 Karan Sachdeva1
Karan Sachdeva1 Danh C. Do
Danh C. Do Yan Zhang
Yan Zhang Xinyue Hu
Xinyue Hu Peisong Gao
Peisong Gao
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol. , 29 November 2019
Sec. Inflammation
Volume 10 - 2019 | https://doi.org/10.3389/fimmu.2019.02787
This article is part of the Research Topic Immune Mechanisms in the Pathologic Response to Particles, Fibers, and Nanomaterials View all 17 articles
Environmental pollutants and allergens induce oxidative stress and mitochondrial dysfunction, leading to key features of allergic asthma. Dysregulations in autophagy, mitophagy, and cellular senescence have been associated with environmental pollutant and allergen-induced oxidative stress, mitochondrial dysfunction, secretion of multiple inflammatory proteins, and subsequently development of asthma. Particularly, particulate matter 2.5 (PM2.5) has been reported to induce autophagy in the bronchial epithelial cells through activation of AMP-activated protein kinase (AMPK), drive mitophagy through activating PTEN-induced kinase 1(PINK1)/Parkin pathway, and induce cell cycle arrest and senescence. Intriguingly, allergens, including ovalbumin (OVA), Alternaria alternata, and cockroach allergen, have also been shown to induce autophagy through activation of different signaling pathways. Additionally, mitochondrial dysfunction can induce cell senescence due to excessive ROS production, which affects airway diseases. Although autophagy and senescence share similar properties, recent studies suggest that autophagy can either accelerate the development of senescence or prevent senescence. Thus, in this review, we evaluated the literature regarding the basic cellular processes, including autophagy, mitophagy, and cellular senescence, explored their molecular mechanisms in the regulation of the initiation and downstream signaling. Especially, we highlighted their involvement in environmental pollutant/allergen-induced major phenotypic changes of asthma such as airway inflammation and remodeling and reviewed novel and critical research areas for future studies. Ultimately, understanding the regulatory mechanisms of autophagy, mitophagy, and cellular senescence may allow for the development of new therapeutic targets for asthma.
Asthma is a leading serious chronic illness of children and adults worldwide, and its prevalence has been increasing over the past few decades (1, 2). Million people worldwide are affected, including 24 million in the United States (3). Asthma is chronic airway inflammation characterized by airway hyper-responsiveness, wheezing, cough, and dyspnea, and has become a major contributing factor to missed time from school and work, and is also a major cause of hospitalization and emergency department visits. It is well-recognized that the increase in asthma prevalence may be mainly attributed to industrialization- and urbanization-generated environmental pollutants (4–10). In China, a study of over 30,000 adults showed that the prevalence of persistent cough, sputum production, and wheezing was associated with major traffic roads, factories, and large smokestacks (11). This was supported by another cross-sectional study of 23,326 Chinese children, which showed that the prevalence of asthma was higher for those residing near areas with serious air pollution (12). Diesel exhaust particles (DEPs) are of particular concern and contributed to more than 90% of the particulate matters (PMs) derived from traffic sources in European and American cities (13). Particulate matter 2.5 (PM2.5), one of the major pollutants in urban areas, accounts for a large proportion of the atmospheric particulate matter and increased prevalence and symptom severity in children and adult patients with asthma (14–16) and other respiratory diseases (17, 18). PM2.5 as a mixture of various chemical constituents has been shown to promote oxidative stress and inflammation (19). Furthermore, concentrated transition metals in the environment have been shown to stimulate the production of reactive oxygen species (ROS) 19, leading to airway injury and inflammation (20).
In addition to environmental pollutants, it is well-known that environmental allergens are also major players in the development of allergic sensitization and asthma. Importantly, recent studies made novel findings that environmental pollutants co-exposure with allergens can lead to increased allergic sensitization and severe asthma (21–23). Particularly, prenatal exposure to DEPs is associated with an increased risk of allergic sensitization, early childhood wheeze, and asthma (24, 25). Of interest, co-exposure to DEP and house dust mite (HDM) can promote allergic sensitization and induce major features of a more severe asthma (9, 26–29). Furthermore, we have recently shown that benzo(a)pyrene (BaP) co-exposure with dermatophagoides group 1 (Der f 1) can activate aryl hydrocarbon receptor (AhR) signaling, which regulates ROS generation and TSLP and IL-33 expression (30). Similarly, a very recent study demonstrated that PM2.5 disturbs the balance of Th17/Treg cells by impairing differentiation of Treg cells and promoting differentiation of Th17 cells through the molecular pathways AhR–HIF-1α (hypoxia-inducible factor-1alpha) and AhR–Got1 (glutamate oxaloacetate transaminase 1) in a cockroach allergen-induced mouse model of asthma (31). Warren et al. reported that acute inhalant exposure to an agriculture acquired organic dust extract (ODE) impacts lung inflammatory responses in a murine model of experimental allergic asthma, suggesting that allergic asthma may prime the lung microenvironment response toward an exaggerated response following exposure to a dusty farm environment (32). Thus, future studies are warranted to identify the underlying mechanisms regarding the co-exposure-induced exacerbation of allergic asthma. In this review, we evaluated the literature regarding the basic cellular processes, including autophagy, mitophagy, and cellular senescence, and discussed their involvement in environmental pollutant/allergen-induced major features of asthma and biological regulation. Additionally, we identified areas of unmet research needed and their potentials as novel therapeutic avenues for the treatment of asthma and allergic diseases.
It has been postulated that dysregulation of basic cellular processes which maintain homeostasis and physiological balance may lead to the key clinical features of asthma. Autophagy, a homeostatic process with multiple effects on immunity, has been shown to play important roles in causing downstream changes initiated by environmental pollutants, allergens, and respiratory tract infections (33–40). Autophagy is a mechanism in which the eukaryotic cell encapsulates damaged proteins or organelles for lysosomal degradation and recycling (41). The autophagic pathway has recently been suggested to be involved in the several key features of asthma pathogenesis, including eosinophilic airway inflammation (42), airway hyper-responsiveness (36), and airway remodeling (43). It has been shown that PM2.5 exposure can induce cell autophagy and airway inflammation through different immunological and molecular mechanisms (44–46). Furthermore, exposure to allergens has also been shown to activate autophagy, as demonstrated in studies with cockroach allergen (47), Alternaria extract (48), and caffeine (49).
Autophagy is a process that has been maintained over ages of evolution, and by which damaged and misfolded proteins along with aged or damaged organelles are transported to lysosomes for elimination and digestion (50). Currently, three major types of autophagy are recognized: macroautophagy, microautophagy, and chaperone-mediated autophagy (51). Of these, macroautophagy is the most extensively studied, which uses autophagosomes, double-membraned vesicles, to engulf cytoplasmic proteins and organelles for delivery to the lysosome for degradation. Autophagosomes fusing with lysosomes are termed autophagolysosomes (52). After fusion with lysosomes, the cargo delivered is degraded by lysosomal enzymes and then transported to the cytoplasm (53–55). The byproducts of lysosomal degradation (e.g., amino acids) are recycled and then used for protein synthesis that enables salvage of energy normally used in de novo synthesis. Microautophagy as a second type of autophagy does not require autophagosomes but involves the direct engulfment of the cargo that may include proteins and lipids by the invagination of the lysosomal membrane (56). Chaperone-mediated autophagy (CMA) as a third type of autophagy is unique to mammalian cells (57). CMA is a highly regulated cellular process that involves the degradation of a selective subset of cytosolic proteins in lysosomes. In contrast to macroautophagy that engulfs and delivers predominantly larger structures for bulk degradation of cargo, CMA delivers individual proteins for lysosomal degradation. CMA involves a co-chaperone complex led by heat shock cognate 70 (HSC70) that recognizes target proteins that have a KFERQ-like pentapeptide sequence (52). Chaperone-bound proteins are transported to lysosomes, in which they are recognized by the lysosome-associated membrane protein type 2a (LAMP2a) receptor, a major regulator of CMA. LAMP2a is a transmembrane protein component that oligomerizes and forms a translocon complex for internalization and degradation of chaperone-delivered cargo in the lysosome (58). In this review, we mainly focused on macroautophagy, the form of autophagy dealing with the destruction and recycling of damaged macromolecules and organelle structures, and highlighted the significance of macroautophagy in the maintenance of cellular energetic balance and homeostasis.
Significant progress has been made in understanding the molecular mechanisms of autophagy and the regulation of autophagy in the past 10 years (59). These studies, together with discoveries of the autophagy-related (ATG) genes and their associations with specific diseases (60, 61), provide a multidimensional perspective of mechanisms by which ATG gene-dependent autophagy pathways are critical in the pathogenesis of human diseases. The autophagy pathway is usually described as involving a set of 16–20 core conserved ATG genes. These core proteins are involved in regulating initiation of autophagy by the UNC51-like kinase (ULK) complex (e.g., ULK1, FIP200, ATG13), autophagosome nucleation (Beclin 1, VPS34, VPS15, and ATG14), autophagosome elongation and maturation (e.g., ATG5, ATG12, ATL16L1, ATG8/microtubule-associated protein 1 light chain 3 [LC3]), and induction of autophagosomes and fusion of autophagosomes with lysosomes (i.e., ATG9/mammalian Atg9 and vacuole membrane protein 1) (59, 62). Amongst these ATG proteins, LC3 is a well-defined protein, which is cleaved from a pro-form by Atg4 and then conjugated with phosphatidyl-ethanolamine by the sequential action of Atg7 and Atg3 (63) to form LC3-II (Figure 1). The conversion of LC3-I (unconjugated cytosolic form) to LC3-II (autophagosomal membrane-associated phosphatidylethanolamine-conjugated form) has been considered as a major feature of autophagosome formation. Additionally, SQSTM1/p62 has an ubiquitin binding domain and an LC3 interaction domain and thus can bring ubiquitinated cargos to the autophagosomes for autophagy.
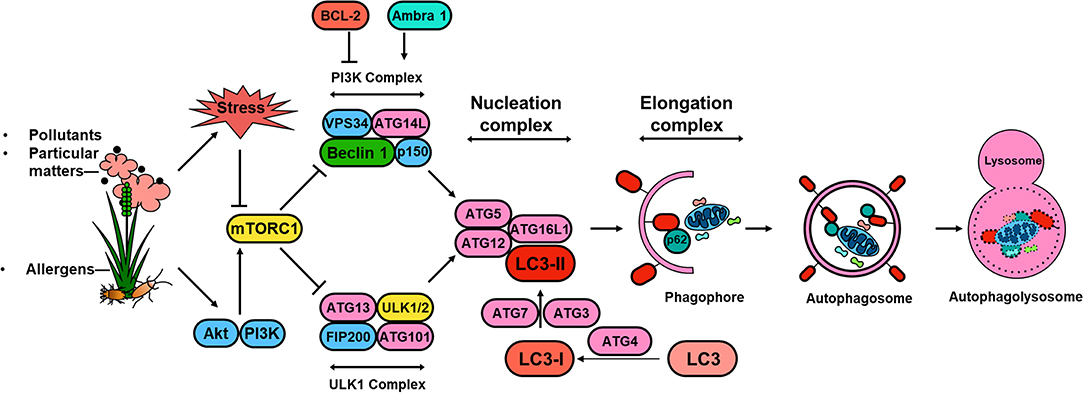
Figure 1. Schematic overview of autophagy regulation. Environmental signals, such as environmental pollutants and allergens, induce cellular stress leading to the activation of the mTOR signaling complex 1 (mTORC1). Induction of autophagy begins with the formation of the phagophore, which is initiated by the ULK complex, consisting of ULK1 (or ULK2), autophagy-related protein 13 (ATG13), FAK family kinase interacting protein of 200 kDa (FIP200) and ATG101. PI3K complex, consisting of the vacuolar protein sorting 34 (VPS34) and the regulator subunits ATG14L, p150 and beclin 1, provides further nucleation signal. Autophagosome formation requires phagophore membrane elongation by a complex composed of ATG5, ATG12, ATG16L, and LC3-II, which are derived from the microtubule-associated protein 1 light chain 3 (LC3) by the activity of ATG4 generating LC3-I and the conjugation C-terminal glycine of LC3-I to phosphatidylethanolamine by ATG7, and ATG3. The formation of the autophagolysosome is a result of the fusion between the autophagosome and lysosomal compartments. Lysosomal hydrolyases degrade the autophagy cargo in all three processes.
Significant numbers of signaling molecules particularly cytokine have been shown to regulate autophagy (52, 64). For example, IL-10 and IL-10 receptor signaling inhibits the starvation induced autophagy of murine macrophages via class I phosphatidylinositol 3-kinase (PI3K) pathway (64), suggesting that IL-10 plays a critical role in the autophagic process of macrophages. Distinct classes of PI3K have previously been shown to be involved in signaling pathways that control macro-autophagy in human colon cancer HT-29 cells (65, 66). Moreover, the Th1 cytokine IFN-gamma induces autophagy in macrophages (67). In contrast, Th2 cytokines, IL-4 and IL-13, inhibit autophagy in macrophages under starvation or IFN-gamma stimulation, and inhibit autophagy-mediated killing of intracellular mycobacteria in murine and human macrophages (68). Intriguingly, recent studies suggest that IL4 can induce autophagy in activated CD4+ Th2 cells (68), primary dendritic cells (DCs) (69), and primary B cells that exacerbates experimental asthma through different mechanisms (70). Similarly, IL-13 alone can activate autophagy in airway epithelial cells and drive the secretion of excess mucus (71). These findings suggest that Th2 cytokines may play a dual role in autophagy induction depending on different cell types. However, further studies are essential to investigate how differential modulation of autophagy by Th1 and Th2 cytokines in different cell types, which may represent a key feature of the host response to environmental stresses. Furthermore, neutralization of the receptors VEGFR, β-integrin or CXCR4, or IL-10 can also regulate autophagy by restoring autophagy in macrophage/monocytic cells exposed to HIV-1-infected cells (72). In contrast, autophagy can also regulate cytokine production (73). For example, Atg16L1 is an essential component of the autophagic machinery responsible for control of the endotoxin-induced IL-1β production (74). It has also been shown that autophagy influences IL-1β secretion by either targeting pro-IL-1β for lysomal degradation or regulating activation of the NLRP3 inflammasome (73). Similarly, autophagy plays a pivotal role in the induction and regulation of IL-23 secretion and innate immune responses through effects on IL-1 secretion (75). Furthermore, autophagy regulates inflammatory cytokine secretion [e.g., macrophage migration inhibitory factor (MIF)] by macrophages through controlling mitochondrial ROS (76). ROS can activate STAT3 transcriptional factor, leading to the secretion of IL-6 in starvation-induced autophagy of cancer cells (77). Interestingly, Alternaria extract as a major outdoor allergen can activate autophagy that subsequently induces IL-18 release from airway epithelial cells (48).
