 Serge Nataf
Serge Nataf Marine Guillen2
Marine Guillen2- 1CarMeN Laboratory, INSERM U1060, INRA U1397, INSA de Lyon, Lyon-Sud Faculty of Medicine, University of Lyon, Pierre-Bénite, France
- 2Faculté de Médecine Lyon-Est, University of Lyon 1, Lyon, France
- 3Banque de Tissus et de Cellules des Hospices Civils de Lyon, Hôpital Edouard Herriot, Lyon, France
There is circumstantial evidence that, under neurodegenerative conditions, peptides deriving from aggregated or misfolded specific proteins elicit adaptive immune responses. On another hand, several genes involved in familial forms of neurodegenerative diseases exert key innate immune functions. However, whether or not such observations are causally linked remains unknown. To start addressing this issue, we followed a systems biology strategy based on the mining of large proteomics and immunopeptidomics databases. First, we retrieved the expression patterns of common neurodegeneration-associated proteins in two professional antigen-presenting cells, namely B lymphocytes and dendritic cells. Surprisingly, we found that under physiological conditions, numerous neurodegeneration-associated proteins are abundantly expressed by human B lymphocytes. A survey of the human proteome allowed us to map a unique protein-protein interaction network linking common neurodegeneration-associated proteins and their first shell interactors in human B lymphocytes. Interestingly, network connectivity analysis identified two major hubs that both relate with inflammation and autophagy, namely TRAF6 (TNF Receptor Associated Factor 6) and SQSTM1 (Sequestosome-1). Moreover, the mapped network in B lymphocytes comprised two additional hub proteins involved in both inflammation and autoimmunity: HSPA8 (Heat Shock Protein Family A Member 8 also known as HSC70) and HSP90AA1 (Heat Shock Protein 90 Alpha Family Class A Member 1). Based on these results, we then explored the Immune Epitope Database “IEDB-AR” and actually found that a large share of neurodegeneration-associated proteins were previously reported to provide endogenous MHC class II-binding peptides in human B lymphocytes. Of note, peptides deriving from amyloid beta A4 protein, sequestosome-1 or profilin-1 were reported to bind multiple allele-specific MHC class II molecules. In contrast, peptides deriving from microtubule-associated protein tau, presenilin 2 and serine/threonine-protein kinase TBK1 were exclusively reported to bind MHC molecules encoded by the HLA-DRB1 1501 allele, a recently-identified susceptibility gene for late onset Alzheimer's disease. Finally, we observed that the whole list of proteins reported to provide endogenous MHC class II-binding peptides in human B lymphocytes is specifically enriched in neurodegeneration-associated proteins. Overall, our work indicates that immunization against neurodegeneration-associated proteins might be a physiological process which is shaped, at least in part, by B lymphocytes.
Introduction
Multiple studies have now established that neurodegenerative disorders are not cell-autonomous. The pathophysiological processes leading to neurodegeneration involve and target not only neurons but also glial cells, including astrocytes, microglia, and oligodendrocytes (1–3). Moreover, beyond central nervous system (CNS) cells, the adaptive immune system has emerged as a potentially important player. When considering only T-cell responses, T-cell reactivity against amyloid beta peptides was already reported more than a decade ago in both patients suffering from Alzheimer's disease (AD) and aged healthy subjects (4). Recent works further demonstrated that alpha-synuclein (SNCA)-derived peptides elicit helper and cytotoxic T-cell responses in a subgroup of Parkinson's disease (PD) patients harboring specific major histocompatibility complex (MHC) alleles (5, 6). Similarly, in animal models of neurodegenerative disorders, T-cell responses against peptides deriving from neurodegeneration-associated proteins were also demonstrated. In particular, in the MPTP (1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine) model of PD, pathogenic brain-infiltrating T-cells directed against SNCA were found to target the substantia nigra of diseased mice (7, 8). Along this line, in a mouse model of tauopathy, CD8 cytotoxic T-cells directed against tau protein were shown to infiltrate the hippocampus and to drive cognitive alterations (9). Finally, besides AD and PD, several studies provided evidence that T-cells are activated during the course of Huntington's disease (HD), amyotrophic lateral sclerosis (ALS) and/or their animal models (10–13). Nevertheless, the putative autoantigens targeted under those conditions remain unknown.
Extending the functional results mentioned above, genome-wide association studies (GWAS) have unraveled the central position of autoimmunity-related genes in the genetic susceptibility landscape of neurodegenerative diseases (14–19). This is notably the case for HLA-DRB1 alleles which were recently demonstrated to confer increased risks of developing PD (15) or late-onset AD (14). Also, two genes involved in familial forms of PD, namely PINK1 (PTEN-induced putative kinase 1) and PRKN (Parkin RBR E3 Ubiquitin Protein Ligase also named PARK2 or Parkin) were shown to regulate the presentation of mitochondria-derived antigens in the context of MHC class I molecules (20). Finally, innate immune functions were demonstrated for several genes causatively-linked to familial forms of PD or ALS. These comprise C9ORF72 (21–23), LRRK2 (Leucine-rich repeat kinase 2) (24–27), GRN (Granulin Precursor) (28, 29) as well as PRKN and PINK1 (30–34).
However, advocating for the role of autoimmunity in neurodegenerative disorders requires yet addressing several important issues. In particular, it appears crucial to determine how T-cells directed against neurodegeneration-associated antigens are primed in the periphery. The extent to which autoimmunity against neurodegeneration-associated antigens might be a physiological event needs also to be assessed. Last but not least, a global view on the expression of neurodegeneration-associated proteins by professional antigen-presenting cells (APCs), in particular B lymphocytes and dendritic cells, is still lacking. In an attempt to address these issues, we used here a systems biology approach embracing a large range of previously-published experimental data. We notably explored in normal human B lymphocytes and dendritic cells the expression patterns of the most common neurodegeneration-associated proteins. Our data mining results indicate that in human B lymphocytes, a large majority of neurodegeneration-associated proteins are expressed at the protein level. Moreover, a survey of the human proteome unravels that neurodegeneration-associated proteins expressed by B lymphocytes may form a complex network centered on the inflammation/autophagy-related molecules SQSTM1 (Sequestosome-1) and TRAF6 (TNF Receptor Associated Factor 6). Finally, the analysis of MHC class II immunopeptidome databases provides evidence that neurodegeneration-associated proteins expressed by human B lymphocytes are a source of endogenous peptides which are presented in the context of HLA class II molecules.
Materials and Methods
Workflow
A scheme summarizing the workflow followed in the present work is shown in Figure 1.
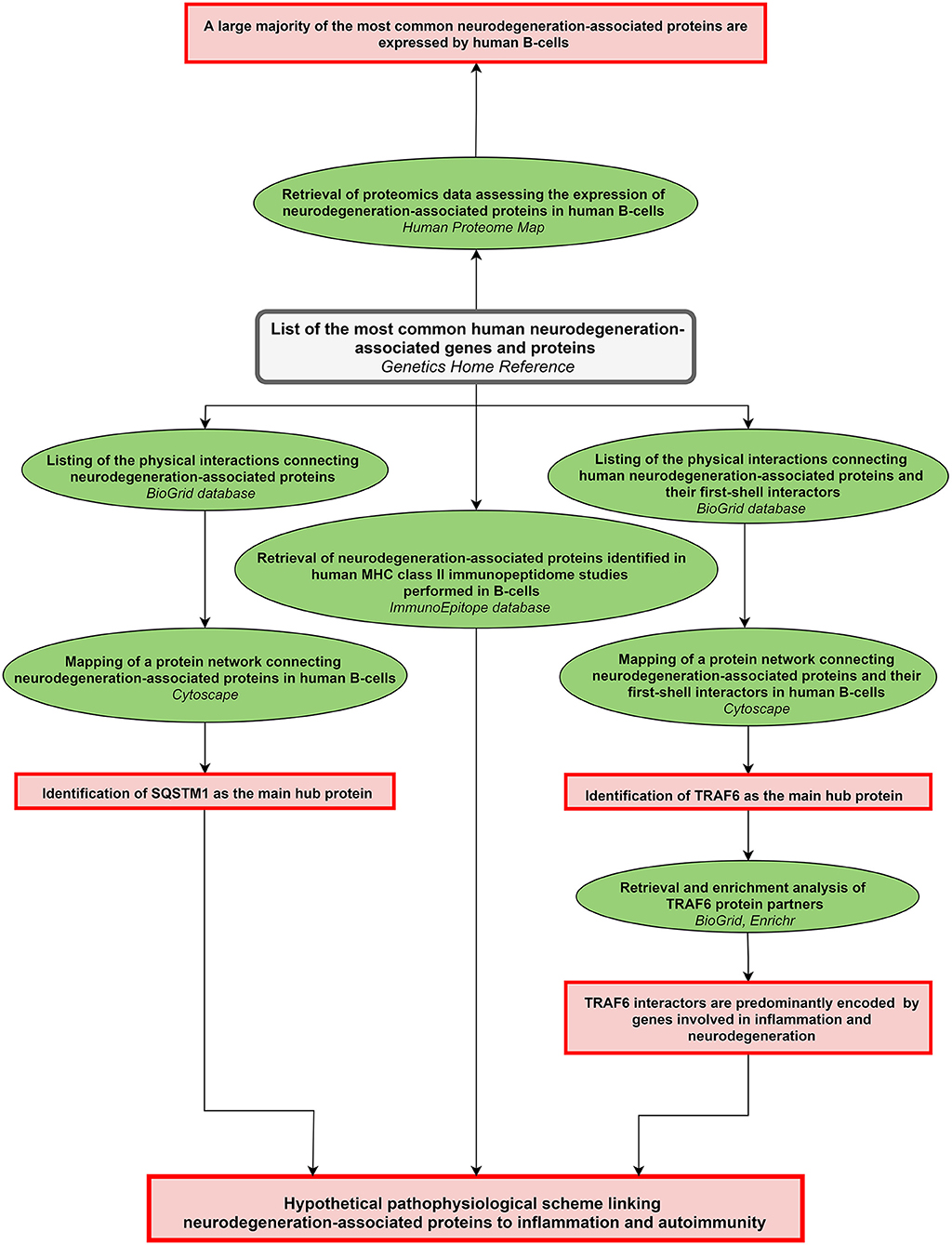
Figure 1. Workflow of the study. The workflow starts from the upper central gray rectangle. Other rectangles (in red) frame the main results obtained following each of the analytical steps which are briefly described in green ellipse shapes. Terms in italics correspond to the name of the bioinformatics tools used for each analytical step. MHC: major histocompatibility complex.
