 Michael D. Howell
Michael D. Howell Fiona I. Kuo†
Fiona I. Kuo† Paul A. Smith
Paul A. Smith- Incyte Corporation, Wilmington, DE, United States
Autoimmune skin diseases are characterized by significant local and systemic inflammation that is largely mediated by the Janus kinase (JAK)–signal transducer and activator of transcription (STAT) pathway. Advanced understanding of this pathway has led to the development of targeted inhibitors of Janus kinases (JAKinibs). As a class, JAK inhibitors effectively treat a multitude of hematologic and inflammatory diseases. Growing evidence suggests that JAK inhibitors are efficacious in atopic dermatitis, alopecia areata, psoriasis, and vitiligo. Additional evidence suggests that JAK inhibition might be broadly useful in dermatology, with early reports of efficacy in several other conditions. JAK inhibitors can be administered orally or used topically and represent a promising new class of medications. Here we review the evolving data on the role of the JAK-STAT pathway in inflammatory dermatoses and the potential therapeutic benefit of JAK-STAT antagonism.
JAK-STAT Signaling Pathway
The mammalian Janus kinase (JAK) family contains three JAKs (JAK1–3) and tyrosine kinase 2 (TYK2), which selectively bind different receptor chains (1). Upon binding of ligand to its cognate cytokine receptor, associated JAKs become activated and undergo autophosphorylation and transphosphorylate the intracellular tail of their receptors. This creates docking sites for the SH2 domain of the cytoplasmic transcription factors termed signal transducers and activators of transcription (STATs). The human STAT family contains seven STATs: STAT1, STAT2, STAT3, STAT4, STAT5A, STAT5B, and STAT6. Following phosphorylation, STATs are translocated to the nucleus, dimerize, and bind to specific DNA sequences to regulate gene transcription (2). The JAK-STAT pathway is pivotal for the downstream signaling of inflammatory cytokines, including interleukins (ILs), interferons (IFNs), and multiple growth factors (3, 4). Overall, the selective use of JAKs by different receptors coupled to downstream STAT signal transduction results in an elegant mechanism to achieve exquisite in vivo specificity for more than 60 cytokines and growth factors (Figure 1).
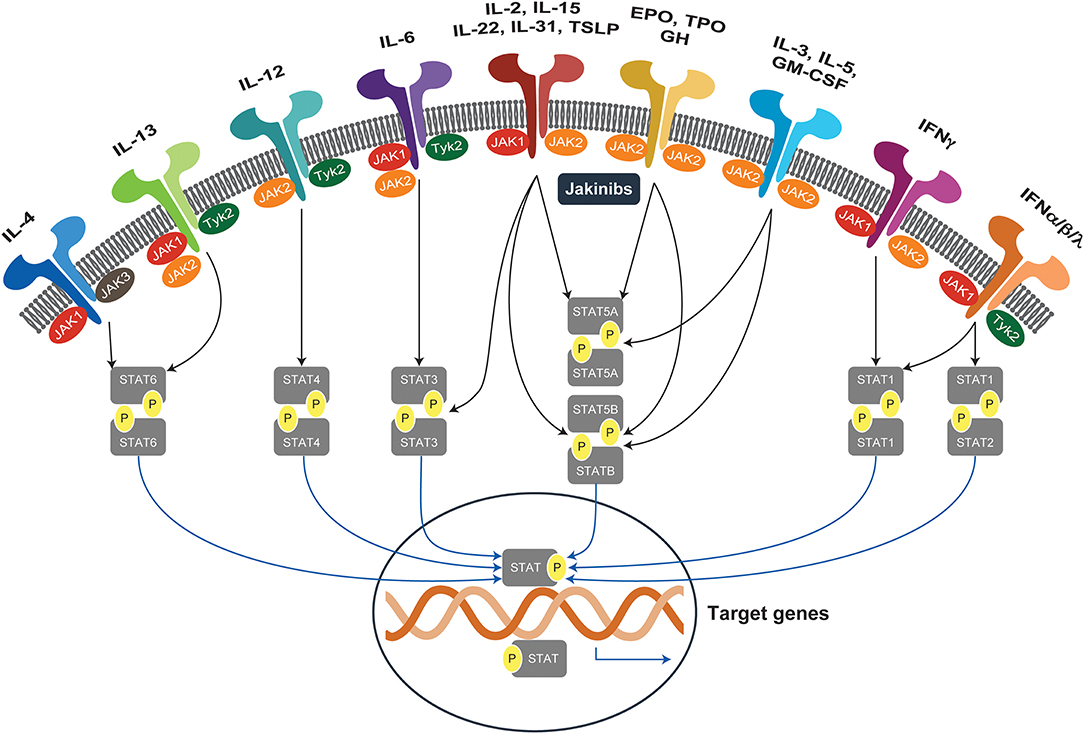
Figure 1. JAK-STAT signaling pathways. Janus kinases (JAK1-3, TYK2) are activated by more than 60 extracellular stimuli and phosphorylate downstream STAT proteins, which translocate to the nucleus and activate target genes. EPO, erythropoietin; GH, growth hormone; GM-CSF, granulocyte-macrophage colony-stimulating factor; IFN, interferon; IL, interleukin; JAK, Janus kinases; JAKinibs, Janus kinase inhibitors; STAT, signal transducer and activator of transcription; TPO, thrombopoietin; TSLP, thymic stromal lymphopoietin; TYK2, tyrosine kinase.
Identification of selective pharmacologic JAK inhibitors (JAKinibs) has been an ongoing research and development goal. The first JAKinib to gain FDA approval in 2011 was ruxolitinib for intermediate or high-risk myelofibrosis, thereby showing that JAK inhibition was not only possible, but safe and effective for its intended uses. More recently, selective JAK inhibitors have been explored for specific inflammatory disease indications (Table 1).
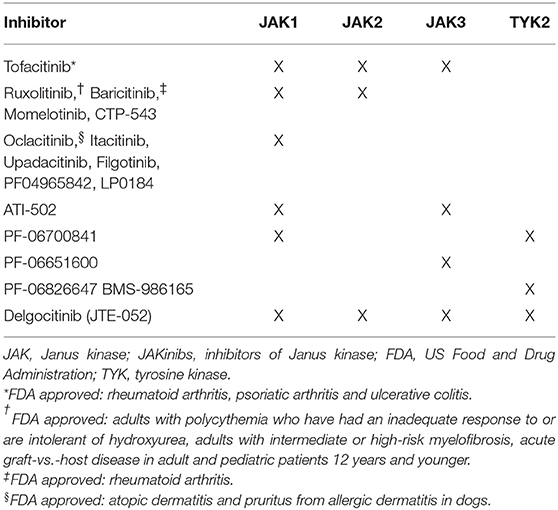
Table 1. Selectivity profiles of clinically active JAKinibs.
The JAK-STAT Pathway and T Helper Subsets
The differential fate of naive T cells into committed T helper (Th) subsets is orchestrated under the instruction of professional antigen-presenting cells within a JAK-STAT–dependent cytokine milieu (Figure 2). In vivo Th1 differentiation depends on JAK-mediated signaling through the IFNγ receptor (IFNGR), the IL-12 receptor (IL-12R), and downstream STAT1/4 phosphorylation culminating with T-bet gene transcription (5). Ultimately, IFNγ signaling initiates the Th1 differentiation program and IL-12 perpetuates it. In contrast, Th2 cells arise after occupancy of the IL-4Rα by its ligands IL-4 and IL-13, triggering JAK1/3 and subsequent activation of STAT6 (6), and leading to transcriptional regulation of the GATA3 target gene (5). More recently, the critical role of IL-17–producing Th cells (termed Th17 cells) in host defense against extracellular bacteria, maintenance of epithelium barrier integrity, and autoimmune pathogenesis has become increasingly clear. Within the immunologic microenvironment, IL-6 produced by activated dendritic cells (DCs) is a key factor in promoting Th17 differentiation via STAT3 and retinoic acid receptor–related orphan receptor γ (RORγt) induction (7) with IL-23 critical for memory Th17 in vivo function (3, 8).
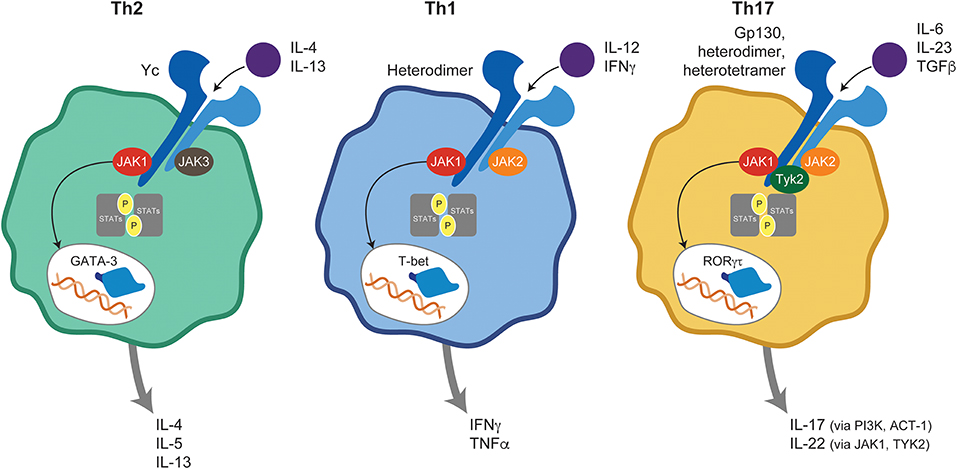
Figure 2. JAK-mediated cytokine signaling in T helper subsets. Ligand binding to its cognate receptor triggers JAK-STAT activation and plays a central role in naive T-cell differentiation into Th1, Th2, and Th17 subsets. ACT, Nuclear factor NF-kappa-B activator 1; GATA, GATA transcription factor 3; IFN, interferon; IL, interleukin; JAK, Janus kinase; PI3K, Phosphoinositide 3-kinases; RORγt, retinoic acid receptor-related orphan receptor γ; STAT, signal transducer and activator of transcription; T-bet, T-box transcription factor TBX21; Th, T helper; TGF, transforming growth factor; TNF, tumor necrosis factor; TYK, tyrosine kinase.
Atopic Dermatitis
Atopic dermatitis (AD) is a chronic, inflammatory skin disease that typically begins in early childhood and occurs more frequently in families with a history of other atopic diseases (bronchial asthma and/or allergic rhinoconjunctivitis). Overall, the prevalence of AD is up to 20% in children and 10% in adults, with rates varying geographically (9, 10). AD clinically manifests as recurrent eczematous lesions that negatively affect quality of life through sleep disturbances due to chronic itch (pruritus) (11, 12), increased likelihood of developing depression (13), and significant economic burden (14).
The cellular infiltrate of AD lesions mainly consist of CD4+ T cells, which are considered key drivers of inflammation (15). Lesional skin is characterized by an overexpression of inflammatory Th2-cytokines (IL-4, IL-13, IL-31), and Th22-cytokines (IL-22) (16). Crucially, the cytokines IL-4, IL-13, IL-31, and IL-22 require JAK-STAT downstream signaling (3) for their biological function (Figure 3). Spontaneous and induced rodent dermatitis models have been extensively used to explore the effectiveness of small-molecule JAK inhibitors on reducing inflammation. Delgocitinib (pan-JAK) inhibited skin inflammation in hapten-induced chronic dermatitis in mice, as evidenced by reduced levels of inflammatory cytokines in the skin and IgE in serum (17). In addition, momelotinib (JAK1/JAK2) downregulated IL-4 expression, reduced the skin severity scores and reduced total serum IgE levels in the 2,4-dinitrochlorobenzene (DNCB)-induced AD mice (18). Similarly, tofacitinib (JAK1/3) and oclacitinib (JAK1) inhibited the production of proinflammatory Th2 cytokines, including IL-4, in the toluene-2,4-diisocyanate (TDI) dermatitis model (19). Moreover, tofacitinib demonstrated anti-inflammatory activity in the oxazolone-induced chronic allergic contact dermatitis model (20).
