
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Immunol. , 06 December 2019
Sec. Cancer Immunity and Immunotherapy
Volume 10 - 2019 | https://doi.org/10.3389/fimmu.2019.02836
This article is part of the Research Topic Molecular Strategies Aimed to Boost NK Cell-based Immunotherapy of Cancer View all 11 articles
 Paola Minetto1,2†
Paola Minetto1,2† Fabio Guolo1,2†
Fabio Guolo1,2† Silvia Pesce3
Silvia Pesce3 Marco Greppi3,4Valentina Obino3,4Elisa Ferretti3,4
Marco Greppi3,4Valentina Obino3,4Elisa Ferretti3,4 Simona Sivori3,4*
Simona Sivori3,4* Carlo Genova5‡Roberto Massimo Lemoli1,2‡
Carlo Genova5‡Roberto Massimo Lemoli1,2‡ Emanuela Marcenaro3,4*‡
Emanuela Marcenaro3,4*‡In the last years, natural killer (NK) cell-based immunotherapy has emerged as a promising therapeutic approach for solid tumors and hematological malignancies. NK cells are innate lymphocytes with an array of functional competences, including anti-cancer, anti-viral, and anti-graft-vs.-host disease potential. The intriguing idea of harnessing such potent innate immune system effectors for cancer treatment led to the development of clinical trials based on the adoptive therapy of NK cells or on the use of monoclonal antibodies targeting the main NK cell immune checkpoints. Indeed, checkpoint immunotherapy that targets inhibitory receptors of T cells, reversing their functional blocking, marked a breakthrough in anticancer therapy, opening new approaches for cancer immunotherapy and resulted in extensive research on immune checkpoints. However, the clinical efficacy of T cell-based immunotherapy presents a series of limitations, including the inability of T cells to recognize and kill HLA-Ineg tumor cells. For these reasons, new strategies for cancer immunotherapy are now focusing on NK cells. Blockade with NK cell checkpoint inhibitors that reverse their functional block may overcome the limitations of T cell-based immunotherapy, mainly against HLA-Ineg tumor targets. Here, we discuss recent anti-tumor approaches based on mAb-mediated blocking of immune checkpoints (either restricted to NK cells or shared with T cells), used either as a single agent or in combination with other compounds, that have demonstrated promising clinical responses in both solid tumors and hematological malignancies.
Immunotherapy is an innovative approach for the treatment of cancer and is based on the idea of harnessing the immune system to target tumors. Recently, immunotherapy, and in particular immune checkpoint (IC) blockade therapy, has represented a significant step forward for cancer treatment (1–5). Two inhibitory ICs, CTLA-4 (6) and PD-1 (7), received great attention, since the inhibition of CTLA-4 or PD-1 signaling significantly improved the survival of patients with metastatic solid cancers. Given the clinical efficacy of PD-1 and/or CTLA-4 blockade in patients with untreatable solid and hematological cancers (1–5, 8), much attention has been given to IC receptors and their cognate ligands.
Currently, one of the major challenges in immune-oncology is the understanding of the mechanisms of IC inhibitor resistance (indeed, only a fraction of patients respond to immunotherapy), to increase the proportion of patients benefitting from such treatment, and to control treatment toxicity.
A critical point is that the clinical effect of the PD-1/PD-Ls blockade has been conventionally attributed to the restoration of cytotoxic lymphocyte activity. However, a partial or complete loss of HLA-I expression is one of the most frequent mechanisms of tumor escape from T-cell control in different human tumor types (9). In this scenario, the role of the “innate counterpart” of cytotoxic T cells, the natural killer (NK) cells, which show the ability to recognize and kill tumor cells regardless of HLA-I expression, appears to be crucial (10–12).
NK cells were first identified in the mid-1970s as a unique lymphocyte subset able to detect and rapidly kill abnormal cells without prior sensitization or recognition of specific tumor antigens, thus preventing the development of many cancers (13, 14). In the late 1980s, the observation that NK cells could kill a lymphoma cell line that had lost MHC class I surface molecules, while the original MHC class I+ cells were resistant to lysis, led to the formulation of the “missing self-hypothesis” that stated that NK cells are able to sense the absence of “self” MHC class-I molecules on target cells (15, 16). In the 1990s, this hypothesis was confirmed by the discovery of inhibitory (17, 18) and activating NK receptors (19). In humans, the main inhibitory receptors are represented by the inhibitory killer Ig-like receptors (KIRs), recognizing allotypic determinants shared by groups of HLA class-I alleles (20, 21) and by the CD94/NKG2A heterodimer (22), specific for the non-classical HLA-E molecule.
Inhibitory KIRs are type I molecules with two (KIR2D) or three (KIR3D) highly polymorphic extracellular Ig-like domains followed by long (L) cytoplasmic tails harboring two ITIMs, able to transduce an inhibitory signal through the recruitment of tyrosine phosphatases. The four main inhibitory KIRs are specific for epitopes shared by distinct groups of HLA class I allotypes. In particular, KIR2DL1 recognizes HLA-C2 epitope, while KIR2DL2/L3 recognizes HLA-C1 epitope. KIR3DL1 is specific for HLA-B or HLA-A molecules sharing the Bw4 public epitope (Bw4I80 or Bw4T80), and KIR3DL2 binds HLA-A*03 and -A*11 allotypes (18, 21).
The activating NK cell receptors include a series of non-HLA-specific receptors and co-receptors able to induce NK cell triggering by directly interacting with ligands overexpressed or expressed de novo on tumor-transformed or virus-infected cells (23–25).
These findings indicate that autologous cells are not killed by NK cells thanks to an appropriate expression of all self-HLA alleles, while a wide spectrum of tumor types can be killed due to the loss of HLA molecules and to the expression/overexpression of ligands for NK cell activating receptors (Figure 1). During NK cell differentiation, CD94/NKG2A is the first HLA-I-specific receptor expressed by appearing on the most immature CD56bright NK cell subset. After several maturation steps, CD56bright cells become CD56dim, lose NKG2A, and acquire KIR receptors (26–28). The most mature NK cells are KIR+ and NKG2A– and express the marker of terminal differentiation CD57 (29).
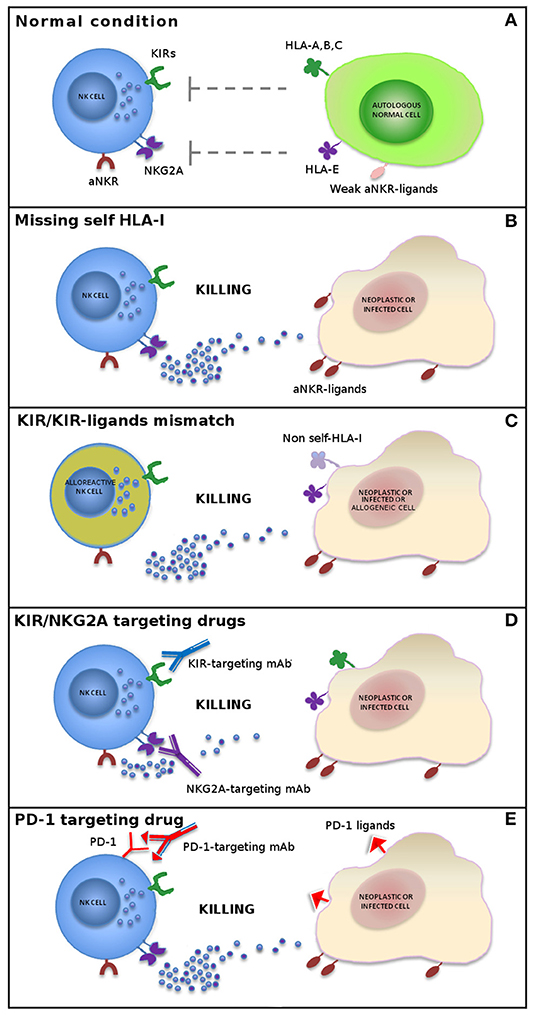
Figure 1. Mechanisms of NK cell-mediated killing. In physiological conditions, NK cell activity is tightly regulated by a complex interplay between inhibitory and activating receptors that prevents killing of normal autologous cells expressing an appropriate level of all self-HLA alleles and low/negative levels of ligands for non-HLA-specific activating receptors (aNKR) (A). Downregulation of HLA-I molecules on neoplastic or infected cells induces NK-mediated killing by a “missing-self” recognition mechanism. NK cell activating receptors are co-responsible in inducing NK cell triggering by interacting with ligands (aNKR-ligands) overexpressed or expressed de novo on tumor-transformed or virus-infected cells (B). Allogeneic (alloreactive) donor NK cells are able to kill neoplastic cells of the recipient expressing non-self allotypic determinants on HLA-I molecules (“KIR/KIR-ligand mismatch”) and to control infections with a limited risk of toxicity (e.g., GvHD and HvG) (C). The use of inhibitors of classical NK cell immune checkpoints (i.e., KIR and NKG2A) (D) or immune checkpoints shared with T cells (e.g., PD-1) (E) or, finally, a combination of these approaches represents new promising strategies in NK cell-based immunotherapy.