In addition to cytokines, several significant molecules have also been identified to regulate autophagy (52). Of these, mTOR (mammalian target of rapamycin) has been shown to regulate cell-signaling pathways after exposure to several major factors including amino acids, oxidative stress, energy levels, and growth factors (78, 79). Particularly, mTORC1 (one of the functional forms of mTOR) regulates autophagy by directly interacting with the ULK complex ULK1-ATG13-FIP200 (80). mTORC1 can suppress autophagy by inhibiting ATG1/ULK complexes under normal physiological conditions (51). In addition, AMP-activated protein kinase (AMPK)/ULK1 pathway mediates autophagy by transmitting stress signals for autophagosome formation, independent of mTOR signaling (80, 81). AMPK is capable of inhibiting non-autophagy VPS34 complexes but activating the proautophagy VPS34 by the phosphorylation of Beclin 1 (Beclin1/VPS34) to initiate phagophore formation (82). In addition to AMPK and mTORC1, calmodulin-dependent protein kinase II (CaMKII) also plays a role in engaging autophagy regulation (83). CaMKII, a serine/threonine-specific protein kinase regulated by the Ca2+/calmodulin complex, can directly phosphorylate Beclin 1 at Ser90 that enhances K63-linked ubiquitination of Beclin 1 and activation of autophagy (84). CaMKII can also stimulate K63-linked ubiquitination of inhibitor of differentiation 1/2 (Id-1/2). Of interest, the increased ubiquitinated Id-1/Id-2 can bind p62 and then be transported to autolysosomes for degradation, which can subsequently promote the differentiation of neuroblastoma cells and suppress the proportion of stem-like cells (84).
Recently, transcriptional regulation of autophagy genes has drawn a lot of attention in autophagic responses to specific stimuli (85). Several transcription factors and histone modifications have been identified to regulate autophagy gene expression. In addition to the well-known two transcription factors, p53 and Forkhead box O3 (FOXO3) (86), Transcription Factor EB (TFEB) is one of the most recently identified transcriptional regulators of autophagy (87). TFEB is highly phosphorylated by various kinases such as AKT, Extracellular Signal-Regulated Kinase 2 (ERK2), and mTORC1, and sequestered in the cytoplasm under nutrient rich conditions. In contrast, TFEB is dephosphorylated by calcineurin (CaN) and translocates to the nucleus where it activates autophagy and lysosome gene transcription upon nutrient deprivation (88). Forkhead box K (FOXK) engages in the transcriptional repression of autophagy gene expression by binding to promoter regions of early-stage autophagy genes (e.g., ULK complex) and recruits the SIN3A-Histone deacetylase (HDAC) repressor complex to these regions under nutrient rich conditions (89). However, most intriguingly, the post-translational modification status on histones is also linked to autophagy gene regulation, including histone H4K16 acetylation, H3K9 dimethylation, and H3K27 trimethylation (90). Of these, H4K16 acetylation suppresses autophagy gene expression through H4K16 acetyltransferase human Males absent On the First (hMOF) degradation and/or Sirtuin1 (SIRT1)-dependent histone deacetylation (91). H3K27 trimethylation catalyzed by Enhancer of Zeste Homolog 2 (EZH2) suppresses the expression of negative regulators of the mTORC1 signaling components and leads to mTORC1 activation and autophagy inhibition (92). Interestingly, many of the transcriptional factors that modulate expression of autophagy genes are regulated by common upstream kinases such as mTORC1 and AMPK. Furthermore, histone modification status is also a significant determinant of transcriptional regulators to autophagic stimuli.
Exposure to traffic and industrial pollution particulate matters, predominantly DEPs, have been shown to increase the risk of asthma (15, 26). Environmental pollutants (e.g., PM2.5) can induce ROS generation and impair lung function in asthmatic patients (93–97). It was well-documented that ROS are key mediators that contribute to oxidative damage and chronic airway inflammation in allergy and asthma (98–101). However, the underlying mechanisms still remain unclear. Recent studies have suggested that autophagy may be a new frontier in human asthma (50) and may play a crucial role in chronic airway inflammation (42). Indeed, higher autophagy levels have been shown in sputum granulocytes, peripheral blood cells and peripheral eosinophils of patients with severe asthma (102). The increased autophagy has been associated with important immune mechanisms and extracellular matrix deposition and fibrosis in airway remodeling in asthma (43). Furthermore, genetic mutations in autophagy genes have been associated with asthma. For example, single nucleotide polymorphisms in Atg5 are correlated with reduced lung function (103). Thus, these cumulative findings raise the possibility that environment/allergen exposure initiates the production of ROS in airway epithelial cells, which serve as “signaling molecules” modulating the process of autophagic cycle through activating signaling molecules and autophagy pathways, thereby leading to the major phenotypic changes of asthma as summarized in Figure 2, including airway inflammation, airway remodeling, and airway hyper-responsiveness.
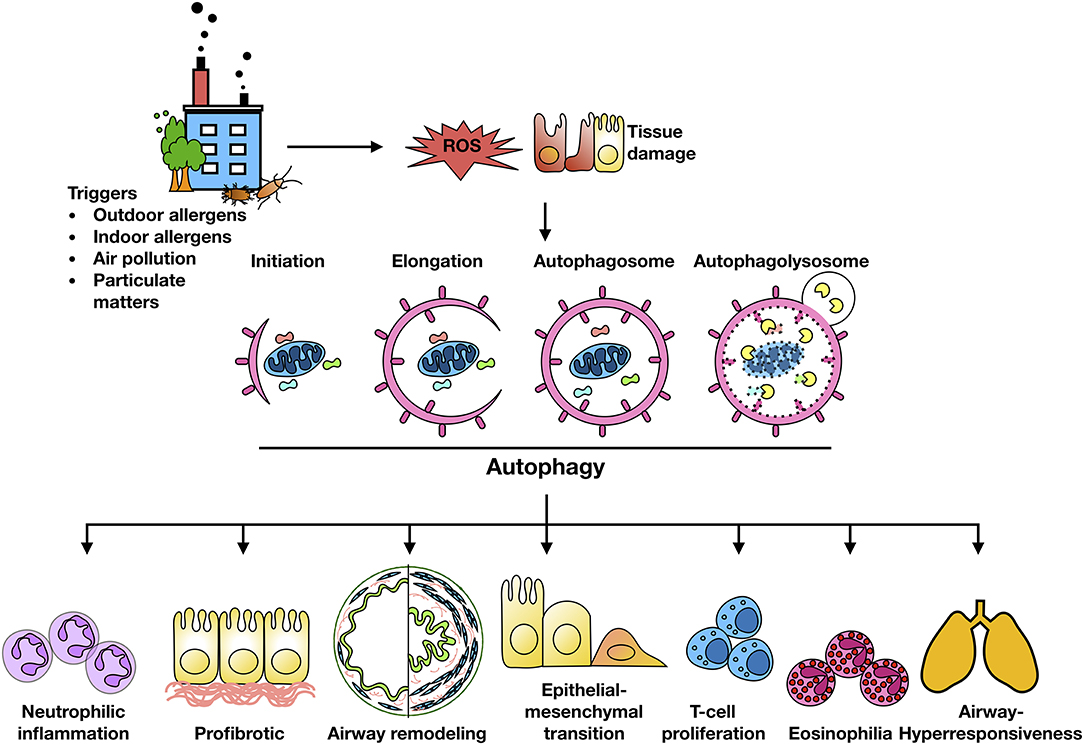
Figure 2. Autophagy and major features of asthma. Environment/allergen exposure initiates ROS generation in airway epithelial cells, which serve as “signaling molecules” modulating the process of autophagic cycle through activating signaling molecules and autophagy pathways, thereby leading to the major phenotypic changes of asthma, including airway inflammation, airway remodeling, and airway hyper-responsiveness.
Autophagy plays important roles in airway inflammation (36, 42). It has been suggested that autophagy plays a critical role in PM-induced inflammation in airway epithelium through the activation of NF-kB1 and activator protein-1 (AP-1) (104). Furthermore, PM2.5 can induce inflammatory cytokine release (e.g., IL-6, IL-8, IL-1β1, and TNFα) and oxidative injury of lung cells (105). Additionally, Long et al. found that PM2.5 can induce cell arrest in the G0/G1 phase and increase mitochondrial membrane potential, ROS generation, and airway epithelial cell apoptosis (106). PM2.5 not only induced the production of pro-inflammatory cytokine IL-6, TNFα, and activation of AMPK, but also promoted the expression of ATG5, Beclin-1 and LC3II in the airway epithelial cells (107). Interestingly, knockdown of ATG5 limited PM2.5 -induced autophagy, ROS generation, cell apoptosis, and production of IL-6 and TNFα. Mechanistically, this study suggests that the activation of AMPK may be critical in autophagy-mediated PM2.5-induced airway inflammation. In addition, allergens have also been shown to induce autophagy. OVA (ovalbumin) used in a murine asthma model can induce autophagy in airway tissues (36). Alternaria alternata as one of the major outdoor allergens that cause allergic airway diseases (108) has been shown to induce IL-18 secretion from airway epithelial cells, and thereby initiate Th2-type responses (109). IL-18 is a pro-inflammatory cytokine that belongs to the IL-1 family (110). Importantly, Alternaria extract stimulation can activate an autophagy-based unconventional secretion pathway and induce airway epithelial cells to release IL-18 via an autophagy dependent, but caspase 1 and 8 independent pathway (48). Studies from our research group showed that cockroach extract can induce autophagy in airway epithelial cells in vitro and in a mouse model of asthma (47). Further studies on the underlying mechanisms demonstrated that ROS and oxidized CaMKII (ox-CaMKII) in airway epithelial cells are critical in regulating cockroach allergen-induced autophagy (111).
Although environmental pollutants/allergens can induce autophagy, its role in airway inflammation remains unclear. It has been suggested that, at baseline, autophagy is critical for inhibiting spontaneous lung inflammation and is fundamental for airway mucus secretion by airway goblet cells. Autophagy deficient mice (Atg5−/− and Atg7−/−) develop spontaneous sterile lung inflammation (110). Similarly, deficiency of CD11c-specific autophagy results in severe IL-17A-mediated neutrophilic lung inflammation and unprovoked spontaneous airway hyperactivity (112). Furthermore, deficiency of ATG5 in airway epithelial cells results in an increased airway inflammation (113), and disruption or deletion of autophagy in airway epithelial cells resulted in airway hyperreactivity (114). Autophagy deficiency (ER-Cre: Atg7fl/fl) in mice after exposure to P. aeruginosa impairs pathogen clearance, increases neutrophilic inflammation, and the production of IL-1β (115). Although autophagy appears to be a protective mechanism, autophagy may also exacerbate airway inflammation. For example, inhibition of autophagy by 3-MA and intranasal knockdown of Atg5 led to marked improvement in AHR, eosinophilia, IL-5 levels in bronchoalveolar lavage fluid, and histological inflammatory features (36). Similarly, autophagy deficiency in macrophages (siRNA targeting PIK3C3) during LPS-induced lung inflammation attenuates lung and bronchoalveolar immune cell infiltration and air space cytokine levels (116). Additionally, IL-4-induced autophagy in B cells exacerbated asthma through an mTOR-independent, PtdIns3K-dependent pathway (70). Thus, autophagy may play diverse roles, either protective or detrimental, in asthma. Although the reason is unknown, it has been suggested that autophagy may represent a protective role in maintaining homeostasis at baseline or during acute infection, but play a detrimental role due to impaired autophagy or a persistent autophagy responses leading to an accumulation of excessive autophagosome in a prolonged exposure of environmental pollutants/allergens or inflammation. Furthermore, autophagy involvement in different cell types may result in different characteristic phenotypic changes. For example, deletion of ATG5 and ATG14 or pharmacological inhibition (e.g., 3-MA, Baf-A1) in cultured airway epithelial cells treated with IL-13 results in less mucus secretion and less CCL26 secretion. In contrast, autophagy deficiency in macrophages (117) or DCs (112) results in the exacerbation of inflammation. Thus, the investigation of the real impact of autophagy, protective or detrimental, is extremely challenging.
Recent studies have linked autophagy to the major features of airway remodeling in asthma, including airway smooth muscle (ASM) 44 (118–120), extracellular matrix (ECM) (121, 122), fibrosis (117), and epithelial-mesenchymal transition (EMT) (123). Particularly, it has been suggested that TGFβ1 induced autophagy is essential for collagen and fibronectin production in human airway smooth muscle cells, and deletion of Atg5 and Atg7 leads to reduction in pro-fibrotic signaling and ECM protein release (50, 124). In turn, autophagy has also been shown to participate in profibrotic changes induced by TGFβ1 (125). Furthermore, McAlinden et al. provided evidence of increased activation of the autophagy pathway in the airways of patients with asthma (43). Especially, they showed an association for TGFβ1 and accumulation of collagen and increased profibrotic signaling in an autophagy-dependent manner in ASM cells (43). Furthermore, inhibition of autophagy in murine model has been shown to attenuate airway inflammation and reduce the concentration of TGFβ1, and subsequently lead to a reduced airway remodeling. However, the critical mechanistic evidence is limited.
