
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Immunol. , 15 November 2019
Sec. Inflammation
Volume 10 - 2019 | https://doi.org/10.3389/fimmu.2019.02519
 Kyle Caution1
Kyle Caution1 Nicholas Young2
Nicholas Young2 Frank Robledo-Avila3
Frank Robledo-Avila3 Kathrin Krause1
Kathrin Krause1 Arwa Abu Khweek1,4
Arwa Abu Khweek1,4 Kaitlin Hamilton1
Kaitlin Hamilton1 Asmaa Badr1Anup Vaidya1
Asmaa Badr1Anup Vaidya1 Kylene Daily1Hawin Gosu1Midhun N. K. Anne1
Kylene Daily1Hawin Gosu1Midhun N. K. Anne1 Mostafa Eltobgy1
Mostafa Eltobgy1 Duaa Dakhlallah5Sudha Argwal2
Duaa Dakhlallah5Sudha Argwal2 Shady Estfanous1
Shady Estfanous1 Xiaoli Zhang6
Xiaoli Zhang6 Santiago Partida-Sanchez3
Santiago Partida-Sanchez3 Mikhail A. Gavrilin7
Mikhail A. Gavrilin7 Wael N. Jarjour2
Wael N. Jarjour2 Amal O. Amer1*
Amal O. Amer1*Gout is characterized by attacks of arthritis with hyperuricemia and monosodium urate (MSU) crystal-induced inflammation within joints. Innate immune responses are the primary drivers for tissue destruction and inflammation in gout. MSU crystals engage the Nlrp3 inflammasome, leading to the activation of caspase-1 and production of IL-1β and IL-18 within gout-affected joints, promoting the influx of neutrophils and monocytes. Here, we show that caspase-11−/− mice and their derived macrophages produce significantly reduced levels of gout-specific cytokines including IL-1β, TNFα, IL-6, and KC, while others like IFNγ and IL-12p70 are not altered. IL-1β induces the expression of caspase-11 in an IL-1 receptor-dependent manner in macrophages contributing to the priming of macrophages during sterile inflammation. The absence of caspase-11 reduced the ability of macrophages and neutrophils to migrate in response to exogenously injected KC in vivo. Notably, in vitro, caspase-11−/− neutrophils displayed random migration in response to a KC gradient when compared to their WT counterparts. This phenotype was associated with altered cofilin phosphorylation. Unlike their wild-type counterparts, caspase-11−/− neutrophils also failed to produce neutrophil extracellular traps (NETs) when treated with MSU. Together, this is the first report demonstrating that caspase-11 promotes neutrophil directional trafficking and function in an acute model of gout. Caspase-11 also governs the production of inflammasome-dependent and -independent cytokines from macrophages. Our results offer new, previously unrecognized functions for caspase-11 in macrophages and neutrophils that may apply to other neutrophil-mediated disease conditions besides gout.
Gout is an ancient disease that is estimated to affect 8.3 million people in the United States, and this number is steadily increasing (1). Gout, in its acute or chronic forms, occurs when the body mounts an inflammatory response against the crystalized form of uric acid, monosodium urate crystals (MSU), within joint spaces (2, 3).
The activation of inflammasomes requires two signals; one for priming and one for activation. This leads to inflammasome assembly, caspase-1 activation, maturation of IL-1β and IL-18, and sometimes pyroptosis via Gasdermin (GSDM) and the P2X7 receptor (4–7). Gout has been identified as a prototype IL-1β-dependent inflammatory disease (8, 9). IL-1β, an endogenous pyrogen, is first synthesized as an inactive precursor and then is cleaved to its bioactive form by caspase-1 that is canonically activated within the inflammasome complex (10–12). An intense inflammatory response is initiated when MSU crystals are phagocytosed by resident, primed macrophages and sensed by the NLR Family Pyrin Domain-Containing 3 (NLRP3), which is followed by the assembly of the inflammasome and caspase-1 activation (11). Inflammasome-dependent cytokines, interleukin-1 beta (IL-1β), and interleukin-18 (IL-18) are then released, activating interstitial cells and promoting influx of polymorphonuclear cells (PMNs—neutrophils), monocytes, and macrophages (11, 13). Release of these cytokines in gout induces the differentiation of osteoclasts from mononuclear precursors and stimulates bone resorption (14, 15). This inflammatory microenvironment also promotes more neutrophil extravasation into the joint space (16, 17). Within the affected joint space, immune cells release other pro-inflammatory cytokines like TNFα, IL-6, and IL-8 (CXCL1/KC); eicosanoid lipid mediators; and production of the enzymes collagenase and stromelysin-1 that degrade the extracellular matrix in articular structures. This causes a cascade of cytotoxic cellular contents and danger-associated molecular patterns (DAMPs) to leak into extracellular venues and recruit other immune cells, producing more inflammation and tissue destruction (11, 18). MSU crystals have been found to activate these infiltrating PMNs, not only by triggering cytokine secretion, but also by inducing neutrophil extracellular traps (NETs) (19, 20). Neutrophils form NETs in response to various proinflammatory, infectious stimuli, or crystals including MSU (21). NET formation results in the release of nuclear DNA coated with neutrophil granule enzymes, to generate NETs (21, 22). NETs orchestrate the initiation and progression of inflammation in gout (8, 20, 23).
The role of caspase-11 is versatile according to the insult. Our group and others have demonstrated several non-apoptotic functions for caspase-11. We found that caspase-11 restricts Legionella pneumophila infection by promoting the activation of the RhoA GTPase–cofilin pathway in order to regulate actin dynamics and phagolysosomal fusion within the cell (24, 25). Caspase-11 also promotes autophagosome formation to control Burkholderia cenocepacia infection via regulation of Rab7 and actin dynamics (26). Yan's group has demonstrated that caspase-11 modulates cofilin via the actin interacting protein 1 (Aip1) to promote migration of T cells (27). We recently found that caspase-11 is actually exploited by methicillin-resistant Staphylococcus aureus (MRSA) to survive in macrophages (28). When endotoxin contaminates the intracellular spaces of macrophages, caspase-11 senses the LPS and promotes downstream activation of caspase-1 and IL-1β (29–31). In other circumstances, caspase-11 mediates the release, but not the activation of IL-1β (30, 32, 33). Interestingly, caspase-11 is also essential for the production of KC in response to infection with B. cenocepacia (26). These data demonstrate that, in macrophages, caspase-11 exerts essential immune functions independently of cell death. However, the role of caspase-11 in neutrophils is still enigmatic.
The role of caspase-11 in gout has not yet been investigated. In this study, we found that caspase-11−/− mice exhibit significantly less signs of joint inflammation, including swelling and tissue damage upon injection of MSU crystals in the tibio-tarsal joint space in vivo. The absence of caspase-11 is associated with significantly less neutrophil influx and lower levels of inflammatory cytokines in the synovial fluid of MSU-injected joints. The reduction in neutrophil migration was not solely due to low cytokine production within the MSU-injected joint, but also due to an innate alteration in the ability of caspase-11−/− neutrophils to migrate toward a chemotactic signal. Particularly, caspase-11−/− neutrophils displayed a random migration pattern in response to a KC gradient when compared to their WT counterparts. In addition, caspase-11−/− neutrophils produced significantly less NETs in response to MSU (16, 34, 35) independently of the receptor-interacting protein kinase 3 (RIPK3) and mixed lineage kinase domain-like (MLKL) pathway. This phenotype is independent of gasdermin (GSDM). In addition, we found that the phosphorylation of cofilin is altered in caspase-11−/− neutrophils in response to MSU. Together, this is the first report demonstrating that caspase-11 promotes neutrophil directional trafficking and NET formation in an acute model of gout. Our results offer caspase-11 and its human orthologs, caspase 4 and 5, as viable targets in gout and other disease conditions that are driven by activated neutrophils.
Gout is due to an acute or chronic inflammatory response to MSU deposition in the joint space (13, 36). This causes the classical signs of inflammation, including swelling, tenderness, heat, and redness. Meanwhile, it has been shown that caspase-11 plays a role in promoting inflammation and cell death in response to PAMPs (pathogen-associated molecular patterns) and DAMPs (37–39). To determine if caspase-11 contributes to the classical signs of inflammation during gout, WT and caspase-11−/− mice were injected with MSU crystals into the tibio-tarsal joint of the right leg. After 24 h, the affected ankle joints were examined. We found that MSU injection promotes acute swelling in joints of WT but not caspase-11−/− mice. Caliper measurements demonstrated that WT mice exhibited significantly increased joint diameter size when compared to caspase-11−/− mice after MSU injection (Figure 1A). In addition, the joint diameter after MSU injection was significantly increased in WT mice, while the joint diameter in the caspase-11−/− mice remained unchanged after MSU injection (Figure 1B). Accordingly, MSU injection caused more punctuated redness in the footpad, ankle, and leg of the WT mice when compared to caspase-11−/− counterparts (Figure 1C). Vehicle (PBS) injection in mice did not induce swelling, as quantified by caliper, or visible signs of inflammation, demonstrating that this response was specific to MSU (Supplementary Figures 1A–C). Real-time quantitative (RT) PCR analysis performed on RNA isolated from joint samples revealed that MSU significantly induced the expression of caspase-11 mRNA in vivo, compared to the vehicle (PBS)-treated joints (Supplementary Figure 2). Taken together, these data indicate that caspase-11 is required for the swelling in the tibio-tarsal joints using an acute gout mouse model.
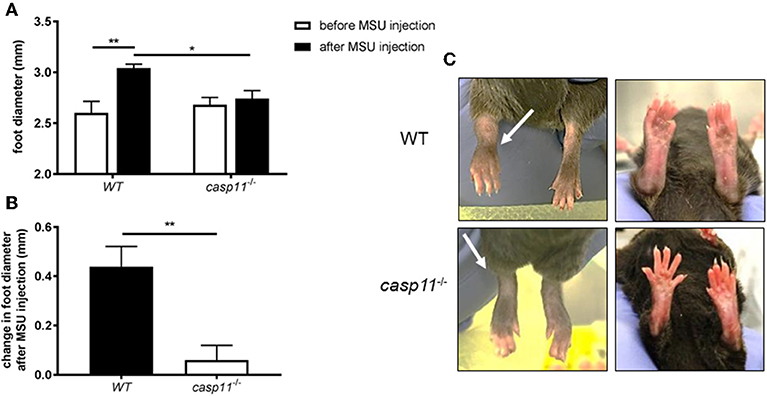
Figure 1. Swelling and increase in ankle diameter during acute gout requires caspase-11 expression. (A) Caliper measurement of WT and caspase-11−/− ankle diameters before and after injection of 0.5 mg monosodium urate (MSU) crystals into the tibio-tarsal joint. A two-way ANOVA with a Holm's Sidak comparison test was performed for statistical analysis. (B) Caliper measurement before MSU injection subtracted from measurement after injection to depict change in ankle diameter. Student's t test analysis was performed. *p <0.05, **p <0.01, n = 5 mice. (C) Representative superior and inferior pictures of WT and caspase-11−/− ankles 24 h post injection.
The expression of caspase-11 mediates swelling of MSU-treated joints (Figure 1). To determine if caspase-11 plays a role in the cellular influx and/or overall structural changes in our acute gout model, we injected MSU crystals into the right tibio-tarsal joint of WT and caspase-11−/− mice. High-resolution images of hematoxylin and eosin (H&E) slides were generated using the Aperio ImageScope™ (Leica Biosystems) technology and assessed by a pathologist in the Ohio State University Comparative Pathology and Mouse Phenotyping core. H&E analysis of the synovial area demonstrated that joints of caspase-11−/− mice contained less immune cells within the deep dermis, peri-articular soft tissue, and areas subjacent to the periosteal bone of the joint (Figures 2A,B). Signs of necrosis and damage were also blunted in the caspase-11−/− mice as tissue organization remained intact, while their WT counterparts displayed greater signs of tissue destruction (Figures 2A,B). Because H&E analysis indicated that caspase-11−/− mice exhibited impairment in cellular recruitment after MSU injection, we then determined the identity of the inflammatory cells extravasating into the MSU-injected joint by immunohistochemical assays on processed ankle joints 24 h after MSU injection. Slides prepared from WT and caspase-11−/− mice were stained for myeloperoxidase (MPO) and F4/80 (Emr1) to assay neutrophil and macrophage influx, respectively, into the joint space. Immunohistological staining demonstrated that caspase-11−/− mice exhibited a marked impairment in MPO+ and F4/80+ cell infiltration into the tibio-tarsal space when compared to their WT counterparts (Figures 3A,C). In addition, using high-resolution images, histological staining was quantified via the Aperio ImageScope software and showed that caspase-11−/− mice displayed significantly less neutrophils and macrophages in comparison to the WT ankle joints (Figures 3B,D). In sum, these data demonstrate that caspase-11 is required for immune cell influx into the synovium that leads to tissue destruction and remodeling in the gout mouse model. This phenotype could be due to reduced production of cytokines in the absence of caspase-11, an inherent trafficking defect of immune cells or both.