Data Mining Methods and Bioinformatics Tools
All the bioinformatics analyses were performed at least 3 times between March 2018 and October 2019. Databases, bioinformatic tools, and corresponding tasks performed in this study are described below.
- The Genetics Home Reference website (35), is a regularly updated consumer health resource from the National Library of Medicine. It provides information to the general public about the effects of genetic variation on human health. In the present paper, we used the “Genetics Home Reference” website to select without a priori the most common genetic variations/alterations that are linked to the following neurodegenerative disorders: Alzheimer's disease, Parkinson's disease, amyotrophic lateral sclerosis, Huntington's disease, fronto-temporal dementia (FTD).
- The enrichment web platform Enrichr (36) performs enrichment analyses from queried lists of genes. The Enrichr website allows surveying simultaneously 132 libraries gathering 245,575 terms and their associated lists of genes or proteins. Enrichment analysis tools provided by the Enrichr bioinformatics platform provides adjusted P-values computed from the Fisher's exact test. We focused our analysis on the “Jensen DISEASES” ontology library (37) which is based exclusively on text-mining and allow determining whether a list of genes is significantly associated with specific disease-related terms.
- The BioGrid database (38) compiles 29 169 experimentally-proven protein-protein interactions connecting 23 098 human proteins. Querying the BioGrid database allows retrieving for any given human protein the current list of published experimentally-identified human protein partners.
- The Human Proteome Map database (39) compiles protein expression data obtained by mass spectrometry from human normal tissues and cells including a total of 85 human blood samples from which the protein profiles of 6 blood-circulating cell types were established. In parallel, we also explored 3 recently-published mass spectrometry datasets reporting on the protemics profiles of blood-derived human B lymphocytes (40), blood-derived human dendritic cells (DCs) (41) and cultured monocyte-derived human dendritic cells (MoDCs) (42).
- The Immune Epitope Database and Analysis Resource (IEDB-AR) (43) compiles experimental data on antibody and T-cell epitopes in humans, non-human primates and other animal species. We followed a 3 step strategy as described below.
Step 1: retrieval of a list of human MHC class II binding peptides and their parent proteins
- in the “Epitope” tab, the “Any Epitopes” item was marked
- in the “Assay” tab, the “MHC Ligand Assays” item was marked
- in the “Antigen” tab, “Homo sapiens” was entered in the “Organism” line
- in the “MHC restriction” tab, the “MHC class II” item was marked
- in the “Host” tab, the “Humans” item was marked
- in the “Disease” tab, the “Any Disease” item was marked.
Search was then launched and, from the results page, the “Assays” tab was selected and a list of currently known ligands (peptides and parent proteins) of human MHC molecules was retrieved.
Step 2: filtering the results
We retained only results which were both: (i) obtained by mass spectrometry analysis of peptides eluted from MHC class II molecules and (ii) generated from cells of the B- cell lineage, in the absence of immunization or stimulation protocols.
Step 3: checking the reference IDs associated to each identified parent protein
For each selected study and/or set of results, the reference ID associated to each parent protein was checked on the UniProt website (44) in order to avoid redundant or obsolete IDs.
- The Cytoscape software (45) is an open source software allowing to visualize complex networks. We used the function “Network analysis” to identify hubs i.e., objects exhibiting the highest number of degrees (connections to other objects) in the generated networks.
Statistics
When not embedded in bioinformatics webtools, statistics for enrichment analyses were performed using the Fisher's exact test. In particular, we assessed for several identified hub proteins whether their retrieved list of partners (obtained via the BioGrid database) was statistically enriched in neurodegeneration-associated proteins. To calculate enrichment factors, we set the expected reference ratio as 0.002 which corresponds to the number of common neurodegeneration-associated proteins (i.e., 48 according to the “Home Genetics Reference” website) over the number of coding genes for which interactors can be retrieved from the BioGrid database (i.e., 23098). The obtained p-values were then adjusted using Bonferroni correction. The same approach was used to determine whether lists of parent proteins from which derive human MHC class II-binding peptides are significantly enriched in neurodegeneration-associated proteins.
Results
Neurodegeneration-Associated Proteins Form a Unique Interaction Network in Which PRKN Is the Main Hub
From the most recently updated version of the human proteome compiled in the “BioGrid” database, we extracted the currently published and experimentally-proven protein-protein interactions connecting the most common neurodegeneration-associated proteins. Self-interactions were excluded from our analysis. From these retrieved interactions we were able to build and visualize a protein network which encompasses 35 (72%) of the 48 most common neurodegeneration-associated proteins (Figure 2). Interestingly, network analysis showed that PRKN harbored the highest number of interacting partners (n = 18). Moreover, when considering the whole list of known PRKN protein partners, the calculated enrichment factor in neurodegeneration associated protein reached 20.86 and was highly significant (Fisher's exact test p-value: 2.77E−16). It should be noticed that the great majority of the protein-protein interactions reported so far between PRKN and other common neurodegeneration-associated proteins were demonstrated via robust low throughput biochemical approaches as detailed in Data Supplement 1. Our data mining result points thus to a yet unnoticed property of PRKN as a major hub protein connecting a large array of proteins involved in not only PD but also ALS, AD, Huntington's disease and/or FTD.
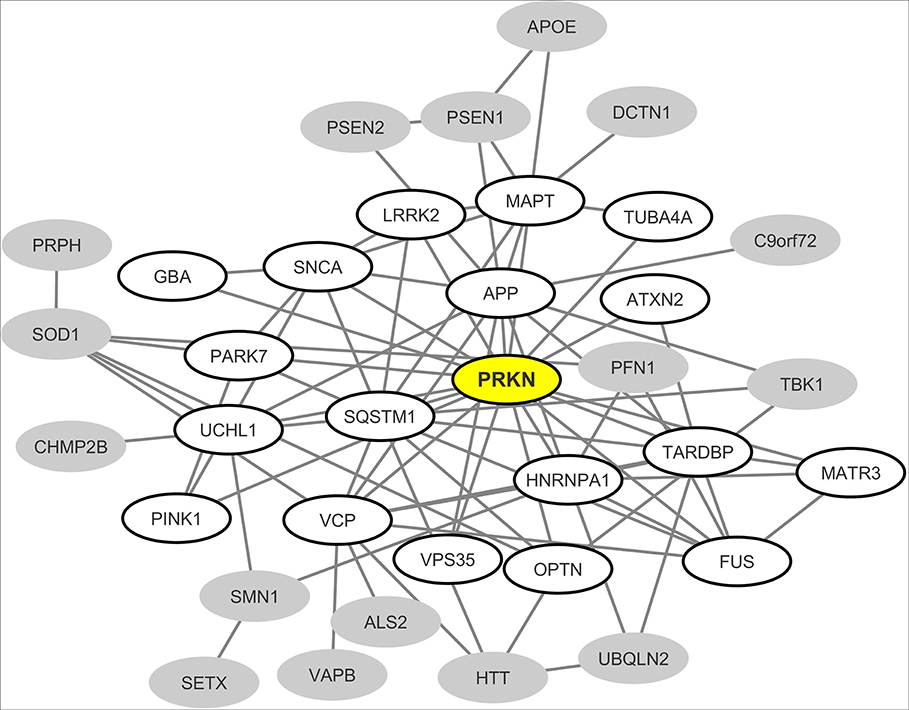
Figure 2. Mapping of the protein-protein interaction network linking common neurodegeneration-associated proteins irrespective of the cell type considered. A survey of the human proteome was performed by querying the protein-protein interaction database BioGRID (38). Each node represents a protein indicated by the corresponding gene symbol and each edge represents an experimentally-demonstrated protein-protein interaction. In this network, the “PRKN” (Parkin) node, highlighted in yellow, exhibits the highest degree (i.e., the highest number of direct interactors). White nodes correspond to the first-shell partners of PRKN. Gray nodes are not direct interactors of PRKN.
A Majority of Common Neurodegeneration-Associated Proteins Are Abundantly Expressed in Human B Lymphocytes
We then explored proteomics databases to determine whether neurodegeneration-associated proteins are expressed by APCs under physiological conditions. Our search was restricted to 2 cell lineages which harbor demonstrated antigen-presenting functions: B lymphocytes (46–48) and dendritic cells (DCs) (49–51). Results were combined with those retrieved from proteomics data obtained in 3 independent studies by the mass spectrometry analysis of human B lymphocytes (40) or human dendritic cells (41, 42). Since mass spectrometry is not a sensitive technique, one may consider that detected proteins are abundantly expressed. As shown in Table 1, it is striking that 34 (70%) of 48 common neurodegeneration-associated proteins are detectable by mass spectrometry in human B lymphocytes (Table 1). In human DCs, 20 (41%) of 48 common neurodegeneration-associated proteins were detected and all of these were also detected in human B lymphocytes. Conversely, 14 common degeneration-associated proteins were reported to be detected in B lymphocytes but not DCs. These include notably apolipoprotein E, amyloid beta A4 protein, presenilin-1 and alpha-synuclein.