Under normal conditions, the HLA-I-specific inhibitory receptors recognize autologous cells and prevent auto-reactive responses. However, under pathological conditions, these receptors function as ICs, by blocking the cytotoxic activity of NK cells against those tumors that maintain the expression of HLA-I molecules (11, 30).
In order to restore NK cell activity against HLA-I+ tumor cells, novel immunotherapies have been developed, based on the use of therapeutic monoclonal antibodies anti-pan-KIR2D (lirilumab) (https://www.innate-pharma.com/en/pipeline/lirilumab-first-class-anti-kir-mab-licensed-bristol-myers-squibb) and anti-NKG2A (monalizumab) (https://www.innate-pharma.com/en/pipeline/monalizumab-anti-nkg2a-mab-partnered-astrazeneca) mimicking “missing-self” response by disrupting the interaction between these ICs and their ligands. Therefore, NK cells can efficiently kill tumor cells that have lost HLA-I expression, thus becoming resistant to T lymphocytes, but also HLA-I+ cancers when blockers of ICs are used (Figure 1). These agents are currently used in phase I/II clinical trials on a range of hematologic and solid tumors as monotherapy or in combination with other agents, including other forms of IC blockade (31–37).
Notably, NK cells may also express non-HLA class I-specific inhibitory receptors such as PD-1 (38). This receptor was originally discovered on T cells and was found to exert a sharp inhibitory effect on their anti-tumor activity. In healthy donors, PD-1 is expressed on a subset of fully mature (KIR+NKG2A–CD57+) NK cells from HCMV+ individuals (38). Higher proportions of PD-1+ NK cells can be detected in patients affected by different types of tumors (36, 38, 39).
The finding that NK cells from cancer patients express PD-1 IC coupled with the observation that the use of anti-PD-1 or anti-PD-L1 monoclonal antibodies improve the anti-tumor activity of NK cells (36, 38, 39) (Figure 1) is clinically relevant for patients with tumors displaying a T-cell-resistant (HLA class Ineg) phenotype.
Recent data strongly suggest a possible role for NK cells in immunotherapeutic strategies targeting the PD-1/PD-L1 axis particularly against HLA-I-deficient tumor cells (40, 41).
NK cells also express additional constitutive or inducible IC shared with T cells, recognizing additional ligands other than HLA class I molecules. These include CTLA-4, T cell immunoglobulin- and mucin-domain-containing molecule 3 (TIM-3), lymphocyte activation gene 3 (LAG-3), T cell immunoreceptor with Ig and immunoreceptor tyrosine-based inhibition motif domains (TIGIT), and CD96 (12, 42–44).
Here, we review recent developments to improve NK cell responses against solid and hematological tumors mainly focusing on NK cell ICs.
Although the ability of NK cells to destroy solid tumors has been questioned, their capacity to prevent metastatic dissemination by killing circulating cancer cells is well-known. However, tumor cells frequently develop strategies to evade NK cell immunosurveillance including changes at the tumor cell level (e.g., abnormal expression of ligands for activating and inhibitory receptors) and changes in tumor microenvironment (e.g., immunosuppressive cytokines), resulting in tumor escape and cancer progression (12, 45–48). Exact mechanisms and manipulation strategies to durably and reproducibly enhance NK cell function in vivo are not known. However, the use of IC blockade, including lirilumab and monalizumab, to create a condition of “missing-self” recognition (consequent to the antibody-mediated disruption of pan-KIR2D or NKG2A/HLA-I interactions) may represent a promising novel therapeutic approach to cure tumor patients (49).
HLA-E is one of the emerging suppressive ligands in human tumors, and its expression negatively correlates with the overall survival (OS) of cancer patients (48). NKG2A is expressed on NK cells but also on T cells infiltrating different types of solid tumors (36, 50, 51). These findings suggested that NKG2A/HLA-E interaction could suppress the cytotoxic lymphocyte functions directly in the tumor microenvironment.
The IgG4 anti-NKG2A antibody monalizumab is currently in clinical development for the treatment of various solid tumors, either as single-agent or in combination with other compounds. In the initial clinical experiences, single-agent intravenous monalizumab was administered to patients affected by advanced gynecologic malignancies (including ovarian, endometrial, and cervical carcinomas), divided into a dose-ranging cohort and an expansion cohort, for a total of 58 patients. The drug was generally well-tolerated but achieved only short-term disease stabilization as best response (NCT02459301) (37).
In another phase II study, monalizumab was administered in combination with the anti-epithelial growth factor receptor (EGFR) antibody cetuximab in patients affected by squamous cell carcinoma of the head and neck (SCCHN) (NCT02643550). Cetuximab represents an established therapeutic approach to SCCHN acting through induction of antibody-dependent cell cytotoxicity through CD16 (FcγRIII) receptor expressed on NK cells (52). The rationale of this combination lies on the evidence that SCCHN tumors were strongly positive for HLA-E and were infiltrated with CD8+ T and NK cells, suggesting a potential sensitivity to NKG2A inhibitors. The regimen was well tolerated, being characterized mostly by grade 1–2 adverse events, and the interim analysis reported an overall response rate (ORR) of 31% and a disease stabilization rate of 54%. Although preliminary, these data appear encouraging (37).
With regard to the combination of anti-NKG2A with PD-1/PD-L1 disrupting agents, a combination of monalizumab and durvalumab has been evaluated in a first-in-human dose-escalation/dose-expansion phase I trial in patients with metastatic microsatellite-stable colorectal cancer (MSS-CRC). The rationale of this study was supported by preclinical models (https://www.innate-pharma.com/sites/default/files/180205asco_15poster_09.pdf) and was based on the hypothesis that the inhibition of NKG2A might improve the efficacy of an anti-PD-L1 antibody in a patient population characterized by poor response to PD-1/PD-L1 antibodies. The study included 40 patients in the MSS-CRC expansion cohort. The treatment was well-tolerated; in the expansion cohort, three responses and 11 disease stabilizations were observed, with a disease control rate of 24% at 16 weeks (https://ascopubs.org/doi/abs/10.1200/JCO.2018.36.15_suppl.3540). Currently, other clinical trials involving the combination of monalizumab with durvalumab in different solid tumors are ongoing (NCT03794544; NCT02671435).
The efficacy and safety of the first-in-class anti-pan-KIR2D agent lirilumab was explored in several clinical trials. In a first-in-human phase I trial, escalating doses of lirilumab were administered to patients with solid tumors (breast, kidney, or ovarian carcinoma) or hematologic malignancies. No dose-limiting toxicities were reported and maximum tolerated dose was not reached for doses up to 10 mg/kg (53). With regard to combinations including lirilumab, a phase I/II trial explored the safety of increasing doses of lirilumab in combination with the anti-PD-1 antibody nivolumab or with the anti-CTLA-4 antibody ipilimumab in a population of patients with solid tumors (136 with nivolumab; 22 with ipilimumab). Both combinations were well-tolerated, encouraging further developments. Although definitive efficacy results are not available yet, data from 29 patients with SCCHN in the lirilumab-nivolumab cohort showed an ORR equal to 24%, with durable responses. Notably, increased PD-L1 expression was strongly associated with improved probability of objective response (https://news.bms.com/press-release/bristolmyers/interim-phase-12-data-show-encouraging-clinical-benefit-lirilumab-combina). Currently, other trials designed to explore the potential role of lirilumab plus nivolumab are being conducted in populations of patients with SCCHN and invasive bladder cancer in the neoadjuvant setting (NCT03532451 and NCT03341936).
