere CB. Differential activation of innate immune pathways by distinct islet amyloid polypeptide (IAPP) aggregates. J Biol Chem. (2016) 291:8908–17. doi: 10.1074/jbc.M115.712455
120. Gorevic PD. Amyloid and inflammation. Proc Natl Acad Sci USA. (2013) 110:16291–2. doi: 10.1073/pnas.1315112110
121. Simons JP, Al-Shawi R, Ellmerich S, Speck I, Aslam S, Hutchinson WL, et al. Pathogenetic mechanisms of amyloid A amyloidosis. Proc Natl Acad Sci USA. (2013) 110:16115–20. doi: 10.1073/pnas.1306621110
122. Tilg H, Dinarello CA, Mier JW. IL-6 and APPs: anti-inflammatory and immunosuppressive mediators. Immunol Today. (1997) 18:428–32. doi: 10.1016/S0167-5699(97)01103-1
123. Pousset F, Cremona S, Dantzer R, Kelley K, Parnet P. Interleukin-4 and interleukin-10 regulate IL1-beta induced mouse primary astrocyte activation: a comparative study. Glia. (1999) 26: 12–21. doi: 10.1002/(sici)1098-1136(199903)26:1<12::aid-glia2>3.0.co;2-s
124. Szczepanik AM, Funes S, Petko W, Ringheim GE. IL-4, IL-10 and IL-13 modulate A beta(1–42)-induced cytokine and chemokine production in primary murine microglia and a human monocyte cell line. J Neuroimmunol. (2001) 113:49–62. doi: 10.1016/S0165-5728(00)00404-5
125. Ledeboer A, Breve JJ, Wierinckx A, van der Jagt S, Bristow AF, Leysen JE, et al. Expression and regulation of interleukin-10 and interleukin-10 receptor in rat astroglial and microglial cells. Eur J Neurosci. (2002) 16:1175–85. doi: 10.1046/j.1460-9568.2002.02200.x
126. Shaftel SS, Kyrkanides S, Olschowka JA, Miller JN, Johnson RE, O'Banion MK. Sustained hippocampal IL-1 beta overexpression mediates chronic neuroinflammation and ameliorates Alzheimer plaque pathology. J Clin Invest. (2007) 117:1595–604. doi: 10.1172/JCI31450
127. Bradshaw EM, Chibnik LB, Keenan BT, Ottoboni L, Raj T, Tang A, et al. CD33 Alzheimer's disease locus: altered monocyte function and amyloid biology. Nat Neurosci. (2013) 16:848–50. doi: 10.1038/nn.3435
128. Griciuc A, Serrano-Pozo A, Parrado AR, Lesinski AN, Asselin CN, Mullin K, et al. Alzheimer's disease risk gene CD33 inhibits microglial uptake of amyloid beta. Neuron. (2013) 78:631–43. doi: 10.1016/j.neuron.2013.04.014
129. Bouchon A, Hernandez-Munain C, Cella M, Colonna M. A DAP12-mediated pathway regulates expression of CC chemokine receptor 7 and maturation of human dendritic cells. J Exp Med. (2001) 194:1111–22. doi: 10.1084/jem.194.8.1111
130. Hamerman JA, Jarjoura JR, Humphrey MB, Nakamura MC, Seaman WE, Lanier LL. Cutting edge: inhibition of TLR and FcR responses in macrophages by triggering receptor expressed on myeloid cells (TREM)-2 and DAP12. J Immunol. (2006) 177:2051–5. doi: 10.4049/jimmunol.177.4.2051
131. Hickman SE, Allison EK, El Khoury J. Microglial dysfunction and defective beta-amyloid clearance pathways in aging Alzheimer's disease mice. J Neurosci. (2008) 28:8354–60. doi: 10.1523/JNEUROSCI.0616-08.2008
132. Kubo M, Hanada T, Yoshimura A. Suppressors of cytokine signaling and immunity. Nat Immunol. (2003) 4:1169–76. doi: 10.1038/ni1012
133. Yoshimura A, Ohishi HM, Aki D, Hanada T. Regulation of TLR signaling and inflammation by SOCS family proteins. J Leukoc Biol. (2004) 75:422–7. doi: 10.1189/jlb.0403194
134. Gourdain P, Ballerini C, Nicot AB, Carnaud C. Exacerbation of experimental autoimmune encephalomyelitis in prion protein (PrPc)-null mice: evidence for a critical role of the central nervous system. J Neuroinflammation. (2012) 9:25. doi: 10.1186/1742-2094-9-25