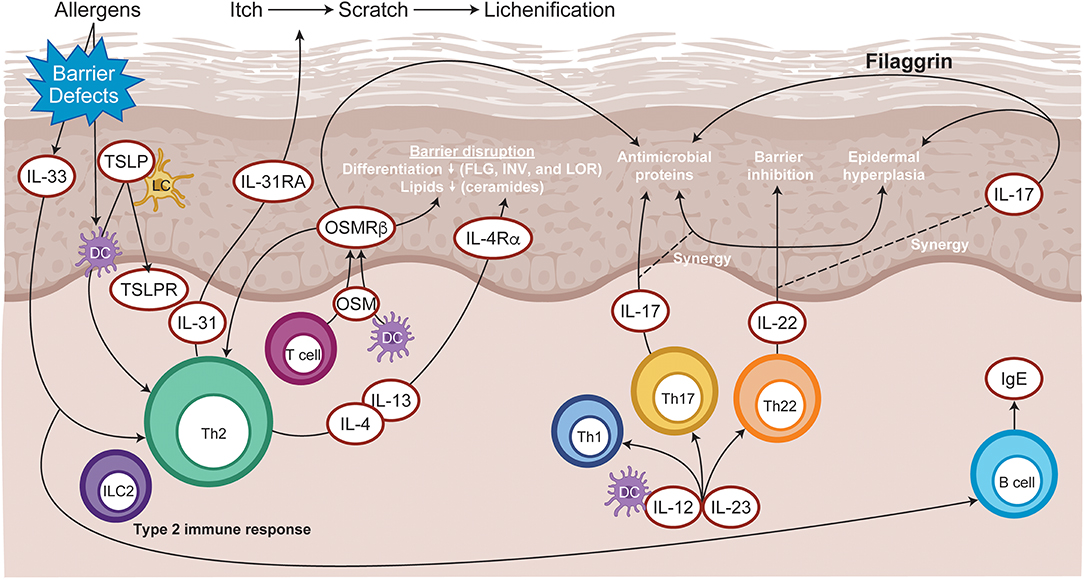
Figure 3. Immunopathogenesis of atopic dermatitis. Allergen entry through the disrupted epidermal barrier stimulates keratinocytes to express cytokines, such as IL-33 and TSLP, which trigger ILC2 and Th2 cell mediated inflammation. Skin-resident dendritic cells take up exogenous and self-antigens released from damaged cells and promote type 2 immunity. CD8+ T cells infiltrate atopic dermatitis skin and activate Th2 cells to further release IL-4 and IL-13, which promotes IgE class switching. Cytokines released from skin infiltrating Th17 and Th22 lymphocytes synergize, leading to further barrier impairment and epidermal hyperplasia. DC, dendritic cell; FLG, Filaggrin; Ig, immunoglobulin; IL, interleukin; ILC2, type 2 innate lymphoid cells; INV, Involucrin; LC, Langerhans cell; LOR, Loricrin; OSM, Oncostatin M; OSMRβ, Oncostatin M receptor β; Th, T helper; TSLP, thymic stromal lymphopoietin.
Interleukin-22 is elevated in AD lesions and is associated with epidermal thickening, skin barrier disruption, and increased expression of thymic stromal lymphopoietin (TSLP) and IL-33 cytokines (21). In addition, IL-22 potently induces the expression of gastrin-releasing peptide, a neuropeptide pruritogen, in dermal cells, dermal afferent fibers, and skin innervating ganglion neurons that positively correlate with the scratching behavior (22). The relevance of IL-22 in AD pathogenesis was emphasized by the observation of sustained clinical improvements in patients with moderate to severe AD receiving anti–IL-22 therapy (23). IL-22 binds its cognate receptor comprising a heterodimeric complex of IL-22RA1 and IL-10R2 subunits, leading to activation of JAK1 and TYK2 and phosphorylation of STAT3 (24).
Thymic stromal lymphopoietin and IL-31 cytokines also significantly contribute to triggering of inflammatory itch, under the control of IL-4, IL-13, and IL-33 (25). Crucially, pruritogenic cytokines IL-31 and TSLP use JAK1 and JAK2 downstream signaling (26, 27). Additionally, preclinical evidence has confirmed that pharmacologic inhibition of the JAK-STAT pathway is sufficient for the amelioration of pruritus-associated dermatitis. Examples include oclacitinib, which is licensed for pruritus associated with allergic dermatitis in dogs (28). Similarly, topical application of ruxolitinib (JAK1/JAK2) ameliorated TSLP-induced inflammation in mice (29). In the TDI-induced mouse model of dermatitis, oclacitinib and tofacitinib inhibited itch symptoms and significantly reduced IL-31, tumor necrosis factor–α (TNFα), and TSLP cytokine secretions (19). Moreover, TSLP can activate tissue resident dendritic cells that promote the transformation of Th-naïve lymphocytes to the Th2 phenotype thereby facilitating tissue inflammation (30). Finally, neuronal IL-4Rα acting via JAK1 signaling can also significantly contribute to chronic itch (31).
Skin barrier disruption and the resulting continuous exposure to allergens are presumed to be responsible for the development of atopic dermatitis (AD). JAK1-mediated Th2 cytokines IL-4 and IL-13 acting in a STAT-dependent manner (32) negatively affect skin barrier integrity by inhibiting the expression of filaggrin, loricrin, and involucrin, resulting in destabilization of tight junctions (33, 34). JAK inhibition restored filaggrin and loricrin expression following in vitro pretreatment with IL-4 /IL-13 cytokines of human keratinocyte. Moreover, mice harboring a point mutation leading to JAK1-specific hyperactivation develop spontaneous skin barrier disruption and a dermatitis phenotype (35). Topical application of delgocitinib ameliorated spontaneous AD-like skin inflammation and barrier disruption in an NC/Nga “dry skin” mouse model and restored filaggrin levels in an experimental human skin graft model leading to improved barrier function (36). Moreover, downstream signaling of IL-4 and IL-13 also suppresses the induction of innate immune response genes, such as β-defensins (33), thereby facilitating skin microbiome dysbiosis, including aberrant Staphylococcus aureus colonization (37).
The role and activation of Th1 and Th17 cell-mediated responses require further elucidation, but these pathways appear to be overexpressed in chronic disease stages, children, and people of Asian ethnicity (38, 39).
Targeting the JAK family of kinases in AD has proven, in recent years, to be therapeutically beneficial. Oral tofacitinib administration was evaluated in 6 patients with moderate to severe AD and showed a promising reduction in skin severity (40). The next generation of orally administered JAKinibs includes baricitinib (JAK1/2) along with two JAK1 selective molecules, upadacitinib (JAK1) and abrocitinib (JAK1). In clinical trials in moderate to severe AD patients, oral administration of these JAKinibs significantly reduced the eczema area severity index (EASI) scores by more than 50%. More specifically, in one clinical trial (ClinicalTrials.gov identifier, NCT02925117), oral administration of upadacitinib (JAK1) resulted in 90% improvement in the eczema area severity index (EASI) score for ~50% of enrolled participants after 16 weeks of treatment (41). Results from two baricitinib phase 3 studies showed that more patients achieved an investigator global assessment (IGA) 0/1 with barcitinib 4 mg once daily (QD) and 2 mg QD than with placebo (42). Significant improvements in EASI and patient-reported outcomes were observed as early as Week 1 (Table 2) (53). In a recent phase 1b study (NCT03139981), ASN002, a pan JAK inhibitor that also inhibits spleen tyrosine kinase (SYK), showed a 50% improvement in EASI in 100% of participants within 4 weeks (56).
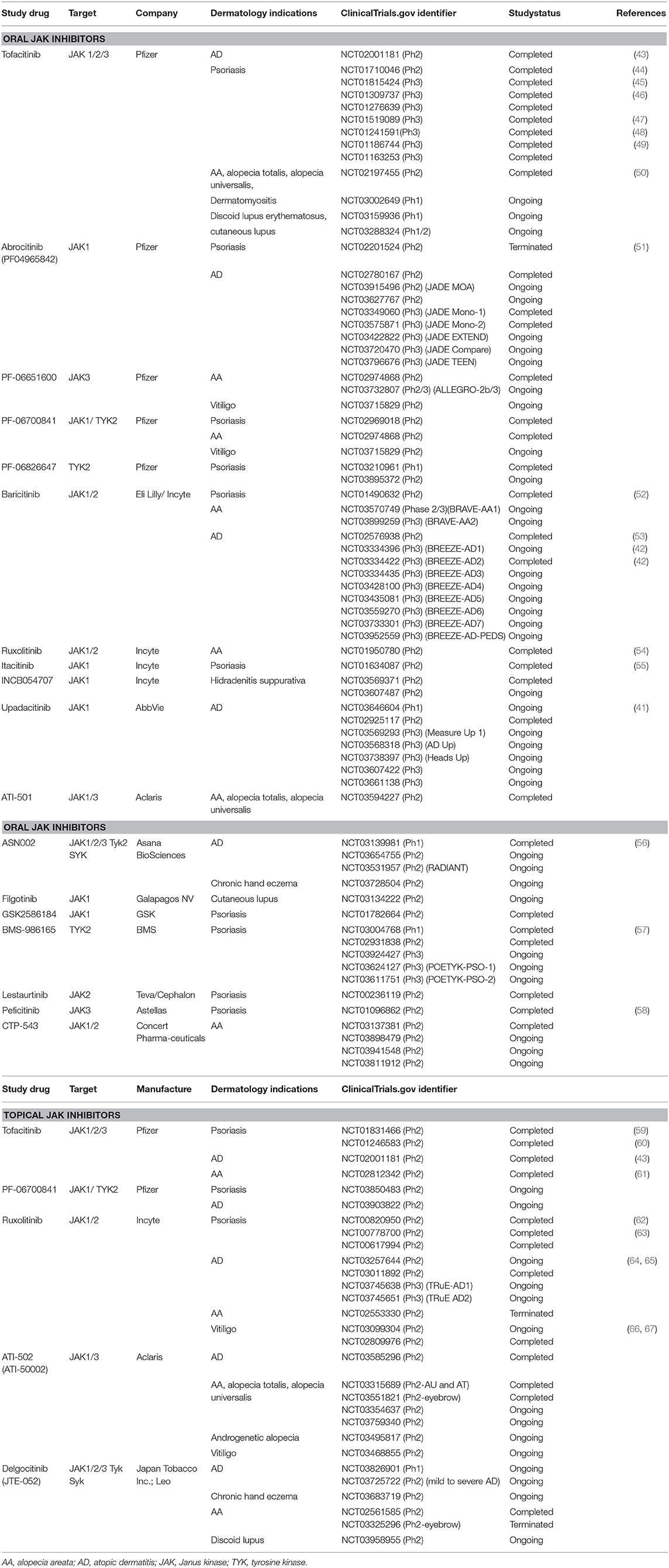
Table 2. Summary of JAK inhibitor use in the treatment of dermatologic conditions.
Topical administration of tofacitinib to patients with mild to moderate AD in a clinical trial (NCT02001181) demonstrated significant improvement in EASI scores at Week 4 (43) and improvement in pruritus as early as Day 2. In a phase 2 study (NCT03011892) involving patients with mild to moderate AD receiving ruxolitinib cream, mean percentage change from baseline at Week 4 in EASI score demonstrated a significant improvement and was non-inferior to triamcinolone. Interestingly, significant reductions in itch were noted as early as 1 day after initiation of therapy (64, 65). More recently, pilot studies of topical ATI-502 (JAK1/3) solution (NCT03585296) and PF-06700841 (JAK1/TYK2) cream (NCT03903822) in AD are ongoing. Given the early successes of JAKinibs in AD, ongoing investigation and evaluation is expected to further elucidate the differential effects of JAK selectivity.