Mitophagy is the selective degradation of mitochondria by autophagy. It often happens to damaged mitochondria following the exposure to environmental pollutants/allergens or stress and plays a critical role in promoting turnover of mitochondria and preventing accumulation of dysfunctional mitochondria (38). Mitochondrial dysfunction and elevated ROS production have been associated with allergic diseases, including atopy, atopic dermatitis, and asthma (126–129). Of interest, a disturbance in the homeostasis of mitochondria leads to ROS generation, which cause weakened barriers and subsequently airway inflammation, epithelial fragility, and impaired secretion capacity (130). Furthermore, PM2.5-exposed rat lung injury is associated with mitochondrial fusion-fission dysfunction, mitochondrial lipid peroxidation and cellular homeostasis imbalance, and ROS generation, leading to the disruption of mitochondrial dynamics (131). PM2.5 can regulate the dynamics of mitochondria via facilitating mitochondrial fission, and the excess ROS induced by PM2.5 can trigger mitophagy by activating PINK1/Parkin pathway (132). Acrolein, an ubiquitous environmental pollutant that is abundant in tobacco smoke, cooking fumes, and automobile fumes (133), has also been reported to induce mitochondrial DNA (mtDNA) damages, mitochondrial fission and mitophagy in human lung cells (134). Of interest, mitophagy was found to prevent mitochondria-induced inflammation (mito-inflammation) (135). Thus, mitophagy may be critical in environmental pollutant/allergen-induced mitochondrial dysfunction and dysregulation of mitochondrial bioenergetics. These may ultimately result in a dysregulated mitophagic cycle and significant phenotypic changes observed in asthma (Figure 3).
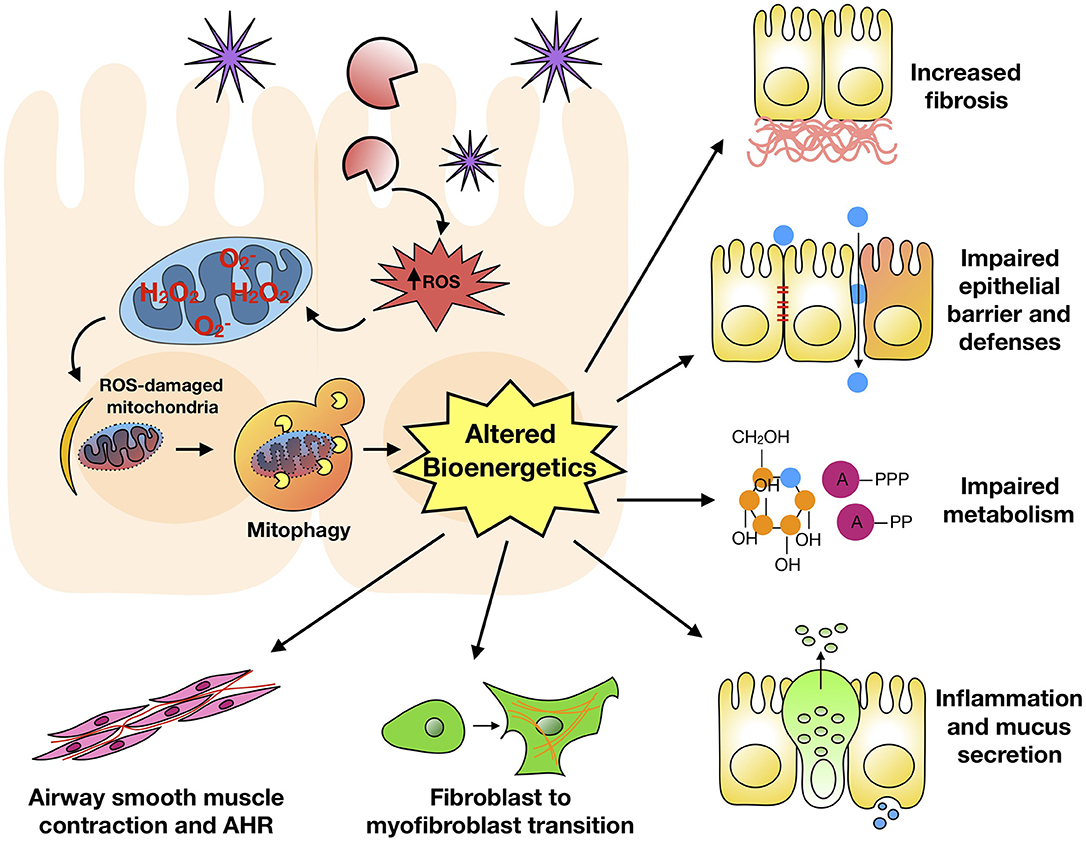
Figure 3. Mitophagy and major features of asthma. Environmental pollutant/allergen induces excessive ROS generation, mitochondrial dysfunction, and mitophagy, subsequently leading to dysregulation of mitochondrial bioenergetics. These may ultimately result in significant phenotypic changes observed in asthma.
Mitophagy is an evolutionarily conserved homeostatic process by which the cells selectively degrade only dysfunctional or damaged mitochondria (136, 137). Mitophagy is a normal physiological process during cell life and functions as surveiling mitochondrial population, eliminating superfluous and/or impaired organelles (137). Defective removal of damaged mitochondria leads to hyper-activation of inflammatory signaling pathways and subsequently to chronic systemic inflammation. However, important questions remain regarding the molecular mechanisms of mitophagy. It has been suggested that mitophagy is regulated by “PTEN-induced kinase 1 (PINK)-Parkin-mediated pathway” and “receptor-mediated pathway” (137). Under physiological conditions, the transport of PINK1 preprotein onto the inner mitochondrial membrane (IMM) is followed by sequential proteolytic cleavage by the mitochondrial processing peptidase and pre-protein-associated rhomboid-like protease (138). Under challenged condition, active PINK1 accumulates on the outer mitochondrial membrane (OMM) through its interaction with the translocations of the outer mitochondrial membrane complex (TOM complex), promoting Parkin recruitment through phosphoralation of both Parkin and ubiquitin (139). In turn, Parkin triggers the polyubiqutination of several OMM proteins, including voltage-dependent-anion-selective channer 1 (VDAC1), mitofusin 1 and 2 (MFN1/2), and mitochondrial import receptor subunit TOM20 homolog (TOMM20) 138. Several adaptor molecules [e.g., p62, optineurin (OPTN), and nuclear domain protein 52 (NDP52)] bindphosphorylated polyubiqutinated proteins and initiate autophagosomal formation through binding with LC3 (140). Recent studies suggest that both NDP5 and OPTN are phosphorylated by the Tank-binding kinase 1 (TBK-1) which enhances their binding affinity to ubiquitin (141). The OPTN-TBK1 complex forms a feed-forward mechanism that speeds up the mitochondrial clearance (142). In contrast, the receptor-mediated mitophagy is dependent on various OMM proteins such as Nip3-like protein X (NIX), BCL2 interacting protein 3 (BNIP3), and Fun14 domain-containing protein 1 (FUNDC 1) (137, 140). These proteins localize to the OMM and interact directly with LC3 to regulate mitochondrial elimination. Cardiolipin and prohibitin 2 (PHB2) are externalized to OMM and interact with LC3 in response to mitochondrial damage to promote the engulfment of defective mitochondria (143, 144).
Similar to autophagy, accumulating evidence suggests that both enhanced and impaired mitophagy has an important role in the pathogenesis of COPD and lung fibrosis (145–149). However, limited studies were found for asthma. Recent studies have shown that PM2.5 can induce increased ROS and mitochondrial damage, which triggers the mitophagy through activating PINK/Parkin pathway (132). In the nucleus, excessive ROS could activate HIF-1 FOXO3, and NRF2, which promote the transcription of BNIP33/NIX, LC3/BNIP3, and p62, thereby facilitate mitophagy (150). Furthermore, the impairment of mitochondrial degradation by mitophagy can lead to the accumulation of fragmented mitochondria and activation of the mitochondrial apoptosis pathway (134). Thus, these studies suggest that environmental pollutants can induce ROS and mitochondrial damage, which triggers mitophagy to maintain stable mitochondrial function in cells by scavenging impaired mitochondria and reducing excessive ROS. Further studies on regulatory mechanisms regarding ROS and mitophagy may provide a new angle on therapies for allergy and asthma.
Cellular senescence is characterized by irreversible cell cycle arrest and triggered by a number of factors such as aging, DNA damage, oxidative stress, mitochondrial dysfunction (151–153), telomere shortening (154, 155), epigenetic modifications (156), and inflammation (157). Senescence arrest occurs mostly in the G1 phase of the cell cycle, distinguishing it from G0-arrested quiescent cells, and is mediated by cyclin-dependent kinase inhibitors (CDKis) (e.g., p21CIP1, p16INK4a) and is dependent on the TP53 and pRB tumor suppressor pathways (158). Also, telomeres and nucleoprotein complexes located at the ends of linear chromosomes (159) are critical to the cellular senescence (160). Furthermore, senescent cells accumulated in tissues secrete a large amount of pro-inflammatory mediators termed the senescence associated secretory phenotype (SASP), which drives chronic inflammation, leading to further senescence (157). The composition of the SASP is stimulus-dependent and includes pro- and anti-inflammatory cytokines, chemokines, matrix metalloproteinases, growth factors, and other factors, and has an important role in the immune-mediated clearance of senescent cells and tissue dysfunction (160). Senescent cells have been found at sites of chronic age-related diseases like osteoarthritis (161), atherosclerosis (162–164), and aging lung (153), highlighting the significant role of senescent cells in the pathogenesis of chronic diseases. Senescent cells exhibit increased protein turnover and massive proteotoxic stress due to augmented autophagy and SASP component synthesis (165). Senescence cells also show increased rates of mitochondrial metabolic activity, including the tricarboxylic acid cycle, oxidative phosphorylation, and glycolytic pathways. Senescent cells have increased AMP/ADP:ATP and NAD+/NADH ratios, activating AMPK, which reinforces a TP53-dependent cell-cycle arrest (166, 167). In addition, senescent cells do not proliferate, but are resistant to autophagy and apoptosis, and are thus long living. Importantly, senescent cells can exacerbate mitochondrial dysfunction, inflammation, and other disease-promoting pathways through SASP (153).
Accumulation of senescent cells may slow or stop cell regeneration and tissue maintenance, thus leading to tissue aging (166). Indeed, clearing senescent cells from tissues of mouse models was shown sufficient to delay, prevent, or alleviate multiple age-related disorders (168). Although the underlying mechanisms regarding the elimination of senescent cells are poorly understood, the immune system has been recognized to be critical (169). Different immune cells have been suggested to be involved in the surveillance of senescent cells, including neutrophils, macrophages, natural killer cells, and CD4+ T cells (170). These immune cell-derived senescent cells can be immunogenic by expressing stimulatory ligands (e.g., MICA/B) that bind to NKG2D and activating their killing by NK cells (171). Furthermore, senescent cells can recruit immune cells to eliminate senescent cells by secreting cytokine and chemokines (172). Interestingly, recent studies support a balance between activating and inhibitory signals that will determine whether NK and T-cells respond to senescent cells. These studies also suggest a novel mechanism whereby the increased expression of HLA-E on senescent fibroblasts reduced the clearance of senescent cells by NK and CD8+ T cells expressing inhibitory receptor NKG2A (157). This represents a novel therapeutic approach to improve the immune clearance of senescent cells by blocking the interaction between HAL-E and NKG2A. In addition, the SASP-related cytokine IL-6 contributes to the increased expression of HAL-E in senescent cells, and that persistent inflammation may result in remaining of senescent cells in tissues, further contributing to the diseases.
Mitochondrial dysfunction has been demonstrated to be able to drive a cell into premature senescence, which affects airway diseases (151, 152). Indeed, the potential role of cell senescence in the pathogenesis of asthma has drawn great attention (162). Studies have implicated that cell senescence in the lung may be an important risk factor for the development of asthma (39, 173). Both COPD and idiopathic pulmonary fibrosis (IPF) are increased in prevalence with age and have been associated with senescence (174, 175). Senescence-related changes are also found in the lungs of adults with asthma, and in the airways of asthmatic children (176). However, mechanistic links between environmental pollutants, allergens, senescence, and pathophysiology of asthma have not been established. Studies have demonstrated that exposure to PM2.5 can induce senescence of human dermal fibroblasts (177). Increased exposure to PM2.5 is correlated with shortened telomeres in placental tissues and umbilical cord blood (178). Similarly, Bisphenol A (BPA) can induce Th2 inflammatory cascade and trigger DSB-ATM-p53 signaling pathway leading to cell cycle arrest, senescence, autophagy, and stress response in human fetal lung fibroblasts (179). Telomeres are critical to the cellular senescence (160), and telomere shortening is a strong indication of cellular senescence. In a study of 730 mother-baby pairs, increased exposure to PM2.5 has been shown to correlate with shortened telomeres in placental tissue and umbilical blood (178). Telomere shortening was also associated with airway hyper-responsiveness and is an inducer of accelerated replicative senescence of bronchial fibroblasts in patients with asthma (154). This was supported by findings in the chronic asthmatic patients who also displayed shorter telomere lengths and suggested that asthma chronicity may be associated with telomere length even at early ages (155). Furthermore, TSLP-induced cellular senescence with elevated p21 and p16 in human epithelial cells was essential for airway remodeling in vitro (39). This was further supported by the fact that inhibition of TSLP signaling attenuates epithelial senescence, airway hyper-reactivity, and airway remodeling in an OVA mouse model (39). Plasminogen activator inhibitor (PAI-1), a well-known cell senescence and fibrosis mediator, could activate p53 and mediate bleomycine- and doxorubicin-induced alveolar type II (ATII) cell senescence (180). Further studies suggest that PAI-1 mediates TGF-β1-induced ATII cell senescence, which may contribute to lung fibrogenesis in part by activating alveolar macrophages via secreting pro-fibrotic and pro-inflammatory mediators. Interestingly, this effect is highly dependent on the target cell, because it seems that PAI-1 has opposite effects on fibroblasts and ATII cells in patients with IPF (181). Together, these findings suggest a significant role of senescence in airway fibrosis and remodeling.
