 Gianluca Scarno1
Gianluca Scarno1 Giuseppe Pietropaolo1
Giuseppe Pietropaolo1 Chiara Di Censo1
Chiara Di Censo1 Massimo Gadina2
Massimo Gadina2 Angela Santoni1,3
Angela Santoni1,3 Giuseppe Sciumè1*
Giuseppe Sciumè1*- 1Department of Molecular Medicine, Laboratory Affiliated to Istituto Pasteur Italia—Fondazione Cenci Bolognetti, Sapienza University of Rome, Rome, Italy
- 2Translational Immunology Section, Office of Science Technology (OST), National Institute of Arthritis and Musculoskeletal and Skin Diseases, NIH, Bethesda, MD, United States
- 3IRCCS Neuromed, Pozzilli, Italy
Differentiation of Natural Killer (NK) cells is a stepwise process having its origin in the bone marrow and proceeding in the periphery, where these cells follow organ specific trajectories. Several soluble factors and cytokines regulate the distinct stages of NK cell differentiation, and ultimately, their functional properties. Cytokines activating the Janus kinases (JAKs) and members of the signal transducer and activator of transcription (STAT) pathway control distinct aspects of NK cell biology, ranging from development, terminal differentiation, activation, and generation of cells with adaptive properties. Here, we discuss how the recent advances of next generation sequencing (NGS) technology have led to unravel novel molecular aspects of gene regulation, with the aim to provide genomic views of how STATs regulate transcriptional and epigenetic features of NK cells during the different functional stages.
Introduction
Natural Killer (NK) cells are the founding members of the ILC family and represent the innate counterpart of cytotoxic T lymphocytes (1, 2). Like CD8+ T cells, NK cells are able to kill infected or transformed cells in a perforin and granzyme dependent manner, as well, these cells are able to mount a rapid type-1 response by releasing the eponymous cytokine, interferon (IFN)-γ (3, 4). NK cells share the ability to produce type-1 cytokines with a distinct “helper” prototypical innate subset, termed ILC1 (5, 6). NK cells differ from ILC1 for their cytotoxic abilities, for a higher propension to circulate in the bloodstream and for the expression of lineage defining transcription factors (LDTFs) (7–9). In this regard, both NK cells and ILC1 are regulated by transcription factors (TFs) of the T-box family; however, while Eomes is expressed and required only by NK cells, T-bet (encoded by Tbx21) is expressed by both prototypical subsets (10–14). Expression of T-bet is fundamental for the generation of ILC1, and it also has non-redundant roles in regulating NK cell turnover, effector functions and egression from bone marrow (10, 11, 15).
Cytokines and other soluble factors regulate several aspects of NK cell biology, acting through signal-dependent TFs (SDTFs). In particular, cytokines activating the Janus kinases (JAKs) and members of the signal transducer and activator of transcription (STAT) pathway control NK cell development, terminal differentiation, acquisition of effector phenotype up to generation of cells with adaptive features able to provide secondary responses (16, 17). Mammalian genomes contain four genes encoding for JAKs, namely JAK1, JAK2, JAK3, and TYK2; and seven genes for STATs, STAT1-4, STAT5A, STAT5B, and STAT6 (18, 19). Activation of the JAK tyrosine kinases occurs upon receptor engagement, and the juxtaposition of JAKs and STATs allows, after phosphorylation, STAT dimers to dissociate from the membrane complex and to migrate into the nucleus, where they bind specific DNA-motifs modulating gene expression (20).
The role of the JAK/STAT dependent signals on NK cells and other ILCs has been discussed in recent reviews (16, 17, 21); herein, we focus on the molecular mechanisms underlying NK cell differentiation in physiological and pathological contexts. We discuss how the advances of next generation sequencing (NGS) technology and the establishment of novel mouse models have led to a better definition of the genes regulated by STATs, and their transcriptional and epigenetic control of NK cells during differentiation and host defense. Finally, we provide an overview of the JAK inhibitors currently approved for the treatment of immune-mediated disorders and their possible implication on NK cells.
STAT5 as a Central Node for Development, Identity and Homeostasis of NK Cells
The bone marrow is the main site for NK cell and ILC development in the adult, containing distinct progenitors and precursors able to give rise to cells having different fates (22, 23). Differentiation proceeds with a pool of circulating progenitors which move to the periphery, where NK cells and other ILCs follow organ specific trajectories and acquire distinct effector functions (24). In the current model, NK cells have a dedicated pathway of differentiation comprising a pool of committed NK cell precursors (NKps) (25–27). Their differentiation follows a stepwise process encompassing distinct developmental and/or functional stages, discriminated through the expression of CD27 and CD11b levels in mice and CD56 and CD16 in humans [redefined recently by single cell RNA-seq approach (28, 29)].
The cytokines IL-7 and IL-15 are critical for lymphoid development by transmitting their signals through the common IL-2 γ-chain receptor (CD132) and by activating JAK3, JAK1, and STAT5 (30). Deletion of Jak3 in mice is associated with reduced numbers of lymphoid and ILC precursors, in contrast to an accumulation of NKp (31). This evidence is in line with previous findings demonstrating that IL-15 was required for the NKp to proceed toward the next maturation stages (25). Similarly, mice carrying conditional deletion of Jak1 in Ncr1-expressing cells (Jak1fl/fl Ncr1Cre) show profound defects in NK cell differentiation and homeostasis; Jak2 deletion, instead, does not affect NK cell development and survival (32).