Alopecia Areata
Alopecia areata (AA) is an autoimmune disease resulting in partial or complete nonscarring hair loss, with a prevalence of ~1.7 to 2.1% (68). Susceptibility to AA is indiscriminate between the sexes and ethnicities, with initial disease onset often occurring before the third decade of life. Early symptoms are typically characterized by small, well-defined patches of hair loss that may spontaneously resolve with time; however subsequent relapses occur in around a third of cases. Spontaneous remission is rare in patients with alopecia totalis or alopecia universalis. To date, no FDA- or European Medicines Agency–approved treatments exist.
Multiple lines of evidence have demonstrated that AA pathogenesis is autoimmune in nature, with loss of immune privilege and associated T cells infiltration selectively attacking growth at the hair follicle (i.e., anagen phase) (69–72). Healthy hair follicles achieve immune privilege at the anagen phase by downregulation of expression of major histocompatibility complex (MHC) class I and class II molecules (70, 73) and by expression of NK and CD8+ cell inhibitors, such as macrophage migration inhibitory factor (MIF) and transforming growth factors (TGF) β1 and β2, which generate an immunosuppressive microenvironment (74–76). Importantly, hair follicle epithelial stem cells are usually spared during the autoimmune attack, which provides a potential mechanism of hair growth recovery with effective anti-inflammatory treatment (70).
Many different mammalian species, including rodents, are susceptible to AA and this has facilitated preclinical models for the elucidation of cellular and molecular immune pathways (77, 78). The inbred C3H/HeJ strain spontaneously develops alopecia in up to 20% of mice via an IFNγ- and inflammasome-sensitive mechanism (79); however recipient C3H/HeJ animals that receive skin grafts from donor alopecic C3H/HeJ mice develop an accelerated phenotype with nearly 100% disease penetrance (80). Transfer or deletion of effector CD8+ T cells is sufficient to induce or block disease in preclinical models (70, 71, 81), which in consistent with the observation that cytotoxic CD8+NKG2D+ T cells expressing granzyme B (82) infiltrate around the hair follicles and are major contributors of hair loss (81, 83).
Global transcriptional analyses of mouse and human affected skin identified expression signatures indicative of cytotoxic T-cell infiltration, such as increased production of IFNγ and γ-chain (γc) cytokines, including IL-15 (81, 84). Furthermore, inhibiting IFNγ either by genetic deletion or neutralizing antibody significantly ameliorates AA development and severity (85), supporting the hypothesis that IFNγ drives AA pathogenesis by inducing ectopic expression of MHC molecules and ligands that stimulate NK-cell receptors (NKG2D) in the anagen hair bulb leading to the collapse of the hair follicle immune privilege (70, 86–88). An important cellular source of IFN is plasmacytoid dendritic cells (pDCs), which are normally absent from healthy skin, but migrate into tissues in response to inflammatory stimuli or infection. Infiltrating pDCs have been identified around hair follicles of patients with AA (89) and, upon activation, produce large quantities of type I IFNs (90).
The IFNγ-induced chemokine receptor CXCR3 and its ligands CXCL9 and CXCL10 are upregulated around hair follicles during early AA pathogenesis (Figure 4), thereby facilitating lymphocyte recruitment (82, 91). CXCR3 is primarily expressed on Th1 CD4+ T cells, CD8+ T cells, NK, and PDCs during skin inflammation (92), whereas CXCR3 ligands are secreted by many tissue resident cells, including dendritic cells.
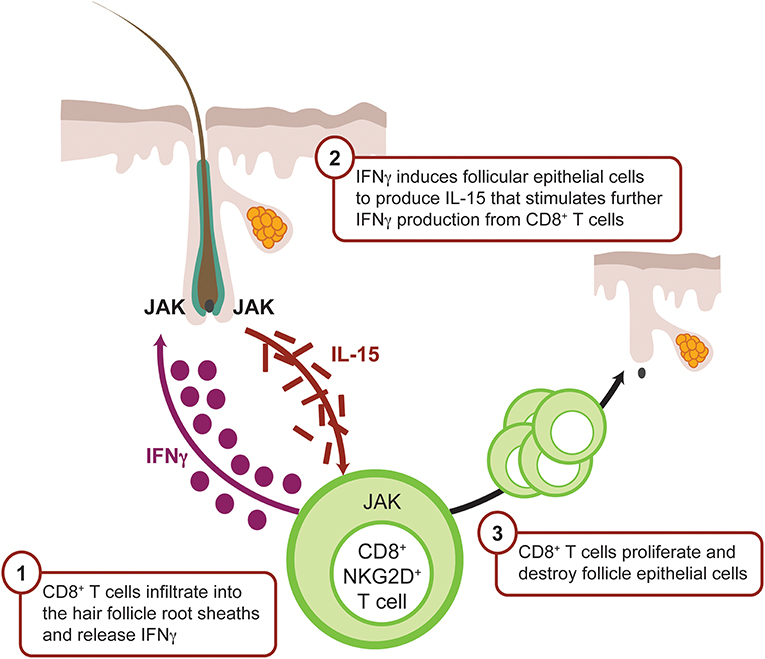
Figure 4. IFNγ-driven inflammation in alopecia areata is JAK mediated. CD8+ T cells infiltrate the dermis, localize to the hair follicle bulb, and release IFNγ. IFNγ binds the IFN receptor on the surface of the follicular epithelial cell, which in turn signals via JAK1 and JAK2 to promote production of IL-15, a mediator of CD8+ T-cell activation. IL-15 binds IL-15 receptor on the CD8+ T cell surface, resulting in signaling via JAK1 and JAK3 to enhance the production of IFNγ and amplify the feedback loop. CD8+ T cells then attack the hair follicle, which causes hair loss. CXCL, chemokine (C-X-C motif) ligand; IFN, interferon; JAK, Janus kinase.
Like IFNγ, IL-15 enhances innate and self-reactive memory T-cell immunity, including autoimmune disease pathogenesis (84, 93), and signals via the JAK1/3 pathway with downstream STAT-5 activation (94). Similarly blocking IL-2 or IL-15 receptor beta (IL-15Rβ) ameliorated disease development by inhibiting CD8+NKG2D+ T-cell accumulation in the skin (81).
The combination of published genome-wide association studies in patients with AA that highlighted JAK signaling (87, 95) and the knowledge that IFNγ primarily signals through JAK1/2 and IL-15 mostly through JAK1/3, provided a compelling rationale for the exploration of small-molecule JAK inhibitors in AA disease (81, 96). Preclinical evaluation of orally administered ruxolitinib and tofacitinib in the skin graft C3H/HeJ mouse model demonstrated disease prevention (81). Marked decreases of CD4, CD8, and MHC class I and II as well as a reduced numbers of CD8+/NKG2D+ cells were observed in the skin (81). Subsequently, prophylactic and therapeutic baricitinib treatment ameliorated disease and normalized the Alopecia Areata Disease Activity Index (ALADIN) IFNγ gene expression signature (80). Topical administration of JAK inhibitors reversed AA in C3H/HeJ mice (81); however, murine skin is significantly thinner and easier to penetrate, and, therefore the translational validity of these data is still unknown. In addition to its proinflammatory activity, IFNγ-induced JAK/STAT signaling and the recruitment of CD8+ T cells through CXCL9 and CXCL10 can directly interfere with the hair growth cycle via suppressed proliferation and activation of hair stem cells (97) and reduction of angiogenesis (98).
Several case studies have reported improvement of AA in patients who received JAK inhibitors for other autoimmune/autoinflammatory disorders or JAK-STAT gain-of-function mutation diseases (99–103).
Oral tofacitinib has been tested in two open-label studies (NCT02197455 and NCT02312882) and several case reports. In one trial, tofacitinib 5 mg twice daily (BID) was given to patients with severe AA, alopecia totalis, or alopecia universalis. After the 12-week treatment period, nearly two-thirds of patients showed some hair regrowth and 32% of patients achieved a 50% improvement in their Severity of Alopecia Tool (SALT) score (50). The second smaller open-label study in moderate to severe AA demonstrated improved results by increasing the dose of tofacitinib to 10 mg BID (104). Recently, two retrospective studies showed successful treatment of severe AA, alopecia totalis, or alopecia universalis for up to 18 months using tofacitinib, with 58% achieving a 50% improvement 20% achieving a 90% improvement in their SALT score (105–107). Oral ruxolitinib was tested in an open-label study in 12 patients with moderate to severe AA and treatment with 20 mg ruxolitinib BID for 6 months was associated with ≥50% improvement in SALT score for 75% of patients (NCT01950780) (54). Regrowth was seen, in patches as soon as 1 month after study medication was initiated. Following cessation of treatment, shedding was observed, suggesting that pharmacologic JAK inhibition suppresses AA pathogenesis but does deplete autoreactive lymphocytes. Topical formulations of ruxolitinib, tofacitinib, ATI-502 (JAK1/3), and delgocitinib have reported mixed efficacy results in case studies and small proof-of-concept clinical trials (61, 108, 109). At present, there are several clinical trials testing topical JAK inhibitors in patients with different forms of AA, but published results are not yet available (Table 2).
Psoriasis
Psoriasis is a chronic, autoimmune, erythematosquamous dermatosis disorder that affects 2 to 3% of the world population. Skin lesions appear with scaling and redness (110) and are characterized by excessive keratinocyte proliferation (acanthosis), as well as retention of nuclei in the stratum corneum owing to aberrant keratinocyte differentiation (parakeratosis). Multiple inflammatory cell populations are observed within the lesions, including T cells, B cells, neutrophils, and DCs (110). Infiltrating autoreactive T lymphocytes, mainly represented by Th17, Th1, and Th22 cells, release IL-17, IFNγ, IL-22, and TNFα to potentiate disease pathogenesis. All of these cytokines induce keratinocyte-mediated recruitment and activation of additional DCs and lymphocytes, thereby perpetuating the pathogenic cycle (111, 112).
Many of the critical pathogenic mediators of psoriasis are linked to the JAK-STAT signaling pathway. For example, IL-23 engagement with its cognate receptor uses JAK1/2/TYK2 signaling, resulting in downstream STAT3 and STAT4 activation. Within psoriatic skin, dendritic cells and macrophages produce IL-23, which promotes Th17 cell expansion and survival (113). Furthermore, IL-23 together with IL-1β activates γδ T cells to amplify IL-17 production (114). Th17 and γδ T cells found in psoriatic skin are the primary source of IL-22, and this cytokine triggers reduced differentiation, increased proliferation, and acanthosis in psoriatic keratinocytes via STAT3 activation (115). IL-22 binds to its IL-10R2 and IL-22R1 heterodimeric cell surface receptor coupled to JAK1/TYK2 and STAT3 signaling (111, 116). Moreover, gene polymorphisms of IL23A, IL23R, STAT3, RUNX3, and TYK2 have also been identified as susceptibility factors for developing psoriasis (117). More recently, JAK1 expression has been reported to positively correlate with disease duration and Psoriasis Area and Severity Index (PASI) (118) score. Within the inflamed tissue psoriatic lesion microenvironment, other cytokines, such as IL-6 and IL-21, can enhance IL-17 production from Th17 cells in a JAK-STAT–dependent manner (119, 120).