Many studies also support the rationale that senescence is associated with a higher pro-inflammatory cytokine profile (182). Of note, higher amounts of IL-6 have been found in patients with asthma and have been shown to trigger or to reinforce premature cellular senescence (183). This IL-6 driven immune-senescence may serve as part of a feed forward loop that drives asthma progression and reduces the efficacy of anti-inflammatory treatments. SASPs released from senescent cells contain inflammatory cytokines that may increase inflammation and impair cellular function in asthma. Furthermore, elevated p21 expression in asthmatic epithelium is not reduced with corticosteroid treatment (40), and in turn, loss of p16INK4a protein results in decreased cell sensitivity to dexamethasone treatment (184), raising the possibility that senescence may play an important role in glucocorticoid resistance in the patients with asthma. Indeed, lymphocyte senescence in COPD has been suggested to be associated with loss of glucocorticoid receptor (GCR) expression by pro-inflammatory/cytotoxic lymphocytes (185). Thus, investigation into the role of senescence in glucocorticoid resistance may provide novel approaches for the treatment of asthma. In addition, although current studies suggest a role of senescence in asthma, little is known about the pathway regarding the environmental pollutants/allergens, ROS, mitochondrial dysfunction, and senescence. Furthermore, it is poorly understood about the mechanistic links between cell senescence and asthma pathophysiology. Additionally, asthma is typically associated with an imbalance between Th1 and Th2 pathways, and over-driven Th2-mediated inflammation can result in airway inflammation and asthma (186). On the other hand, immune-senescence has been associated with lung aging, and that altered Th1/Th2 imbalance may contribute to the process of accelerated lung aging and immune-senescence (187). Indeed, with aging, mouse lungs showed typically increased Th1 cells with increased levels of IFN-gamma. However, the link between Th1/Th2 cells and senescence remains largely un-explored. Thus, further research is needed to establish the mechanistic links between increased cytokines with aging and senescent cell induction, and asthma pathophysiology.
Autophagy plays a role in homeostatic energy supply and elimination of aggregate-prone proteins, damaged organelles, and intracellular microbes. Autophagy also plays a critical role in the regulation of innate and adaptive immune responses in response to environmental stresses. In contrast, cellular senescence is caused by insufficient regulatory mechanisms of homeostasis. One of the most common causes for cellular senescence is that mitochondrial dysfunction results in cellular senescence due to excessive ROS production (153). Although autophagy and senescence are known to share similar properties, recent studies suggest a “double-edged” sword that autophagy can either accelerate the development of senescence or prevent senescence (188, 189). Especially, autophagy can produce large amounts of recycled amino acids, which trigger the production of SASP (e.g., IL-6, IL-8) through the activation of mTOR, thereby leading to senescence (190). By contrast, inhibition of autophagy or insufficient autophagy may promote cell senescence (190). Recent studies suggest that autophagy could be either pro-senescent or anti-senescent, depending on the type of autophagy (general or selective), stimulatory signals, and can be cell-type specific (189). Although it seems that autophagy and senescence are highly related, a great deal of questions remain unanswered regarding the mutual relationship between autophagy and senescence at both molecular and cellular levels in diseases like asthma.
Autophagy, mitophagy, and cellular senescence are potential targets that can be manipulated at various levels, and inhibition of these processes have been considered as potential therapeutic strategies. For example, Liu et al. found that 3-methyladenine (3-MA), an inhibitor of autophagy, suppresses the formation of autophagosomes through the inhibition of PI3K (36). Chloroquine (CQ), another inhibitor of autophagy, also has the ability to inhibit HDM-induced airway remodeling through modulating autophagy pathways (191). In addition, bafilomycine (Baf-A), a macrolide antibiotic derived from Streptomyces griseus, can block late-phase autophagy through significant cytosolic acidification (192). In addition, administration of drugs currently in use for asthma (e.g., dexamethasone, montelukast, anti-IL-5, and anti-IgE antibody) can also inhibit autophagy (36). However, these current drugs for autophagy are far from specific, and may play a dual role in modulation of autophagy. Thus, addressing the real impact of autophagy in activation or inhibition of inflammation in disease models is challenging.
It has also been suggested that targeting mitophagy may possess therapeutic potential. Rapamycin and metformin as general autophagy-inducing drugs have been shown to attenuate AMPK and mTOR activity, and preserve energy metabolism through regulating mitophagy and mitochondrial biogenesis stimulation (193, 194). Of note, administration of metformin can induce mitophagy by promoting Parkin activity through p53 downregulation. In addition, several naturally occurring compounds, such as spermidine, resveratrol, urolithin A and antibiotics, have been demonstrated to maintain mitochondrial integrity by the induction of mitophagy and promotion of mitochondrial biogenesis (137). However, the therapeutic potential in human diseases still remains to be determined. Thus, identification of mitophagy modulators may result in therapeutic intervention strategies by targeting mitochondrial-associated pathologenesis of diseases.
Lastly, there is ongoing research to target senescence in cases of pulmonary fibrosis and asthma (195). Specifically, patients with age-related lung diseases (such as COPD and asthma) showed high levels of oxidative stress in the lung tissues, Thus, patients with COPD or asthma could benefit from the use of antioxidants (e.g., NAC, Nrf2 activators, NOX-4 inhibitors, MitoQ), which suppress inflammation and reduce the progression of senescence-associated pathways (196). Moreover, orchestrating SASP modulation could be a better strategy. Indeed, rapamycin, metformin, sirtuin activators, or PAI-1 inhibitor have been suggested to have beneficial effects due to their ability to act as a SASP suppressor (160, 181). For example, mTOR activation has been shown to be essential for asthma onset (197), and inhibition of mTOR with rapamycin can suppress IL-1 translation and reduce mRNA stability of SASP factors (198). Similarly, metformin can also inhibit mTOR and could have the potential to inhibit SASP in asthma. Furthermore, specific induction of apoptosis in senescent cells using senolytics could also lead to beneficial effects (175). Additionally, inducing senescence cell clearance [e.g., ABT-263, also known as navitoclax (199)] by manipulating the immune system to recognize and clear these cells is also an important therapeutic approach. However, these current drugs are far from specific, and some of them may have off-target effects. For example, metformin and sirt1 are also complex I inhibitor, which can promote mitochondrial fission and ROS production and subsequently cell senescence. Furthermore, although SASP is critical for the immune-mediated clearance of senescent cells, it also contributes to tissue dysfunction. Similarly, accumulation of senescent cells with time may lead to age-related loss of structure and tissue function (160, 200). By contrast, senescence can be beneficial in inhibiting the proliferation of transformed cells, and in some of key biological processes such as tissue repairing and wound healing (164). Because of these opposing effects, further studies are clearly needed to understand the exact role of senescence in diseases. Particularly, understanding of the molecular mechanisms regarding the processes involved in senescence will be helpful for the identification of modulators of cellular senescence, which could serve as therapeutic targets for senescence-associated diseases in the future.
Dysregulations in autophagy, mitophagy, and cellular senescence have been associated with environmental pollutant/allergen-induced oxidative stress, mitochondrial dysfunction, secretion of multiple inflammatory proteins known as SASP, and development of asthma. PM2.5 was reported to induce autophagy through activating AMPK (107), and to drive the mitophagy through activating PINK/Parkin pathway (132). PM2.5 was also shown to induce senescence of human dermal fibroblasts (177), while increased PM2.5 exposure was correlated with shortened telomeres in placental tissue and umbilical blood (178). Intriguingly, allergens, including OVA (36), A. alternata (48), and cockroach allergen (47, 111), have also been shown to induce autophagy through activating different signaling pathways. Thus, as hypothesized in Figure 4, environmental triggers, e.g., environmental pollutants or allergens, can induce ROS generation, which serve as “signaling molecules” modulating the process of autophagy through activating downstream signaling molecules and autophagy, thereby leading to the major phenotypic changes of asthma, including airway inflammation, airway remodeling, and airway hyper-responsiveness. These elevated ROS levels can also induce mitochondrial damage, thereby leading to the mitochondria-induced inflammation (mito-inflammation) and major features of asthma. Furthermore, oxidative stress can also cause DNA damage, telomere shortening, and epigenomic disruption, all of which induce cell cycle arrest and cellular senescence. Senescent cells can secrete SASP, which contains multiple inflammatory cytokines, chemokines, mtrix metalloproteinases (MMPs), and growth factors. The SASP leads to airway inflammation and remodeling. In turn, the SASP can also induce senescence and senescent cells can secrete ROS, which further promote the process of senescence. In addition, Th1 or Th2 cytokines may induce autophagy/mitophagy/senescence, and in turn, this autophagy/mitophagy/ senescence may also regulate the balance of Th1 and Th2 responses in asthma. Although comprehensive studies have been focused on investigating the role of autophagy, mitophagy and cellular senescence in the pathogenesis of diseases (e.g., beneficial or detrimental), many questions still remain untouched and unanswered. Thus, future studies are clearly needed to better understand these cellular processes, particularly after exposure to environmental pollutants and allergens, and to identify the therapeutic targets to regulate the autophagy/mitophagy/senescence-associated asthma.
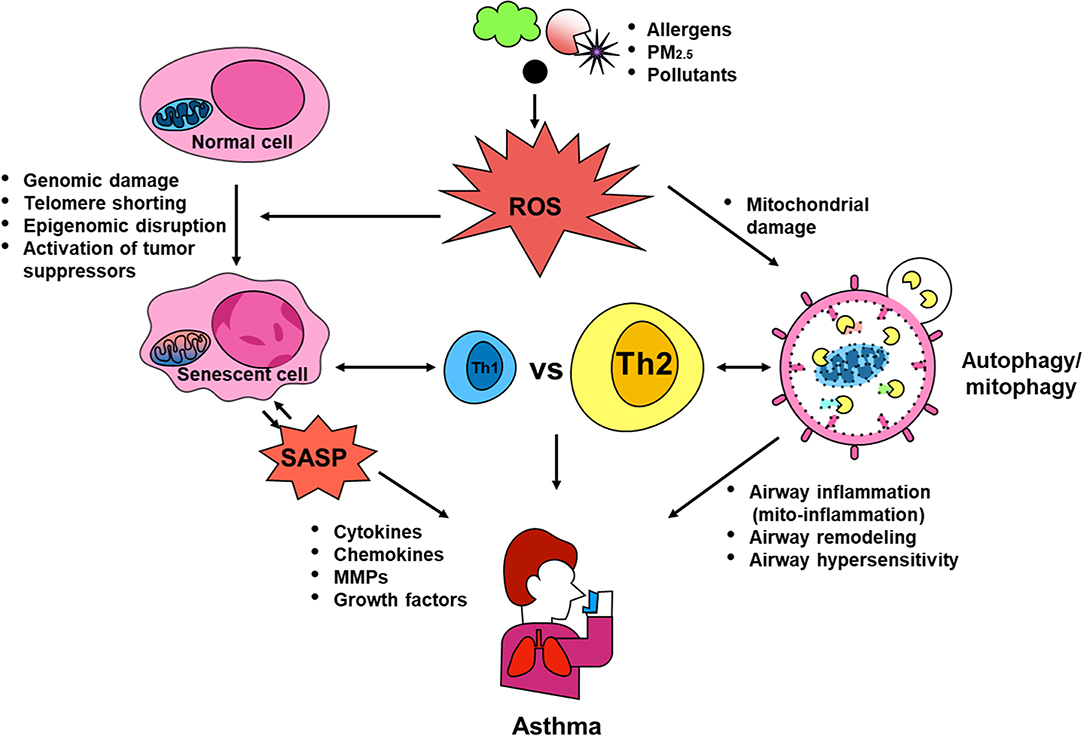
Figure 4. Contribution of autophagy, mitophagy, and senescence to asthma. Environmental triggers, such as environmental pollutants and allergens, can induce excessive ROS generation, which serve as “signaling molecules” modulating the process of autophagic cycle through activating signaling molecules and autophagy, thereby leading to the major phenotypic changes of asthma, including airway inflammation, airway remodeling, and airway hyper-responsiveness. These elevated ROS levels can also induce mitochondrial damage and mitophagy, thereby leading to the mitochondria-induced inflammation (mito-inflammation). Oxidative stress can cause DNA damage, telomere shortening, and epigenomic disruption, which convert normal cells into senescent cells, leading to secretion of SASP. SASP can regulate sensecent cells and induce airway inflammation and remodeling thought the secretion of cytokines, chemokines, MMPs, and growth factors. Additionally, Th1 or Th2 cytokines may induce autophagy/mitophagy/senescence, and in turn, these may also regulate the balance of Th1 and Th2 responses in asthma.
KS, YZ, XH, and JC wrote the manuscript. DD and PG reviewed the manuscript.