JAK3 and JAK1 mainly activate STAT5, which represents a key multi-lineage TF (MLTF) controlling development of both adaptive and innate lymphocytes (33, 34). Ablation of the entire Stat5 locus, comprising both Stat5a and Stat5b, results in a high perinatal lethality, due to the pleiotropic role of this TF; however, the few viable Stat5−/− mice show absence of NK cells (35). Conditional deletion of Stat5 in Ncr1-expressing cells allows to eliminate the confounding effects related to lymphopenia and inflammation observed in mice carrying germline ablation; in these settings, both development and survival of NK cells remain highly impaired (36).
Due to the massive effect of STAT5 deletion on NK cells, our understanding of how this SDTF works at the molecular level has remained elusive; the use of mice bearing only one allele of STAT5 has helped to clarify this aspect. Between the two paralogs, Stat5b is more expressed than Stat5a in innate and adaptive lymphocytes, and its deletion has broad effects on NK cell differentiation (37–39). Transcriptomic analyses performed on NK cells retaining only one Stat5 allele (Stat5a−/−Stat5b+/−) have shed light on the homeostatic impact of this TF on NK cells, which consists on regulation of over 400 genes (39). The residual NK cells present in these mice show a developmental block associated with an accumulation of CD11blow cells, and a drastic decrease of the expression of the anti-apoptotic gene, Bcl2. Along with defects in development and survival, STAT5 sustains the expression of most of the genes (52 out of 76) defining NK cell identity, including NKG2D, perforin and granzymes, and the LDTF T-bet (39). These findings have helped to discriminate between the instructive role of STAT5 during NK cell differentiation and its permissive function in regulating survival.
Upon activation, STAT5 can form dimers but also tetramers having distinct ability to interact with DNA-regulatory elements (40, 41). While STAT5 dimers bind to canonical GAS (IFN-γ activation site, TTCN3GAA) motif, STAT5 tetramers bind to divergent motifs having an optimal spacing of 2–27 base pairs between GAS and GAS-like sequences. The relative importance of STAT5 dimers vs. tetramers in NK cells has been evaluated by the generation of a mouse model carrying genes encoding for tetramer defective mutant STAT5 proteins (40, 42). In these mice, the impaired STAT5 binding to the Bcl2 locus, and the consequent lower mRNA and protein expression, leads to a more rapid cell death of NK cells compared to wild type cells (40). Interestingly, transgenic expression of Bcl2 is able to rescue the effect of Stat5 deficiency on the homeostatic pool of NK cells (43). These “Bcl2-rescued” NK cells undergo a functional switch from tumor-suppressive to tumor-promoting cells, since loss of STAT5 determines upregulation of the pro-angiogenic factor VEGFA, which sustains tumor growth (43). Thus, while STAT5 represents a central node in NK cell development, acquisition of cell identity, and homeostasis (Figure 1), the involvement of other STATs in regulating these processes appears limited. Of note, type I IFNs and STAT1 can have distinct indirect effects on NK cell homeostasis: including the regulation of MHC class I expression (44), as well as the regulation of the production and trans-presentation of IL-15 on accessory cells (45–47).
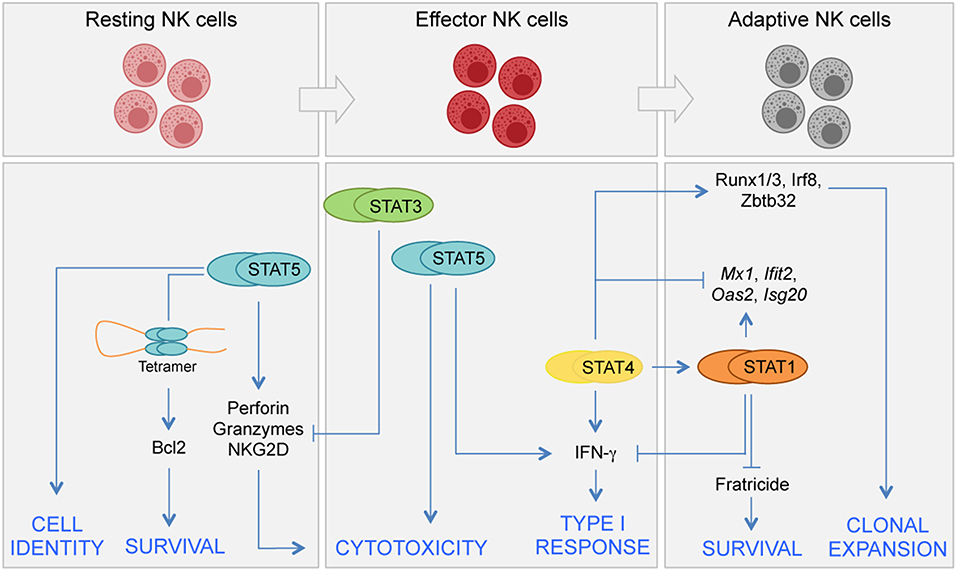
Figure 1. Distinct requirements for STATs in NK cell differentiation. JAK/STAT signals control several aspects of NK cell biology, including development, terminal differentiation, acquisition of effector functions, and generation of adaptive NK cells. NK cell development begins in the bone marrow from committed precursors and it is driven by signals inducing STAT5 activation. In homeostatic conditions STAT5 sustains NK cell survival by direct regulation of Bcl2 expression. STAT5 is also required for terminal differentiation and acquisition of NK cell identity. STAT4 and STAT1 have both specific and shared roles during viral infection. STAT4 controls a network of TFs required for clonal expansion of NK cells during proliferation. STAT4 and STAT1 compete at genomic level for the expression of IFN-γ and other genes. STAT3 has a role in restraining NK cell effector functions by inhibiting perforin, granzyme B, and NKG2D expression.
