
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Immunol. , 24 October 2019
Sec. Alloimmunity and Transplantation
Volume 10 - 2019 | https://doi.org/10.3389/fimmu.2019.02511
This article is part of the Research Topic Kidney Transplantation and Innate Immunity View all 17 articles
 Rosa G. M. Lammerts1*
Rosa G. M. Lammerts1* Michele F. Eisenga1
Michele F. Eisenga1 Mohammed Alyami1
Mohammed Alyami1 Mohamed R. Daha1
Mohamed R. Daha1 Marc A. Seelen1
Marc A. Seelen1 Robert A. Pol2
Robert A. Pol2 Jacob van den Born1
Jacob van den Born1 Jan-Stephan Sanders1
Jan-Stephan Sanders1 Stephan J. L. Bakker1
Stephan J. L. Bakker1 Stefan P. Berger1 on behalf of the COMBAT Consortium
Stefan P. Berger1 on behalf of the COMBAT ConsortiumThe pathophysiology of late kidney-allograft failure remains complex and poorly understood. Activation of filtered or locally produced complement may contribute to the progression of renal failure through tubular C5b-9 formation. This study aimed to determine urinary properdin and sC5b-9 excretion and assess their association with long-term outcome in renal transplant recipients (RTR).
Methods: We measured urinary properdin and soluble C5b-9 in a well-defined cross-sectional cohort of RTR. Urinary specimens were taken from a morning urine portion, and properdin and sC5b-9 were measured using an enzyme-linked-immunosorbent assay (ELISA). Cox proportional hazard regression analyses were used to investigate prospective associations with death-censored graft failure.
Results: We included 639 stable RTR at a median [interquartile range] 5.3 (1.8–12.2) years after transplantation. Urinary properdin and sC5b-9 excretion were detectable in 161 (27%) and 102 (17%) RTR, respectively, with a median properdin level of 27.6 (8.6–68.1) ng/mL and a median sC5b-9 level of 5.1 (2.8–12.8) ng/mL. In multivariable-adjusted Cox regression analyses, including adjustment for proteinuria, urinary properdin (HR, 1.12; 95% CI 1.02–1.28; P = 0.008) and sC5b-9 excretion (HR, 1.34; 95% CI 1.10–1.63; P = 0.003) were associated with an increased risk of graft failure. If both urinary properdin and sC5b-9 were detectable, the risk of graft failure was further increased (HR, 3.12; 95% CI 1.69–5.77; P < 0.001).
Conclusions: Our findings point toward a potential role for urinary complement activation in the pathogenesis of chronic allograft failure. Urinary properdin and sC5b-9 might be useful biomarkers for complement activation and chronic kidney allograft deterioration, suggesting a potential role for an alternative pathway blockade in RTR.
Despite improvements in immunosuppressive therapy over the last decades, chronic and irreversible deterioration of a transplanted kidney graft remains a major problem and is responsible for disappointing outcomes in long-term graft survival (1). Even though registry data can be used to define risk factors, chronic allograft failure pathophysiology remains complex and poorly understood, due to difficulty in distinguishing the contribution of several immunological and non-immunological factors (2). Interstitial fibrosis/tubular atrophy (IFTA), presents itself as renal allograft dysfunction (occurring at least 3 months post-transplant) in the absence of active acute rejection, drug toxicity, or other diseases. Due to its multiple possible causes and complex etiology, classification of IFTA is still an ongoing process (3, 4). The clinical diagnosis is usually suggested by gradual deterioration of allograft function, manifested by a slowly rising serum creatinine concentration, worsening hypertension, and increasing proteinuria. Proteinuria is known to be a progression marker and a predictor for renal failure (5, 6). It is thought that proteinuria contributes to the progression of renal failure by various mechanisms. One of these mechanisms is suggested to be leakage of albumin-bound lipids across the damaged glomerular filtration barrier, leading to lipoapoptosis after reabsorption by the downstream proximal tubule (7, 8). Alongside this, activation of filtered or locally produced complement may be harmful to renal tubular cells and contribute to the progression of renal failure by initiating interstitial fibrosis (9, 10). Complement activation leads to the formation of C5b-9 (11), which can be used as a clinical indicator of complement activation in native kidney diseases (12, 13).
Renal proximal tubular cells are known to activate complement via the alternative pathway (AP) (14). Gaarkeuken et al. showed that complement activation on tubular cells is mediated by properdin binding on the tubular brush border (15). Our group identified tubular heparan sulfate as the docking platform for properdin and the consequent AP activation on tubular cells (9). In proteinuric patients, urinary properdin excretion is associated with intrarenal complement activation and poor renal function (16, 17).
Although it has been established that there is a strong relationship between proteinuria, tubulo-interstitial injury and a poor prognosis in kidney disease, to our knowledge no studies have examined the role of urinary complement activation products in kidney transplantation outcomes.
We hypothesized that the AP regulator properdin and the terminal complement complex sC5b-9 play an important role in graft failure and could serve as early biomarkers for late graft failure. Hence, the aim of the present study is to investigate the role of properdin and sC5b9 in renal transplant recipients (RTR) in relation to the development of graft failure over time.
The study population consisted of a well-characterized and previously described cohort of 707 RTR (18). In short, this cohort comprised RTR (aged ≥ 18 years) who visited the outpatient clinic of the University Medical Center Groningen (UMCG), Groningen, The Netherlands, between November 2008 and June 2011, and who had a functional graft for at least 1 year after transplantation. All patients provided written informed consent. Urinary morning samples were collected at inclusion in the study and immediately placed on ice. The samples were centrifuged at 4°C at 4,000 RPM for 15 min to remove components and debris, and the supernatants were stored at −80°C. They were not subjected to freeze/thaw cycles before analysis. There were 639 patients eligible for analysis after we excluded 67 patients with missing urinary samples which precluded the measurement of urinary properdin and sC5b-9 levels. Death-censored graft failure was defined as return to dialysis or re-transplantation. Kidney function was assessed by estimating glomerular filtration rate (eGFR) by applying the Chronic Kidney Disease Epidemiology Collaboration equation (19). Protein excretion of ≥0.5 g per day was defined as proteinuria. The study was approved by the UMCG institutional review board (METc 2008/186), adheres to the Declarations of Helsinki and Istanbul and has NCT02811835 as ClinicalTrials.gov identifier.