This work was supported by grants from the US National Institutes of Health (NIH) (R56 AI143668, R21 AI109062, R21 AI137547, and R01AI141642).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
CRE, Cockroach extract; HDM, House dust mite; PM2.5, Particulate matter 2.5; SASP, Senescence-associated secretory phenotype; ROS, Reactive oxygen species; NAC, N-Acetyl Cysteine; AMPK, AMP-activated protein kinase; PINK1, PTEN-induced kinase; PI3K, Phosphatidylinositol-4,5-bisphosphate 3-kinases; mTOR, Mechanistic target of rapamycin; ATG, Autophagy-related gene; CaMKII, Calmodulin-dependent protein kinase II; CQ, Chloroquine; BAL, Bronchoalveolar lavage; AHR, Airway hyperresponsiveness; COPD, Chronic obstructive pulmonary disease; IPF, Idiopathic pulmonary fibrosis; 3-MA, 3-Methyladenine.
1. Akinbami LJ, Moorman JE, Bailey C, Zahran HS, King M, Johnson CA, et al. Trends in asthma prevalence, health care use, and mortality in the United States, 2001-2010. NCHS Data Brief. (2012):1–8.
2. World Health Organisation. Asthma. (2017). Available online at:https://www.who.int/news-room/fact-sheets/detail/asthma
3. Masoli M, Fabian D, Holt S, Beasley R. The global burden of asthma: executive summary of the GINA Dissemination Committee report. Allergy. (2004) 59:469–78. doi: 10.1111/j.1398-9995.2004.00526.x
4. Eder W, Ege MJ, von Mutius E. The asthma epidemic. N Engl J Med. (2006) 355:2226–35. doi: 10.1056/NEJMra054308
5. Hansel NN, McCormack MC, Belli AJ, Matsui EC, Peng RD, Aloe C, et al. In-home air pollution is linked to respiratory morbidity in former smokers with chronic obstructive pulmonary disease. Am J Respir Crit Care Med. (2013) 187:1085–90. doi: 10.1164/rccm.201211-1987OC
6. Hao Y, Balluz L, Strosnider H, Wen XJ, Li C, Qualters JR. Ozone, fine particulate matter, and chronic lower respiratory disease mortality in the United States. Am J Respir Crit Care Med. (2015) 192:337–41. doi: 10.1164/rccm.201410-1852OC
7. Mukherjee A, Agrawal M. Use of GLM approach to assess the responses of tropical trees to urban air pollution in relation to leaf functional traits and tree characteristics. Ecotoxicol Environ Saf. (2018) 152:42–54. doi: 10.1016/j.ecoenv.2018.01.038
8. Mukherjee A, Agrawal M. Global perspective of fine particulate matter pollution and its health effects. Rev Environ Contam Toxicol. (2018) 244:5–51. doi: 10.1007/398_2017_3
9. Wu JZ, Ge DD, Zhou LF, Hou LY, Zhou Y, Li QY. Effects of particulate matter on allergic respiratory diseases. Chronic Dis Transl Med. (2018) 4:95–102. doi: 10.1016/j.cdtm.2018.04.001
10. Murrison LB, Brandt EB, Myers JB, Hershey GKK. Environmental exposures and mechanisms in allergy and asthma development. J Clin Invest. (2019) 129:1504–15. doi: 10.1172/JCI124612
11. Wilson D, Takahashi K, Pan G, Chan CC, Zhang S, Feng Y, et al. Respiratory symptoms among residents of a heavy-industry province in China: prevalence and risk factors. Respir Med. (2008) 102:1536–44. doi: 10.1016/j.rmed.2008.06.010
12. Liu F, Zhao Y, Liu YQ, Liu Y, Sun J, Huang MM, et al. Asthma and asthma related symptoms in 23,326 Chinese children in relation to indoor and outdoor environmental factors: the Seven Northeastern Cities (SNEC) study. Sci Total Environ. (2014) 497–8:10–7. doi: 10.1016/j.scitotenv.2014.07.096
13. Brandt EB, Myers JM, Ryan PH, Hershey GK. Air pollution and allergic diseases. Curr Opin Pediatr. (2015) 27:724–35. doi: 10.1097/MOP.0000000000000286
14. Gowers AM, Cullinan P, Ayres JG, Anderson HR, Strachan DP, Holgate ST, et al. Does outdoor air pollution induce new cases of asthma? Biological plausibility and evidence; a review. Respirology. (2012) 17:887–98. doi: 10.1111/j.1440-1843.2012.02195.x
15. Guarnieri M, Balmes JR. Outdoor air pollution and asthma. Lancet. (2014) 383:1581–92. doi: 10.1016/S0140-6736(14)60617-6
16. Berhane K, Chang CC, McConnell R, Gauderman WJ, Avol E, Rapapport E, et al. Association of changes in air quality with bronchitic symptoms in children in California, 1993–2012. JAMA. (2016) 315:1491–501. doi: 10.1001/jama.2016.3444
17. Li T, Hu R, Chen Z, Li Q, Huang S, Zhu Z, et al. Fine particulate matter (PM2.5): the culprit for chronic lung diseases in China. Chronic Dis Transl Med. (2018) 4:176–86. doi: 10.1016/j.cdtm.2018.07.002
18. Chen F, Lin Z, Chen R, Norback D, Liu C, Kan H, et al. The effects of PM2.5 on asthmatic and allergic diseases or symptoms in preschool children of six Chinese cities, based on China, Children, Homes and Health (CCHH) project. Environ Pollut. (2018) 232:329–37. doi: 10.1016/j.envpol.2017.08.072
19. Jin L, Xie J, Wong CKC, Chan SKY, Abbaszade G, Schnelle-Kreis J, et al. Contributions of city-specific fine particulate matter (PM2.5) to differential in vitro oxidative stress and toxicity implications between Beijing and Guangzhou of China. Environ Sci Technol. (2019) 53:2881–91. doi: 10.1021/acs.est.9b00449
20. Jung KH, Torrone D, Lovinsky-Desir S, Perzanowski M, Bautista J, Jezioro JR, et al. Short-term exposure to PM2.5 and vanadium and changes in asthma gene DNA methylation and lung function decrements among urban children. Respir Res. (2017) 18:63. doi: 10.1186/s12931-017-0550-9
21. Miller RL, Peden DB. Environmental effects on immune responses in patients with atopy and asthma. J Allergy Clin Immunol. (2014) 134:1001–8. doi: 10.1016/j.jaci.2014.07.064
22. Kanchongkittiphon W, Mendell MJ, Gaffin JM, Wang G, Phipatanakul W. Indoor environmental exposures and exacerbation of asthma: an update to the 2000 review by the institute of medicine. Environ Health Perspect. (2015) 123:6–20. doi: 10.1289/ehp.1307922
23. Kanchongkittiphon W, Gaffin JM, Phipatanakul W. The indoor environment and inner-city childhood asthma. Asian Pac J Allergy Immunol. (2014) 32:103–10.
24. Finkelman FD. Diesel exhaust particle exposure during pregnancy promotes development of asthma and atopy. J Allergy Clin Immunol. (2014) 134:73–4. doi: 10.1016/j.jaci.2014.04.002
25. Jedrychowski WA, Perera FP, Maugeri U, Mrozek-Budzyn D, Mroz E, Klimaszewska-Rembiasz M, et al. Intrauterine exposure to polycyclic aromatic hydrocarbons, fine particulate matter and early wheeze. Prospective birth cohort study in 4-year olds. Pediatr Allergy Immunol. (2010) 21(4 Pt 2):e723–32. doi: 10.1111/j.1399-3038.2010.01034.x
26. Acciani TH, Brandt EB, Khurana Hershey GK, Le Cras TD. Diesel exhaust particle exposure increases severity of allergic asthma in young mice. Clin Exp Allergy. (2013) 43:1406–18. doi: 10.1111/cea.12200
27. Brandt EB, Biagini Myers JM, Acciani TH, Ryan PH, Sivaprasad U, Ruff B, et al. Exposure to allergen and diesel exhaust particles potentiates secondary allergen-specific memory responses, promoting asthma susceptibility. J Allergy Clin Immunol. (2015) 136:295–303.e7. doi: 10.1016/j.jaci.2014.11.043
28. Zhang X, Chen X, Weirauch MT, Zhang X, Burleson JD, Brandt EB, et al. Diesel exhaust and house dust mite allergen lead to common changes in the airway methylome and hydroxymethylome. Environ Epigenet. (2018) 4:dvy020. doi: 10.1093/eep/dvy020
29. Bolcas PE, Brandt EB, Zhang Z, Biagini Myers JM, Ruff BP, Khurana Hershey GK. Vitamin D supplementation attenuates asthma development following traffic-related particulate matter exposure. J Allergy Clin Immunol. (2019) 143:386–94.e3. doi: 10.1016/j.jaci.2018.04.042
30. Wang E, Liu X, Tu W, Do DC, Yu H, Yang L, et al. Benzo(a)pyrene facilitates dermatophagoides group 1 (Der f 1)-induced epithelial cytokine release through aryl hydrocarbon receptor in asthma. Allergy. (2019) 74:1675–90. doi: 10.1111/all.13784
31. Sun L, Fu J, Lin SH, Sun JL, Xia L, Lin CH, et al. PM2.5 disturbs the balance of Th17/Treg cells by targeting Got1 and HIF-1alpha in an asthma model. J Allergy Clin Immunol. (2019) doi: 10.1016/j.jaci.2019.10.008
32. Warren KJ, Dickinson JD, Nelson AJ, Wyatt TA, Romberger DJ, Poole JA. Ovalbumin-sensitized mice have altered airway inflammation to agriculture organic dust. Respir Res. (2019) 20:51. doi: 10.1186/s12931-019-1015-0
33. Miraglia del Giudice M, Allegorico A, Parisi G, Galdo F, Alterio E, Coronella A, et al. Risk factors for asthma. Ital J Pediatr. (2014) 40(Suppl 1):A77. doi: 10.1186/1824-7288-40-S1-A77
34. Do DC, Zhao Y, Gao P. Cockroach allergen exposure and risk of asthma. Allergy. (2016) 71:463–74. doi: 10.1111/all.12827
35. Poon AH, Chouiali F, Tse SM, Litonjua AA, Hussain SN, Baglole CJ, et al. Genetic and histologic evidence for autophagy in asthma pathogenesis. J Allergy Clin Immunol. (2012) 129:569–71. doi: 10.1016/j.jaci.2011.09.035
36. Liu JN, Suh DH, Trinh HK, Chwae YJ, Park HS, Shin YS. The role of autophagy in allergic inflammation: a new target for severe asthma. Exp Mol Med. (2016) 48:e243. doi: 10.1038/emm.2016.38
37. Rowlands DJ. Mitochondria dysfunction: a novel therapeutic target in pathological lung remodeling or bystander? Pharmacol Ther. (2016) 166:96–105. doi: 10.1016/j.pharmthera.2016.06.019
38. Prakash YS, Pabelick CM, Sieck GC. Mitochondrial dysfunction in airway disease. Chest. (2017) 152:618–26. doi: 10.1016/j.chest.2017.03.020
39. Wu J, Dong F, Wang RA, Wang J, Zhao J, Yang M, et al. Central role of cellular senescence in TSLP-induced airway remodeling in asthma. PLoS ONE. (2013) 8:e77795. doi: 10.1371/journal.pone.0077795
40. Puddicombe SM, Torres-Lozano C, Richter A, Bucchieri F, Lordan JL, Howarth PH, et al. Increased expression of p21(waf) cyclin-dependent kinase inhibitor in asthmatic bronchial epithelium. Am J Respir Cell Mol Biol. (2003) 28:61–8. doi: 10.1165/rcmb.4715
41. Deretic V, Klionsky DJ. Autophagy and inflammation: a special review issue. Autophagy. (2018) 14:179–80. doi: 10.1080/15548627.2017.1412229
42. Lee J, Kim HS. The role of autophagy in eosinophilic airway inflammation. Immune Netw. (2019) 19:e5. doi: 10.4110/in.2019.19.e5
43. McAlinden KD, Deshpande DA, Ghavami S, Xenaki D, Sohal SS, Oliver BG, et al. Autophagy activation in asthma airways remodeling. Am J Respir Cell Mol Biol. (2018) 60:541–53. doi: 10.1165/rcmb.2018-0169OC
44. Liu T, Wu B, Wang Y, He H, Lin Z, Tan J, et al. Particulate matter 2.5 induces autophagy via inhibition of the phosphatidylinositol 3-kinase/Akt/mammalian target of rapamycin kinase signaling pathway in human bronchial epithelial cells. Mol Med Rep. (2015) 12:1914–22. doi: 10.3892/mmr.2015.3577
45. Zhu XM, Wang Q, Xing WW, Long MH, Fu WL, Xia WR, et al. PM2.5 induces autophagy-mediated cell death via NOS2 signaling in human bronchial epithelium cells. Int J Biol Sci. (2018) 14:557–64. doi: 10.7150/ijbs.24546
46. Li R, Zhou R, Zhang J. Function of PM2.5 in the pathogenesis of lung cancer and chronic airway inflammatory diseases. Oncol Lett. (2018) 15:7506–14. doi: 10.3892/ol.2018.8355
47. Zhao Y, Luo X, Qiu L, Do DC, Ke X, Zaccone E, et al. Aryl hydrocarbon receptor in airway epithelium exacerbates cockroach allergen-induced asthma through autophagy. J Allergy Clin Immunol. (2017) 139:AB268. doi: 10.1016/j.jaci.2016.12.863