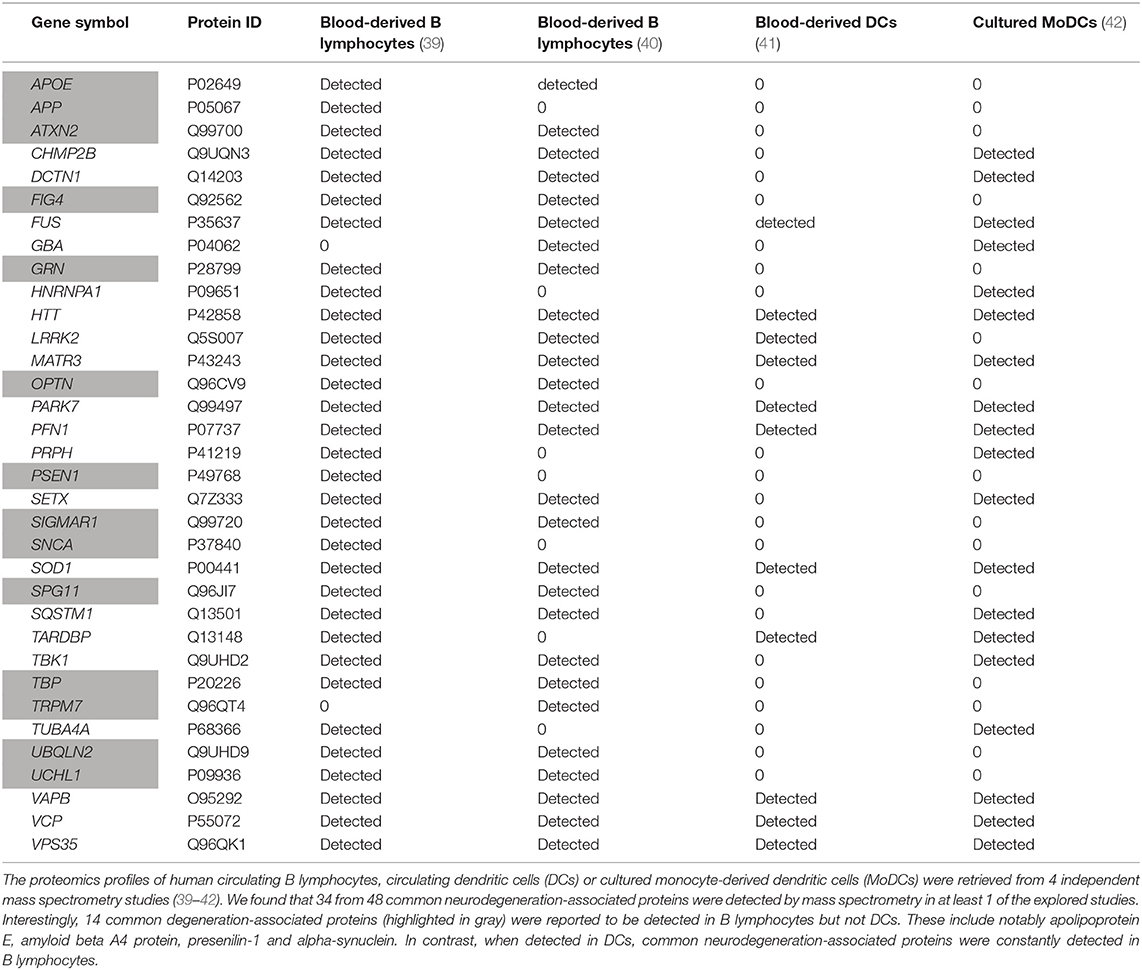
Table 1. Expression patterns of human neurodegeneration-associated proteins in B-cells vs. dendritic cells.
Of note, in both human B lymphocytes and DCs, parkin, the PRKN-encoded protein, was undetectable as assessed by mass spectrometry. This finding urged us to identify the main hub protein(s) which may interconnect neurodegeneration-associated proteins that are abundantly expressed in human B lymphocytes.
Neurodegeneration-Associated Proteins Expressed by Human B Lymphocytes Form a Unique Interaction Network in Which the Inflammation/Autophagy-Related Protein SQSTM1 Is the Main Hub
From the interaction network depicted in Figure 2 we only retained nodes corresponding to neurodegeneration-associated genes that are detectable by mass spectrometry in human B lymphocytes. We observed that in this cell type, 24 neurodegeneration-associated proteins are predicted to form a tight interaction network which is centered on the inflammation/autophagy-related protein SQSTM1 (Figure 3). Again, it should be noticed that the great majority of the protein-protein interactions reported so far between SQSTM1 and other common neurodegeneration-associated proteins were demonstrated via robust low throughput biochemical approaches as detailed in Data Supplement 1.
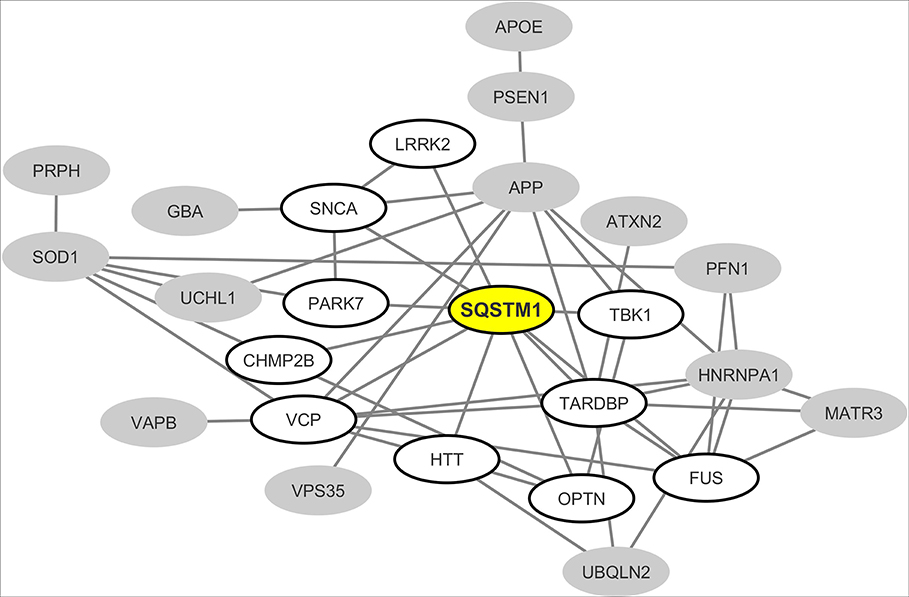
Figure 3. Mapping of the protein-protein interaction network linking common neurodegeneration-associated proteins which are expressed in human B lymphocytes. A survey of the human proteome was performed by querying the protein-protein interaction database BioGRID (38). Each node represents a protein indicated by the corresponding gene symbol and each edge represents an experimentally-demonstrated protein-protein interaction. In this network, the “SQSTM1” (Sequestosome-1) node, highlighted in yellow, exhibits the highest degree (i.e., the highest number of direct interactors). White nodes correspond to the first-shell partners of SQSTM1 in human B lymphocytes. Gray nodes are expressed by B lymphocytes but are not direct interactors of SQSTM1.
The Protein Network Formed by Neurodegeneration-Associated Proteins Connects With a Set of First Shell Interactors Among Which the B-Cell Inflammation/Autophagy-Related Protein TRAF6 Is the Main Hub
To extend these data, we then retrieved from the “BioGrid” database the currently known and experimentally-demonstrated interactors of common neurodegeneration-associated proteins, irrespective of their levels of expression in human B lymphocytes. From this list, we extracted candidate hub proteins interacting with more than 10 neurodegeneration-associated proteins. For each of these candidate hubs, total lists of protein interactors were then retrieved from BioGrid and a Fisher's exact test was applied so to determine if neurodegeneration-associated proteins were actually significantly enriched in each established list. By this mean, we were able to assign an enrichment factor and an associated p-value to each candidate hub. This approach allowed us to identify 20 hub proteins (Table 2) with which common neurodegeneration-associated proteins are specifically connected. Among these 20 hub proteins, 10 are expressed at the protein level as assessed via the “Human Proteome Map” database (39). Moreover, from the 10 hub proteins exhibiting the most significant and highest enrichment factors, 8 are abundantly expressed by human B lymphocytes. These include: (i) the heat shock proteins HSPA8 (also named HSC70), HSPA4 (also named HSP70) and HSP90AA1 (also named HSP90) which are all involved in both antigen presentation by MHC class II molecules (52–54) and the modulation of T-cell responses (55–57) and (ii) YWHAZ and YWHAQ proteins also known as 14-3-3 protein zeta and theta respectively, which bind MHC class II molecules (58, 59) and are implicated in various neurodegenerative diseases (60–62). Finally, unexpectedly, the most significant hub we identified was TRAF6 (TNF receptor associated factor 6), an inflammation/autoimmunity-related molecule (63, 64) playing a major role in the control of B-cell activation (65, 66). Indeed, the whole list of TRAF6 interactors comprises 17 neurodegeneration-associated proteins i.e., 5.3%, which corresponds to an enrichment factor of 27.68 and an adjusted p-value of 1.03E−17 (Fisher's exact test). The robustness of the biochemical techniques which were used to identify TRAF6 neurodegeneration-associated protein partners was checked as detailed in Data Supplement 1.
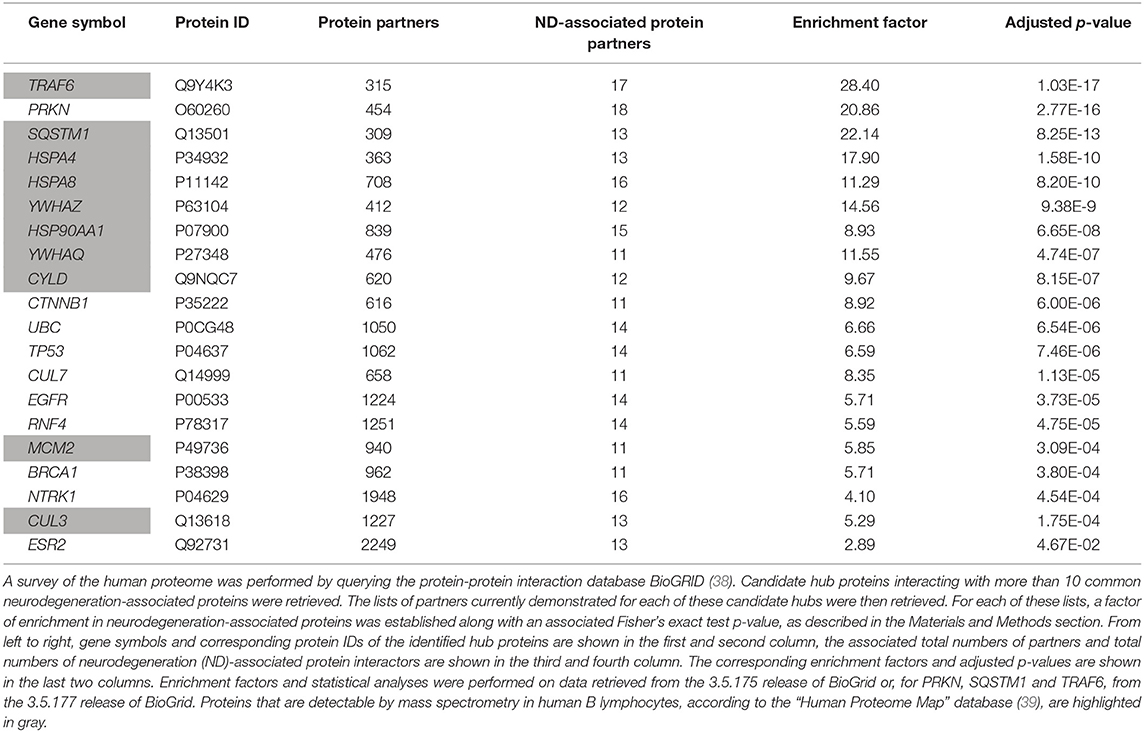
Table 2. Main hub partners of common neurodegeneration-associated proteins.