Various rodent models have mechanistically evaluated the importance of JAK-STAT signaling in psoriasis-like lesion formation and disrupted barrier function. Intradermal injection of IL-23 induces a psoriasis-like pathophysiology in mice (121). Oral administration of delgocitinib or topical administration of ruxolitinib significantly inhibited ear swelling (29, 122), and efficacy was associated with reduced IL-22 expression (29). Tofacitinib, modulates both innate and adaptive immunity leading to inhibited pathogenic Th17 cell differentiation via reduced IL-23 expression (123). In human keratinocyte cultures activated with psoriasis-relevant proinflammatory cytokines, tofacitinib suppressed expression of IFNγ-dependent inflammatory genes and normalized keratinocyte responses. Similarly, in the imiquimod-induced psoriasis model that is IL-23/IL-17/IL-22–dependent (121), tofacitinib significantly reduced epidermal thickening and IL-17+ or IL-22+ lymphocyte infiltration into the dermis (124). Furthermore, a small molecule JAK3 / SYK inhibitor (R348, Rigel Pharmaceuticals), attenuated T-cell–dependent psoriasiform skin lesions in the CD18 mutant PL/J mouse model, including significant reductions in epidermal and dermal lesion scores (125). In T cells, IL-12 induces IFNγ production, IL-23 enhances the differentiation of Th17 cells, and both require TYK2 signaling (126, 127). TYK2 knockout reduced inflammatory response and limited epidermal hyperplasia in the intradermal IL-23 model (128). Furthermore, TYK2-deficient mice were more resistant to several Th1 and Th17 cells autoimmune disorders, including imiquimod-induced psoriasis-like dermatitis (128). The combined JAK1/TYK2 inhibitor, SAR-20347, demonstrated in vitro and in vivo concentration-dependent reduction of IL-12, IL-22, and IFNγ-mediated inflammation and tissue pathology in the imiquimod-induced psoriasis model (129). Finally, experimentally induced skin trauma in the keratin5.Stat3C transgenic mice (130), which constitutively overexpresses active STAT3 in keratinocytes, develops T-cell– and IL-23–dependent psoriasis-like lesions. Topical administration of a STAT3 inhibitor prevented disease symptoms (130–132). These preclinical findings are consistent with the postulate that the JAK-STAT pathway plays a central role in psoriasis pathogenesis.
Clinically, the efficacy of oral tofacitinib in moderate to severe plaque psoriasis was demonstrated in two phase 3 randomized controlled trials (46). Tofacitinib at 10 mg BID was determined to be non-inferior to etanercept (50 mg subcutaneously twice weekly) (47). Baricitinib was reported to be efficacious in moderate to severe psoriasis in a phase 2 trial (NCT01490632). In this 12-week dose-ranging study, a 75% reduction in PASI was achieved by 43% and 54% of patients treated with baricitinib 8 and 10 mg QD, respectively (52). Itacitinib (JAK1) was evaluated in a phase 2 dose-escalation study in which patients experienced a significant improvement in the Physician Global Assessment (PGA) score at Week 4 with itacitinib 600 mg QD vs. placebo (NCT01634087) (55). Peficitinib (JAK1/3) reported improvements in PASI, PGA, and body surface area at higher dose (50 mg QD) at Day 42 in a phase 2 study (58). In another phase 2 study, 57% of patients treated with GSK2586184 (JAK1) 400 mg QD achieved a 75% reduction in PASI at Week 12 (133).
Topical tofacitinib has been tested in patients with psoriasis, with conflicting results (52, 60, 134). Three psoriasis clinical trials have been completed using topical ruxolitinib cream. In a phase 2 vehicle-controlled study in mild to moderate psoriasis (NCT00778700), ruxolitinib reported PASI reduction, although no clear dose-response was observed. A subsequent trial in 29 patients with psoriasis compared ruxolitinib cream to two active comparators (calcipotriene 0.005% cream and betamethasone dipropionate 0.05% cream; NCT00820950). Both ruxolitinib 1% QD and 1.5% BID achieved clinical efficacy, with 1.5% BID topical ruxolitinib cream being non-inferior to active comparators (62). Finally, a third study conducted in 25 patients with limited psoriasis (covering <20% of the body surface area; NCT00617994) showed that epidermal hyperplasia and dermal inflammation were reduced with ruxolitinib in most patients, along with immunohistochemical markers of inflammation (CD3, CD11c, Ki67, and K16). No significant inhibition of phosphorylated STAT3 in peripheral blood cells was observed, suggesting limited systemic exposure (63). A number of other JAK inhibitors have been studied in psoriasis (Table 2).
Vitiligo
Vitiligo is a chronic, autoimmune depigmenting disorder that results from destruction of melanocytes, causing white spots on the affected skin. The global vitiligo prevalence is ~0.5 to 2.0% and varies geographically, with no epidemiologic differences between sexes or races (135, 136). Vitiligo can be stigmatized by society, resulting in a significant impact to patient quality of life (137, 138). It is therefore inappropriate to categorize vitiligo as simply a cosmetic problem.
In vitiligo, the frequency of anti-melanocyte CD8+ T cells in the blood and skin correlates with disease severity, and lesional CD8+ T cells in vitro induce melanocyte apoptosis in unaffected skin (139, 140). These data support the rationale that cytotoxic T lymphocytes are directly responsible for melanocyte destruction in human vitiligo (Figure 5). Expression analysis reveals an IFNγ-specific signature that is associated with infiltrating autoreactive CD8+ T cells (140, 141). Transcriptome analysis on the skin and blood of patients with vitiligo revealed IFNγ-induced chemokines CXCL10 and CXCL9 were increased (142, 143), which is consistent with the observed abundance of autoreactive T cells expressing the cognate CXCR3 receptor (144). Furthermore, serum CXCL10 levels were associated with Vitiligo Area Scoring Index (VASI) of patients with progressive vitiligo, suggesting that the CXCL10/CXCR3 axis mediates T-cell recruitment into the skin of progressive vitiligo.
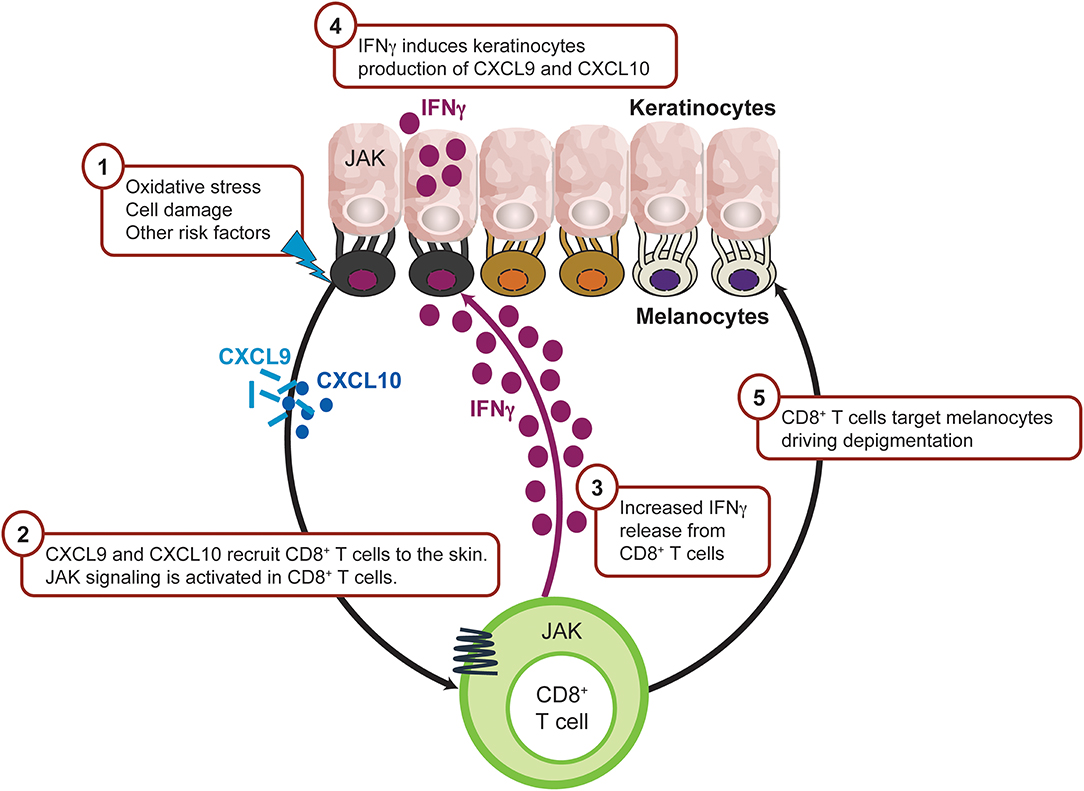
Figure 5. IFNγ-driven inflammation in vitiligo is JAK mediated. Intrinsic and/or extrinsic factors induce the cellular stress response in melanocytes, which then activates innate immunity within the skin to trigger the initial inflammation that leads to autoimmunity. As a result, CXCL9 and CXCL10 are released from keratinocytes leading to recruitment of CD8+ T cells. Activated CD8+T cells produce IFNγ which trigger more CXCL9 and CXCL10 production from keratinocyte through JAK1 and JAK2 signaling and recruit more CD8+ T cells to the inflamed sites. CD8+ T cells then destruct melanocytes and lead to depigmentation. CXCL, chemokine (C-X-C motif) ligand; IFN, interferon; IL, interleukin; JAK, Janus kinase; NKD2D, natural killer group 2D.
Consistent with active human vitiligo reports, an adoptive transfer of melanocyte-specific CD8+ mouse model shows epidermal depigmentation but sparing of the hair follicle. Mechanistic studies, including neutralizing antibodies, have demonstrated that depigmentation is IFNγ-dependent via the local accumulation of melanocyte-specific CD8+ T cells within the skin. Adoptive transfer of CXCR3-deficient T cells or inhibition of CXCL10 signaling ameliorated overall disease phenotype, whereas CXCL9 promoted autoreactive T-cell recruitment to the skin but did not significantly contribute to effector function (141, 145). Keratinocytes appear to significantly contribute to the disease process as major chemokine producers via IFN signaling, resulting in augmented autoreactive T-cell homing to the epidermis (146).
Given the apparent critical role for IFNγ in driving vitiligo inflammation and its downstream signaling dependent on the JAK1-JAK2 heterodimer, it is perhaps not surprising that intense and diffuse JAK1 expression is more present within vitiliginous skin compared with healthy tissue. Moreover, high JAK1 expression was associated with short disease duration and a lower percentage of surviving melanocytes (118, 147, 148).
Multiple case reports suggested that orally administered JAK inhibition significantly modulated the vitiligo autoimmune response and facilitated repigmentation (102, 108, 149). Another possible approach to diminish local inflammation and promote repigmentation in vitiligo, but minimize systemic drug exposures, is the use of topical JAK inhibitors. Recently, Rothstein et al. reported a very small open-label trial without placebo control (NCT02809976) in which nine patients completed the 20 week study period. Twice daily ruxolitinib cream demonstrated time-dependent improvement in facial VASI (F-VASI) in the majority of the enrolled vitiligo patients (67). Recently, ruxolitinib cream was tested in a randomized, double-blinded, dose-ranging, vehicle-controlled, phase 2 study in 157 adult patients with vitiligo (NCT03099304). The results show that significantly more patients treated with ruxolitinib cream for 24 weeks achieved a ≥50% percent improvement from baseline in the facial VASI score compared with patients treated with a control vehicle [(66); World Congress of Dermatology; June 2019; Milan, Italy].
Conclusions
Despite phenotypic differences in the inflammatory mediators responsible for driving disease pathogenesis, these aforementioned dermatoses are characterized by increased inflammatory mediators that signal through the JAK-STAT pathway.
JAK inhibitors are emerging as an exciting class of treatments in the field of dermatology. In murine models of skin inflammation, JAK inhibitors significantly modulated key mechanistic phenotypes that correspond with clinical readouts, such as acanthosis and pruritus. Early phase clinical reports confirmed the positive concept of JAK-STAT antagonism in dermatology, and randomized clinical trials have shown promising results in AD, psoriasis, and vitiligo. Encouraging data were observed in a proportion of AA participants; however, additional studies are needed to fully elucidate the disease pathophysiology and the role for JAK-STAT inhibition.
Larger clinical studies of oral and topical JAK inhibitors in AD, psoriasis, and vitiligo are currently ongoing. These pivotal trials are expected to provide additional insight into the efficacy and safety of JAK inhibitors in dermatology. Safety information for Jakinibs in inflammatory disease indications is mostly based on randomized clinical trials for investigational uses and extension studies. Recent, comprehensive, summaries of the key laboratory changes and clinical adverse events have been reported (150, 151).