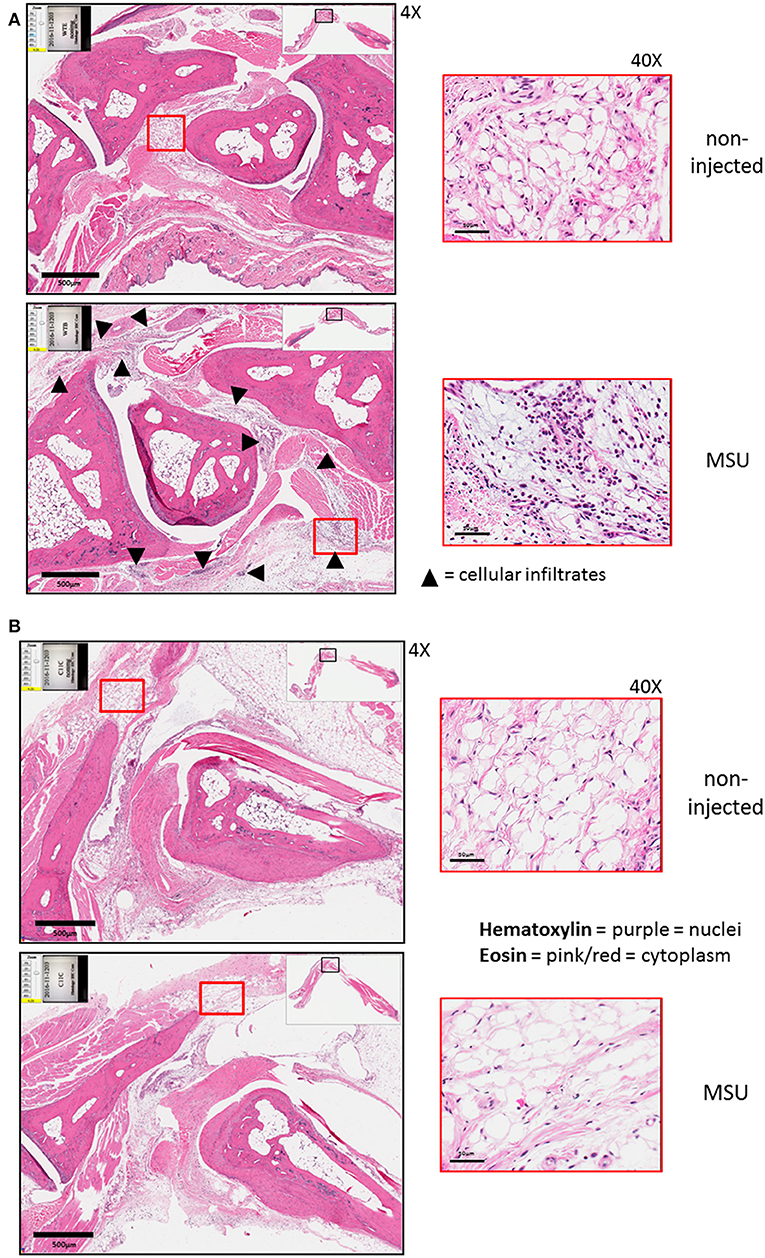
Figure 2. Histological tissue changes in response to MSU injection require the expression of caspase-11. (A) Aperio images of hematoxylin and eosin (H&E) staining of WT ankle joints 24 h post injection with PBS or MSU. Arrowheads on 4 × images indicate cellular infiltrates. 40 × magnifications depict markedly more hematoxylin staining. (B) Aperio images of H&E staining of caspase-11−/− ankle joints 24 h post injection with PBS or MSU. 4 × and 40 × magnifications show lack of cellular infiltrates (arrowheads), as PBS and MSU images look histologically similar. Images are representative from each group of five mice.
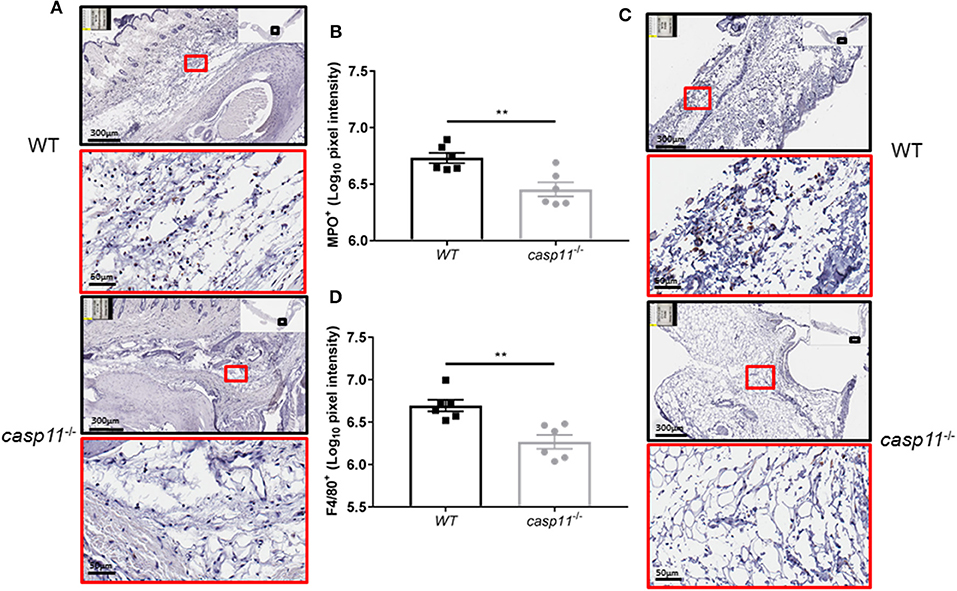
Figure 3. Caspase-11 promotes neutrophil and macrophage influx in response to MSU in vivo. (A) Representative Aperio images of myeloperoxidase (MPO) DAB staining of WT and caspase-11−/− ankle joints 24 h post injection with MSU. Black boxes are 8 × magnification and red boxes are 40 × magnification. Brown staining indicates infiltrating neutrophils (PMNs) within the joint space. (B) Quantification of WT and caspase-11−/− MPO DAB staining of 40 × magnification via Aperio analysis software. **p <0.01, n = 5 mice. (C) Representative Aperio images of F4/80 DAB staining of WT and caspase-11−/− ankle joints 24 h post injection with MSU. Black boxes are 8 × magnification and red boxes are 40 × magnification. Brown staining indicates infiltrating macrophages within the joint space. (D) Quantification of WT and caspase-11−/− MPO DAB staining of 40 × magnification via Aperio analysis software. **p <0.01, n = 5 mice. (B,D) Student's t test analysis was performed.
MSU elicits a strong immune response via a host of pro-inflammatory cytokines. IL-1β plays a central role in the pathogenesis of gout, in addition to the other pro-inflammatory cytokines that are produced within the tissue microenvironment during a gout attack (40, 41). These cytokines, such as TNFα, IL-6, and CXCL1 (KC), are important in activating resident cells, inducing endothelial permeability and cellular infiltration, and promoting overall tissue remodeling (42). To determine if caspase-11 contributes to the production of inflammatory cytokines, WT and caspase-11−/− mice were injected with MSU into the tibio-tarsal joint and synovial fluid and serum were collected after 24 h. Using Meso Scale Discovery's (MSD) electro-chemiluminescence (ECL) V-PLEX platform, we found that TNFα, IL-6, and KC were significantly reduced within the synovium of caspase-11−/− mice when compared to that of WT (Figures 4A–C). Analysis from the sera of the mice, however, indicated that TNFα, IL-6, and KC levels were comparable between the two strains (Figures 4A–C). Therefore, the inflammatory response observed in the WT mice demonstrated that caspase-11 is needed for their secretion within MSU-injected joints. Conversely, non-gout cytokines, IFN-γ and IL-12p70 were similarly secreted into the synovial fluid of WT and caspase-11−/− mice and equally low in the sera in both strains (Figures 4D,E). Since anti-inflammatory mechanisms are usually engaged whenever an inflammatory response occurs, we also tested levels of the classic anti-inflammatory mediator, IL-10. We found that IL-10 was reduced within the synovial fluid of caspase-11−/− mice compared to that of WT mice and was comparable between the sera of the two strains (Figure 4F). Altogether, these data designate caspase-11 as a contributor to the release of TNFα, IL-6, and KC but not IFN-γ and IL-12p70 in the synovial fluid.
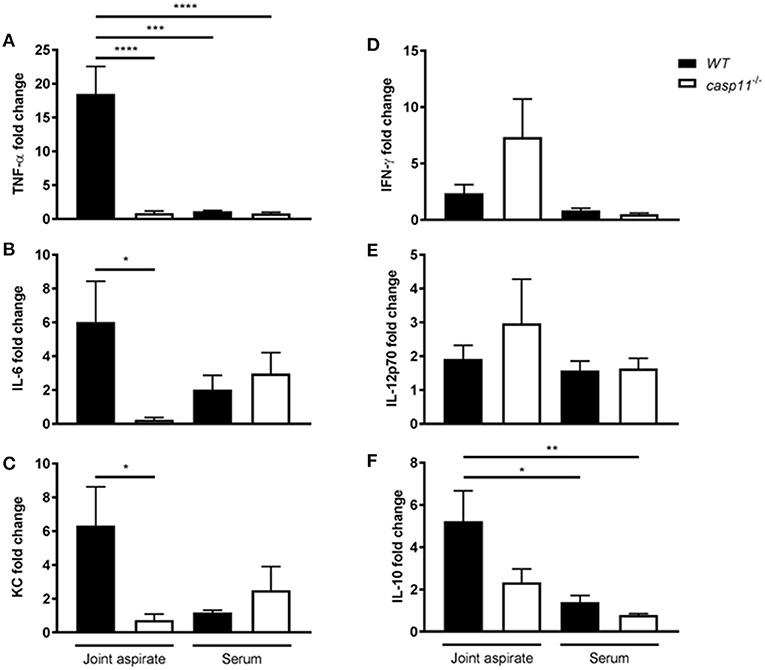
Figure 4. Gout-specific cytokines TNFα, IL-6, and KC are significantly upregulated in joints of WT but not caspase-11−/− mice during acute gout. In vivo MULTI-ARRAY electrochemiluminescence ELISA of WT and caspase-11−/− joint aspirate and serum cytokine fold change levels compared to PBS-treated controls. (A) TNFα, (B) IL-6, (C) KC, (D) IFNγ, (E) IL-12p70, and (F) IL-10. One-way ANOVA with Tukey's multi-comparison performed. *p <0.05, **p <0.01, ***p <0.001, ****p <0.0001, n = 5 mice.