Urinary properdin levels were assessed by a previously described sandwich enzyme-linked-immunosorbent assay (ELISA) (9, 17), with a detection limit of 1.2 ng/mL, a plasma intra-variation of <17% and an inter-variation of <20%. In brief, 96-well ELISA plates (NUNC MaxiSorpTM, Sigma-Aldrich, Saint Louis, MO, USA) were coated overnight at 4°C with monoclonal anti-human properdin (Hycult HM2282, Uden, the Netherlands). Urinary samples were diluted 5 times in DPBS with 0.1% Tween and bovine serum albumin (PTB) and incubated for 1 h at 37°C, followed by secondary antibody; polyclonal rabbit anti-human properdin-biotin (kindly provided by M. R. Daha, Leiden, The Netherlands) and detection with Streptavidin-HRP (Dako P0397, Glostrup, Denmark). Enzyme activity was detected using 2,2′azino-bis (3-ethylbenzo-thiazoline-6-sulphonic acid) (A1888, Sigma-Aldrich, Saint Louis, MO, USA). The optical density was measured at 415 nm using a microplate ELISA reader (Benchmark Plus, Bio-Rad, Veenendaal, The Netherlands). A standard curve was prepared using a serial dilution of zymosan activated serum in PTB with a known concentration of properdin. A reference sample, diluted in PTB with a known concentration of properdin was included as positive control. Potential background signal was assessed and corrected for, with PTB functioning as blank.
Urinary sC5b-9 levels were assessed by a previously standardized and validated sandwich ELISA (17, 20), with a detection limit of 2.1 ng/mL, a plasma intra-variation of <13% and an inter-variation of <19%. In brief, 96-well ELISA plates (NUNC MaxiSorpTM, Sigma-Aldrich) were coated overnight at 4°C with monoclonal mouse anti-human C5b-9 (Dako M0777). Urinary samples were diluted 1.25 times and incubated for 1 h at 37°C. Secondary antibody polyclonal goat anti-human C5 (Quidel Ca92121, San Diego, CA, USA), followed by tertiary antibody polyclonal mouse anti-goat IgG HRP (Jackson 205-035-108) were added. Enzyme activity was detected using 3,3′,5,5′-tetramethylbenzidine. The optical density was measured at 450 nm using a microplate ELISA reader. The standard curve, reference sample, and the assessment of a potential background signal was prepared in the same way as the properdin ELISA, with a known concentration of sC5b-9.
Data were analyzed using IBM SPSS software, version 23.0 (SPSS Inc., Chicago, IL, USA) and R version 3.2.3 (Vienna, Austria). Data are expressed as mean ± SD for normally distributed variables and as median [25th−75th interquartile range (IQR)] for variables with a skewed distribution. Categorical data are expressed as number (percentage). Under normal conditions complement factors are not present in the urine. Therefore, we defined urinary properdin and sC5b9 as a negative test when undetectable in the urine and as a positive test when detectable.
We evaluated between-group differences at baseline, comparing RTRs with vs. without detectable properdin and sC5b-9 using Student t-test, Mann-Whitney U-test, or Chi square test, as appropriate. To visualize the association between urinary properdin and urinary sC5b-9 excretion, we generated a restricted cubic spline plot based on linear regression analyses. Knots were placed on the 10th, 50th, and 90th percentile of ln properdin. To visualize the association between urinary properdin and urinary sC5b-9 excretion with proteinuria, we generated restricted cubic spline plots based on linear regression analyses, with knots placed on the 10th, 50th, and 90th percentile of ln proteinuria. Further, Kaplan Meier curves were used to depict the effect of the presence of urinary properdin and/or sC5b-9 on graft failure and all-cause mortality. Differences in survival rates were tested using the Cox-Mantel log-rank test. To study the prospective association with death-censored graft failure and all-cause mortality, we used Cox proportional hazards regression analysis. Prior to analyses, we first adjusted for statistically significant different parameters at baseline and for other known predictors of graft failure like HLA mismatches. First, death-censored graft failure was adjusted for age, sex, primary renal disease, time since transplantation at inclusion, eGFR, HLA mismatches, and donor type (model 1). Additionally, adjustment was made for high sensitive-CRP (hs-CRP) (model 2); further adjustment for systolic blood pressure, and smoking (model 3); and final adjustment for proteinuria (model 4). Due to skewed distribution, hs-CRP, properdin, and sC5b-9 were natural log-transformed. To determine the optimal cut off value of urinary properdin and sC5b-9 for prediction of graft failure in RTR, the Youden index was used. Finally, we performed mediation analyses to assess whether sC5b-9 was a mediator in the association between properdin and graft failure. For this purpose, we used the method as stated by Preacher and Hayes, which is based on logistic regression (21, 22). These analyses allow for testing significance and magnitude of mediation. For all analyses, a two-sided P < 0.05 was considered significant.
We included 639 RTR (age 53 ± 13 years; 58% males at 5.3 (1.8–12.2) years after transplantation). Mean eGFR was 52.2 ± 20.1 ml/min/1.73 m2, and urinary properdin excretion was detectable in 161 (27%) RTR with a median [interquartile range] properdin level of 27.6 (8.7–68.1) ng/mL. Urinary sC5b-9 excretion was detectable in 102 (17%) RTR with median sC5b-9 levels of 5.1 (2.8–12.8) ng/mL.
RTR with detectable urinary properdin were more frequently females (P < 0.001), had significantly higher: body surface area (m2) (P = 0.004), creatinine (P = 0.003), hs-CRP (P < 0.001), frequency of proteinuria (≥0.5 g/24 h) (P < 0.001), and received a deceased—donor kidney transplant (P = 0.02). RTR with detectable urinary sC5b-9 were more frequently males (P = 0.01), had higher levels of creatinine (P < 0.001), a higher frequency of proteinuria (P < 0.001), and a deceased-donor kidney transplant (P = 0.02). An inverse association between eGFR and detectable properdin (P < 0.001) and sC5b-9 levels (P < 0.001) was detected at baseline. No significant differences were found at baseline in HLA mismatches, primary renal disease, history of delayed graft function, and rejection between patients with and without detectable urinary properdin or sC5b-9.
Detectable urinary properdin excretion was present in 11 and 16% of RTR with and without proteinuria, respectively. Detectable urinary sC5b-9 excretion was present in 9 and 8% of RTR with and without proteinuria, respectively (Figure 1). Urinary properdin was significantly associated with urinary sC5b-9 excretion in RTR in whom both complement products were detectable (β = 0.25; P < 0.001) (Figure 2). Urinary properdin and urinary sC5b-9 excretion were both significantly associated with proteinuria (β = 0.26; P < 0.001 and β = 0.36; P < 0.001, respectively) (Supplementary Figures 1, 2). Further demographics and clinical characteristics dichotomized into detectable or undetectable urinary properdin and sC5b-9 are specified in Table 1.