Interestingly, in those tumors resistant to anti-PD-1 immune-therapies, an up-regulation of alternative immune checkpoints, including TIM-3, has been observed. In this context, therapeutic approaches combining the administration of anti-TIM-3 and anti-PD-1 antibodies showed that the adaptive resistance to PD-1 blockade can be overcome (54).
TIM-3 is a checkpoint receptor that binds several ligands including galectin-9 (Gal-9) (55), phosphatidylserine on apoptotic cells (56), high mobility group box 1 (56), and CEA-related cell adhesion molecule-1 (57). High frequencies of circulating and/or tumor infiltrating TIM-3+ NK cells have been found in different types of malignant tumors (58–60). The increased surface levels of TIM-3 on NK cells in cancers induce NK cell impairments (61), while TIM-3 blockade results in increased NK cell cytotoxicity both in vitro and ex vivo (59, 62, 63).
TIM-3 functions as a potential prognostic marker in several tumor types, such as lung adenocarcinoma, gastric cancer, bladder cancer, and esophageal cancer (58, 59, 62, 63).
On the other hand, contradictorily, studies have also reported stimulatory functions of TIM-3 (64). These divergent functions are likely associated with the existence of multiple and different TIM-3 ligands.
An anti-TIM-3 (Sym023) antibody has been developed and is currently being tested in phase I clinical trials in patients with advanced, unresectable, and metastatic solid tumor malignancies or lymphomas that are refractory to currently available therapies, in monotherapy or in combination with anti-PD-1 or anti-LAG-3 antibodies (NCT03489343 and NCT03311412). Additional phase I studies of anti-TIM-3 antibodies have been activated in patients with advanced solid tumors, as a monotherapy or in combination with an anti-PD-1 antibody (NCT02817633, NCT03680508, NCT04139902, and NCT03744468).
LAG-3 is a negative co-inhibitory receptor expressed on T cells and NK cells that binds MHC class II (MHC-II) molecules, the C-type lectin receptor LSECtin, and a fibrinogen-like protein 1 (FGL1) on target cells (65–68). LAG-3 has been shown to suppress immune responses in several tumors, including Hodgkin's lymphoma, gastric cancer, breast cancer, and other solid tumors (69). Thus, the use of anti-LAG-3 antibodies in combination with anti-PD-1 immunotherapy has been proposed to restore T cell function (70). Although the specific role of LAG-3 on NK cells remains to be fully clarified, this inhibitory immune checkpoint is currently considered a good target for immunotherapy because of its potential to activate both T and NK cells. In this context, different anti-LAG-3 antibodies are currently being used in phase I and phase II clinical trials as single drugs in metastatic cancer, solid tumor, and lymphoma (NCT03489369 and NCT03250832) or in association with other immune checkpoints inhibitors, including anti-TIM-3 antibody, in multiple myeloma patients (NCT04150965), with anti-PD-1 antibodies in treating patients with glioblastoma (NCT02658981), solid tumors (NCT01968109), advanced malignancies including lymphoma (NCT03005782), and SCCHN (NCT04080804) or other anti-PD-1 agents in solid tumor patients (NCT02676869). A number of additional LAG-3 antibodies are currently in preclinical development.
TIGIT and CD96 are co-inhibitory receptors expressed on both T and NK cells and compete with the activating NK cell receptor DNAM-1 for binding to PVR (CD155) and Nectin-2 (CD112) (71). These receptors participate in a balanced system to control NK cell effector functions. Indeed, following interaction with their ligands, TIGIT inhibits NK cell cytotoxicity directly through its ITIM domain, whereas CD96 hampers NK cell IFN-γ production (72), thus counterbalancing DNAM-1-mediated activation. The expression of TIGIT is highly variable among different cancer types. It has been recently demonstrated that TIGIT is highly expressed on tumor-infiltrating NK cells and associated with NK cell exhaustion in different tumor models and patients with colon cancer (73). Notably, the therapeutic effects of anti-TIGIT, anti-PD-L1, or anti-TIGIT antibodies combined with anti-PD-L1 antibodies depended on the presence of NK cells (73), indicating the importance of NK cells in checkpoint-targeted immunotherapy. Currently, several ongoing clinical trials (phase I and phase II) focus on testing the feasibility of targeting TIGIT pathway and improving therapeutic effects through combination with existing immunotherapies, including anti-PD-1 agents (NCT04150965, NCT03119428, NCT04047862, and NCT03563716), mainly in solid tumor patients.
Recently, results from clinical studies have demonstrated the safety of the infusion of allogeneic NK cells for immunotherapy of both hematological malignancies and solid tumors (74) despite the microenvironment rich of NK-inhibiting factors and the limited ability of infiltration of immune cells. While generally safe, allogenic NK cell infusions generally showed poor anti-tumor activity.
Therefore, strategies designed to improve the efficacy of adoptive transfer of NK cells are currently being explored and include associations with IC inhibitors or chemotherapy, induction of chemokine expression that can improve NK cell migration and trafficking into tumors, as well as concurrent administration of cytokines with activating effect on NK cells. However, while some of such approaches achieved encouraging results, none of these has evolved into an established protocol; hence, additional efforts in this setting are warranted (75–79). Relevant trials are reported in Table 1.
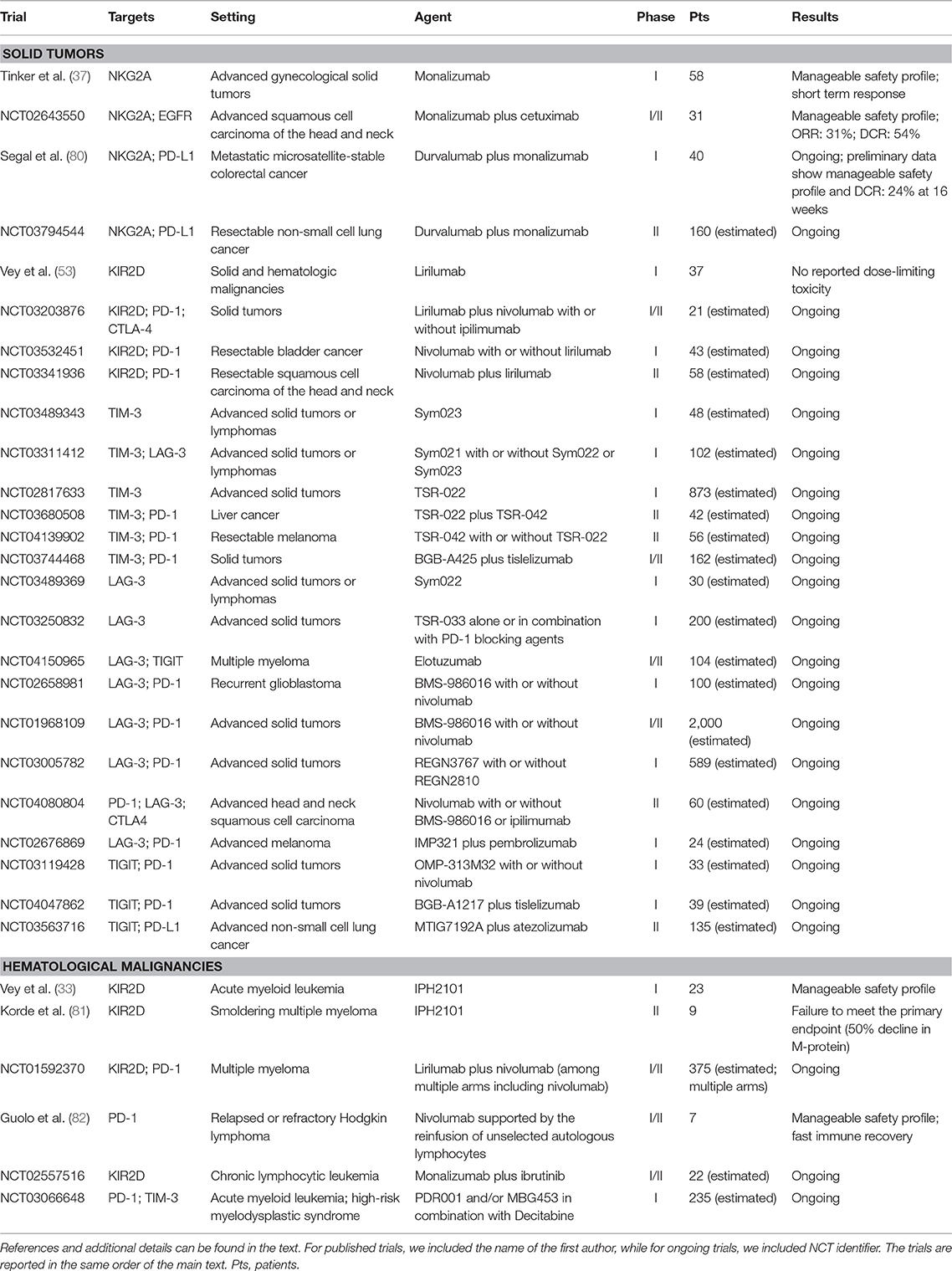
Table 1. Relevant clinical trials involving immune checkpoint blockade in NK cells.