Because IL-1β is a pivotal cytokine in the pathogenesis of gout and its presence designates the activation of the inflammasome, we determined the role of caspase-11 in the production of IL-1β within the synovium. We injected MSU into the tibio-tarsal joints of WT and caspase-11−/− mice and isolated joint tissue after 24 h. Real-time quantitative PCR analysis performed on RNA isolated from joint samples revealed that Il1b expression was upregulated in WT mouse joints, whereas caspase-11−/− mice expressed significantly less (Figure 5A). Furthermore, synovial fluid and serum were collected from the mice and analyzed for IL-1β secretion into the joint space and the circulation, respectively. In response to MSU injection, IL-1β secretion was significantly increased in the synovial fluid of the WT mice, but severely hindered in the caspase-11−/− mice (Figure 5B). IL-1β levels from the sera of both mice were not significantly increased. Moreover, we conducted immuno-histochemical assays on ankle joints 24 h after MSU injection. Slides prepared from WT and caspase-11−/− mice were stained for IL-1β to examine in vivo production of this key gout cytokine within the joint space. Images quantified via the Aperio ImageScope™ demonstrated that caspase-11−/− mice exhibited a significant defect in production of IL-1β protein in cells of the synovium compared to that of WT mice (Figures 5C,D). However, this difference is not due to differential cell death, since the treatment of macrophages with MSU with or without IL-1β produces minimal cytotoxicity (Supplementary Figure 4). The difference is also not due to difference in MSU phagocytosis (Supplementary Figures 5A,B). In addition, because the recognition and uptake of MSU contributes to the inflammatory response during an acute gouty attack, we determined if the absence of caspase-11 affected the phagocytosis of MSU crystals. Flow cytometric analysis from WT and caspase-11−/− macrophages treated with MSU revealed that levels of crystal uptake were comparable between the two strains of macrophages. These data indicate that the reduction in inflammatory cytokine production was not due to an innate deficit in phagocytic capability of the caspase-11−/− cells (Supplementary Figures 5A,B). Taken together, these data demonstrate that caspase-11 promotes the production and maturation of IL-1β during MSU injection in our gout model in a localized fashion and does not affect the phagocytosis of MSU.
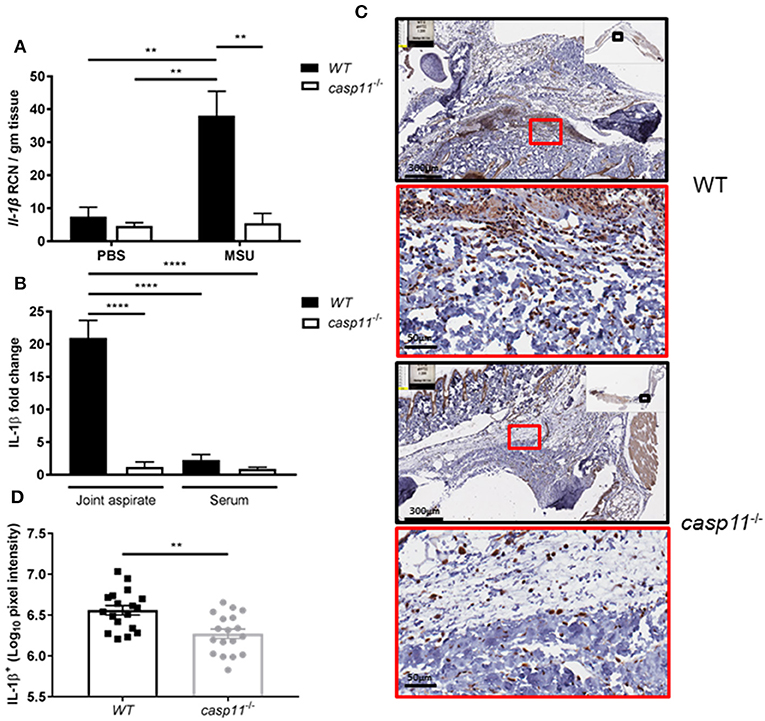
Figure 5. IL-1β is induced and secreted in a caspase-11-dependent manner during acute gout in vivo. (A) Relative copy number (RCN) per gram of tissue of Il-1β from WT and caspase-11−/− joint tissue treated with vehicle or MSU. Two-way ANOVA with a Holm's Sidak post-hoc test was used for statistical analysis. **p <0.01, n = 5 mice. (B) In vivo MULTI-ARRAY electrochemiluminescence ELISA of joint aspirate and serum fold change levels of IL-1β from WT and caspase-11−/− mice in response to MSU and compared to PBS-treated controls. Statistical analysis performed using a one-way ANOVA with Tukey's multiple comparisons. ****p <0.0001, n = 5 mice. (C) Representative Aperio images of IL-1β DAB staining of WT and caspase-11−/− ankle joints 24 h post injection with MSU. Black boxes are 8 × magnification and red boxes are 40 × magnification. Brown staining indicates IL-1β-positive cells within the joint space. (D) Quantification of WT and caspase-11−/− IL-1β DAB staining of 40 × magnification via Aperio analysis software. Student's t test was performed. **p <0.01, n = 5 mice.
Unlike caspase-1, caspase-11 is expressed at low levels in resting immune cells, and is induced following stimulation with various PAMPs or DAMPs (Supplementary Figures 3A,B) (43, 44). MSU treatment is accompanied by the activation of the inflammasome only in primed immune cells (11). MSU treatment alone does not induce the expression of caspase-11 (Supplementary Figures 3C,D) and is not contaminated with LPS (Supplementary Figure 6). Thus, macrophages require lipopolysaccharide (LPS) priming before MSU treatment in order to induce the expression of caspase-11 (Supplementary Figures 3C,D). Since LPS does not play a role in gout, we determined if other agents, such as cytokines, mediate the priming of immune cells in gout. To determine if the inflammatory environment of gout, specifically IL-1 cytokines, promotes caspase-11 expression, we treated macrophages with IL-1α, IL-1β, and HMGB1. Because IL-1 cytokines signal through the IL-1 receptor (R), Il-1r1−/− macrophages were used as a control. IL-1α and IL-1β significantly upregulated caspase-11 expression in an IL-1R-dependent manner, while HMGB1 and LPS did so independently of the IL-1 receptor (Figures 6A,B). Together, these data show that the IL-1 cytokines induce caspase-11 expression via IL-1R. Surface cell receptors transmit signals from extracellular stimuli intracellularly to the nucleus via the signaling adaptors myeloid differentiation factor 88 (MYD88) and TIR domain-containing adapter-inducing interferon-β (TRIF) (45, 46). The IL-1R employs the MYD88 adaptor to transduce signals (47). To determine the requirement of MYD88 for caspase-11 expression in the presence of IL-1 cytokines, WT, myd88−/−, trif−/−, and myd88/trif−/− macrophages were treated with IL-1α, IL-1β, IL-18, and HMGB1, and then caspase-11 expression was assessed. RT-qPCR and Western blot analysis revealed that caspase-11 mRNA and protein levels were significantly reduced in the myd88−/− and myd88/trif−/− cells when treated with IL-1α and IL-1β, or in combination, respectively (Figures 7A,B). HMGB1 utilizes a variety of receptors to promote its signaling events and LPS is sensed by TLR4, which uses both adaptors to signal (48–50). Subsequently, caspase-11 expression was not significantly hampered in the WT or the single knockout macrophages during stimulation with either HMGB1 or LPS, except when both MYD88 and TRIF were absent, indicating that either adapter was able to countervail for the deficient one in order to induce caspase-11 (Figures 7A,B). Together, these data indicate that IL-1α and IL-1β are able to upregulate caspase-11 via IL-1R dependent on the MYD88 adaptor. These data also suggest that IL-1α and IL-1β contribute to priming of immune cells under sterile inflammatory conditions.
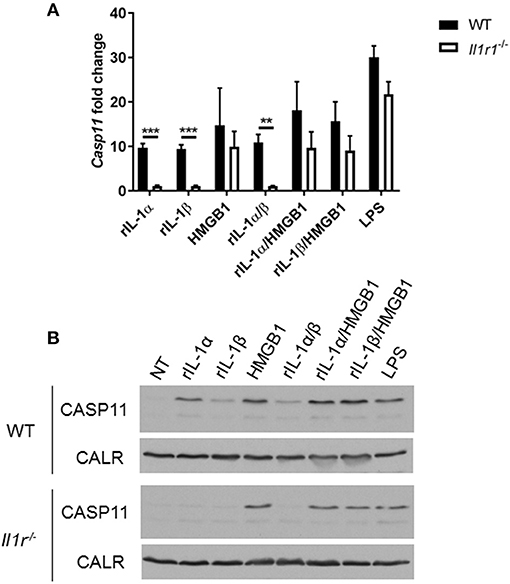
Figure 6. IL-1R-mediated signaling promotes caspase-11 induction in vitro. WT and Il-1r1−/− macrophages stimulated with IL-1α, IL-1β, HMGB1, and LPS and in combination for 4 h. Induction of caspase-11 assayed via (A) RT-qPCR relative copy number and (B) Western with housekeeping protein calreticulin (CALR) (representative). Statistical significance was determined by using multiple t test with a Holm's Sidak correction. **p <0.01, ***p <0.001, n = 3 independent experiments.
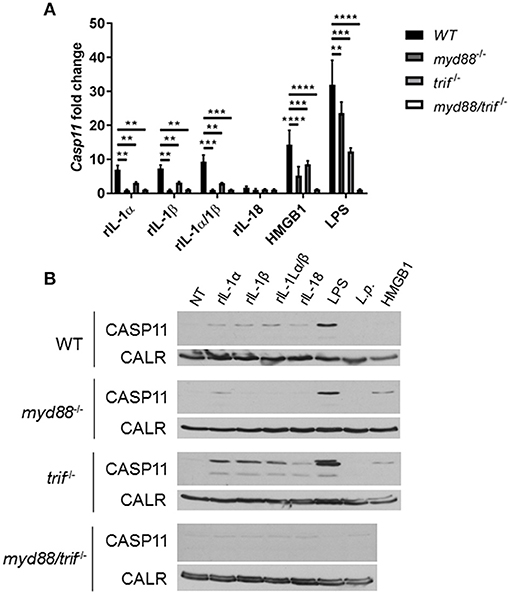
Figure 7. Caspase-11 induction via IL-1/IL-1R is Myd88-dependent in vitro. WT, myd88−/−, trif−/−, and myd88/trif−/− macrophages stimulated with IL-1α, IL-1β, IL-18, HMGB1, LPS, or in combination for 4 h. Induction of caspase-11 gene expression assayed via (A) RT-qPCR via relative copy number and (B) Western with loading control calreticulin (CALR) (representative). Two-way ANOVA using a Tukey's multiple comparison test was performed. **p <0.01, ***p <0.001, ****p <0.0001, n = 3 independent experiments.
A characteristic feature of crystal-induced gouty arthritis is the rapid accumulation of neutrophils (8, 16, 17). Chemokines that bind the IL-8 receptor CXCR-2 are essential for the development of acute neutrophilic inflammation in response to MSU in the subcutaneous air pouch model (51). Our histological data above demonstrate that significantly less inflammatory cells infiltrate the gout-afflicted joint in caspase-11−/− mice. It is possible that the lack of inflammatory cell migration in the absence of caspase-11 is due to low cytokine and chemokine production (Figure 4 and Supplementary Figure 7). It is also possible that the lack of caspase-11 expression leads to an intrinsic defect in cell migration even in the presence of chemo-attractants. To differentiate between these two possibilities, WT and caspase-11−/− mice were injected with KC, CXCL2 (MIP-2), and thioglycollate into the peritoneal cavity. Flow cytometry analysis of peritoneal lavages revealed that the ability of caspase-11−/− neutrophils to migrate in response to KC was significantly reduced after 4 h of KC stimulation (Figure 8). Although the recruitment of neutrophils in response to MIP-2 was reduced in the caspase-11−/− mice, as compared to WT mice, the difference was not statistically significant. In contrast, chemical stimulation with thioglycollate led to similar recruitment of inflammatory cells in the peritoneal cavity of caspase-11−/− and WT mice (Figures 8A,B). It was also revealed that Ly6Chi monocytes infiltrated the peritoneal cavity in significantly lower numbers in the caspase-11−/− mouse when injected with either MIP2 or KC (Supplementary Figure 9). These results indicate that caspase-11−/− neutrophils and monocytes are less capable to migrate in response to chemokines in vivo.
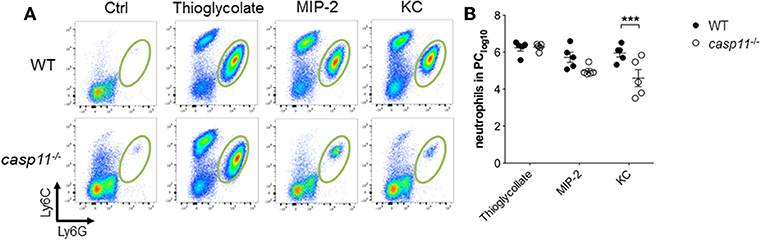
Figure 8. Caspase-11 promotes cellular migration in vivo. WT and caspase-11−/− mice were treated with PBS, thioglycolate, CXCL1 (KC), or CXCL2 (MIP-2). At 4 h post injection, cells infiltrating into the peritoneum were isolated and analyzed via flow cytometry. (A) Flow plots represent the distribution of neutrophils of each group (Ly6Ghigh/Ly6Cint neutrophils). (B) Graphs show total numbers of neutrophils in peritoneal cavity. A Student's t test was performed for statistical analysis. ***p <0.001, n = 5 mice.