Multiple STATs Underlie Effector Functions of NK Cells
Effector functions of NK cells depend both on cytokines and on a complex equilibrium between activating and inhibitory receptors, which bind molecules present on healthy and stressed cells including MHC class I and adhesion molecules (48–52). The ability of NK cells to sense environmental changes and rapidly release their effector potentials is favored by a primed epigenetic and transcriptional state, leading to high basal expression of cytokine receptors, LDTFs, and SDTFs, including STATs (53). Although distinct STATs can be easily linked to particular effector and helper functions, it is now clear that activation of NK cells, like the other ILCs, can be influenced by complementary actions of multiple STATs (54–56).
Acting downstream of IL-12, STAT4 is necessary to mount a proper innate response against pathogens by actively regulating NK effector functions, including both IFN-γ production and cytotoxic response (57). The global impact of STAT4 in NK cell activation has been recently tackled using transcriptomic and epigenetic approaches (58, 59). Upon cytokine stimulation, over 300 differentially expressed genes are bound by STAT4 within or in proximity of the locus (59). Along with direct regulation of key effector genes, STAT4 controls the expression of several TFs required for a proper antiviral response, including Zbtb32, Runx1, Runx3, and Irf8. At molecular level, STAT4 binds to the promoter and intergenic regions of the gene locus of these TFs, leading to an increase of the permissiveness of the transcription through modification of the chromatin state, via trimethylation of histone H3 lysine 4 (59). Mouse models carrying selective deletion of these TFs have helped to unravel their impact on the cell cycle program of NK cells during viral infection. The effects of Zbtb32 on the proliferative burst and protective ability of NK cells are mediated by antagonizing the anti-proliferative effects of the TF Blimp-1 (encoded by Prdm1) (60); Irf8, instead, regulates proliferation acting upstream of Zbtb32 (61). During the course of viral infection, the expression of STAT4 and STAT1 follows an opposite fate. Indeed, while STAT4 expression is down-regulated, STAT1 results progressively up-regulated (58, 62). This differential expression pattern affects the signaling downstream of type I IFNs, which mainly activates STAT4 in the early phases of infection and STAT1 in later phases. The increased levels of STAT1 cause a displacement of STAT4 from type I IFN receptors, this switch induces a STAT1 dependent down-regulation of IFN-γ production in NK cells (62).
The role of STAT3 on NK cells has been dissected by employing distinct mouse models, showing differential effects whether deletion of Stat3 gene occurs before or after NK cell development (63, 64). When Stat3fl/fl mice are crossed with Tie2-Cre mice, the effects of Stat3 deletion extend to the whole hematopoietic compartment. In these settings, NK cells show a decreased expression of NKG2D and impaired effector functions (63). In line with these findings, NK cells from subjects with dominant-negative STAT3 mutations show an impaired expression of NKG2D both at steady state and after cytokine stimulation (63). On the other hand, specific deletion of Stat3 in differentiated NK cells, using Ncr1iCre Stat3fl/fl mice, leads to an increased expression of DNAM-1, Perforin, and Granzyme B, and enhanced anti-tumor activity, as the result of the possible repressive functions of STAT3 on these cells (64). Considering these conflicting findings, genome-wide studies aimed at dissecting the transcriptomic impact of Stat3 deletion on NK cells would be particularly relevant to discriminate between the direct and indirect roles of this TF in regulating differentiation and effector functions.
Beyond the homeostatic requirement in sustaining the expression of NK effector molecules, cytokines activating STAT5 have been used to stimulate NK cell functions in vitro, for decades (65, 66). Genomic maps of STAT5 distribution obtained by ChIP-seq analysis have revealed a widespread DNA binding in untreated and IL-15-treated NK cells. However, the acute stimulation with IL-15 induces a redistribution of this TF to a new set of DNA regulatory elements. In these settings, STAT5 binding occurs on almost half of the differentially expressed genes. Gene set enrichment analysis (GSEA) have confirmed a positive enrichment for IL-2/STAT5 signaling in STAT5 bound genes (39). In contrast, unbound genes show a positive enrichment for downstream targets of the mTOR pathway, which has been shown to mediate IL-15-dependent functions in NK cells, including proliferation and terminal differentiation, by regulating CD122 (IL-2Rβ) and CD132 (IL-2Rγ) expression; as well as metabolism, and acquisition of cytolytic features (67, 68).
Specific Roles for STATs During Formation of Adaptive NK Cells
In the context of viral infection, NK cells are able to provide secondary immune responses by following a differentiation path which leads to generation of long-lived cells, named “memory” or “adaptive” NK cells (69, 70). Changes of chromatin accessibility of NK cells have been tracked in vivo up to 35 days after MCMV infection, by ATAC-seq (58). This analysis has revealed that the epigenetic landscape of NK cells is highly dynamic, with the majority of chromatin remodeling occurring in the first 2 weeks. These modifications pave the way for a further acquisition of the transcriptional adaptive state, observed at later time points (58). Genomic maps of STAT4 and STAT1 distribution in cytokine-stimulated NK cells have shown a differential DNA occupancy, being STAT4 mainly localized at putative enhancer sites and STAT1 at promoter regions (58). In line with these results, during MCMV infection the chromatin accessibility of putative enhancer sites and promoters remains less accessible in NK cells deficient for STAT4 and STAT1, respectively. Moreover, due to the existing competitive effects between STAT4 and STAT1, deletion of Stat1 in NK cells leads to an increased DNA accessibility of non-promoter regions; as well as, to an increased expression of selected STAT4 regulated genes, such as Ifng. Conversely, the expression of several STAT1 targets, including Mx1, Ifit2, Oas2, and Isg20, is upregulated in absence of Stat4 (58).
