 Charlène Niogret
Charlène Niogret Walter Birchmeier2
Walter Birchmeier2 Greta Guarda
Greta Guarda- 1Department of Biochemistry, University of Lausanne, Épalinges, Switzerland
- 2Max-Delbrueck-Center for Molecular Medicine (MDC) in the Helmholtz Society, Berlin, Germany
- 3Institute for Research in Biomedicine, Università della Svizzera italiana, Bellinzona, Switzerland
Somewhat counterintuitively, the tyrosine phosphatase SHP-2 (SH2 domain-containing protein tyrosine phosphatase-2) is crucial for the activation of extracellular signal-regulated kinase (ERK) downstream of various growth factor receptors, thereby exerting essential developmental functions. This phosphatase also deploys proto-oncogenic functions and specific inhibitors have recently been developed. With respect to the immune system, the role of SHP-2 in the signaling of cytokines relevant for myelopoiesis and myeloid malignancies has been intensively studied. The function of this phosphatase downstream of cytokines important for lymphocytes is less understood, though multiple lines of evidence suggest its importance. In addition, SHP-2 has been proposed to mediate the suppressive effects of inhibitory receptors (IRs) that sustain a dysfunctional state in anticancer T cells. Molecules involved in IR signaling are of potential pharmaceutical interest as blockade of these inhibitory circuits leads to remarkable clinical benefit. Here, we discuss the dichotomy in the functions ascribed to SHP-2 downstream of cytokine receptors and IRs, with a focus on T and NK lymphocytes. Further, we highlight the importance of broadening our understanding of SHP-2′s relevance in lymphocytes, an essential step to inform on side effects and unanticipated benefits of its therapeutic blockade.
Introduction
Protein phosphorylation is a post-translational modification fundamental for intracellular signaling cascades and is therefore tightly regulated by kinases and phosphatases. SHP-2 (SH2 domain-containing protein tyrosine phosphatase-2, encoded by the PTPN11 gene) is a broadly expressed, cytoplasmic phosphatase highly relevant for human health (1–4). In fact, PTPN11 mutations cause the polymalformative Noonan and LEOPARD syndromes, two developmental disorders characterized by manifestations such as craniofacial abnormalities, growth defects, cardiac malformations, and—in some cases—mental retardation (5, 6). To understand the biological function of SHP-2, genetic mouse models have been generated. Full-body deletion of Shp-2 resulted in embryonic lethality due to multiple defects in mesoderm patterning (7), whereas inducible Shp-2 deletion in adult mice led to death within 6–8 weeks and was accompanied by bone marrow aplasia and anemia (8). Further, conditional Shp-2 deletion revealed the role of this phosphatase in the development of various organs and tissues, including in the nervous system, the heart, the mammary gland, the kidney, and the intestine (8–14). In most instances, the effects of SHP-2 have been ascribed to its positive function in regulating extracellular signal-regulated kinase (ERK) signaling downstream of a number of growth factor receptors (1–4). Overactivation of SHP-2 is also involved in multiple cancers, a notion that encouraged the development of small molecule inhibitors (2, 15–20). As discussed later, SHP-2 blockade markedly suppressed cancer growth in preclinical models and specific inhibitors are currently tested in clinical studies (19, 21–26).
In this review, we focus on the role of SHP-2 in T and natural killer (NK) lymphocytes, which are crucial players in immunity and in anticancer immunotherapy. Regrettably, the role of SHP-2 in these immune subsets remains incompletely understood. Whereas, SHP-2's function in activating ERK downstream of multiple growth factors has been firmly established, it is less well-characterized downstream of cytokines relevant for lymphoid cells. Further, a role for this phosphatase in “immune checkpoint” signaling cascades has been reported. Here, we discuss recent advances in the understanding of how SHP-2 shapes these pathways and highlight open questions that—with the advent of inhibitors for clinical use—are becoming increasingly pressing.
Molecular Function of SHP-2
SHP-2 possesses two N-terminal SH2 domains (N-SH2 and C-SH2) and a central protein tyrosine phosphatase (PTP) core (Figure 1) (3, 4, 27–30). The PTP domain is highly conserved among classical PTP phosphatases and is responsible for the catalytic activity of these enzymes. It is characterized by the [I/V]HCSXGXGR[S/T] sequence, with the invariant cysteine being responsible for the nucleophilic attack of the phosphate group to be removed (31, 32). The C-terminal tail of SHP-2 contains tyrosine residues that can become phosphorylated and modulate the phosphatase activity (3).
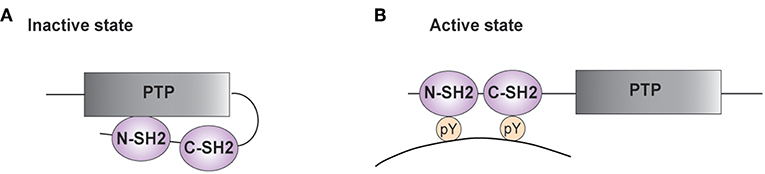
Figure 1. Structure of SHP-2. (A,B) A schematic representation of the phosphatase SHP-2 (SH2 domain-containing protein tyrosine phosphatase-2) is illustrated. The functional domains of SHP-2 comprise two SH2 domains [N-terminal SH2 (N-SH2) and C-terminal SH2 (C-SH2)] and a protein tyrosine phosphatase (PTP) domain. (A) In the absence of a tyrosine-phosphorylated substrate, the N-SH2 domain interacts with the PTP domain and blocks the catalytic site. (B) Interaction of SH2 domains with tyrosine-phosphorylated (pY) residues on targets enables phosphatase activity.
In the inactive state, the N-SH2 domain interacts with the PTP region, limiting access of substrates into the active site (Figure 1A) (33–35). The auto-inhibition is relieved upon SH2 binding to phosphotyrosine residues on targets (Figure 1B). The importance of this autoinhibitory mechanism is confirmed by studies on the mutations of PTPN11 associated to LEOPARD and Noonan Syndromes. The latter genetic disorder is caused by PTPN11 gain of function mutations, whereas the clinically similar LEOPARD Syndrome is linked to mutations reducing the catalytic activity of SHP-2. Recent findings started unraveling this paradox, showing that mutations found in LEOPARD Syndrome, besides decreasing the phosphatase activity, affect the intramolecular interaction between the N-SH2 and the PTP domain, favoring the transition to its active conformation and producing a gain of function-like phenotype (36, 37).
Through the interaction of the SH2 domains with phosphotyrosine residues on targets, SHP-2 is recruited to various receptors, directly or indirectly through docking proteins such as Insulin Receptor Substrate 1 (IRS1) and GRB2-associated-binding protein 1 or 2 (GAB1/2) (Figure 2) (3, 38, 39). Upon recruitment, SHP-2 is found in a signaling complex comprising growth factor receptor-bound protein 2 (GRB2) and the associated Son of Sevenless (SOS) (38, 40–43). By promoting the conversion of RAS-bound GDP to GTP, SOS activates the mitogen-activated protein kinase (MAPK) pathway involving RAF-MEK (mitogen-activated protein kinase kinase or MAPKK)-ERK. The expression of a catalytically-inactive SHP-2 and the use of specific inhibitors demonstrated the importance of the phosphatase activity for ERK activation (16, 25, 44–46). Thus, SHP-2 is an atypical phosphatase involved in positively regulating intracellular signaling pathways through its catalytic function.
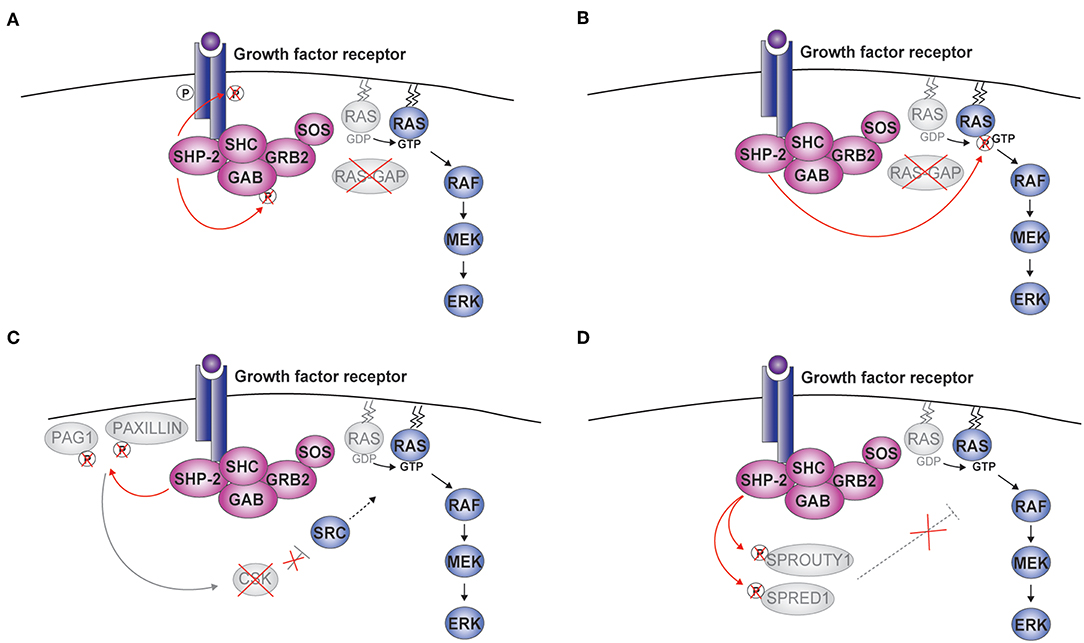
Figure 2. SHP-2-mediated activation of ERK. Upon cytokine binding, a complex including SHP-2, growth factor receptor-bound protein 2 (GRB2), and Son of Sevenless (SOS) is formed at the receptor. Four molecular mechanisms linking the phosphatase activity of SHP-2 to the activation of the RAS-RAF-MEK (mitogen-activated protein kinase kinase or MAPKK)-extracellular signal-regulated kinase (ERK) pathway are schematically illustrated (A–D). CSK, c-src tyrosine kinase; GAB, GRB2-associated-binding protein; PAG1, phosphoprotein associated with glycosphingolipid microdomains; RAS-GAP, RAS-GTPase activating protein; SHC, Src homology 2 domain containing; SPRED1, Sprouty-related ena/vasodilator-stimulated phosphoprotein homology 1 domain-containing protein1.