Based on the promising results so far and the large number of ongoing clinical trials, it is possible that JAK inhibitors will become an important part of the dermatologist's treatment armamentarium in the future.
Author Contributions
All authors listed have made a substantial, direct and intellectual contribution to the work, and approved it for publication.
Funding
The authors declare that this study received funding from Incyte Corporation. The funder was not involved in the study design, collection, analysis, interpretation of data, the writing of this article or the decision to submit it for publication.
Conflict of Interest
MH, FK, and PS are employees of Incyte Corporation.
Acknowledgments
Editorial and graphical support was provided by Complete Healthcare Communications (North Wales, PA) and was funded by Incyte Corporation.
Abbreviations
AA, Alopecia areata; ACT, Nuclear factor NF-kappa-B activator 1; AD, Atopic dermatitis; BID, Twice daily; CD, Cluster of differentiation; CXCL, Chemokine (C-X-C motif) ligand; CXCR, Chemokine (C-X-C motif) receptor; DC, Dendritic cell; DNCB, 2,4-dinitrochlorobenzene; EASI, Eczema Area Severity Index; EPO, Erythropoietin; FDA, US Food and Drug Administration; FLG, Filaggrin; GATA-3, GATA transcription factor 3; GH, Growth hormone; GM-CSF, Granulocyte-macrophage colony-stimulating factor; IFN, Interferon; Ig, Immunoglobulin; IL, Interleukin; ILC2, Type 2 innate lymphoid cells; IGA, Investigator global assessment; INV, Involucrin; JAK, Janus kinase; JAKinibs, Janus kinase inhibitors; LOR, Loricrin; MHC, Major histocompatibility complex; NK, Natural killer cell; NKD2D, Natural Killer Group 2D; NKD2DL3, Natural Killer Group 2D ligand 3; PASI, Psoriasis Area and Severity Index; pDC, Plasmacytoid dendritic cell; PGA, Physician Global Assessment; QD, Once daily; OSM, Oncostatin M; OSMRβ, Oncostatin M receptor β; PI3K, Phosphoinositide 3-kinases; RAET1L, Retinoic acid early transcript 1L; RORγt, Retinoic acid receptor-related orphan receptor γ; SALT, Severity of Alopecia Tool; STAT, Signal transducer and activator of transcription; T-bet, T-box transcription factor TBX21; TGF, Transforming growth factor; Th, T helper; TNFα, Tumor necrosis factor alpha; TPO, Thrombopoietin; TSLP, Thymic stromal lymphopoietin; TYK2, Tyrosine kinase 2; VASI, Vitiligo Area Scoring Index.
References
1. Villarino AV, Kanno Y, O'shea JJ. Mechanisms and consequences of Jak-STAT signaling in the immune system. Nat Immunol. (2017) 18:374–84. doi: 10.1038/ni.3691
2. Rawlings JS, Rosler KM, Harrison DA. The JAK/STAT signaling pathway. J Cell Sci. (2004) 117:1281–3. doi: 10.1242/jcs.00963
3. Howell MD, Fitzsimons C, Smith PA. JAK/STAT inhibitors and other small molecule cytokine antagonists for the treatment of allergic disease. Ann Allergy Asthma Immunol. (2018) 120:367–75. doi: 10.1016/j.anai.2018.02.012
4. Stark GR, Cheon H, Wang Y. Responses to cytokines and interferons that depend upon JAKs and STATs. Cold Spring Harb Perspect Biol. (2018) 10:a028555. doi: 10.1101/cshperspect.a028555
5. Maldonado RA, Soriano MA, Perdomo LC, Sigrist K, Irvine DJ, Decker T, et al. Control of T helper cell differentiation through cytokine receptor inclusion in the immunological synapse. J Exp Med. (2009) 206:877–92. doi: 10.1084/jem.20082900
6. Mowen KA, Glimcher LH. Signaling pathways in Th2 development. Immunol Rev. (2004) 202:203–22. doi: 10.1111/j.0105-2896.2004.00209.x
7. Chen Z, Laurence A, O'shea JJ. Signal transduction pathways and transcriptional regulation in the control of Th17 differentiation. Semin Immunol. (2007) 19:400–8. doi: 10.1016/j.smim.2007.10.015
8. O'shea JJ, Steward-Tharp SM, Laurence A, Watford WT, Wei L, Adamson AS, et al. Signal transduction and Th17 cell differentiation. Microbes Infect. (2009) 11:599–611. doi: 10.1016/j.micinf.2009.04.007
9. Wuthrich B, Schmid-Grendelmeier P. The atopic eczema/dermatitis syndrome. Epidemiology, natural course, and immunology of the IgE-associated (“extrinsic”) and the nonallergic (“intrinsic”). AEDS J Investig Allergol Clin Immunol. (2003) 13:1–5. doi: 10.3109/9780203908877-2
10. Deckers IA, Mclean S, Linssen S, Mommers M, Van Schayck CP, Sheikh A. Investigating international time trends in the incidence and prevalence of atopic eczema 1990-2010: a systematic review of epidemiological studies. PLoS ONE. (2012) 7:e39803. doi: 10.1371/journal.pone.0039803
11. Zuberbier T, Orlow SJ, Paller AS, Taieb A, Allen R, Hernanz-Hermosa JM, et al. Patient perspectives on the management of atopic dermatitis. J Allergy Clin Immunol. (2006) 118:226–32. doi: 10.1016/j.jaci.2006.02.031
12. Wollenberg A, Oranje A, Deleuran M, Simon D, Szalai Z, Kunz B, et al. ETFAD/EADV Eczema task force 2015 position paper on diagnosis and treatment of atopic dermatitis in adult and paediatric patients. J Eur Acad Dermatol Venereol. (2016) 30:729–47. doi: 10.1111/jdv.13599
13. Thyssen JP, Hamann CR, Linneberg A, Dantoft TM, Skov L, Gislason GH, et al. Atopic dermatitis is associated with anxiety, depression, and suicidal ideation, but not with psychiatric hospitalization or suicide. Allergy. (2018) 73:214–20. doi: 10.1111/all.13231
14. Hebert AA, Stingl G, Ho LK, Lynde C, Cappelleri JC, Tallman AM, et al. Patient impact and economic burden of mild-to-moderate atopic dermatitis. Curr Med Res Opin. (2018) 34:2177–85. doi: 10.1080/03007995.2018.1498329
15. Weidinger S, Novak N. Atopic dermatitis. Lancet. (2016) 387:1109–22. doi: 10.1016/S0140-6736(15)00149-X
16. Neis MM, Peters B, Dreuw A, Wenzel J, Bieber T, Mauch C, et al. Enhanced expression levels of IL-31 correlate with IL-4 and IL-13 in atopic and allergic contact dermatitis. J Allergy Clin Immunol. (2006) 118:930–7. doi: 10.1016/j.jaci.2006.07.015
17. Amano W, Nakajima S, Yamamoto Y, Tanimoto A, Matsushita M, Miyachi Y, et al. JAK inhibitor JTE-052 regulates contact hypersensitivity by downmodulating T cell activation and differentiation. J Dermatol Sci. (2016) 84:258–65. doi: 10.1016/j.jdermsci.2016.09.007
18. Jin W, Huang W, Chen L, Jin M, Wang Q, Gao Z, et al. Topical application of JAK1/JAK2 inhibitor momelotinib exhibits significant anti-inflammatory responses in DNCB-induced atopic dermatitis model mice. Int J Mol Sci. (2018) 19:3973. doi: 10.3390/ijms19123973
19. Fukuyama T, Ehling S, Cook E, Baumer W. Topically administered janus-kinase inhibitors tofacitinib and oclacitinib display impressive antipruritic and anti-inflammatory responses in a model of allergic dermatitis. J Pharmacol Exp Ther. (2015) 354:394–405. doi: 10.1124/jpet.115.223784
20. Fujii Y, Sengoku T. Effects of the Janus kinase inhibitor CP-690550 (tofacitinib) in a rat model of oxazolone-induced chronic dermatitis. Pharmacology. (2013) 91:207–13. doi: 10.1159/000347184
21. Boniface K, Bernard FX, Garcia M, Gurney AL, Lecron JC, Morel F. IL-22 inhibits epidermal differentiation and induces proinflammatory gene expression and migration of human keratinocytes. J Immunol. (2005) 174:3695–702. doi: 10.4049/jimmunol.174.6.3695
22. Lou H, Lu J, Choi EB, Oh MH, Jeong M, Barmettler S, et al. Expression of IL-22 in the Skin Causes Th2-Biased Immunity, Epidermal Barrier Dysfunction, and Pruritus via Stimulating Epithelial Th2 Cytokines and the GRP Pathway. J Immunol. (2017) 198:2543–55. doi: 10.4049/jimmunol.1600126
23. Guttman-Yassky E, Brunner PM, Neumann AU, Khattri S, Pavel AB, Malik K, et al. Efficacy and safety of fezakinumab (an IL-22 monoclonal antibody) in adults with moderate-to-severe atopic dermatitis inadequately controlled by conventional treatments: a randomized, double-blind, phase 2a trial. J Am Acad Dermatol. (2018) 78:872–81 e876. doi: 10.1016/j.jaad.2018.01.016
24. Lejeune D, Dumoutier L, Constantinescu S, Kruijer W, Schuringa JJ, Renauld JC. Interleukin-22 (IL-22) activates the JAK/STAT, ERK, JNK, and p38 MAP kinase pathways in a rat hepatoma cell line. Pathways that are shared with and distinct from IL-10. J Biol Chem. (2002) 277:33676–82. doi: 10.1074/jbc.M204204200
25. Klonowska J, Glen J, Nowicki RJ, Trzeciak M. New cytokines in the pathogenesis of atopic dermatitis-new therapeutic targets. Int J Mol Sci. (2018) 19:E3086. doi: 10.3390/ijms19103086
26. Zhang Q, Putheti P, Zhou Q, Liu Q, Gao W. Structures and biological functions of IL-31 and IL-31 receptors. Cytokine Growth Factor Rev. (2008) 19:347–56. doi: 10.1016/j.cytogfr.2008.08.003
27. Zhong J, Sharma J, Raju R, Palapetta SM, Prasad TS, Huang TC, et al. TSLP signaling pathway map: a platform for analysis of TSLP-mediated signaling. Database. (2014) 2014:bau007. doi: 10.1093/database/bau007
28. Gonzales AJ, Bowman JW, Fici GJ, Zhang M, Mann DW, Mitton-Fry M. Oclacitinib (APOQUEL®) is a novel Janus kinase inhibitor with activity against cytokines involved in allergy. J Vet Pharmacol Ther. (2014) 37:317–24. doi: 10.1111/jvp.12101
29. Fridman JS, Scherle PA, Collins R, Burn T, Neilan CL, Hertel D, et al. Preclinical evaluation of local JAK1 and JAK2 inhibition in cutaneous inflammation. J Invest Dermatol. (2011) 131:1838–44. doi: 10.1038/jid.2011.140
30. Leyva-Castillo JM, Hener P, Jiang H, Li M. TSLP produced by keratinocytes promotes allergen sensitization through skin and thereby triggers atopic march in mice. J Invest Dermatol. (2013) 133:154–63. doi: 10.1038/jid.2012.239
31. Oetjen LK, Mack MR, Feng J, Whelan TM, Niu H, Guo CJ, et al. Sensory neurons co-opt classical immune signaling pathways to mediate chronic itch. Cell. (2017) 171:217–28 e213. doi: 10.1016/j.cell.2017.08.006
32. Kim BE, Leung DY, Boguniewicz M, Howell MD. Loricrin and involucrin expression is down-regulated by Th2 cytokines through STAT-6. Clin Immunol. (2008) 126:332–7. doi: 10.1016/j.clim.2007.11.006
33. Albanesi C, Fairchild HR, Madonna S, Scarponi C, De Pita O, Leung DY, et al. IL-4 and IL-13 negatively regulate TNF-alpha- and IFN-gamma-induced beta-defensin expression through STAT-6, suppressor of cytokine signaling (SOCS)-1, and SOCS-3. J Immunol. (2007) 179:984–92. doi: 10.4049/jimmunol.179.2.984
34. Sehra S, Yao Y, Howell MD, Nguyen ET, Kansas GS, Leung DY, et al. IL-4 regulates skin homeostasis and the predisposition toward allergic skin inflammation. J Immunol. (2010) 184:3186–90. doi: 10.4049/jimmunol.0901860
35. Yasuda T, Fukada T, Nishida K, Nakayama M, Matsuda M, Miura I, et al. Hyperactivation of JAK1 tyrosine kinase induces stepwise, progressive pruritic dermatitis. J Clin Invest. (2016) 126:2064–76. doi: 10.1172/JCI82887
36. Amano W, Nakajima S, Kunugi H, Numata Y, Kitoh A, Egawa G, et al. The Janus kinase inhibitor JTE-052 improves skin barrier function through suppressing signal transducer and activator of transcription 3 signaling. J Allergy Clin Immunol. (2015) 136:667–77 e667. doi: 10.1016/j.jaci.2015.03.051
37. Brauweiler AM, Goleva E, Leung DYM. Th2 cytokines increase Staphylococcus aureus alpha toxin-induced keratinocyte death through the signal transducer and activator of transcription 6 (STAT6). J Invest Dermatol. (2014) 134:2114–21. doi: 10.1038/jid.2014.43
38. Koga C, Kabashima K, Shiraishi N, Kobayashi M, Tokura Y. Possible pathogenic role of Th17 cells for atopic dermatitis. J Invest Dermatol. (2008) 128:2625–30. doi: 10.1038/jid.2008.111
39. Noda S, Suarez-Farinas M, Ungar B, Kim SJ, De Guzman Strong C, Xu H, et al. The Asian atopic dermatitis phenotype combines features of atopic dermatitis and psoriasis with increased TH17 polarization. J Allergy Clin Immunol. (2015) 136:1254–64. doi: 10.1016/j.jaci.2015.08.015
40. Levy LL, Urban J, King BA. Treatment of recalcitrant atopic dermatitis with the oral Janus kinase inhibitor tofacitinib citrate. J Am Acad Dermatol. (2015) 73:395–9. doi: 10.1016/j.jaad.2015.06.045
41. Guttman-Yassky E. Primary results from a phase 2b, randomized, placebo-controlled trial of upadacitinib for patients with atopic dermatitis. In: Presented at: The American Academy of Dermatology (AAD) Annual Meeting, February 16-20. San Diego, CA (2018).