135. Weinger JG, Davies P, Acker CM, Brosnan CF, Tsiperson V, Bayewitz A, et al. Mice devoid of Tau have increased susceptibility to neuronal damage in myelin oligodendrocyte glycoprotein-induced experimental autoimmune encephalomyelitis. J Neuropathol Exp Neurol. (2012) 71:422–33. doi: 10.1097/NEN.0b013e3182540d2e
136. Ji Z, Ke ZJ, Geng JG. SAP suppresses the development of experimental autoimmune encephalomyelitis in C57BL/6 mice. Immunol Cell Biol. (2012) 90:388–95. doi: 10.1038/icb.2011.51
137. Zhang XX, Qiao YC, Li W, Zou X, Chen YL, Shen J, et al. Human amylin induces CD4+Foxp3+ regulatory T cells in the protection from autoimmune diabetes. Immunol Res. (2018) 66:179–86. doi: 10.1007/s12026-017-8956-5
138. Kurnellas MP, Ghosn EE, Schartner JM, Baker J, Rothbard JJ, Negrin RS, et al. Amyloid fibrils activate B-1a lymphocytes to ameliorate inflammatory brain disease. Proc Natl Acad Sci USA. (2015) 112:15016–23. doi: 10.1073/pnas.1521206112
139. Kurnellas MP, Schartner JM, Fathman CG, Jagger A, Steinman L, Rothbard JB. Mechanisms of action of therapeutic amyloidogenic hexapeptides in amelioration of inflammatory brain disease. J Exp Med. (2014) 211:1847–56. doi: 10.1084/jem.20140107
140. Kurnellas MP, Rothbard JB, Steinman L. Self-assembling peptides form immune suppressive amyloid fibrils effective in autoimmune encephalomyelitis. Curr Top Behav Neurosci. (2015) 26:221–32. doi: 10.1007/7854_2015_377
141. Kurnellas MP, Brownell SE, Su L, Malkovskiy AV, Rajadas J, Dolganov G, et al. Chaperone activity of small heat shock proteins underlies therapeutic efficacy in experimental autoimmune encephalomyelitis. J Biol Chem. (2012) 287:36423–34. doi: 10.1074/jbc.M112.371229
142. Kraus A, Race B, Phillips K, Winkler C, Saturday G, Kurnellas M, et al. Genetic background modulates outcome of therapeutic amyloid peptides in treatment of neuroinflammation. J Neuroimmunol. (2016) 298:42–50. doi: 10.1016/j.jneuroim.2016.06.010
143. Ousman SS, Tomooka BH, van Noort JM, Wawrousek EF, O'Connor KC, Hafler DA, et al. Protective and therapeutic role for alphaB-crystallin in autoimmune demyelination. Nature. (2007) 448:474–9. doi: 10.1038/nature05935
144. Han MH, Hwang SI, Roy DB, Lundgren DH, Price JV, Ousman SS, et al. Proteomic analysis of active multiple sclerosis lesions reveals therapeutic targets. Nature. (2008) 451:1076–81. doi: 10.1038/nature06559
145. van Noort JM, van Sechel AC, Bajramovic JJ, el Ouagmiri M, Polman CH, Lassmann H, et al. The small heat-shock protein alpha B-crystallin as candidate autoantigen in multiple sclerosis. Nature. (1995) 375:798–801. doi: 10.1038/375798a0
146. Tanaka N, Tanaka R, Tokuhara M, Kunugi S, Lee YF, Hamada D. Amyloid fibril formation and chaperone-like activity of peptides from alphaA-crystallin. Biochemistry. (2008) 47:2961–7. doi: 10.1021/bi701823g
147. Soscia SJ, Kirby JE, Washicosky KJ, Tucker SM, Ingelsson M, Hyman B, et al. The Alzheimer's disease-associated amyloid β-protein is an antimicrobial peptide. PLoS ONE. (2010) 5:e9505. doi: 10.1371/journal.pone.0009505
148. Bourgade K, Garneau H, Giroux G, Le Page AY, Bocti C, Dupuis G, et al. beta-Amyloid peptides display protective activity against the human Alzheimer's disease-associated herpes simplex virus-1. Biogerontology. (2015) 16:85–98. doi: 10.1007/s10522-014-9538-8
