 Michael Olbrich
Michael Olbrich Axel Künstner1,2
Axel Künstner1,2 Mareike Witte
Mareike Witte Hauke Busch
Hauke Busch Anke Fähnrich
Anke Fähnrich- 1Medical Systems Biology, Institute of Experimental Dermatology, University of Lübeck, Lübeck, Germany
- 2Institute of Cardiogenetics, University of Lübeck, Lübeck, Germany
- 3Department of Dermatology, University of Lübeck, Lübeck, Germany
Autoimmune blistering diseases (AIBDs) of the skin are characterized by autoantibodies against different intra-/extracellular structures within the epidermis and at the basement membrane zone (BMZ). Binding of the antibodies to their target antigen leads to inflammation at the respective binding site and degradation of these structures, resulting in the separation of the affected skin layers. Clinically, blistering, erythema and lesions of the skin and/or mucous membranes can be observed. Based on the localization of the autoantigen, AIBDs can be divided into pemphigus (intra-epidermal blistering diseases) and pemphigoid diseases (sub-epidermal blistering diseases), respectively. Although autoantigens have been extensively characterized, the underlying causes that trigger the diseases are still poorly understood. Besides the environment, genetic factors seem to play an important role in a predisposition to AIBDs. Here, we review currently known genetic and immunological mechanisms that contribute to the pathogenesis of AIBDs. Among the most commonly encountered genetic predispositions for AIBDs are the HLA gene region, and deleterious mutations of key genes for the immune system. Particularly, HLA class II genes such as the HLA-DR and HLA-DQ alleles have been shown to be prevalent in patients. This has prompted further epidemiological studies as well as unbiased Omics approaches on the transcriptome, microbiome, and proteome level to elucidate common and individual genetic risk factors as well as the molecular pathways that lead to the pathogenesis of AIBDs.
1. Introduction
Autoimmune blistering diseases (AIBDs) of the skin are rare, yet potentially fatal autoimmune disorders. The autoantibodies are directed against distinct molecules expressed in the epidermis and at the dermal-epidermal junction (DEJ) of skin and/or mucous membranes. Binding of these autoantibodies ultimately leads to loss of cell-cell and cell-matrix adhesion in the skin and/or mucous membranes, which results in erosions and/or blister formation (1, 2). Antigens are presented as a cleaved fragment via the major histocompatibility complex (MHC). MHC, also known as the human leukocyte antigen (HLA) region in humans, comprises a region of 7.6 megabases (Mb) on chromosome 6p21. It is the most gene-dense region of the human genome, encoding 252 expressed loci, of which 40% are thought to play a key role in the immune system (3). The HLA region is furthermore characterized by an extraordinarily high degree of polymorphisms with more than 1000 known alleles for HLA-A and -B. The HLA class I and class II gene clusters comprise the isotypes HLA-A/-B/-C as well as HLA-DPA1, HLA-DPB1, HLA-DQA1, HLA-DQB1, HLA-DRA, and HLA-DRB1), respectively. They are involved in antigen processing and presentation, and usually show highly significant associations with autoimmune diseases, representing the strongest predisposing genetic factors (3, 4).
The development of autoimmune diseases is generally multi-factorial. Factors involved are a genetic predisposition, ethnicity, age, the environment, and gender. Autoimmune diseases show a prevalence and age of onset bias toward females (5, 6), which is particularly strong in systemic lupus erythematosus, Sjogren's syndrome, and autoimmune thyroiditis with females representing over 85% of all cases. In rheumatoid arthritis (RA) and multiple sclerosis, 60–75% of patients are female (5, 7). Sex-specific immune responses were also observed in mice. Female mice produced more antibodies and showed a stronger T cell activation than male mice after immunization (7–9). Yet, similar approaches in humans, in which responses to vaccination were analyzed, showed mixed results, with either no differences between males and females or an increased antibody response in females (9). It was found that females have a higher absolute number of CD4+ T lymphocytes than men (10) and produce more Th1 cytokines after vaccination. While these observations are still not completely understood, recent studies suggest that differences in the sex hormone composition, like progesterone and testosterone, may explain the differences in the immune mechanisms and autoimmune disease prevalence in females (5, 9, 11). Sex steroids may directly influence the immune system and affect components of antigen presentation, lymphocyte activation, cytokine gene expression, and/or homing of immune cells. They may also have indirect effects on corticosterone-cortisol concentrations, which are higher in females than in males. Additionally, glucocorticoids suppress the production of sex hormones and their mechanism of action in tissues (5, 11).
So far, more than 80 autoimmune diseases are known, ranging from familiar types like RA to rare forms like myasthenia gravis. Likewise, AIBDs can be subdivided into two major groups: pemphigus and pemphigoid diseases, based on the autoantigen localization. In the following sections we provide a brief description on the different AIBDs, together with their clinical manifestation and known genetic predispositions. An overview over the most common AIBDs is provided in Table 1.
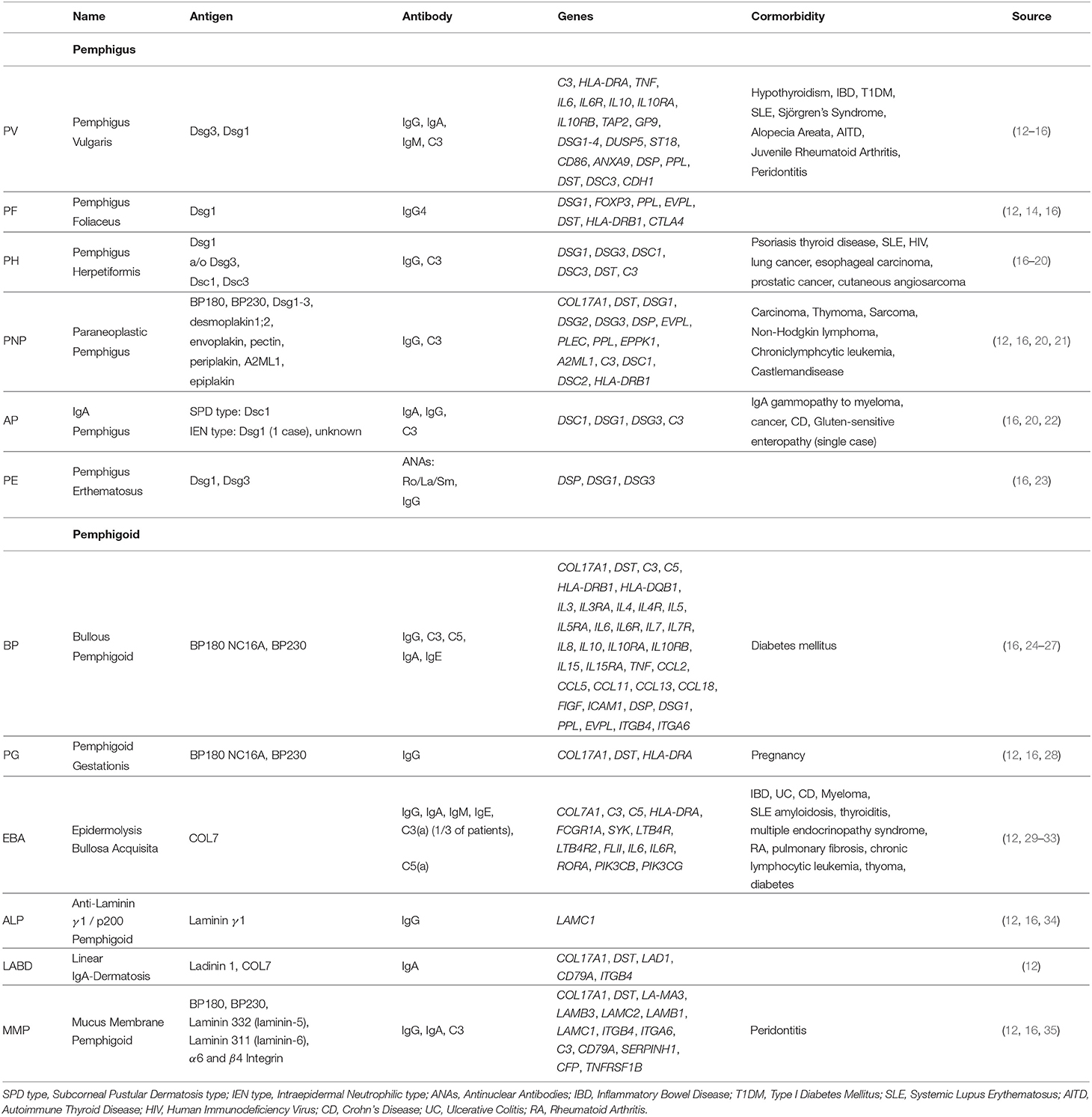
Table 1. Autoimmune skin blistering diseases: summary of targeted antigens, produced antibodies and associated genetics of common phemphigus and pemphigoid disease.
1.1. Pemphigus Diseases
Pemphigus diseases are characterized by intraepidermal autoantibody binding. The two major pemphigus types are pemphigus vulgaris (PV) and pemphigus foliaceus (PF), accounting for 70% and 15–20% of all pemphigus cases, respectively. Rarer forms of pemphigus diseases include pemephigus herpetiformis (PH), paraneoplastic pemphigus (PNP) and IgA pemphigus (AP). The incidence of pemphigus diseases is population-dependent (36) and ranges between 0.7 and 5 per million/year. It is highest in Central Europe and the United States with an estimated range for new cases between 1 and 7 per million/year. PV is between 4- and 10-fold more common among the Jewish population as compared to other Caucasian populations (37). In contrast, PF is not particularly prevalent among the Jewish population, but endemic PF-variants have been described in South America and Tunisia (38). Another endemic form of PF, namely fogo selvagem, occurs in rural areas of Brazil with a prevalence of 3.4% on certain Amerindian link reservations and an incidence of 1–4 cases per 1,200 persons per year (38). Depending on the subtype, pemphigus antibodies are mostly directed against desmoglein 3 (Dsg3) and desmoglein 1 (Dsg1). In some cases, antibodies against other antigens, such as desmocollins (Dsc) and plakins can be found. The abundance of transmembrane glycoproteins varies with the different skin layers (cf. Figure 1A); Dsg1 is predominantly expressed in the upper layers, while Dsg3 is expressed in the lower layers of the epidermis (39). Degradation of these structures leads to a loss of cell-cell adhesion and formation of intra-epidermal blisters. Accordingly, this AIBD subtype is also referred to as intra-epidermal blistering disorders.