42. Simpson EL, Lacour E-P, Spelman L, Galimberti R, Eichenfield L, Bissonnette R, et al. Efficacy and safety of baricitinib in moderate to severe atopic dermatitis: results of two phase 3 monotherapy randomized, double-blind, placebo-controlled 16-week trials (BREEZE-AD1 and BREEZA-AD2). In: Presented at: The 24th World Congress of Dermatology (WCD). Milan (2019).
43. Bissonnette R, Papp KA, Poulin Y, Gooderham M, Raman M, Mallbris L, et al. Topical tofacitinib for atopic dermatitis: a phase IIa randomized trial. Br J Dermatol. (2016) 175:902–11. doi: 10.1111/bjd.14871
44. Krueger J, Clark JD, Suarez-Farinas M, Fuentes-Duculan J, Cueto I, Wang CQ, et al. Tofacitinib attenuates pathologic immune pathways in patients with psoriasis: a randomized phase 2 study. J Allergy Clin Immunol. (2016) 137:1079–90. doi: 10.1016/j.jaci.2015.12.1318
45. Zhang J, Tsai TF, Lee MG, Zheng M, Wang G, Jin H, et al. The efficacy and safety of tofacitinib in Asian patients with moderate to severe chronic plaque psoriasis: a Phase 3, randomized, double-blind, placebo-controlled study. J Dermatol Sci. (2017) 88:36–45. doi: 10.1016/j.jdermsci.2017.05.004
46. Merola JF, Elewski B, Tatulych S, Lan S, Tallman A, Kaur M. Efficacy of tofacitinib for the treatment of nail psoriasis: two 52-week, randomized, controlled phase 3 studies in patients with moderate-to-severe plaque psoriasis. J Am Acad Dermatol. (2017) 77:79–87 e71. doi: 10.1016/j.jaad.2017.01.053
47. Bachelez H, Van De Kerkhof PC, Strohal R, Kubanov A, Valenzuela F, Lee JH, et al. Tofacitinib versus etanercept or placebo in moderate-to-severe chronic plaque psoriasis: a phase 3 randomised non-inferiority trial. Lancet. (2015) 386:552–61. doi: 10.1016/S0140-6736(14)62113-9
48. Bissonnette R, Iversen L, Sofen H, Griffiths CE, Foley P, Romiti R, et al. Tofacitinib withdrawal and retreatment in moderate-to-severe chronic plaque psoriasis: a randomized controlled trial. Br J Dermatol. (2015) 172:1395–406. doi: 10.1111/bjd.13551
49. Valenzuela F, Korman NJ, Bissonnette R, Bakos N, Tsai TF, Harper MK, et al. Tofacitinib in patients with moderate-to-severe chronic plaque psoriasis: long-term safety and efficacy in an open-label extension study. Br J Dermatol. (2018) 179:853–62. doi: 10.1111/bjd.16798
50. Kennedy Crispin M, Ko JM, Craiglow BG, Li S, Shankar G, Urban JR, et al. Safety and efficacy of the JAK inhibitor tofacitinib citrate in patients with alopecia areata. JCI Insight. (2016) 1:e89776. doi: 10.1172/jci.insight.89776
51. Schmieder GJ, Draelos ZD, Pariser DM, Banfield C, Cox L, Hodge M, et al. Efficacy and safety of the Janus kinase 1 inhibitor PF-04965842 in patients with moderate-to-severe psoriasis: phase II, randomized, double-blind, placebo-controlled study. Br J Dermatol. (2018) 179:54–62. doi: 10.1111/bjd.16004
52. Papp KA, Menter MA, Raman M, Disch D, Schlichting DE, Gaich C, et al. A randomized phase 2b trial of baricitinib, an oral Janus kinase (JAK) 1/JAK2 inhibitor, in patients with moderate-to-severe psoriasis. Br J Dermatol. (2016) 174:1266–76. doi: 10.1111/bjd.14403
53. Guttman-Yassky E, Silverberg JI, Nemoto O, Forman SB, Wilke A, Prescilla R, et al. Baricitinib in adult patients with moderate-to-severe atopic dermatitis: a phase 2 parallel, double-blinded, randomized placebo-controlled multiple-dose study. J Am Acad Dermatol. (2019) 80:913–21 e919. doi: 10.1016/j.jaad.2018.01.018
54. Mackay-Wiggan J, Jabbari A, Nguyen N, Cerise JE, Clark C, Ulerio G, et al. Oral ruxolitinib induces hair regrowth in patients with moderate-to-severe alopecia areata. JCI Insight. (2016) 1:e89790. doi: 10.1172/jci.insight.89790
55. Bissonnette R, Luchi M, Fidelus-Gort R, Jackson S, Zhang H, Flores R, et al. A randomized, double-blind, placebo-controlled, dose-escalation study of the safety and efficacy of INCB039110, an oral janus kinase 1 inhibitor, in patients with stable, chronic plaque psoriasis. J Dermatolog Treat. (2016) 27:332–8. doi: 10.3109/09546634.2015.1115819
56. Bissonnette R, Maari C, Forman S, Bhatia N, Lee M, Fowler J, et al. The oral Janus kinase/spleen tyrosine kinase inhibitor ASN002 demonstrates efficacy and improves associated systemic inflammation in patients with moderate-to-severe atopic dermatitis: results from a randomized double-blind placebo-controlled study. Br J Dermatol. (2019). doi: 10.1111/bjd.17932. [Epub ahead of print].
57. Papp K, Gordon K, Thaci D, Morita A, Gooderham M, Foley P, et al. Phase 2 trial of selective tyrosine Kinase 2 inhibition in psoriasis. N Engl J Med. (2018) 379:1313–21. doi: 10.1056/NEJMoa1806382
58. Papp K, Pariser D, Catlin M, Wierz G, Ball G, Akinlade B, et al. A phase 2a randomized, double-blind, placebo-controlled, sequential dose-escalation study to evaluate the efficacy and safety of ASP015K, a novel Janus kinase inhibitor, in patients with moderate-to-severe psoriasis. Br J Dermatol. (2015) 173:767–76. doi: 10.1111/bjd.13745
59. Papp KA, Bissonnette R, Gooderham M, Feldman SR, Iversen L, Soung J, et al. Treatment of plaque psoriasis with an ointment formulation of the Janus kinase inhibitor, tofacitinib: a Phase 2b randomized clinical trial. BMC Dermatol. (2016) 16:15. doi: 10.1186/s12895-016-0051-4
60. Ports WC, Khan S, Lan S, Lamba M, Bolduc C, Bissonnette R, et al. A randomized phase 2a efficacy and safety trial of the topical Janus kinase inhibitor tofacitinib in the treatment of chronic plaque psoriasis. Br J Dermatol. (2013) 169:137–45. doi: 10.1111/bjd.12266
61. Liu LY, Craiglow BG, King BA. Tofacitinib 2% ointment, a topical Janus kinase inhibitor, for the treatment of alopecia areata: a pilot study of 10 patients. J Am Acad Dermatol. (2018) 78:403–4 e401. doi: 10.1016/j.jaad.2017.10.043
62. Punwani N, Scherle P, Flores R, Shi J, Liang J, Yeleswaram S, et al. Preliminary clinical activity of a topical JAK1/2 inhibitor in the treatment of psoriasis. J Am Acad Dermatol. (2012) 67:658–64. doi: 10.1016/j.jaad.2011.12.018
63. Punwani N, Burn T, Scherle P, Flores R, Shi J, Collier P, et al. Downmodulation of key inflammatory cell markers with a topical Janus kinase 1/2 inhibitor. Br J Dermatol. (2015) 173:989–97. doi: 10.1111/bjd.13994
64. Kim B, Nasir A, Papp K, Parish LC, Kuligowski M, Venturanza M, et al. A phase 2, randomized, dose-ranging, vehicle- and active-controlled study to evaluate the safety and efficacy of topical ruxolitinib in adult patients with atopic dermatitis. In: Presented at: The 27th European Academy of Dermatology and Venereology (EADV) Congress. Paris (2018).
65. Raoof T, Kircik L, Kuligowski M, Venturanza M, Sun K, Tan J. 12-Week efficacy and safety data of ruxolitinib cream in adult patients with atopic dermatitis: results from a phase 2 study. In: Presented at: The 24th World Congress of Dermatology (WCD). Milan (2019).
66. Rosmarin D, Pandya A, Lebwohl M, Grimes P, Hamzavi I, Gottlieb AB, et al. Efficacy and safety of ruxolitinib cream for the treatment of Vitiligo: results of a 24-week randomized, double-blind, dose-ranging, vehicle-controlled study. In: Presented at the 24th World Congress of Dermatology. Milan (2019).