To explain how the phosphatase activity of SHP-2 stimulates the RAS-ERK pathway, five mechanisms have been proposed. First, SHP-2 was shown to dephosphorylate specific positions of the receptor (e.g., PDGFR) or GAB thus preventing the recruitment of the RAS-GTPase activating protein RAS-GAP (Figure 2A) (47–51). Opposite to SOS, RAS-GAP terminates the activation of the MAPK signaling pathway by inducing hydrolysis of RAS-bound GTP. Second, RAS tyrosine phosphorylation at position 32 negatively impacts on downstream signaling, possibly by favoring the interaction with RAS-GAP; by removing this modification, SHP-2 promotes ERK activation (Figure 2B) (52). Third, SHP-2 was found to eliminate phosphorylated docking sites on the scaffolding proteins Paxillin (PXN) and PAG1 (phosphoprotein associated with glycosphingolipid microdomains 1) (Figure 2C). These phosphorylation sites are involved in recruiting/modulating the activity of CSK (c-src tyrosine kinase), which suppresses receptor tyrosine kinase (RTK)-activated Src kinases and, indirectly, ERK signaling (53, 54). Fourth, Sprouty (SPRY) 1 and SPRED1 (Sprouty-related ena/vasodilator-stimulated phosphoprotein homology 1-domain-containing protein1) are known to inhibit ERK signaling and have been proposed to do so by multiple mechanisms acting at the level, downstream, or upstream of RAS (55). Interestingly, the function of SPRY1 and SPRED1 requires specific phosphorylations, which can be removed by SHP-2 (Figure 2D) (13, 56–58). Finally, two recent publications support a model whereby SHP-2's catalytic function is necessary for the assembly of the complex including SHP-2 itself, GAB, and GRB2 at the receptor. This model is attractive, as it suggests that the action of SHP-2 might involve more general mechanisms than interfering with specific inhibitory proteins. However, the underlying molecular events remain to be defined and might integrate the mechanisms described above (25, 59).
In addition to the ERK cascade, SHP-2 has been involved in the Phosphoinositide 3-kinase (PI3K)-AKT pathway. The adaptor GAB has been found to associate with SHP-2 and the PI3K p85 regulatory subunit, indirectly modulating PI3K signaling in response to selected cytokines (Figure 3) (38, 60–64). However, studies assessing PI3K/AKT activity or the phosphorylation of AKT at position 308, which is controlled by the PI3K-phosphoinositide-dependent kinase 1 (PDK1) axis (65), show negative as well as positive roles for SHP-2 on this pathway. For example, insulin- and epidermal growth factor (EGF)-dependent PI3K activation were found to be negatively influenced by SHP-2, most likely through the dephosphorylation of the p85 binding sites on the adaptor proteins GAB or IRS1 (64, 66–68). Conversely, SHP-2 interaction with p85 has been shown to be required for the association of the PI3K catalytic subunit p110 and for full PI3K activity downstream of insulin-like growth factor 1(IGF-1) (69). Similar effects were observed downstream of additional growth factors including insulin, PDGF, and granulocyte-macrophage colony-stimulating factor (GM-CSF) (63, 69–72). We therefore lack a unified view on the effects of SHP-2 on the PI3K pathway.
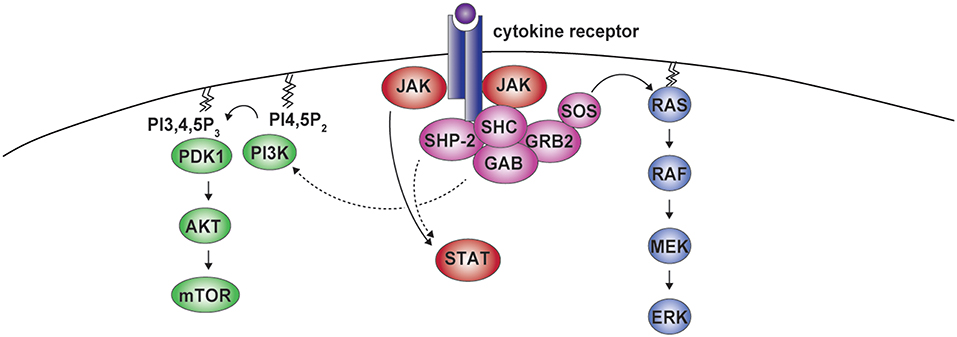
Figure 3. SHP-2 in cytokine receptor signaling. Cytokine binding to the receptor induces formation of the SHP-2-containing complex. Besides being involved in the activation of ERK, SHP-2 can modulate phosphoinositide 3-kinase (PI3K) activity. PI3K mediates the conversion of phosphatidylinositol 4,5-bisphosphate (PI4,5P2) into phosphatidylinositol 3,4,5-trisphosphate (PI3,4,5P3), which leads to phosphoinositide-dependent kinase 1 (PDK1) recruitment, AKT phosphorylation, and mammalian target of rapamycin (mTOR) activation. Residues phosphorylated by janus kinases (JAKs) in the cytoplasmic portion of the receptor act as binding sites for signal transducers and activators of transcription (STAT) proteins, that are further phosphorylated by JAKs, allowing dimerization and nuclear translocation. This pathway can also be modulated by SHP-2.
Along the same lines, SHP-2 has been reported to modulate the phosphorylation of signal transducers and activators of transcription (STAT) transcription factors downstream of various cytokines (Figure 3) (73). Upon engagement, cytokine receptors initiate signaling through Janus kinases (JAKs), which phosphorylate multiple residues in their cytoplasmic portions forming docking sites for STATs, that are themselves phosphorylated by JAKs to translocate to the nucleus and exert central transcriptional functions (73). On the one hand, SHP-2 was found to promote the dephosphorylation of different STATs, including downstream of interleukin (IL)-3, leukemia inhibitory factor (LIF), or IL-10 in cells of various origin (73–76). On the other hand, no effect or even the opposite outcome has been observed, as for instance in response to transforming growth factor-β (9, 73, 77–79). SHP-2 acts therefore downstream of several receptors to activate the ERK pathway and can modulate PI3K-AKT and JAK-STAT axes.
Small Molecular Weight Inhibitors of SHP-2 for Cancer Treatment
Several cancers rely on overactive MAPK signaling. Indeed, activating mutations of PTPN11 have been identified in juvenile myelomonocytic leukemia (JMML) (2, 20, 80–82). In many other cancer types, enhanced MAPK signaling is achieved through alternative mechanisms, such as alterations of RTKs like EGFR. Despite mutation of SHP-2 in tumors, particularly in solid ones, is an infrequent event, its key role in RTK-triggered signaling cascades renders it an attractive target for pharmacological intervention.
The identification of small molecule inhibitors for SHP-2 has however been a challenging endeavor, and no SHP-2 inhibitor has yet reached advanced stages of clinical trials (19, 83). A SHP-2 inhibitor, PHPS1, has been identified early and further developed into GS493 (17, 84). GS493 acts on purified SHP-2 in the nanomolar range, and was shown to inhibit breast cancer upon administration in mice (21). This and other SHP-2 inhibitors bind to or close to the active site of the enzyme. This straightforward approach is however potentially complicated by the high degree of homology across PTP catalytic domains, in particular with respect to Src Homology 2 (SH2) domain-containing tyrosine phosphatase 1 (SHP-1), the closest homolog of SHP-2 (85–87). More recently, inhibitors of SHP-2 have been reported, which act by new allosteric mechanisms (16, 25, 88). Two such compounds are SHP099, which stabilizes the inactive conformation of SHP-2 by occupying a tunnel-like binding site between the two SH2 and the PTP domain, and RMC-4550, which inhibits by a similar mode of action. SHP099 blocks SHP-2 in the nanomolar range, whereas RMC-4550 acts at even lower doses, with an IC50 of 0.58 nM. Both drugs were shown to limit the growth of xenografted cancers driven by oncogenic mutations of the RAF kinase and RAS member BRAF and KRAS, respectively (16, 22–26). Taken together, these data indicate that SHP-2 inhibition can be of use as a monotherapy.
However, cancer drug resistance is a massive clinical problem (89). Tumor cells often evade inhibition of proteins targeted by molecular therapies by re-activation of the signaling pathways via elaborate feedback mechanisms. This is the case for KRAS- or BRAF-driven cancers treated with MEK and BRAF inhibitors. The phosphatase SHP-2, being a crucial component in the signal transduction cascade between growth factor receptors and these downstream pathways, is an excellent potential target to battle drug resistance mediated by such cascades. This principle has been shown to work for BRAF inhibitor-resistant BRAF-mutant colon cancers (90). Treatment with BRAF inhibitor concomitant with genetic ablation or pharmacological inhibition of SHP-2 by the inhibitor GS493 prevented re-activation of MAPK signaling by feedback activation of the EGF receptor, inducing synthetic lethality of the transformed cells. In addition, in MEK inhibitor-resistant KRAS-mutant pancreatic, lung epithelial, and gastric cancer cell lines, simultaneous blocking of MEK and SHP2 acted synergistically, substantially hindering cell proliferation in vitro and tumor growth in xenograft models (23–26). Importantly, the combination treatment was well-tolerated, as evidenced by the similar body mass these mice maintained over time compared to vehicle-treated animals. Collectively, these works have provided proof-of-principle that small molecule inhibitors of SHP-2 can prevent resistance to MAPK pathway-targeting drugs in BRAF and KRAS mutant tumor cells. Together, these results establish SHP-2 blockade as a potentially powerful option to treat inhibitor refractory tumors in human patients.
Whereas SHP-2 is a central node in the commonly altered RTK/MAPK pathways, this phosphatase is mutated in few cancers, such as JMML (2). Nearly half of patients with SHP-2-mutated cancers bear strongly activating mutations that are thought to perturb its autoinhibited conformation, such as the common mutation of the position D61 and E76 in the N-SH2 domain. As the currently available allosteric inhibitors interact simultaneously with the C-SH2, N-SH2, and PTP domains, it is uncertain that successful suppression of such SHP-2 mutants is achievable in a clinical setting. This encourages further investigation to develop inhibitors targeting the catalytic site or the most common mutants, which might find broader application in patients with activating SHP-2 mutations (16, 25, 26, 91).