Current approaches of NK cell immunotherapy for hematological malignancies involve methods for in vivo potentiation of NK cell proliferation and activity; adoptive transfer of NK cells from autologous and allogeneic sources, and NK cell lines; genetic modification of NK cells; and, similar to solid tumor, the use of IC blockade (12, 83).
Following the introduction of haploidentical hematopoietic stem cell transplantation (haplo-HSCT), the potential anti-leukemic activity of donor-derived alloreactive NK cells has been observed (84–87). After engraftment, NK cells are the first lymphocyte subset that appears in peripheral blood, suggesting their role as graft-vs.-leukemia effector cells, in the absence of GvHD (84, 88–90). The generation of alloreactive NK cells (i.e., NK cells expressing inhibitory KIRs that are not engaged by any of the HLA-I alleles present on allogeneic target cells) results in improved clinical outcome in haplo-HSCT thanks to their ability to kill not only leukemic blasts (GvL) but also patient dendritic cells (DCs) (91) and T cells, thus preventing GvHD and HvG reaction, respectively (92–95).
Given the crucial role of alloreactive NK cells in mediating safe and durable anti-tumor immunity in patients receiving KIR/HLA-C-mismatched transplantation, different strategies, including miRNA targeting the expression of HLA-C-specific KIRs (i.e., KIR2DL1, KIR2DL2/L3) (96) or therapeutic antibody blocking these inhibitory KIRs (lirilumab) for HLA-C, were generated to simulate the mechanism of “missing self” condition.
The IgG4 antibody lirilumab prevents inhibitory signals from pan-KIR2D, increasing NK cell-mediated tumor killing of AML in vitro and in vivo (31). This anti-pan-KIR2D antibody had acceptable safety without significant toxicity or autoimmunity in AML and multiple myeloma (MM) patients (33, 97). Although lirilumab failed to show significant efficacy as a monotherapy in MM, dual immune therapy with lenalidomide showed a good response on relapsed/refractory (R/R) MM in clinical trials (81, 97). Lirilumab is currently being widely tested in combination with other therapeutics, including rituximab (an anti-CD20 antibody), and other forms of IC blockade, such as nivolumab in R/R non-Hodgkin's lymphoma, Hodgkin's lymphoma (HL), and MM patients (NCT01592370) (34, 97).
In R/R HL patients (98–102), IC inhibitors have shown good activity; indeed, nivolumab is currently approved for this indication by the Food and Drug Administration and the European Medicines Agency (103). Interestingly, the Hodgkin neoplastic cells (Reed Sternberg cells) hyper-express PD-L1 but have low/negative expression of HLA-I molecules. This means that NK cells that exert their killing activity mainly against HLA-Ineg targets, but not cytotoxic T CD8+ cells, may be the primary effectors in the immune response induced by nivolumab in HL patients. In this view, an increase in cytotoxic NK cell population during IC blockade treatment in HL patients has been recently observed (104). In order to better elucidate the exact mechanism of action of nivolumab in HL and to improve the efficacy in terms of complete response (CR) in R/R HL, an innovative clinical protocol based on the combined application of high-dose chemotherapy with autologous stem cell transplant (ASCT) and early post-transplant administration of nivolumab, supported by autologous lymphocytes re-infusions (ALI), has been recently proposed (https://doi.org/10.1182/blood-2018-99-118901). Preliminary observations support the hypothesis that NK cells play a primary role in response to nivolumab in HL patients (82, 104). Furthermore, from a clinical point of view, preliminary data are encouraging, as all six R/R patients treated so far achieved CR (82).
Leukemic cells have been shown to overexpress HLA-E, a ligand for NKG2A. This provides the rationale for the use of monalizumab for the treatment of leukemia. The use of monalizumab in HSCT has recently also been proposed to induce NK cell alloreactivity in the first weeks after transplant when almost all reconstituting NK cells are NKG2Apos, in order to limit opportunistic infection and leukemia relapse (105). Moreover, monalizumab is currently under evaluation in several phase I/II clinical trials in monotherapy (35) or in combination with other therapeutic antibodies, such as the Bruton's tyrosine kinase inhibitor ibrutinib, in patients with R/R or previously untreated chronic lymphocytic leukemia (NCT02557516).
A phase I clinical trial evaluating the hypomethylating agent decitabine together with either PDR001 (anti-PD-1 antibody), MBG453 (anti-TIM-3 antibody), or their combination is currently recruiting patients with R/R AML patients not eligible for intensive therapy, as well as high-risk myelodysplastic patients (NCT03066648).
Another anti-TIM-3 antibody (Sym023) is currently being tested in phase I clinical trials in patients with lymphoma refractory to currently available therapies, in monotherapy or in combination with anti-PD-1 (Sym021) or anti-LAG-3 (Sym022) antibodies (NCT03489343 and NCT03311412).
The anti-LAG-3 antibody BMS-986016 is currently being used in phase I and phase II clinical trials in combination with nivolumab in subjects with relapsed or refractory HL, and relapsed or refractory diffuse large B cell lymphoma (DLBCL) (NCT02061761) or as a single drug in lymphoma (NCT03489369). Relevant trials are reported in Table 1.
Several molecular mechanisms regulating the anti-tumor activity of NK cells have been discovered over the last decades. However, further characterization of the main immunosuppressive pathways developed by tumor cells to evade NK cell recognition is still needed. Analysis of NK cells and tumor cells and their relationship is necessary to define personalized immunotherapy procedures in cancer patients.
The combined blockade of checkpoint molecules expressed by T cells and NK cells could trigger antitumor immunity mediated by innate and adaptive populations, allowing the two approaches to complement each other. NK cell-targeted immunotherapy may provide an alternative, or a complementary approach, to overcome the limitations of T-cell immunotherapy. In addition, combination with NK cell immunotherapy could increase the response rate of treatments targeting T cells (Figure 1 and Table 1).
In conclusion, considering the excellent outcome of some patients, future efforts should be addressed to identify the best inhibitory pathways to target for future clinical applications. Moreover, further studies should aim at improving NK cell-based immunotherapy by targeting the tumor-induced NK cell inhibition, thus promoting the maximal anti-tumor effect of these innate effectors.
All authors listed have made a substantial, direct and intellectual contribution to the work, and approved it for publication.