Successful chemotaxis requires not only increased motility but also sustained directionality (52, 53). In order to determine whether caspase-11 controls motility and/or directionality in response to KC, we performed time-lapse and trajectory analyses of chemotactic neutrophils in vitro. Bacterial peptide N-formyl-methionylleucyl- phenylalanine (fMLP) was used as a control. Neutrophils were seeded into a chemotaxis iBidi chamber that allows free cell migration in a 360° field and maintains a stable chemotactic gradient (34, 54). The migration tracks of neutrophils moving toward fMLP and KC (Figures 9A–I) were recorded, and directionality and mean velocity were analyzed (Figures 9G–I). The accumulated distance and velocity of caspase-11−/− and WT neutrophils were similar in response to KC. However, Euclidean distance (shortest distance between start and end of migration) traveled in response to KC is significantly reduced in caspase-11−/− neutrophils when compared to WT cells (Figure 9H). The lack of linear displacement and lack of directionality of caspase-11−/− neutrophils, despite normal velocity in response to KC, are clear indications of a loss of efficiency in guided motility. However, all parameters including accumulated distance, Euclidean distance, and velocity are significantly reduced in caspase-11−/− neutrophils in response to fMLP (Figures 9G–I). Together, these results demonstrate that caspase-11 is required for neutrophils to reach the site of inflammation.
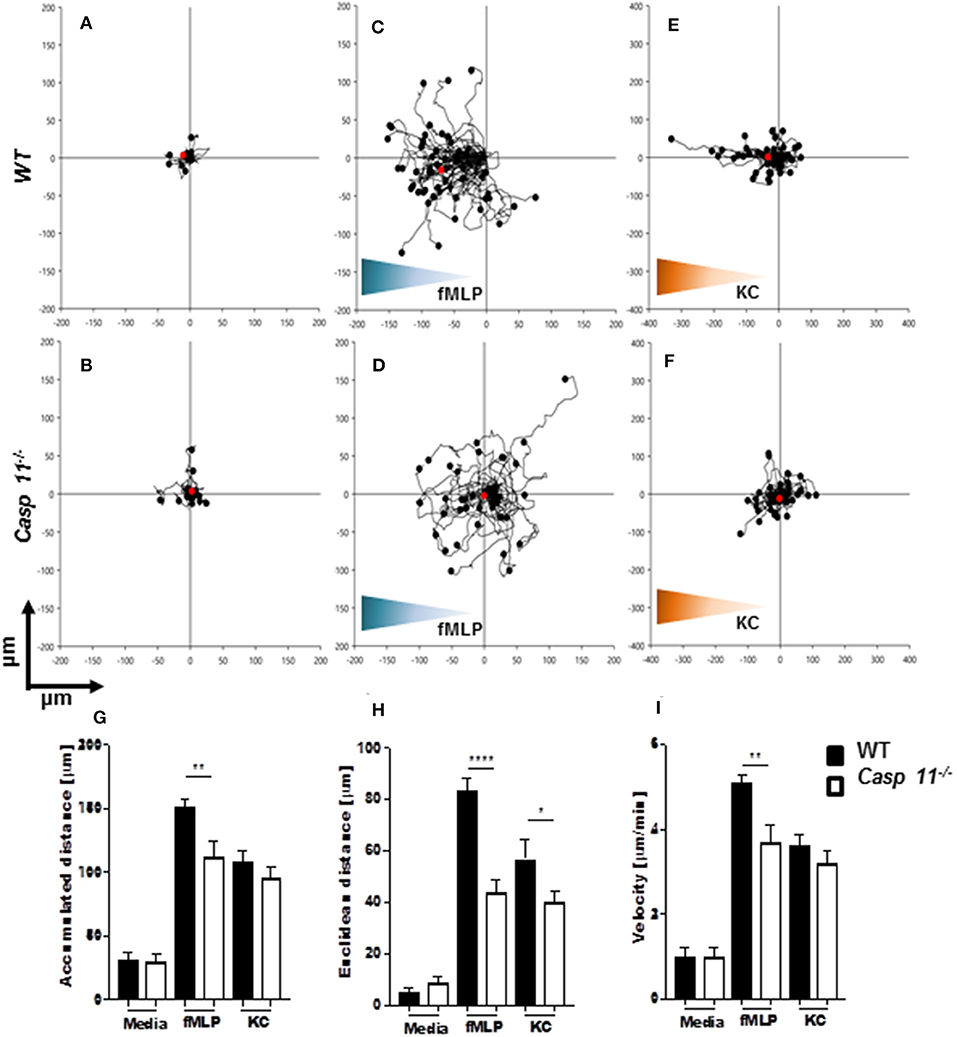
Figure 9. Caspase-11−/− neutrophils display an intrinsic defect in chemotaxis in vitro. WT and caspase-11−/− neutrophils were seeded in μ-slide chemotaxis chamber for 20 min, followed by the addition of the respective chemoattractant. Cell motility was recorded by video microscopy for 30 min. The start point of each track was normalized to x = 0 and y = 0, and positive x-axis values represent movement in the direction of the source of chemoattractant. The polar plots show the patterns of migration of WT (upper panel) or caspase-11−/− (lower panel) neutrophils in the presence of (A,B) media, (C,D) 5 μM fMLP gradient (blue gradient), and (E,F) 500 ng/ml mouse recombinant KC (green gradient). The red dots indicate the average motility center of mass, (A and B, n = 40 cells; C and F, n = 60). The motility parameters of accumulated distance (G), Euclidean distance (H), or velocity (I) for WT and caspase-11−/− neutrophils were calculated using the tracking tool PRO v2.1 software. Images were analyzed with the iBidi Chemotaxis and Migration Tool 2.0 software. The trajectories of 40 migrating cells were plotted (per treatment) and represented as rose diagrams to indicate directional integrity. The accumulated distance (μm), Euclidean distance (μm), and cellular velocity (μm/min) were calculated. Student's t test was performed. *p <0.05, **p <0.01, ****p <0.0001, n = 3 mice.
NETs are web-like structures formed by the mixture of chromatin with the contents of neutrophil granules including myeloperoxidase, elastase, and cathelicidins (55, 56). To determine if caspase-11 is required for NET formation by neutrophils in response to MSU, WT and caspase-11−/− neutrophils were treated with MSU and NET structures were determined by fluorescent co-staining of extracellular DNA and neutrophil elastase (NE). Caspase-11−/− neutrophils produced drastically less extracellular DNA and NET formation upon stimulation with MSU alone or with ATP (Figures 10A–C). Both WT and caspase-11−/− neutrophils did not produce significant NETs when treated with ATP or IL-1β alone (Figures 10A,C). Although phorbol 12-myristate 13-acetate (PMA) is a strong inducer of NET formation, we found that NET formation by caspase-11−/− neutrophils was reduced (Figures 10B,C). Therefore, the lack of caspase-11 reduces the production of NETs in response to different stimuli. To understand the mechanism linking caspase-11 with NET formation, we examined the phosphorylation of RIPK3 since the MLKL/RIPK3 pathway was shown to promote NET formation and necroptosis (57, 58). RIPK3 phosphorylation was not significantly reduced between WT and caspase-11−/− neutrophils in response to MSU (Figures 10D,E). Therefore, caspase-11 but not MLKL/RIPK3 is needed for NETosis in response to MSU.
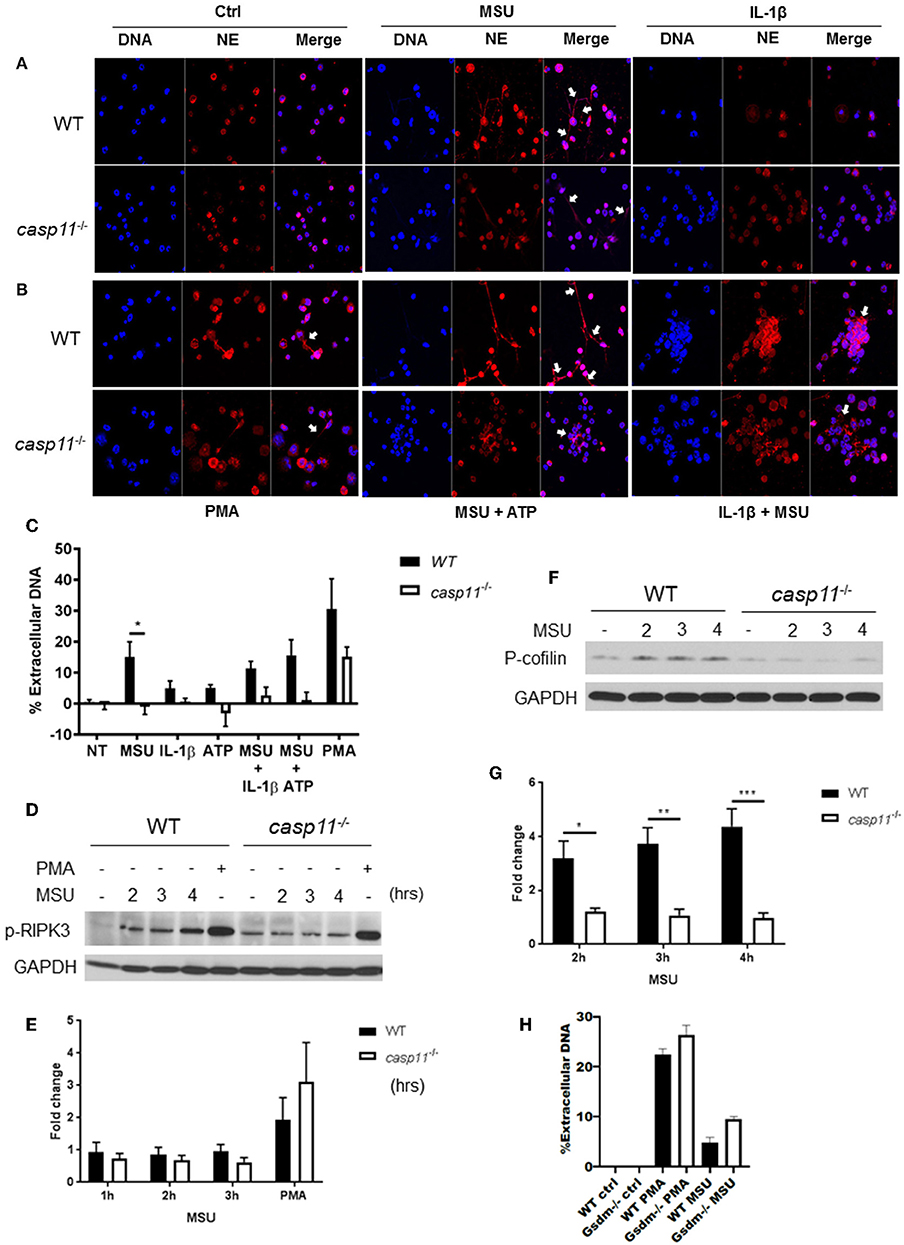
Figure 10. Caspase-11−/− neutrophils produce less NETs independently of Rip3 phosphorylation and associated with altered cofilin phosphorylation in response to MSU in vitro. (A,B) Immunofluorescence assay of WT and caspase-11−/− neutrophils treated with MSU, MSU + ATP, IL-1β, IL-1β + MSU, and PMA for 3 h. Cells were stained with neutrophil elastase (NE) and DAPI (DNA) to visualize NET formation, n = 3. (C) Quantification of NET formation in vitro. Response measured from WT and caspase-11−/− neutrophils stimulated with MSU, ATP, IL-1β, combinations of treatments, and PMA. Cells were stained with Sytox green, n = 2 independent experiments (3 mice per group, total 6 mice), *p <0.05. (D) Representative Western blot for phospho-RIPK3 (p-RIPK3) during MSU treatment for 2, 3, and 4 h, n = 3 experiments. (E) Densitometry analysis of phosphorylation of Ripk3, n = 5. (F) Representative Western blot for phospho-cofilin (p-cofilin) and (G) densitometry analysis, n = 5. (H) Quantification of NET formation in vitro in response to MSU and PMA in WT and gsdm-11−/− neutrophils. (C,E,G,H) Student's t test was performed for statistical analysis. n = 3 mice. *p <0.05, **p <0.01, ***p <0.001.