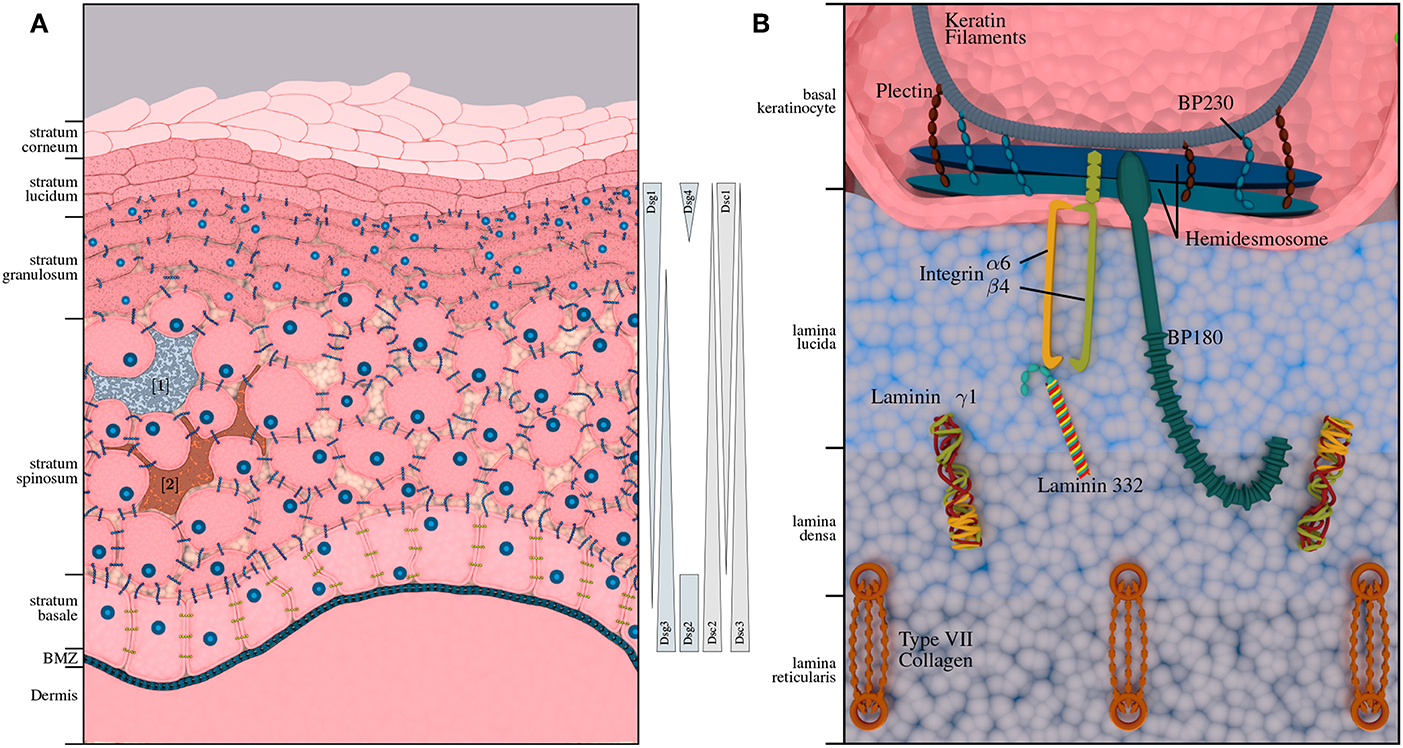
Figure 1. Structural composition of human epidermis and basement membrane zone (BMZ). (A) Shows the composition of the human skin, including melanocytes [1] (3) and immunocompetent Langerhans cells [2] (4). The approximate distribution of pemphigus antigens within the layers of the epidermis is depicted on the right-hand side. While desmogleins 1 and 4 and desmocollin 1 are expressed in the upper layers of the epidermis; desmogleins 2 and 3 as well as desmocollin 2 and 3 are expressed in the lower layers of the epidermis. (B) Depicts the BMZ with its cellular adhesion proteins, connecting epidermis and dermis, which are the main autoantigens in pemphigoid diseases.
1.1.1. Pemphigus Vulgaris
PV is a pemphigus disease with autoantibodies against Dsg3 and, in some cases, additionally against Dsg1. Due to the expression of Dsg3 in the lower layers of the epidermis and in the epithelium, PV mainly affects mucous membranes. Skin involvement is determined by the presence of Dsg1 autoantibodies: In mucocutaneus PV, both Dsg1 and Dsg3 autoantibodies can be observed (40).
The age of onset is around 50–60 years. However, early and even childhood cases of PV have been reported. PV predominantly affects females (female-to-male ratio of 1.5:1) (41). Additionally, the outcome of PV may be worse in females, as a recent analysis showed that the HLA alleles DRB1*04:02 and DQB1*03:02 were associated with severe PV, and DQB1*03:02 were found more frequently in female as compared to male patients (42, 43).
Search strategy and selection criteria
Literature: We searched the public literature databases PubMed, ResearchGate and Google Scholar, using the terms AIBD, autoimmune blistering disorders of the skin, pemphigus and pemphigoid. The search covered articles in English published between Jan 1, 2015, and March 31, 2019. Further publications, beyond the denoted selection criteria, were selected from the reference lists of the retrieved articles.
Data repositories: The NCBI databases, particularly Gene Expression Omnibus (GEO), and European Nucleotide Archive (ENA) were searched for high-throughput data studies of the outlined phenotypes.
So far, the genetic association between PV and HLA class II genes remains the strongest and the most reported. While some of the HLA types are more population specific, others are associated with PV across different ethnicities. Several studies have found associations between PV and HLA class I alleles including HLA-A3, HLA-A26, and HLA-B60 in the Han Chinese population; HLA-B38, HLA-C12, HLA-B57, and HLA-C15 in the Brazilian population; HLA-A10, and HLA-B15 in the Japanese population; HLA-B35 and HLA-B44 in the Turkish population; HLA-B38 in the Jewish and Spanish population; and HLA-B4402, HLA-C0401, and HLA-C1502 in the Iranian population (13, 44–46), respectively. Population studies have shown an association between certain class II alleles and PV in different ethnic groups. For example, HLA-DRB1*0402 is associated with PV in over 90% of Ashkenazi Jews, and HLA-DQB1*0503 is associated in non-Jewish populations. Likewise, HLA-DRB1*1404 is the most important risk factor in an Indo-Asian population. The two most common PV-associated alleles are HLA-DQB1*0503 and HAL-DRB1*0402, both of which were found to be associated with the disease in the Spanish, French, Italian, Slovak, North American and Brazilian population (47). In addition, several studies have shown an association between PV and non-classic HLA class Ib alleles (HLA-E, HLA-F, and HLA-G). HLA-G polymorphisms were found to have a significant association with Jewish PV patients, while HLA-E, previously demonstrated to play a role in multiple autoimmune conditions, was found in association with Caucasian and Ashkenazi Jewish patients and was suggested to be involved in the disruption of immune tolerance in PV (44–46, 48).
1.1.2. Pemphigus Foliaceus
In PF, the autoantibodies are directed against Dsg1, while Dsg3 antibodies cannot be detected. Thus, PF affects only the skin, while mucosal lesions are completely absent (39, 49). Skin lesions are also more superficial than in PV, with desquamation/scaling rather than erosions involving the deeper skin layers. The average age of onset for sporadic PF is between 50 and 60 years with no reported gender bias. There are endemic forms of PF in Tunisia, Brazil, Peru and Colombia, which diverge in rate of incidence and observed sexual predisposition. For example, the Brazilian and Tunisian forms of PF present with higher incident rates (50, 51). A male prevalence is observed in Colombian PF, where about 95% of the cases are reported in males (52). The endemic subtypes in particular, indicate the role of environmental factors in their pathogenesis. The hotspot regions for endemic PF are characterized by poor living standards and hygienic conditions, low age of onset (around 20 years), and a seasonally varying incidence rate, which is highest at the end of the rainy season and lowest in dry summers.
Previous studies have shown that the DSG1 gene is polymorphic and that a coding synonymous T/C single nucleotide polymorphism at position 809 is associated with PF. To determine whether the disease occurs due to complex genetic interactions, it was tested whether MHC class II genes and DSG1 polymorphisms contribute to PF sensitivity. An analysis performed in 31 PF patients and 84 healthy controls first confirmed the previously reported common DRB1*04 and DRB1*14 genetic background in PF and individualized DRB1*0102, DRB1*0402 and DRB1*0406, and DRB1*1404 as susceptibility MHC class II alleles in French Caucasian PF patients (47, 53).
1.1.3. Pemphigus Herpetiformis
Pemphigus herpetiformis (PH), also known as mixed bullous disease, eosinophilic spongiosis in pemphigus or acantholytic herpetiform dermatitis, is considered a clinical variant of pemphigus that combines the clinical features of dermatitis herpetiformis with the immunopathologic features of pemphigus. It accounts for 6–7.3% of all pemphigus patients. Clinically, PH is characterized by erythematous, itchy blisters and hive like swellings on several areas of the body. In contrast to PV and PF, the characteristic intense inflammation may not be associated with acantholysis (54, 55). Even though the phenotype closely resembles the features presented in dermatitis herpetiformis, its immunologic features conform to pemphigus (19). Autoantibodies in PH mainly target Dsg1 and, less commonly, Dsg3. Recently, several cases of PH without anti-Dsg1 or anti-Dsg3 autoantibodies have been reported with reactivity against other antigens such as desmocollin (Dsc) (56). It is currently unclear why the same autoantibodies result in a different clinical representation for PH and PF/PV. One explanation could be preferential binding to different epitopes on the same antigen molecule.