SHP-2 in Cytokines' Signaling in T and NK Lymphocytes
PTPN11 mutations found in JMML confer increased sensitivity to the growth factors GM-CSF and IL-3 (2, 15, 20, 92). These two cytokines share the β subunit of the receptor, which is common also to the receptor for IL-5, a cytokine important for the B cell and the eosinophil lineages. Upon cytokine stimulation, this receptor subunit recruits SHP-2, leading to the activation of the MAPK pathway and the interaction with the p85 subunit of PI3K (93–98). Lending support to the role of SHP-2 in these signaling cascades, a recent study demonstrated that Shp-2-deficient eosinophils failed to induce ERK activation upon IL-5 exposure, exhibiting reduced airway hyper-responsiveness in allergic models (99). Further to its role in pathways favoring myelogenous leukemias and normal myelopoiesis, SHP-2 is involved in the signaling by cytokines promoting hematopoiesis more broadly. Its function in the maintenance of hematopoietic stem cells and lineage progenitors has been attributed to the signaling downstream of multiple growth factors including stem cell factor (SCF), thrombopoietin (TPO), Fms-like tyrosine kinase 3 ligand (FLT3L), and interleukin (IL)-3 (8, 94, 95, 100–104). SHP-2 has also been implicated downstream of the receptors for cytokines important in mature immune cells, including lymphocytes (8).
Two decades ago, SHP-2 has been found to participate in the signaling induced by IL-6, a pleiotropic cytokine regulating inflammation, B cell responses, and T cell differentiation (105). An interaction between SHP-2 and the IL-6 receptor (IL-6R) subunit gp130 has been reported and mutation of the SHP-2 recruitment site suggested that this phosphatase was important to engage ERK and dampen STAT3 activation, limiting autoimmunity (106–113). Later studies showed that the same gp130 binding site recruited the JAK inhibitor Suppressor of cytokine signaling 3 (SOCS3), attributing to the latter the antagonism with STAT3, and confounding the role of SHP-2 (113, 114). These data indicate that, whereas SHP-2's function in activating the ERK pathway is widely accepted, the mechanisms underlying its effects on STAT activation shall be carefully evaluated. Therefore, the function of SHP-2 downstream of IL-6R and other less characterized gp130-containing receptors, such as the ones of IL-11, LIF, oncostatin M (OSM), and IL-27, which is of great relevance for T cells, await further experimental investigation.
SHP-2 has also been implicated in the response to IL-2 and IL-15 (60–62, 115–117). IL-2 is essential for regulatory, effector CD4+, and effector CD8+ T cells. IL-15 is important for the survival of memory CD8+ T cells and for development, survival, and activation of NK cells, two cytotoxic subsets which are central to immunity against intracellular pathogens and cancers. The receptors for IL-2 and IL-15 share the γc and the CD122 subunits (also known as IL-2 receptor β subunit). Phosphorylation of SHP-2, a phenomenon occurring upon receptor recruitment, was found to be largely dependent on the latter receptor subunit (62, 116). In agreement with what has been observed for other growth factor receptors, IL-2 and IL-15 stimulation led to the formation of a complex comprising SHP-2, GAB2, GRB2, and the PI3K p85 subunit (60, 61). Downstream of the IL-2R in T cells, SHP-2 has been involved in ERK engagement, while no or a positive effect was observed on STAT5 activation (79, 117, 118). Recently, we investigated the role of Shp-2 downstream of IL-15 stimulation in primary murine NK cells. While STAT5 phosphorylation was largely unaffected, Shp-2 was essential for ERK engagement (78). Interestingly, genetic ablation of Shp-2 also impaired phosphorylation of AKT (position 308), metabolic raise, and NK cell expansion in response to IL-15, suggesting a significant connection to cell metabolism (78). The family of γc-dependent cytokines comprises other members, which are instrumental for the lymphocytic compartment and have receptor subunits different from CD122. While stimulation with IL-4 and IL-7 did not induce phosphorylation of GAB2 or SHP-2 itself, IL-21 and the related thymic stromal lymphopoietin (TSLP) were shown to engage ERK and AKT and lead to phosphorylation of SHP-2 or other components typical of the SHP-2-containing complex (119–121). These results reveal therefore a role for SHP-2 in regulating the response to several cytokines and suggest a broader involvement, encouraging future studies.
SHP-2 and Inhibitory Receptor Signaling in T and NK Cells
SHP-2 is considered a central molecule downstream of inhibitory receptors (IRs). IRs are expressed by immune cells and regulate their function in diverse contexts. The cytoplasmic portion of IRs contains inhibitory motifs, such as immunoreceptor tyrosine-based inhibition motifs (ITIMs) and tyrosine-based switch motifs (ITSMs). Both motifs bear tyrosine residues that are phosphorylated upon IR engagement and recruit SH2 domain-containing phosphatases to antagonize activating cascades (122). During NK cell development, specific IRs interact with major histocompatibility complex (MHC) class I molecules, preventing a state of anergy (123–127). This process known as “NK cell education” mainly depends on SHP-1, the closest homolog of SHP-2. Biochemical evidence demonstrated SHP-1 recruitment to the ITIMs in the cytoplasmic portion of these IR, whereas elegant genetic approaches showed its essential function in maintaining NK cell responsiveness (126, 127). Notably, SHP-2 has also been shown to interact with NK cell IRs, suggesting a role in these suppressive signals (123, 124, 128). Through in vivo genetic approaches, we could however rule out a major role for this phosphatase in this pathway (78).
On T cells, transient IR expression is observed upon T cell receptor (TCR) triggering. Instead, constitutive IR display is associated and contributes to a dysfunctional state—known as “exhaustion”—that impairs T cell proliferative and effector capacities in cases of chronic antigen exposure (129–131). In particular, T cell exhaustion has been described in the context of chronic infections and cancer. Blockade of IR-mediated inhibitory circuits has recently transformed cancer immunotherapy, enabling to reactivate anti-tumoral T cell responses, and better control disease (131–133). Therefore, it is important to define the molecular events mediating IR effects, which might represent novel targets for pharmacological intervention.
Earlier studies showed interaction of SHP-2 with the intracellular tail of IRs and this correlated with the inhibition of T cell activation pathways (134–141). For instance, SHP-2 has been shown to interact with the cytoplasmic tail of the IR B- and T-lymphocyte attenuator (BTLA), whose blockade shows promise in immunogenic cancer treatment (142–144). Further, one of the most relevant IR is programmed cell death 1 (PD-1), whose blockade reinvigorates T cells against various cancer types. Its engagement has been shown to affect both TCR and co-stimulatory signaling (136, 141, 145). SHP-2 has been reported to robustly interact with the cytoplasmic tail of PD-1 and to exert a negative effect on interleukin (IL)-2 production, a surrogate read-out for TCR signaling (135, 138, 146, 147). This was observed in T cell hybridomas and in the Jurkat T cell line upon TCR and PD-1 engagement (138, 146). Of note, one of the effects of PD-1 engagement is the impairment of ERK activation (136, 138, 146, 148). The possibility that SHP-2 inhibits this cascade downstream of PD-1 is difficult to reconcile with its well-documented role in promoting it downstream of growth factor receptors. Moreover, despite the role of SHP-2 in TCR signaling remains controversial (79, 149–157), a positive effect on ERK engagement has been observed also in this context (149, 152, 153). The dichotomy in the effects of SHP-2 could be explained by a model in which IRs reduce the availability of the phosphatase, thus preventing its contribution to the ERK cascade, or by a very distinct regulation of SHP-2 activity downstream of IRs and growth factor receptors.
To evaluate whether the absence of SHP-2 reverted T cell exhaustion in more physiological conditions, we generated mice lacking this phosphatase in T cells. In the context of chronic viral infection, we found that antiviral Ptpn11-knockout T cells presented typical signs of exhaustion, exhibiting compromised cytokine production and tolerable immunopathology (156, 158, 159). In addition, immunogenic cancers developed in these mice with kinetics similar to the ones observed in the control groups (156). Along these lines, studies by others showed that the growth of immunogenic tumors in mice lacking Shp-2 in T cells was moderately retarded or accelerated, but even in the former case the effects on tumor growth were distant from the ones of PD-1-deficiency (154, 160, 161). One interpretation is that the therapeutic effects of PD-1 blockade are not largely mediated by T cells, a quite unlikely hypothesis in light of cytotoxic T cell depletion results (131, 162). With respect to this question, conditional PD-1 deletion will be informative. Most importantly, genetic deletion or pharmacological inhibition of Shp-2 did not prevent the therapeutic benefit of antibody-mediated PD-1 blockade (156, 161). These results challenge the possibility that IRs antagonize TCR and co-stimulatory signaling by reducing the availability of this phosphatase and imply that PD-1 signaling occurs in the absence of Shp-2 activity.
Intriguingly, SHP-2 has been shown to dephosphorylate the cytoplasmic tail of PD-1 as part of a feedback loop (138, 141), even suggesting a role in the termination of the inhibitory function of this IR and the possible accumulation of other SH-containing proteins and phosphatases in its absence. In addition to SHP-2, SHP-1 has been shown to interact with PD-1 and other IRs, albeit to a lesser extent (136, 137, 143, 144). Given their homology, a recent study explored the possibility that these two phosphatases exert redundant functions in PD-1 signaling. This work showed how only the abrogation of both phosphatases robustly relieved the inhibitory effects of PD-1 on TCR- and CD28-induced signaling, including ERK, in Jurkat T cells (Figure 4) (147). This important study paves the road to evaluate SHP-1 and SHP-2 redundancy in anticancer T cells in vivo.
Figure 4. SHP-2 and redundant mechanisms in T cell inhibitory receptor signaling. Recent in vitro data indicate that SHP-2 and SHP-1 are engaged by programmed cell death 1 (PD-1) and possibly other inhibitory receptors involved in T cell exhaustion. Notably, these two phosphatases exert redundant functions in limiting T cell receptor (TCR)/CD28 signaling and interleukin-2 (IL-2) production in Jurkat T cells.