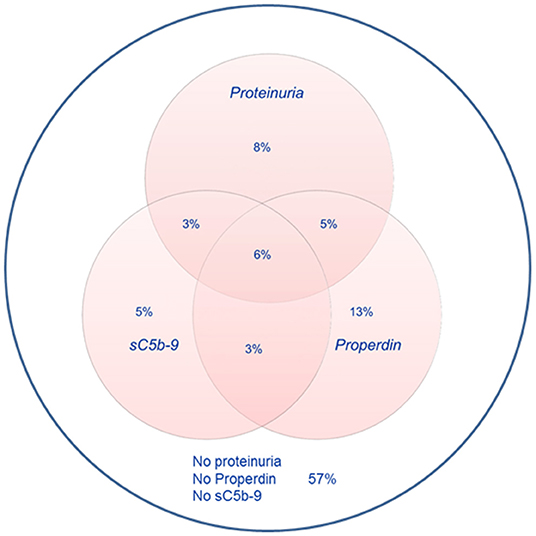
Figure 1. Prevalences of urinary properdin, urinary sC5b-9, and proteinuria.
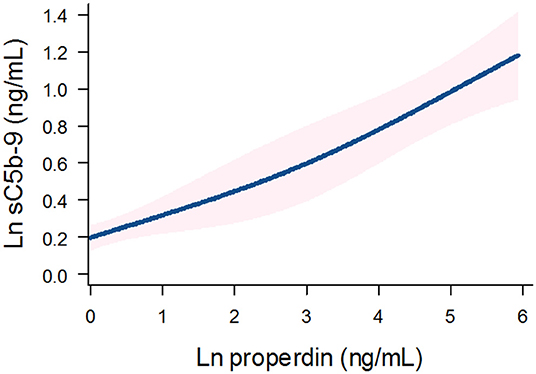
Figure 2. Association between urinary properdin and urinary sC5b-9 excretion in the RTR. A restricted cubic spline is generated based on linear regression analyses. Knots are placed on 10th, 50th, and 90th percentile of ln properdin. Blue line represents the coefficient, and pink band represents the 95% confidence interval.
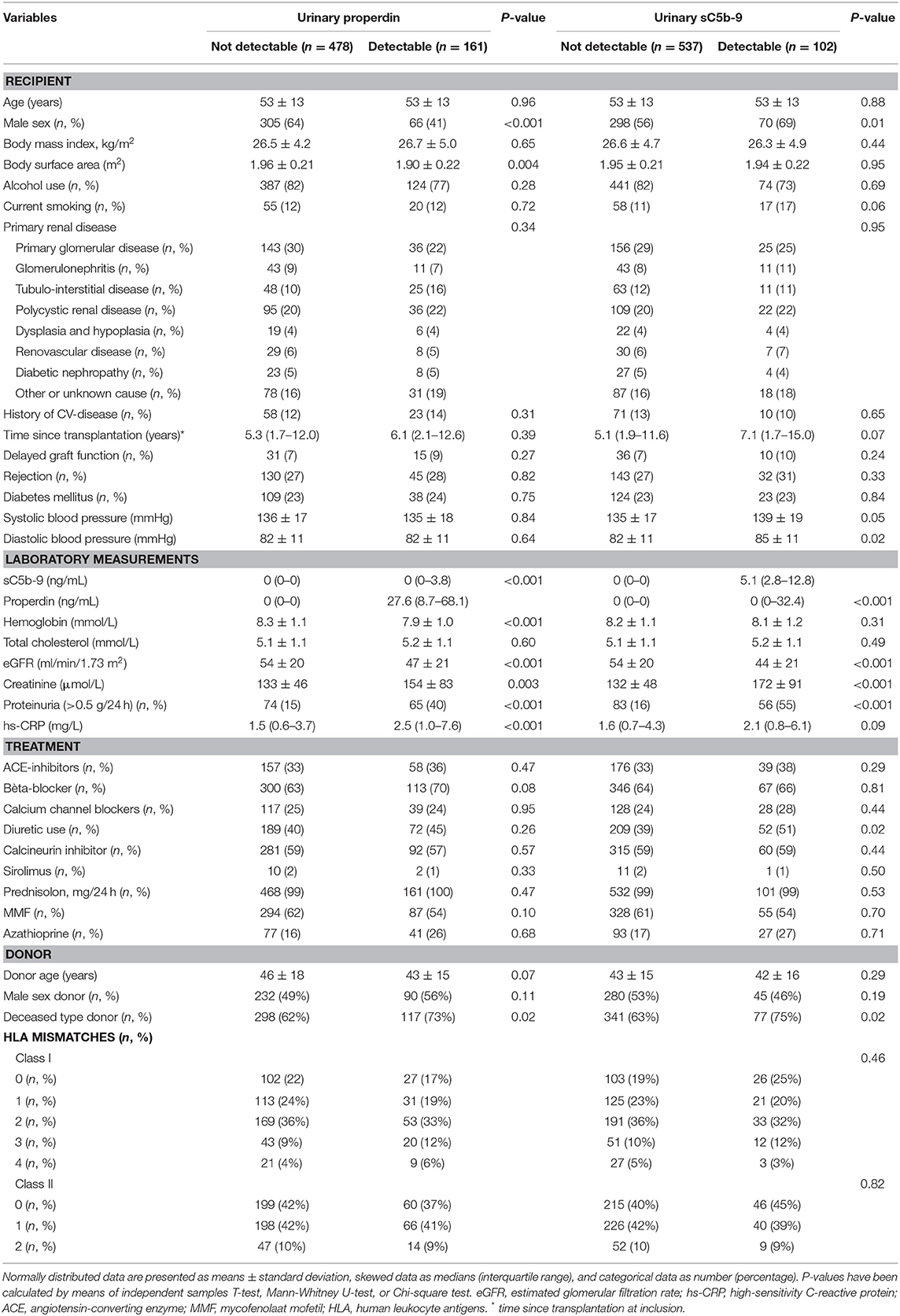
Table 1. Baseline characteristics according to detectable urinary properdin urinary sC5b-9 levels.