This work was supported by Fondazione Associazione Italiana per la Ricerca sul Cancro (IG 2017-20312 and 5x1000-21147); Fondazione Roche (Progetto Roche per la Ricerca 2017); Fondazione Beat Leukemia Dr. Alessandro Cevenini Onlus, via Bellini 27, 20900 Monza (MB) (http://www.beat-leukemia.org, mail: Z3JvLmFpbWVrdWVsLXRhZWJAb2ZuaQ==); and an Italian Society of Experimental Hematology (SIES) fellowship to PM.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
2. Sharma P, Wagner K, Wolchok JD, Allison JP. Novel cancer immunotherapy agents with survival benefit: recent successes and next steps. Nat Rev Cancer. (2011) 11:805–12. doi: 10.1038/nrc3153
3. Topalian SL, Hodi FS, Brahmer JR, Gettinger SN, Smith DC, McDermott DF, et al. Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. N Engl J Med. (2012) 366:2443–54. doi: 10.1056/NEJMoa1200690
4. Pardoll DM. The blockade of immune checkpoints in cancer immunotherapy. Nat Rev Cancer. (2012) 12:252–64. doi: 10.1038/nrc3239
5. Simsek M, Tekin SB, Bilici M. Immunological agents used in cancer treatment. Eurasian J Med. (2019) 51:90–4. doi: 10.5152/eurasianjmed.2018.18194
6. Tivol EA, Borriello F, Schweitzer AN, Lynch WP, Bluestone JA, Sharpe AH. Loss of CTLA-4 leads to massive lymphoproliferation and fatal multiorgan tissue destruction, revealing a critical negative regulatory role of CTLA-4. Immunity. (1995) 3:541–7. doi: 10.1016/1074-7613(95)90125-6
7. Nishimura H, Nose M, Hiai H, Minato N, Honjo T. Development of lupus-like autoimmune diseases by disruption of the PD-1 gene encoding an ITIM motif-carrying immunoreceptor. Immunity. (1999) 11:141–51. doi: 10.1016/S1074-7613(00)80089-8
8. Wang Y, Wu L, Tian C, Zhang Y. PD-1-PD-L1 immune-checkpoint blockade in malignant lymphomas. Ann Hematol. (2018) 97:229–37. doi: 10.1007/s00277-017-3176-6
9. Smahel M. PD-1/PD-L1 blockade therapy for tumors with downregulated MHC class I expression. Int J Mol Sci. (2017) 18:E1331. doi: 10.3390/ijms18061331
10. Fang F, Xiao W, Tian Z. NK cell-based immunotherapy for cancer. Semin Immunol. (2017) 31:37–54. doi: 10.1016/j.smim.2017.07.009
11. Pesce S, Greppi M, Grossi F, Del Zotto G, Moretta L, Sivori S, et al. PD/1-PD-Ls checkpoint: insight on the potential role of NK cells. Front Immunol. (2019) 10:1242. doi: 10.3389/fimmu.2019.01242
12. Di Vito C, Mikulak J, Zaghi E, Pesce S, Marcenaro E, Mavilio D. NK cells to cure cancer. Semin Immunol. (2019) 41:101272. doi: 10.1016/j.smim.2019.03.004
13. Kiessling R, Klein E, Pross H, Wigzell H. “Natural” killer cells in the mouse. II. Cytotoxic cells with specificity for mouse Moloney leukemia cells. Characteristics of the killer cell. Eur J Immunol. (1975) 5:117–21. doi: 10.1002/eji.1830050209
14. Herberman RB, Nunn ME, Lavrin DH. Natural cytotoxic reactivity of mouse lymphoid cells against syngeneic acid allogeneic tumors. I. Distribution of reactivity and specificity. Int J Cancer. (1975) 16:216–29. doi: 10.1002/ijc.2910160204
15. Ljunggren HG, Karre K. Experimental strategies and interpretations in the analysis of changes in MHC gene expression during tumour progression. Opposing influences of T cell and natural killer mediated resistance? J Immunogenet. (1986) 13:141–51. doi: 10.1111/j.1744-313X.1986.tb01095.x
16. Ljunggren HG, Karre K. In search of the 'missing self': MHC molecules and NK cell recognition. Immunol Today. (1990) 11:237–44. doi: 10.1016/0167-5699(90)90097-S
17. Yokoyama WM, Seaman WE. The Ly-49 and NKR-P1 gene families encoding lectin-like receptors on natural killer cells: the NK gene complex. Annu Rev Immunol. (1993) 11:613–35. doi: 10.1146/annurev.iy.11.040193.003145
18. Moretta A, Bottino C, Vitale M, Pende D, Biassoni R, Mingari MC, et al. Receptors for HLA class-I molecules in human natural killer cells. Annu Rev Immunol. (1996) 14:619–48. doi: 10.1146/annurev.immunol.14.1.619
19. Moretta A, Bottino C, Vitale M, Pende D, Cantoni C, Mingari MC, et al. Activating receptors and coreceptors involved in human natural killer cell-mediated cytolysis. Annu Rev Immunol. (2001) 19:197–223. doi: 10.1146/annurev.immunol.19.1.197
20. Moretta A, Tambussi G, Bottino C, Tripodi G, Merli A, Ciccone E, et al. A novel surface antigen expressed by a subset of human CD3- CD16+ natural killer cells. Role in cell activation and regulation of cytolytic function. J Exp Med. (1990) 171:695–714. doi: 10.1084/jem.171.3.695
21. Moretta A, Bottino C, Pende D, Tripodi G, Tambussi G, Viale O, et al. Identification of four subsets of human CD3-CD16+ natural killer (NK) cells by the expression of clonally distributed functional surface molecules: correlation between subset assignment of NK clones and ability to mediate specific alloantigen recognition. J Exp Med. (1990) 172:1589–98. doi: 10.1084/jem.172.6.1589
22. Braud VM, Allan DS, O'Callaghan CA, Soderstrom K, D'Andrea A, Ogg GS, et al. HLA-E binds to natural killer cell receptors CD94/NKG2A, B and C. Nature. (1998) 391:795–9. doi: 10.1038/35869
23. Sivori S, Carlomagno S, Pesce S, Moretta A, Vitale M, Marcenaro E. TLR/NCR/KIR: which one to use and when? Front Immunol. (2014) 5:105. doi: 10.3389/fimmu.2014.00105
24. Vivier E, Raulet DH, Moretta A, Caligiuri MA, Zitvogel L, Lanier LL, et al. Innate or adaptive immunity? The example of natural killer cells. Science. (2011) 331:44–9. doi: 10.1126/science.1198687
25. Marcenaro E, Dondero A, Moretta A. Multi-directional cross-regulation of NK cell function during innate immune responses. Transpl Immunol. (2006) 17:16–9. doi: 10.1016/j.trim.2006.09.019
26. Di Santo JP. Natural killer cell developmental pathways: a question of balance. Annu Rev Immunol. (2006) 24:257–86. doi: 10.1146/annurev.immunol.24.021605.090700
27. Freud AG, Caligiuri MA. Human natural killer cell development. Immunol Rev. (2006) 214:56–72. doi: 10.1111/j.1600-065X.2006.00451.x
28. Romagnani C, Juelke K, Falco M, Morandi B, D'Agostino A, Costa R, et al. CD56brightCD16- killer Ig-like receptor- NK cells display longer telomeres and acquire features of CD56dim NK cells upon activation. J Immunol. (2007) 178:4947–55. doi: 10.4049/jimmunol.178.8.4947
29. Bjorkstrom NK, Riese P, Heuts F, Andersson S, Fauriat C, Ivarsson MA, et al. Malmberg. expression patterns of NKG2A, KIR, and CD57 define a process of CD56dim NK-cell differentiation uncoupled from NK-cell education. Blood. (2010) 116:3853–64. doi: 10.1182/blood-2010-04-281675
30. Moretta L, Bottino C, Pende D, Vitale M, Mingari MC, Moretta A. Different checkpoints in human NK-cell activation. Trends Immunol. (2004) 25:670–6. doi: 10.1016/j.it.2004.09.008