Discussion
On the one hand, detailing IR signaling in exhausted T cells is of high clinical value. Yet, the lack of definite knowledge on the molecular events downstream of IRs delays the design of small molecule inhibitor-based interventions. Better understanding the mechanism of action of SHP-2 in these cascades is therefore relevant and timely. On the other hand, our understanding of SHP-2 function downstream of important growth factor receptors remains incomplete from a mechanistic viewpoint and in immune cells, lymphocytes in particular. Investigation in this direction would help answer the long-standing question of how a phosphatase enhances selected signaling and suggest novel targets for immunomodulation. Furthermore, currently available genetic models allow detailing the physiological contribution of SHP-2 in vivo with unprecedented accuracy. In the future, the study of tissue-specific and inducible knockout mice will be essential to define the immune subset-specific functions of SHP-2, while limiting the confounding effects of compensatory mechanisms.
Preclinical and clinical studies assessing the efficacy of SHP-2 inhibitors in cancer therapies raise the question on possible side effects, and immune cells shall be carefully examined in this respect. We deem that studies mapping SHP-2's functions are a prerequisite for evaluating these aspects, which will be highly relevant if immunotherapeutic approaches would be used in complement to SHP-2 inhibitors. Besides, these investigations might suggest unanticipated benefits of SHP-2 inhibitor therapies, as for instance in normalizing deregulated immune responses, such as in autoimmunity, and atopy.
Author Contributions
CN, WB, and GG wrote the manuscript.
Funding
Studies in the group of GG are funded by the Swiss National Science Foundation (PP00P3_165833 and 310030_185185), the European Research Council (ERC-2012-StG310890), the Novartis Foundation, the San Salvatore Foundation, and OM Pharma (Vifor Pharma).
Conflict of Interest
Unrelated projects in GG laboratory are supported by OM Pharma (Vifor Pharma) and Novartis Foundation. CN is now a clinical affairs specialist at Bio-Rad laboratories and works on unrelated projects.
The remaining author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank L. Brossay, Brown University, Providence, and Pedro Ventura, IRB, Bellinzona, for critical reading of the manuscript.
References
1. Neel BG, Gu H, Pao L. The 'Shp'ing news: SH2 domain-containing tyrosine phosphatases in cell signaling. Trends Biochem Sci. (2003) 28:284–93. doi: 10.1016/S0968-0004(03)00091-4
2. Grossmann KS, Rosario M, Birchmeier C, Birchmeier W. The tyrosine phosphatase Shp2 in development and cancer. Adv Cancer Res. (2010) 106:53–89. doi: 10.1016/S0065-230X(10)06002-1
3. Tajan M, de Rocca Serra A, Valet P, Edouard T, Yart A. SHP2 sails from physiology to pathology. Eur J Med Genet. (2015) 58:509–25. doi: 10.1016/j.ejmg.2015.08.005
4. Qu CK. The SHP-2 tyrosine phosphatase: signaling mechanisms and biological functions. Cell Res. (2000) 10:279–88. doi: 10.1038/sj.cr.7290055
5. Tartaglia M, Mehler EL, Goldberg R, Zampino G, Brunner HG, Kremer H, et al. Mutations in PTPN11, encoding the protein tyrosine phosphatase SHP-2, cause Noonan syndrome. Nat Genet. (2001) 29:465–8. doi: 10.1038/ng772
6. De Rocca Serra-Nedelec A, Edouard T, Treguer K, Tajan M, Araki T, Dance M, et al. Noonan syndrome-causing SHP2 mutants inhibit insulin-like growth factor 1 release via growth hormone-induced ERK hyperactivation, which contributes to short stature. Proc Natl Acad Sci USA. (2012) 109:4257–62. doi: 10.1073/pnas.1119803109
7. Saxton TM, Henkemeyer M, Gasca S, Shen R, Rossi DJ, Shalaby F, et al. Abnormal mesoderm patterning in mouse embryos mutant for the SH2 tyrosine phosphatase Shp-2. EMBO J. (1997) 16:2352–64. doi: 10.1093/emboj/16.9.2352
8. Chan G, Cheung LS, Yang W, Milyavsky M, Sanders AD, Gu S, et al. Essential role for Ptpn11 in survival of hematopoietic stem and progenitor cells. Blood. (2011) 117:4253–61. doi: 10.1182/blood-2010-11-319517
9. Ke Y, Lesperance J, Zhang EE, Bard-Chapeau EA, Oshima RG, Muller WJ, et al. Conditional deletion of Shp2 in the mammary gland leads to impaired lobulo-alveolar outgrowth and attenuated Stat5 activation. J Biol Chem. (2006) 281:34374–80. doi: 10.1074/jbc.M607325200
10. Ke Y, Zhang EE, Hagihara K, Wu D, Pang Y, Klein R, et al. Deletion of Shp2 in the brain leads to defective proliferation and differentiation in neural stem cells and early postnatal lethality. Mol Cell Biol. (2007) 27:6706–17. doi: 10.1128/MCB.01225-07
11. Kontaridis MI, Yang W, Bence KK, Cullen D, Wang B, Bodyak N, et al. Deletion of Ptpn11 (Shp2) in cardiomyocytes causes dilated cardiomyopathy via effects on the extracellular signal-regulated kinase/mitogen-activated protein kinase and RhoA signaling pathways. Circulation. (2008) 117:1423–35. doi: 10.1161/CIRCULATIONAHA.107.728865
12. Grossmann KS, Wende H, Paul FE, Cheret C, Garratt AN, Zurborg S, et al. The tyrosine phosphatase Shp2 (PTPN11) directs Neuregulin-1/ErbB signaling throughout Schwann cell development. Proc Natl Acad Sci USA. (2009) 106:16704–9. doi: 10.1073/pnas.0904336106
13. Willecke R, Heuberger J, Grossmann K, Michos O, Schmidt-Ott K, Walentin K, et al. The tyrosine phosphatase Shp2 acts downstream of GDNF/Ret in branching morphogenesis of the developing mouse kidney. Dev Biol. (2011) 360:310–7. doi: 10.1016/j.ydbio.2011.09.029
14. Heuberger J, Kosel F, Qi J, Grossmann KS, Rajewsky K, Birchmeier W. Shp2/MAPK signaling controls goblet/paneth cell fate decisions in the intestine. Proc Natl Acad Sci USA. (2014) 111:3472–7. doi: 10.1073/pnas.1309342111
15. Zhang J, Zhang F, Niu R. Functions of Shp2 in cancer. J Cell Mol Med. (2015) 19:2075–83. doi: 10.1111/jcmm.12618
16. Chen YN, LaMarche MJ, Chan HM, Fekkes P, Garcia-Fortanet J, Acker MG, et al. Allosteric inhibition of SHP2 phosphatase inhibits cancers driven by receptor tyrosine kinases. Nature. (2016) 535:148–52. doi: 10.1038/nature18621
17. Hellmuth K, Grosskopf S, Lum CT, Wurtele M, Roder N, von Kries JP, et al. Specific inhibitors of the protein tyrosine phosphatase Shp2 identified by high-throughput docking. Proc Natl Acad Sci USA. (2008) 105:7275–80. doi: 10.1073/pnas.0710468105
18. Chen L, Sung SS, Yip ML, Lawrence HR, Ren Y, Guida WC, et al. Discovery of a novel shp2 protein tyrosine phosphatase inhibitor. Mol Pharmacol. (2006) 70:562–70. doi: 10.1124/mol.106.025536
19. Mullard A. Phosphatases start shedding their stigma of undruggability. Nat Rev Drug Discov. (2018) 17:847–9. doi: 10.1038/nrd.2018.201
20. Chan G, Kalaitzidis D, Neel BG. The tyrosine phosphatase Shp2 (PTPN11) in cancer. Cancer Metastasis Rev. (2008) 27:179–92. doi: 10.1007/s10555-008-9126-y
21. Lan L, Holland JD, Qi J, Grosskopf S, Rademann J, Vogel R, et al. Shp2 signaling suppresses senescence in PyMT-induced mammary gland cancer in mice. EMBO J. (2015) 34:1493–508. doi: 10.15252/embj.201489004
22. Dardaei L, Wang HQ, Singh M, Fordjour P, Shaw KX, Yoda S, et al. SHP2 inhibition restores sensitivity in ALK-rearranged non-small-cell lung cancer resistant to ALK inhibitors. Nat Med. (2018) 24:512–7. doi: 10.1038/nm.4497
23. Wong GS, Zhou J, Liu JB, Wu Z, Xu X, Li T, et al. Targeting wild-type KRAS-amplified gastroesophageal cancer through combined MEK and SHP2 inhibition. Nat Med. (2018) 24:968–77. doi: 10.1038/s41591-018-0022-x
24. Mainardi S, Mulero-Sanchez A, Prahallad A, Germano G, Bosma A, Krimpenfort P, et al. SHP2 is required for growth of KRAS-mutant non-small-cell lung cancer in vivo. Nat Med. (2018) 24:961–7. doi: 10.1038/s41591-018-0023-9
25. Nichols RJ, Haderk F, Stahlhut C, Schulze CJ, Hemmati G, Wildes D, et al. RAS nucleotide cycling underlies the SHP2 phosphatase dependence of mutant BRAF-, NF1- and RAS-driven cancers. Nat Cell Biol. (2018) 20:1064–73. doi: 10.1038/s41556-018-0169-1