However, one may argue that the list of currently-known TRAF6 partners might be enriched in not only neurodegeneration-associated proteins but also many other sets of proteins which do not relate with neurodegeneration. To address this issue, we performed on the whole list of TRAF6 partners a non a priori enrichment analysis using the “JENSEN Disease” text-mining webtool (37) embedded in the “Enrichr” analysis platform (36). Results shown in Table 3 indicate that the 10 disease-related terms with which TRAF6 partners are the most significantly associated comprise the terms “Frontotemporal dementia,” “Neurodegenerative disease” and “Pick's disease.” This finding points to a specific link between TRAF6 and neurodegeneration.
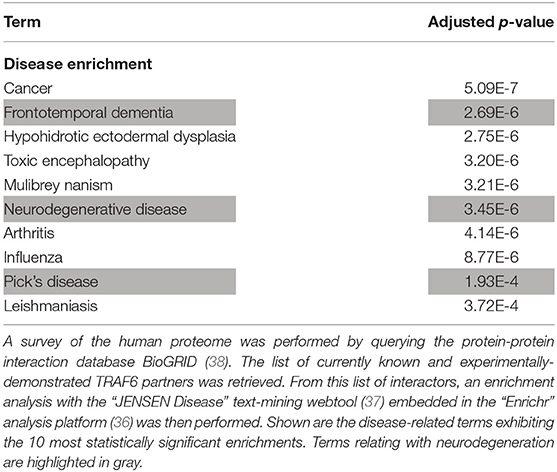
Table 3. Enrichment analysis of TRAF6 partners list.
As a control and to further establish the specificity of our findings, we assessed whether similar results would be obtained from a list of common demyelination-associated proteins i.e., proteins commonly considered as candidate autoantigens in multiple sclerosis (67). We found that neither TRAF6 nor the other hubs linking common neurodegeneration-associated proteins exhibited lists of protein partners which were significantly enriched in common demyelination-associated proteins (Data Supplement 2).
From these results we then built and visualized a B-cell protein-protein interaction network encompassing the most significant hub proteins and their neurodegeneration-associated interactors expressed in B lymphocytes (Figure 4). We observed that from the 32 neurodegeneration-associated proteins expressed by B lymphocytes, 22 (68%) are first shell interactors of HSPA4, HSPA8, TRAF6, or SQSTM1. These results unravel a yet unknown function of these molecules as major hub proteins connecting in B lymphocytes a large array of proteins involved in PD, ALS, AD, Huntington's disease and/or FTD.
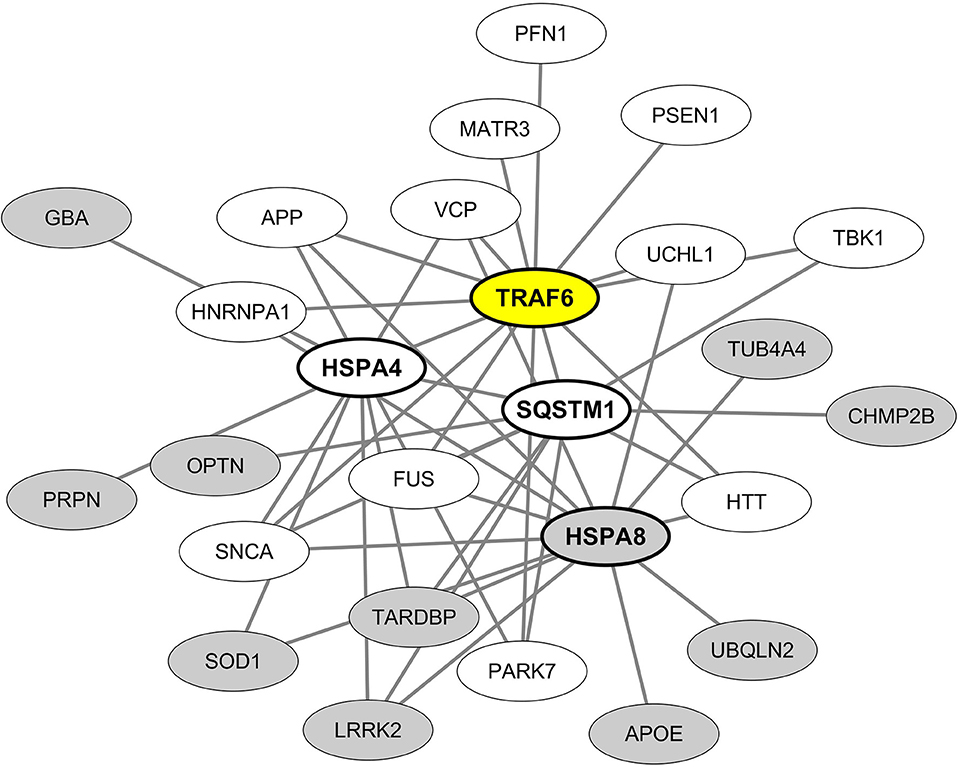
Figure 4. Mapping of the protein-protein interaction network linking common neurodegeneration-associated proteins expressed in B lymphocytes and their hub protein partners. A survey of the human proteome was performed by querying the protein-protein interaction database BioGRID (38). Only proteins expressed by human B lymphocytes according to the “Human Proteome Map” database (39) were taken into account. Each node represents a protein indicated by its corresponding gene symbol. The 4 hub proteins expressed by B lymphocytes and whose partners exhibit the most significant and highest enrichment factors in neurodegeneration-associated proteins are indicated in bold characters. White nodes correspond to neurodegeneration-associated proteins expressed by B lymphocytes and interacting with TRAF6. Gray nodes correspond to common neurodegeneration-associated proteins expressed by B lymphocytes but not reported to interact with TRAF6.
Common Neurodegeneration-Associated Proteins Provide a Source of Endogenous MHC Class II-Binding Peptides in Human B Lymphocytes
While peptides presented by MHC class II molecules are classically generated by the proteolysis of phagocytized exogenous antigens, the presentation of endogenous peptides by MHC class II molecules is an alternate pathway which has been robustly documented (68–71). We thus surveyed the Immune Epitope DataBase (IEDB-AR) (43) to assess whether peptides derived from neurodegeneration-associated proteins had been previously identified as binding MHC class II molecules in immunopeptidome studies which used human cells of the B-cell lineage as a source of endogenous peptides. Of note, we excluded studies assessing the MHC binding of exogenously-provided specific peptides and retained only works relying on the systematic mass spectrometry-based identification of peptides eluted from MHC class II molecules. In addition, we excluded experiments in which immunization or stimulation protocols were applied to B lymphocytes. On this basis, we retained 19 studies (Data Supplement 3) which were performed on cells of the B-cell lineage including predominantly Epstein-Barr virus (EBV)-transformed B lymphocytes. When screening these studies, we found that 23 out of 48 common neurodegeneration-associated proteins were reported to provide endogenous MHC class II-binding peptides in B lymphocytes (Table 4 and Data Supplement 3). The most frequently identified neurodegeneration-associated parent proteins are abundantly expressed in B lymphocytes and comprise notably the proteins encoded by: (i) the AD-related genes APP and PSEN1, (ii) the ALS/FTD-related genes PFN1, SQSTM1, GRN, SOD1, and VCP and (iii) the PD-related proteins PARK7 and GBA (Figure 1).

Table 4. List of neurodegeneration-associated proteins from which derive MHC class II-binding endogenous peptides in the human B-cell lineage.
Hub Molecules Linking Common Neurodegeneration-Associated Proteins Provide a Source of Endogenous MHC Class II-Binding Peptides in Human B Lymphocytes
From the 19 relevant B-cell studies we retained on IEDB-AR, we also attempted to determine whether the hub molecules we identified as linking neurodegeneration-associated proteins in B lymphocytes (Table 2) were, in parallel, reported to provide endogenous ligands for MHC class II molecules in B lymphocytes. We found that from 10 candidate hubs abundantly expressed by B lymphocytes, 8 were reported to provide endogenous ligands for MHC class II molecules in human B lymphocytes (Table 5 and Data Supplement 3).

Table 5. List of the main hub partners linking neurodegeneration-associated proteins and giving rise to MHC class II-binding endogenous peptides in the human B-cell lineage.
Among these 8 hubs, HSPA8 was the parent protein being the most frequently identified as providing endogenous peptides which bind MHC class II molecules in human B lymphocytes (Table 5).
In Human B Lymphocytes, the List of Parent Proteins From Which Derive Endogenous MHC Class II-Binding Peptides Is Specifically Enriched in Neurodegeneration-Associated Proteins
The whole list of parent proteins identified as providing MHC class II-binding endogenous peptides in B lymphocytes was retrieved from IEDB-AR and an enrichment analysis was performed to determine whether such a list was significantly enriched in neurodegeneration-associated proteins. We found that genes coding for neurodegeneration-associated proteins encompassed 0.62% (22 out of 3523) of the whole genes coding for such parent proteins (Data Supplement 4), which corresponds to an enrichment of 2.58 (p-value = 0.0006, Fisher's exact test) when considering the whole number of human protein-coding genes as roughly 20 000 (72). To confirm these results and assess their level of specificity, we used the “JENSEN Disease” text-mining webtool (37) and performed an unsupervised enrichment analysis from the whole list of genes coding for parent proteins previously identified as providing MHC class II-binding peptides in human B lymphocytes (Data Supplement 4). From this list of 3522 genes, 96 (2.72%) were annotated with the term “Neurodegenerative disease” (Data Supplement 4) which corresponds to the second most significant enrichment, after the term “Arthritis” (Table 6).