During a median follow-up of 5.3 (4.5–6.0) years, 75 (12%) RTRs developed death-censored graft failure. As depicted in the Kaplan Meier curves shown in Figure 3, RTR with both detectable urinary properdin and sC5b-9 had the highest risk of developing graft failure (P < 0.001). RTR with urine in which either properdin or sC5b-9 was detectable, showed an intermediate risk with worse graft survival compared to RTR without detectable urinary properdin or sC5b-9 (Figure 3).
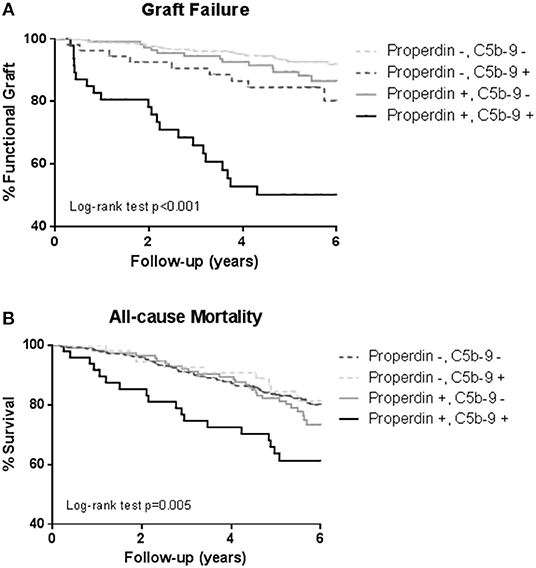
Figure 3. Kaplan-Meier analyses for percentage graft failure (A) and survival (B) according to no sC5b-9/no properdin, sC5b-9/no properdin, no sC5b-9/properdin, sC5b-9/properdin. Log-rank tests showed that the prevalence of graft failure and survival were significantly higher in the patients with urinary properdin and sC5b-9. Associations between survival and urinary properdin and sC5b-9 did not remain significant after adjustment for potential confounders.
In unadjusted Cox regression analysis, detectable urinary properdin was significantly associated with development of death-censored graft failure (HR, 3.08; 95% CI 1.95–4.85; P < 0.001), in patients with neither urinary properdin or sC5b-9 as the reference group. In multivariable analyses, detectable urinary properdin remained associated with development of graft failure (HR, 2.30; 95% CI 1.37–3.82; P < 0.001, Table 2), independent of adjustment for age, sex, primary renal disease, time since transplantation, eGFR, HLA mismatches, donor type, hs-CRP, systolic blood pressure, and smoking. However, the association between detectable properdin and graft failure became borderline significant after further adjustment for proteinuria (HR, 1.47; 95% CI 0.85–2.54; P = 0.05).
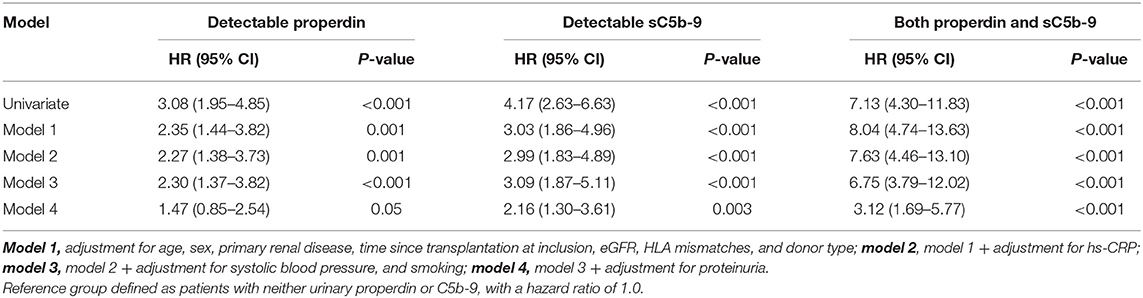
Table 2. Association of detectable urinary properdin and detectable urinary sC5b-9 with graft failure in renal transplant recipients.
When we assessed the association between properdin as a continuous variable and graft failure, findings were similar. After adjustment for potential confounders, urinary properdin as a continuous variable was significantly associated with graft failure (HR, 1.25; 95% CI 1.10–1.42; P < 0.001) (Table 3). After adjustment for proteinuria, the association of properdin as a continuous variable with graft failure remained significant (HR, 1.12; 95% CI 1.02–1.38; P = 0.008). The optimal cut-off (Youden index) of urinary properdin for prediction of graft failure was 2.35 ng/mL. At this cut-off value, there was a sensitivity of 59% and a specificity of 79% for prediction of graft failure.
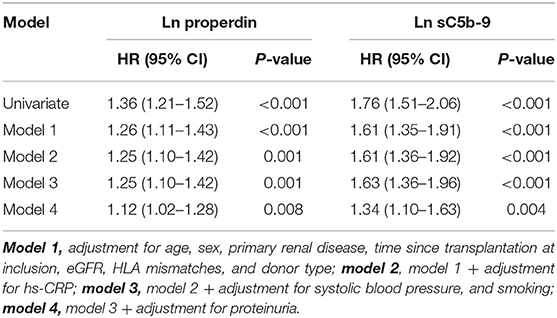
Table 3. Association of continuous natural log transformed urinary properdin and urinary sC5b-9 with graft failure in renal transplant recipients.
In unadjusted analysis, detectable urinary sC5b-9 was significantly associated with development of death-censored graft failure (HR, 4.17; 95% CI 2.63–6.63; P < 0.001). In multivariable analyses, detectable sC5b-9 remained associated with the development of graft failure (HR, 3.09; 95% CI 1.87–5.11; P < 0.001), independent of age, sex, primary renal disease, time since transplantation, eGFR, HLA mismatches, donor type, hs-CRP, systolic blood pressure, and smoking (Table 2). The association between detectable sC5b-9 and graft failure also remained after further adjustment for proteinuria (HR, 2.16; 95% CI 1.30–3.61; P = 0.003).
When we assessed the association between sC5b-9 as a continuous variable and graft failure, findings were similar. sC5b-9 as a continuous variable was associated with risk of developing graft failure in the unadjusted analysis and in multivariable analyses, after adjustment for potential confounders, including proteinuria (HR, 1.34; 95% CI 1.10–1.63; P = 0.004) (Table 3). The optimal cut-off (Youden index) of urinary sC5b-9 for prediction of graft failure was 2.88 ng/mL, there was a sensitivity of 48% and a specificity of 91% for prediction of graft failure.