48. Murai H, Okazaki S, Hayashi H, Kawakita A, Hosoki K, Yasutomi M, et al. Alternaria extract activates autophagy that induces IL-18 release from airway epithelial cells. Biochem Biophys Res Commun. (2015) 464:969–74. doi: 10.1016/j.bbrc.2015.05.076
49. Liu HX, Yan HY, Qu W, Wen X, Hou LF, Zhao WH, et al. Inhibition of thymocyte autophagy-associated CD4(+)T thymopoiesis is involved in asthma susceptibility in mice exposed to caffeine prenatally. Arch Toxicol. (2019) 93:1323–35. doi: 10.1007/s00204-019-02418-5
50. Zeki AA, Yeganeh B, Kenyon NJ, Post M, Ghavami S. Autophagy in airway diseases: a new frontier in human asthma? Allergy. (2016) 71:5–14. doi: 10.1111/all.12761
51. Glick D, Barth S, Macleod KF. Autophagy: cellular and molecular mechanisms. J Pathol. (2010) 221:3–12. doi: 10.1002/path.2697
52. Ghosh R, Pattison JS. Macroautophagy and chaperone-mediated autophagy in heart failure: the known and the unknown. Oxid Med Cell Longev. (2018) 2018:8602041. doi: 10.1155/2018/8602041
53. Schulze H, Kolter T, Sandhoff K. Principles of lysosomal membrane degradation: cellular topology and biochemistry of lysosomal lipid degradation. Biochim Biophys Acta. (2009) 1793:674–83. doi: 10.1016/j.bbamcr.2008.09.020
54. Ryter SW, Choi AM. Autophagy in lung disease pathogenesis and therapeutics. Redox Biol. (2015) 4:215–25. doi: 10.1016/j.redox.2014.12.010
55. Klionsky DJ, Abdelmohsen K, Abe A, Abedin MJ, Abeliovich H, Acevedo Arozena A, et al. Guidelines for the use and interpretation of assays for monitoring autophagy (3rd edition). Autophagy. (2016) 12:1–222. doi: 10.1080/15548627.2015
56. Li WW, Li J, Bao JK. Microautophagy: lesser-known self-eating. Cell Mol Life Sci. (2012) 69:1125–36. doi: 10.1007/s00018-011-0865-5
57. Kaushik S, Cuervo AM. Chaperone-mediated autophagy: a unique way to enter the lysosome world. Trends Cell Biol. (2012) 22:407–17. doi: 10.1016/j.tcb.2012.05.006
58. Cuervo AM, Wong E. Chaperone-mediated autophagy: roles in disease and aging. Cell Res. (2014) 24:92–104. doi: 10.1038/cr.2013.153
59. Levine B, Kroemer G. Biological functions of autophagy genes: a disease perspective. Cell. (2019) 176:11–42. doi: 10.1016/j.cell.2018.09.048
60. Jiang P, Mizushima N. Autophagy and human diseases. Cell Res. (2014) 24:69–79. doi: 10.1038/cr.2013.161
61. van Beek N, Klionsky DJ, Reggiori F. Genetic aberrations in macroautophagy genes leading to diseases. Biochim Biophys Acta Mol Cell Res. (2018) 1865:803–16. doi: 10.1016/j.bbamcr.2018.03.002
62. Qian M, Fang X, Wang X. Autophagy and inflammation. Clin Transl Med. (2017) 6:24. doi: 10.1186/s40169-017-0154-5
63. He C, Klionsky DJ. Regulation mechanisms and signaling pathways of autophagy. Annu Rev Genet. (2009) 43:67–93. doi: 10.1146/annurev-genet-102808-114910
64. Park HJ, Lee SJ, Kim SH, Han J, Bae J, Kim SJ, et al. IL-10 inhibits the starvation induced autophagy in macrophages via class I phosphatidylinositol 3-kinase (PI3K) pathway. Mol Immunol. (2011) 48:720–7. doi: 10.1016/j.molimm.2010.10.020
65. Petiot A, Ogier-Denis E, Blommaart EF, Meijer AJ, Codogno P. Distinct classes of phosphatidylinositol 3'-kinases are involved in signaling pathways that control macroautophagy in HT-29 cells. J Biol Chem. (2000) 275:992–8. doi: 10.1074/jbc.275.2.992
66. Arico S, Petiot A, Bauvy C, Dubbelhuis PF, Meijer AJ, Codogno P, et al. The tumor suppressor PTEN positively regulates macroautophagy by inhibiting the phosphatidylinositol 3-kinase/protein kinase B pathway. J Biol Chem. (2001) 276:35243–6. doi: 10.1074/jbc.C100319200
67. Rovetta AI, Peña D, Hernández Del Pino RE, Recalde GM, Pellegrini J, Bigi F, et al. IFNG-mediated immune responses enhance autophagy against Mycobacterium tuberculosis antigens in patients with active tuberculosis. Autophagy. (2014) 10:2109–21. doi: 10.4161/15548627.2014.981791
68. Botbol Y, Patel B, Macian F. Common gamma-chain cytokine signaling is required for macroautophagy induction during CD4+ T-cell activation. Autophagy. (2015) 11:1864–77. doi: 10.1080/15548627.2015.1089374
69. Terawaki S, Camosseto V, Prete F, Wenger T, Papadopoulos A, Rondeau C, et al. RUN and FYVE domain-containing protein 4 enhances autophagy and lysosome tethering in response to Interleukin-4. J Cell Biol. (2015) 210:1133–52. doi: 10.1083/jcb.201501059
70. Xia F, Deng C, Jiang Y, Qu Y, Deng J, Cai Z, et al. IL4 (interleukin 4) induces autophagy in B cells leading to exacerbated asthma. Autophagy. (2018) 14:450–64. doi: 10.1080/15548627.2017.1421884
71. Dickinson JD, Alevy Y, Malvin NP, Patel KK, Gunsten SP, Holtzman MJ, et al. IL13 activates autophagy to regulate secretion in airway epithelial cells. Autophagy. (2016) 12:397–409. doi: 10.1080/15548627.2015.1056967
72. Van Grol J, Subauste C, Andrade RM, Fujinaga K, Nelson J, Subauste CS. HIV-1 inhibits autophagy in bystander macrophage/monocytic cells through Src-Akt and STAT3. PLoS ONE. (2010) 5:e11733. doi: 10.1371/journal.pone.0011733
73. Harris J, Hartman M, Roche C, Zeng SG, O'Shea A, Sharp FA, et al. Autophagy controls IL-1beta secretion by targeting pro-IL-1beta for degradation. J Biol Chem. (2011) 286:9587–97. doi: 10.1074/jbc.M110.202911
74. Saitoh T, Fujita N, Jang MH, Uematsu S, Yang BG, Satoh T, et al. Loss of the autophagy protein Atg16L1 enhances endotoxin-induced IL-1beta production. Nature. (2008) 456:264–8. doi: 10.1038/nature07383
75. Peral de Castro C, Jones SA, Ní Cheallaigh C, Hearnden CA, Williams L, Winter J, et al. Autophagy regulates IL-23 secretion and innate T cell responses through effects on IL-1 secretion. J Immunol. (2012) 189:4144–53. doi: 10.4049/jimmunol.1201946
76. Lee JP, Foote A, Fan H, Peral de Castro C, Lang T, Jones SA, et al. Loss of autophagy enhances MIF/macrophage migration inhibitory factor release by macrophages. Autophagy. (2016) 12:907–16. doi: 10.1080/15548627.2016.1164358
77. Yoon S, Woo SU, Kang JH, Kim K, Kwon MH, Park S, et al. STAT3 transcriptional factor activated by reactive oxygen species induces IL6 in starvation-induced autophagy of cancer cells. Autophagy. (2010) 6:1125–38. doi: 10.4161/auto.6.8.13547
78. Kiffin R, Christian C, Knecht E, Cuervo AM. Activation of chaperone-mediated autophagy during oxidative stress. Mol Biol Cell. (2004) 15:4829–40. doi: 10.1091/mbc.e04-06-0477
79. Laplante M, Sabatini DM. mTOR signaling in growth control and disease. Cell. (2012) 149:274–93. doi: 10.1016/j.cell.2012.03.017
80. Kim J, Kundu M, Viollet B, Guan KL. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat Cell Biol. (2011) 13:132–41. doi: 10.1038/ncb2152
81. Jang JE, Eom JI, Jeung HK, Cheong JW, Lee JY, Kim JS, et al. AMPK-ULK1-mediated autophagy confers resistance to BET inhibitor JQ1 in acute myeloid leukemia stem cells. Clin Cancer Res. (2017) 23:2781–94. doi: 10.1158/1078-0432.CCR-16-1903
82. Kim J, Kim YC, Fang C, Russell RC, Kim JH, Fan W, et al. Differential regulation of distinct Vps34 complexes by AMPK in nutrient stress and autophagy. Cell. (2013) 152:290–303. doi: 10.1016/j.cell.2012.12.016
83. Green DR, Levine B. To be or not to be? How selective autophagy and cell death govern cell fate. Cell. (2014) 157:65–75. doi: 10.1016/j.cell.2014.02.049
84. Li X, Wu XQ, Deng R, Li DD, Tang J, Chen WD, et al. CaMKII-mediated Beclin 1 phosphorylation regulates autophagy that promotes degradation of Id and neuroblastoma cell differentiation. Nat Commun. (2017) 8:1159. doi: 10.1038/s41467-017-01272-2
85. Sakamaki JI, Long JS, New M, Van Acker T, Tooze SA, Ryan KM. Emerging roles of transcriptional programs in autophagy regulation. Transcription. (2018) 9:131–6. doi: 10.1080/21541264.2017.1372045
86. Zhao J, Brault JJ, Schild A, Cao P, Sandri M, Schiaffino S, et al. FoxO3 coordinately activates protein degradation by the autophagic/lysosomal and proteasomal pathways in atrophying muscle cells. Cell Metab. (2007) 6:472–83. doi: 10.1016/j.cmet.2007.11.004
87. Napolitano G, Ballabio A. TFEB at a glance. J Cell Sci. (2016) 129:2475–81. doi: 10.1242/jcs.146365
88. Medina DL, Di Paola S, Peluso I, Armani A, De Stefani D, Venditti R, et al. Lysosomal calcium signalling regulates autophagy through calcineurin and TFEB. Nat Cell Biol. (2015) 17:288–99. doi: 10.1038/ncb3114
89. Bowman CJ, Ayer DE, Dynlacht BD. Foxk proteins repress the initiation of starvation-induced atrophy and autophagy programs. Nat Cell Biol. (2014) 16:1202–14. doi: 10.1038/ncb3062
90. Baek SH, Kim KI. Epigenetic control of autophagy: nuclear events gain more attention. Mol Cell. (2017) 65:781–5. doi: 10.1016/j.molcel.2016.12.027
91. Füllgrabe J, Klionsky DJ, Joseph B. The return of the nucleus: transcriptional and epigenetic control of autophagy. Nat Rev Mol Cell Biol. (2014) 15:65–74. doi: 10.1038/nrm3716
92. Wei FZ, Cao Z, Wang X, Wang H, Cai MY, Li T, et al. Epigenetic regulation of autophagy by the methyltransferase EZH2 through an MTOR-dependent pathway. Autophagy. (2015) 11:2309–22. doi: 10.1080/15548627.2015.1117734
93. Sierra-Vargas MP, Guzman-Grenfell AM, Blanco-Jimenez S, Sepulveda-Sanchez JD, Bernabe-Cabanillas RM, Cardenas-Gonzalez B, et al. Airborne particulate matter PM2.5 from Mexico City affects the generation of reactive oxygen species by blood neutrophils from asthmatics: an in vitro approach. J Occup Med Toxicol. (2009) 4:17. doi: 10.1186/1745-6673-4-17
94. Prieto-Parra L, Yohannessen K, Brea C, Vidal D, Ubilla CA, Ruiz-Rudolph P. Air pollution, PM2.5 composition, source factors, and respiratory symptoms in asthmatic and nonasthmatic children in Santiago, Chile. Environ Int. (2017) 101:190–200. doi: 10.1016/j.envint.2017.01.021
95. Bleck B, Tse DB, Curotto de Lafaille MA, Zhang F, Reibman J. Diesel exhaust particle-exposed human bronchial epithelial cells induce dendritic cell maturation and polarization via thymic stromal lymphopoietin. J Clin Immunol. (2008) 28:147–56. doi: 10.1007/s10875-007-9149-0
96. Bleck B, Tse DB, Gordon T, Ahsan MR, Reibman J. Diesel exhaust particle-treated human bronchial epithelial cells upregulate Jagged-1 and OX40 ligand in myeloid dendritic cells via thymic stromal lymphopoietin. J Immunol. (2010) 185:6636–45. doi: 10.4049/jimmunol.1000719
97. Nakamura Y, Miyata M, Ohba T, Ando T, Hatsushika K, Suenaga F, et al. Cigarette smoke extract induces thymic stromal lymphopoietin expression, leading to T(H)2-type immune responses and airway inflammation. J Allergy Clin Immunol. (2008) 122:1208–14. doi: 10.1016/j.jaci.2008.09.022
98. Qu J, Do DC, Zhou Y, Luczak E, Mitzner W, Anderson ME, et al. Oxidized CaMKII promotes asthma through the activation of mast cells. JCI Insight. (2017) 2:e90139. doi: 10.1172/jci.insight.90139
99. Qu J, Li Y, Zhong W, Gao P, Hu C. Recent developments in the role of reactive oxygen species in allergic asthma. J Thorac Dis. (2017) 9:E32–43. doi: 10.21037/jtd.2017.01.05
100. Sanders PN, Koval OM, Jaffer OA, Prasad AM, Businga TR, Scott JA, et al. CaMKII is essential for the proasthmatic effects of oxidation. Sci Transl Med. (2013) 5:195ra97. doi: 10.1126/scitranslmed.3006135