1.1.4. Paraneoplastic Pemphigus
Paraneoplastic pemphigus (PNP) is an AIBD that may be accompanied by both malignant and benign neoplasms which are often hematologic and lymphomatoid. The most frequently associated malignancies are chronic lymphocytic leukemia, B cell lymphoma, Castleman's disease, thymoma, and Waldenstrom's macroglobulinemia (21). Autoantibodies in paraneoplastic pemphigus typically target Dsg3 and proteins of the plakin family, including periplakin, envoplakin, plectin, desmoplakin 1 and 2, BP230, and the protease inhibitor alpha-2-macroglobulin-like-1 (57). The average age of onset for PNP is 51 years with no reported gender preference. Due to the association with neoplasms, PNP is hypothesized to be a side-effect of an antitumor response that cross-reacts with epithelial cells, either because the tumor is comprised of epithelial tissue or anomalously produces desmosome-like junctions (21). However, it should be considered that other pemphigus diseases may also be associated with malignancy (58).
1.1.5. IgA Pemphigus
IgA pemphigus is characterized by IgA autoantibodies against desmosomal and non-desmosomal keratinocyte cell surface components. The two major types of IgA pemphigus are subcorneal pustular dermatosis (SPD) and intraepidermal neutrophilic IgA dermatosis (IEN). The autoantigen of the SPD type was identified as Dsc1, while the antigen of the IEN type is variable (59, 60). However, in some reported cases of IEN type IgA pemphigus, IgA autoantibodies reacted with Dsg1 or Dsg3 (20, 59, 60). IgA pemphigus may be associated with monoclonal IgA gammopathy, multiple myeloma, HIV infection, Sjogren's syndrome, RA, and Crohn's disease. It is still unclear whether these diseases precede or follow IgA pemphigus. As one of the rarest AIBDs, the knowledge on IgA pemphigus is limited. No evident gender prevalence has been reported so far and the disorder may affect all age groups (22).
1.1.6. Pemphigus Erythematosus
Pemphigus erythematosus, also known as Senear-Usher syndrome, was originally described as a variant of pemphigus with features of lupus erythematosus but is today regarded as a localized form of PF and is considered an AIBD in it's own right. Autoantibodies target Dsg1, but may further target Ro, La, Sm, and double-stranded DNA antigens (23, 61). Clinically, blistering coincides with a seborrheic erythematous rash resembling the rash associated with lupus (61).
1.2. Pemphigoid Diseases
Pemphigoid diseases are characterized by autoantibodies against connective molecules at the DEJ, which is shown in Figure 1B. Binding of the autoantibodies leads to inflammation at the DEJ and degradation of the anchoring filaments and fibrils, resulting in sub-epidermal blistering. Accordingly, this group is also referred to as sub-epidermal blistering disorders. The most common pemphigoid diseases include bullous pemphigoid (BP), and mucous membrane pemphigoid (MMP). Other, less common types include pemphigoid gestationis (PG), epidermolysis bullosa acquisita (EBA), linear IgA dermatosis (LABD), and anti-laminin γ1 / p200 pemphigoid.
1.2.1. Bullous Pemphigoid
Bullous pemphigoid (BP) is the most common pemphigoid disease in Central Europe, with an incidence rate of about 10–20 per million/year. It is characterized by sub-epidermal blistering accompanied by inflammatory cell infiltration in the upper dermis (62). There are two major target antigens in BP patients: Bullous Pemphigoid Antigen 2 (BPAG2 also known as BP180 or type XVII collagen), and the Bullous Pemphigoid Antigen 1 (BPAG1, also known as BP230), a cytoplasmic plakin protein family member that links the hemidesmosome to the keratin of intermediate filaments. BP180 is a transmembrane glycoprotein that extends from the intracellular domain of basal keratinocytes to the lamina densa. The immunodominant region of BP180 is the noncollagenous domain 16A (NC16A). BP230 is a 230-kDa protein with an intracellular component associated with the hemidesmosome plate belonging to the family of plakin proteins [Figure 1B; (63, 64)]. The major immunoglobulin class in BP is IgG. It has been shown however, that some patients also develop anti-BP180 IgA and IgE autoantibodies. In fact, most of the BP sera contain both IgG and IgA autoantibodies to BP180 (65). Autoreactive CD4+ T lymphocytes recognize unique epitopes within the extracellular region of BP180. BP180-reactive Th cells and IgG autoantibodies recognize similar or identical epitopes clustered in distinct regions of the BP180 ectodomain and BP230. Many polymorphisms of HLA-II class alleles have been identified in patients with BP in several populations, especially HLA-DQ alleles. These polymorphic HLA class II alleles are likely to occur due to changes in the active binding site on the HLA molecules for binding autoantigenic peptides. A common HLA class II allele, HLA-DQB1*0301, is positively associated with BP in multiple populations and also appears to be associated with distinct clinical pemphigoid variants. Computer-based models demonstrate that the HLA-DQB1*0301 allele is capable of binding to multiple T cell epitopes within BP230 and BP180 for BP and α6 integrin, and β4 integrin for MMP. Binding leads to the activation of antigen specific T cells interacting with B cells through CD40/CD40L interaction, to produce four distinct anti-BMZ antibodies with different specificities. These antibodies bind to their specific target antigen resulting in the production of subepidermal blisters. In addition, the activation of BP180-autoreactive T cells from a cohort of BP patients with HLA-DQB1*03:01, was found to be restricted by this BP-associated HLA class II allele (46, 66–70).
1.2.2. Pemphigoid Gestationis
Pemphigoid gestationis (PG) is a rare dermatosis that occurs during pregnancy with a reported incidence rate between 0.5 and 2 per million/year (71–73). It usually affects pregnant women during the third trimenon and, less commonly, during the second trimenon or post-partum period (28, 74, 75). When occurring during the first pregnancy, the disease reoccurs in following pregnancies in 90% of the cases. PG persists and converts to BP in less than 5% of patients. In contrast to BP, blisters are infrequent and usually small in size with predominating urticarial erythema that first affects the periumbilical region. Autoantibodies are mainly directed against BP180 NC16A and in 10% of the cases against BP230. The main IgG subclasses are IgG1 and IgG3 and a strong association with maternal HLA-DR3 and HLA-DR4 exists (74, 75).
1.2.3. Epidermolysis Bullosa Acquisita
In epidermolysis bullosa acquisita (EBA), the autoantibodies are directed against type VII collagen, an anchoring fibril at the BMZ. Both skin and mucous membranes can be affected by EBA, albeit the latter to a lesser extent. The EBA incidence rate has been reported to be between 0.2 and 0.5 new cases per million/year (76). Clinically, mechanobullous (classical) EBA and inflammatory EBA can be distinguished. A characterizing feature of the classical variant is fragility of the skin that usually affects the trauma-prone areas, such as the extensor side of joints. The inflammatory subtype can resemble other AIBDs in clinical presentation as well as in serologic and histologic findings. Common to both types is the scarring and formation of milia. The scarring is particularly problematic in mucous membranes as it reduces the tissue function, even after successful suppression of the disorder (73). Inflammatory bowel disease (IBD) has been reported in 20% of the EBA patients. A link between IBD and EBA is strengthened by the presence of type VII collagen in the colon and the finding of its respective autoantibodies in IBD patients (33). EBA has been associated with environmental factors and genetically with the MHC locus HLA-DR2 notably the DRB1*15:03 allele in patients of African descent (73).
1.2.4. Anti-p200/Anti-Laminin γ1 Pemphigoid
Anti-p200 pemphigoid is a pemphigoid disease with IgG autoantibodies against a 200 kDa protein at the DEJ (77), while IgA reactivity has also been reported (78). In 90% of anti-p200 pemphigoid cases, laminin γ1 is the target antigen (34). Therefore, anti-p200 pemphigoid is also known as anti-laminin γ1 pemphigoid. However, because reactivity with laminin γ1 cannot be demonstrated in all patients with anti-p200 pemphigoid, it is recommended to restrict the term “anti-laminin γ1 pemphigoid” to those patients with reactivity against laminin γ1 (40). The clinical presentation is highly variable, often resembling BP or the inflammatory variant of EBA, and lesions heal without scarring or milia formation (77). Mucous membranes may be affected (79) and one third of the patients present with psoriasis as a co-morbidity (73). Due to variable clinical representation, histopathology, serological, and direct immunofluorescence microscopy findings, the diagnosis requires specialized assays. Therefore, anti-p200/ anti-laminin γ1 pemphigoid might be underdiagnosed (73).
1.2.5. Linear IgA Bullous Dermatosis
Linear IgA bullous dermatosis (LABD) is a pemphigoid disease with an incidence rate of about 0.2–2.3 per million/year. The autoantibodies (mainly IgA) are directed against antigens with various molecular weights, including 97-, 120-, 180-, 200-, 230-, 280-, 285-, and 290-kDa proteins (80–82). In the majority of patients, the autoantibodies target the soluble ectodomain of BP180, LAD-1, and 20% of the sera recognize BP180 NC16A (83). The disorder can affect both children and adults. The age of onset for the adult variant shows two peaks in the teenage years and around 60 years, respectively. The childhood variant has an age onset of 4.5 years (84). A significant association between the HLA locus and LABD has been reported. In particular, the haplotypes B8, DR3 and DQ2 increase the likelihood of an early onset and are thus commonly seen in the chronic bullous disease of childhood (CBDC) variant. In both children and adults alike, the tumor necrosis factor-2 (TNF-2) serves as an indicator of the increased duration of the disorder, whereas TNF-1 indicates a reduced duration and an overall better prognosis (85).