31. Romagne F, Andre P, Spee P, Zahn S, Anfossi N, Gauthier L, et al. Preclinical characterization of 1-7F9, a novel human anti-KIR receptor therapeutic antibody that augments natural killer-mediated killing of tumor cells. Blood. (2009) 114:2667–77. doi: 10.1182/blood-2009-02-206532
32. Zaghi E, Calvi M, Marcenaro E, Mavilio D, Di Vito C. Targeting NKG2A to elucidate natural killer cell ontogenesis and to develop novel immune-therapeutic strategies in cancer therapy. J Leukoc Biol. (2019) 105:1243–51. doi: 10.1002/JLB.MR0718-300R
33. Vey N, Bourhis JH, Boissel N, Bordessoule D, Prebet T, Charbonnier A, et al. A phase 1 trial of the anti-inhibitory KIR mAb IPH2101 for AML in complete remission. Blood. (2012) 120:4317–23. doi: 10.1182/blood-2012-06-437558
34. Kohrt HE, Thielens A, Marabelle A, Sagiv-Barfi I, Sola C, Chanuc F, et al. Anti-KIR antibody enhancement of anti-lymphoma activity of natural killer cells as monotherapy and in combination with anti-CD20 antibodies. Blood. (2014) 123:678–86. doi: 10.1182/blood-2013-08-519199
35. McWilliams EM, Mele JM, Cheney C, Timmerman EA, Fiazuddin F, Strattan EJ, et al. Therapeutic CD94/NKG2A blockade improves natural killer cell dysfunction in chronic lymphocytic leukemia. Oncoimmunology. (2016) 5:e1226720. doi: 10.1080/2162402X.2016.1226720
36. Andre P, Denis C, Soulas C, Bourbon-Caillet C, Lopez J, Arnoux T, et al. Anti-NKG2A mAb is a checkpoint inhibitor that promotes anti-tumor immunity by unleashing both T and NK cells. Cell. (2018) 175:1731–43 e13. doi: 10.1016/j.cell.2018.10.014
37. Tinker AV, Hirte HW, Provencher DM, Butler MO, Ritter H, Tu D, et al. Dose-ranging and cohort-expansion study of monalizumab(IPH2201) in advanced gynecologic malignancies: A trial of the Canadian Cancer Trials Group (CCTG):IND221. Clin Cancer Res. (2019) 25:6052–60. doi: 10.1158/1078-0432.CCR-19-0298
38. Pesce S, Greppi M, Tabellini G, Rampinelli F, Parolini S, Olive D, et al. Identification of a subset of human natural killer cells expressing high levels of programmed death 1: a phenotypic and functional characterization. J Allergy Clin Immunol. (2017) 139:335–46 e3. doi: 10.1016/j.jaci.2016.04.025
39. Beldi-Ferchiou A, Lambert M, Dogniaux S, Vely F, Vivier E, Olive D, et al. PD-1 mediates functional exhaustion of activated NK cells in patients with Kaposi sarcoma. Oncotarget. (2016) 7:72961–77. doi: 10.18632/oncotarget.12150
40. Chen Z, Yang Y, Liu LL, Lundqvist A. Strategies to augment Natural Killer (NK) cell activity against solid tumors. Cancers. (2019) 11:E1040. doi: 10.3390/cancers11071040
41. Hsu J, Hodgins JJ, Marathe M, Nicolai CJ, Bourgeois-Daigneault MC, Trevino TN, et al. Contribution of NK cells to immunotherapy mediated by PD-1/PD-L1 blockade. J Clin Invest. (2018) 128:4654–68. doi: 10.1172/JCI99317
42. Sun H, Sun C. The rise of NK cell checkpoints as promising therapeutic targets in cancer immunotherapy. Front Immunol. (2019) 10:2354. doi: 10.3389/fimmu.2019.02354
43. Sivori S, Vacca P, Del Zotto G, Munari E, Mingari MC, Moretta L. Human NK cells: surface receptors, inhibitory checkpoints, and translational applications. Cell Mol Immunol. (2019) 16:430–41. doi: 10.1038/s41423-019-0206-4
44. Del Zotto G, Marcenaro E, Vacca P, Sivori S, Pende D, Della Chiesa M, et al. Markers and function of human NK cells in normal and pathological conditions. Cytometry B Clin Cytom. (2017) 92:100–14. doi: 10.1002/cyto.b.21508
45. Pesce S, Tabellini G, Cantoni C, Patrizi O, Coltrini D, Rampinelli F, et al. B7-H6-mediated downregulation of NKp30 in NK cells contributes to ovarian carcinoma immune escape. Oncoimmunology. (2015) 4:e1001224. doi: 10.1080/2162402X.2014.1001224
46. Isidori A, Salvestrini V, Ciciarello M, Loscocco F, Visani G, Parisi S, et al. The role of the immunosuppressive microenvironment in acute myeloid leukemia development and treatment. Expert Rev Hematol. (2014) 7:807–18. doi: 10.1586/17474086.2014.958464
47. Noguchi T, Ward JP, Gubin MM, Arthur CD, Lee SH, Hundal J, et al. Temporally distinct PD-L1 expression by tumor and host cells contributes to immune escape. Cancer Immunol Res. (2017) 5:106–17. doi: 10.1158/2326-6066.CIR-16-0391
48. Chiossone L, Vienne M, Kerdiles YM, Vivier E. Natural killer cell immunotherapies against cancer: checkpoint inhibitors and more. Semin Immunol. (2017) 31:55–63. doi: 10.1016/j.smim.2017.08.003
49. Muntasell A, Ochoa MC, Cordeiro L, Berraondo P, Lopez-Diaz de Cerio A, Cabo M, et al. Targeting NK-cell checkpoints for cancer immunotherapy. Curr Opin Immunol. (2017) 45:73–81. doi: 10.1016/j.coi.2017.01.003
50. Malmberg KJ, Levitsky V, Norell H, de Matos CT, Carlsten M, Schedvins K, et al. IFN-gamma protects short-term ovarian carcinoma cell lines from CTL lysis via a CD94/NKG2A-dependent mechanism. J Clin Invest. (2002) 110:1515–23. doi: 10.1172/JCI15564
51. Mamessier E, Sylvain A, Thibult ML, Houvenaeghel G, Jacquemier J, Castellano R, et al. Human breast cancer cells enhance self tolerance by promoting evasion from NK cell antitumor immunity. J Clin Invest. (2011) 121:3609–22. doi: 10.1172/JCI45816
52. van Sorge NM, van der Pol WL, van de Winkel JG. FcgammaR polymorphisms: implications for function, disease susceptibility and immunotherapy. Tissue Antigens. (2003) 61:189–202. doi: 10.1034/j.1399-0039.2003.00037.x
53. Vey N, Karlin L, Sadot-Lebouvier S, Broussais F, Berton-Rigaud D, Rey J, et al. A phase 1 study of lirilumab (antibody against killer immunoglobulin-like receptor antibody KIR2D; IPH2102) in patients with solid tumors and hematologic malignancies. Oncotarget. (2018) 9:17675–88. doi: 10.18632/oncotarget.24832
54. Koyama S, Akbay EA, Li YY, Herter-Sprie GS, Buczkowski KA, Richards WG, et al. Adaptive resistance to therapeutic PD-1 blockade is associated with upregulation of alternative immune checkpoints. Nat Commun. (2016) 7:10501. doi: 10.1038/ncomms10501
55. Zhu C, Anderson AC, Schubart A, Xiong H, Imitola J, Khoury SJ, et al. The Tim-3 ligand galectin-9 negatively regulates T helper type 1 immunity. Nat Immunol. (2005) 6:1245–52. doi: 10.1038/ni1271
56. Nakayama M, Akiba H, Takeda K, Kojima Y, Hashiguchi M, Azuma M, et al. Tim-3 mediates phagocytosis of apoptotic cells and cross-presentation. Blood. (2009) 113:3821–30. doi: 10.1182/blood-2008-10-185884
57. Huang YH, Zhu C, Kondo Y, Anderson AC, Gandhi A, Russell A, et al. CEACAM1 regulates TIM-3-mediated tolerance and exhaustion. Nature. (2015) 517:386–90. doi: 10.1038/nature13848