The interplay between STATs and LDTFs is a further mechanism underlying acquisition of specific functions in innate lymphocytes, including the generation of the adaptive phenotype in NK cells. This is the case for the cross-regulation occurring between STATs and T-bet (39, 71, 72); while STAT5 induces T-bet expression in homeostatic conditions (39), STAT4 binds to Tbx21 locus at a distal enhancer site and promotes T-bet expression during MCMV infection (72). T-bet and Eomes are both necessary for NK cell proliferation; however, the IL-12/STAT4/T-bet axis plays a non-redundant role for the maintenance of adaptive NK cells (72). We have discussed in the previous section the network of TFs induced by STAT4, namely Zbtb32, Runx1, Runx3, and Irf8, which are all necessary to enhance proliferation and clonal expansion of NK cells (59–61). As well, expression of STAT1 has a non-redundant role for survival, regulating a Bcl2-independent mechanism enabling NK cells to evade cell death after viral infection. In particular, type 1 IFNs and STAT1 are required to prevent a mechanism of NK cell mediated fratricide, occurring via NKG2D and perforin (73). Overall, these findings shed light on the complex network of TFs and molecules regulated by STATs, required for the acquisition of the adaptive traits by NK cells.
Conclusion: Translational Relevance of Targeting the JAK/STAT Pathway in Inflammation and Cancer
Manipulation of cytokine signaling in NK cells and other ILCs is drawing a growing interest for the treatment of inflammatory diseases and cancer (74, 75). In particular, harnessing NK cell effector functions against cancer by interfering with cytokine signaling has led to promising results in several mouse models (76–79). In this context, the suppressor of cytokine signaling (SOCS) proteins are a class of natural regulators of the activity of STATs. The SOCS protein CIS (encoded by Cish) is at the top among the genes induced by STAT5 activation, and acts as a negative regulator of IL-15 signaling, preventing excessive activation (77). Targeting Cish has a huge impact in enhancing NK cell dependent tumor immunity in several mouse models (77, 79); thus, given its primary role in restraining NK cell functions, CIS represents a novel immune checkpoint for these cells.
On the other hand, several small molecules capable to inhibit JAKs enzymatic activity have been recently developed. At least five JAK inhibitors (also known as JAKinibs) are now approved by various regulatory agencies to treat immune-mediated disorders. These first-generation JAKinibs comprise ruxolitinib, a JAK1 and JAK2 inhibitor, approved for myeloproliferative malignancies; tofacitinib, a JAK1, JAK2, JAK3 inhibitor, approved for rheumatoid arthritis, psoriatic arthritis, and ulcerative colitis; baricitinib, a JAK1 and JAK2 inhibitor, approved for rheumatoid arthritis; peficitinib, a pan-JAK inhibitor approved (only in Japan) for the treatment of rheumatoid arthritis; and oclacitinib, a JAK1 and JAK2 inhibitor, approved for allergic dermatitis in dogs (80).
The impact of ruxolitinib in NK cell homeostasis and functions has been evaluated in humans in distinct contexts. Myelofibrosis patients undergoing ruxolitinib treatment show a defect in NK cell number and differentiation, as well as, impaired functions upon cytokine stimulation; these effects have been related to the increased rates of infection observed in these patients (81). Ruxolitinib also inhibits the generation and functions of cytokine-induced memory-like NK cells by interfering with both IL-15 and IL-12 signaling (82). Finally, Ruxolitinib administration can limit STAT1 activation in patients carrying STAT1 gain of function mutations. In these patients, the prolonged STAT1 activation leads to an impaired NK cell maturation and function, associated with lower STAT5 phosphorylation downstream of IL-15 stimulation, and with lower levels of perforin. These defects are partially reverted by ruxolitinib administration (83).
More selective agents have been developed and are currently being tested. These next-generation inhibitors may possess the advantage of a reduced toxicity. For example, selective targeting of JAK1 would spare interfering with many of JAK2-dependent cytokines involved in hematopoiesis, including Epo, Tpo, G-CSF, GM-CSF, IL-3, and IL-11. Conversely, their efficacy could also be limited. Recently, immunogenomic analysis of mice administered with several JAKinibs, including both first- and second-generation inhibitors, have highlighted the impact of blocking either one or both JAK1 and JAK3 on NK cell homeostasis. Moreover, the JAK1-specific inhibitor (PF-02384554) was more efficient than the JAK3-specific (PF-06651600) in blocking the secondary autocrine response to IFN-γ induced in IL-2 activated NK cells (84).
The optimal degree of JAK inhibition required for an individual cell type in any given tissue remains unknown. To this end, selective JAKinibs may be the key to provide new mechanistic insights in the modulation of the JAK/STAT pathway in NK cells. This approach could be more effective than the use of JAK-deficient mice, in which developmental defects can mask the functional relevance of each JAK. Finally, we are now aware that JAKinibs can affect the structure of the epigenome and preferentially impact genes with super-enhancer structure (85). Notably, several genes encoding for cytokines or their cognate receptors are located within loci with super-enhancer architecture. Therapeutically, it will be important to understand how these drugs, alone or in combination with other chemotherapeutic agents, can be used to effectively, and safely, regulate these critical loci and, in turn, immune as well as non-immune cells.
Author Contributions
GSca, GP, CD, MG, AS and GSci wrote the manuscript. CD designed the figure and made the necessary edits. The final manuscript was a result of the joint efforts of all the authors.
Funding
GSci and AS were supported by the Italian Association for Cancer Research (AIRC), MFAG-21311; 5x1000-21147, respectively; by Institut Pasteur (France), Transversal Research Program, PTR-113-17 program. GSci was supported by Istituto Pasteur Italia—Fondazione Cenci Bolognetti (Under 45-213).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We would like to thank Dr. Lorenzo Cuollo and Dr. Silvia Piconese for the helpful criticism and discussion.
Abbreviations
ILC, innate lymphoid cell; IFN, interferon; LDTF, lineage defining transcription factor; SDTF, signal dependent transcription factor; NK, natural killer; STAT, Signal Transducer and Activator of Transcription.