26. Ruess DA, Heynen GJ, Ciecielski KJ, Ai J, Berninger A, Kabacaoglu D, et al. Mutant KRAS-driven cancers depend on PTPN11/SHP2 phosphatase. Nat Med. (2018) 24:954–60. doi: 10.1038/s41591-018-0024-8
27. Freeman RM Jr, Plutzky J, Neel BG. Identification of a human src homology 2-containing protein-tyrosine-phosphatase: a putative homolog of Drosophila corkscrew. Proc Natl Acad Sci USA. (1992) 89:11239–43. doi: 10.1073/pnas.89.23.11239
28. Ahmad S, Banville D, Zhao Z, Fischer EH, Shen SH. A widely expressed human protein-tyrosine phosphatase containing src homology 2 domains. Proc Natl Acad Sci USA. (1993) 90:2197–201. doi: 10.1073/pnas.90.6.2197
29. Feng GS, Hui CC, Pawson T. SH2-containing phosphotyrosine phosphatase as a target of protein-tyrosine kinases. Science. (1993) 259:1607–11. doi: 10.1126/science.8096088
30. Vogel W, Lammers R, Huang J, Ullrich A. Activation of a phosphotyrosine phosphatase by tyrosine phosphorylation. Science. (1993) 259:1611–4. doi: 10.1126/science.7681217
31. Andersen JN, Mortensen OH, Peters GH, Drake PG, Iversen LF, Olsen OH, et al. Structural and evolutionary relationships among protein tyrosine phosphatase domains. Mol Cell Biol. (2001) 21:7117–36. doi: 10.1128/MCB.21.21.7117-7136.2001
32. Alonso A, Pulido R. The extended human PTPome: a growing tyrosine phosphatase family. FEBS J. (2016) 283:2197–201. doi: 10.1111/febs.13748
33. Barford D, Neel BG. Revealing mechanisms for SH2 domain mediated regulation of the protein tyrosine phosphatase SHP-2. Structure. (1998) 6:249–54. doi: 10.1016/S0969-2126(98)00027-6
34. Hof P, Pluskey S, Dhe-Paganon S, Eck MJ, Shoelson SE. Crystal structure of the tyrosine phosphatase SHP-2. Cell. (1998) 92:441–50. doi: 10.1016/S0092-8674(00)80938-1
35. Cunnick JM, Mei L, Doupnik CA, Wu J. Phosphotyrosines 627 and 659 of Gab1 constitute a bisphosphoryl tyrosine-based activation motif (BTAM) conferring binding and activation of SHP2. J Biol Chem. (2001) 276:24380–7. doi: 10.1074/jbc.M010275200
36. Yu ZH, Xu J, Walls CD, Chen L, Zhang S, Zhang R, et al. Structural and mechanistic insights into LEOPARD syndrome-associated SHP2 mutations. J Biol Chem. (2013) 288:10472–82. doi: 10.1074/jbc.M113.450023
37. Qiu W, Wang X, Romanov V, Hutchinson A, Lin A, Ruzanov M, Battaile KP, et al. Structural insights into Noonan/LEOPARD syndrome-related mutants of protein-tyrosine phosphatase SHP2 (PTPN11). BMC Struct Biol. (2014) 14:10. doi: 10.1186/1472-6807-14-10
38. Schaeper U, Gehring NH, Fuchs KP, Sachs M, Kempkes B, Birchmeier W. Coupling of Gab1 to c-Met, Grb2, and Shp2 mediates biological responses. J Cell Biol. (2000) 149:1419–32. doi: 10.1083/jcb.149.7.1419
39. Schaeper U, Vogel R, Chmielowiec J, Helene J, Rosario M, Birchmeier W. Distinct requirements for Gab1 in Met and EGF receptor signaling in vivo. Proc Natl Acad Sci USA. (2007) 104:15376–81. doi: 10.1073/pnas.0702555104
40. Wohrle FU, Daly RJ, Brummer T. Function, regulation and pathological roles of the Gab/DOS docking proteins. Cell Commun Signal. (2009) 7:22. doi: 10.1186/1478-811X-7-22
41. Bennett AM, Tang TL, Sugimoto S, Walsh CT, Neel BG. Protein-tyrosine-phosphatase SHPTP2 couples platelet-derived growth factor receptor beta to Ras. Proc Natl Acad Sci USA. (1994) 91:7335–9. doi: 10.1073/pnas.91.15.7335
42. Li W, Nishimura R, Kashishian A, Batzer AG, Kim WJ, Cooper JA, et al. A new function for a phosphotyrosine phosphatase: linking GRB2-Sos to a receptor tyrosine kinase. Mol Cell Biol. (1994) 14:509–17. doi: 10.1128/MCB.14.1.509
43. Araki T, Nawa H, Neel BG. Tyrosyl phosphorylation of Shp2 is required for normal ERK activation in response to some, but not all, growth factors. J Biol Chem. (2003) 278:41677–84. doi: 10.1074/jbc.M306461200
44. Yamauchi K, Milarski KL, Saltiel AR, Pessin JE. Protein-tyrosine-phosphatase SHPTP2 is a required positive effector for insulin downstream signaling. Proc Natl Acad Sci USA. (1995) 92:664–8. doi: 10.1073/pnas.92.3.664
45. Deb TB, Wong L, Salomon DS, Zhou G, Dixon JE, Gutkind JS, et al. A common requirement for the catalytic activity and both SH2 domains of SHP-2 in mitogen-activated protein (MAP) kinase activation by the ErbB family of receptors. A specific role for SHP-2 in map, but not c-Jun amino-terminal kinase activation. J Biol Chem. (1998) 273:16643–6. doi: 10.1074/jbc.273.27.16643
46. Maroun CR, Naujokas MA, Holgado-Madruga M, Wong AJ, Park M. The tyrosine phosphatase SHP-2 is required for sustained activation of extracellular signal-regulated kinase and epithelial morphogenesis downstream from the met receptor tyrosine kinase. Mol Cell Biol. (2000) 20:8513–25. doi: 10.1128/MCB.20.22.8513-8525.2000
47. Agazie YM, Hayman MJ. Molecular mechanism for a role of SHP2 in epidermal growth factor receptor signaling. Mol Cell Biol. (2003) 23:7875–86. doi: 10.1128/MCB.23.21.7875-7886.2003
48. Ekman S, Kallin A, Engstrom U, Heldin CH, Ronnstrand L. SHP-2 is involved in heterodimer specific loss of phosphorylation of Tyr771 in the PDGF beta-receptor. Oncogene. (2002) 21:1870–5. doi: 10.1038/sj.onc.1205210
49. Montagner A, Yart A, Dance M, Perret B, Salles JP, Raynal P. A novel role for Gab1 and SHP2 in epidermal growth factor-induced Ras activation. J Biol Chem. (2005) 280:5350–60. doi: 10.1074/jbc.M410012200
50. Cleghon V, Feldmann P, Ghiglione C, Copeland TD, Perrimon N, Hughes DA, et al. Opposing actions of CSW and RasGAP modulate the strength of Torso RTK signaling in the Drosophila terminal pathway. Mol Cell. (1998) 2:719–27. doi: 10.1016/S1097-2765(00)80287-7
51. Klinghoffer RA, Kazlauskas A. Identification of a putative Syp substrate, the PDGF beta receptor. J Biol Chem. (1995) 270:22208–17. doi: 10.1074/jbc.270.38.22208
52. Bunda S, Burrell K, Heir P, Zeng L, Alamsahebpour A, Kano Y, et al. Inhibition of SHP2-mediated dephosphorylation of Ras suppresses oncogenesis. Nat Commun. (2015) 6:8859. doi: 10.1038/ncomms9859
53. Zhang SQ, Yang W, Kontaridis MI, Bivona TG, Wen G, Araki T, et al. Shp2 regulates SRC family kinase activity and Ras/Erk activation by controlling Csk recruitment. Mol Cell. (2004) 13:341–55. doi: 10.1016/S1097-2765(04)00050-4
54. Ren Y, Meng S, Mei L, Zhao ZJ, Jove R, Wu J. Roles of Gab1 and SHP2 in paxillin tyrosine dephosphorylation and Src activation in response to epidermal growth factor. J Biol Chem. (2004) 279:8497–505. doi: 10.1074/jbc.M312575200
55. Kawazoe T, Taniguchi K. The Sprouty/Spred family as tumor suppressors: coming of age. Cancer Sci. (2019) 110:1525–35. doi: 10.1111/cas.13999
56. Jarvis LA, Toering SJ, Simon MA, Krasnow MA, Smith-Bolton RK. Sprouty proteins are in vivo targets of Corkscrew/SHP-2 tyrosine phosphatases. Development. (2006) 133:1133–42. doi: 10.1242/dev.02255
57. Hanafusa H, Torii S, Yasunaga T, Matsumoto K, Nishida E. Shp2, an SH2-containing protein-tyrosine phosphatase, positively regulates receptor tyrosine kinase signaling by dephosphorylating and inactivating the inhibitor Sprouty. J Biol Chem. (2004) 279:22992–5. doi: 10.1074/jbc.M312498200
58. Quintanar-Audelo M, Yusoff P, Sinniah S, Chandramouli S, Guy GR. Sprouty-related Ena/vasodilator-stimulated phosphoprotein homology 1-domain-containing protein (SPRED1), a tyrosine-protein phosphatase non-receptor type 11 (SHP2) substrate in the Ras/extracellular signal-regulated kinase (ERK) pathway. J Biol Chem. (2011) 286:23102–12. doi: 10.1074/jbc.M110.212662
59. Batth TS, Papetti M, Pfeiffer A, Tollenaere MAX, Francavilla C, Olsen JV. Large-scale phosphoproteomics reveals Shp-2 phosphatase-dependent regulators of pdgf receptor signaling. Cell Rep. (2018) 22:2784–96. doi: 10.1016/j.celrep.2018.02.038
60. Gesbert F, Guenzi C, Bertoglio J. A new tyrosine-phosphorylated 97-kDa adaptor protein mediates interleukin-2-induced association of SHP-2 with p85-phosphatidylinositol 3-kinase in human T lymphocytes. J Biol Chem. (1998) 273:18273–81. doi: 10.1074/jbc.273.29.18273
61. Gadina M, Sudarshan C, O'Shea JJ. IL-2, but not IL-4 and other cytokines, induces phosphorylation of a 98-kDa protein associated with SHP-2, phosphatidylinositol 3'-kinase, and Grb2. J Immunol. (1999) 162:2081–6.
