 Sietse Q. Nagelkerke
Sietse Q. Nagelkerke David E. Schmidt
David E. Schmidt Masja de Haas4,5,6,7
Masja de Haas4,5,6,7- 1Sanquin Research and Landsteiner Laboratory, Department of Blood Cell Research, Amsterdam UMC, University of Amsterdam, Amsterdam, Netherlands
- 2Pediatric Hematology, Immunology and Infectious Diseases, Emma Children's Hospital, Amsterdam UMC, University of Amsterdam, Amsterdam, Netherlands
- 3Sanquin Research and Landsteiner Laboratory, Department of Experimental Immunology, Amsterdam UMC, University of Amsterdam, Amsterdam, Netherlands
- 4Sanquin Diagnostic Services, Department of Immunohematology Diagnostics, Amsterdam, Netherlands
- 5Sanquin Research, Center for Clinical Transfusion Research, Leiden, Netherlands
- 6Jon J. van Rood Center for Clinical Transfusion Science, Leiden University Medical Center, Leiden, Netherlands
- 7Department of Immunohematology and Blood Transfusion, Leiden University Medical Center, Leiden, Netherlands
Fc-gamma receptors (FcγR) are the cellular receptors for Immunoglobulin G (IgG). Upon binding of complexed IgG, FcγRs can trigger various cellular immune effector functions, thereby linking the adaptive and innate immune systems. In humans, six classic FcγRs are known: one high-affinity receptor (FcγRI) and five low-to-medium-affinity FcγRs (FcγRIIA, -B and -C, FcγRIIIA and -B). In this review we describe the five genes encoding the low-to-medium -affinity FcγRs (FCGR2A, FCGR2B, FCGR2C, FCGR3A, and FCGR3B), including well-characterized functionally relevant single nucleotide polymorphisms (SNPs), haplotypes as well as copy number variants (CNVs), which occur in distinct copy number regions across the locus. The evolution of the locus is also discussed. Importantly, we recommend a consistent nomenclature of genetic variants in the FCGR2/3 locus. Next, we focus on the relevance of genetic variation in the FCGR2/3 locus in auto-immune and auto-inflammatory diseases, highlighting pathophysiological insights that are informed by genetic association studies. Finally, we illustrate how specific FcγR variants relate to variation in treatment responses and prognosis amongst autoimmune diseases, cancer and transplant immunology, suggesting novel opportunities for personalized medicine.
Introduction
Fc-gamma receptors (FcγR) are cellular receptors for Immunoglobulin G (IgG) and upon binding of complexed IgG can trigger various cellular immune effector functions that can destroy and eliminate the opsonized target. These responses are only initiated when multiple IgG molecules are bound simultaneously; a single IgG does not activate FcγRs. When multiple IgG molecules are fixed close to each other, as is the case in opsonized bacteria or in an immune complex, this results in cross-linking of several FcγRs in the cell membrane, which leads to their activation. In this way, FcγRs play an important role in immunity by linking the adaptive and innate immune systems.
FcγRs are differentially expressed on a range of immune cells (Figure 1) (3). In humans, six classic FcγRs are known, which can be distinguished into one high-affinity receptor (FcγRI) and five low-to-medium-affinity FcγRs (FcγRIIA, -B and -C, FcγRIIIA and -B). The low-to-medium-affinity FcγRs are the focus of this review. On a functional level, most of the FcγRs are activating receptors that can induce the cellular responses mentioned above, but one, FcγRIIB, is an inhibitory receptor.