149. Bourgade K, Le Page A, Bocti C, Witkowski JM, Dupuis G, Frost EH, et al. Protective effect of amyloid-beta peptides against herpes simplex virus-1 infection in a neuronal cell culture model. J Alzheimers Dis. (2016) 50:1227–41. doi: 10.3233/JAD-150652
150. Kumar DK, Choi SH, Washicosky KJ, Eimer WA, Tucker S, Ghofrani J, et al. Amyloid-beta peptide protects against microbial infection in mouse and worm models of Alzheimer's disease. Sci Transl Med. (2016) 8:340ra372. doi: 10.1126/scitranslmed.aaf1059
151. Last NB, Miranker AD. Common mechanism unites membrane poration by amyloid and antimicrobial peptides. Proc Natl Acad Sci USA. (2013) 110:6382–7. doi: 10.1073/pnas.1219059110
152. Park SC, Moon JC, Shin SY, Son H, Jung YJ, Kim NH, et al. Functional characterization of alpha-synuclein protein with antimicrobial activity. Biochem Biophys Res Commun. (2016) 478:924–8. doi: 10.1016/j.bbrc.2016.08.052
153. Hou F, Sun L, Zheng H, Skaug B, Jiang QX, Chen ZJ. MAVS forms functional prion-like aggregates to activate and propagate antiviral innate immune response. Cell. (2011) 146:448–61. doi: 10.1016/j.cell.2011.06.041
154. Nijnik A, Pistolic J, Wyatt A, Tam S, Hancock RE. Human cathelicidin peptide LL-37 modulates the effects of IFN-gamma on APCs. J Immunol. (2009) 183:5788–98. doi: 10.4049/jimmunol.0901491
155. Nishimori JH, Newman TN, Oppong GO, Rapsinski GJ, Yen JH, Biesecker SG, et al. Microbial amyloids induce interleukin 17A (IL-17A) and IL-22 responses via Toll-like receptor 2 activation in the intestinal mucosa. Infect Immun. (2012) 80:4398–408. doi: 10.1128/IAI.00911-12
156. Romero D, Aguilar C, Losick R, Kolter R. Amyloid fibers provide structural integrity to Bacillus subtilis biofilms. Proc Natl Acad Sci USA. (2010) 107:2230–4. doi: 10.1073/pnas.0910560107
157. Blanco LP, Evans ML, Smith DR, Badtke MP, Chapman MR. Diversity, biogenesis and function of microbial amyloids. Trends Microbiol. (2012) 20:66–73. doi: 10.1016/j.tim.2011.11.005
158. Oppong GO, Rapsinski GJ, Newman TN, Nishimori JH, Biesecker SG, Tukel C. Epithelial cells augment barrier function via activation of the Toll-like receptor 2/phosphatidylinositol 3-kinase pathway upon recognition of Salmonella enterica serovar Typhimurium curli fibrils in the gut. Infect Immun. (2013) 81:478–86. doi: 10.1128/IAI.00453-12
159. Oppong GO, Rapsinski GJ, Tursi SA, Biesecker SG, Klein-Szanto AJ, Goulian M, et al. Biofilm-associated bacterial amyloids dampen inflammation in the gut: oral treatment with curli fibres reduces the severity of hapten-induced colitis in mice. NPJ Biofilms Microb. (2015) 1:1–8. doi: 10.1038/npjbiofilms.2015.19
160. Sakaguchi S, Sakaguchi N, Shimizu J, Yamazaki S, Sakihama T, Itoh M, et al. Immunologic tolerance maintained by CD25+ CD4+ regulatory T cells: their common role in controlling autoimmunity, tumor immunity, and transplantation tolerance. Immunol Rev. (2001) 182:18–32. doi: 10.1034/j.1600-065X.2001.1820102.x
161. Rosenkranz D, Weyer S, Tolosa E, Gaenslen A, Berg D, Leyhe T, et al. Higher frequency of regulatory T cells in the elderly and increased suppressive activity in neurodegeneration. J Neuroimmunol. (2007) 188:117–27. doi: 10.1016/j.jneuroim.2007.05.011
162. Saresella M, Calabrese E, Marventano I, Piancone F, Gatti A, Calvo MG, et al. PD1 negative and PD1 positive CD4+ T regulatory cells in mild cognitive impairment and Alzheimer's disease. J Alzheimers Dis. (2010) 21:927–38. doi: 10.3233/JAD-2010-091696