To form NETs, neutrophils release a DNA scaffold through a process that requires rearrangements in the microtubule network and F-actin. Inflammatory mediators, such as IL-8, fMLP, LPS, and TNF, can trigger NET formation (59–61). Because the actin cytoskeleton plays a major role in neutrophil migration (54), we investigated the activation status of cofilin in WT and caspase-11−/− neutrophils in response to MSU. Notably, WT neutrophils elicit low levels of phospho-cofilin expression when unstimulated (Figures 10F,G). Then, upon treatment with MSU, the phosphorylation increased significantly (Figure 10G). On the other hand, caspase-11−/− neutrophils did not elicit a change in cofilin phosphorylation in response to MSU treatment when compared to WT. Of note, there was no difference in NET formation between WT and GSDM−/− neutrophils in response to MSU (Figure 10H). Therefore, the lack of caspase-11 expression alters neutrophil's ability to phosphorylate cofilin, which is associated with reduced neutrophil migration to specific chemokine.
Innate immune cell recruitment, inflammatory cytokine release, tissue damage, and remodeling in gout have been linked to the activation of the Nlrp3 inflammasome in response to MSU (11). Plasma concentrations of IL-1β, IL-18, IL-6, and IL-8 are elevated in the gout patients (9, 13), but little is known about the role of these cytokines in the progression of gout. Additionally, many reports have focused on the role of caspase-1 in gout; however, little is known to date about the role of caspase-11 in this sterile, inflammatory disease condition. The role of caspase-11 is versatile according to the insult. It has been described to be upstream, independent, or parallel to caspase-1 activation (26, 26, 30, 62–66). When macrophages are treated with intracellular LPS, caspase-11 senses this complex molecule leading to downstream activation of caspase-1 and IL-1β (32, 67). However, when macrophages face an organism such as L. pneumophila, caspase-1 activation is independent of caspase-11 (25). Several reports demonstrated that caspase-11 mediates the release but not the activation of IL-1β (66). Furthermore, we demonstrated that caspase-11 modulates vesicular trafficking through altering the activation of cofilin via the Rho GTPase (24, 25). We demonstrated that after phagocytosis of L. pneumophila, caspase-11 modulates the phosphorylation of cofilin through upstream effector molecules. This balance in its activation promotes actin polymerization and, consequently, efficient phagolysosomal fusion and destruction of the intracellular pathogen (24, 25). Without affecting the initial uptake, the absence of caspase-11 allowed greater intracellular growth because of the defect in actin dynamics (24, 25). Because of this, we believe that caspase-11 regulates actin polymerization through cofilin, affecting subcellular organization of vesicles and organelles without affecting phagocytic events. In addition, we found that caspase-11 promotes the formation and maturation of autophagosomes (26). Recently, it was shown that caspase-11 responds to lipotechoic acid and Gram-positive bacteria, leading to activation of the inflammasome (68). In the case of methicillin-resistant Staphylococcus aureus (MRSA), caspase-11 is actually exploited to actively prevent the recruitment of mitochondria to the vicinity of the bacteria-containing vacuoles to avoid intracellular killing (28).
It is still not clear what specifically induces the expression of caspase-11 in immune cells during sterile inflammation. Here, we found that IL-1β and IL-1α induce caspase-11 expression via the IL-1R/MYD88 pathway. It is possible that IL-1β and IL-1α are released from dead cells with the gout-inflicted joint leading to the induction of caspase-11 in an IL-1R-dependent manner, priming immune cells for inflammasome activation by subsequent insults including MSU. Therefore, the expression of caspase-11 is induced not only by microbes but also by specific cytokines. It would be interesting to examine if other conditions where caspase-11 was induced were not due to IL-1β-mediated induction rather than directly by the PAMP or DAMP molecules.
Several prominent studies have demonstrated that caspase-11-mediated functions, including the release of IL-1β, are attributed to pyroptosis (66, 69, 70). Notably, the release of IL-1β from macrophages in response to MSU was not accompanied by cell death, as shown in our study. Our findings are corroborated by recent reports describing how Gsdm forms pores within the intact plasma membrane, allowing the release of IL-1β independently of cell death (4, 71). Additionally, we found that caspase-11 promotes the secretion of inflammasome-independent cytokines/chemokines from macrophages such as TNFα, IL-6, and KC without provoking cell death as demonstrated by the scarce amounts of LDH. Therefore, caspase-11 plays a major role in gout pathogenesis through the regulation of cytokine production independently of cell death.
Gout has been classified as an IL-1β prototypic condition since neutralizing antibodies to IL-1β or the caspase-1 inhibitor z-YVAD significantly reduce inflammation and the production of other cytokines within the joints (9, 11, 13, 18). Nevertheless, these approaches were not effective in most gout patients and could not prevent joint damage since they target extracellular IL-1β after tissue damage has occurred. Alternatively, preventing the expression of caspase-11 in gout-prone individuals may prevent the instigation of tissue damage.
Neutrophils dramatically contribute to joint injury once they migrate to the MSU deposition site (8, 16, 17). Neutrophil remnants may amplify the inflammatory response beyond their short lifespan in the tissues, contributing to chronic inflammation. Neutrophil influx is impaired in an in vivo model of crystal-induced peritonitis in Nlrp3−/−, Asc−/−, Casp-1/Caspase-11−/−, and Il-1r−/− mice (11). Asc−/− and Casp-1/Caspase-11−/− neutrophils migrated similar to WT neutrophils in response to zymosan, but not MSU (11). The main mechanism of reduced migration has been attributed to low production of cytokines. However, the role of caspase-11 in neutrophil migration and function in gout has not been previously studied. Here, we show that neutrophil migration to MSU-injected joints is significantly reduced in the absence of caspase-11 expression. Indeed, the absence of caspase-11 is also accompanied by reduced KC production, which plays an important role in neutrophil chemotaxis. Therefore, it is possible that the reduced migration of caspase-11−/− neutrophils is due to reduced KC production. It is also probable that the lack of caspase-11 expression in neutrophils exerts an inherent defect in the migration machinery, which was not previously tested. We demonstrate here that neutrophils lacking caspase-11 expression migrate similar to their WT counterparts in response to intraperitoneal injection of PMA or MIP2 but not KC. Nevertheless, iBidi chamber analysis showed that the accumulated distance, velocity, and directionality in response to fMLP require the expression of caspase-11. Yet, caspase-11 was essential for the directionality of neutrophils toward KC but not velocity or total distance traveled. Together these data describe a novel role for caspase-11 in neutrophil migration that is not universal for all chemoattractants, which warrant further studies.
Notably, macrophage-derived IL-1β enhances MSU crystal-induced NET formation in neutrophils (72). Neutrophils were thought to release NETs during a distinct form of cell death, named NETosis (20, 59). Current evidence, however, suggests that neutrophils may remain viable and functional even after NET extrusion and that NET formation is independent of neutrophil death (22, 73). Discovering here that caspase-11 is required for NET formation in response to MSU and to a lesser extent to PMA, IL-1β, and ATP points to the distinct and specific roles of caspase-11 in different cell types and disease conditions. It has been reported that necroptosis and NETosis require the functions of RIPK3 and MLKL protein. However, we found that this pathway is not engaged in response to MSU. Our findings support a recent report showing that NET formation occurs independently of the MLKL/RIPK3 pathway in response to PMA, LPS, and Gram-negative bacteria (74). Autophagy has also been implicated in NETs (8, 55). Of note, we have demonstrated that caspase-11 promotes autophagy in response to bacterial infection (26). Yet, reports have demonstrated that NET formation can still occur in autophagy-deficient and RIPK3 knockout neutrophils. It is clear that MSU and maybe other crystals induce NETs through a molecular pathway that is distinct from that of PMA. Interestingly, the lack of NETs in caspase-11−/− neutrophils in response to MSU is also accompanied by lack of cofilin phosphorylation, supporting a role for caspase-11-mediated actin polymerization in NET formation. It has also been shown that NET formation plays a role in the resolution of inflammation. Aggregated NETs sequester and degrade inflammatory mediators (75). The authors point to the importance of ROS production as a main factor for NET aggregation that in turn resolve neutrophil-mediated cytokines and chemokines. In our model, caspase-11−/− produced less NETs when treated with PMA, a strong ROS inducer. Thus, we postulate that NET formation is not solely reliant on ROS and that caspase-11-mediated regulation of the actin cytoskeleton via cofilin plays a major role. In addition, the reduction of NETs from caspase-11 PMNs would suggest an increase of inflammatory mediators. Conversely, significantly lower levels of gout cytokines were present within the articular joints of caspase-11−/− mice, indicating that NETs could be involved in the reduction of inflammatory cytokines, but are not needed for their production. Accordingly, it is likely that caspase-11 is needed for the inflammatory cytokine production and chemotaxis in response to MSU. Additionally, it is possible that the caspase-11 downstream effector GSDM is required for NET formation in response to MSU. A recent report demonstrated that this is the case during infection with the Gram-negative bacteria Salmonella (76). Together, these findings demonstrate a distinct physiological NET pathway in response to MSU crystals that is independent of MLKL/RIPK3 and dependent on caspase-11.
Our findings can apply to other sterile inflammatory conditions where neutrophil migration and NET formation is significantly involved in the pathogenesis of the disease such as thrombosis, systemic lupus erythematosus (SLE) ANCA vasculitis, and other crystallopathies (73). Targeting caspase-11 is a viable possibility during or before acute gout attacks, but further studies are needed to test this option in chronic gout. Targeting caspase-11 can also be used to avert early steps of gout pathobiology before the onset of symptoms and before tissue damage occurs in prone individuals. Cumulatively, these results offer previously unrecognized functions for caspase-11 that adds to its reported diverse roles. Concurrently, we provide mechanistic evidence that caspase-11 is a viable therapeutic target in gout and other disease conditions that are inflicted by activated neutrophils.
Wild-type (WT) C57BL/6 mice were purchased from The Jackson Laboratory (Bar Harbor, ME). Caspase-11−/− mice were generously donated by Dr. Yuan at Harvard Medical School (43). Caspase-1−/− (Caspase-1−/−/Caspase-11Tg) mice were a gift from Dr. Vishva Dixit at Genentech (77). All mice were housed in a pathogen-free facility and experiments were performed with approval and in accordance with regulations and guidelines from the Institutional Animal Care and Use Committee (IACUC 2010A00000066-R3) at The Ohio State University (Columbus, OH).
MSU crystals were purchased from Invivogen and verified to be endotoxin free by LAL assay from Pierce (Supplementary Figure 6). Using Hamilton syringes, 0.5 mg of MSU crystals (tlrl-msu) was injected into the tibio-tarsal joints of mice under light anesthesia according to a previously characterized model of MSU crystal-induced ankle arthritis (78). After 24 h, mice were anesthetized and ankle joints were photographed and measured via caliper. Ankle joint aspirates were collected by flushing with 20 μl of PBS and blood was taken for serum isolation by axillary vessel incision of the right subclavian vein at the time of sacrifice. Cytokine analysis on collected serum was performed using electrochemiluminescence detection (V-PLEX Proinflammatory Panel 1 mouse kit; Meso Scale Diagnostics, Rockville, MD) per manufacturer's protocol. Tibio-tarsal joints were isolated and cut in half. One half was for histological analysis and the other half was stored in TRIzol™ reagent (Life Technologies, 15596018) for RNA/protein analysis.
Hind limbs of mice were dissected at experimental endpoint and fixed in 10% neutral buffered formalin for 24 h. Tissue was then decalcified in TBD-2 (ThermoFisher, 6764003) according to the manufacturer's protocol and transferred to 70% ethanol. Following paraffin processing, blocks were cut at 4 microns, placed on positively charged slides, and fixed in cold acetone. Serial paraffin sections were used for immunohistochemistry and H&E staining as previously described (24, 79–81). Briefly, all slides were stained in Richard Allan Scientific Hematoxylin (Thermo Scientific, 7211L) and Eosin-Y (Thermo Scientific, 71211) with the Leica Autostainer ST5020 (Leica Biosystems, Buffalo Grove, IL). Immunohistochemistry/DAB staining was performed with MPO, F4/80, and IL-1β (1:200, Abcam, ab9787) with specific secondary (Abcam, ab6721). The Aperio ScanScope XT eSlide capture device (Aperio, Vista, CA) was used to digitally scan slides at 40 × magnification and Aperio Digital Image Analysis software (v9.1) was used to analyze and measure pixel intensities according to previously described methods (26, 82).