67. Rothstein B, Joshipura D, Saraiya A, Abdat R, Ashkar H, Turkowski Y, et al. Treatment of vitiligo with the topical Janus kinase inhibitor ruxolitinib. J Am Acad Dermatol. (2017). 76:1054–60 e1051. doi: 10.1016/j.jaad.2017.02.049
68. Strazzulla LC, Wang EHC, Avila L, Lo Sicco K, Brinster N, Christiano AM, et al. Alopecia areata: Disease characteristics, clinical evaluation, and new perspectives on pathogenesis. J Am Acad Dermatol. (2018) 78:1–12. doi: 10.1016/j.jaad.2017.04.1141
69. Paus R, Nickoloff BJ, Ito T. A 'hairy' privilege. Trends Immunol. (2005) 26:32–40. doi: 10.1016/j.it.2004.09.014
70. Gilhar A, Etzioni A, Paus R. Alopecia areata. N Engl J Med. (2012) 366:1515–25. doi: 10.1056/NEJMra1103442
71. Mcelwee KJ, Gilhar A, Tobin DJ, Ramot Y, Sundberg JP, Nakamura M, et al. What causes alopecia areata? Exp Dermatol. (2013) 22:609–26. doi: 10.1111/exd.12209
72. Guo H, Cheng Y, Shapiro J, Mcelwee K. The role of lymphocytes in the development and treatment of alopecia areata. Expert Rev Clin Immunol. (2015) 11:1335–51. doi: 10.1586/1744666X.2015.1085306
73. Paus R, Christoph T, Muller-Rover S. Immunology of the hair follicle: a short journey into terra incognita. J Investig Dermatol Symp Proc. (1999) 4:226–34. doi: 10.1038/sj.jidsp.5640217
74. Kang H, Wu WY, Lo BK, Yu M, Leung G, Shapiro J, et al. Hair follicles from alopecia areata patients exhibit alterations in immune privilege-associated gene expression in advance of hair loss. J Invest Dermatol. (2010) 130:2677–80. doi: 10.1038/jid.2010.180
75. Breitkopf T, Lo BK, Leung G, Wang E, Yu M, Carr N, et al. Somatostatin expression in human hair follicles and its potential role in immune privilege. J Invest Dermatol. (2013) 133:1722–30. doi: 10.1038/jid.2013.53
76. Wang X, Marr AK, Breitkopf T, Leung G, Hao J, Wang E, et al. Hair follicle mesenchyme-associated PD-L1 regulates T-cell activation induced apoptosis: a potential mechanism of immune privilege. J Invest Dermatol. (2014) 134:736–45. doi: 10.1038/jid.2013.368
77. Mcelwee KJ, Boggess D, King LEJr, Sundberg JP. Experimental induction of alopecia areata-like hair loss in C3H/HeJ mice using full-thickness skin grafts. J Invest Dermatol. (1998) 111:797–803. doi: 10.1046/j.1523-1747.1998.00380.x
78. Mcelwee KJ, Boggess D, Olivry T, Oliver RF, Whiting D, Tobin DJ, et al. Comparison of alopecia areata in human and nonhuman mammalian species. Pathobiology. (1998) 66:90–107. doi: 10.1159/000028002
79. Shin JM, Choi DK, Sohn KC, Koh JW, Lee YH, Seo YJ, et al. Induction of alopecia areata in C3H/HeJ mice using polyinosinic-polycytidylic acid (poly[I:C]) and interferon-gamma. Sci Rep. (2018) 8:12518. doi: 10.1038/s41598-018-30997-3
80. Jabbari A, Dai Z, Xing L, Cerise JE, Ramot Y, Berkun Y, et al. Reversal of alopecia areata following treatment with the JAK1/2 inhibitor baricitinib. EBioMedicine. (2015) 2:351–5. doi: 10.1016/j.ebiom.2015.02.015
81. Xing L, Dai Z, Jabbari A, Cerise JE, Higgins CA, Gong W, et al. Alopecia areata is driven by cytotoxic T lymphocytes and is reversed by JAK inhibition. Nat Med. (2014) 20:1043–9. doi: 10.1038/nm.3645
82. Ghoreishi M, Martinka M, Dutz JP. Type 1 interferon signature in the scalp lesions of alopecia areata. Br J Dermatol. (2010) 163:57–62. doi: 10.1111/j.1365-2133.2010.09775.x
83. De Jong A, Jabbari A, Dai Z, Xing L, Lee D, Li MM, et al. High-throughput T cell receptor sequencing identifies clonally expanded CD8+ T cell populations in alopecia areata. JCI Insight. (2018) 3:121949. doi: 10.1172/jci.insight.121949
84. Fuentes-Duculan J, Gulati N, Bonifacio KM, Kunjravia N, Zheng X, Suarez-Farinas M, et al. Biomarkers of alopecia areata disease activity and response to corticosteroid treatment. Exp Dermatol. (2016) 25:282–6. doi: 10.1111/exd.12918
85. Freyschmidt-Paul P, Mcelwee KJ, Hoffmann R, Sundberg JP, Vitacolonna M, Kissling S, et al. Interferon-gamma-deficient mice are resistant to the development of alopecia areata. Br J Dermatol. (2006) 155:515–21. doi: 10.1111/j.1365-2133.2006.07377.x
86. Ito T, Ito N, Bettermann A, Tokura Y, Takigawa M, Paus R. Collapse and restoration of MHC class-I-dependent immune privilege: exploiting the human hair follicle as a model. Am J Pathol. (2004) 164:623–34. doi: 10.1016/S0002-9440(10)63151-3
87. Petukhova L, Duvic M, Hordinsky M, Norris D, Price V, Shimomura Y, et al. Genome-wide association study in alopecia areata implicates both innate and adaptive immunity. Nature. (2010) 466:113–7. doi: 10.1038/nature09114
88. Pratt CH, King LE Jr, Messenger AG, Christiano AM, Sundberg JP. Alopecia areata. Nat Rev Dis Primers. (2017) 3:17011. doi: 10.1038/nrdp.2017.11
89. Abou Rahal J, Kurban M, Kibbi AG, Abbas O. Plasmacytoid dendritic cells in alopecia areata: missing link? J Eur Acad Dermatol Venereol. (2016) 30:119–23. doi: 10.1111/jdv.12932
90. Charles J, Chaperot L, Salameire D, Di Domizio J, Aspord C, Gressin R, et al. Plasmacytoid dendritic cells and dermatological disorders: focus on their role in autoimmunity and cancer. Eur J Dermatol. (2010) 20:16–23. doi: 10.1684/ejd.2010.0816
91. Mcphee CG, Duncan FJ, Silva KA, King LEJr, Hogenesch H, Roopenian DC, et al. Increased expression of Cxcr3 and its ligands, Cxcl9 and Cxcl10, during the development of alopecia areata in the mouse. J Invest Dermatol. (2012) 132:1736–8. doi: 10.1038/jid.2012.17
92. Kuo PT, Zeng Z, Salim N, Mattarollo S, Wells JW, Leggatt GR. The role of CXCR3 and its chemokine ligands in skin disease and cancer. Front Med. (2018) 5:271. doi: 10.3389/fmed.2018.00271
93. Jabri B, Abadie V. IL-15 functions as a danger signal to regulate tissue-resident T cells and tissue destruction. Nat Rev Immunol. (2015) 15:771–83. doi: 10.1038/nri3919
94. Cooley ID, Read KA, Oestreich KJ. Trans-presentation of IL-15 modulates STAT5 activation and Bcl-6 expression in TH1 cells. Sci Rep. (2015) 5:15722. doi: 10.1038/srep15722
95. Betz RC, Petukhova L, Ripke S, Huang H, Menelaou A, Redler S, et al. Genome-wide meta-analysis in alopecia areata resolves HLA associations and reveals two new susceptibility loci. Nat Commun. (2015) 6:5966. doi: 10.1038/ncomms6966
96. O'shea JJ, Plenge R. JAK and STAT signaling molecules in immunoregulation and immune-mediated disease. Immunity. (2012) 36:542–50. doi: 10.1016/j.immuni.2012.03.014
97. Harel S, Higgins CA, Cerise JE, Dai Z, Chen JC, Clynes R, et al. Pharmacologic inhibition of JAK-STAT signaling promotes hair growth. Sci Adv. (2015) 1:e1500973. doi: 10.1126/sciadv.1500973
98. Meephansan J, Thummakriengkrai J, Ponnikorn S, Yingmema W, Deenonpoe R, Suchonwanit P. Efficacy of topical tofacitinib in promoting hair growth in non-scarring alopecia: possible mechanism via VEGF induction. Arch Dermatol Res. (2017) 309:729–38. doi: 10.1007/s00403-017-1777-5
99. Craiglow BG, King BA. Killing two birds with one stone: oral tofacitinib reverses alopecia universalis in a patient with plaque psoriasis. J Invest Dermatol. (2014) 134:2988–90. doi: 10.1038/jid.2014.260
100. Higgins E, Al Shehri T, Mcaleer MA, Conlon N, Feighery C, Lilic D, et al. Use of ruxolitinib to successfully treat chronic mucocutaneous candidiasis caused by gain-of-function signal transducer and activator of transcription 1 (STAT1) mutation. J Allergy Clin Immunol. (2015) 135:551–3. doi: 10.1016/j.jaci.2014.12.1867
101. Pieri L, Guglielmelli P, Vannucchi AM. Ruxolitinib-induced reversal of alopecia universalis in a patient with essential thrombocythemia. Am J Hematol. (2015) 90:82–3. doi: 10.1002/ajh.23871
102. Harris JE, Rashighi M, Nguyen N, Jabbari A, Ulerio G, Clynes R, et al. Rapid skin repigmentation on oral ruxolitinib in a patient with coexistent vitiligo and alopecia areata (AA). J Am Acad Dermatol. (2016) 74:370–1. doi: 10.1016/j.jaad.2015.09.073
103. Boyadzhiev M, Marinov L, Boyadzhiev V, Iotova V, Aksentijevich I, Hambleton S. Disease course and treatment effects of a JAK inhibitor in a patient with CANDLE syndrome. Pediatr Rheumatol Online J. (2019) 17:19. doi: 10.1186/s12969-019-0322-9
104. Jabbari A, Sansaricq F, Cerise J, Chen JC, Bitterman A, Ulerio G, et al. An Open-Label Pilot Study to Evaluate the Efficacy of Tofacitinib in Moderate to Severe Patch-Type Alopecia Areata, Totalis, and Universalis. J Invest Dermatol. (2018) 138:1539–45. doi: 10.1016/j.jid.2018.01.032
105. Craiglow BG, Liu LY, King BA. Tofacitinib for the treatment of alopecia areata and variants in adolescents. J Am Acad Dermatol. (2017) 76:29–32. doi: 10.1016/j.jaad.2016.09.006
106. Liu LY, Craiglow BG, Dai F, King BA. Tofacitinib for the treatment of severe alopecia areata and variants: a study of 90 patients. J Am Acad Dermatol. (2017) 76:22–8. doi: 10.1016/j.jaad.2016.09.007
107. Liu LY, Strassner JP, Refat MA, Harris JE, King BA. Repigmentation in vitiligo using the Janus kinase inhibitor tofacitinib may require concomitant light exposure. J Am Acad Dermatol. (2017) 77:675–82 e671. doi: 10.1016/j.jaad.2017.05.043
108. Craiglow BG, King BA. Tofacitinib citrate for the treatment of vitiligo: a pathogenesis-directed therapy. JAMA Dermatol. (2015) 151:1110–2. doi: 10.1001/jamadermatol.2015.1520
109. Bayart CB, Deniro KL, Brichta L, Craiglow BG, Sidbury R. Topical Janus kinase inhibitors for the treatment of pediatric alopecia areata. J Am Acad Dermatol. (2017) 77:167–70. doi: 10.1016/j.jaad.2017.03.024
110. Nestle FO, Kaplan DH, Barker J. Psoriasis. N Engl J Med. (2009) 361:496–509. doi: 10.1056/NEJMra0804595
111. Afzali B, Lombardi G, Lechler RI, Lord GM. The role of T helper 17 (Th17) and regulatory T cells (Treg) in human organ transplantation and autoimmune disease. Clin Exp Immunol. (2007) 148:32–46. doi: 10.1111/j.1365-2249.2007.03356.x