References
1. Cichocki F, Sitnicka E, Bryceson YT. NK cell development and function–plasticity and redundancy unleashed. Semin Immunol. (2014) 26:114–26. doi: 10.1016/j.smim.2014.02.003
2. Sun JC, Lanier LL. NK cell development, homeostasis and function: parallels with CD8+ T cells. Nat Rev Immunol. (2011) 11:645–57. doi: 10.1038/nri3044
3. Spits H, Bernink JH, Lanier L. NK cells and type 1 innate lymphoid cells: partners in host defense. Nat Immunol. (2016) 17:758–64. doi: 10.1038/ni.3482
4. Geiger TL, Sun JC. Development and maturation of natural killer cells. Curr Opin Immunol. (2016) 39:82–9. doi: 10.1016/j.coi.2016.01.007
5. Spits H, Artis D, Colonna M, Diefenbach A, Di Santo JP, Eberl G, et al. Innate lymphoid cells–a proposal for uniform nomenclature. Nat Rev Immunol. (2013) 13:145–9. doi: 10.1038/nri3365
6. Vivier E, Artis D, Colonna M, Diefenbach A, Di Santo JP, Eberl G, et al. Innate lymphoid cells: 10 years on. Cell. (2018) 174:1054–66. doi: 10.1016/j.cell.2018.07.017
7. Cortez VS, Colonna M. Diversity and function of group 1 innate lymphoid cells. Immunol Lett. (2016) 179:19–24. doi: 10.1016/j.imlet.2016.07.005
8. Zhang J, Marotel M, Fauteux-Daniel S, Mathieu A-L, Viel S, Marçais A, et al. T-bet and Eomes govern differentiation and function of mouse and human NK cells and ILC1. Eur J Immunol. (2018) 48:738–50. doi: 10.1002/eji.201747299
9. Sojka DK, Tian Z, Yokoyama WM. Tissue-resident natural killer cells and their potential diversity. Semin Immunol. (2014) 26:127–31. doi: 10.1016/j.smim.2014.01.010
10. Townsend MJ, Weinmann AS, Matsuda JL, Salomon R, Farnham PJ, Biron CA, et al. T-bet regulates the terminal maturation and homeostasis of NK and Valpha14i NKT cells. Immunity. (2004) 20:477–94. doi: 10.1016/S1074-7613(04)00076-7
11. Gordon SM, Chaix J, Rupp LJ, Wu J, Madera S, Sun JC, et al. The transcription factors T-bet and Eomes control key checkpoints of natural killer cell maturation. Immunity. (2012) 36:55–67. doi: 10.1016/j.immuni.2011.11.016
12. Sciumé G, Hirahara K, Takahashi H, Laurence A, Villarino AV, Singleton KL, et al. Distinct requirements for T-bet in gut innate lymphoid cells. J Exp Med. (2012) 209:2331–8. doi: 10.1084/jem.20122097
13. Klose CSN, Flach M, Möhle L, Rogell L, Hoyler T, Ebert K, et al. Differentiation of type 1 ILCs from a common progenitor to all helper-like innate lymphoid cell lineages. Cell. (2014) 157:340–56. doi: 10.1016/j.cell.2014.03.030
14. Daussy C, Faure F, Mayol K, Viel S, Gasteiger G, Charrier E, et al. T-bet and Eomes instruct the development of two distinct natural killer cell lineages in the liver and in the bone marrow. J Exp Med. (2014) 211:563–77. doi: 10.1084/jem.20131560
15. Jenne CN, Enders A, Rivera R, Watson SR, Bankovich AJ, Pereira JP, et al. T-bet-dependent S1P5 expression in NK cells promotes egress from lymph nodes and bone marrow. J Exp Med. (2009) 206:2469–81. doi: 10.1084/jem.20090525
16. Gotthardt D, Sexl V. STATs in NK-cells: the good, the bad, and the ugly. Front Immunol. (2016) 7:694. doi: 10.3389/fimmu.2016.00694
17. Stabile H, Scarno G, Fionda C, Gismondi A, Santoni A, Gadina M, et al. JAK/STAT signaling in regulation of innate lymphoid cells: the gods before the guardians. Immunol Rev. (2018) 286:148–59. doi: 10.1111/imr.12705
18. Liongue C, Ward AC. Evolution of the JAK-STAT pathway. JAK-STAT. (2013) 2:e22756. doi: 10.4161/jkst.22756
19. Wang Y, Levy DE. Comparative evolutionary genomics of the STAT family of transcription factors. JAK-STAT. (2012) 1:23–33. doi: 10.4161/jkst.19418
20. Reich NC. STAT dynamics. Cytokine Growth Factor Rev. (2007) 18:511–8. doi: 10.1016/j.cytogfr.2007.06.021
21. Vargas-Hernández A, Forbes LR. The Impact of Immunodeficiency on NK cell maturation and function. Curr Allergy Asthma Rep. (2019) 19:2. doi: 10.1007/s11882-019-0836-8
22. Diefenbach A, Colonna M, Koyasu S. Development, differentiation, and diversity of innate lymphoid cells. Immunity. (2014) 41:354–65. doi: 10.1016/j.immuni.2014.09.005
23. Lim AI, Di Santo JP. ILC-poiesis: ensuring tissue ILC differentiation at the right place and time. Eur J Immunol. (2018) 49:11–8. doi: 10.1002/eji.201747294
24. Cherrier DE, Serafini N, Di Santo JP. Innate lymphoid cell development: a T cell perspective. Immunity. (2018) 48:1091–103. doi: 10.1016/j.immuni.2018.05.010
25. Vosshenrich CAJ, Ranson T, Samson SI, Corcuff E, Colucci F, Rosmaraki EE, et al. Roles for common cytokine receptor gamma-chain-dependent cytokines in the generation, differentiation, and maturation of NK cell precursors and peripheral NK cells in vivo. J Immunol. (2005) 174:1213–21. doi: 10.4049/jimmunol.174.3.1213