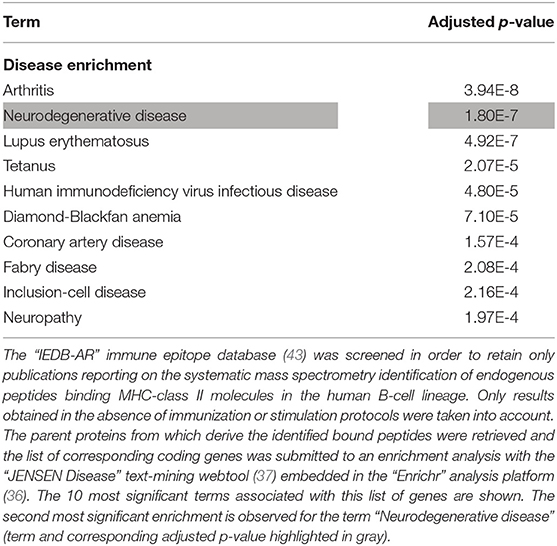
Table 6. Enrichment analysis of the whole list of parent proteins giving rise to MHC class II-binding endogenous peptides in the human B-cell lineage.
Since the HLA-DRB1 1501 allele (corresponding to the HLA-DR15 serotype) was recently identified as a risk factor for sporadic forms of late onset AD (14), we retrieved the whole list of parent proteins (and corresponding coding genes) which, in human B lymphocytes, were previously reported to provide peptides that bind HLA-DRB1 1501-encoded MHC class II molecules. Importantly, such a list was significantly enriched in neurodegeneration-associated proteins (enrichment factor: 5.31; p-value = 0.0001, Fisher exact test). Moreover, when this list of parent proteins was submitted to an unsupervised enrichment analysis, the term “Neurodegenerative disease” was found to reach the highest level of statistical significance (Table 7).
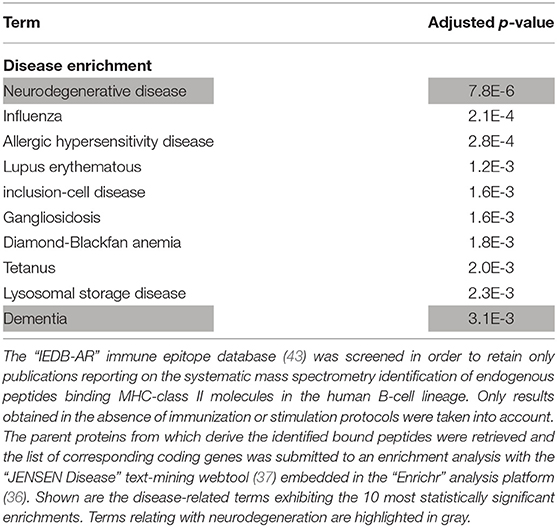
Table 7. Enrichment analysis of the whole list of parent proteins giving rise to endogenous peptides binding HLA-DRB1 1501-encoded MHC class II molecules in the human B-cell lineage.
Finally, based on the IEDB-AR survey we performed, HLA-DRB1 1501-encoded MHC class II molecules are the only HLA class II molecules which, in human B lymphocytes, were reported to bind endogenous peptides deriving from microtubule-associated protein tau, presenilin-2 or serine/threonine-protein kinase TBK1 (Table 4 and Data Supplement 3).
Discussion
In the present work, we mined large publically-available databases to provide experiment-based evidence of a link between neurodegeneration and autoimmunity. Using a systems biology approach we report that a large range of common neurodegeneration-associated proteins: (i) are expressed by human B lymphocytes under physiological conditions, (ii) form a comprehensive and functionally-relevant protein-protein interaction network and (iii) provide endogenous peptides which bind MHC class-II molecules in human B lymphocytes. Patients suffering from neurodegenerative conditions exhibit T cell- and/or antibody-mediated responses directed against major neurodegeneration-associated proteins such as amyloid beta A4 protein, alpha-synuclein and tau protein (4–6, 73). However, naturally-occurring antibodies against amyloid-beta (74), alpha-synuclein (75–77) and tau protein (73, 77, 78) were also demonstrated in cohorts of healthy subjects. Similarly, apart from any pathological context, robust T-cell responses against peptides deriving from tau protein were recently demonstrated to widely occur in the general population (79). These findings suggest that the autoimmune processes described in patients with neurodegenerative conditions might be shaped by pre-existing physiological autoimmune responses directed against common neurodegeneration-associated proteins. It is worth noting that, while physiological autoimmunity was firmly demonstrated nearly 50 years ago (80–82), the intimate nature of the links bridging physiological autoimmunity to its pathological counterpart is still matter of debate. Numerous functions have been assigned to physiological autoimmunity (83, 84), including, more recently, a support to cognition (85–90). In line with these findings, our data indicate that a specific set of brain antigens expressed by B lymphocytes and involved in neurodegenerative diseases might prime a neuroprotective and, possibly, cognition-promoting T-cell response under physiological conditions. Of note, B lymphocytes are now recognized as professional APCs (46–48) and, most interestingly, memory B lymphocytes were demonstrated to trigger the activation of autoreactive T-cells in an MHC-class II-dependent manner (91). Indeed, the autophagy of cytosolic and nuclear proteins in B lymphocytes provide a continuous source of endogenous MHC class-II ligands (92) and such autophagosome-derived peptides induce the proliferation of autologous T-cells under in vitro conditions (93). However, that neurodegeneration-associated proteins provide MHC class II-binding endogenous peptides in B lymphocytes neither prove that T-cells are actually primed against such peptides in vivo nor that peptides deriving from misfolded neurodegeneration-associated proteins are presented by B lymphocytes under neurodegenerative conditions. Furthermore, even if it was actually the case, the phenotype of autoreactive T-cells generated via such a mechanism would need to be explored. More generally, one has to keep in mind that no consensus has been currently reached regarding the phenotype of physiological autoreactive T-cells. Thus, both autoreactive Tregs and autoreactive pro-inflammatory T-cells belong to the physiological T-cell repertoire and were both found to exert neuroprotective effects (94–97).
Several genes involved in familial forms of neurodegenerative disorders exert key functions in the autophagy pathway. These notably comprise PRKN, PINK1, and SQSTM1 (98–101). In the recent years, major works provided evidence that, in neurons and immune cells, functional defects in such genes hamper mitophagy (a specialized form of autophagy), stimulate the inflammasome pathway and foster the presentation of mitochondrial antigens by MHC class I molecules (33, 102, 103). However, these findings do not render account for the existence of HLA class II-restricted T-cell responses against neurodegeneration-associated proteins. Moreover, antigens targeted by autoimmunity during neurodegenerative conditions are far from deriving only from the mitochondrial compartment. In this regard, our work suggests that in B lymphocytes, the inflammation/autophagy-related molecules SQSTM1 and TRAF6 are crucially involved in the presentation of neurodegeneration-associated antigens by MHC class II molecules. However, while our data mining approach was restricted to the retrieval of experimentally-demonstrated protein-protein interactions, whether such interactions actually occur in human B lymphocytes remains to be ascertained. This issue clearly requires to be addressed in future studies.
The mining of previously published mass spectrometry analyses performed on MHC class II-eluted peptides showed that, in human B lymphocytes, several neurodegeneration-associated proteins provide endogenous peptides which bind a large range of MHC class II alleles. This is notably the case for amyloid beta A4 protein, sequestosome-1 and profilin-1. Similarly, peptides deriving from HSPA8, a hub molecule which links a high number of neurodegeneration-associated proteins, bind multiple MHC class II alleles. Our observations suggest that these 4 molecules are likely to elicit immune responses in a large range of the human population. Determining whether or not such antigens trigger cognition-promoting autoimmunity is a potentially important issue. On another hand, in human B lymphocytes, several neurodegeneration-associated antigens appear to provide endogenous peptides harboring an allele-specific MHC class II restriction in B lymphocytes. For e.g., endogenous peptides deriving from microtubule-associated protein tau, PSEN2 and Serine/threonine-protein kinase TBK1 were exclusively reported to bind HLA-DRB1 1501-encoded MHC class II molecules under the experimental conditions described above. Since the HLA-DRB1 1501 allele is a susceptibility gene for late onset AD (14), this may prove to be of interest in the context of AD pathophysiology. In particular, although a widespread T-cell reactivity against Tau peptides was recently demonstrated in the general human population (79), this finding indicates that individuals bearing the HLA-DRB1 1501 allele may exhibit qualitative and/or quantitative specificities regarding their anti-Tau T-cell responses. Finally, it should be noticed that in subjects bearing the HLA-DRB1 1501 allele, B lymphocytes were shown to express membrane-anchored HLA-DRB1 1501-encoded MHC molecules (104) and to efficiently present autoantigens to CD4 T-cells via such MHC class II molecules (91).
We previously proposed that the genetic polymorphism of the HLA-DRB1 locus, which, among primates, is extremely high in the human species (105), might allow the allele-specific presentation of distinct sets of “brain superautoantigens” (106, 107) leading, in turn, to the development and maintenance of distinct sets of cognition-promoting T-cells. The present work indicates that common neurodegeneration-associated proteins might represent an important share of brain superautoantigens. Interestingly, recent magnetic resonance imaging studies reported that, in healthy subjects, specific HLA-DR alleles correlate with the volume ranges of specific brain structures (108, 109). Future studies should be designed to determine whether HLA-DR polymorphism might match both brain structural features and the diversity of T-cell responses against common neurodegeneration-associated proteins. Finally, in the human B-cell lineage, MHC class II-binding endogenous peptides are highly significantly and specifically enriched in peptides deriving from common neurodegeneration-associated proteins. This result raises the intriguing possibility that a main function of physiological autoimmunity could be to control the blood-circulating levels of aggregated forms of amyloid beta A4 protein, alpha-synuclein, tau protein and possibly other neurodegeneration-associated proteins.