62. Gadina M, Sudarshan C, Visconti R, Zhou YJ, Gu H, Neel BG, et al. The docking molecule gab2 is induced by lymphocyte activation and is involved in signaling by interleukin-2 and interleukin-15 but not other common gamma chain-using cytokines. J Biol Chem. (2000) 275:26959–66. doi: 10.1074/jbc.M004021200
63. Wu CJ, O'Rourke DM, Feng GS, Johnson GR, Wang Q, Greene MI. The tyrosine phosphatase SHP-2 is required for mediating phosphatidylinositol 3-kinase/Akt activation by growth factors. Oncogene. (2001) 20:6018–25. doi: 10.1038/sj.onc.1204699
64. Zhang SQ, Tsiaras WG, Araki T, Wen G, Minichiello L, Klein R. Neel BG, Receptor-specific regulation of phosphatidylinositol 3'-kinase activation by the protein tyrosine phosphatase Shp2. Mol Cell Biol. (2002) 22:4062–72. doi: 10.1128/MCB.22.12.4062-4072.2002
65. Ross SH, Cantrell DA. Signaling and function of interleukin-2 in T lymphocytes. Annu Rev Immunol. (2018) 36:411–33. doi: 10.1146/annurev-immunol-042617-053352
66. Myers MG Jr, Mendez R, Shi P, Pierce JH, Rhoads R, White MF. The COOH-terminal tyrosine phosphorylation sites on IRS-1 bind SHP-2 and negatively regulate insulin signaling. J Biol Chem. (1998) 273:26908–14. doi: 10.1074/jbc.273.41.26908
67. Ouwens DM, van der Zon GC, Maassen JA. Modulation of insulin-stimulated glycogen synthesis by src homology phosphatase 2. Mol Cell Endocrinol. (2001) 175:131–40. doi: 10.1016/S0303-7207(01)00389-6
68. Marin TM, Keith K, Davies B, Conner DA, Guha P, Kalaitzidis D, et al. Rapamycin reverses hypertrophic cardiomyopathy in a mouse model of LEOPARD syndrome-associated PTPN11 mutation. J Clin Invest. (2011) 121:1026–43. doi: 10.1172/JCI44972
69. Kwon M, Ling Y, Maile LA, Badley-Clark J, Clemmons DR. Recruitment of the tyrosine phosphatase Src homology 2 domain tyrosine phosphatase-2 to the p85 subunit of phosphatidylinositol-3 (PI-3) kinase is required for insulin-like growth factor-I-dependent PI-3 kinase activation in smooth muscle cells. Endocrinology. (2006) 147:1458–65. doi: 10.1210/en.2005-1115
70. Goodwin CB, Yang Z, Yin F, Yu M, Chan RJ. Genetic disruption of the PI3K regulatory subunits, p85alpha, p55alpha, and p50alpha, normalizes mutant PTPN11-induced hypersensitivity to GM-CSF. Haematologica. (2012) 97:1042–7. doi: 10.3324/haematol.2011.046896
71. Ugi S, Maegawa H, Kashiwagi A, Adachi M, Olefsky JM, Kikkawa R. Expression of dominant negative mutant SHPTP2 attenuates phosphatidylinositol 3'-kinase activity via modulation of phosphorylation of insulin receptor substrate-1. J Biol Chem. (1996) 271:12595–602. doi: 10.1074/jbc.271.21.12595
72. Ivins Zito C, Kontaridis MI, Fornaro M, Feng GS, Bennett AM. SHP-2 regulates the phosphatidylinositide 3'-kinase/Akt pathway and suppresses caspase 3-mediated apoptosis. J Cell Physiol. (2004) 199:227–36. doi: 10.1002/jcp.10446
73. Xu D, Qu CK. Protein tyrosine phosphatases in the JAK/STAT pathway. Front Biosci. (2008) 13:4925–32. doi: 10.2741/3051
74. Chen Y, Wen R, Yang S, Schuman J, Zhang EE, Yi T, et al. Identification of Shp-2 as a Stat5A phosphatase. J Biol Chem. (2003) 278:16520–7. doi: 10.1074/jbc.M210572200
75. You M, Yu DH, Feng GS. Shp-2 tyrosine phosphatase functions as a negative regulator of the interferon-stimulated Jak/STAT pathway. Mol Cell Biol. (1999) 19:2416–24. doi: 10.1128/MCB.19.3.2416
76. Xiao P, Zhang H, Zhang Y, Zheng M, Liu R, Zhao Y, et al. Phosphatase Shp2 exacerbates intestinal inflammation by disrupting macrophage responsiveness to interleukin-10. J Exp Med. (2019) 216:337–49. doi: 10.1084/jem.20181198
77. Zehender A, Huang J, Gyorfi AH, Matei AE, Trinh-Minh T, Xu X, et al. The tyrosine phosphatase SHP2 controls TGFbeta-induced STAT3 signaling to regulate fibroblast activation and fibrosis. Nat Commun. (2018) 9:3259. doi: 10.1038/s41467-018-05768-3
78. Niogret C, Miah MMS, Rota G, Fonta NP, Wang H, Held W, et al. Shp-2 is critical for ERK and metabolic engagement downstream of IL-15 receptor in NK cells. Nat Commun. (2019) 10:1444. doi: 10.1038/s41467-019-09431-3
79. Salmond RJ, Huyer G, Kotsoni A, Clements L, Alexander DR. The src homology 2 domain-containing tyrosine phosphatase 2 regulates primary T-dependent immune responses and Th cell differentiation. J Immunol. (2005) 175:6498–508. doi: 10.4049/jimmunol.175.10.6498
80. Tartaglia M, Martinelli S, Iavarone I, Cazzaniga G, Spinelli M, Giarin E, et al. Somatic PTPN11 mutations in childhood acute myeloid leukaemia. Br J Haematol. (2005) 129:333–9. doi: 10.1111/j.1365-2141.2005.05457.x
81. Loh ML, Vattikuti S, Schubbert S, Reynolds MG, Carlson E, Lieuw KH, et al. Mutations in PTPN11 implicate the SHP-2 phosphatase in leukemogenesis. Blood. (2004) 103:2325–31. doi: 10.1182/blood-2003-09-3287
82. Bentires-Alj M, Paez JG, David FS, Keilhack H, Halmos B, Naoki K, et al. Activating mutations of the noonan syndrome-associated SHP2/PTPN11 gene in human solid tumors and adult acute myelogenous leukemia. Cancer Res. (2004) 64:8816–20. doi: 10.1158/0008-5472.CAN-04-1923
83. Butterworth S, Overduin M, Barr AJ. Targeting protein tyrosine phosphatase SHP2 for therapeutic intervention. Future Med Chem. (2014) 6:1423–37. doi: 10.4155/fmc.14.88
84. Grosskopf S, Eckert C, Arkona C, Radetzki S, Bohm K, Heinemann U, et al. Selective inhibitors of the protein tyrosine phosphatase SHP2 block cellular motility and growth of cancer cells in vitro and in vivo. ChemMedChem. (2015) 10:815–26. doi: 10.1002/cmdc.201500015
85. Liu W, Yu B, Xu G, Xu WR, Loh ML, Tang LD, et al. Identification of cryptotanshinone as an inhibitor of oncogenic protein tyrosine phosphatase SHP2 (PTPN11). J Med Chem. (2013) 56:7212–21. doi: 10.1021/jm400474r
86. Zeng LF, Zhang RY, Yu ZH, Li S, Wu L, Gunawan AM, et al. Therapeutic potential of targeting the oncogenic SHP2 phosphatase. J Med Chem. (2014) 57:6594–609. doi: 10.1021/jm5006176
87. Duan YQ, Ma Y, Wang XJ, Jin YY, Wang RL, Dong WL, et al. Design potential selective inhibitors for treating cancer by targeting the Src homology 2 (SH2) domain-containing phosphatase 2 (Shp2) with core hopping approach. Protein Pept Lett. (2014) 21:556–63. doi: 10.2174/0929866521666131223143913
88. Chio CM, Lim CS, Bishop AC. Targeting a cryptic allosteric site for selective inhibition of the oncogenic protein tyrosine phosphatase Shp2. Biochemistry. (2015) 54:497–504. doi: 10.1021/bi5013595
89. Groenendijk FH, Bernards R. Drug resistance to targeted therapies: deja vu all over again. Mol Oncol. (2014) 8:1067–83. doi: 10.1016/j.molonc.2014.05.004
90. Prahallad A, Heynen GJ, Germano G, Willems SM, Evers B, Vecchione L, et al. PTPN11 is a central node in intrinsic and acquired resistance to targeted cancer drugs. Cell Rep. (2015) 12:1978–85. doi: 10.1016/j.celrep.2015.08.037
91. LaRochelle JR, Fodor M, Vemulapalli V, Mohseni M, Wang P, Stams T, et al. Structural reorganization of SHP2 by oncogenic mutations and implications for oncoprotein resistance to allosteric inhibition. Nat Commun. (2018) 9:4508. doi: 10.1038/s41467-018-06823-9
92. Chan RJ, Leedy MB, Munugalavadla V, Voorhorst CS, Li Y, Yu M, et al. Human somatic PTPN11 mutations induce hematopoietic-cell hypersensitivity to granulocyte-macrophage colony-stimulating factor. Blood. (2005) 105:3737–42. doi: 10.1182/blood-2004-10-4002
93. Bone H, Dechert U, Jirik F, Schrader JW, Welham MJ. SHP1 and SHP2 protein-tyrosine phosphatases associate with betac after interleukin-3-induced receptor tyrosine phosphorylation. Identification of potential binding sites and substrates. J Biol Chem. (1997) 272:14470–6. doi: 10.1074/jbc.272.22.14470
94. Gu H, Pratt JC, Burakoff SJ, Neel BG. Cloning of p97/Gab2, the major SHP2-binding protein in hematopoietic cells, reveals a novel pathway for cytokine-induced gene activation. Mol Cell. (1998) 2:729–40. doi: 10.1016/S1097-2765(00)80288-9
95. Wheadon H, Edmead C, Welham MJ. Regulation of interleukin-3-induced substrate phosphorylation and cell survival by SHP-2 (Src-homology protein tyrosine phosphatase 2). Biochem J. (2003) 376:147–57. doi: 10.1042/bj20031160
96. Pazdrak K, Adachi T, Alam R. Src homology 2 protein tyrosine phosphatase (SHPTP2)/Src homology 2 phosphatase 2 (SHP2) tyrosine phosphatase is a positive regulator of the interleukin 5 receptor signal transduction pathways leading to the prolongation of eosinophil survival. J Exp Med. (1997) 186:561–8. doi: 10.1084/jem.186.4.561
97. Itoh T, Muto A, Watanabe S, Miyajima A, Yokota T, Arai K. Granulocyte-macrophage colony-stimulating factor provokes RAS activation and transcription of c-fos through different modes of signaling. J Biol Chem. (1996) 271:7587–92. doi: 10.1074/jbc.271.13.7587
98. Welham MJ, Dechert U, Leslie KB, Jirik F, Schrader JW. Interleukin (IL)-3 and granulocyte/macrophage colony-stimulating factor, but not IL-4, induce tyrosine phosphorylation, activation, and association of SHPTP2 with Grb2 and phosphatidylinositol 3'-kinase. J Biol Chem. (1994) 269:23764–8.