1.2.6. Mucous Membrane Pemphigoid
Mucous membrane pemphigoid (MMP) is a pemphigoid disease which predominantly affects the mucous membranes. It is also known as cicatricial pemphigoid and has an incidence of 0.5–2 new cases per million/year. In contrast to other pemphigoid diseases, a common characteristic of MMP is scarring, which may cause functional limitations of the affected tissue and adds to the severity of the disease. The age of onset varies between 60–65 years of age (71–73). The target antigens are hemidesmosome proteins, such as BP180, BP230, laminin 332, α6, β4 integrin, and collagen VII (63, 73).
2. Omics Approaches to AIBD Diagnosis and Disease Etiology
Clinical diagnosis of AIBD relies on the combination of clinical representation, i.e., mapping of a patients' symptoms to the aforementioned phenotypic characteristics and detection of tissue-bound auto-antibodies through direct immunofluorescence (DIF) microscopy as the de facto standard. Although a robust and successful approach, DIF microscopy requires cryosections of perilesional biopsies and only offers limited information about the target antigen. Circulating autoantibodies can be detected via indirect immunofluorescence (IIF) microscopy, incubating tissue substrates, e.g., monkey, rabbit or human esophagus with patient serum. Procedures that offer specific information about the autoantigen are, for example, enzyme-linked immunosorbent assays (ELISA), IIF microscopy assays, and immunoblotting/-precipitation (40).
Individual molecules and genetic associations have been unraveled in the context of AIBD pathogenesis. However, AIBDs are complex diseases, with many associations and factors contributing to their etiology (cf. Table 1). As such they should be investigated in light of a systems medicine approach on different, feedback-entangled regulatory layers such as the genome, transcriptome, epigenome, proteome, or microbiome (86–88).
However, despite recent developments in next generation sequencing and OMICS technologies, AIBDs are only now becoming the focus of genetic and high-throughput data studies, and are reviewed below.
2.1. OMICS Studies of AIBD
Beyond the directly affected genes and proteins listed in Table 1, there are few additional studies addressing unbiased, exploratory approaches to AIBD. We searched for AIBD OMICS datasets using Gene Expression Omnibus (GEO), European Nucleotide Archive (ENA), and the NCBI databases. Additionally, we searched for microbiome studies because of growing evidence on the microbiome-immune system crosstalk (89). All identified datasets are shown in Table 2.
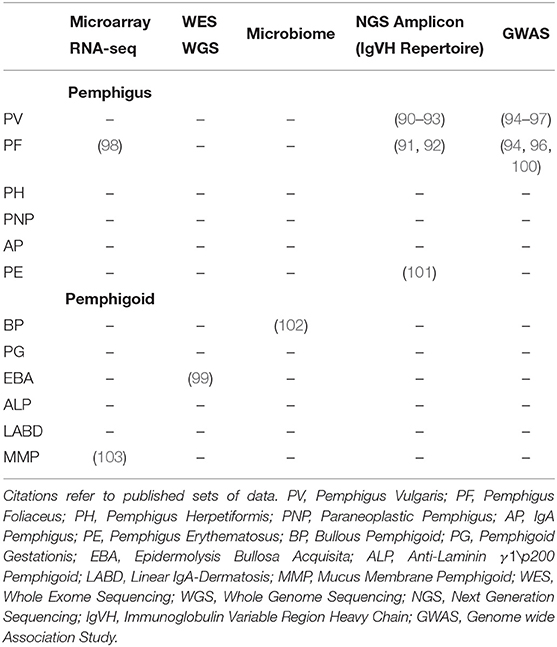
Table 2. Available AIBD microarray or next generation sequencing datasets covering more than the known marker genes.
Two whole transcriptome studies are currently publicly available. In both studies, microarrays were used to quantify transcriptome differences between patients with AIBD and the controls. Malheiros et al. (98) compared gene expression in isolated CD4+ T cells from 15 PF patients to five controls (Gene Expression Omnibus ID GSE53873). A patient subgroup comparison revealed few (up to 135) differentially regulated genes related to lymphocytes, apoptosis, proliferation and antigen presentation, which may however, be due to the small cohort size and inter-patient heterogeneity. A second, unpublished study quantified gene expression profiles in 12 patients with ocular MMP and 12 controls (Gene Expression Omnibus ID GSE77361).
More recently, the immune repertoire of PV, PF, and PE patients was classified using high-throughput sequencing methods. Two studies investigated four PV patients each (90–93). Additionally, two PF patients were investigated in (91). A third dataset is available that classifies the immune repertoire of two patients with pemphigus, without providing any further details [NCBI Bioproject ID (101)].
During the last decade, genome-wide association studies (GWAS) have become a popular approach to find genetic alterations, and so-called single nucleotide polymorphisms (SNPs) linked to diseases. Such studies compare the genotypes, i.e., a selected set of about 1-2 million SNPs, of large population cohorts in search of point mutations that significantly differ in the trait of interest. So far there are about 6,000 association studies that found more than 70,000 variant-trait associations (104). Further, association studies with gene expression as the trait (eQTL studies) (105) or methylation as the trait (meQTL) have been conducted (106). More recently, GWAS data have also been used for the computation of polygenic risk scores (107). Interestingly, the number of risk loci detected by GWAS studies scales linearly with the cohort size with no sign of saturation (108). Extrapolating on this circumstance, it is most likely that the whole genome of a person contributes to the individual disease risk. In fact, recent perspectives deny the existence of a few core genes that are causing adult-onset of disease. Instead, they argue for polygenicity or an “omnigenic” gene model, in which all mutations contribute with small effect sizes (109, 110). This is due to the robustness of biological systems, which can buffer many deleterious effects such as mutations through multiple back-up mechanisms, redundancy or feedback (111–113).
A number of GWAS for PV and PF were published recently for the Jewish (97) and Han Chinese population (94–96). Besides associations within the HLA locus, all studies found one or multiple significantly associated non-HLA SNPs, however, the associated genes differed between the studies. Another study found long non-coding RNA (lncRNA) polymorphisms associated with PF (100) using SNP data from 229 cases.
Besides the availability of just a small number of datasets for AIBDs, all studies were performed on a rather limited number of cases. Even the number of patients in the GWAS publications is low (between 100 and 365) compared to genome-wide association studies performed in other autoimmune disorders in which tens of thousands of patients were investigated [for review see e.g., (114)]. The small sample sizes lead to a small number of associated variants and genes identified by the use of GWAS.
2.2. Novel Approaches and Technologies in AIBD Research
Recent advances in mass spectrometry technology allows the unbiased quantification of protein abundance in an unprecedented way (115). Alongside proteomics, the field of degradomics has been established, which identifies proteases together with their cleaved substrates, or “degradomes” in vivo (116–118). Granzyme serine proteases in particular play important roles in the context of tissue injury and repair (119, 120). Granzyme B (GzmB) is involved in both intracellular and extracellular processes in immune cell-mediated apoptosis and extracellular proteolysis, respectively, which suggests a proteolytic role of GzmB in the pathogenesis of AIBD including BP (121). GzmB accumulated in the DEJ and blister fluid of AIBD, where it cleaved key anchoring proteins in a murine model of EBA. In line with this, GzmB deficiency reduced blistering (121).
A recent field of investigation is concerned with the organisms living in a state of symbiosis on and inside the human body. Only about 43% of cells in the human body are human, while the remaining 57% are comprised of bacterial (microbiome), fungal (mycobiome) and viral (virome) communities (122). Indeed, the total number of genes of the microbiome is ten times larger compared to the roughly 22,000 human genes. Physiological structures have adapted to the interplay between resident communities and the host, e.g., the vagus nerve represents an interface between the gut-microbiome and brain (123). The gut microbiome exists synergistically with its host to shape both metabolism and immunity. Details of this inter-dependency are still largely unknown, and it is unclear whether dysbiosis is an inducing factor leading to the exacerbation of symptoms or an epiphenomenon (124). There is also little understanding of the relationship between the skin microbiome and host defense. The communities populating the stratum corneum contribute to the first defensive line against outside influences. Furthermore, a connection between host genetics and microbial composition has been shown (125).
So far, one study investigated the role of the skin microbiota and BP with a rather limited number of patients and controls [12 per group; (102)], using a variety of skin locations (five different sampling sites). Despite the small sample size and the large heterogeneity of the skin microbiome, a significantly shifted microbiome at perilesional sites was identified.
Apart from detrimental environmental influences or lifestyle choices, e.g., injuries, pathogens and drug use, it is reasonable to assume that the respective diet has an impact. In their work Fedeles et al. (126) summarized research on the influence that dietary factors exert on bullous skin disorders. Both detrimental and advantageous effects of nutritional contents are presented, i.e., associations to exacerbation or even triggering of symptoms are drawn and protective effects are pointed out as well. This linkage is of particular interest with regards to the human microbiome.
A comparable line of investigation is the effect of the mitochondria on cellular processes. As the main source of cellular energy, mitochondria are essential constituents in signaling processes, cellular metabolism as well as inflammatory responses. It has been reported that changes in the mitochondrial genome can lead to pathological conditions and are implicated in various immune diseases. In particular, the mitochondrially encoded MT-ATP8 gene has been linked to BP susceptibility (127).
Single-cell RNA-sequencing (scRNA-seq) is another recent technology that will have a major impact on elucidating the molecular processes in AIBD. The technology allows the characterization of individual cells, leading to the discovery of new cell types and cellular states that echo the underlying heterogeneity and plasticity of the immune system. scRNA-seq opens up new possibilities to analyze the immune repertoire and its effects on immune cells while in parallel recording both the quantitative gene expression and the repertoire sequence information for both chains (α/β, light/heavy) of the receptor. This additional feature would identify subpopulations of immune cells, that drive the disease to paracrine stimulation or recruitment of further immune cells. Although recent progress in scRNA-sequencing has been achieved, there are currently only a few studies that have investigated the human epidermis. To date, there is still no approach for BP, only for psoriasis, where Cheng et al. (128) analyzed the scRNA-seq profiles of 92,889 human epidermal cells from nine normal and three inflamed skin samples. The analysis of transcriptome levels of keratinocyte subpopulations reflects classical epidermal layers, but also strongly segmented epithelial functions such as cell-cell communication, inflammation, and WNT pathway modulation. The identification of molecular fingerprints of inflammatory skin states, including the enrichment of the CD1C+CD301A+ myeloid dendritic cell population in psoriatic epidermis, provides a critical step toward elucidating epidermal diseases of development, differentiation and inflammation.