Since properdin is involved in sC5b-9 complex formation via the alternative complement pathway (9), we aimed to assess whether the association between properdin and graft failure was mediated by sC5b-9. In mediation analyses, sC5b-9 was found to be a significant mediator of the association between properdin and graft failure, 31% of the association between properdin and graft failure was explained by sC5b-9, the P value for indirect effect is <0.05 (Table 4).
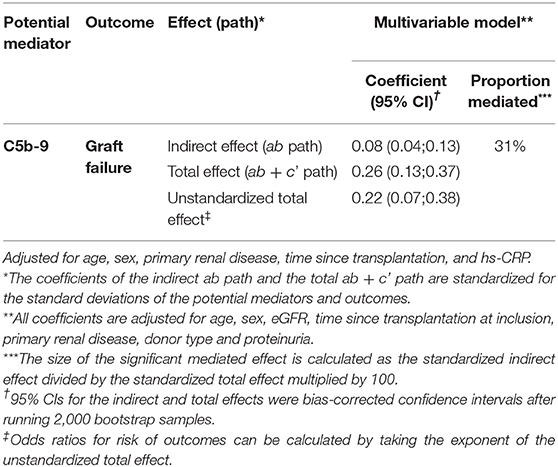
Table 4. Mediation analyses of the impact of sC5b-9 on the association between properdin and graft failure.
In an unadjusted Cox regression analysis, detectable urinary properdin was significantly associated with an increased risk of mortality (HR, 1.58; 95% CI 1.11–2.25; P = 0.01), whereas detectable urinary sC5b-9 was not significantly associated with increased risk of mortality(HR, 1.39; 95% CI 0.92–2.11; P = 0.12). After adjustment for potential confounders, the association between properdin and mortality was abrogated, and the association between sC5b-9 and mortality remained non-significant (Supplementary Table 1).
In unadjusted Cox regression analyses, both urinary properdin and sC5b-9 as continuous variables were significantly associated with an increased risk of mortality (HR, 1.16; 95% CI 1.05–1.27; P = 0.003 and HR, 1.26; 95% CI 1.08–1.48; P = 0.004, respectively). However, after adjustment for potential confounders, the associations between properdin and mortality, and between sC5b-9 and mortality were lost (Supplementary Table 2).
In this study, we show that the alternative pathway complement factor properdin and the terminal sC5b-9 complex are detectable in the urine of patients after kidney transplantation and are independently associated with chronic allograft failure. To our knowledge, this is the first report indicating urinary alternative complement pathway involvement in chronic renal allograft failure, independent of potential cofounders including eGFR and proteinuria.
It has long been recognized that patients with high-grade proteinuria are more likely to develop chronic renal failure than patients without proteinuria (23, 24). Urinary proteins elicit pro-inflammatory and pro-fibrotic effects that directly contribute to chronic tubulo-interstitial damage. Additionally, among multiple other pathways complement activation may be an important component leading to fibrogenesis in the kidney. In physiological conditions complement components are not filtered through the glomerular barrier, however complement components are present in the urine of patients with non-selective proteinuria (25, 26). Properdin positively regulates the AP of the complement system and is also a pattern recognition molecule for C3b that subsequently stabilizes the C3bBb complex and thus contributes to C5b-9 formation (27–30). Tubular epithelial cells are especially susceptible to the effects of C5b-9 formation because they lack the membrane-bound complement regulators on the apical cell surface (31). Recently, urinary complement measurements and their clinical value are of increasing interest in transplant medicine. Schröppel et al. showed not long ago the importance of anaphylatoxins C3a and C5a in donor urine and their association with delayed graft function (32), and van Essen et al. recently reviewed the detection of complement biomarkers in urine to monitor local injury in renal diseases, including properdin (33).
In this study, we have shown a potential role of urinary properdin and sC5b-9 in the pathogenesis of chronic allograft failure. Our data show that graft survival is reduced in patients in whom properdin is present in the urine together with sC5b-9. Remarkably, in patients without overt proteinuria, we identified that properdin, sC5b-9 or both properdin and sC5b-9, were also associated with a worse graft survival. More importantly, not only the presence of properdin and sC5b-9 was significantly associated with graft failure, but also properdin and sC5b-9 were robustly associated with graft survival when analyzed as continuous parameters, pointing toward a dose-dependent effect. There are several possible explanations for this association. Properdin is the only known complement protein that is not produced in the liver, but synthesized by various other cell types like monocytes, primary T cells, granulocytes, and endothelial cells (34–38). Therefore, it is possible that locally produced properdin and/or filtered properdin with other filtered small complement components, causes intratubular C5b-9 activation leading to progressive renal disease without manifest proteinuria, defined as proteinuria >0.5 g/24 h (39, 40). In kidney transplant patients it is generally believed that small amounts of proteinuria, defined as <0.5 g/24 h, are harmless (41, 42). Only persistent proteinuria, >0.5 g/24 h for at least 3–6 months is considered significant according to American Society of Transplantation guidelines, and low-grade proteinuria is often referred to as “subclinical” (43). However, low grade proteinuria may be less harmless than originally described. Halimi et al. showed a dose-dependent effect in transplant patients with low grade proteinuria (<0.5 g/24 h) in whom each 0.1 g/24 h difference in proteinuria increased the risk of graft loss by 25% (44). In line with our findings in transplanted patients, Siezenga et al. showed an association between urinary properdin and worse renal function in patients with diabetic nephropathy or glomerular disease. Furthermore, the association of urinary properdin with urinary sC5b-9 was independent of the degree of proteinuria (17).
The fact that adjustment for proteinuria > 0.5 g/24 h did not materially alter the prospective association in the prospective analysis of continuous properdin and sC5b-9 measurements, is supportive of our hypothesis that alternative pathway complement activation might be one of the driving forces of chronic graft failure. More importantly, after correction for other well-known predictors of graft failure, like HLA mismatches and donor type, the association remains. In mediation analysis, we showed that the association between properdin and graft failure was mediated to a considerable extent by urinary sC5b-9.
This may explain why RTR with properdin alone, or sC5b-9 alone in the urine have a better allograft survival compared to both properdin and sC5b9 in the urine. Therefore, it seems that AP complement activation plays an important role in the loss of allograft function of RTR. The possible mechanism of this effect at a tubular level is illustrated by the scheme presented in Figure 4. Interestingly, urinary properdin was more frequently detected in females and urinary sC5b-9 was more frequently detected in males. We can only speculate on the causes of these differences. Innate immune function may vary between males and females (45), however a limited number of studies have investigated the influence of sex on the complement system (46–49). Properdin is encoded on the short arm of the X chromosome, and together with hormonal differences between males and females this could be explanations of the sexual differences in properdin (50, 51). However, in a healthy Caucasian population, Gaya da Costa et al. recently found decreased serum properdin and serum C9 in healthy human females compared to males (49). In contrast, animal studies have shown that female mice have a similar serum complement cascade functionality at the level of C3 activation compared to male mice, but a strongly reduced level of serum C9, leading to an inability of female mice to promote inflammation through C5b-9 (46).