99. Xia LX, Hua W, Jin Y, Tian BP, Qiu ZW, Zhang C, et al. Eosinophil differentiation in the bone marrow is promoted by protein tyrosine phosphatase SHP2. Cell Death Dis. (2016) 7:e2175. doi: 10.1038/cddis.2016.74
100. Zhu HH, Ji K, Alderson N, He Z, Li S, Liu W, et al. Kit-Shp2-Kit signaling acts to maintain a functional hematopoietic stem and progenitor cell pool. Blood. (2011) 117:5350–61. doi: 10.1182/blood-2011-01-333476
101. Mali RS, Ma P, Zeng LF, Martin H, Ramdas B, He Y, et al. Role of SHP2 phosphatase in KIT-induced transformation: identification of SHP2 as a druggable target in diseases involving oncogenic KIT. Blood. (2012) 120:2669–78. doi: 10.1182/blood-2011-08-375873
102. Zhang S, Broxmeyer HE. p85 subunit of PI3 kinase does not bind to human Flt3 receptor, but associates with SHP2, SHIP, and a tyrosine-phosphorylated 100-kDa protein in Flt3 ligand-stimulated hematopoietic cells. Biochem Biophys Res Commun. (1999) 254:440–5. doi: 10.1006/bbrc.1998.9959
103. Zhang S, Broxmeyer HE. Flt3 ligand induces tyrosine phosphorylation of gab1 and gab2 and their association with shp-2, grb2, and PI3 kinase. Biochem Biophys Res Commun. (2000) 277:195–9. doi: 10.1006/bbrc.2000.3662
104. Qu CK, Nguyen S, Chen J, Feng GS. Requirement of Shp-2 tyrosine phosphatase in lymphoid and hematopoietic cell development. Blood. (2001) 97:911–4. doi: 10.1182/blood.V97.4.911
105. Kopf M, Ramsay A, Brombacher F, Baumann H, Freer G, Galanos C, et al. Pleiotropic defects of IL-6-deficient mice including early hematopoiesis, T and B cell function, and acute phase responses. Ann N Y Acad Sci. (1995) 762:308–18. doi: 10.1111/j.1749-6632.1995.tb32335.x
106. Fukada T, Hibi M, Yamanaka Y, Takahashi-Tezuka M, Fujitani Y, Yamaguchi T, et al. Two signals are necessary for cell proliferation induced by a cytokine receptor gp130: involvement of STAT3 in anti-apoptosis. Immunity. (1996) 5:449–60. doi: 10.1016/S1074-7613(00)80501-4
107. Stahl N, Farruggella TJ, Boulton TG, Zhong Z, Darnell JE Jr, Yancopoulos GD. Choice of STATs and other substrates specified by modular tyrosine-based motifs in cytokine receptors. Science. (1995) 267:1349–53. doi: 10.1126/science.7871433
108. Ohtani T, Ishihara K, Atsumi T, Nishida K, Kaneko Y, Miyata T, et al. Dissection of signaling cascades through gp130 in vivo: reciprocal roles for STAT3- and SHP2-mediated signals in immune responses. Immunity. (2000) 12:95–105. doi: 10.1016/S1074-7613(00)80162-4
109. Atsumi T, Ishihara K, Kamimura D, Ikushima H, Ohtani T, Hirota S, et al. A point mutation of Tyr-759 in interleukin 6 family cytokine receptor subunit gp130 causes autoimmune arthritis. J Exp Med. (2002) 196:979–90. doi: 10.1084/jem.20020619
110. Symes A, Stahl N, Reeves SA, Farruggella T, Servidei T, Gearan T, et al. The protein tyrosine phosphatase SHP-2 negatively regulates ciliary neurotrophic factor induction of gene expression. Curr Biol. (1997) 7:697–700. doi: 10.1016/S0960-9822(06)00298-3
111. Kim H, Hawley TS, Hawley RG, Baumann H. Protein tyrosine phosphatase 2 (SHP-2) moderates signaling by gp130 but is not required for the induction of acute-phase plasma protein genes in hepatic cells. Mol Cell Biol. (1998) 18:1525–33.
112. Schaper F, Gendo C, Eck M, Schmitz J, Grimm C, Anhuf D, et al. Activation of the protein tyrosine phosphatase SHP2 via the interleukin-6 signal transducing receptor protein gp130 requires tyrosine kinase Jak1 and limits acute-phase protein expression. Biochem J. (1998) 335(Pt 3):557–65. doi: 10.1042/bj3350557
113. Lehmann U, Schmitz J, Weissenbach M, Sobota RM, Hortner M, Friederichs K, et al. SHP2 and SOCS3 contribute to Tyr-759-dependent attenuation of interleukin-6 signaling through gp130. J Biol Chem. (2003) 278:661–71. doi: 10.1074/jbc.M210552200
114. Fairlie WD, De Souza D, Nicola NA, Baca M. Negative regulation of gp130 signalling mediated through tyrosine-757 is not dependent on the recruitment of SHP2. Biochem J. (2003) 372:495–502. doi: 10.1042/bj20030104
115. Nelson BH, McIntosh BC, Rosencrans LL, Greenberg PD. Requirement for an initial signal from the membrane-proximal region of the interleukin 2 receptor gamma(c) chain for Janus kinase activation leading to T cell proliferation. Proc Natl Acad Sci USA. (1997) 94:1878–83. doi: 10.1073/pnas.94.5.1878
116. Adachi M, Ishino M, Torigoe T, Minami Y, Matozaki T, Miyazaki T, et al. Interleukin-2 induces tyrosine phosphorylation of SHP-2 through IL-2 receptor beta chain. Oncogene. (1997) 14:1629–33. doi: 10.1038/sj.onc.1200981
117. Gadina M, Stancato LM, Bacon CM, Larner AC, O'Shea JJ. Involvement of SHP-2 in multiple aspects of IL-2 signaling: evidence for a positive regulatory role. J Immunol. (1998) 160:4657–61.
118. Arnaud M, Mzali R, Gesbert F, Crouin C, Guenzi C, Vermot-Desroches C, et al. Interaction of the tyrosine phosphatase SHP-2 with Gab2 regulates Rho-dependent activation of the c-fos serum response element by interleukin-2. Biochem J. (2004) 382:545–56. doi: 10.1042/BJ20040103
119. Zeng R, Spolski R, Casas E, Zhu W, Levy DE, Leonard WJ. The molecular basis of IL-21-mediated proliferation. Blood. (2007) 109:4135–42. doi: 10.1182/blood-2006-10-054973
120. Zhong J, Kim MS, Chaerkady R, Wu X, Huang TC, Getnet D, et al. TSLP signaling network revealed by SILAC-based phosphoproteomics. Mol Cell Proteomics. (2012) 11:M112 017764. doi: 10.1074/mcp.M112.017764
121. Jin L, Nara H, Rahman M, Takeda Y, Araki A, Asao H. Protein tyrosine phosphatase SHP-2 is involved in the interleukin-21-induced activation of extracellular signal-regulated kinase 1/2. Tohoku J Exp Med. (2018) 244:187–93. doi: 10.1620/tjem.244.187
122. Salmond RJ, Alexander DR. SHP2 forecast for the immune system: fog gradually clearing. Trends Immunol. (2006) 27:154–60. doi: 10.1016/j.it.2006.01.007
123. Olcese L, Lang P, Vely F, Cambiaggi A, Marguet D, Blery M, et al. Human and mouse killer-cell inhibitory receptors recruit PTP1C and PTP1D protein tyrosine phosphatases. J Immunol. (1996) 156:4531–4.
124. Le Drean E, Vely F, Olcese L, Cambiaggi A, Guia S, Krystal G, et al. Inhibition of antigen-induced T cell response and antibody-induced NK cell cytotoxicity by NKG2A: association of NKG2A with SHP-1 and SHP-2 protein-tyrosine phosphatases. European J Immunol. (1998) 28:264–76. doi: 10.1002/(SICI)1521-4141(199801)28:01<264::AID-IMMU264>3.0.CO;2-O
125. Brodin P, Karre K, Hoglund P. NK cell education: not an on-off switch but a tunable rheostat. Trends Immunol. (2009) 30:143–9. doi: 10.1016/j.it.2009.01.006
126. Lowin-Kropf B, Kunz B, Beermann F, Held W. Impaired natural killing of MHC class I-deficient targets by NK cells expressing a catalytically inactive form of SHP-1. J Immunol. (2000) 165:1314–21. doi: 10.4049/jimmunol.165.3.1314
127. Viant C, Fenis A, Chicanne G, Payrastre B, Ugolini S, Vivier E. SHP-1-mediated inhibitory signals promote responsiveness and anti-tumour functions of natural killer cells. Nat Commun. (2014) 5:5108. doi: 10.1038/ncomms6108
128. Cheng H, Schwell V, Curtis BR, Fazlieva R, Roder H, Campbell KS. Conformational changes in the cytoplasmic region of KIR3DL1 upon Interaction with SHP-2. Structure. (2019) 27:639–50.e2.