2.3. A Computational Approach to AIBD
Since the number of available genetic and/or OMICS datasets is limited and hitherto provides narrow insights into the disease etiology, one can ask whether it is possible to derive phenotype-specific pathways from given gene-sets, i.e., disease specific transcriptome patterns that allow patient stratification and also provide insight about the molecular processes involved. The exploratory approach of choice was a network diffusion in order to determine the impact of each gene-set on its biological neighborhood (129). It was followed by Gene Set Variation Analysis (GSVA) (130), which enables the assignment of sample-wise pathway enrichments on the basis of the calculated diffusion scores.
The inferred pathways do, however, not enable a clear separation of either phenotypes or clinical subclasses. It can be argued that the number of known associated genes is too small for an accurate inference of protein-protein interactions and related pathways, without a significant amount of noise. A closer investigation is warranted merely on account of the heterogeneity of phenotypes and the presumed multifactorial framework of disease genesis.
3. Summary and Outlook
Autoimmune skin blistering diseases (AIBDs) are complex diseases, mostly with a late onset in life. They are driven by both genetic and environmental factors. So far, most of the research on AIBDs has been conducted on clinical and diagnostic aspects of the diseases. In AIBDs, autoreactive antibodies are generated that target proteins involved in cell-to-cell and cell-to-matrix adhesion in the epidermis/epithelium and at the dermal-epidermal junction. Binding of those autoantibodies leads to inflammation and the loss of function of those proteins. Depending on the autoantigen, AIBDs can be divided into pemphigus and pemphigoid diseases. However, knowledge on detailed disease mechanisms leading to the development of AIBDs is still scarce.
The limited availability of OMICS data-sets in AIBDs (cf. Table 2) illustrates the long road ahead in elucidating the causes of AIBDs. Limiting factors include low prevalence and high age of onset. Those factors hinder the acquisition of sufficient suitable test-subjects and also limit perceived importance of this topic, hence lowering the impact of scientific research in this field.
In recent years, significant efforts have been made to remedy the problem of cohort size by establishing programs that advance regional, national, and international cooperation between medical facilities and research institutions. European Reference Networks (ERN) for instance are part of the Directive on Patient Rights in Cross-Border Healthcare of 2011. They are oriented toward the handling of rare disorders through the interconnection of specialists and resources. An example, with regards to AIBDs, is the “Dimethyl fumarate for the treatment of bullous pemphigoid” (DPem) research network that connects study groups from four European countries. This particular initiative is funded by the ERA-Net for Research Programmes on Rare Diseases, an initiative with the expressed purpose of facilitating cooperative research into rare disorders (131, 132). Observance of incidence and distribution of AIBDs is the objective of the Regibul Register (133).
One way to make the best use of the limited patient data is to use theoretical concepts and test the idea of core vs. peripheral genes. Initial genome-wide association studies on bullous pemphigoid have predicted HLA genes as risk factors. These genes are common to autoimmune diseases but not specific to AIBDs and might be considered as core gene candidates. Alternatively, core genes might be those that are strongly linked to the disease phenotype. Therefore, one might expect immune system and cell adhesion related genes to be critical to AIBD. In addition to identifying core genes it will be important to find tissue specific regulatory networks that mediate the effect of the core genes to the phenotype. To study these networks, it might be important to develop further in vitro and in vivo models, be it active or passive mouse models or organotypic skin models. The latter might be generated from fibroblasts and keratinocytes of AIBD patients or derived from induced pluripotent stem cells, thereby harboring a disease genome. A third approach to better understand disease etiology is the integration of environmental factors through microbiome studies. The effect of microbiome- immune cell interaction is well established but has never been studied in detail in AIBDs. First studies indicate a dysbiosis of the skin microbiome in lesional skin, a mechanistic link to increased immune reaction or decreased barrier function of the skin which still needs to be established.
In summary, an integrated OMICS approach to study skin blistering diseases, comprising genomics, transcriptomics, and proteomics together with the microbiome and novel methods such as single cell RNAseq, is highly warranted. Research in this direction has only recently begun and much work and opportunities still remain. Besides increasing the numbers for further GWAS, we advocate for high-throughput deep phenotyping and whole genome sequencing and investigation of tissue specific gene expression to characterize cellular properties associated with disease-associated genomes. Further, the use of disease models may elucidate the polygenic complexity of the diseases.
Author Contributions
All authors reviewed the literature and collected the data, contributed to the manuscript, read, and approved it.
Funding
This work was supported by the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) under Germany's Excellence Strategy – EXC 22167-390884018 and the clinical research unit CRU 303 Pemphigoid Diseases.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors gratefully acknowledge the support and advice by colleagues of the Institute of Dermatology and the Institute for Experimental Dermatology, Lübeck. Furthermore, the authors acknowledge computational support from the OMICS compute cluster at the University of Lübeck. We acknowledge financial support by Land Schleswig-Holstein within the funding programme Open Access Publikationsfonds.
References
1. Jordon RE, Beutner EH, Witebsky E, Blumental G, Hale WL, Lever WF. Basement zone antibodies in bullous pemphigoid. JAMA. (1967) 200:751–6.
3. Sequencing Consortium TM. Complete sequence and gene map of a human major histocompatibility complex. Nature. (1999) 401:921–23. doi: 10.1038/44853
4. Gough SCL, Simmonds MJ. The HLA region and autoimmune disease: associations and mechanisms of action. Curr Genom. (2007) 8:453–65. doi: 10.2174/138920207783591690
5. Whitacre CC. Sex differences in autoimmune disease. Nat Immunol. (2001) 2:777 EP. doi: 10.1038/ni0901-777
6. Markle JG, Fish EN. SeXX matters in immunity. Trends Immunol. (2014) 35:97–104. doi: 10.1016/j.it.2013.10.006
7. Whitacre CC, Reingold SC, O'Looney PA, Blankenhorn E, Brinley F, Collier E, et al. A gender gap in autoimmunity. Science. (1999) 283:1277. doi: 10.1126/science.283.5406.1277
8. Eidinger D, Garrett TJ. Studies of the regulatory effects of the sex hormones on antibody formation and stem cell differentiation. J Exp Med. (1972) 136:1098. doi: 10.1084/jem.136.5.1098
9. Weinstein Y, Ran S, Segal S. Sex-associated differences in the regulation of immune responses controlled by the MHC of the mouse. J Immunol. (1984) 132:656.
10. Amadori A, Zamarchi R, De Silvestro G, Forza G, Cavatton G, Danieli GA, et al. Genetic control of the CD4/CD8 T-cell ratio in humans. Nat Med. (1995) 1:1279–83. doi: 10.1038/nm1295-1279
11. Griffin AC, Whitacre CC. Sex and strain differences in the circadian rhythm fluctuation of endocrine and immune function in the rat: implications for rodent models of autoimmune disease. J Neuroimmunol. (1991) 35:53–64. doi: 10.1016/0165-5728(91)90161-Y
12. Schmidt E, Zillikens D. Diagnostik und Therapie bull∧ser Autoimmundermatosen. Dtsch Arztebl Int. (2011) 108:399–405.
13. Vodo D, Sarig O, Sprecher E. The genetics of pemphigus vulgaris. Front Med. (2018) 5:226. doi: 10.3389/fmed.2018.00226
14. Buonavoglia A, Leone P, Dammacco R, Di Lernia G, Petruzzi M, Bonamonte D, et al. Pemphigus and mucous membrane pemphigoid: an update from diagnosis to therapy. Autoimmun Rev. (2019) 18:349–58. doi: 10.1016/j.autrev.2019.02.005
15. Miyagawa S, Amagai M, Iida T, Yamamoto Y, Nishikawa T, Shirai T. Late development of antidesmoglein 1 antibodies in pemphigus vulgaris: correlation with disease progression. Brit J Dermatol. (2001) 141:1084–7. doi: 10.1046/j.1365-2133.1999.03209.x
16. Espe S. MalaCards: the human disease database. J Med Library Assoc. (2018) 1:106. doi: 10.5195/JMLA.2018.253
17. Borroni G, Biagi F, Ciocca O, Vassallo C, Carugno A, Cananzi R, et al. IgA anti–epidermal transglutaminase autoantibodies: a sensible and sensitive marker for diagnosis of dermatitis herpetiformis in adult patients. J Eur Acad Dermatol Venereol. (2012) 27:836–41. doi: 10.1111/j.1468-3083.2012.04586.x
18. Sárdy M, Kárpáti S, Merkl B, Paulsson M, Smyth N. Epidermal transglutaminase (TGase 3) is the autoantigen of dermatitis herpetiformis. J Exp Med. (2002) 195:747–57. doi: 10.1084/jem.20011299
19. Mehta V, Balachandran C, Nayak S. Herpetiform pemphigus clinically resembling bullous pemphigoid. Ind J Dermatol. (2008) 53:158–9. doi: 10.4103/0019-5154.43218
20. Porro AM, Caetano LV, Maehara LS, Enokihara MM. Non-classical forms of pemphigus: pemphigus herpetiformis, IgA pemphigus, paraneoplastic pemphigus and IgG/IgA pemphigus. Anais Brasileiros de Dermatologia. (2014) 89:96–106. doi: 10.1590/abd1806-4841.20142459
21. Wieczorek M, Czernik A. Paraneoplastic pemphigus: a short review. Clin Cosmet Investigat Dermatol. (2016) 9:291–5. doi: 10.2147/CCID.S100802
22. Aslanova M, Zito PM. IgA Pemphigus. NCBI Bookshelf. (2019). Available online at: https://www.ncbi.nlm.nih.gov/books/NBK519063/
23. Pérez-Pérez ME, Avalos-Dìaz E, Herrera-Esparza R. Autoantibodies in senear-usher syndrome: cross-reactivity or multiple autoimmunity? Autoimmune Dis. (2012) 2012:7. doi: 10.1155/2012/296214
24. Liu Y, Li L, Xia Y. BP180 is critical in the autoimmunity of bullous pemphigoid. Front Immunol. (2017) 8:1752. doi: 10.3389/fimmu.2017.01752
25. Fang H, Zhang Y, Li L, Wang G, Liu Z. The autoimmune skin disease bullous pemphigoid: the role of mast cells in autoantibody-induced tissue injury. Front Immunol. (2018) 9:407. doi: 10.3389/fimmu.2018.00407
26. Iranzo P, López-Lerma I, Teresa Robles M. Bullous pemphigoid associated with mantle cell lymphoma. Arch Dermatol. (2005) 140:1496–9. doi: 10.1001/archderm.140.12.1496
27. Baigrie D, Nookala V. Bullous Pemphigoid (2018). Available online at: https://www.ncbi.nlm.nih.gov/books/NBK535374/
28. Sävervall C, Thomsen SF. Pemphigoid gestationis: current perspectives. Clin Cosmet Investigat Dermatol. (2017) 10:441–9. doi: 10.2147/CCID.S128144
29. Iwata H, Vorobyev A, Koga H, Ludwig RJ, Zillikens D, Prost-Squarcioni C, et al. Meta-analysis of the clinical and immunopathological characteristics and treatment outcomes in epidermolysis bullosa acquisita patients. Orphanet J Rare Dis. (2018) 13:153–62. doi: 10.1186/s13023-018-0896-1