112. Albanesi C, De Pita O, Girolomoni G. Resident skin cells in psoriasis: a special look at the pathogenetic functions of keratinocytes. Clin Dermatol. (2007) 25:581–8. doi: 10.1016/j.clindermatol.2007.08.013
113. Lee E, Trepicchio WL, Oestreicher JL, Pittman D, Wang F, Chamian F, et al. Increased expression of interleukin 23 p19 and p40 in lesional skin of patients with psoriasis vulgaris. J Exp Med. (2004) 199:125–30. doi: 10.1084/jem.20030451
114. Sutton CE, Lalor SJ, Sweeney CM, Brereton CF, Lavelle EC, Mills KH. Interleukin-1 and IL-23 induce innate IL-17 production from gammadelta T cells, amplifying Th17 responses and autoimmunity. Immunity. (2009) 31:331–41. doi: 10.1016/j.immuni.2009.08.001
115. Zheng Y, Danilenko DM, Valdez P, Kasman I, Eastham-Anderson J, Wu J, et al. Interleukin-22, a T(H)17 cytokine, mediates IL-23-induced dermal inflammation and acanthosis. Nature. (2007) 445:648–51. doi: 10.1038/nature05505
116. Boutet MA, Nerviani A, Gallo Afflitto G, Pitzalis C. Role of the IL-23/IL-17 axis in psoriasis and psoriatic arthritis: the clinical importance of its divergence in skin and joints. Int J Mol Sci. (2018) 19:E530. doi: 10.3390/ijms19020530
117. Belge K, Bruck J, Ghoreschi K. Advances in treating psoriasis. F1000Prime Rep. (2014) 6:4. doi: 10.12703/P6-4
118. Nada HR, El Sharkawy DA, Elmasry MF, Rashed LA, Mamdouh S. Expression of Janus Kinase 1 in vitiligo and psoriasis before and after narrow band UVB: a case-control study. Arch Dermatol Res. (2018) 310:39–46. doi: 10.1007/s00403-017-1792-6
119. Wei L, Laurence A, Elias KM, O'shea JJ. IL-21 is produced by Th17 cells and drives IL-17 production in a STAT3-dependent manner. J Biol Chem. (2007) 282:34605–10. doi: 10.1074/jbc.M705100200
120. Camporeale A, Poli V. IL-6, IL-17 and STAT3: a holy trinity in auto-immunity? Front Biosci. (2012) 17:2306–26. doi: 10.2741/4054
121. Van Der Fits L, Mourits S, Voerman JS, Kant M, Boon L, Laman JD, et al. Imiquimod-induced psoriasis-like skin inflammation in mice is mediated via the IL-23/IL-17 axis. J Immunol. (2009) 182:5836–45. doi: 10.4049/jimmunol.0802999
122. Tanimoto A, Shinozaki Y, Yamamoto Y, Katsuda Y, Taniai-Riya E, Toyoda K, et al. A novel JAK inhibitor JTE-052 reduces skin inflammation and ameliorates chronic dermatitis in rodent models: comparison with conventional therapeutic agents. Exp Dermatol. (2018) 27:22–9. doi: 10.1111/exd.13370
123. Ghoreschi K, Jesson MI, Li X, Lee JL, Ghosh S, Alsup JW, et al. Modulation of innate and adaptive immune responses by tofacitinib (CP-690,550). J Immunol. (2011) 186:4234–43. doi: 10.4049/jimmunol.1003668
124. Morelli M, Scarponi C, Mercurio L, Facchiano F, Pallotta S, Madonna S, et al. Selective immunomodulation of inflammatory pathways in keratinocytes by the Janus Kinase (JAK) inhibitor tofacitinib: implications for the employment of JAK-targeting drugs in psoriasis. J Immunol Res. (2018) 2018:7897263. doi: 10.1155/2018/7897263
125. Chang BY, Zhao F, He X, Ren H, Braselmann S, Taylor V, et al. JAK3 inhibition significantly attenuates psoriasiform skin inflammation in CD18 mutant PL/J mice. J Immunol. (2009) 183:2183–92. doi: 10.4049/jimmunol.0804063
126. Karaghiosoff M, Neubauer H, Lassnig C, Kovarik P, Schindler H, Pircher H, et al. Partial impairment of cytokine responses in Tyk2-deficient mice. Immunity. (2000) 13:549–60. doi: 10.1016/S1074-7613(00)00054-6
127. Shimoda K, Kato K, Aoki K, Matsuda T, Miyamoto A, Shibamori M, et al. Tyk2 plays a restricted role in IFN alpha signaling, although it is required for IL-12-mediated T cell function. Immunity. (2000) 13:561–71. doi: 10.1016/S1074-7613(00)00055-8
128. Ishizaki M, Muromoto R, Akimoto T, Sekine Y, Kon S, Diwan M, et al. Tyk2 is a therapeutic target for psoriasis-like skin inflammation. Int Immunol. (2014) 26:257–67. doi: 10.1093/intimm/dxt062
129. Works MG, Yin F, Yin CC, Yiu Y, Shew K, Tran TT, et al. Inhibition of TYK2 and JAK1 ameliorates imiquimod-induced psoriasis-like dermatitis by inhibiting IL-22 and the IL-23/IL-17 axis. J Immunol. (2014) 193:3278–87. doi: 10.4049/jimmunol.1400205
130. Sano S, Chan KS, Carbajal S, Clifford J, Peavey M, Kiguchi K, et al. Stat3 links activated keratinocytes and immunocytes required for development of psoriasis in a novel transgenic mouse model. Nat Med. (2005) 11:43–9. doi: 10.1038/nm1162
131. Miyoshi K, Takaishi M, Nakajima K, Ikeda M, Kanda T, Tarutani M, et al. Stat3 as a therapeutic target for the treatment of psoriasis: a clinical feasibility study with STA-21, a Stat3 inhibitor. J Invest Dermatol. (2011) 131:108–17. doi: 10.1038/jid.2010.255
132. Nakajima K, Kanda T, Takaishi M, Shiga T, Miyoshi K, Nakajima H, et al. Distinct roles of IL-23 and IL-17 in the development of psoriasis-like lesions in a mouse model. J Immunol. (2011) 186:4481–9. doi: 10.4049/jimmunol.1000148
133. Ludbrook VJ, Hicks KJ, Hanrott KE, Patel JS, Binks MH, Wyres MR, et al. Investigation of selective JAK1 inhibitor GSK2586184 for the treatment of psoriasis in a randomized placebo-controlled phase IIa study. Br J Dermatol. (2016) 174:985–95. doi: 10.1111/bjd.14399
134. Ports WC, Feldman SR, Gupta P, Tan H, Johnson TR, Bissonnette R. Randomized pilot clinical trial of tofacitinib solution for plaque psoriasis: challenges of the intra-subject study design. J Drugs Dermatol. (2015) 14:777–84.
135. Kruger C, Schallreuter KU. A review of the worldwide prevalence of vitiligo in children/adolescents and adults. Int J Dermatol. (2012) 51:1206–12. doi: 10.1111/j.1365-4632.2011.05377.x
136. Boniface K, Seneschal J, Picardo M, Taieb A. Vitiligo: focus on clinical aspects, immunopathogenesis, and therapy. Clin Rev Allergy Immunol. (2018) 54:52–67. doi: 10.1007/s12016-017-8622-7
137. Radtke MA, Schafer I, Gajur A, Langenbruch A, Augustin M. Willingness-to-pay and quality of life in patients with vitiligo. Br J Dermatol. (2009) 161:134–9. doi: 10.1111/j.1365-2133.2009.09091.x
138. Talsania N, Lamb B, Bewley A. Vitiligo is more than skin deep: a survey of members of the Vitiligo Society. Clin Exp Dermatol. (2010) 35:736–9. doi: 10.1111/j.1365-2230.2009.03765.x
139. Ogg GS, Rod Dunbar P, Romero P, Chen JL, Cerundolo V. High frequency of skin-homing melanocyte-specific cytotoxic T lymphocytes in autoimmune vitiligo. J Exp Med. (1998) 188:1203–8. doi: 10.1084/jem.188.6.1203
140. Van Den Boorn JG, Konijnenberg D, Dellemijn TA, Van Der Veen JP, Bos JD, Melief CJ, et al. Autoimmune destruction of skin melanocytes by perilesional T cells from vitiligo patients. J Invest Dermatol. (2009) 129:2220–32. doi: 10.1038/jid.2009.32
141. Rashighi M, Agarwal P, Richmond JM, Harris TH, Dresser K, Su MW, et al. CXCL10 is critical for the progression and maintenance of depigmentation in a mouse model of vitiligo. Sci Transl Med. (2014) 6:223ra223. doi: 10.1126/scitranslmed.3007811
142. Regazzetti C, Joly F, Marty C, Rivier M, Mehul B, Reiniche P, et al. Transcriptional analysis of vitiligo skin reveals the alteration of WNT pathway: a promising target for repigmenting vitiligo patients. J Invest Dermatol. (2015) 135:3105–14. doi: 10.1038/jid.2015.335
143. Abdallah M, El-Mofty M, Anbar T, Rasheed H, Esmat S, Al-Tawdy A, et al. CXCL-10 and Interleukin-6 are reliable serum markers for vitiligo activity: a multicenter cross-sectional study. Pigment Cell Melanoma Res. (2018) 31:330–6. doi: 10.1111/pcmr.12667
144. Strassner JP, Rashighi M, Ahmed Refat M, Richmond JM, Harris JE. Suction blistering the lesional skin of vitiligo patients reveals useful biomarkers of disease activity. J Am Acad Dermatol. (2017) 76:847–55 e845. doi: 10.1016/j.jaad.2016.12.021
145. Richmond JM, Masterjohn E, Chu R, Tedstone J, Youd ME, Harris JE. CXCR3 depleting antibodies prevent and reverse vitiligo in mice. J Invest Dermatol. (2017) 137:982–5. doi: 10.1016/j.jid.2016.10.048
146. Richmond JM, Bangari DS, Essien KI, Currimbhoy SD, Groom JR, Pandya AG, et al. Keratinocyte-derived chemokines orchestrate T-cell positioning in the epidermis during vitiligo and may serve as biomarkers of disease. J Invest Dermatol. (2017) 137:350–8. doi: 10.1016/j.jid.2016.09.016
147. Abdou AG, Maraee A, Yassien H, Sarhan M. Immunohistochemistry of Janus Kinase 1 (JAK1) Expression in Vitiligo. J Pathol Transl Med. (2018) 52:363–8. doi: 10.4132/jptm.2018.09.18
148. Relke N, Gooderham M. The use of janus kinase inhibitors in Vitiligo: a review of the literature. J Cutan Med Surg. (2019) 23:298–306. doi: 10.1177/1203475419833609
149. Kim SR, Heaton H, Liu LY, King BA. Rapid repigmentation of vitiligo using tofacitinib plus low-dose, narrowband UV-B phototherapy. JAMA Dermatol. (2018) 154:370–1. doi: 10.1001/jamadermatol.2017.5778
150. Choy EH. Clinical significance of Janus Kinase inhibitor selectivity. Rheumatology. (2019) 58:953–62. doi: 10.1093/rheumatology/key339
Keywords: Janus kinase, cytokines, skin barrier, dermatology, autoimmunity
Citation: Howell MD, Kuo FI and Smith PA (2019) Targeting the Janus Kinase Family in Autoimmune Skin Diseases. Front. Immunol. 10:2342. doi: 10.3389/fimmu.2019.02342
Received: 07 August 2019; Accepted: 17 September 2019;
Published: 09 October 2019.
Edited by:
Ralf J. Ludwig, Universität zu Lübeck, GermanyReviewed by:
Massimo Gadina, National Institute of Arthritis and Musculoskeletal and Skin Diseases (NIAMS), United StatesKunihiro Yamaoka, Keio University School of Medicine, Japan
Copyright © 2019 Howell, Kuo and Smith. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Paul A. Smith, cHNtaXRoQGluY3l0ZS5jb20=
†These authors have contributed equally to this work