Bone marrow-derived macrophages (BMDMs) were cultured as previously described (25, 80, 81, 83–85). Caspase-11 induction was measured by treating WT macrophages with various inflammatory mediators for 4 h. 20 ng/ml IL-1α, IL-1β, IL-18, and KC (R&D Systems, 400-ML-005, 401-ML-005, 9139-IL-050, and 453-KC-050), HMGB1 (eBioscience, 34-8401-82), 2.5 μg/ml Pam3CSK4 (tlrl-pms), 5 μg/ml poly(I:C) (tlrl-picw), 100 ng/ml LPS (tlrl-eklps), 500 ng/ml flagellin (tlrl-stfla), 100 ng/ml FSL-1 (tlrl-fsl), 5 μg/ml ssRNA (tlrl-lrna40), 5 μg/ml Imiquimod (tlrl-imqs), and 100 ng/ml bacterial DNA (tlrl-ssec) (InvivoGen). Interferon induction of caspase-11 was achieved using 200 ng/ml IFNα and β (BioLegend, 752806, 581306), 50 ng/ml IFNγ (R&D Systems, 485-MI), and 100 ng/ml TNFα (R&D Systems, 410-MT-050) and LPS for 4 h, respectively. In vitro stimulation with MSU was accomplished with 100 μg/ml (Invivogen, tlrl-msu). ATP (Sigma-Aldrich) was used for 30 min at the final concentration of 5 mM. All cells were lysed and stored in TRIzol™.
In vivo samples were sent to the Analytical & Development Lab in the Ohio State University Clinical Research Center Core. Meso Scale Discovery V-PLEX mouse Pro-Inflammatory Panel 1 (K15048G) was used to analyze joint aspirate and serum samples. Fold change of cytokines from synovial fluid or that of serum was achieved by dividing cytokine levels obtained from MSU-injected ankles by those injected with PBS (24, 37).
In vitro supernatants were assessed for pyroptosis by measuring LDH release following the manufacturer's instructions (Promega CytoTox-ONE™ Homogenous Membrane Integrity Assay #G7891) (24, 25).
Total RNA was isolated from cells lysed in TRIzol™ via phenol-chloroform extraction. Caspase-11 and Il-1b mRNA expression was assessed using iQ SYBR Green Supermix (Bio Rad, 64105458). Briefly, Ct values of each target gene were subtracted from the average Ct of the housekeeping genes, Gapdh and Cap-1, and the resulting ΔCt was used to calculate gene expression in relative copy numbers (RCN) as we described earlier (37, 83, 86, 87).
Protein extraction from macrophages was performed using TRIzol™ reagent according to the manufacturer's instructions. Briefly, after phase separation using chloroform, 100% ethanol was added to the interphase/phenol-chloroform layer to precipitate genomic DNA. Subsequently, the phenol-ethanol supernatant was mixed with isopropanol in order to precipitate out proteins. Isolated protein was then denatured in a urea-based lysis buffer. The Bradford method was used to determine protein concentrations. Equal amounts of protein were separated by 12% SDS-PAGE (Biorad, 161-0158) and transferred to a polyvinylidene fluoride (PVDF) membrane (Biorad, 162-0177). Membranes were incubated overnight with antibodies against caspase-11 (Sigma, C1354-.2ML), calreticulin (Enzo Life Sciences, ADI-SPA-601-D), phospho-cofilin (Cell Signaling, #3313S), and RIP3. Corresponding secondary antibodies conjugated with horseradish peroxidase and in combination with enhanced chemiluminescence reagent were used to visualize protein bands. Densitometry analyses were performed by normalizing target protein bands to the loading control (calreticulin) using ImageJ software as previously described (24, 25, 87).
Bone marrow was isolated from the femur and tibia of 6- to 8-week-old WT and caspase-11−/− mice (88). Neutrophils were negatively selected by using the EasySep™ mouse neutrophil enrichment kit (STEM cell technologies, #19762A), and 105 neutrophils were plated in each well of a 96-well black plate (Corning, #3603). PMNs were stimulated for 3 h with 100 μg/ml MSU (Invivogen, tlrl-msu), 100 μM ATP (Roche, 10 519 987 001), 25 ng/ml IL-1β (R&D systems, 401-ML-005/CF), and 100 nM PMA (Sigma-Aldrich, #P8139-10MG). To remove background fluorescence, designated wells were treated with DNAse I (Sigma, #AMPD1-KT). One percent of Triton X-100 (Fisher Scientific, #BP151-100) was added to some wells to adjust the DNA staining to 100%. Before end of assay, wells were treated with 4 μM Sytox green (Life Technologies, #S7020) to stain extracellular DNA. Fluorescence was assayed on a 96-well plate reader (Spectra Max i3X, Molecular Devices). The quantification of extracellular DNA in neutrophils has previously been described (19, 88).
PMNs were negatively selected by using the EasySep™ mouse neutrophil enrichment kit (STEM cell technologies, #19762A); 2 ×105 cells were plated in a 24-well plate on poly-L-lysine coverslips and allowed to equilibrate/adhere for 10 min. PMNs were then stimulated for 3 h with 200 nM PMA (Sigma-Aldrich, #P8139-10MG), 100 μg/ml MSU (Invivogen, tlrl-msu) alone or in combination with 100 μM ATP (Roche, 10 519 987 001), or 25 ng/ml IL-1β (R&D Systems, 401-ML-005/CF) to induce NET formation. Cells were then fixed with 10% neutral buffered formalin (NBF, Sigma, #HT501128-4L) overnight. The next day, the coverslips were blocked and permeabilized with 5% goat serum and 0.1% Triton X-100 for 1 h. Anti-neutrophil elastase antibody (Abcam, #ab21595) was used to visualize NETs (1:200 dilution), and DNA was stained with 1 μg/ml 4′-6′diamino-2-phenylindole (DAPI, Molecular Probes, #D1306). Goat anti-rabbit Alexa 594 (ThermoFisher, #R37117) was used as the secondary. Images were captured using a laser scanning confocal fluorescence microscope (Olympus Fluoview FV10i) using a 60 × objective as we previously describe (24–26).
The peritoneal cavity model of PMN migration has previously been described (89). Briefly, CXCL1/KC (R&D, 453-KC-050), CXCL2/MIP-2 (R&D, 452-M2-050), and 4% of thioglycolate (Fluka Analytical, 70157) were injected intraperitoneally into 8- to 10-week-old WT and caspase-11−/− mice. Four hours after treatment, mice were euthanized and their peritoneal cavities were lavaged with 10 ml PBS + 1 mM EGTA (PBS—Life Technologies, #14190-144; EGTA—Boston Bioproducts, #BM-151). Next, red blood cells were lysed (MACS, #130-094-183), and cells were washed and stained for various cellular, surface markers including anti-CD45 (BioLegend, #103138), anti-CD11b (BioLegend, #101243), anti-Ly6G (BioLegend, #127639), anti-Ly6C (BioLegend, #128031), anti-F4/80 (BioLegend, #123147), and LIVE/Dead™ Fixable Blue dead cell stain (ThermoFisher, #L34962). Relative cell counts were assessed via flow cytometry using countBright absolute counting beads (ThermoFisher, #C36950). Gating strategy for neutrophils: single cells/live cells/CD45+/CD11bhigh/Ly6Ghigh Ly6Cint.
Neutrophils were isolated from WT and caspase-11−/− mice and resuspended in HBSS plus 2% FBS media. Cells were seeded into the narrow channel of μ-slide chemotaxis chamber 2D (Ibidi) for 20 min at 37°C, and then non-adherent cells were washed twice with media. The narrow channel (observation area) connects two 40-μl reservoirs. To form the chemotaxis gradient, the reservoir at the right of the chamber was filled with media and the left reservoir was filled with 5 μM fMLP (Sigma-Aldrich) or 500 ng/ml KC (R&D). The cell migration was video recorded using bright-field microscopy (Nikon Eclipse Ti). Images were captured every 30 s for 30 min, maintaining the chamber at 37°C. The chemotaxis and migration tracks analyses were performed using the Tracking Tool Pro v 2.1 software (Gradientech). Forty to 60 randomly selected cells were manually tracked per condition in each chemotaxis experiment.
BMDMs from WT and caspase-11−/− mice were seeded 1 ×106 cells in a 6-well plate. Macrophages were allowed to rest overnight and then treated for 16 h with 100 μg/ml MSU to allow for uptake of crystals. Cells were isolated and then stained with F4/80 (Invitrogen # 12-4801-82, clone BM8) to confirm macrophage marker expression and also with LIVE/DEAD Fixable Near-IR Dead Cell stain (Invitrogen #L10119) to differentiate live and dead cells. For phagocytosis, cells were assayed on BD FACS Canto II for increase in side scatter (i.e., internal complexity/granularity) and analyzed using FlowJo™ v10.
MSU was purchased from InvivoGen and tested for the absence of endotoxin contamination by LAL Chromogenic Endotoxin Quantitation kit (Pierce) according to the manufacturer's suggestions. Briefly, standard curve was created using E. coli endotoxin at 1, 0.5, 0.25, and 0.1 EU/ml. Endotoxin-free water was used as a negative control and MSU was used as an unknown sample. Samples were measured on a microplate absorbance reader at 405 nm.
Data in figures are presented as mean± SE (standard error). At least three independent experiments were used as listed in the figure legends. Two sample t tests (for two group comparisons such as experiments in Figures 1B, 3B,D, 5D, 8B, 9, 10) or ANOVA (for multiple group comparisons such as experiments in Figures 1A, 4, 5A,B, 7) were used for most of the experiments as detailed in the figure legends. Tukey's or Holm's Sidak methods were used to adjust for multiple comparisons to control Type I error at α = 0.05. p-values ≤ 0.05 after adjustment for multiple comparisons were considered as significant. Data were analyzed using GraphPad Prism 7 software.
All datasets generated for this study are included in the article/Supplementary Material.
The animal study was reviewed and approved by OSU IACUC.
The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Center for Advancing Translational Sciences or the National Institutes of Health.