Data Availability Statement
Publicly available datasets were analyzed in this study. This data can be found here: https://thebiogrid.org/, https://haemosphere.org/, http://www.humanproteomemap.org/, https://www.iedb.org/.
Author Contributions
SN performed the bioinformatics analyses and wrote the paper. MG and LP performed quality controls of bioinformatics analyses and wrote the paper.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank Céline Auxenfans and Pascale Pascal from the Bank of Tissues and Cells of the Hospices Civils de Lyon for leaving us the opportunity to develop a bioinformatics research group. This manuscript was released as a preprint at BioRxiv (110).
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2019.02704/full#supplementary-material
References
1. Ilieva H, Polymenidou M, Cleveland DW. Non–cell autonomous toxicity in neurodegenerative disorders: ALS and beyond. J Cell Biol. (2009) 187:761–72. doi: 10.1083/jcb.200908164
2. Scekic-Zahirovic J, El Oussini H, Mersmann S, Drenner K, Wagner M, Sun Y, et al. Motor neuron intrinsic and extrinsic mechanisms contribute to the pathogenesis of FUS-associated amyotrophic lateral sclerosis. Acta Neuropathol. (2017) 133:887–906. doi: 10.1007/s00401-017-1687-9
3. Boillee S, Yamanaka K, Lobsiger CS, Copeland NG, Jenkins NA, Kassiotis G, et al. Onset and progression in inherited ALS determined by motor neurons and microglia. Science. (2006) 312:1389–92. doi: 10.1126/science.1123511
4. Monsonego A, Zota V, Karni A, Krieger JI, Bar-Or A, Bitan G, et al. Increased T cell reactivity to amyloid β protein in older humans and patients with Alzheimer disease. J Clin Invest. (2003) 112:415–22. doi: 10.1172/JCI200318104
5. Sulzer D, Alcalay RN, Garretti F, Cote L, Kanter E, Agin-Liebes J, et al. T cells from patients with Parkinson's disease recognize α-synuclein peptides. Nature. (2017) 546:656–61. doi: 10.1038/nature22815
6. Garretti F, Agalliu D, Lindestam Arlehamn CS, Sette A, Sulzer D. Autoimmunity in Parkinson's Disease: the role of α-synuclein-specific T cells. Front Immunol. (2019) 10:303. doi: 10.3389/fimmu.2019.00303
7. Benner EJ, Banerjee R, Reynolds AD, Sherman S, Pisarev VM, Tsiperson V, et al. Nitrated α-synuclein immunity accelerates degeneration of nigral dopaminergic neurons. PLoS ONE. (2008) 3:e1376. doi: 10.1371/journal.pone.0001376
8. Brochard V, Combadière B, Prigent A, Laouar Y, Perrin A, Beray-Berthat V, et al. Infiltration of CD4+ lymphocytes into the brain contributes to neurodegeneration in a mouse model of Parkinson disease. J Clin Invest. (2008) 119:182–92. doi: 10.1172/JCI36470
9. Laurent C, Dorothée G, Hunot S, Martin E, Monnet Y, Duchamp M, et al. Hippocampal T cell infiltration promotes neuroinflammation and cognitive decline in a mouse model of tauopathy. Brain. (2017) 140:184–200. doi: 10.1093/brain/aww270
10. Sheean RK, McKay FC, Cretney E, Bye CR, Perera ND, Tomas D, et al. Association of regulatory T-cell expansion with progression of amyotrophic lateral sclerosis. JAMA Neurol. (2018) 75:681. doi: 10.1001/jamaneurol.2018.0035
11. Beers DR, Zhao W, Appel SH. The role of regulatory T lymphocytes in amyotrophic lateral sclerosis. JAMA Neurol. (2018) 75:656. doi: 10.1001/jamaneurol.2018.0043
12. Coque E, Salsac C, Espinosa-Carrasco G, Varga B, Degauque N, Cadoux M, et al. Cytotoxic CD8 + T lymphocytes expressing ALS-causing SOD1 mutant selectively trigger death of spinal motoneurons. Proc Natl Acad Sci U.S.A. (2019) 116:2312–7. doi: 10.1073/pnas.1815961116
13. Niemelä V, Burman J, Blennow K, Zetterberg H, Larsson A, Sundblom J. Cerebrospinal fluid sCD27 levels indicate active T cell-mediated inflammation in premanifest Huntington's disease. PLoS ONE. (2018) 13:e0193492. doi: 10.1371/journal.pone.0193492
14. Kunkle BW, Grenier-Boley B, Sims R, Bis JC, Damotte V, Naj AC, et al. Genetic meta-analysis of diagnosed Alzheimer's disease identifies new risk loci and implicates Aβ, tau, immunity and lipid processing. Nat Genet. (2019) 51:414–30. doi: 10.1038/s41588-019-0358-2
15. Hollenbach JA, Norman PJ, Creary LE, Damotte V, Montero-Martin G, Caillier S, et al. A specific amino acid motif of HLA-DRB1 mediates risk and interacts with smoking history in Parkinson's disease. Proc Natl Acad Sci USA. (2019) 116:7419–24. doi: 10.1073/pnas.1821778116
16. Witoelar A, Jansen IE, Wang Y, Desikan RS, Gibbs JR, Blauwendraat C, et al. Genome-wide pleiotropy between Parkinson Disease and Autoimmune Diseases. JAMA Neurol. (2017) 74:780. doi: 10.1001/jamaneurol.2017.0469
17. Broce I, Karch CM, Wen N, Fan CC, Wang Y, Hong Tan C, et al. Immune-related genetic enrichment in frontotemporal dementia: an analysis of genome-wide association studies. PLOS Med. (2018) 15:e1002487. doi: 10.1371/journal.pmed.1002487
18. Pottier C, Ren Y, Perkerson RB, Baker M, Jenkins GD, van Blitterswijk M, et al. Genome-wide analyses as part of the international FTLD-TDP whole-genome sequencing consortium reveals novel disease risk factors and increases support for immune dysfunction in FTLD. Acta Neuropathol. (2019) 137:879–99. doi: 10.1007/s00401-019-01962-9
19. Raj T, Rothamel K, Mostafavi S, Ye C, Lee MN, Replogle JM, et al. Polarization of the effects of autoimmune and neurodegenerative risk alleles in leukocytes. Science. (2014) 344:519–23. doi: 10.1126/science.1249547
20. Matheoud D, Sugiura A, Bellemare-Pelletier A, Laplante A, Rondeau C, Chemali M, et al. Parkinson's Disease-related proteins PINK1 and parkin repress mitochondrial antigen presentation. Cell. (2016) 166:314–27. doi: 10.1016/j.cell.2016.05.039
21. Lall D, Baloh RH. Microglia and C9orf72 in neuroinflammation and ALS and frontotemporal dementia. J Clin Invest. (2017) 127:3250–8. doi: 10.1172/JCI90607
22. ORourke JG, Bogdanik L, Yanez A, Lall D, Wolf AJ, Muhammad AKMG, et al. C9orf72 is required for proper macrophage and microglial function in mice. Science.(2016) 351:1324–9. doi: 10.1126/science.aaf1064
23. Nataf S, Pays L. Gene co-expression analysis unravels a link between C9orf72 and RNA metabolism in myeloid cells. Acta Neuropathol Commun. (2015) 3:64. doi: 10.1186/s40478-015-0242-y
24. Fava VM, Manry J, Cobat A, Orlova M, Van Thuc N, Ba NN, et al. A missense LRRK2 variant is a risk factor for excessive inflammatory responses in leprosy. PLoS Negl Trop Dis. (2016) 10:e0004412. doi: 10.1371/journal.pntd.0004412
25. Wong AYW, Oikonomou V, Paolicelli G, De Luca A, Pariano M, Fric J, et al. Leucine-Rich Repeat Kinase 2 Controls the Ca2+/Nuclear Factor of Activated T Cells/IL-2 pathway during aspergillus non-canonical autophagy in dendritic cells. Front Immunol. (2018) 9:210. doi: 10.3389/fimmu.2018.00210
26. Härtlova A, Herbst S, Peltier J, Rodgers A, Bilkei-Gorzo O, Fearns A, et al. LRRK2 is a negative regulator of Mycobacterium tuberculosis phagosome maturation in macrophages. EMBO J. (2018) 37:e98694. doi: 10.15252/embj.201798694
27. Kozina E, Sadasivan S, Jiao Y, Dou Y, Ma Z, Tan H, et al. Mutant LRRK2 mediates peripheral and central immune responses leading to neurodegeneration in vivo. Brain. (2018) 141:1753–69. doi: 10.1093/brain/awy077
28. Nielsen SR, Quaranta V, Linford A, Emeagi P, Rainer C, Santos A, et al. Macrophage-secreted granulin supports pancreatic cancer metastasis by inducing liver fibrosis. Nat Cell Biol. (2016) 18:549–60. doi: 10.1038/ncb3340
29. Nguyen AD, Nguyen TA, Singh RK, Eberlé D, Zhang J, Abate JP, et al. Progranulin in the hematopoietic compartment protects mice from atherosclerosis. Atherosclerosis. (2018) 277:145–54. doi: 10.1016/j.atherosclerosis.2018.08.042
30. Carneiro FRG, Lepelley A, Seeley JJ, Hayden MS, Ghosh S. An essential role for ECSIT in mitochondrial complex I assembly and mitophagy in macrophages. Cell Rep. (2018) 22:2654–66. doi: 10.1016/j.celrep.2018.02.051
31. Zhou J, Yang R, Zhang Z, Liu Q, Zhang Y, Wang Q, et al. Mitochondrial protein PINK1 positively regulates RLR signaling. Front Immunol. (2019) 10:1069. doi: 10.3389/fimmu.2019.01069
32. Xu Y, Shen J, Ran Z. Emerging views of mitophagy in immunity and autoimmune diseases. Autophagy. (2019) 15:1–15. doi: 10.1080/15548627.2019.1603547
33. Zhong Z, Umemura A, Sanchez-Lopez E, Liang S, Shalapour S, Wong J, et al. NF-κB restricts inflammasome activation via elimination of damaged mitochondria. Cell. (2016) 164:896–910. doi: 10.1016/j.cell.2015.12.057