129. Pauken KE, Wherry EJ. Overcoming T cell exhaustion in infection and cancer. Trends Immunol. (2015) 36:265–76. doi: 10.1016/j.it.2015.02.008
130. Speiser DE, Utzschneider DT, Oberle SG, Munz C, Romero P, Zehn D. T cell differentiation in chronic infection and cancer: functional adaptation or exhaustion? Nature reviews. Immunology. (2014) 14:768–74. doi: 10.1038/nri3740
131. Wei SC, Duffy CR, Allison JP. Fundamental mechanisms of immune checkpoint blockade therapy. Cancer Discov. (2018) 8:1069–86. doi: 10.1158/2159-8290.CD-18-0367
132. Khalil DN, Smith EL, Brentjens RJ, Wolchok JD. The future of cancer treatment: immunomodulation, CARs and combination immunotherapy. Nat Rev Clin Oncol. (2016) 13:273–90. doi: 10.1038/nrclinonc.2016.25
133. Topalian SL, Drake CG, Pardoll DM. Immune checkpoint blockade: a common denominator approach to cancer therapy. Cancer Cell. (2015) 27:450–61. doi: 10.1016/j.ccell.2015.03.001
134. Lee KM, Chuang E, Griffin M, Khattri R, Hong DK, Zhang W, et al. Molecular basis of T cell inactivation by CTLA-4. Science. (1998) 282:2263–6. doi: 10.1126/science.282.5397.2263
135. Okazaki T, Maeda A, Nishimura H, Kurosaki T, Honjo T. PD-1 immunoreceptor inhibits B cell receptor-mediated signaling by recruiting src homology 2-domain-containing tyrosine phosphatase 2 to phosphotyrosine. Proc Natl Acad Sci USA. (2001) 98:13866–71. doi: 10.1073/pnas.231486598
136. Sheppard KA, Fitz LJ, Lee JM, Benander C, George JA, Wooters J, et al. PD-1 inhibits T-cell receptor induced phosphorylation of the ZAP70/CD3zeta signalosome and downstream signaling to PKCtheta. FEBS Lett. (2004) 574:37–41. doi: 10.1016/j.febslet.2004.07.083
137. Chemnitz JM, Parry RV, Nichols KE, June CH, Riley JL. SHP-1 and SHP-2 associate with immunoreceptor tyrosine-based switch motif of programmed death 1 upon primary human T cell stimulation, but only receptor ligation prevents T cell activation. J Immunol. (2004) 173:945–54. doi: 10.4049/jimmunol.173.2.945
138. Yokosuka T, Takamatsu M, Kobayashi-Imanishi W, Hashimoto-Tane A, Azuma M, Saito T. Programmed cell death 1 forms negative costimulatory microclusters that directly inhibit T cell receptor signaling by recruiting phosphatase SHP2. J Exp Med. (2012) 209:1201–17. doi: 10.1084/jem.20112741
139. Latchman Y, Wood CR, Chernova T, Chaudhary D, Borde M, Chernova I, et al. PD-L2 is a second ligand for PD-1 and inhibits T cell activation. Nat Immunol. (2001) 2:261–8. doi: 10.1038/85330
140. Yamamoto R, Nishikori M, Kitawaki T, Sakai T, Hishizawa M, Tashima M, et al. PD-1-PD-1 ligand interaction contributes to immunosuppressive microenvironment of Hodgkin lymphoma. Blood. (2008) 111:3220–4. doi: 10.1182/blood-2007-05-085159
141. Hui E, Cheung J, Zhu J, Su X, Taylor MJ, Wallweber HA, et al. T cell costimulatory receptor CD28 is a primary target for PD-1-mediated inhibition. Science. (2017) 355:1428–33. doi: 10.1126/science.aaf1292
142. Fourcade J, Sun Z, Pagliano O, Guillaume P, Luescher IF, Sander C, et al. CD8(+) T cells specific for tumor antigens can be rendered dysfunctional by the tumor microenvironment through upregulation of the inhibitory receptors BTLA and PD-1. Cancer Res. (2012) 72:887–96. doi: 10.1158/0008-5472.CAN-11-2637
143. Gavrieli M, Watanabe N, Loftin SK, Murphy TL, Murphy KM. Characterization of phosphotyrosine binding motifs in the cytoplasmic domain of B and T lymphocyte attenuator required for association with protein tyrosine phosphatases SHP-1 and SHP-2. Biochem Biophys Res Commun. (2003) 312:1236–43. doi: 10.1016/j.bbrc.2003.11.070
144. Watanabe N, Gavrieli M, Sedy JR, Yang J, Fallarino F, Loftin SK, et al. BTLA is a lymphocyte inhibitory receptor with similarities to CTLA-4 and PD-1. Nat Immunol. (2003) 4:670–9. doi: 10.1038/ni944
145. Kamphorst AO, Wieland A, Nasti T, Yang S, Zhang R, Barber DL, et al. Rescue of exhausted CD8 T cells by PD-1-targeted therapies is CD28-dependent. Science. (2017) 355:1423–7. doi: 10.1126/science.aaf0683
146. Peled M, Tocheva AS, Sandigursky S, Nayak S, Philips EA, Nichols KE, et al. Affinity purification mass spectrometry analysis of PD-1 uncovers SAP as a new checkpoint inhibitor. Proc Natl Acad Sci USA. (2017) 115:E468–77. doi: 10.1073/pnas.1710437115
147. Celis-Gutierrez J, Blattmann P, Zhai Y, Jarmuzynski N, Ruminski K, Gregoire C, et al. Quantitative interactomics in primary T cells provides a rationale for concomitant PD-1 and BTLA coinhibitor blockade in cancer immunotherapy. Cell Rep. (2019) 27:3315–30 e7. doi: 10.1016/j.celrep.2019.05.041
148. Patsoukis N, Brown J, Petkova V, Liu F, Li L, Boussiotis VA. Selective effects of PD-1 on Akt and Ras pathways regulate molecular components of the cell cycle and inhibit T cell proliferation. Sci Signal. (2012) 5:ra46. doi: 10.1126/scisignal.2002796
149. Frearson JA, Alexander DR. The phosphotyrosine phosphatase SHP-2 participates in a multimeric signaling complex and regulates T cell receptor (TCR) coupling to the Ras/mitogen-activated protein kinase (MAPK) pathway in Jurkat T cells. J Exp Med. (1998) 187:1417–26. doi: 10.1084/jem.187.9.1417
150. Kwon J, Qu CK, Maeng JS, Falahati R, Lee C, Williams MS. Receptor-stimulated oxidation of SHP-2 promotes T-cell adhesion through SLP-76-ADAP. EMBO J. (2005) 24:2331–41. doi: 10.1038/sj.emboj.7600706
151. Liu W, Guo W, Shen L, Chen Z, Luo Q, Luo X, et al. T lymphocyte SHP2-deficiency triggers anti-tumor immunity to inhibit colitis-associated cancer in mice. Oncotarget. (2017) 8:7586–97. doi: 10.18632/oncotarget.13812
152. Nguyen TV, Ke Y, Zhang EE, Feng GS. Conditional deletion of Shp2 tyrosine phosphatase in thymocytes suppresses both pre-TCR and TCR signals. J Immunol. (2006) 177:5990–6. doi: 10.4049/jimmunol.177.9.5990
153. Dong B, Gao Y, Zheng X, Gao G, Gu H, Chen X, et al. T cell activation is reduced by the catalytically inactive form of protein tyrosine phosphatase SHP-2. Int J Clin Exp Med. (2015) 8:6568–77.
154. Zhang T, Guo W, Yang Y, Liu W, Guo L, Gu Y, et al. Loss of SHP-2 activity in CD4+ T cells promotes melanoma progression and metastasis. Scient Rep. (2013) 3:2845. doi: 10.1038/srep02845
155. S.Miah MS, Jayasuriya CT, Salter AI, Reilly EC, Fugere C, Yang W, et al. Ptpn11 deletion in CD4+ cells does not affect T cell development and functions but causes cartilage tumors in a T cell-independent manner. Front Immunol. (2017) 8:1326. doi: 10.3389/fimmu.2017.01326
156. Rota G, Niogret C, Dang AT, Barros CR, Fonta NP, Alfei F, et al. Shp-2 is dispensable for establishing T cell exhaustion and for PD-1 signaling in vivo. Cell Rep. (2018) 23:39–49. doi: 10.1016/j.celrep.2018.03.026
157. Yamasaki S, Nishida K, Hibi M, Sakuma M, Shiina R, Takeuchi A, et al. Docking protein Gab2 is phosphorylated by ZAP-70 and negatively regulates T cell receptor signaling by recruitment of inhibitory molecules. J Biol Chem. (2001) 276:45175–83. doi: 10.1074/jbc.M105384200
158. Frebel H, Nindl V, Schuepbach RA, Braunschweiler T, Richter K, Vogel J, et al. Programmed death 1 protects from fatal circulatory failure during systemic virus infection of mice. J Exp Med. (2012) 209:2485–99. doi: 10.1084/jem.20121015
159. Odorizzi PM, Pauken KE, Paley MA, Sharpe A, Wherry EJ. Genetic absence of PD-1 promotes accumulation of terminally differentiated exhausted CD8+ T cells. J Exp Med. (2015) 212:1125–37. doi: 10.1084/jem.20142237
160. Juneja VR, McGuire KA, Manguso RT, LaFleur MW, Collins N, Haining WN, et al. PD-L1 on tumor cells is sufficient for immune evasion in immunogenic tumors and inhibits CD8 T cell cytotoxicity. J Exp Med. (2017) 214:895–904. doi: 10.1084/jem.20160801
161. Zhao M, Guo W, Wu Y, Yang C, Zhong L, Deng G, et al. SHP2 inhibition triggers anti-tumor immunity and synergizes with PD-1 blockade. Acta Pharmaceutica Sinica B. (2018) 9:304–15. doi: 10.1016/j.apsb.2018.08.009
162. Selby MJ, Engelhardt JJ, Johnston RJ, Lu LS, Han M, Thudium K, et al. Preclinical development of ipilimumab and nivolumab combination immunotherapy: mouse tumor models, in vitro functional studies, and cynomolgus macaque toxicology. PLoS ONE. (2016) 11:e0161779. doi: 10.1371/journal.pone.0161779
Keywords: SHP-2 phosphatase, SHP-2 inhibitors, PTPN11 gene, lymphocytes, cytokine, inhibitory receptors of lymphocytes, PD-1, cancer
Citation: Niogret C, Birchmeier W and Guarda G (2019) SHP-2 in Lymphocytes' Cytokine and Inhibitory Receptor Signaling. Front. Immunol. 10:2468. doi: 10.3389/fimmu.2019.02468
Received: 03 July 2019; Accepted: 03 October 2019;
Published: 25 October 2019.
Edited by:
Michael Loran Dustin, University of Oxford, United KingdomReviewed by:
Christopher E. Rudd, Université de Montréal, CanadaPaul E. Love, National Institutes of Health (NIH), United States
Copyright © 2019 Niogret, Birchmeier and Guarda. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Greta Guarda, Z3JldGEuZ3VhcmRhQGlyYi51c2kuY2g=