26. Fathman JW, Bhattacharya D, Inlay MA, Seita J, Karsunky H, Weissman IL. Identification of the earliest natural killer cell-committed progenitor in murine bone marrow. Blood. (2011) 118:5439–47. doi: 10.1182/blood-2011-04-348912
27. Renoux VM, Zriwil A, Peitzsch C, Michaëlsson J, Friberg D, Soneji S, et al. Identification of a human natural killer cell lineage-restricted progenitor in fetal and adult tissues. Immunity. (2015) 43:394–407. doi: 10.1016/j.immuni.2015.07.011
28. Crinier A, Milpied P, Escalière B, Piperoglou C, Galluso J, Balsamo A, et al. High-dimensional single-cell analysis identifies organ-specific signatures and conserved NK cell subsets in humans and mice. Immunity. (2018) 49:971–86.e5. doi: 10.1016/j.immuni.2018.09.009
29. Collins PL, Cella M, Porter SI, Li S, Gurewitz GL, Hong HS, et al. Gene regulatory programs conferring phenotypic identities to human NK cells. Cell. (2019) 176:348–60.e12. doi: 10.1016/j.cell.2018.11.045
30. Leonard WJ, Lin J-X, O'Shea JJ. The γc family of cytokines: basic biology to therapeutic ramifications. Immunity. (2019) 50:832–50. doi: 10.1016/j.immuni.2019.03.028
31. Robinette ML, Cella M, Telliez JB, Ulland TK, Barrow AD, Capuder K, et al. Jak3 deficiency blocks innate lymphoid cell development. Mucosal Immunol. (2018) 11:50–60. doi: 10.1038/mi.2017.38
32. Witalisz-Siepracka A, Klein K, Prinz D, Leidenfrost N, Schabbauer G, Dohnal A, et al. Loss of JAK1 drives innate immune deficiency. Front Immunol. (2018) 9:3108. doi: 10.3389/fimmu.2018.03108
33. Villarino AV, Kanno Y, O'Shea JJ. Mechanisms and consequences of Jak-STAT signaling in the immune system. Nat Immunol. (2017) 18:374–84. doi: 10.1038/ni.3691
34. Heltemes-Harris LM, Farrar MA. The role of STAT5 in lymphocyte development and transformation. Curr Opin Immunol. (2012) 24:146–52. doi: 10.1016/j.coi.2012.01.015
35. Yao Z, Cui Y, Watford WT, Bream JH, Yamaoka K, Hissong BD, et al. Stat5a/b are essential for normal lymphoid development and differentiation. Proc Natl Acad Sci USA. (2006) 103:1000–5. doi: 10.1073/pnas.0507350103
36. Eckelhart E, Warsch W, Zebedin E, Simma O, Stoiber D, Kolbe T, et al. A novel Ncr1-Cre mouse reveals the essential role of STAT5 for NK-cell survival and development. Blood. (2011) 117:1565–73. doi: 10.1182/blood-2010-06-291633
37. Imada K, Bloom ET, Nakajima H, Horvath-Arcidiacono JA, Udy GB, Davey HW, et al. Stat5b is essential for natural killer cell-mediated proliferation and cytolytic activity. J Exp Med. (1998) 188:2067–74. doi: 10.1084/jem.188.11.2067
38. Villarino A, Laurence A, Robinson GW, Bonelli M, Dema B, Afzali B, et al. Signal transducer and activator of transcription 5 (STAT5) paralog dose governs T cell effector and regulatory functions. Elife. (2016) 5:e08384. doi: 10.7554/eLife.08384
39. Villarino AV, Sciumè G, Davis FP, Iwata S, Zitti B, Robinson GW, et al. Subset- and tissue-defined STAT5 thresholds control homeostasis and function of innate lymphoid cells. J Exp Med. (2017) 214:2999–3014. doi: 10.1084/jem.20150907
40. Lin J-X, Du N, Li P, Kazemian M, Gebregiorgis T, Spolski R, et al. Critical functions for STAT5 tetramers in the maturation and survival of natural killer cells. Nat Commun. (2017) 8:1320. doi: 10.1038/s41467-017-01477-5
41. Soldaini E, John S, Moro S, Bollenbacher J, Schindler U, Leonard WJ. DNA binding site selection of dimeric and tetrameric Stat5 proteins reveals a large repertoire of divergent tetrameric Stat5a binding sites. Mol Cell Biol. (2000) 20:389–401. doi: 10.1128/MCB.20.1.389-401.2000
42. Lin J-X, Li P, Liu D, Jin HT, He J, Ata Ur Rasheed M, et al. Critical Role of STAT5 transcription factor tetramerization for cytokine responses and normal immune function. Immunity. (2012) 36:586–99. doi: 10.1016/j.immuni.2012.02.017
43. Gotthardt D, Putz EM, Grundschober E, Prchal-Murphy M, Straka E, Kudweis P, et al. STAT5 is a key regulator in nk cells and acts as a molecular switch from tumor surveillance to tumor promotion. Cancer Discov. (2016) 6:414–29. doi: 10.1158/2159-8290.CD-15-0732
44. Robbins SH, Tessmer MS, Van Kaer L, Brossay L. Direct effects of T-bet and MHC class I expression, but not STAT1, on peripheral NK cell maturation. Eur J Immunol. (2005) 35:757–65. doi: 10.1002/eji.200425797