30. Shaffer BR. Epidermolysis bullosa acquisita in association with mantle cell lymphoma. Cutis. (2018) 101:E13–5.
31. Koga H, Prost-Squacioni C, Iwata H, Jonkman MF, Ludwig RJ, Bieber K. Epidermolysis bullosa acquisita: the 2019 update. Front Med. (2019) 5:362. doi: 10.3389/fmed.2018.00362
32. Koga H, Kasprick A, López R, Aulí M, Pont M, Godessart N. Therapeutic effect of a novel phosphatidylinositol-3-kinase δ inhibitor in experimental epidermolysis bullosa acquisita. Front Immunol. (2018) 9:1558. doi: 10.3389/fimmu.2018.01558
33. Woodley DT, Chen M, Kim G. Epidermolysis Bullosa Acquisita. UpToDate Inc. (2019). Available online at: https://www.uptodate.com/contents/epidermolysis-bullosa-acquisita#H306466519
34. Dainichi T, Kurono S, Zillikens D. Anti-laminin gamma-1 pemphigoid. In: Hakomori S, editor. Proceedings of the National Academy of Sciences of the United States of America. Fukuoka: US National Library of Medicine National Institutes of Health (2009). p. 2800–5.
35. Zumelzu C, Alexandre M, Le Roux C, Weber P, Guyot A, Levy A, et al. Mucous membrane pemphigoid, bullous pemphigoid, and anti-programmed death-1/ programmed death-ligand 1: a case report of an elderly woman with mucous membrane pemphigoid developing after pembrolizumab therapy for metastatic melanoma and review of the literature. Front Med. (2018) 5:268. doi: 10.3389/fmed.2018.00268
36. Meyer N, Misery L. Geoepidemiologic considerations of auto-immune pemphigus. Autoimmun Rev. (2010) 9:A379–A382. doi: 10.1016/j.autrev.2009.10.009
37. Gazit E, Loewenthal R. The immunogenetics of pemphigus vulgaris. Autoimmun Rev. (2005) 4:16–20. doi: 10.1016/j.autrev.2004.05.002
38. Aoki V, Millikan RC, Rivitti EA, Hans-Filho G, Eaton DP, Warren SJP, et al. Environmental risk factors in endemic pemphigus foliaceus (fogo selvagem). J Investigat Dermatol Symp Proc. (2004) 9:34–40. doi: 10.1111/j.1087-0024.2004.00833.x
39. Shirakata Y, Amagai M, Hanakawa Y, Nishikawa T, Hashimoto K. Lack of mucosal involvement in pemphigus foliaceus may be due to low expression of desmoglein 1. J Investigat Dermatol. (1998) 110:76–8. doi: 10.1046/j.1523-1747.1998.00085.x
40. Witte M, Zillikens D. Diagnosis of autoimmune blistering diseases. Front Med. (2018) 5:296. doi: 10.3389/fmed.2018.00296
41. Mimouni D, Bar H, Gdalevich M, Katzenelson V, David M. Pemphigus—analysis of epidemiological factors in 155 patients. J Eur Acad Dermatol Venereol. (2008) 22:1232–5. doi: 10.1111/j.1468-3083.2008.02786.x
42. McPherson T, Venning VV. Management of autoimmune blistering diseases in pregnancy. Dermatol Clin. (2011) 29:585–90. doi: 10.1016/j.det.2011.06.008
43. Svecova D, Parnicka Z, Pastyrikova L, Urbancek S, Luha J, Buc M. HLA DRB1* and DQB1* alleles are associated with disease severity in patients with pemphigus vulgaris. Int J Dermatol. (2015) 54:168–73. doi: 10.1111/ijd.12418
44. Ahmed AR, Yunis EJ, Khatri K, Wagner R, Notani G, Awdeh Z, et al. Major histocompatibility complex haplotype studies in Ashkenazi Jewish patients with pemphigus vulgaris. Proc Natl Acad Sci USA. (1990) 87:7658–62. doi: 10.1073/pnas.87.19.7658
45. Gazit E, Slomov Y, Goldberg I, Brenner S, Loewenthal R. HLA-G is associated with pemphigus vulgaris in jewish patients. Hum Immunol. (2004) 65:39–46. doi: 10.1016/j.humimm.2003.09.019
46. Delgado JC, Turbay D, Yunis EJ, Yunis JJ, Morton ED, Bhol K, et al. A common major histocompatibility complex class II allele HLA-DQB1* 0301 is present in clinical variants of pemphigoid. Proc Natl Acad Sci USA. (1996) 93:8569–71. doi: 10.1073/pnas.93.16.8569
47. Loiseau P, Lecleach L, Prost C, Lepage V, Busson M, Bastuji-Garin S, et al. HLA class II polymorphism contributes to specify desmoglein derived peptides in pemphigus vulgaris and pemphigus foliaceus. J Autoimmun. (2000) 15:67–73. doi: 10.1006/jaut.2000.0388
48. Scharf SJ, Friedmann A, Brautbar C, Szafer F, Steinman L, Horn G, et al. HLA class II allelic variation and susceptibility to pemphigus vulgaris. Proc Natl Acad Sci USA. (1988) 85:3504–8. doi: 10.1073/pnas.85.10.3504
49. Cirillo N, Al-Jandan BA. Desmosomal adhesion and pemphigus vulgaris: the first half of the story. Cell Commun Adhesion. (2013) 20:1–10. doi: 10.3109/15419061.2013.763799
50. Culton DA, Qian Y, Li N, Rubenstein D, Aoki V, Filhio GH, et al. Advances in pemphigus and its endemic pemphigus foliaceus (Fogo Selvagem) phenotype: a paradigm of human autoimmunity. J Autoimmun. (2008) 31:311–24. doi: 10.1016/j.jaut.2008.08.003
51. Bastuji-Garin S, Souissi R, Blum L, Turki H, Nouira R, Jomaa B, et al. Comparative epidemiology of pemphigus in tunisia and France: unusual incidence of pemphigus foliaceus in young tunisian women. J Investigat Dermatol. (1995) 104:302–5. doi: 10.1111/1523-1747.ep12612836
52. Kridin K. Pemphigus group: overview, epidemiology, mortality, and comorbidities. Immunol Res. (2018) 66:255–70. doi: 10.1007/s12026-018-8986-7
53. Martel P, Gilbert D, Busson M, Loiseau P, Lepage V, Drouot L, et al. Epistasis between DSG1 and HLA class II genes in pemphigus foliaceus. Genes Immun. (2002) 3:205–10. doi: 10.1038/sj.gene.6363839
54. Kasperkiewicz M, Kowalewski C, Jabłoǹska S. Pemphigus herpetiformis: From first description until now. J Am Acad Dermatol. (2014) 70:780–7. doi: 10.1016/j.jaad.2013.11.043
55. Kozlowska A, Hashimoto T, Jarzabek-Chorzelska M, Amagai A, Nagata Y, Strasz Z, et al. Pemphigus herpetiformis with IgA and IgG antibodies to desmoglein 1 and IgG antibodies to desmocollin 3. J Am Acad Dermatol. (2003) 48:117–22. doi: 10.1067/mjd.2003.23
56. Ishii N, Teye K, Fukuda S, Uehara R, Hachiya T, Koga H, et al. Anti-desmocollin autoantibodies in nonclassical pemphigus. Brit J Dermatol. (2015) 173:59–68. doi: 10.1111/bjd.13711
57. Schepens I, Jaunin F, Begre N, Läderach U, Marcus K, Hashimoto T, et al. The protease inhibitor alpha-2-macroglobuline-like-1 is the p170 antigen recognized by paraneoplastic pemphigus autoantibodies in human. PLoS ONE. (2010) 5:e12250. doi: 10.1371/journal.pone.0012250
58. Kridin K, Zelber-Sagi S, Comaneshter D, Cohen AD. Coexistent solid malignancies in pemphigus: a population-based study. JAMA Dermatol. (2018) 154:435–40. doi: 10.1001/jamadermatol.2017.6334
59. Tajima M, Mitsuhashi Y, Irisawa R, Amagai M, Hashimoto T, Tsuboi R. IgA pemphigus reacting exclusively to desmoglein 3. Eur J Dermatol. (2011) 20:626–9. doi: 10.1684/ejd.2010.1021
60. Kárpáti S, Amagai M, Liu WL, Dmochowski M, Hashimoto T, Horváth A. Identification of desmoglein 1 as autoantigen in a patient with intraepidermal neutrophilic IgA dermatosis type of IgA pemphigus. Exp Dermatol. (2000) 9:224–8. doi: 10.1034/j.1600-0625.2000.009003224.x
61. Sánchez-Pérez J, Garcìa-Dìez A. Pemphigus. Actas Dermo-Sifiliograficas. (2005) 96:329–56. doi: 10.1016/S0001-7310(05)73090-8
62. Jordon RE, Beutner EH, Witebsky E, Blumental G, Hale WL, Lever WF. Basement zone antibodies in bullous pemphigoid. JAMA. (1967) 200:751–6. doi: 10.1001/jama.200.9.751
63. Di Zenzo G, della Torre R, Zambruno G, Borradori L. Bullous pemphigoid: from the clinic to the bench. Clin Dermatol. (2012) 30:3–16. doi: 10.1016/j.clindermatol.2011.03.005
64. Nishie W. Update on the pathogenesis of bullous pemphigoid: an autoantibody-mediated blistering disease targeting collagen XVII. J Dermatol Sci. (2014) 73:179–86. doi: 10.1016/j.jdermsci.2013.12.001
65. Horváth B, Niedermeier A, Podstawa E, Müller R, Hunzelmann N, Kárpáti S, et al. IgA autoantibodies in the pemphigoids and linear IgA bullous dermatosis. Exp Dermatol. (2010) 19:648–53. doi: 10.1111/j.1600-0625.2010.01080.x