163. Kohm AP, Carpentier PA, Anger HA, Miller SD. Cutting edge: CD4+CD25+ regulatory T cells suppress antigen-specific autoreactive immune responses and central nervous system inflammation during active experimental autoimmune encephalomyelitis. J Immunol. (2002) 169:4712–6. doi: 10.4049/jimmunol.169.9.4712
164. Toly-Ndour C, Lui G, Nunes MM, Bruley-Rosset M, Aucouturier P, Dorothee G. MHC-independent genetic factors control the magnitude of CD4+ T cell responses to amyloid-beta peptide in mice through regulatory T cell-mediated inhibition. J Immunol. (2011) 187:4492–500. doi: 10.4049/jimmunol.1003953
165. Dansokho C, Ait Ahmed D, Aid S, Toly-Ndour C, Chaigneau T, Calle V, et al. Regulatory T cells delay disease progression in Alzheimer-like pathology. Brain. (2016) 139(Pt 4):1237–51. doi: 10.1093/brain/awv408
166. Yang H, Yang H, Xie Z, Wei L, Bi J. Systemic transplantation of human umbilical cord derived mesenchymal stem cells-educated T regulatory cells improved the impaired cognition in AbetaPPswe/PS1dE9 transgenic mice. PLoS ONE. (2013) 8:e69129. doi: 10.1371/journal.pone.0069129
167. Ye M, Chung HS, Lee C, Yoon MS, Yu AR, Kim JS, et al. Neuroprotective effects of bee venom phospholipase A2 in the 3xTg AD mouse model of Alzheimer's disease. J Neuroinflammation. (2016) 13:10. doi: 10.1186/s12974-016-0476-z
168. Britton S. Experimental amyloidosis: the inducer is a polyclonal B-cell activator to which susceptibility is under genetic control. J Exp Med. (1975) 142:1564–9. doi: 10.1084/jem.142.6.1564
169. Scheinberg MA. Immunology of amyloid disease: a review. Semin Arthritis Rheum. (1977) 7:133–40. doi: 10.1016/0049-0172(77)90019-1
170. Marsh SE, Abud EM, Lakatos A, Karimzadeh A, Yeung ST, Davtyan H, et al. The adaptive immune system restrains Alzheimer's disease pathogenesis by modulating microglial function. Proc Natl Acad Sci USA. (2016) 113:E1316–25. doi: 10.1073/pnas.1525466113
171. Spani C, Suter T, Derungs R, Ferretti MT, Welt T, Wirth F, et al. Reduced beta-amyloid pathology in an APP transgenic mouse model of Alzheimer's disease lacking functional B and T cells. Acta Neuropathol Commun. (2015) 3:71. doi: 10.1186/s40478-015-0251-x
172. Togo T, Akiyama H, Iseki E, Kondo H, Ikeda K, Kato M, et al. Occurrence of T cells in the brain of Alzheimer's disease and other neurological diseases. J Neuroimmunol. (2002) 124:83–92. doi: 10.1016/S0165-5728(01)00496-9
173. Town T, Tan J, Flavell RA, Mullan M. T-cells in Alzheimer's disease. Neuromolecular Med. (2005) 7:255–64. doi: 10.1385/NMM:7:3:255
174. Yamada M, Itoh Y, Shintaku M, Kawamura J, Jensson O, Thorsteinsson L, et al. Immune reactions associated with cerebral amyloid angiopathy. Stroke. (1996) 27:1155–62. doi: 10.1161/01.STR.27.7.1155
175. Ferretti MT, Merlini M, Spani C, Gericke C, Schweizer N, Enzmann G, et al. T-cell brain infiltration and immature antigen-presenting cells in transgenic models of Alzheimer's disease-like cerebral amyloidosis. Brain Behav Immun. (2016) 54:211–25. doi: 10.1016/j.bbi.2016.02.009
176. Monsonego A, Maron R, Zota V, Selkoe DJ, Weiner HL. Immune hyporesponsiveness to amyloid beta-peptide in amyloid precursor protein transgenic mice: implications for the pathogenesis and treatment of Alzheimer's disease. Proc Natl Acad Sci USA. (2001) 98:10273–8. doi: 10.1073/pnas.191118298