101. Abdala-Valencia H, Earwood J, Bansal S, Jansen M, Babcock G, Garvy B, et al. Nonhematopoietic NADPH oxidase regulation of lung eosinophilia and airway hyperresponsiveness in experimentally induced asthma. Am J Physiol Lung Cell Mol Physiol. (2007) 292:L1111–25. doi: 10.1152/ajplung.00208.2006
102. Ban GY, Pham DL, Trinh TH, Lee SI, Suh DH, Yang EM, et al. Autophagy mechanisms in sputum and peripheral blood cells of patients with severe asthma: a new therapeutic target. Clin Exp Allergy. (2016) 46:48–59. doi: 10.1111/cea.12585
103. Martin LJ, Gupta J, Jyothula SS, Butsch Kovacic M, Biagini Myers JM, Patterson TL, et al. Functional variant in the autophagy-related 5 gene promotor is associated with childhood asthma. PLoS ONE. (2012) 7:e33454. doi: 10.1371/journal.pone.0033454
104. Chen ZH, Wu YF, Wang PL, Wu YP, Li ZY, Zhao Y, et al. Autophagy is essential for ultrafine particle-induced inflammation and mucus hyperproduction in airway epithelium. Autophagy. (2016) 12:297–311. doi: 10.1080/15548627.2015.1124224
105. Li Y, Qian W, Wang D, Meng Y, Wang X, Chen Y, et al. Resveratrol relieves particulate matter (mean diameter <2.5 mum)-induced oxidative injury of lung cells through attenuation of autophagy deregulation. J Appl Toxicol JAT. (2018) 38:1251–61. doi: 10.1002/jat.3636
106. Long F, Jiang H, Yi H, Su L, Sun J. Particulate matter 2.5 induced bronchial epithelial cell injury via activation of 5'-adenosine monophosphate-activated protein kinase-mediated autophagy. J Cell Biochem. (2019) 120:3294–305. doi: 10.1002/jcb.27597
107. Fu Q, Lyu D, Zhang L, Qin Z, Tang Q, Yin H, et al. Airborne particulate matter (PM2.5) triggers autophagy in human corneal epithelial cell line. Environ Pollut. (2017) 227:314–22. doi: 10.1016/j.envpol.2017.04.078
108. Bush RK, Prochnau JJ. Alternaria-induced asthma. J Allergy Clin Immunol. (2004) 113:227–34. doi: 10.1016/j.jaci.2003.11.023
109. Murai H, Qi H, Choudhury B, Wild J, Dharajiya N, Vaidya S, et al. Alternaria-induced release of IL-18 from damaged airway epithelial cells: an NF-kappaB dependent mechanism of Th2 differentiation? PLoS ONE. (2012) 7:e30280. doi: 10.1371/journal.pone.0030280
110. Abdel Fattah E, Bhattacharya A, Herron A, Safdar Z, Eissa NT. Critical role for IL-18 in spontaneous lung inflammation caused by autophagy deficiency. J Immunol. (2015) 194:5407–16. doi: 10.4049/jimmunol.1402277
111. Zhang YDD, Zhang X, Wang J, Anderson M, Gao P. Autophagy participates in cockroach allergen-induced lung inflammation through ROS and oxidized CaMKII. J Allergy Clin Immunol. (2019) 143:AB218. doi: 10.1016/j.jaci.2018.12.665
112. Suzuki Y, Maazi H, Sankaranarayanan I, Lam J, Khoo B, Soroosh P, et al. Lack of autophagy induces steroid-resistant airway inflammation. J Allergy Clin Immunol. (2016) 137:1382–9.e9. doi: 10.1016/j.jaci.2015.09.033
113. Wang SB, Wu YF, Chen ZH, Li W, Shen HH. [Airway epithelial ATG5 suppresses asthmatic inflammation in mice]. Zhonghua Jie He He Hu Xi Za Zhi. (2018) 41:873–7. doi: 10.3760/cma.j.issn.1001-0939.2018.11.009
114. Inoue D, Kubo H, Taguchi K, Suzuki T, Komatsu M, Motohashi H, et al. Inducible disruption of autophagy in the lung causes airway hyper-responsiveness. Biochem Biophys Res Commun. (2011) 405:13–8. doi: 10.1016/j.bbrc.2010.12.092
115. Pu Q, Gan C, Li R, Li Y, Tan S, Li X, et al. Atg7 deficiency intensifies inflammasome activation and pyroptosis in pseudomonas sepsis. J Immunol. (2017) 198:3205–13. doi: 10.4049/jimmunol.1601196
116. Guo L, Stripay JL, Zhang X, Collage RD, Hulver M, Carchman EH, et al. CaMKIalpha regulates AMP kinase-dependent, TORC-1-independent autophagy during lipopolysaccharide-induced acute lung neutrophilic inflammation. J Immunol. (2013) 190:3620–8. doi: 10.4049/jimmunol.1102975
117. Jessop F, Hamilton RF, Rhoderick JF, Shaw PK, Holian A. Autophagy deficiency in macrophages enhances NLRP3 inflammasome activity and chronic lung disease following silica exposure. Toxicol Appl Pharmacol. (2016) 309:101–10. doi: 10.1016/j.taap.2016.08.029
118. Cheng Z, Wang X, Dai L, Jia L, Jing X, Liu Y, et al. Suppression of microRNA-384 enhances autophagy of airway smooth muscle cells in asthmatic mouse. Oncotarget. (2017) 8:67933–41. doi: 10.18632/oncotarget.18913
119. Ghavami S, Mutawe MM, Schaafsma D, Yeganeh B, Unruh H, Klonisch T, et al. Geranylgeranyl transferase 1 modulates autophagy and apoptosis in human airway smooth muscle. Am J Physiol Lung Cell Mol Physiol. (2012) 302:L420–8. doi: 10.1152/ajplung.00312.2011
120. Pan S, Sharma P, Shah SD, Deshpande DA. Bitter taste receptor agonists alter mitochondrial function and induce autophagy in airway smooth muscle cells. Am J Physiol Lung Cell Mol Physiol. (2017) 313:L154–65. doi: 10.1152/ajplung.00106.2017
121. Neill T, Schaefer L, Iozzo RV. Instructive roles of extracellular matrix on autophagy. Am J Pathol. (2014) 184:2146–53. doi: 10.1016/j.ajpath.2014.05.010
122. Poon AH, Choy DF, Chouiali F, Ramakrishnan RK, Mahboub B, Audusseau S, et al. Increased autophagy-related 5 gene expression is associated with collagen expression in the airways of refractory asthmatics. Front Immunol. (2017) 8:355. doi: 10.3389/fimmu.2017.00355
123. Liu T, Liu Y, Miller M, Cao L, Zhao J, Wu J, et al. Autophagy plays a role in FSTL1-induced epithelial mesenchymal transition and airway remodeling in asthma. Am J Physiol Lung Cell Mol Physiol. (2017) 313:L27–40. doi: 10.1152/ajplung.00510.2016
124. Kota A, Deshpande DA, Haghi M, Oliver B, Sharma P. Autophagy and airway fibrosis: is there a link? F1000Res. (2017) 6:409. doi: 10.12688/f1000research.11236.1
125. Ghavami S, Yeganeh B, Serebrin A, et al. Autophagy regulates Tgf-beta1 induced fibrosis in human airway smooth muscle cells. Proc Am Thorac Soc. (2011) 183:A2110. doi: 10.1164/ajrccm-conference.2011.183.1_MeetingAbstracts.A2110
126. Aguilera-Aguirre L, Bacsi A, Saavedra-Molina A, Kurosky A, Sur S, Boldogh I. Mitochondrial dysfunction increases allergic airway inflammation. J Immunol. (2009) 183:5379–87. doi: 10.4049/jimmunol.0900228
127. Iyer D, Mishra N, Agrawal A. Mitochondrial function in allergic disease. Curr Allergy Asthma Rep. (2017) 17:29. doi: 10.1007/s11882-017-0695-0
128. Pattnaik B, Bodas M, Bhatraju NK, Ahmad T, Pant R, Guleria R, et al. IL-4 promotes asymmetric dimethylarginine accumulation, oxo-nitrative stress, and hypoxic response-induced mitochondrial loss in airway epithelial cells. J Allergy Clin Immunol. (2016) 138:130–41.e9. doi: 10.1016/j.jaci.2015.11.036
129. Reddy PH. Mitochondrial dysfunction and oxidative stress in asthma: implications for mitochondria-targeted antioxidant therapeutics. Pharmaceuticals. (2011) 4:429–56. doi: 10.3390/ph4030429
130. Wang A, Keita ÅV, Phan V, McKay CM, Schoultz I, Lee J, et al. Targeting mitochondria-derived reactive oxygen species to reduce epithelial barrier dysfunction and colitis. Am J Pathol. (2014) 184:2516–27. doi: 10.1016/j.ajpath.2014.05.019
131. Li R, Kou X, Geng H, Xie J, Yang Z, Zhang Y, et al. Effect of ambient PM(2.5) on lung mitochondrial damage and fusion/fission gene expression in rats. Chem Res Toxicol. (2015) 28:408–18. doi: 10.1021/tx5003723
132. Qiu YN, Wang GH, Zhou F, Hao JJ, Tian L, Guan LF, et al. PM2.5 induces liver fibrosis via triggering ROS-mediated mitophagy. Ecotoxicol Environ Saf. (2019) 167:178–87. doi: 10.1016/j.ecoenv.2018.08.050
133. Stevens JF, Maier CS. Acrolein: sources, metabolism, and biomolecular interactions relevant to human health and disease. Mol Nutr Food Res. (2008) 52:7–25. doi: 10.1002/mnfr.200700412
134. Wang HT, Lin JH, Yang CH, Haung CH, Weng CW, Maan-Yuh Lin A, et al. Acrolein induces mtDNA damages, mitochondrial fission and mitophagy in human lung cells. Oncotarget. (2017) 8:70406–21. doi: 10.18632/oncotarget.19710
135. Sliter DA, Martinez J, Hao L, Chen X, Sun N, Fischer TD, et al. Parkin and PINK1 mitigate STING-induced inflammation. Nature. (2018) 561:258–62. doi: 10.1038/s41586-018-0448-9
136. Youle RJ, Narendra DP. Mechanisms of mitophagy. Nat Rev Mol Cell Biol. (2011) 12:9–14. doi: 10.1038/nrm3028
137. Palikaras K, Lionaki E, Tavernarakis N. Mechanisms of mitophagy in cellular homeostasis, physiology and pathology. Nat Cell Biol. (2018) 20:1013–22. doi: 10.1038/s41556-018-0176-2
138. Jin SM, Lazarou M, Wang C, Kane LA, Narendra DP, Youle RJ. Mitochondrial membrane potential regulates PINK1 import and proteolytic destabilization by PARL. J Cell Biol. (2010) 191:933–42. doi: 10.1083/jcb.201008084
139. Pickles S, Vigié P, Youle RJ. Mitophagy and quality control mechanisms in mitochondrial maintenance. Curr Biol CB. (2018) 28:R170–85. doi: 10.1016/j.cub.2018.01.004
140. Lazarou M, Sliter DA, Kane LA, Sarraf SA, Wang C, Burman JL, et al. The ubiquitin kinase PINK1 recruits autophagy receptors to induce mitophagy. Nature. (2015) 524:309–14. doi: 10.1038/nature14893
141. Moore AS, Holzbaur EL. Dynamic recruitment and activation of ALS-associated TBK1 with its target optineurin are required for efficient mitophagy. Proc Natl Acad Sci USA. (2016) 113:E3349–58. doi: 10.1073/pnas.1523810113
142. Richter B, Sliter DA, Herhaus L, Stolz A, Wang C, Beli P, et al. Phosphorylation of OPTN by TBK1 enhances its binding to Ub chains and promotes selective autophagy of damaged mitochondria. Proc Natl Acad Sci USA. (2016) 113:4039–44. doi: 10.1073/pnas.1523926113
143. Shen Z, Li Y, Gasparski AN, Abeliovich H, Greenberg ML. Cardiolipin regulates mitophagy through the protein kinase C pathway. J Biol Chem. (2017) 292:2916–23. doi: 10.1074/jbc.M116.753574
144. Wei Y, Chiang WC, Sumpter R Jr., Mishra P, Levine B. Prohibitin 2 is an inner mitochondrial membrane mitophagy receptor. Cell. (2017) 168:224–38.e10. doi: 10.1016/j.cell.2016.11.042
145. Tsubouchi K, Araya J, Kuwano K. PINK1-PARK2-mediated mitophagy in COPD and IPF pathogeneses. Inflamm Regen. (2018) 38:18. doi: 10.1186/s41232-018-0077-6
146. Bueno M, Lai YC, Romero Y, Brands J, St Croix CM, Kamga C, et al. PINK1 deficiency impairs mitochondrial homeostasis and promotes lung fibrosis. J Clin Invest. (2015) 125:521–38. doi: 10.1172/JCI74942
147. Kobayashi K, Araya J, Minagawa S, Hara H, Saito N, Kadota T, et al. Involvement of PARK2-mediated mitophagy in idiopathic pulmonary fibrosis pathogenesis. J Immunol. (2016) 197:504–16. doi: 10.4049/jimmunol.1600265
148. Sosulski ML, Gongora R, Danchuk S, Dong C, Luo F, Sanchez CG. Deregulation of selective autophagy during aging and pulmonary fibrosis: the role of TGFβ1. Aging Cell. (2015) 14:774–83. doi: 10.1111/acel.12357
149. Patel AS, Song JW, Chu SG, Mizumura K, Osorio JC, Shi Y, et al. Epithelial cell mitochondrial dysfunction and PINK1 are induced by transforming growth factor-beta1 in pulmonary fibrosis. PLoS ONE. (2015) 10:e0121246. doi: 10.1371/journal.pone.0121246
150. Fan P, Xie XH, Chen CH, Peng X, Zhang P, Yang C, et al. Molecular regulation mechanisms and interactions between reactive oxygen species and mitophagy. DNA Cell Biol. (2019) 38:10–22. doi: 10.1089/dna.2018.4348
151. Wiley CD, Velarde MC, Lecot P, Liu S, Sarnoski EA, Freund A, et al. Mitochondrial dysfunction induces senescence with a distinct secretory phenotype. Cell Metab. (2016) 23:303–14. doi: 10.1016/j.cmet.2015.11.011