34. Sliter DA, Martinez J, Hao L, Chen X, Sun N, Fischer TD, et al. Parkin and PINK1 mitigate STING-induced inflammation. Nature. (2018) 561:258–62. doi: 10.1038/s41586-018-0448-9
35. National Library of Medicine (US). Genetics Home Reference. Bethesda, MD: The Library. Available online at: https://ghr.nlm.nih.gov
36. Kuleshov MV, Jones MR, Rouillard AD, Fernandez NF, Duan Q, Wang Z, et al. Enrichr: a comprehensive gene set enrichment analysis web server 2016 update. Nucleic Acids Res. (2016) 44:W90–7. doi: 10.1093/nar/gkw377
37. Pletscher-Frankild S, Pallejà A, Tsafou K, Binder JX, Jensen LJ. DISEASES: text mining and data integration of disease–gene associations. Methods. (2015) 74:83–9. doi: 10.1016/j.ymeth.2014.11.020
38. Oughtred R, Stark C, Breitkreutz B-J, Rust J, Boucher L, Chang C, et al. The BioGRID interaction database: 2019 update. Nucleic Acids Res. (2019) 47:D529–41. doi: 10.1093/nar/gky1079
39. Kim M-S, Pinto SM, Getnet D, Nirujogi RS, Manda SS, Chaerkady R, et al. A draft map of the human proteome. Nature. (2014) 509:575–81. doi: 10.1038/nature13302
40. Johnston HE, Carter MJ, Larrayoz M, Clarke J, Garbis SD, Oscier D, et al. Proteomics profiling of CLL versus healthy B-cells identifies putative therapeutic targets and a subtype-independent signature of spliceosome dysregulation. Mol Cell Proteomics. (2018) 17:776–91. doi: 10.1074/mcp.RA117.000539
41. Worah K, Mathan TSM, Vu Manh TP, Keerthikumar S, Schreibelt G, Tel J, et al. Proteomics of human dendritic cell subsets reveals subset-specific surface markers and differential inflammasome function. Cell Rep. (2016) 16:2953–66. doi: 10.1016/j.celrep.2016.08.023
42. Strasser L, Dang H-H, Schwarz H, Asam C, Ferreira F, Horejs-Hoeck J, et al. Unbiased quantitative proteomics reveals a crucial role of the allergen context for the activation of human dendritic cells. Sci Rep. (2017) 7:16638. doi: 10.1038/s41598-017-16726-2
43. Dhanda SK, Mahajan S, Paul S, Yan Z, Kim H, Jespersen MC, et al. IEDB-AR: immune epitope database—analysis resource in 2019. Nucleic Acids Res. (2019) 47:W502–6. doi: 10.1093/nar/gkz452
44. UniProt Consortium. UniProt: a worldwide hub of protein knowledge. Nucleic Acids Res. (2019) 47:D506–15. doi: 10.1093/nar/gky1049
45. Shannon P, Markiel A, Ozier O, Baliga NS, Wang JT, Ramage D, et al. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. (2003) 13:2498–504. doi: 10.1101/gr.1239303
46. Bretou M, Kumari A, Malbec O, Moreau HD, Obino D, Pierobon P, et al. Dynamics of the membrane-cytoskeleton interface in MHC class II-restricted antigen presentation. Immunol Rev. (2016) 272:39–51. doi: 10.1111/imr.12429
47. Yuseff M-I, Pierobon P, Reversat A, Lennon-Duménil A-M. How B cells capture, process and present antigens: a crucial role for cell polarity. Nat Rev Immunol. (2013) 13:475–86. doi: 10.1038/nri3469
48. Abbas AK. Antigen presentation by B lymphocytes: mechanisms and functional significance. Semin Immunol. (1989) 1:5–12.
49. Villadangos JA, Schnorrer P. Intrinsic and cooperative antigen-presenting functions of dendritic-cell subsets in vivo. Nat Rev Immunol. (2007) 7:543–55. doi: 10.1038/nri2103
50. Guermonprez P, Valladeau J, Zitvogel L, Théry C, Amigorena S. Antigen presentation and T cell stimulation by dendritic cells. Annu Rev Immunol. (2002) 20:621–67. doi: 10.1146/annurev.immunol.20.100301.064828
51. Steinman RM, Hawiger D, Nussenzweig MC. Tolerogenic dendritic cells. Annu Rev Immunol. (2003) 21:685–711. doi: 10.1146/annurev.immunol.21.120601.141040
52. Zhou D, Li P, Lin Y, Lott JM, Hislop AD, Canaday DH, Brutkiewicz RR, et al. −2a facilitates MHC class II presentation of cytoplasmic antigens. Immunity. (2005) 22:571–81. doi: 10.1016/j.immuni.2005.03.009
53. Mycko MP, Cwiklinska H, Szymanski J, Szymanska B, Kudla G, Kilianek L, et al. Inducible heat shock protein 70 promotes myelin autoantigen presentation by the HLA class II. J Immunol. (2004) 172:202–13. doi: 10.4049/jimmunol.172.1.202
54. Houlihan JL, Metzler JJ, Blum JS. HSP90alpha and HSP90beta isoforms selectively modulate MHC class II antigen presentation in B cells. J Immunol. (2009) 182:7451–8. doi: 10.4049/jimmunol.0804296
55. van Eden W, Jansen MAA, Ludwig I, van Kooten P, van der Zee R, Broere F. The Enigma of heat shock proteins in immune tolerance. Front Immunol. (2017) 8:1599. doi: 10.3389/fimmu.2017.01599
56. Cohen IR. Autoantibody repertoires, natural biomarkers, and system controllers. Trends Immunol. (2013) 34:620–5. doi: 10.1016/j.it.2013.05.003
57. Quintana FJ, Cohen IR. Heat shock proteins as endogenous adjuvants in sterile and septic inflammation. J Immunol. (2005) 175:2777–82. doi: 10.4049/jimmunol.175.5.2777
58. Kuwana T, Peterson PA, Karlsson L. Exit of major histocompatibility complex class II-invariant chain p35 complexes from the endoplasmic reticulum is modulated by phosphorylation. Proc Natl Acad Sci USA. (1998) 95:1056–61. doi: 10.1073/pnas.95.3.1056
59. Buschow SI, van Balkom BWM, Aalberts M, Heck AJR, Wauben M, Stoorvogel W. MHC class II-associated proteins in B-cell exosomes and potential functional implications for exosome biogenesis. Immunol Cell Biol. (2010) 88:851–6. doi: 10.1038/icb.2010.64
60. Umahara T, Uchihara T, Tsuchiya K, Nakamura A, Iwamoto T, Ikeda K, et al. 14-3-3 proteins and zeta isoform containing neurofibrillary tangles in patients with Alzheimer's disease. Acta Neuropathol. (2004) 108:279–86. doi: 10.1007/s00401-004-0885-4
61. Kawamoto Y, Akiguchi I, Nakamura S, Honjyo Y, Shibasaki H, Budka H. 14-3-3 proteins in Lewy bodies in Parkinson disease and diffuse Lewy body disease brains. J Neuropathol Exp Neurol. (2002) 61:245–53. doi: 10.1093/jnen/61.3.245
62. Jia B, Wu Y, Zhou Y. 14-3-3 and aggresome formation: implications in neurodegenerative diseases. Prion. (2014) 8:173–7. doi: 10.4161/pri.28123
63. Magilnick N, Reyes EY, Wang W-L, Vonderfecht SL, Gohda J, Inoue J, et al. miR-146a – Traf6 regulatory axis controls autoimmunity and myelopoiesis, but is dispensable for hematopoietic stem cell homeostasis and tumor suppression. Proc Natl Acad Sci USA. (2017) 114:E7140–9. doi: 10.1073/pnas.1706833114
64. Brenke JK, Popowicz GM, Schorpp K, Rothenaigner I, Roesner M, Meininger I, et al. Targeting TRAF6 E3 ligase activity with a small-molecule inhibitor combats autoimmunity. J Biol Chem. (2018) 293:13191–203. doi: 10.1074/jbc.RA118.002649
65. Ahonen CL, Manning EM, Erickson LD, O'Connor BP, Lind EF, Pullen SS, et al. The CD40-TRAF6 axis controls affinity maturation and the generation of long-lived plasma cells. Nat Immunol. (2002) 3:451–6. doi: 10.1038/ni792
66. Jalukar SV, ostager BS, Bishop GA. Characterization of the roles of TNF receptor-associated factor 6 in CD40-Mediated B lymphocyte effector functions. J Immunol. (2000) 164:623–30. doi: 10.4049/jimmunol.164.2.623
67. Ben-Nun A, Kaushansky N, Kawakami N, Krishnamoorthy G, Berer K, Liblau R, et al. From classic to spontaneous and humanized models of multiple sclerosis: Impact on understanding pathogenesis and drug development. J Autoimmun. (2014) 54:33–50. doi: 10.1016/j.jaut.2014.06.004
68. Adorini L, Moreno J, Momburg F, Hämmerling GJ, Guéry JC, Valli A, et al. Exogenous peptides compete for the presentation of endogenous antigens to major histocompatibility complex class II-restricted T cells. J Exp Med. (1991) 174:945–8. doi: 10.1084/jem.174.4.945
69. Leung CSK. Endogenous antigen presentation of MHC class II epitopes through non-autophagic pathways. Front Immunol. (2015) 6:464. doi: 10.3389/fimmu.2015.00464
70. Coulon P-G, Richetta C, Rouers A, Blanchet FP, Urrutia A, Guerbois M, et al. HIV-infected dendritic cells present endogenous MHC class II-restricted antigens to HIV-specific CD4+ T cells. J Immunol. (2016) 197:517–32. doi: 10.4049/jimmunol.1600286
71. Rudensky AY, Maric M, Eastman S, Shoemaker L, DeRoos PC, Blum JS. Intracellular assembly and transport of endogenous peptide-MHC class II complexes. Immunity. (1994) 1:585–94. doi: 10.1016/1074-7613(94)90048-5
72. Pertea M, Shumate A, Pertea G, Varabyou A, Breitwieser FP, Chang Y-C, et al. CHESS: a new human gene catalog curated from thousands of large-scale RNA sequencing experiments reveals extensive transcriptional noise. Genome Biol. (2018) 19:208. doi: 10.1186/s13059-018-1590-2