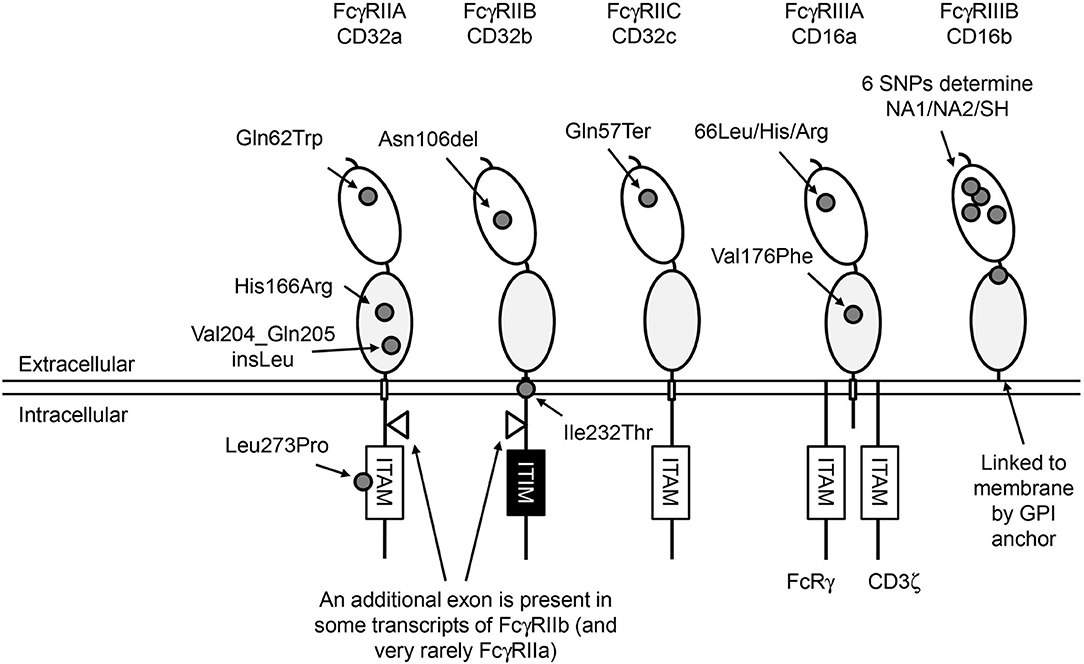
Figure 1. Human FcγRs. Overview of the structure of human low-to-medium-affinity FcγRs. The oval shapes in the extracellular part of the FcγRs represent the different extracellular domains; the light gray domains are the domains where IgG molecules bind. All FcγRs except FcγRIIIB are linked to the plasma membrane by transmembrane (TM) domains indicated by small rectangles. FcγRIIIB is linked to the plasma membrane through a GPI anchor. FcγRIIIA has a small intracellular domain that associates with adaptor molecules that can initiate an intracellular signaling cascade when multiple FcγRs are cross-linked, which ultimately leads to activation of the cell on which the FcγRs are expressed. FcγRII receptors have a much larger intracellular domain, and contain a signaling motif to start this cascade in their own polypeptide chain. Signaling by activating FcγRs is mediated by immunoreceptor tyrosine-based activating motifs (ITAM) that are present either in the cytoplasmic tail of the receptor itself or in non-covalently associated signaling adaptor proteins, such as the common γ-chain (FcRγ). Aggregation of activating FcγR by binding of multivalent ligands, such as an opsonized pathogen or blood cell or an immune complex, results in the phosphorylation of ITAM tyrosine residues by Src family protein tyrosine kinases (PTKs), and ultimately leads to activation of cellular responses (1). Aggregation of the inhibitory FcγRIIB, which contains an immunoreceptor tyrosine-based inhibitory motif (ITIM), also results in phosphorylation of tyrosine residues by Src family PTKs. In contrast to ITAMs, phosphorylated ITIMs serve as binding sites for phosphotyrosine phosphatases (PTPs) which dephosphorylate other proteins resulting in inhibition of activating pathways (2). Approximate location of functional SNPs in the FcγRs are indicated by small gray circles, SNPs are indicated by 3-letter amino-acid codes. ITAM, immunoreceptor tyrosine-based activating motif; ITIM, immunoreceptor tyrosine-based inhibitory motif.
The five genes for the low-to-medium-affinity FcγRs are located in a single cluster on chromosome 1q23.3 (the FCGR2/3 locus) and several genetic variations resulting in functional changes have been found in all of the genes in this locus. These variations are associated with auto-immune, auto-inflammatory, and infectious diseases and with efficacy of immunotherapy in cancer patients, but genetic analysis of the variants at the locus is hampered by the genetic complexity deriving from a segmental duplication, inconsistent nomenclature, and a high degree of linkage disequilibrium.
The gene encoding FcγRI, FCGR1A, is also located at chromosome 1 (1q21.2), and has two presumed pseudogenes (FCGR1B at 1p11.2 and FCGR1C at 1q21.1) that have stop codons in the third extracellular domain and theoretically cannot be expressed as transmembrane receptors (4). Recently, some functional SNPs that occur at low frequency in the population were discovered in FCGR1A (5, 6), but because this gene lies far outside the complex FCGR2/3 locus and no disease associations have been described yet, these SNPs are beyond the scope of this review.
We provide an overview of the currently known genetic variation in low-to-medium-affinity FcγRs, with a focus on the genetic challenges in characterizing this locus, nomenclature of the variations, functional consequences, disease associations with specific diseases and in general, and will discuss the potential of FCGR2/3 genotyping for personalized medicine.
Low-to-Medium-Affinity Fc-Gamma Receptors
IgG-FcγR interactions depend on the IgG subclass (IgG1, IgG2, IgG3, and IgG4) and IgG-Fc glycosylation structure of p.Asn297 in the IgG protein, as well as on the specific FcγR and variation within its amino acid sequence by genetic polymorphisms (7, 8). A schematic representation of the low-to-medium-affinity Fc-gamma receptors and the approximate location of the genetic variants is provided in Figure 1.