66. Wojnarowska F, Venning VA. Immunobullous diseases. In: Rook's Textbook of Dermatology, 7th ed. Blackwell Science. (2004). p. 1–41. Available online at: https://doi.org/10.1002/9780470750520.ch41
67. Zakka LR, Reche P, Ahmed AR. Role of MHC Class II genes in the pathogenesis of pemphigoid. Autoimmun Rev. (2011) 11:40–7. doi: 10.1016/j.autrev.2011.07.002
68. Büdinger L, Borradori L, Yee C, Eming R, Ferencik S, Grosse-Wilde H, et al. Identification and characterization of autoreactive T cell responses to bullous pemphigoid antigen 2 in patients and healthy controls. J Clin Investigat. (1998) 102:2082–9. doi: 10.1172/JCI3335
69. Büdinger L, Borradori L, Yee C, Eming R, Ferencik S, Grosse-Wilde H, et al. T cell control in autoimmune bullous skin disorders. J Clin Investigat. (2006) 116:1159–66. doi: 10.1172/JCI28547
70. Lin M, Diaz LA, Fu C, Giudice GJ, Olague-Marchan M, Lazaro AM, et al. Epitopes targeted by bullous pemphigoid T lymphocytes and autoantibodies map to the same sites on the bullous pemphigoid 180 ectodomain. J Investigat Dermatol. (2000) 115:955–61. doi: 10.1046/j.1523-1747.2000.00153.x
71. Bertram F, Bröcker E, Zillikens D, Schmidt E. Prospective analysis of the incidence of autoimmune bullous disorders in Lower Franconia, Germany. J Deutschen Dermatologischen Gesellschaft. (2009) 7:434–9. doi: 10.1111/j.1610-0387.2008.06976.x
72. Joly P, Baricault S, Sparsa A, Bernard P, Bédane C, Duvert-Lehembre S, et al. Incidence and mortality of bullous pemphigoid in France. J Investigat Dermatol. (2012) 132:1998–2004. doi: 10.1038/jid.2012.35
73. Schmidt E, Zillikens D. Pemphigoid diseases. Lancet. (2013) 381:320–32. doi: 10.1016/S0140-6736(12)61140-4
74. Cobo MF, Santi CG, Maruta CW, Aoki V. Pemphigoid gestationis: clinical and laboratory evaluation. Clinics (São Paulo, Brazil). (1999) 64:1043–7. doi: 10.1590/S1807-59322009001100002
75. Jenkins RE, Hern S, Black MM. Clinical features and management of 87 patients with pemphigoid gestationis. Clin Exp Dermatol. (2009) 24:255–9. doi: 10.1046/j.1365-2230.1999.00472.x
76. Vorobyev A, Ludwig RJ, Schmidt E. Clinical features and diagnosis of epidermolysis bullosa acquisita. Exp Rev Clin Immunol. (2017) 13:157–69. doi: 10.1080/1744666X.2016.1221343
77. Dilling A, Rose C, Hashimoto T, Zillikens D, Shimanovich I. Anti-p200 pemphigoid: a novel autoimmune subepidermal blistering disease. J Dermatol. (2007) 34:1–8. doi: 10.1111/j.1346-8138.2007.00208.x
78. Wozniak K, Hashimoto T, Fukuda S, Ohyama B, Ishii N, Koga H, et al. IgA Anti-p200 pemphigoid. JAMA Dermatol. (2011) 147:1306–10. doi: 10.1001/archdermatol.2011.303
79. Goletz S, Hashimoto T, Zillikens D, Schmidt E. Anti-p200 pemphigoid. J Am Acad Dermatol. (2014) 71:185–91. doi: 10.1016/j.jaad.2014.02.036
80. Wojnarowska F, Whitehead P, Leigh IM, Bhogal BS, Black MM. Identification of the target antigen in chronic bullous disease of childhood and linear IgA disease of adults. Brit J Dermatol. (1991) 124:157–62. doi: 10.1111/j.1365-2133.1991.tb00425.x
81. Marinkovich MP, Taylor TB, Keene DR, Burgeson RE, Zone JJ. LAD-1, the linear IgA bullous dermatosis autoantigen, is a novel 120-kDa anchoring filament protein synthesized by epidermal cells. J Investigat Dermatol. (1996) 106:734–8. doi: 10.1111/1523-1747.ep12345782
82. Zone JJ, Taylor TB, Meyer LJ, Petersen MJ. The 97kDa linear IgA bullous disease antigen is identical to a portion of the extracellular domain of the 180kDa bullous pemphigoid antigen, BPAg2. J Investigat Dermatol. (1998) 110:207–10. doi: 10.1046/j.1523-1747.1998.00129.x
83. Zillikens D, Herzele K, Georgi M, Schmidt E, Chimanovitch I, Bröcker E, et al. Autoantibodies in a subgroup of patients with linear IgA disease react with the NC16A domain of BP1801. J Investigat Dermatol. (1999) 113:947–53. doi: 10.1046/j.1523-1747.1999.00808.x
84. Chaudhari S, Mobini N. Linear IgA bullous dermatosis: a rare clinicopathologic entity with an unusual presentation. J Clin Aesthet Dermatol. (2015) 8:43–6.
85. Collier PM, Wojnarowska F, Welsh K, McGuire W, Black MM. Adult linear IgA disease and chronic bullous disease of childhood: the association with human lymphocyte antigens Cw7, B8, DR3 and tumour necrosis factor influences disease expression. Brit J Dermatol. (1999) 141:867–75. doi: 10.1046/j.1365-2133.1999.03110.x
86. Civelek M, Lusis AJ. Systems genetics approaches to understand complex traits. Nat Rev Genet. (2014) 15:34–48. doi: 10.1038/nrg3575
87. Yugi K, Kubota H, Hatano A, Kuroda S. Trans-omics: how to reconstruct biochemical networks across multiple ‘omic' layers. Trends Biotechnol. (2016) 34:276–90. doi: 10.1016/j.tibtech.2015.12.013
88. Durmaz AA, Karaca E, Demkow U, Toruner G, Schoumans J, Cogulu O. Evolution of genetic techniques: past, present, and beyond. BioMed Res Int. (2015) 2015:461524–31. doi: 10.1155/2015/461524
89. De Luca F, Shoenfeld Y. The microbiome in autoimmune diseases. Clin Exp Immunol. (2019) 195:74–85. doi: 10.1111/cei.13158
90. University of Pennsylvania and University of Maryland Homo sapiens isolate: Human IgG isolate Targeted Locus (Loci). Data-set: Characterization of Autoantibody Repertoires in Antibody-Mediated Diseases Is Necessary to Understand Pathophysiology and Therapeutic Approaches (2016). Available online at: https://www.ncbi.nlm.nih.gov/bioproject/309663
91. Chen J, Zheng Q, Hammers CM, Ellebrecht CT, Mukherjee EM, Tang H, et al. Proteomic analysis of pemphigus autoantibodies indicates a larger, more diverse, and more dynamic repertoire than determined by B cell genetics. Cell Rep. (2017) 18:237–47. doi: 10.1016/j.celrep.2016.12.013
92. Mukherjee EM, Millar SE, Payne AS, Stanley JR. Distinct Clonal Relationships Among Autoreactive IgG and IgA B-Cells in Pemphigus Vulgaris. Data-set: Lineage Analysis of Autoreactive B-Cells Can Reveal the Origins of Autoimmunity. (2018). Available online at: https://www.ncbi.nlm.nih.gov/bioproject/?term=PRJNA437136
93. Ellebrecht CT, Mukherjee EM, Zheng Q, Choi EJ, Reddy SG, Mao X, et al. Autoreactive IgG and IgA B cells evolve through distinct subclass switch pathways in the autoimmune disease pemphigus vulgaris. Cell Rep. (2018) 24:2370–80. doi: 10.1016/j.celrep.2018.07.093
94. Sun Y, Liu H, Yang B, Wang C, Foo JN, Bao F, et al. Investigation of the predisposing factor of pemphigus and its clinical subtype through a genome-wide association and next generation sequence analysis. J Eur Acad Dermatol Venereol. (2019) 33:410–5. doi: 10.1111/jdv.15227
95. Gao J, Zhu C, Zhang Y, Sheng Y, Yang F, Wang W, et al. Association study and fine-mapping major histocompatibility complex analysis of pemphigus vulgaris in a han chinese population. J Investigat Dermatol. (2018) 138:2307–14. doi: 10.1016/j.jid.2018.05.011
96. Zhang SY, Zhou XY, Zhou XL, Zhang Y, Deng Y, Liao F, et al. Subtype-specific inherited predisposition to pemphigus in the Chinese population. Brit J Dermatol. (2019) 180:828–35. doi: 10.1111/bjd.17191
97. Sarig O, Bercovici S, Zoller L, Goldberg I, Indelman M, Nahum S, et al. Population-specific association between a polymorphic variant in ST18, encoding a pro-apoptotic molecule, and pemphigus vulgaris. J Investigat Dermatol. (2012) 132:1798–805. doi: 10.1038/jid.2012.46
98. Malheiros D, Panepucci RA, Roselino AM, Araújo AG, Zago MA, Petzl–Erler ML. Genome-wide gene expression profiling reveals unsuspected molecular alterations in pemphigus foliaceus. Immunology. (2014) 143:381–95. doi: 10.1111/imm.12315