73. Bartos A, Fialová L, Švarcová J. Lower serum antibodies against tau protein and heavy neurofilament in Alzheimer's Disease. J Alzheimers Dis. (2018) 64:751–60. doi: 10.3233/JAD-180039
74. Taguchi H, Planque S, Nishiyama Y, Szabo P, Weksler ME, Friedland RP, et al. Catalytic antibodies to amyloid beta peptide in defense against Alzheimer disease. Autoimmun Rev. (2008) 7:391–7. doi: 10.1016/j.autrev.2008.03.004
75. Huang Y, Xie X, Ji M, Yu X, Zhu J, Zhang L, et al. Naturally occurring autoantibodies against α-synuclein rescues memory and motor deficits and attenuates α-synuclein pathology in mouse model of Parkinson's disease. Neurobiol Dis. (2019) 124:202–17. doi: 10.1016/j.nbd.2018.11.024
76. Albus A, Gold M, Bach J-P, Burg-Roderfeld M, Jördens M, Kirchhein Y, et al. Extending the functional characteristics of naturally occurring autoantibodies against β-Amyloid, Prion Protein and α-Synuclein. PLoS ONE. (2018) 13:e0202954. doi: 10.1371/journal.pone.0202954
77. Kuhn I, Rogosch T, Schindler TI, Tackenberg B, Zemlin M, Maier RF, et al. Serum titers of autoantibodies against α-synuclein and tau in child- and adulthood. J Neuroimmunol. (2018) 315:33–9. doi: 10.1016/j.jneuroim.2017.12.003
78. Krestova M, Hromadkova L, Ricny J. Purification of natural antibodies against tau protein by affinity chromatography. Methods Mol Biol. (2017) 1643:33–44. doi: 10.1007/978-1-4939-7180-0_4
79. Lindestam Arlehamn CS, Pham J, Alcalay RN, Frazier A, Shorr E, Carpenter C, et al. Widespread Tau-specific CD4 T cell reactivity in the general population. J Immunol. (2019) 203:84–92. doi: 10.4049/jimmunol.1801506
80. Cohen IR, Wekerle H. Regulation of autosensitization: the immune activation and specific inhibition of self-recognizing thymus-derived lymphocytes. J Exp Med. (1973) 137:224–38. doi: 10.1084/jem.137.2.224
81. Cohen IR, Wekerle H. Autosensitization of lymphocytes against thymus reticulum cells. Science. (1972) 176:1324–5. doi: 10.1126/science.176.4041.1324
82. Cohen IR, Globerson A, Feldman M. Autosensitization in vitro. J Exp Med. (1971) 133:834–45. doi: 10.1084/jem.133.4.834
83. Coutinho A, Kazatchkine MD, Avrameas S. Natural autoantibodies. Curr Opin Immunol. (1995) 7:812–8. doi: 10.1016/0952-7915(95)80053-0
84. Schwartz M, Cohen IR. Autoimmunity can benefit self-maintenance. Immunol Today. (2000) 21:265–8. doi: 10.1016/S0167-5699(00)01633-9
85. Schwartz M, Baruch K. Breaking peripheral immune tolerance to CNS antigens in neurodegenerative diseases: boosting autoimmunity to fight-off chronic neuroinflammation. J Autoimmun. (2014) 54:8–14. doi: 10.1016/j.jaut.2014.08.002
86. Kipnis J, Cardon M, Strous RD, Schwartz M. Loss of autoimmune T cells correlates with brain diseases: possible implications for schizophrenia? Trends Mol Med. (2006) 12:107–12. doi: 10.1016/j.molmed.2006.01.003
87. Radjavi A, Smirnov I, Derecki N, Kipnis J. Dynamics of the meningeal CD4+ T-cell repertoire are defined by the cervical lymph nodes and facilitate cognitive task performance in mice. Mol Psychiatry. (2014) 19:531–2. doi: 10.1038/mp.2013.79
88. Derecki NC, Cardani AN, Yang CH, Quinnies KM, Crihfield A, Lynch KR, et al. Regulation of learning and memory by meningeal immunity: a key role for IL-4. J Exp Med. (2010) 207:1067–80. doi: 10.1084/jem.20091419
89. Brynskikh A, Warren T, Zhu J, Kipnis J. Adaptive immunity affects learning behavior in mice. Brain Behav Immun. (2008) 22:861–9. doi: 10.1016/j.bbi.2007.12.008
90. Kipnis J, Cohen H, Cardon M, Ziv Y, Schwartz M. T cell deficiency leads to cognitive dysfunction: Implications for therapeutic vaccination for schizophrenia and other psychiatric conditions. Proc Natl Acad Sci USA. (2004) 101:8180–5. doi: 10.1073/pnas.0402268101
91. Jelcic I, Al Nimer F, Wang J, Lentsch V, Planas R, Jelcic I, et al. Memory B cells activate brain-homing, autoreactive CD4+ T cells in multiple sclerosis. Cell. (2018) 175:85–100.e23. doi: 10.1016/j.cell.2018.08.011
92. Schmid D, Münz C. Innate and adaptive immunity through autophagy. Immunity. (2007) 27:11–21. doi: 10.1016/j.immuni.2007.07.004
93. Mohme M, Hotz C, Stevanović S, Binder T, Lee J-H, Okoniewski M, et al. HLA-DR15-derived self-peptides are involved in increased autologous T cell proliferation in multiple sclerosis. Brain. (2013) 136:1783–98. doi: 10.1093/brain/awt108
94. Kipnis J, Avidan H, Caspi RR, Schwartz M. Dual effect of CD4+CD25+ regulatory T cells in neurodegeneration: a dialogue with microglia. Proc Natl Acad Sci USA. (2004) 101:14663–14669. doi: 10.1073/pnas.0404842101
95. Baruch K, Rosenzweig N, Kertser A, Deczkowska A, Sharif AM, Spinrad A, et al. Breaking immune tolerance by targeting Foxp3+ regulatory T cells mitigates Alzheimer's disease pathology. Nat Commun. (2015) 6:7967. doi: 10.1038/ncomms8967
96. Baek H, Ye M, Kang G-H, Lee C, Lee G, Bin D, et al. Neuroprotective effects of CD4+CD25+Foxp3+ regulatory T cells in a 3xTg-AD Alzheimer's disease model. Oncotarget. (2016) 7:69347–57. doi: 10.18632/oncotarget.12469
97. Dansokho C, Ait Ahmed D, Aid S, Toly-Ndour C, Chaigneau T, Calle V, et al. Regulatory T cells delay disease progression in Alzheimer-like pathology. Brain. (2016) 139:1237–51. doi: 10.1093/brain/awv408
98. Majcher V, Goode A, James V, Layfield R. Autophagy receptor defects and ALS-FTLD. Mol Cell Neurosci. (2015) 66:43–52. doi: 10.1016/j.mcn.2015.01.002
99. Ciechanover A, Kwon YT. Protein quality control by molecular chaperones in neurodegeneration. Front Neurosci. (2017) 11:185. doi: 10.3389/fnins.2017.00185
100. Springer W, Kahle PJ. Regulation of PINK1-parkin-mediated mitophagy. Autophagy. (2011) 7:266–78. doi: 10.4161/auto.7.3.14348
101. Vives-Bauza C, Przedborski S. Mitophagy: the latest problem for Parkinson's disease. Trends Mol Med. (2011) 17:158–65. doi: 10.1016/j.molmed.2010.11.002
102. Baden P, Deleidi M. Mitochondrial antigen presentation: a vacuolar path to autoimmunity in Parkinson's Disease. Trends Immunol. (2016) 37:719–21. doi: 10.1016/j.it.2016.08.016
103. Roberts RF, Fon EA. Presenting mitochondrial antigens: PINK1, Parkin and MDVs steal the show. Cell Res. (2016) 26:1180–1. doi: 10.1038/cr.2016.104
104. Prat E, Tomaru U, Sabater L, Park DM, Granger R, Kruse N, et al. HLA-DRB5*0101 and -DRB1*1501 expression in the multiple sclerosis-associated HLA-DR15 haplotype. J Neuroimmunol. (2005) 167:108–19. doi: 10.1016/j.jneuroim.2005.04.027
105. Bergström TF, Josefsson A, Erlich HA, Gyllensten U. Recent origin of HLA-DRB1 alleles and implications for human evolution. Nat Genet. (1998) 18:237–42. doi: 10.1038/ng0398-237
106. Nataf S. Evolution, immunity and the emergence of brain superautoantigens. F1000Research. (2017) 6:171. doi: 10.12688/f1000research.10950.1
107. Nataf S. Autoimmunity as a driving force of cognitive evolution. Front Neurosci. (2017) 11:582. doi: 10.3389/fnins.2017.00582
108. James LM, Christova P, Lewis SM, Engdahl BE, Georgopoulos A, Georgopoulos AP. Protective effect of Human Leukocyte Antigen (HLA) Allele DRB1*13:02 on age-related brain gray matter volume reduction in healthy women. EBioMedicine. (2018) 29:31–7. doi: 10.1016/j.ebiom.2018.02.005
109. Wang Z-X, Wan Y, Tan L, Liu J, Wang H-F, Sun F-R, et al. Genetic association of HLA gene variants with MRI brain structure in Alzheimer's Disease. Mol Neurobiol. (2017) 54:3195–204. doi: 10.1007/s12035-016-9889-z
Keywords: neurodegeneration, bioinformatics, autoimmunity, B-lymphocytes, amyloid-beta-protein, tau & phospho-tau protein, synuclein, prion
Citation: Nataf S, Guillen M and Pays L (2019) Common Neurodegeneration-Associated Proteins Are Physiologically Expressed by Human B Lymphocytes and Are Interconnected via the Inflammation/Autophagy-Related Proteins TRAF6 and SQSTM1. Front. Immunol. 10:2704. doi: 10.3389/fimmu.2019.02704
Received: 03 July 2019; Accepted: 04 November 2019;
Published: 25 November 2019.
Edited by:
Robert Weissert, University of Regensburg, GermanyReviewed by:
Hans-Georg Rammensee, University of Tübingen, GermanyUmesh K. Jinwal, University of South Florida, United States
Copyright © 2019 Nataf, Guillen and Pays. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Serge Nataf, serge.nataf@inserm.fr