FcγRIIA (CD32a) consists of a single polypeptide chain which contains an immunoreceptor tyrosine-based activating motif (ITAM) in the intracellular domain. FcγRIIA is the most widely expressed isoform of FcγRII and is found on monocytes, macrophages, dendritic cells, neutrophils, and platelets. It can induce many different cellular defense mechanisms such as phagocytosis of IgG-opsonized targets, antibody-dependent cellular cytotoxicity (ADCC), production of reactive oxygen species (ROS), and cytokine production.
FcγRIIB (CD32b) is the only FcγR that results in an inhibitory signal to the cell, which is transferred by the immunoreceptor tyrosine-based inhibitory motif (ITIM) on its intracellular signaling domain. FcγRIIB is found in two isoforms deriving from two different transcripts (Figure 1), FcγRIIB-1 and FcγRIIB-2, with FcγRIIB-1 having an additional intracellular exon in between the transmembrane and signaling domains. FcγRIIB-1 is highly expressed on B cells, where it constitutes the only surface-expressed FcγR, and co-crosslinking of FcγRIIB-1 with the B cell receptor (BCR) inhibits activating signals induced by the BCR. Other cell types also express FcγRIIB, albeit at much lower levels, and on these cells FcγRIIB-2 is the main transcript expressed. These cells include a subset of monocytes, macrophages, and dendritic cells. Expression of FcγRIIB can also be detected on neutrophils and NK cells, but only in individuals with certain genotypes (9–11). When transfected in COS-1 cells, FcγRIIB can inhibit pro-phagocytic signals induced by activating FcγRs, balancing the immune response against IgG-opsonized targets (12), but it remains currently unknown if this mechanism is also involved in myeloid cells. Interestingly, at phagocytic cups, FcγRIIB may be relatively excluded whereas FcγRIIA is enriched, likely due to their difference in IgG affinity, which may affect the ability of FcγRIIB to exert inhibitory signals (13).
FcγRIIC (CD32c) has long been considered not to be expressed at all, as its gene (FCGR2C) was thought to be a pseudogene (14, 15), and therefore relatively little was known about the expression pattern of this receptor. In 1998, FcγRIIC was first found on NK cells of individuals that carry an open reading frame (ORF) of this receptor (p.57Gln, FCGR2C-ORF), as opposed to the majority of individuals in which this receptor is indeed a pseudogene and cannot be expressed as a result of a stop codon in exon3 (p.57Ter, FCGR2C-Stop) (16). Determining the cellular expression pattern of FcγRIIC has long been complicated because the extracellular domains are identical to FcγRIIB, but specific detection of FcγRIIC is possible by comparison of cellular expression between individuals that can or cannot express FcγRIIC as a result of the stop codon, detection of FCGR2C mRNA and western blots of immunoprecipitated FcγRIIC. We now know that FcγRIIC can be expressed on NK cells, neutrophils, monocytes (9), and macrophages (17). This receptor has also been reported to be expressed on B cells (18) although expression on B cells could not be reproduced in our own laboratory (17). Obviously, FcγRIIC can only be functional in individuals with an FCGR2C-ORF. Although expression on NK cells is relatively low, it has been shown to be capable of inducing killing of target cells in a redirected ADCC assay (19), functioning as an activating receptor.
FcγRIIIA (CD16a) has two extracellular (EC) Ig-like domains, involved in binding of IgG, a transmembrane (TM) domain and a short intracellular (IC) domain. The TM domain associates with adaptor proteins containing an immunoreceptor tyrosine-based activating motif (ITAM) to induce intracellular signaling. In monocytes and macrophages, this receptor associates with the FcRγ-chain, while in NK cells it associates with the CD3 ζ-chain (20–22). Association with these adaptor proteins is not only essential for signaling and maintaining stable expression, but also for targeting the receptor to the cell membrane (22). FcγRIIIA expressed on NK cells can induce ADCC by these cells (23), and on phagocytes it can induce phagocytosis (24). Recently, it was suggested that FcγRIIIA is also expressed in low levels on neutrophils (25), which is surprising since it was never found before and contradicts the finding that two donors completely deficient for FcγRIIIB did not show any staining on neutrophils with a MoAb (3G8) that recognizes both FcγRIIIA and FcγRIIIB (11).