KC designed, performed, analyzed, and interpreted data and wrote, edited, and reviewed the manuscript. NY assisted in performing in vivo experiments and contributed to writing and reviewing the manuscript. FR-A, KK, AA, KH, AB, AV, KD, HG, MA, ME, DD, SA, SE, SP-S, and MG contributed by performing experiments and editing the manuscript. WJ helped conceptualize experiments and interpreted data. XZ performed statistical analysis. AOA obtained funding and resources, assisted in the experimental design and implementation, interpretation of data, as well as the writing and editing of the manuscript.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We thank Dr. Sue E. Knoblaugh at the College of Veterinary Medicine/Department of Veterinary Biosciences at the Ohio State University, Columbus OH, USA, for the histological evaluation of tibio-tarsal joint tissue. We thank J. Yuan at Harvard Medical School for providing the breeding pairs for the caspase-11-deficient (caspase-11−/−) mice used in these studies. Studies in the Amer laboratory are supported by R21 AI113477, R01 AI124121, and R01 HL094586. Cytokine analysis using the V-PLEX Proinflammatory Panel 1 (mouse) Kit was supported by Award Number Grant UL1TR001070 from the National Center for Advancing Translational Sciences. KC was supported by a Cystic Fibrosis Foundation Post-doctoral Research Fellowship. KK has been supported by Deutsche Forschungsgemeinschaft (DFG—German Research Foundation) and Cystic Fibrosis Foundation (CFF). KH is supported by a Cure Cystic Fibrosis Columbus Training grant. AB and SE were supported by the Egyptian Cultural and Educational bureau. AA is supported by Taawon Welfare Association, West Bank, Palestine and Bank of Palestine. MG was supported in part by R01 HL076278. FR-A and SP-S were supported by the Cystic Fibrosis Foundation grant PARTID18P0.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2019.02519/full#supplementary-material
1. Rymal E, Rizzolo D. Gout: a comprehensive review. JAAPA. (2014) 27:26–31. doi: 10.1097/01.JAA.0000453233.24754.ec
2. McGill NW, Dieppe PA. Evidence for a promoter of urate crystal formation in gouty synovial fluid. Ann Rheum Dis. (1991) 50:558–61. doi: 10.1136/ard.50.8.558
3. Perl-Treves D, Addadi L. A structural approach to pathological crystallizations. Gout: the possible role of albumin in sodium urate crystallization. Proc R Soc Lond B Biol Sci. (1988) 235:145–59. doi: 10.1098/rspb.1988.0069
4. Evavold CL, Ruan J, Tan Y, Xia S, Wu H, Kagan JC. The pore-forming protein gasdermin D regulates interleukin-1 secretion from living macrophages. Immunity. (2018) 48, 35–44.e6. doi: 10.1016/j.immuni.2017.11.013
5. Shi J, Gao W, Shao F. Pyroptosis: gasdermin-mediated programmed necrotic cell death. Trends Biochem Sci. (2017) 42:245–54. doi: 10.1016/j.tibs.2016.10.004
6. Aglietti RA, Estevez A, Gupta A, Ramirez MG, Liu PS, Kayagaki N, et al. GsdmD p30 elicited by caspase-11 during pyroptosis forms pores in membranes. Proc Natl Acad Sci U.S.A. (2016) 113:7858–63. doi: 10.1073/pnas.1607769113
7. He WT, Wan H, Hu L, Chen P, Wang X, Huang Z, et al. Gasdermin D is an executor of pyroptosis and required for interleukin-1beta secretion. Cell Res. (2015) 25:1285–98. doi: 10.1038/cr.2015.139
8. Mitroulis I, Kambas K, Chrysanthopoulou A, Skendros P, Apostolidou E, Kourtzelis I, et al. Neutrophil extracellular trap formation is associated with IL-1beta and autophagy-related signaling in gout. PLoS ONE. (2011) 6:e29318. doi: 10.1371/journal.pone.0029318
9. So A, Dumusc A, Nasi S. The role of IL-1 in gout: from bench to bedside. Rheumatology. (2018) 57:i12–9. doi: 10.1093/rheumatology/kex449
10. Mariathasan S, Newton K, Monack DM, Vucic D, French DM, Lee WP, et al. Differential activation of the inflammasome by caspase-1 adaptors ASC and Ipaf. Nature. (2004) 430:213–8. doi: 10.1038/nature02664
11. Martinon F, Petrilli V, Mayor A, Tardivel A, Tschopp J. Gout-associated uric acid crystals activate the NALP3 inflammasome. Nature. (2006) 440:237–41. doi: 10.1038/nature04516
12. Lamkanfi M, Kanneganti TD, Franchi L, Nunez G. Caspase-1 inflammasomes in infection and inflammation. J Leukoc Biol. (2007) 82:220–5. doi: 10.1189/jlb.1206756
13. Busso N, So A. Mechanisms of inflammation in gout. Arthritis Res Ther. (2010) 12:206. doi: 10.1186/ar2952
14. Gowen M, Wood DD, Ihrie EJ, McGuire MK, Russell RG. An interleukin 1 like factor stimulates bone resorption in vitro. Nature. (1983) 306:378–80. doi: 10.1038/306378a0
15. Wei S, Kitaura H, Zhou P, Ross FP, Teitelbaum SL. IL-1 mediates TNF-induced osteoclastogenesis. J Clin Invest. (2005) 115:282–90. doi: 10.1172/JCI200523394
16. Maueroder C, Kienhofer D, Hahn J, Schauer C, Manger B, Schett G, et al. How neutrophil extracellular traps orchestrate the local immune response in gout. J Mol Med. (2015) 93:727–34. doi: 10.1007/s00109-015-1295-x
17. Martin WJ, Grainger R, Harrison A, Harper JL. Differences in MSU-induced superoxide responses by neutrophils from gout subjects compared to healthy controls and a role for environmental inflammatory cytokines and hyperuricemia in neutrophil function and survival. J Rheumatol. (2010) 37:1228–35. doi: 10.3899/jrheum.091080
18. Kingsbury SR, Conaghan PG, McDermott MF. The role of the NLRP3 inflammasome in gout. J Inflamm Res. (2011) 4:39–49. doi: 10.2147/JIR.S11330
19. Vong L, Sherman PM, Glogauer M. Quantification and visualization of neutrophil extracellular traps (NETs) from murine bone marrow-derived neutrophils. Methods Mol Biol. (2013) 1031:41–50. doi: 10.1007/978-1-62703-481-4_5
20. Hasler P, Giaglis S, Hahn S. Neutrophil extracellular traps in health and disease. Swiss Med Wkly. (2016) 146:w14352. doi: 10.4414/smw.2016.14352
21. Galluzzi L, Vitale I, Aaronson SA, Abrams JM, Adam D, Agostinis P, et al. Molecular mechanisms of cell death: recommendations of the Nomenclature Committee on Cell Death 2018. Cell Death Differ. (2018) 25:486–541. doi: 10.1038/s41418-017-0012-4
22. Boeltz S, Amini P, Anders HJ, Andrade F, Bilyy R, Chatfield S, et al. To NET or not to NET:current opinions and state of the science regarding the formation of neutrophil extracellular traps. Cell Death Differ. (2019) 26:395–408. doi: 10.1038/s41418-018-0261-x
23. Rada B. Neutrophil extracellular traps and microcrystals. J Immunol Res. (2017) 2017:2896380. doi: 10.1155/2017/2896380
24. Caution K, Gavrilin MA, Tazi M, Kanneganti A, Layman D, Hoque S, et al. Caspase-11 and caspase-1 differentially modulate actin polymerization via RhoA and Slingshot proteins to promote bacterial clearance. Sci Rep. (2015) 5:18479. doi: 10.1038/srep18479
25. Akhter A, Caution K, Abu Khweek A, Tazi M, Abdulrahman BA, Abdelaziz DH, et al. Caspase-11 promotes the fusion of phagosomes harboring pathogenic bacteria with lysosomes by modulating actin polymerization. Immunity. (2012) 37:35–47. doi: 10.1016/j.immuni.2012.05.001
26. Krause K, Caution K, Badr A, Hamilton K, Saleh A, Patel K, et al. CASP4/caspase-11 promotes autophagosome formation in response to bacterial infection. Autophagy. (2018) 14:1928–42. doi: 10.1080/15548627.2018.1491494
27. Li J, Brieher WM, Scimone ML, Kang SJ, Zhu H, Yin H, et al. Caspase-11 regulates cell migration by promoting Aip1-Cofilin-mediated actin depolymerization. Nat Cell Biol. (2007) 9:276–86. doi: 10.1038/ncb1541
28. Krause K, Daily K, Estfanous S, Hamilton K, Badr A, Abu Khweek A, et al. Caspase-11 counteracts mitochondrial ROS-mediated clearance of Staphylococcus aureus in macrophages. EMBO Rep. (2019). doi: 10.15252/embr.201948109. [Epub ahead of print].
29. Lagrange B, Benaoudia S, Wallet P, Magnotti F, Provost A, Michal F, et al. Human caspase-4 detects tetra-acylated LPS and cytosolic Francisella and functions differently from murine caspase-11. Nat Commun. (2018) 9:242. doi: 10.1038/s41467-017-02682-y
30. Yi YS. Caspase-11 non-canonical inflammasome: a critical sensor of intracellular lipopolysaccharide in macrophage-mediated inflammatory responses. Immunology. (2017) 152:207–17. doi: 10.1111/imm.12787
31. Vanaja SK, Russo AJ, Behl B, Banerjee I, Yankova M, Deshmukh SD, et al. Bacterial outer membrane vesicles mediate cytosolic localization of LPS and caspase-11 activation. Cell. (2016) 165:1106–19. doi: 10.1016/j.cell.2016.04.015
32. Man SM, Karki R, Briard B, Burton A, Gingras S, Pelletier S, et al. Differential roles of caspase-1 and caspase-11 in infection and inflammation. Sci Rep. (2017) 7:45126. doi: 10.1038/srep45126
33. Py BF, Jin M, Desai BN, Penumaka A, Zhu H, Kober M, et al. Caspase-11 controls interleukin-1beta release through degradation of TRPC1. Cell Rep. (2014) 6:1122–8. doi: 10.1016/j.celrep.2014.02.015
34. He Y, Li D, Cook SL, Yoon MS, Kapoor A, Rao CV, et al. Mammalian target of rapamycin and Rictor control neutrophil chemotaxis by regulating Rac/Cdc42 activity and the actin cytoskeleton. Mol Biol Cell. (2013) 24:3369–80. doi: 10.1091/mbc.e13-07-0405
35. Campa CC, Germena G, Ciraolo E, Copperi F, Sapienza A, Franco I, et al. Rac signal adaptation controls neutrophil mobilization from the bone marrow. Sci Signal. (2016) 9:ra124. doi: 10.1126/scisignal.aah5882
36. Ragab G, Elshahaly M, Bardin T. Gout: An old disease in new perspective - A review. J Adv Res. (2017) 8:495–511. doi: 10.1016/j.jare.2017.04.008
37. Krause K, Kopp BT, Tazi MF, Caution K, Hamilton K, Badr A, et al. The expression of Mirc1/Mir17-92 cluster in sputum samples correlates with pulmonary exacerbations in cystic fibrosis patients. J Cyst Fibros. (2017). 17:454–61. doi: 10.1016/j.jcf.2017.11.005
38. Stowe I, Lee B, Kayagaki N. Caspase-11: arming the guards against bacterial infection. Immunol Rev. (2015) 265:75–84. doi: 10.1111/imr.12292
39. Kayagaki N, Wong MT, Stowe IB, Ramani SR, Gonzalez LC, Akashi-Takamura S, et al. Noncanonical inflammasome activation by intracellular LPS independent of TLR4. Science. (2013) 341:1246–9. doi: 10.1126/science.1240248
40. Dinarello CA, Joosten LA. Inflammation in rheumatology in 2015: New tools to tackle inflammatory arthritis. Nat Rev Rheumatol. (2016) 12:78–80. doi: 10.1038/nrrheum.2015.180
41. Dinarello CA. How interleukin-1beta induces gouty arthritis. Arthritis Rheum. (2010) 62:3140–4. doi: 10.1002/art.27663
42. Busso N, Ea HK. The mechanisms of inflammation in gout and pseudogout (CPP-induced arthritis). Reumatismo. (2012) 63:230–7. doi: 10.4081/reumatismo.2011.230
43. Wang S, Miura M, Jung YK, Zhu H, Li E, Yuan J. Murine caspase-11, an ICE-interacting protease, is essential for the activation of ICE. Cell. (1998) 92:501–9. doi: 10.1016/S0092-8674(00)80943-5
44. Schauvliege R, Vanrobaeys J, Schotte P, Beyaert R. Caspase-11 gene expression in response to lipopolysaccharide and interferon-gamma requires nuclear factor-kappa B and signal transducer and activator of transcription (STAT) 1. J Biol Chem. (2002) 277:41624–30. doi: 10.1074/jbc.M207852200