45. Lucas M, Schachterle W, Oberle K, Aichele P, Diefenbach A. Dendritic cells prime natural killer cells by trans-presenting interleukin 15. Immunity. (2007) 26:503–17. doi: 10.1016/j.immuni.2007.03.006
46. Baranek T, Manh T-PV, Alexandre Y, Maqbool MA, Cabeza JZ, Tomasello E, et al. Differential responses of immune cells to type I interferon contribute to host resistance to viral infection. Cell Host Microbe. (2012) 12:571–84. doi: 10.1016/j.chom.2012.09.002
47. Beuneu H, Deguine J, Bouvier I, Di Santo JP, Albert ML, Bousso P. Cutting edge: a dual role for type I IFNs during polyinosinic-polycytidylic acid-induced NK cell activation. J Immunol. (2011) 187:2084–8. doi: 10.4049/jimmunol.1004210
48. Guia S, Fenis A, Vivier E, Narni-Mancinelli E. Activating and inhibitory receptors expressed on innate lymphoid cells. Semin Immunopathol. (2018) 40:331–41. doi: 10.1007/s00281-018-0685-x
49. Kruse PH, Matta J, Ugolini S, Vivier E. Natural cytotoxicity receptors and their ligands. Immunol Cell Biol. (2014) 92:221–9. doi: 10.1038/icb.2013.98
50. Lanier LL. NK cell receptors. Annu Rev Immunol. (1998) 16:359–93. doi: 10.1146/annurev.immunol.16.1.359
51. Raulet DH, Gasser S, Gowen BG, Deng W, Jung H. Regulation of ligands for the NKG2D activating receptor. Annu Rev Immunol. (2013) 31:413–41. doi: 10.1146/annurev-immunol-032712-095951
52. Sciumè G, Fionda C, Stabile H, Gismondi A, Santoni A. Negative regulation of innate lymphoid cell responses in inflammation and cancer. Immunol Lett. (in press) doi: 10.1016/j.imlet.2019.01.011
53. Sciumè G, Shih H-Y, Mikami Y, O'Shea JJ. Epigenomic views of innate lymphoid cells. Front Immunol. (2017) 8:1579. doi: 10.3389/fimmu.2017.01579
54. O'Shea JJ, Paul WE. Mechanisms underlying lineage commitment and plasticity of helper CD4+ T cells. Science. (2010) 327:1098–102. doi: 10.1126/science.1178334
55. Zhu J, Paul WE. Peripheral CD4+ T-cell differentiation regulated by networks of cytokines and transcription factors. Immunol Rev. (2010) 238:247–62. doi: 10.1111/j.1600-065X.2010.00951.x
56. Rochman Y, Spolski R, Leonard WJ. New insights into the regulation of T cells by gamma(c) family cytokines. Nat Rev Immunol. (2009) 9:480–90. doi: 10.1038/nri2580
57. Watford WT, Hissong BD, Bream JH, Kanno Y, Muul L, O'Shea JJ. Signaling by IL-12 and IL-23 and the immunoregulatory roles of STAT4. Immunol Rev. (2004) 202:139–56. doi: 10.1111/j.0105-2896.2004.00211.x
58. Lau CM, Adams NM, Geary CD, Weizman O-E, Rapp M, Pritykin Y, et al. Epigenetic control of innate and adaptive immune memory. Nat Immunol. (2018) 19:963–72. doi: 10.1038/s41590-018-0176-1
59. Rapp M, Lau CM, Adams NM, Weizman O-E, O'Sullivan TE, Geary CD, et al. Core-binding factor β and Runx transcription factors promote adaptive natural killer cell responses. Sci Immunol. (2017) 2:eaan3796. doi: 10.1126/sciimmunol.aan3796
60. Beaulieu AM, Zawislak CL, Nakayama T, Sun JC. The transcription factor Zbtb32 controls the proliferative burst of virus-specific natural killer cells responding to infection. Nat Immunol. (2014) 15:546–53. doi: 10.1038/ni.2876
61. Adams NM, Lau CM, Fan X, Rapp M, Geary CD, Weizman O-E, et al. Transcription factor IRF8 orchestrates the adaptive natural killer cell response. Immunity. (2018) 48:1172–82.e6. doi: 10.1016/j.immuni.2018.04.018
62. Miyagi T, Gil MP, Wang X, Louten J, Chu W-M, Biron CA. High basal STAT4 balanced by STAT1 induction to control type 1 interferon effects in natural killer cells. J Exp Med. (2007) 204:2383–96. doi: 10.1084/jem.20070401
63. Zhu S, Phatarpekar PV, Denman CJ, Senyukov VV, Somanchi SS, Nguyen-Jackson HT, et al. Transcription of the activating receptor NKG2D in natural killer cells is regulated by STAT3 tyrosine phosphorylation. Blood. (2014) 124:403–11. doi: 10.1182/blood-2013-05-499707
64. Gotthardt D, Putz EM, Straka E, Kudweis P, Biaggio M, Poli V, et al. Loss of STAT3 in murine NK cells enhances NK cell-dependent tumor surveillance. Blood. (2014) 124:2370–9. doi: 10.1182/blood-2014-03-564450
65. Rautela J, Huntington ND. IL-15 signaling in NK cell cancer immunotherapy. Curr Opin Immunol. (2017) 44:1–6. doi: 10.1016/j.coi.2016.10.004
66. Romee R, Leong JW, Fehniger TA. Utilizing cytokines to function-enable human NK cells for the immunotherapy of cancer. Scientifica. (2014) 2014:205796. doi: 10.1155/2014/205796
67. Marçais A, Cherfils-Vicini J, Viant C, Degouve S, Viel S, Fenis A, et al. The metabolic checkpoint kinase mTOR is essential for IL-15 signaling during the development and activation of NK cells. Nat Immunol. (2014) 15:749–57. doi: 10.1038/ni.2936