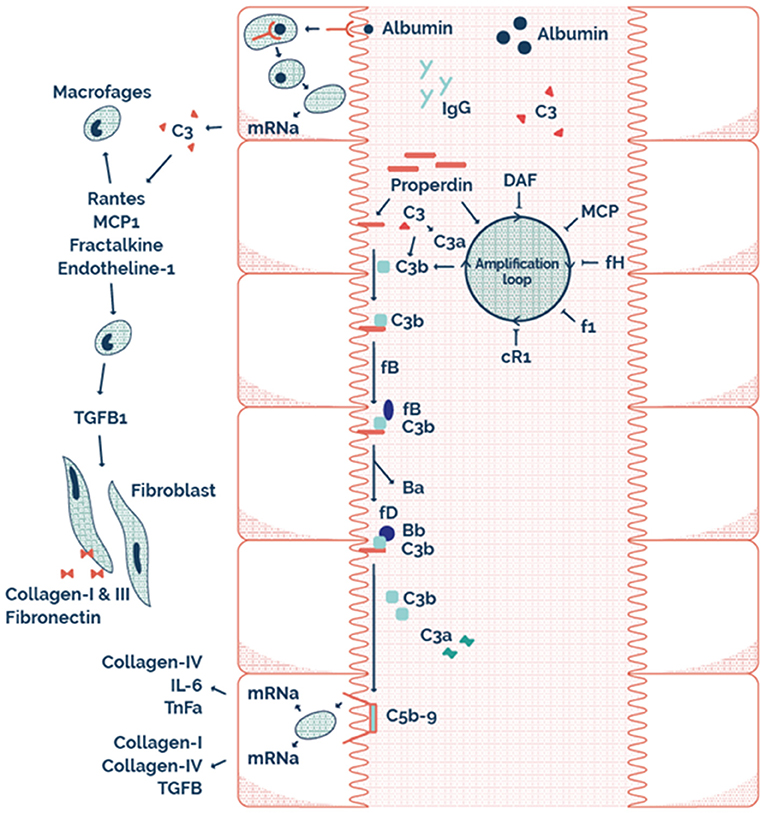
Figure 4. The possible mechanism on a tubular level illustrating tubular alternative pathway complement activation via properdin as pattern recognition molecule.
Multiple therapeutic modalities to inhibit complement pathway intervention are currently being developed. Our study points toward the potential for complement inhibition at the tubular level in proteinuric patients, which may improve long term outcome in patients with chronic allograft nephropathy.
The main strength of our study is that it comprises a large prospective cohort of stable RTR, in which several renal parameters as well as both urinary properdin and sC5b-9 were measured. In addition, end-point evaluation was complete in all participants despite the long follow-up period. We acknowledge several limitations of the study. First, no gold standard exists for the definition of urinary properdin and sC5b-9. In our study, we defined the detectability of properdin and sC5b-9 as urinary properdin and urinary sC5b-9. Second, complement activation may only be partially reflected by urinary properdin and sC5b-9 excretion, since the excretion may be altered by tubular complement binding and fixation. Third, possible residual confounding in this study cannot be excluded due to the observational status of this single center study. Furthermore, we do not have data on the presence of donor specific antibodies or protocol biopsies in this cohort. Thus, we cannot differentiate between general effects of glomerular filtration of complement products and a specific contribution of alloantibody mediated complement activation. Unfortunately our prospective cohort contained too few events of graft failure to perform analysis for the underlying cause of graft loss and their relation to urinary complement.
We identified that the presence of urinary properdin and sC5b-9 is independently associated with increased risk of late graft failure in RTR, compared to RTR without urinary properdin and sC5b-9. This suggests that urinary properdin and sC5b-9 can serve as useful biomarkers of immunological injury and kidney allograft deterioration. Importantly, urinary properdin and sC5b-9 was associated with graft failure independently of eGFR and significant proteinuria. We suggest that an important part of proteinuria mediated toxicity, is caused by the presence of complement in the primary urine and subsequent activation at the tubular surface. Further studies are needed to unravel the exact interplay between urinary properdin, sC5b-9 and the development of fibrosis, and moreover the potential for therapeutic interventions.
The datasets generated for this study are available on request to the corresponding author.
The study was approved by the University Medical Center Groningen institutional review board (METc 2008/186), adheres to the Declarations of Helsinki and Istanbul and has NCT02811835 as ClinicalTrials.gov identifier. The patients/participants provided their written informed consent to participate in this study.
RL was involved in study design, carrying out assays, interpreting data, statistical analysis, creating tables and figures, and writing of the manuscript. ME was involved in interpreting data, statistical analysis, figure and table design, and manuscript editing. MA was involved in carrying out assays and interpreting data. MD, MS, RP, JB, and J-SS were all involved in interpreting data and manuscript editing. SB and SPB were involved in study design, interpreting data, statistical analysis, and manuscript editing.