58. Wang Z, Zhu J, Gu H, Yuan Y, Zhang B, Zhu D, et al. The clinical significance of abnormal Tim-3 expression on NK cells from patients with gastric cancer. Immunol Invest. (2015) 44:578–89. doi: 10.3109/08820139.2015.1052145
59. da Silva IP, Gallois A, Jimenez-Baranda S, Khan S, Anderson AC, Kuchroo VK, et al. Reversal of NK-cell exhaustion in advanced melanoma by Tim-3 blockade. Cancer Immunol Res. (2014) 2:410–22. doi: 10.1158/2326-6066.CIR-13-0171
60. Komita H, Koido S, Hayashi K, Kan S, Ito M, Kamata Y, et al. Expression of immune checkpoint molecules of T cell immunoglobulin and mucin protein 3/galectin-9 for NK cell suppression in human gastrointestinal stromal tumors. Oncol Rep. (2015) 34:2099–105. doi: 10.3892/or.2015.4149
61. Gallois A, Silva I, Osman I, Bhardwaj N. Reversal of natural killer cell exhaustion by TIM-3 blockade. Oncoimmunology. (2014) 3:e946365. doi: 10.4161/21624011.2014.946365
62. Ju Y, Hou N, Meng J, Wang X, Zhang X, Zhao D, et al. T cell immunoglobulin- and mucin-domain-containing molecule-3 (Tim-3) mediates natural killer cell suppression in chronic hepatitis B. J Hepatol. (2010) 52:322–9. doi: 10.1016/j.jhep.2009.12.005
63. Xu L, Huang Y, Tan L, Yu W, Chen D, Lu C, et al. Increased Tim-3 expression in peripheral NK cells predicts a poorer prognosis and Tim-3 blockade improves NK cell-mediated cytotoxicity in human lung adenocarcinoma. Int Immunopharmacol. (2015) 29:635–41. doi: 10.1016/j.intimp.2015.09.017
64. De Sousa Linhares A, Leitner J, Grabmeier-Pfistershammer K, Steinberger P. Not all immune checkpoints are created equal. Front Immunol. (2018) 9:1909. doi: 10.3389/fimmu.2018.01909
65. Baixeras E, Huard B, Miossec C, Jitsukawa S, Martin M, Hercend T, et al. Characterization of the lymphocyte activation gene 3-encoded protein. A new ligand for human leukocyte antigen class II antigens. J Exp Med. (1992) 176:327–37. doi: 10.1084/jem.176.2.327
66. Sun H, Sun C, Xiao W. Expression regulation of co-inhibitory molecules on human natural killer cells in response to cytokine stimulations. Cytokine. (2014) 65:33–41. doi: 10.1016/j.cyto.2013.09.016
67. Xu F, Liu J, Liu D, Liu B, Wang M, Hu Z, et al. LSECtin expressed on melanoma cells promotes tumor progression by inhibiting antitumor T-cell responses. Cancer Res. (2014) 74:3418–28. doi: 10.1158/0008-5472.CAN-13-2690
68. Wang J, Sanmamed MF, Datar I, Su TT, Ji L, Sun J, et al. Fibrinogen-like protein 1 is a major immune inhibitory ligand of LAG-3. Cell. (2019) 176:334–47 e12. doi: 10.1016/j.cell.2018.11.010
69. Anderson AC, Joller N, Kuchroo VK. Lag-3, Tim-3, and TIGIT: co-inhibitory receptors with specialized functions in immune regulation. Immunity. (2016) 44:989–1004. doi: 10.1016/j.immuni.2016.05.001
70. Blackburn SD, Shin H, Haining WN, Zou T, Workman CJ, Polley A, et al. Coregulation of CD8+ T cell exhaustion by multiple inhibitory receptors during chronic viral infection. Nat Immunol. (2009) 10:29–37. doi: 10.1038/ni.1679
71. Martinet L, Smyth MJ. Balancing natural killer cell activation through paired receptors. Nat Rev Immunol. (2015) 15:243–54. doi: 10.1038/nri3799
72. Nold-Petry CA, Lo CY, Rudloff I, Elgass KD, Li S, Gantier MP, et al. IL-37 requires the receptors IL-18Ralpha and IL-1R8 (SIGIRR) to carry out its multifaceted anti-inflammatory program upon innate signal transduction. Nat Immunol. (2015) 16:354–65. doi: 10.1038/ni.3103
73. Zhang Q, Bi J, Zheng X, Chen Y, Wang H, Wu W, et al. Blockade of the checkpoint receptor TIGIT prevents NK cell exhaustion and elicits potent anti-tumor immunity. Nat Immunol. (2018) 19:723–32. doi: 10.1038/s41590-018-0132-0
74. Lupo KB, Matosevic S. Natural Killer cells as allogeneic effectors in adoptive cancer immunotherapy. Cancers. (2019) 11:E769. doi: 10.3390/cancers11060769
75. Oyer JL, Gitto SB, Altomare DA, Copik AJ. PD-L1 blockade enhances anti-tumor efficacy of NK cells. Oncoimmunology. (2018) 7:e1509819. doi: 10.1080/2162402X.2018.1509819
76. Matosevic S. Viral and nonviral engineering of natural killer cells as emerging adoptive cancer immunotherapies. J Immunol Res. (2018) 2018:4054815. doi: 10.1155/2018/4054815
77. Kremer V, Ligtenberg MA, Zendehdel R, Seitz C, Duivenvoorden A, Wennerberg E, et al. Genetic engineering of human NK cells to express CXCR2 improves migration to renal cell carcinoma. J Immunother Cancer. (2017) 5:73. doi: 10.1186/s40425-017-0275-9
78. Marcenaro E, Pesce S, Sivori S, Carlomagno S, Moretta L, Moretta A. KIR2DS1-dependent acquisition of CCR7 and migratory properties by human NK cells interacting with allogeneic HLA-C2+ DCs or T-cell blasts. Blood. (2013) 121:3396–401. doi: 10.1182/blood-2012-09-458752
79. Marcenaro E, Cantoni C, Pesce S, Prato C, Pende D, Agaugue S, et al. Uptake of CCR7 and acquisition of migratory properties by human KIR+ NK cells interacting with monocyte-derived DC or EBV cell lines: regulation by KIR/HLA-class I interaction. Blood. (2009) 114:4108–16. doi: 10.1182/blood-2009-05-222265
80. Segal NH, Naidoo J, Curigliano G, Patel S, Sahebjam S, Papadopoulos K, et al. First-in-Human Dose Escalation of Monalizumab Plus Durvalumab With Expansion in Patients With Metastatic Microsatellite-Stable Colorectal Cancer. Available online at: https://www.innate-pharma.com/sites/default/files/180205asco_15poster_09.pdf
81. Korde N, Carlsten M, Lee MJ, Minter A, Tan E, Kwok M, et al. A phase II trial of pan-KIR2D blockade with IPH2101 in smoldering multiple myeloma. Haematologica. (2014) 99:e81–3. doi: 10.3324/haematol.2013.103085
82. Guolo F, Minetto P, Ballerini F, Canale FA, Frello M, Coviello E, et al. Feasibility and efficacy of post-transplant consolidation immunotherapy with nivolumab supported by the reinfusion of unselected autologous lymphocytes in patients affected by relapsed/refractory Hodgkin lymphoma. Blood. (2018) 132(Suppl. 1):4598. doi: 10.1182/blood-2018-99-118901
83. Sivori S, Meazza R, Quintarelli C, Carlomagno S, Della Chiesa M, Falco M, et al. NK cell-based immunotherapy for hematological malignancies. J Clin Med. (2019) 8:E1702. doi: 10.3390/jcm8101702
84. Ruggeri L, Capanni M, Urbani E, Perruccio K, Shlomchik WD, Tosti A, et al. Effectiveness of donor natural killer cell alloreactivity in mismatched hematopoietic transplants. Science. (2002) 295:2097–100. doi: 10.1126/science.1068440
85. Pende D, Marcenaro S, Falco M, Martini S, Bernardo ME, Montagna D, et al. Anti-leukemia activity of alloreactive NK cells in KIR ligand-mismatched haploidentical HSCT for pediatric patients: evaluation of the functional role of activating KIR and redefinition of inhibitory KIR specificity. Blood. (2009) 113:3119–29. doi: 10.1182/blood-2008-06-164103