99. Takeichi T, Liu L, Fong K, Ozoemena L, McMillan JR, Salam A, et al. Whole-exome sequencing improves mutation detection in a diagnostic epidermolysis bullosa laboratory. Brit J Dermatol. (2015) 172:94–100. doi: 10.1111/bjd.13190
100. Lobo–Alves SC, Augusto DG, Magalhães WCS, Tarazona–Santos E, Lima–Costa MF, Barreto ML, et al. Long non-coding RNA polymorphisms influence susceptibility to endemic pemphigus foliaceus. Brit J Dermatol. (2019) 181:324–31. doi: 10.1111/bjd.17640
101. University E. Homo Sapiens Targeted Locus (Loci). Data-set: Accession: PRJNA260985; ID: 260985 (2014). Available online at: https://www.ncbi.nlm.nih.gov/bioproject/?term=PRJNA260985
102. Miodovnik M, Künstner A, Langan EA, Zillikens D, Gläser R, Sprecher E, et al. A distinct cutaneous microbiota profile in autoimmune bullous disease patients. Exp Dermatol. (2017) 11:839–7. doi: 10.1111/exd.13357
103. Ahadome S, Dart J. Genome Wide Analysis of Gene Expression in Conjunctiva Whole Tissue and Primary Conjunctival Fibroblasts From Patients With Ocular Mucous Membrane Pemphigoid and Control Patients. (2016). Available online at: https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE77361
104. Richardson TG, Harrison S, Hemani G, Davey-Smith G. An atlas of polygenic risk score associations to highlight putative causal relationships across the human phenome. eLife. (2019) 8:e43657. doi: 10.7554/eLife.43657
105. Gilad Y, Rifkin SA, Pritchard JK. Revealing the architecture of gene regulation: the promise of eQTL studies. Trends Genet. (2008) 24:408–15. doi: 10.1016/j.tig.2008.06.001
106. Smith AK, Kilaru V, Kocak M, Almli LM, Mercer KB, Ressler KJ, et al. Methylation quantitative trait loci (meQTLs) are consistently detected across ancestry, developmental stage, and tissue type. BMC Genom. (2014) 15:145. doi: 10.1186/1471-2164-15-145
107. Torkamani A, Wineinger NE, Topol EJ. The personal and clinical utility of polygenic risk scores. Nat Rev Genet. (2018) 19:581. doi: 10.1038/s41576-018-0018-x
108. Canela-Xandri O, Rawlik K, Tenesa A. An atlas of genetic associations in UK Biobank. Nat Genet. (2018) 50:1593–9. doi: 10.1038/s41588-018-0248-z
109. Wray NR, Wijmenga C, Sullivan PF, Yang J, Visscher PM. Common disease is more complex than implied by the core gene omnigenic model. Cell. (2018) 173:1573–80. doi: 10.1016/j.cell.2018.05.051
110. Boyle EA, Li YI, Pritchard JK. An expanded view of complex traits: from polygenic to omnigenic. Cell. (2017) 169:1177–86. doi: 10.1016/j.cell.2017.05.038
111. Kitano H. Cancer as a robust system: implications for anticancer therapy. Nat Rev Cancer. (2004) 4:227–35. doi: 10.1038/nrc1300
112. Masel J, Siegal ML. Robustness: mechanisms and consequences. Trends Genet. (2009) 25:395–403. doi: 10.1016/j.tig.2009.07.005
113. Wang X, Goldstein DB. Enhancer redundancy predicts gene pathogenicity and informs complex disease gene discovery. bioRxiv 459123. (2018). Available online at: https://www.biorxiv.org/content/10.1101/459123v1
114. Inshaw JRJ, Cutler AJ, Burren OS, Stefana MI, Todd JA. Approaches and advances in the genetic causes of autoimmune disease and their implications. Nat Immunol. (2018) 19:674–84. doi: 10.1038/s41590-018-0129-8
115. Aebersold R, Mann M. Mass-spectrometric exploration of proteome structure and function. Nature. (2016) 537:347–55. doi: 10.1038/nature19949
116. Savickas S, Auf dem Keller U. Targeted degradomics in protein terminomics and protease substrate discovery. Biol Chem. (2017) 399:47–54. doi: 10.1515/hsz-2017-0187
117. Klein T, Eckhard U, Dufour A, Solis N, Overall CM. Proteolytic cleavage-mechanisms, function, and "omic" approaches for a near-ubiquitous posttranslational modification. Chem Rev. (2018) 118:1137–68. doi: 10.1021/acs.chemrev.7b00120
118. López-Otín C, Overall CM. Protease degradomics: a new challenge for proteomics. Nat Rev Mol Cell Biol. (2002) 3:509–19. doi: 10.1038/nrm858
119. Granville DJ. Granzymes in disease: bench to bedside. Cell Death Different. (2010) 17:565–6. doi: 10.1038/cdd.2009.218
120. Turner CT, Lim D, Granville DJ. Granzyme B in skin inflammation and disease. Matrix Biol. (2019) 75–76:126–40. doi: 10.1016/j.matbio.2017.12.005
121. Russo V, Klein T, Lim DJ, Solis N, Machado Y, Hiroyasu S, et al. Granzyme B is elevated in autoimmune blistering diseases and cleaves key anchoring proteins of the dermal-epidermal junction. Sci Rep. (2018) 8:1–11. doi: 10.1038/s41598-018-28070-0
122. Sender R, Fuchs S, Milo R. Revised estimates for the number of human and bacteria cells in the body. PLoS Biol. (2016) 14:e1002533. doi: 10.1371/journal.pbio.1002533
123. Carabotti M, Scirocco A, Maselli MA, Severia C. The gut-brain axis: interactions between enteric microbiota, central and enteric nervous systems. Ann Gastroenterol. (2015) 28:203–9.
124. Li B, Selmi C, Tang R, Gershwin ME, Ma X. The microbiome and autoimmunity: a paradigm from the gut-liver axis. Cell Mol Immunol. (2018) 15:595–609. doi: 10.1038/cmi.2018.7
125. Ross AA, Müller KM, Weese JS, Neufeld JD. Comprehensive skin microbiome analysis reveals the uniqueness of human skin and evidence for phylosymbiosis within the class Mammalia. Proc Natl Acad Sci USA. (2018) 115:E5786–95. doi: 10.1073/pnas.1801302115
126. Fedeles F, Murphy M, Rothe MJ, Grant-Kels JM. Nutrition and bullous skin diseases. Clin Dermatol. (2010) 28:627–43. doi: 10.1016/j.clindermatol.2010.03.036
127. Hirose M, Schilf P, Benoit S, Eming R, Gläser R, Homey B, et al. Polymorphisms in the mitochondrially encoded ATP synthase 8 gene are associated with susceptibility to bullous pemphigoid in the German population. Exp Dermatol. (2015) 24:715–7. doi: 10.1111/exd.12732
128. Cheng JB, Sedgewick AJ, Finnegan AI, Harirchian P, Lee J, Kwon S, et al. Transcriptional programming of normal and inflamed human epidermis at single-cell resolution. Cell Rep. (2018) 25:871–83. doi: 10.1016/j.celrep.2018.09.006
129. Picart-Armada S, Thompson WK, Buil A, Perera-Lluna A. An R package to compute diffusion-based scores on biological networks: diffuStats. Bioinformatics. (2017) 34:533–4. doi: 10.1093/bioinformatics/btx632
130. Hänzelmann S, Castelo R, Guinney J. GSVA: gene set variation analysis for microarray and RNA-Seq data. BMC Bioinformatics. (2013) 14:7. doi: 10.1186/1471-2105-14-7
131. Ludwig RJ, Martinez A, Bernard P, Savas Y, Kowalewski C. DPem: Dimethylfumarate for the Treatment of Bullous Pemphigoid. (2011). Available online at: https://www.orpha.net/consor/cgi-bin/ResearchTrials_Networks.php?lng=EN&data_id=119994&title=DPem—Dimethylfumarate-for-the-treatment-of-bullous-pemphigoid&search=Disease_Search_Simple
132. Julkowska D, Austin CP, Cutillo CM, Gancberg D, Hager C, Halftermeyer J, et al. The importance of international collaboration for rare diseases research: a European perspective. Nature. (2017) 24:562–71. doi: 10.1038/gt.2017.29
Keywords: autoimmune bullous diseases, autoantigens, HLA class II genes, systems medicine, genetics, transcriptomics
Citation: Olbrich M, Künstner A, Witte M, Busch H and Fähnrich A (2019) Genetics and Omics Analysis of Autoimmune Skin Blistering Diseases. Front. Immunol. 10:2327. doi: 10.3389/fimmu.2019.02327
Received: 29 May 2019; Accepted: 16 September 2019;
Published: 15 October 2019.
Edited by:
Karin Loser, University of Münster, GermanyReviewed by:
Takashi Hashimoto, Graduate School of Medicine, Osaka City University, JapanCristina Has, University of Freiburg, Germany
Teruki Dainichi, Kyoto University, Japan
Copyright © 2019 Olbrich, Künstner, Witte, Busch and Fähnrich. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Anke Fähnrich, anke.faehnrich@uksh.de