This study was supported by the Dutch Kidney Foundation (Grant 13OCA27).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We thank H. M. van der Lugt for her contribution for the preparation of this manuscript, H. L. Lancaster for critical and careful reading of the manuscript, and J. Potma for assistance in preparing Figure 4.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2019.02511/full#supplementary-material
1. Gaston RS, Fieberg A, Hunsicker L, Kasiske BL, Leduc R, Cosio FG, et al. Late graft failure after kidney transplantation as the consequence of late versus early events. Am J Transplant. (2018) 18:1158–67. doi: 10.1111/ajt.14590
2. Nankivell BJ, Borrows RJ, Fung CL, O'Connell PJ, Allen RD, Chapman JR. The natural history of chronic allograft nephropathy. N Engl J Med. (2003) 349:2326–33. doi: 10.1056/NEJMoa020009
3. Solez K, Axelsen RA, Benediktsson H, Burdick JF, Cohen AH, Colvin RB, et al. International standardization of criteria for the histologic diagnosis of renal allograft rejection: the Banff working classification of kidney transplant pathology. Kidney Int. (1993) 44:411–22. doi: 10.1038/ki.1993.259
4. Nankivell BJ, Shingde M, Keung KL, Fung CL, Borrows RJ, O'Connell PJ, et al. The causes, significance and consequences of inflammatory fibrosis in kidney transplantation: the Banff i-IFTA lesion. Am J Transplant. (2018) 18:364–76. doi: 10.1111/ajt.14609
5. Hallan SI, Ritz E, Lydersen S, Romundstad S, Kvenild K, Orth SR. Combining GFR and albuminuria to classify CKD improves prediction of ESRD. J Am Soc Nephrol. (2009) 20:1069–77. doi: 10.1681/ASN.2008070730
6. Nauta FL, Bakker SJ, van Oeveren W, Navis G, van der Heide JJ, van Goor H, et al. Albuminuria, proteinuria, and novel urine biomarkers as predictors of long-term allograft outcomes in kidney transplant recipients. Am J Kidney Dis. (2011) 57:733–43. doi: 10.1053/j.ajkd.2010.12.022
7. Khan S, Cabral PD, Schilling WP, Schmidt ZW, Uddin AN, Gingras A, et al. Kidney proximal tubule lipoapoptosis is regulated by fatty acid transporter-2 (FATP2). J Am Soc Nephrol. (2018) 29:81–91. doi: 10.1681/ASN.2017030314
8. Ishola DA, Post JA, van Timmeren MM, Bakker SJ, Goldschmeding R, Koomans HA, et al. Albumin-bound fatty acids induce mitochondrial oxidant stress and impair antioxidant responses in proximal tubular cells. Kidney Int. (2006) 70:724–31. doi: 10.1038/sj.ki.5001629
9. Zaferani A, Vivès RR, van der Pol P, Hakvoort JJ, Navis GJ, van Goor H, et al. Identification of tubular heparan sulfate as a docking platform for the alternative complement component properdin in proteinuric renal disease. J Biol Chem. (2011) 286:5359–67. doi: 10.1074/jbc.M110.167825
10. Tang S, Lai KN, Sacks SH. Role of complement in tubulointerstitial injury from proteinuria. Kidney Blood Press Res. (2002) 25:120–6. doi: 10.1159/000063520
12. He C, Imai M, Song H, Quigg RJ, Tomlinson S. Complement inhibitors targeted to the proximal tubule prevent injury in experimental nephrotic syndrome and demonstrate a key role for C5b-9. J Immunol. (2005) 174:5750–7. doi: 10.4049/jimmunol.174.9.5750
13. Nangaku M, Pippin J, Couser WG. C6 mediates chronic progression of tubulointerstitial damage in rats with remnant kidneys. J Am Soc Nephrol. (2002) 13:928–36.
14. Biancone L, David S, Della Pietra V, Montrucchio G, Cambi V, Camussi G. Alternative pathway activation of complement by cultured human proximal tubular epithelial cells. Kidney Int. (1994) 45:451–60. doi: 10.1038/ki.1994.59
15. Gaarkeuken H, Siezenga MA, Zuidwijk K, van Kooten C, Rabelink TJ, Daha MR, et al. Complement activation by tubular cells is mediated by properdin binding. Am J Physiol Renal Physiol. (2008) 295:F1397–403. doi: 10.1152/ajprenal.90313.2008
16. Gou SJ, Yuan J, Wang C, Zhao MH, Chen M. Alternative complement pathway activation products in urine and kidneys of patients with ANCA-associated GN. Clin J Am Soc Nephrol. (2013) 8:1884–91. doi: 10.2215/CJN.02790313
17. Siezenga MA, Van Der Geest RN, Mallat MJK, Rabelink TJ, Daha MR, Berger SP. Urinary properdin excretion is associated with intrarenal complement activation and poor renal function. Nephrol Dial Transplant. (2010) 25:1157–61. doi: 10.1093/ndt/gfp630
18. Van Den Berg E, Engberink MF, Brink EJ, van Baak MA, Gans RO, Navis G, et al. Dietary protein, blood pressure and renal function in renal transplant recipients. Br J Nutr. (2013) 109:1463–70. doi: 10.1017/S0007114512003455
19. Levey AS, Stevens LA, Schmid CH, Zhang YL, Castro AF, Feldman HI, et al. A new equation to estimate glomerular filtration rate. Ann Intern Med. (2009) 150:604–12. doi: 10.7326/0003-4819-150-9-200905050-00006
20. Seelen MA, Roos A, Wieslander J, Mollnes TE, Sjöholm AG, Wurzner R, et al. Functional analysis of the classical, alternative, and MBL pathways of the complement system: standardization and validation of a simple ELISA. J Immunol Methods. (2005) 296:187–98. doi: 10.1016/j.jim.2004.11.016
21. Preacher K, Hayes A. SPSS and SAS procedures for estimating indirect effects in simple mediation models. Behav Res Methods Instrum Comput. (2004) 36:717–31. doi: 10.3758/BF03206553
22. Hayes AF. Beyond Baron and Kenny: statistical mediation analysis in the new millennium. Commun Monogr. (2009) 76:408–20. doi: 10.1080/03637750903310360
23. D'Amico G. Natural history of idiopathic IgA nephropathy: role of clinical and histological prognostic factors. Am J Kidney Dis. (2000) 36:227–37. doi: 10.1053/ajkd.2000.8966
24. Abbate M, Zoja C, Remuzzi G. How does proteinuria cause progressive renal damage? J Am Soc Nephrol. (2006) 17:2974–84. doi: 10.1681/ASN.2006040377
25. Nangaku M, Pippin J, Couser WG. Complement membrane attack complex (C5b-9) mediates interstitial disease in experimental nephrotic syndrome. J Am Soc Nephrol. (1999) 10:2323–31.