45. Akira S, Takeda K. Toll-like receptor signalling. Nat Rev Immunol. (2004) 4:499–511. doi: 10.1038/nri1391
46. Akira S, Takeda K. Functions of toll-like receptors: lessons from KO mice. C R Biol. (2004) 327:581–9. doi: 10.1016/j.crvi.2004.04.002
47. Chen CJ, Shi Y, Hearn A, Fitzgerald K, Golenbock D, Reed G, et al. MyD88-dependent IL-1 receptor signaling is essential for gouty inflammation stimulated by monosodium urate crystals. J Clin Invest. (2006) 116:2262–71. doi: 10.1172/JCI28075
48. Kokkola R, Andersson A, Mullins G, Ostberg T, Treutiger CJ, Arnold B, et al. RAGE is the major receptor for the proinflammatory activity of HMGB1 in rodent macrophages. Scand J Immunol. (2005) 61:1–9. doi: 10.1111/j.0300-9475.2005.01534.x
49. Yang H, Hreggvidsdottir HS, Palmblad K, Wang H, Ochani M, Li J, et al. A critical cysteine is required for HMGB1 binding to Toll-like receptor 4 and activation of macrophage cytokine release. Proc Natl Acad Sci U.S.A. (2010) 107:11942–7. doi: 10.1073/pnas.1003893107
50. Yanai H, Ban T, Wang Z, Choi MK, Kawamura T, Negishi H, et al. HMGB proteins function as universal sentinels for nucleic-acid-mediated innate immune responses. Nature. (2009) 462:99–103. doi: 10.1038/nature08512
51. Terkeltaub R, Baird S, Sears P, Santiago R, Boisvert W. The murine homolog of the interleukin-8 receptor CXCR-2 is essential for the occurrence of neutrophilic inflammation in the air pouch model of acute urate crystal-induced gouty synovitis. Arthritis Rheum. (1998) 41:900–93. doi: 10.1002/1529-0131(199805)41:5<900::AID-ART18>3.0.CO;2-K
52. Petrie RJ, Doyle AD, Yamada KM. Random versus directionally persistent cell migration. Nat Rev Mol Cell Biol. (2009) 10:538–49. doi: 10.1038/nrm2729
53. Stephens L, Milne L, Hawkins P. Moving towards a better understanding of chemotaxis. Curr Biol. (2008) 18:R485–94. doi: 10.1016/j.cub.2008.04.048
54. Wille C, Eiseler T, Langenberger ST, Richter J, Mizuno K, Radermacher P, et al. PKD regulates actin polymerization, neutrophil deformability, and transendothelial migration in response to fMLP and trauma. J Leukoc Biol. (2018) 104:615–30. doi: 10.1002/JLB.4A0617-251RR
55. Remijsen Q, Vanden Berghe T, Wirawan E, Asselbergh B, Parthoens E, De Rycke R, et al. Neutrophil extracellular trap cell death requires both autophagy and superoxide generation. Cell Res. (2011) 21:290–304. doi: 10.1038/cr.2010.150
56. Remijsen Q, Kuijpers TW, Wirawan E, Lippens S, Vandenabeele P, Vanden Berghe T. Dying for a cause: NETosis, mechanisms behind an antimicrobial cell death modality. Cell Death Differ. (2011) 18:581–8. doi: 10.1038/cdd.2011.1
57. Wang X, Yousefi S, Simon HU. Necroptosis and neutrophil-associated disorders. Cell Death Dis. (2018) 9:111. doi: 10.1038/s41419-017-0058-8
58. Desai J, Kumar SV, Mulay SR, Konrad L, Romoli S, Schauer C, et al. PMA and crystal-induced neutrophil extracellular trap formation involves RIPK1-RIPK3-MLKL signaling. Eur J Immunol. (2016) 46:223–9. doi: 10.1002/eji.201545605
59. Yousefi S, Stojkov D, Germic N, Simon D, Wang X, Benarafa C, et al. Untangling “NETosis” from NETs. Eur J Immunol. (2019) 49:221–7. doi: 10.1002/eji.201747053
60. Stojkov D, Amini P, Oberson K, Sokollik C, Duppenthaler A, Simon HU, et al. ROS and glutathionylation balance cytoskeletal dynamics in neutrophil extracellular trap formation. J Cell Biol. (2017) 216:4073–90. doi: 10.1083/jcb.201611168
61. Cooper PR, Palmer LJ, Chapple IL. Neutrophil extracellular traps as a new paradigm in innate immunity: friend or foe? Periodontol 2000. (2013) 63:165–97. doi: 10.1111/prd.12025
62. Lee BL, Stowe IB, Gupta A, Kornfeld OS, Roose-Girma M, Anderson K, et al. Caspase-11 auto-proteolysis is crucial for noncanonical inflammasome activation. J Exp Med. (2018) 215:2279–88. doi: 10.1084/jem.20180589
63. Lacey CA, Mitchell WJ, Dadelahi AS, Skyberg JA. Caspases-1 and caspase-11 mediate pyroptosis, inflammation, and control of Brucella joint infection. Infect. Immun. (2018) 86:1–18. doi: 10.1128/IAI.00361-18
64. Zanoni I, Tan Y, Di Gioia M, Broggi A, Ruan J, Shi J, et al. An endogenous caspase-11 ligand elicits interleukin-1 release from living dendritic cells. Science. (2016) 352:1232–6. doi: 10.1126/science.aaf3036
65. Napier BA, Brubaker SW, Sweeney TE, Monette P, Rothmeier GH, Gertsvolf NA, et al. Complement pathway amplifies caspase-11-dependent cell death and endotoxin-induced sepsis severity. J Exp Med. (2016) 213:2365–82. doi: 10.1084/jem.20160027
66. Caution K, Amer AO. Caspase-11: mediator of pyroptotic and non-pyroptotic host responses. Trends Cell Mol Biol. (2016) 11:19–33.
67. Baker PJ, Boucher D, Bierschenk D, Tebartz C, Whitney PG, D'Silva DB, et al. NLRP3 inflammasome activation downstream of cytoplasmic LPS recognition by both caspase-4 and caspase-5. Eur J Immunol. (2015) 45:2918–26. doi: 10.1002/eji.201545655
68. Hara H, Seregin SS, Yang D, Fukase K, Chamaillard M, Alnemri ES, et al. The NLRP6 inflammasome recognizes lipoteichoic acid and regulates gram-positive pathogen infection. Cell. (2018) 175, 1651–64.e14. doi: 10.1016/j.cell.2018.09.047
69. de Gassart A, Martinon F. Pyroptosis: caspase-11 unlocks the gates of death. Immunity. (2015) 43:835–7. doi: 10.1016/j.immuni.2015.10.024
70. Man SM, Kanneganti TD. Regulation of inflammasome activation. Immunol Rev. (2015) 265:6–21. doi: 10.1111/imr.12296
71. Kuriakose T, Kanneganti TD. Gasdermin D Flashes an Exit Signal for IL-1. Immunity. (2018) 48:1–3. doi: 10.1016/j.immuni.2018.01.003
72. Sil P, Wicklum H, Surell C, Rada B. Macrophage-derived IL-1beta enhances monosodium urate crystal-triggered NET formation. Inflamm Res. (2017) 66:227–37. doi: 10.1007/s00011-016-1008-0
73. Frangou E, Vassilopoulos D, Boletis J, Boumpas DT. An emerging role of neutrophils and NETosis in chronic inflammation and fibrosis in systemic lupus erythematosus (SLE) and ANCA-associated vasculitides (AAV): implications for the pathogenesis and treatment. Autoimmun Rev. (2019) 18:751–60. doi: 10.1016/j.autrev.2019.06.011
74. Amini P, Stojkov D, Wang X, Wicki S, Kaufmann T, Wong WW, et al. NET formation can occur independently of RIPK3 and MLKL signaling. Eur J Immunol. (2016) 46:178–84. doi: 10.1002/eji.201545615
75. Schauer C, Janko C, Munoz LE, Zhao Y, Kjenhöfer D, Frey B, et al. Aggregated neutrophil extracellular traps limit inflammation by degrading cytokines and chemokines. Nat Med. (2014) 20:511–7. doi: 10.1038/nm.3547.
76. Chen KW, Monteleone M, Boucher D, Sollberger G, Ramnath D, Condon ND, et al. Noncanonical inflammasome signaling elicits gasdermin D-dependent neutrophil extracellular traps. Sci Immunol. (2018) 3:eaar6676. doi: 10.1126/sciimmunol.aar6676
77. Kayagaki N, Warming S, Lamkanfi M, Vande Walle L, Louie S, Dong J, et al. Non-canonical inflammasome activation targets caspase-11. Nature. (2011) 479:117–21. doi: 10.1038/nature10558
78. Torres R, Macdonald L, Croll SD, Reinhardt J, Dore A, Stevens S, et al. Hyperalgesia, synovitis and multiple biomarkers of inflammation are suppressed by interleukin 1 inhibition in a novel animal model of gouty arthritis. Ann Rheum Dis. (2009) 68:1602–8. doi: 10.1136/ard.2009.109355
79. Young G, Wang K, He J, Otto G, Hawryluk M, Zwirco Z, et al. Clinical next-generation sequencing successfully applied to fine-needle aspirations of pulmonary and pancreatic neoplasms. Cancer Cytopathol. (2013) 121:688–94. doi: 10.1002/cncy.21338
80. Abdulrahman BA, Khweek AA, Akhter A, Caution K, Kotrange S, Abdelaziz DH, et al. Autophagy stimulation by rapamycin suppresses lung inflammation and infection by Burkholderia cenocepacia in a model of cystic fibrosis. Autophagy. (2011) 7:1359–70. doi: 10.4161/auto.7.11.17660
81. Abdulrahman BA, Khweek AA, Akhter A, Caution K, Tazi M, Hassan H, et al. Depletion of the ubiquitin-binding adaptor molecule SQSTM1/p62 from macrophages harboring cftr DeltaF508 mutation improves the delivery of Burkholderia cenocepacia to the autophagic machinery. J Biol Chem. (2013) 288:2049–58. doi: 10.1074/jbc.M112.411728
82. Young NA, Wu LC, Bruss M, Kaffenberger BH, Hampton J, Bolon B, et al. A chimeric human-mouse model of Sjogren's syndrome. Clin Immunol. (2014) 156:1–8. doi: 10.1016/j.clim.2014.10.004
83. Abdelaziz DH, Gavrilin MA, Akhter A, Caution K, Kotrange S, Khweek AA, et al. Asc-dependent and independent mechanisms contribute to restriction of legionella pneumophila infection in murine macrophages. Front Microbiol. (2011) 2:18. doi: 10.3389/fmicb.2011.00018
84. Abu Khweek A, Kanneganti A, Guttridge DD, Amer AO. The Sphingosine-1-Phosphate Lyase (LegS2) Contributes to the Restriction of Legionella pneumophila in Murine Macrophages. PLoS ONE. (2016) 11:e0146410. doi: 10.1371/journal.pone.0146410
85. Amer A, Franchi L, Kanneganti TD, Body-Malapel M, Ozoren N, Brady G, et al. Regulation of Legionella phagosome maturation and infection through flagellin and host Ipaf. J Biol Chem. (2006) 281:35217–23. doi: 10.1074/jbc.M604933200
86. Akhter A, Gavrilin MA, Frantz L, Washington S, Ditty C, Limoli D, et al. Caspase-7 activation by the Nlrc4/Ipaf inflammasome restricts Legionella pneumophila infection. PLoS Pathog. (2009) 5:e1000361. doi: 10.1371/journal.ppat.1000361
87. Tazi MF, Dakhlallah DA, Caution K, Gerber MM, Chang SW, Khalil H, et al. Elevated Mirc1/Mir17-92 cluster expression negatively regulates autophagy and CFTR (cystic fibrosis transmembrane conductance regulator) function in CF macrophages. Autophagy. (2016) 12:2026–37. doi: 10.1080/15548627.2016.1217370
88. Robledo-Avila FH, Ruiz-Rosado JD, Brockman KL, Kopp BT, Amer AO, McCoy K, et al. Dysregulated calcium homeostasis in cystic fibrosis neutrophils leads to deficient antimicrobial responses. J Immunol. (2018) 201:2016–27. doi: 10.4049/jimmunol.1800076
Keywords: caspase-11, gout, neutrophils, macrophages, NETosis, IL-1β, cell migration, inflammasome
Citation: Caution K, Young N, Robledo-Avila F, Krause K, Abu Khweek A, Hamilton K, Badr A, Vaidya A, Daily K, Gosu H, Anne MNK, Eltobgy M, Dakhlallah D, Argwal S, Estfanous S, Zhang X, Partida-Sanchez S, Gavrilin MA, Jarjour WN and Amer AO (2019) Caspase-11 Mediates Neutrophil Chemotaxis and Extracellular Trap Formation During Acute Gouty Arthritis Through Alteration of Cofilin Phosphorylation. Front. Immunol. 10:2519. doi: 10.3389/fimmu.2019.02519
Received: 16 July 2019; Accepted: 09 October 2019;
Published: 15 November 2019.
Edited by:
Guochang Hu, University of Illinois at Chicago, United StatesReviewed by:
Izabela Galvao, Centenary Institute Australia, AustraliaCopyright © 2019 Caution, Young, Robledo-Avila, Krause, Abu Khweek, Hamilton, Badr, Vaidya, Daily, Gosu, Anne, Eltobgy, Dakhlallah, Argwal, Estfanous, Zhang, Partida-Sanchez, Gavrilin, Jarjour and Amer. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Amal O. Amer, YW1hbC5hbWVyQG9zdW1jLmVkdQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.