177. Speciale L, Calabrese E, Saresella M, Tinelli C, Mariani C, Sanvito L, et al. Lymphocyte subset patterns and cytokine production in Alzheimer's disease patients. Neurobiol Aging. (2007) 28:1163–9. doi: 10.1016/j.neurobiolaging.2006.05.020
178. Stowe AM, Ireland SJ, Ortega SB, Chen D, Huebinger RM, Tarumi T, et al. Adaptive lymphocyte profiles correlate to brain Aβ burden in patients with mild cognitive impairment. J Neuroinflammation. (2017) 14:149. doi: 10.1186/s12974-017-0910-x
179. Lombardi VR, Garcia M, Rey L, Cacabelos R. Characterization of cytokine production, screening of lymphocyte subset patterns and in vitro apoptosis in healthy and Alzheimer's Disease (AD) individuals. J Neuroimmunol. (1999) 97:163–71. doi: 10.1016/S0165-5728(99)00046-6
180. Richartz-Salzburger E, Batra A, Stransky E, Laske C, Kohler N, Bartels M, et al. Altered lymphocyte distribution in Alzheimer's disease. J Psychiatr Res. (2007) 41:174–8. doi: 10.1016/j.jpsychires.2006.01.010
181. Lueg G, Gross CC, Lohmann H, Johnen A, Kemmling A, Deppe M, et al. Clinical relevance of specific T-cell activation in the blood and cerebrospinal fluid of patients with mild Alzheimer's disease. Neurobiol Aging. (2015) 36:81–9. doi: 10.1016/j.neurobiolaging.2014.08.008
182. Baruch K, Deczkowska A, Rosenzweig N, Tsitsou-Kampeli A, Sharif AM, Matcovitch-Natan O, et al. PD-1 immune checkpoint blockade reduces pathology and improves memory in mouse models of Alzheimer's disease. Nat Med. (2016) 22:135–7. doi: 10.1038/nm.4022
183. Shalit F, Sredni B, Brodie C, Kott E, Huberman M. T lymphocyte subpopulations and activation markers correlate with severity of Alzheimer's disease. Clin Immunol Immunopathol. (1995) 75:246–50. doi: 10.1006/clin.1995.1078
184. Kipnis J. Multifaceted interactions between adaptive immunity and the central nervous system. Science. (2016) 353:766–71. doi: 10.1126/science.aag2638
185. Orgogozo JM, Gilman S, Dartigues JF, Laurent B, Puel M, Kirby LC, et al. Subacute meningoencephalitis in a subset of patients with AD after Abeta42 immunization. Neurology. (2003) 61:46–54. doi: 10.1212/01.WNL.0000073623.84147.A8
186. Gelinas DS, DaSilva K, Fenili D, St George-Hyslop P, McLaurin J. Immunotherapy for Alzheimer's disease. Proc Natl Acad Sci USA. (2004) 101(Suppl. 2):14657–62. doi: 10.1073/pnas.0404866101
187. Nicoll JA, Wilkinson D, Holmes C, Steart P, Markham H, Weller RO. Neuropathology of human Alzheimer disease after immunization with amyloid-beta peptide: a case report. Nat Med. (2003) 9:448–52. doi: 10.1038/nm840
188. Robinson SR, Bishop GM, Lee HG, Munch G. Lessons from the AN 1792 Alzheimer vaccine: lest we forget. Neurobiol Aging. (2004) 25:609–15. doi: 10.1016/j.neurobiolaging.2003.12.020
189. Lemere CA, Masliah E. Can Alzheimer disease be prevented by amyloid-beta immunotherapy? Nat Rev Neurol. (2010) 6:108–19. doi: 10.1038/nrneurol.2009.219
190. Pepys MB, Booth D, Hutchinson W, Gallimore J, Collins I, Hohenester E. Amyloid P component. A critical review. Amyloid. (1997) 4:274–95. doi: 10.3109/13506129709003838
191. Botto M, Hawkins PN, Bickerstaff MC, Herbert J, Bygrave AE, McBride A, et al. Amyloid deposition is delayed in mice with targeted deletion of the serum amyloid P component gene. Nat Med. (1997) 3:855–9. doi: 10.1038/nm0897-855
192. Burlingame RW, Boey ML, Starkebaum G, Rubin RL. The central role of chromatin in autoimmune responses to histones and DNA in systemic lupus erythematosus. J Clin Invest. (1994) 94:184–92. doi: 10.1172/JCI117305