152. Schuliga M, Pechkovsky DV, Read J, Waters DW, Blokland KEC, Reid AT, et al. Mitochondrial dysfunction contributes to the senescent phenotype of IPF lung fibroblasts. J Cell Mol Med. (2018) 22:5847–61. doi: 10.1111/jcmm.13855
153. Parikh P, Wicher S, Khandalavala K, Pabelick CM, Britt RD Jr., Prakash YS. Cellular senescence in the lung across the age spectrum. Am J Physiol Lung Cell Mol Physiol. (2019) 316:L826–42. doi: 10.1152/ajplung.00424.2018
154. Hadj Salem I, Dubé J, Boulet LP, Chakir J. Telomere shortening correlates with accelerated replicative senescence of bronchial fibroblasts in asthma. Clin Exp Allergy. (2015) 45:1713–5. doi: 10.1111/cea.12611
155. Belsky DW, Shalev I, Sears MR, Hancox RJ, Lee Harrington H, Houts R, et al. Is chronic asthma associated with shorter leukocyte telomere length at midlife? Am J Respir Crit Care Med. (2014) 190:384–91. doi: 10.1164/rccm.201402-0370OC
156. Sidler C, Kovalchuk O, Kovalchuk I. Epigenetic regulation of cellular senescence and aging. Front Genet. (2017) 8:138. doi: 10.3389/fgene.2017.00138
157. Pereira BI, Devine OP, Vukmanovic-Stejic M, Chambers ES, Subramanian P, Patel N, et al. Senescent cells evade immune clearance via HLA-E-mediated NK and CD8(+) T cell inhibition. Nat Commun. (2019) 10:2387. doi: 10.1038/s41467-019-10335-5
158. Campisi J, d'Adda di Fagagna F. Cellular senescence: when bad things happen to good cells. Nat Rev Mol Cell Biol. (2007) 8:729–40. doi: 10.1038/nrm2233
159. Krtolica A, Parrinello S, Lockett S, Desprez PY, Campisi J. Senescent fibroblasts promote epithelial cell growth and tumorigenesis: a link between cancer and aging. Proc Natl Acad Sci USA. (2001) 98:12072–7. doi: 10.1073/pnas.211053698
160. Birch J, Barnes PJ, Passos JF. Mitochondria, telomeres and cell senescence: implications for lung ageing and disease. Pharmacol Ther. (2018) 183:34–49. doi: 10.1016/j.pharmthera.2017.10.005
161. Li C, Chai Y, Wang L, Gao B, Chen H, Gao P, et al. Programmed cell senescence in skeleton during late puberty. Nat Commun. (2017) 8:1312. doi: 10.1038/s41467-017-01509-0
162. Childs BG, Baker DJ, Wijshake T, Conover CA, Campisi J, van Deursen JM. Senescent intimal foam cells are deleterious at all stages of atherosclerosis. Science. (2016) 354:472–7. doi: 10.1126/science.aaf6659
163. Jeon OH, Kim C, Laberge RM, Demaria M, Rathod S, Vasserot AP, et al. Local clearance of senescent cells attenuates the development of post-traumatic osteoarthritis and creates a pro-regenerative environment. Nat Med. (2017) 23:775. doi: 10.1038/nm.4324
164. van Deursen JM. The role of senescent cells in ageing. Nature. (2014) 509:439–46. doi: 10.1038/nature13193
165. Lee S, Schmitt CA. The dynamic nature of senescence in cancer. Nat Cell Biol. (2019) 21:94–101. doi: 10.1038/s41556-018-0249-2
166. Martínez-Zamudio RI, Robinson L, Roux PF, Bischof O. SnapShot: cellular senescence pathways. Cell. (2017) 170:816–e1. doi: 10.1016/j.cell.2017.07.049
167. Hardie DG, Ross FA, Hawley SA. AMPK: a nutrient and energy sensor that maintains energy homeostasis. Nat Rev Mol Cell Biol. (2012) 13:251–62. doi: 10.1038/nrm3311
168. Ogrodnik M, Zhu Y, Langhi LGP, Tchkonia T, Krüger P, Fielder E, et al. Obesity-induced cellular senescence drives anxiety and impairs neurogenesis. Cell Metab. (2019) 29:1061–77.e8. doi: 10.1016/j.cmet.2018.12.008
169. Burton DGA, Stolzing A. Cellular senescence: immunosurveillance and future immunotherapy. Ageing Res Rev. (2018) 43:17–25. doi: 10.1016/j.arr.2018.02.001
170. Krizhanovsky V, Yon M, Dickins RA, Hearn S, Simon J, Miething C, et al. Senescence of activated stellate cells limits liver fibrosis. Cell. (2008) 134:657–67. doi: 10.1016/j.cell.2008.06.049
171. Sagiv A, Burton DG, Moshayev Z, Vadai E, Wensveen F, Ben-Dor S, et al. NKG2D ligands mediate immunosurveillance of senescent cells. Aging. (2016) 8:328–44. doi: 10.18632/aging.100897
172. Iannello A, Thompson TW, Ardolino M, Lowe SW, Raulet DH. p53-dependent chemokine production by senescent tumor cells supports NKG2D-dependent tumor elimination by natural killer cells. J Exp Med. (2013) 210:2057–69. doi: 10.1084/jem.20130783
173. Kang JY, Lee SY, Rhee CK, Kim SJ, Kwon SS, Kim YK. Effect of aging on airway remodeling and muscarinic receptors in a murine acute asthma model. Clin Interv Aging. (2013) 8:1393–403. doi: 10.2147/CIA.S50496
174. Amsellem V, Gary-Bobo G, Marcos E, Maitre B, Chaar V, Validire P, et al. Telomere dysfunction causes sustained inflammation in chronic obstructive pulmonary disease. Am J Respir Crit Care Med. (2011) 184:1358–66. doi: 10.1164/rccm.201105-0802OC
175. Schafer MJ, White TA, Iijima K, Haak AJ, Ligresti G, Atkinson EJ, et al. Cellular senescence mediates fibrotic pulmonary disease. Nat Commun. (2017) 8:14532. doi: 10.1038/ncomms14532
176. Fedorov IA, Wilson SJ, Davies DE, Holgate ST. Epithelial stress and structural remodelling in childhood asthma. Thorax. (2005) 60:389–94. doi: 10.1136/thx.2004.030262
177. Jedrychowski WA, Perera FP, Maugeri U, Mroz E, Klimaszewska-Rembiasz M, Flak E, et al. Effect of prenatal exposure to fine particulate matter on ventilatory lung function of preschool children of non-smoking mothers. Paediatr Perinat Epidemiol. (2010) 24:492–501. doi: 10.1111/j.1365-3016.2010.01136.x
178. Martens DS, Cox B, Janssen BG, Clemente DBP, Gasparrini A, Vanpoucke C, et al. Prenatal air pollution and newborns' predisposition to accelerated biological aging. JAMA Pediatr. (2017) 171:1160–7. doi: 10.1001/jamapediatrics.2017.3024
179. Mahemuti L, Chen Q, Coughlan MC, Qiao C, Chepelev NL, Florian M, et al. Bisphenol A induces DSB-ATM-p53 signaling leading to cell cycle arrest, senescence, autophagy, stress response, and estrogen release in human fetal lung fibroblasts. Arch Toxicol. (2018) 92:1453–69. doi: 10.1007/s00204-017-2150-3
180. Jiang C, Liu G, Luckhardt T, Antony V, Zhou Y, Carter AB, et al. Serpine 1 induces alveolar type II cell senescence through activating p53-p21-Rb pathway in fibrotic lung disease. Aging Cell. (2017) 16:1114–24. doi: 10.1111/acel.12643
181. Rana T, Jiang C, Liu G, Miyata T, Antony V, Thannickal VJ, et al. PAI-1 regulation of TGF-beta1-induced ATII Cell senescence, SASP secretion, and SASP-mediated Activation of Alveolar Macrophages. Am J Respir Cell Mol Biol. (2019). doi: 10.1165/rcmb.2019-0071OC. [Epub ahead of print].
182. Rodier F, Coppé JP, Patil CK, Hoeijmakers WA, Muñoz DP, Raza SR, et al. Persistent DNA damage signalling triggers senescence-associated inflammatory cytokine secretion. Nat Cell Biol. (2009) 11:973–9. doi: 10.1038/ncb1909
183. Loubaki L, Semlali A, Boisvert M, Jacques E, Plante S, Aoudjit F, et al. Crosstalk between T cells and bronchial fibroblasts obtained from asthmatic subjects involves CD40L/alpha 5 beta 1 interaction. Mol Immunol. (2010) 47:2112–8. doi: 10.1016/j.molimm.2010.03.011
184. Roca R, Kypta RM, Vivanco Md. Loss of p16INK4a results in increased glucocorticoid receptor activity during fibrosarcoma development. Proc Natl Acad Sci USA. (2003) 100:3113–8. doi: 10.1073/pnas.0634912100
185. Hodge G, Jersmann H, Tran HB, Holmes M, Reynolds PN, Hodge S. Lymphocyte senescence in COPD is associated with loss of glucocorticoid receptor expression by pro-inflammatory/cytotoxic lymphocytes. Respir Res. (2015) 16:2. doi: 10.1186/s12931-015-0287-2
186. KleinJan A. Airway inflammation in asthma: key players beyond the Th2 pathway. Curr Opin Pulm Med. (2016) 22:46–52. doi: 10.1097/MCP.0000000000000224
187. Huang J, Li Z, Yao X, Li Y, Reng X, Li J, et al. Altered Th1/Th2 commitment contributes to lung senescence in CXCR3-deficient mice. Exp Gerontol. (2013) 48:717–26. doi: 10.1016/j.exger.2013.04.001
188. Kuwano K, Araya J, Hara H, Minagawa S, Takasaka N, Ito S, et al. Cellular senescence and autophagy in the pathogenesis of chronic obstructive pulmonary disease (COPD) and idiopathic pulmonary fibrosis (IPF). Respir Investig. (2016) 54:397–406. doi: 10.1016/j.resinv.2016.03.010
189. Rajendran P, Alzahrani AM, Hanieh HN, Kumar SA, Ben Ammar R, Rengarajan T, et al. Autophagy and senescence: a new insight in selected human diseases. J Cell Physiol. (2019) 234:21485–92. doi: 10.1002/jcp.28895
190. Narita M, Young AR, Arakawa S, Samarajiwa SA, Nakashima T, Yoshida S, et al. Spatial coupling of mTOR and autophagy augments secretory phenotypes. Science. (2011) 332:966–70. doi: 10.1126/science.1205407
191. Suzuki T, Nakagawa M, Yoshikawa A, Sasagawa N, Yoshimori T, Ohsumi Y, et al. The first molecular evidence that autophagy relates rimmed vacuole formation in chloroquine myopathy. J Biochem. (2002) 131:647–51. doi: 10.1093/oxfordjournals.jbchem.a003147
192. Werner G, Hagenmaier H, Drautz H, Baumgartner A, Zähner H. Metabolic products of microorganisms. 224. Bafilomycins, a new group of macrolide antibiotics. Production, isolation, chemical structure and biological activity. J Antibiot. (1984) 37:110–7. doi: 10.7164/antibiotics.37.110
193. Hardie DG. AMPK: a target for drugs and natural products with effects on both diabetes and cancer. Diabetes. (2013) 62:2164–72. doi: 10.2337/db13-0368
194. Kim J, Yang G, Kim Y, Kim J, Ha J. AMPK activators: mechanisms of action and physiological activities. Exp Mol Med. (2016) 48:e224. doi: 10.1038/emm.2016.16
195. Mora AL, Rojas M, Pardo A, Selman M. Emerging therapies for idiopathic pulmonary fibrosis, a progressive age-related disease. Nat Rev Drug Discov. (2017) 16:755–72. doi: 10.1038/nrd.2017.170
196. Barnes PJ. Senescence in COPD and its comorbidities. Annu Rev Physiol. (2017) 79:517–39. doi: 10.1146/annurev-physiol-022516-034314
197. Zhang Y, Jing Y, Qiao J, Luan B, Wang X, Wang L, et al. Activation of the mTOR signaling pathway is required for asthma onset. Sci Rep. (2017) 7:4532. doi: 10.1038/s41598-017-04826-y
198. Herranz N, Gallage S, Mellone M, Wuestefeld T, Klotz S, Hanley CJ, et al. mTOR regulates MAPKAPK2 translation to control the senescence-associated secretory phenotype. Nat Cell Biol. (2015) 17:1205–17. doi: 10.1038/ncb3225
199. Chang J, Wang Y, Shao L, Laberge RM, Demaria M, Campisi J, et al. Clearance of senescent cells by ABT263 rejuvenates aged hematopoietic stem cells in mice. Nat Med. (2016) 22:78–83. doi: 10.1038/nm.4010
Keywords: oxidative stress, autophagy, mitophagy, senescence, asthma
Citation: Sachdeva K, Do DC, Zhang Y, Hu X, Chen J and Gao P (2019) Environmental Exposures and Asthma Development: Autophagy, Mitophagy, and Cellular Senescence. Front. Immunol. 10:2787. doi: 10.3389/fimmu.2019.02787
Received: 07 August 2019; Accepted: 13 November 2019;
Published: 29 November 2019.
Edited by:
Paola Italiani, Italian National Research Council (CNR), ItalyReviewed by:
James Harris, Monash University, AustraliaCopyright © 2019 Sachdeva, Do, Zhang, Hu, Chen and Gao. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Peisong Gao, cGdhbzFAamhtaS5lZHU=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.