68. Mao Y, van Hoef V, Zhang X, Wennerberg E, Lorent J, Witt K, et al. IL-15 activates mTOR and primes stress-activated gene expression leading to prolonged antitumor capacity of NK cells. Blood. (2016) 128:1475–89. doi: 10.1182/blood-2016-02-698027
69. Sun JC, Beilke JN, Lanier LL. Adaptive immune features of natural killer cells. Nature. (2009) 457:557–61. doi: 10.1038/nature07665
70. Holmes TD, Bryceson YT. Natural killer cell memory in context. Semin Immunol. (2016) 28:368–76. doi: 10.1016/j.smim.2016.05.008
71. Mikami Y, Scarno G, Zitti B, Shih H-Y, Kanno Y, Santoni A, et al. NCR+ ILC3 maintain larger STAT4 reservoir via T-BET to regulate type 1 features upon IL-23 stimulation in mice. Eur J Immunol. (2018) 48:1174–80. doi: 10.1002/eji.201847480
72. Madera S, Geary CD, Lau CM, Pikovskaya O, Reiner SL, Sun JC. Cutting edge: divergent requirement of T-box transcription factors in effector and memory NK cells. J Immunol. (2018) 200:1977–81. doi: 10.4049/jimmunol.1700416
73. Madera S, Rapp M, Firth MA, Beilke JN, Lanier LL, Sun JC. Type I IFN promotes NK cell expansion during viral infection by protecting NK cells against fratricide. J Exp Med. (2016) 213:225–33. doi: 10.1084/jem.20150712
74. Sciumè G, Le MT, Gadina M. HiJAKing innate lymphoid cells? Front Immunol. (2017) 8:438. doi: 10.3389/fimmu.2017.00438
75. O'Shea JJ, Gadina M. Selective Janus kinase inhibitors come of age. Nat Rev Rheumatol. (2019) 15:74–5. doi: 10.1038/s41584-018-0155-9
76. Molgora M, Bonavita E, Ponzetta A, Riva F, Barbagallo M, Jaillon S, et al. IL-1R8 is a checkpoint in NK cells regulating anti-tumour and anti-viral activity. Nature. (2017) 551:110–4. doi: 10.1038/nature24293
77. Delconte RB, Kolesnik TB, Dagley LF, Rautela J, Shi W, Putz EM, et al. CIS is a potent checkpoint in NK cell-mediated tumor immunity. Nat Immunol. (2016) 17:816–24. doi: 10.1038/ni.3470
78. Gao Y, Souza-Fonseca-Guimaraes F, Bald T, Ng SS, Young A, Ngiow SF, et al. Tumor immunoevasion by the conversion of effector NK cells into type 1 innate lymphoid cells. Nat Immunol. (2017) 18:1004–15. doi: 10.1038/ni.3800
79. Putz EM, Guillerey C, Kos K, Stannard K, Miles K, Delconte RB, et al. Targeting cytokine signaling checkpoint CIS activates NK cells to protect from tumor initiation and metastasis. Oncoimmunology. (2017) 6:e1267892. doi: 10.1080/2162402X.2016.1267892
80. Gadina M, Johnson C, Schwartz D, Bonelli M, Hasni S, Kanno Y, et al. Translational and clinical advances in JAK-STAT biology: the present and future of jakinibs. J Leukoc Biol. (2018) 104:499–514. doi: 10.1002/JLB.5RI0218-084R
81. Schönberg K, Rudolph J, Vonnahme M, Parampalli Yajnanarayana S, Cornez I, Hejazi M, et al. JAK inhibition impairs NK cell function in myeloproliferative neoplasms. Cancer Res. (2015) 75:2187–99. doi: 10.1158/0008-5472.CAN-14-3198
82. Terrén I, Mikelez I, Odriozola I, Gredilla A, González J, Orrantia A, et al. Implication of interleukin-12/15/18 and ruxolitinib in the phenotype, proliferation, and polyfunctionality of human cytokine-preactivated natural killer cells. Front Immunol. (2018) 9:737. doi: 10.3389/fimmu.2018.00737
83. Vargas-Hernández A, Mace EM, Zimmerman O, Zerbe CS, Freeman AF, Rosenzweig S, et al. Ruxolitinib partially reverses functional natural killer cell deficiency in patients with signal transducer and activator of transcription 1 (STAT1) gain-of-function mutations. J Allergy Clin Immunol. (2018) 141:2142–55.e5. doi: 10.1016/j.jaci.2017.08.040
84. Moodley D, Yoshida H, Mostafavi S, Asinovski N, Ortiz-Lopez A, Symanowicz P, et al. Network pharmacology of JAK inhibitors. Proc Natl Acad Sci USA. (2016) 113:9852–7. doi: 10.1073/pnas.1610253113
Keywords: NK cells, innate lymphoid cells, JAK, STAT, cytokine, transcriptome, transcription factor
Citation: Scarno G, Pietropaolo G, Di Censo C, Gadina M, Santoni A and Sciumè G (2019) Transcriptional, Epigenetic and Pharmacological Control of JAK/STAT Pathway in NK Cells. Front. Immunol. 10:2456. doi: 10.3389/fimmu.2019.02456
Received: 23 July 2019; Accepted: 01 October 2019;
Published: 17 October 2019.
Edited by:
Ewa Sitnicka, Lund University, SwedenReviewed by:
Lisa Renee Forbes, Baylor College of Medicine, United StatesFrancisco Borrego, Biocruces Bizkaia Health Research Institute, Spain
Copyright © 2019 Scarno, Pietropaolo, Di Censo, Gadina, Santoni and Sciumè. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Giuseppe Sciumè, Z2l1c2VwcGUuc2NpdW1lQHVuaXJvbWExLml0