193. Casciola-Rosen LA, Anhalt G, Rosen A. Autoantigens targeted in systemic lupus erythematosus are clustered in two populations of surface structures on apoptotic keratinocytes. J Exp Med. (1994) 179:1317–30. doi: 10.1084/jem.179.4.1317
194. Mattson MP. Apoptosis in neurodegenerative disorders. Nat Rev Mol Cell Biol. (2000) 1:120–9. doi: 10.1038/35040009
195. Li S, Sun Y, Liang CP, Thorp EB, Han S, Jehle AW, et al. Defective phagocytosis of apoptotic cells by macrophages in atherosclerotic lesions of ob/ob mice and reversal by a fish oil diet. Circ Res. (2009) 105:1072–82. doi: 10.1161/CIRCRESAHA.109.199570
196. Kolstoe SE, Ridha BH, Bellotti V, Wang N, Robinson CV, Crutch SJ, et al. Molecular dissection of Alzheimer's disease neuropathology by depletion of serum amyloid P component. Proc Natl Acad Sci USA. (2009) 106:7619–23. doi: 10.1073/pnas.0902640106
197. Zhang W, Xu W, Xiong S. Macrophage differentiation and polarization via phosphatidylinositol 3-kinase/Akt-ERK signaling pathway conferred by serum amyloid P component. J Immunol. (2011) 187:1764–77. doi: 10.4049/jimmunol.1002315
198. Bickerstaff MC, Botto M, Hutchinson WL, Herbert J, Tennent GA, Bybee A, et al. Serum amyloid P component controls chromatin degradation and prevents antinuclear autoimmunity. Nat Med. (1999) 5:694–7. doi: 10.1038/9544
199. Mold C, Baca R, Du Clos TW. Serum amyloid P component and C-reactive protein opsonize apoptotic cells for phagocytosis through Fcgamma receptors. J Autoimmun. (2002) 19:147–54. doi: 10.1006/jaut.2002.0615
200. Zhao HL, Lai FM, Tong PC, Zhong DR, Yang D, Tomlinson B, et al. Prevalence and clinicopathological characteristics of islet amyloid in chinese patients with type 2 diabetes. Diabetes. (2003) 52:2759–66. doi: 10.2337/diabetes.52.11.2759
201. Clark A, Nilsson MR. Islet amyloid: a complication of islet dysfunction or an aetiological factor in Type 2 diabetes? Diabetologia. (2004) 47:157–69. doi: 10.1007/s00125-003-1304-4
202. Somers EC, Thomas SL, Smeeth L, Hall AJ. Are individuals with an autoimmune disease at higher risk of a second autoimmune disorder? Am J Epidemiol. (2009) 169:749–55. doi: 10.1093/aje/kwn408
203. Hughes JW, Riddlesworth TD, DiMeglio LA, Miller KM, Rickels MR, McGill JB, et al. Autoimmune diseases in children and adults with type 1 diabetes from the T1D exchange clinic registry. J Clin Endocrinol Metab. (2016) 101:4931–7. doi: 10.1210/jc.2016-2478
Keywords: amyloid, amyloid conformation, autoimmune, proinflammation, immunomodulation, homeostasis, Alzheimer's disease, diabetes
Citation: Huang Y-M, Hong X-Z, Shen J, Geng L-J, Pan Y-H, Ling W and Zhao H-L (2020) Amyloids in Site-Specific Autoimmune Reactions and Inflammatory Responses. Front. Immunol. 10:2980. doi: 10.3389/fimmu.2019.02980
Received: 07 May 2019; Accepted: 04 December 2019;
Published: 09 January 2020.
Edited by:
Stefania Gallucci, Lewis Katz School of Medicine, Temple University, United StatesReviewed by:
Çagla Tükel, Temple University, United StatesCopyright © 2020 Huang, Hong, Shen, Geng, Pan, Ling and Zhao. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Hai-Lu Zhao, zhaohailu9@126.com; zhaohailu@glmc.edu.cn
†Present address: Yan-Mei Huang, Department of Geriatrics, Zhongshan Hospital, Fudan University, Shanghai, China
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.