86. Giebel S, Locatelli F, Lamparelli T, Velardi A, Davies S, Frumento G, et al. Survival advantage with KIR ligand incompatibility in hematopoietic stem cell transplantation from unrelated donors. Blood. (2003) 102:814–9. doi: 10.1182/blood-2003-01-0091
87. Locatelli F, Pende D, Mingari MC, Bertaina A, Falco M, Moretta A, et al. Cellular and molecular basis of haploidentical hematopoietic stem cell transplantation in the successful treatment of high-risk leukemias: role of alloreactive NK cells. Front Immunol. (2013) 4:15. doi: 10.3389/fimmu.2013.00015
88. Handgretinger R, Lang P, Andre MC. Exploitation of natural killer cells for the treatment of acute leukemia. Blood. (2016) 127:3341–9. doi: 10.1182/blood-2015-12-629055
89. Curti A, Ruggeri L, D'Addio A, Bontadini A, Dan E, Motta MR, et al. Successful transfer of alloreactive haploidentical KIR ligand-mismatched natural killer cells after infusion in elderly high risk acute myeloid leukemia patients. Blood. (2011) 118:3273–9. doi: 10.1182/blood-2011-01-329508
90. Curti A, Ruggeri L, Parisi S, Bontadini A, Dan E, Motta MR, et al. Larger size of donor alloreactive NK cell repertoire correlates with better response to NK cell immunotherapy in elderly acute myeloid leukemia patients. Clin Cancer Res. (2016) 22:1914–21. doi: 10.1158/1078-0432.CCR-15-1604
91. Marcenaro E, Ferranti B, Moretta A. NK-DC interaction: on the usefulness of auto-aggression. Autoimmun Rev. (2005) 4:520–5. doi: 10.1016/j.autrev.2005.04.015
92. Ruggeri L, Mancusi A, Burchielli E, Aversa F, Martelli MF, Velardi A. Natural killer cell alloreactivity and haplo-identical hematopoietic transplantation. Cytotherapy. (2006) 8:554–8. doi: 10.1080/14653240601078721
93. Moretta L, Locatelli F, Pende D, Marcenaro E, Mingari MC, Moretta A. Killer Ig-like receptor-mediated control of natural killer cell alloreactivity in haploidentical hematopoietic stem cell transplantation. Blood. (2011) 117:764–71. doi: 10.1182/blood-2010-08-264085
94. Parham P, McQueen KL. Alloreactive killer cells: hindrance and help for haematopoietic transplants. Nat Rev Immunol. (2003) 3:108–22. doi: 10.1038/nri999
95. Marcenaro E, Carlomagno S, Pesce S, Della Chiesa M, Moretta A, Sivori S. Role of alloreactive KIR2DS1(+) NK cells in haploidentical hematopoietic stem cell transplantation. J Leukoc Biol. (2011) 90:661–7. doi: 10.1189/jlb.0311137
96. Pesce S, Squillario M, Greppi M, Loiacono F, Moretta L, Moretta A, et al. New miRNA signature heralds human NK cell subsets at different maturation steps: involvement of miR-146a-5p in the regulation of KIR expression. Front Immunol. (2018) 9:2360. doi: 10.3389/fimmu.2018.02360
97. Benson DM Jr, Bakan CE, Mishra A, Hofmeister CC, Efebera Y, Becknell B, et al. The PD-1/PD-L1 axis modulates the natural killer cell versus multiple myeloma effect: a therapeutic target for CT-011, a novel monoclonal anti-PD-1 antibody. Blood. (2010) 116:2286–94. doi: 10.1182/blood-2010-02-271874
98. Stiff PJ, Unger JM, Forman SJ, McCall AR, LeBlanc M, Nademanee AP, et al. Southwest oncology: the value of augmented preparative regimens combined with an autologous bone marrow transplant for the management of relapsed or refractory Hodgkin disease: a Southwest Oncology Group phase II trial. Biol Blood Marrow Transplant. (2003) 9:529–39. doi: 10.1016/S1083-8791(03)00205-2
99. Schmitz N, Pfistner B, Sextro M, Sieber M, Carella AM, Haenel M, et al. Aggressive conventional chemotherapy compared with high-dose chemotherapy with autologous haemopoietic stem-cell transplantation for relapsed chemosensitive Hodgkin's disease: a randomised trial. Lancet. (2002) 359:2065–71. doi: 10.1016/S0140-6736(02)08938-9
100. Spaepen K, Stroobants S, Dupont P, Vandenberghe P, Maertens J, Bormans G, et al. Prognostic value of pretransplantation positron emission tomography using fluorine 18-fluorodeoxyglucose in patients with aggressive lymphoma treated with high-dose chemotherapy and stem cell transplantation. Blood. (2003) 102:53–9. doi: 10.1182/blood-2002-12-3842
101. Fanale MA, Forero-Torres A, Rosenblatt JD, Advani RH, Franklin AR, Kennedy DA, et al. A phase I weekly dosing study of brentuximab vedotin in patients with relapsed/refractory CD30-positive hematologic malignancies. Clin Cancer Res. (2012) 18:248–55. doi: 10.1158/1078-0432.CCR-11-1425
102. Younes A, Gopal AK, Smith SE, Ansell SM, Rosenblatt JD, Savage KJ, et al. Results of a pivotal phase II study of brentuximab vedotin for patients with relapsed or refractory Hodgkin's lymphoma. J Clin Oncol. (2012) 30:2183–9. doi: 10.1200/JCO.2011.38.0410
103. Ansell SM, Lesokhin AM, Borrello I, Halwani A, Scott EC, Gutierrez M, et al. PD-1 blockade with nivolumab in relapsed or refractory Hodgkin's lymphoma. N Engl J Med. (2015) 372:311–9. doi: 10.1056/NEJMoa1411087
104. Armand P, Chen YB, Redd RA, Joyce RM, Bsat J, Jeter E, et al. PD-1 blockade with pembrolizumab for classical Hodgkin lymphoma after autologous stem cell transplantation. Blood. (2019) 134:22–9. doi: 10.1182/blood.2019000215
105. Roberto A, Di Vito C, Zaghi E, Mazza EMC, Capucetti A, Calvi M, et al. The early expansion of anergic NKG2A(pos)/CD56(dim)/CD16(neg) natural killer represents a therapeutic target in haploidentical hematopoietic stem cell transplantation. Haematologica. (2018) 103:1390–402. doi: 10.3324/haematol.2017.186619
Keywords: NK cells, NK cell receptors, immune checkpoint blockade, immunotherapy, solid tumors, hematological malignancies, adoptive NK cell therapy
Citation: Minetto P, Guolo F, Pesce S, Greppi M, Obino V, Ferretti E, Sivori S, Genova C, Lemoli RM and Marcenaro E (2019) Harnessing NK Cells for Cancer Treatment. Front. Immunol. 10:2836. doi: 10.3389/fimmu.2019.02836
Received: 14 August 2019; Accepted: 18 November 2019;
Published: 06 December 2019.
Edited by:
Loredana Cifaldi, Bambino Gesù Children Hospital (IRCCS), ItalyReviewed by:
Dr. Kawaljit Kaur, University of California, Los Angeles, United StatesCopyright © 2019 Minetto, Guolo, Pesce, Greppi, Obino, Ferretti, Sivori, Genova, Lemoli and Marcenaro. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Simona Sivori, c2ltb25hLnNpdm9yaUB1bmlnZS5pdA==; Emanuela Marcenaro, ZW1hbnVlbGEubWFyY2VuYXJvQHVuaWdlLml0
†These authors share first authorship
‡These authors share last authorship
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.