26. Spitzer D, Mitchell LM, Atkinson JP, Hourcade DE. Properdin can initiate complement activation by binding specific target surfaces and providing a platform for de novo convertase assembly. J Immunol. (2007) 179:2600–8. doi: 10.4049/jimmunol.179.4.2600
27. Kemper C, Mitchell LM, Zhang L, Hourcade DE. The complement protein properdin binds apoptotic T cells and promotes complement activation and phagocytosis. Proc Natl Acad Sci USA. (2008) 105:9023–8. doi: 10.1073/pnas.0801015105
28. Austen KF, Fearon DT. Properdin: binding to C3b and stabilization of the C3b-dependent C3 convertase. J Exp Med. (1975) 142:856. doi: 10.1084/jem.142.4.856
29. Medicus RG, Götze O, Müller-Eberhard HJ. Alternative pathway of complement: recruitment of precursor properdin by the labile C3/C5 convertase and the potentiation of the pathway. J Exp Med. (1976) 144:1076–93. doi: 10.1084/jem.144.4.1076
30. Xu W, Berger SP, Trouw LA, de Boer HC, Schlagwein N, Mutsaers C, et al. Properdin binds to late apoptotic and necrotic cells independently of C3b and regulates alternative pathway complement activation. J Immunol. (2008) 180:7613–21. doi: 10.4049/jimmunol.180.11.7613
31. Nangaku M. Complement regulatory proteins in glomerular diseases. Kidney Int. (1998) 54:1419–28. doi: 10.1046/j.1523-1755.1998.00130.x
32. Schröppel B, Heeger PS, Thiessen-Philbrook H, Hall IE, Doshi MD, Weng FL, et al. Donor urinary C5a levels independently correlate with post-transplant delayed graft function. Transplantation. (2018) 103:e29–35. doi: 10.1097/TP.0000000000002494
33. van Essen MF, Ruben JM, de Vries APJ, van Kooten C. Role of properdin in complement-mediated kidney diseases. Nephrol Dial Transplant. (2019) 34:742–50. doi: 10.1093/ndt/gfy233
34. Cortes C, Ohtola JA, Saggu G, Ferreira VP. Local release of properdin in the cellular microenvironment: role in pattern recognition and amplification of the alternative pathway of complement. Front Immunol. (2013) 3:412. doi: 10.3389/fimmu.2012.00412
35. Schwaeble W, Huemer HP, Möst J, Dierich MP, Ströbel M, Claus C, et al. Expression of properdin in human monocytes. Eur J Biochem. (1994) 219:759–64. doi: 10.1111/j.1432-1033.1994.tb18555.x
36. Nolan KF, Reid KBM. [2] Properdin. Methods Enzymol. (1993) 223:35–46. doi: 10.1016/0076-6879(93)23036-M
37. Pangburn MK. Analysis of the natural polymeric forms of human properdin and their functions in complement activation. J Immunol. (1989) 142:202–7.
38. Wirthmueller U, Dewald B, Thelen M, Schäfer MK, Stover C, Whaley K, et al. Properdin, a positive regulator of complement activation, is released from secondary granules of stimulated peripheral blood neutrophils. J Immunol. (1997) 158:4444–51.
39. Hadders MA, Beringer DX, Gros P. Structure of C8 -MACPF reveals mechanism of membrane attack in complement immune defense. Science. (2007) 317:1552–4. doi: 10.1126/science.1147103
40. Hobart MJ, Fernie BA, DiScipio RG. Structure of the human C7 gene and comparison with the C6, C8A, C8B, and C9 genes. J Immunol. (1995) 154:5188–94.
41. Yildiz A, Erkoç R, Sever MS, Türkmen A, Ecder ST, Türk S, et al. The prognostic importance of severity and type of post-transplant proteinuria. Clin Transplant. (1999) 13:241–4. doi: 10.1034/j.1399-0012.1999.130304.x
42. D'Cunha PT, Parasuraman R, Venkat KK. Rapid resolution of proteinuria of native kidney origin following live donor renal transplantation. Am J Transplant. (2005) 5:351–5. doi: 10.1111/j.1600-6143.2004.00665.x
43. Kasiske BL, Vazquez MA, Harmon WE, Brown RS, Danovitch GM, Gaston RS, et al. Recommendations for the outpatient surveillance of renal transplant recipients. Am Soc Transplant. (2000) 11 (Suppl 1):S1–86.
44. Halimi JM, Laouad I, Buchler M, Al-Najjar A, Chatelet V, Houssaini TS, et al. Early low-grade proteinuria: causes, short-term evolution and long-term consequences in renal transplantation. Am J Transplant. (2005) 5:2281–8. doi: 10.1111/j.1600-6143.2005.01020.x
45. Libert C, Dejager L, Pinheiro I. The X chromosome in immune functions: when a chromosome makes the difference. Nat Rev Immunol. (2010) 10:594–604. doi: 10.1038/nri2815
46. Kotimaa J, Klar-Mohammad N, Gueler F, Schilders G, Jansen A, Rutjes H, et al. Sex matters: systemic complement activity of female C57BL/6J and BALB/cJ mice is limited by serum terminal pathway components. Mol Immunol. (2016) 76:13–21. doi: 10.1016/j.molimm.2016.06.004
47. Weintraub RM, Churchill WHJ, Crisler C, Rapp HJ, Borsos T. Mouse complement: influence of sex hormones on its activity. Science. (1966) 152:783–5. doi: 10.1126/science.152.3723.783
48. Roach B, Kim Y, Jerome E, Michael AF. Influence of age and sex on serum complement components in children. Am J Dis Child. (1981) 135:918–20. doi: 10.1001/archpedi.1981.02130340030011
49. Gaya da Costa M, Poppelaars F, van Kooten C, Mollnes TE, Tedesco F, Würzner R, et al. Age and sex-associated changes of complement activity and complement levels in a healthy caucasian population. Front Immunol. (2018) 9:2664. doi: 10.3389/fimmu.2018.02664
50. Coleman MP, Murray JC, Willard HF, Nolan KF, Reid KB, Blake DJ, et al. Genetic and physical mapping around the properdin P gene. Genomics. (1991) 11:991–6. doi: 10.1016/0888-7543(91)90024-9
Keywords: transplantation, chronic renal failure, properdin, C5b-9, complement activation
Citation: Lammerts RGM, Eisenga MF, Alyami M, Daha MR, Seelen MA, Pol RA, van den Born J, Sanders J-S, Bakker SJL and Berger SP (2019) Urinary Properdin and sC5b-9 Are Independently Associated With Increased Risk for Graft Failure in Renal Transplant Recipients. Front. Immunol. 10:2511. doi: 10.3389/fimmu.2019.02511
Received: 12 August 2019; Accepted: 08 October 2019;
Published: 24 October 2019.
Edited by:
Guseppe Grandaliano, University of Foggia, ItalyReviewed by:
Paolo Cravedi, Icahn School of Medicine at Mount Sinai, United StatesCopyright © 2019 Lammerts, Eisenga, Alyami, Daha, Seelen, Pol, van den Born, Sanders, Bakker and Berger. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Rosa G. M. Lammerts, ci5nLm0ubGFtbWVydHNAdW1jZy5ubA==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.