FcγRIIIB (CD16b) is a GPI-anchored protein, expressed in high numbers on neutrophils, and sometimes on eosinophils. As it does not have a transmembrane domain, it cannot associate with FcRγ or the ζ-chain. FcγRIIIB is not capable of IgG-induced production of ROS (26). However, it does contribute in in vitro experiments to the exocytosis of neutrophil granule proteins (27) and Ca2+ influx (28), and may also cooperate with FcγRIIA on the same neutrophil to induce such responses (29). Because FcγRIIIB can induce these responses, FcγRIIIB has often been classified as an activating receptor, although the exact mechanism(s) by which FcγRIIIB activates cells are still unclear (30, 31). Nowadays FcγRIIIB is mainly seen as a decoy receptor (32, 33) as it has a clear role in binding to—and mediating internalization of—soluble immune complexes in neutrophils (34, 35). In fact, FcγRIIIB can also decrease antibody-mediated trogocytosis by neutrophils (36). In FcγR-deficient mice transgenic for human FcyRIIIB, FcyRIIIB mediates the interaction of neutrophils with soluble immune complexes and render the neutrophil susceptible to tissue adhesion and capillary transmigration (37). FcyRIIIB also allows flowing neutrophils to tether to IgG and immune complexes (38).
The FCGR2/3 Locus
The low-to-medium-affinity FcγRs; FcγRIIA, FcγRIIB, FcγRIIC, FcγRIIIA and FcγRIIIB are encoded respectively by FCGR2A, FCGR2B, FCGR2C, FCGR3A, and FCGR3B. All these genes are located in a cluster at 1q23.3 in the FCGR2/3 locus (Figure 2). The locus consists of two 82 kb paralogous repeats with >98% sequence homology, that were formed as the result of an unequal crossover event (15). This unequal crossover event between FCGR2A and FCGR2B, the two genes that flank the region, has led to a segmental duplication in which FCGR2C was formed (15), with the resulting FCGR2C gene being highly homologous to FCGR2B in the first six exons and highly homologous to FCGR2A in the last 2 exons. Figure 3 provides an overview of the differences between the three FCGR2 genes. Furthermore, the segmental duplication created the two different FCGR3 genes, FCGR3A and FCGR3B, which are also highly homologous in sequence (Figure 4). The genes encoding the classic FcγRs are highly polymorphic and functionally relevant genetic variations have been described for all low-to-medium-affinity FcγRs. An overview of the functionally relevant SNPs, is given in Table 1, and the approximate locations within the FcγRs are shown in Figure 1. The functional consequences of the SNPs are discussed below.
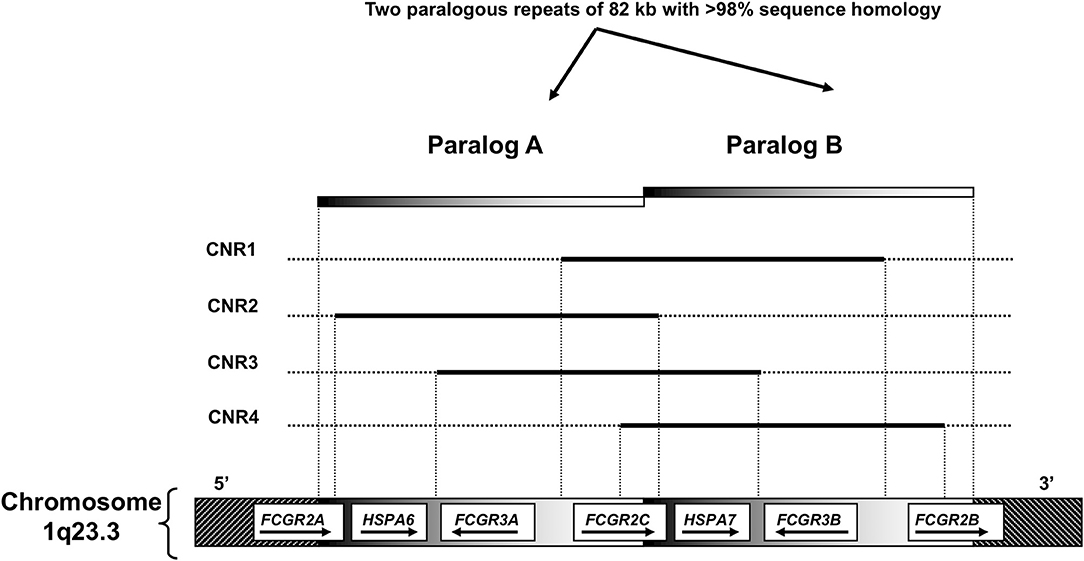
Figure 2. Overview of the FCGR2/3 locus. A Structural overview of the locus, with the orientation of the genes indicated by arrows. B Overview of copy number variation at the locus. Four combinations (copy number variable regions, CNRs) of FcγR genes have been shown to occur in duplication/deletion. Black lines indicate which genes are involved in copy number variation (CNV).
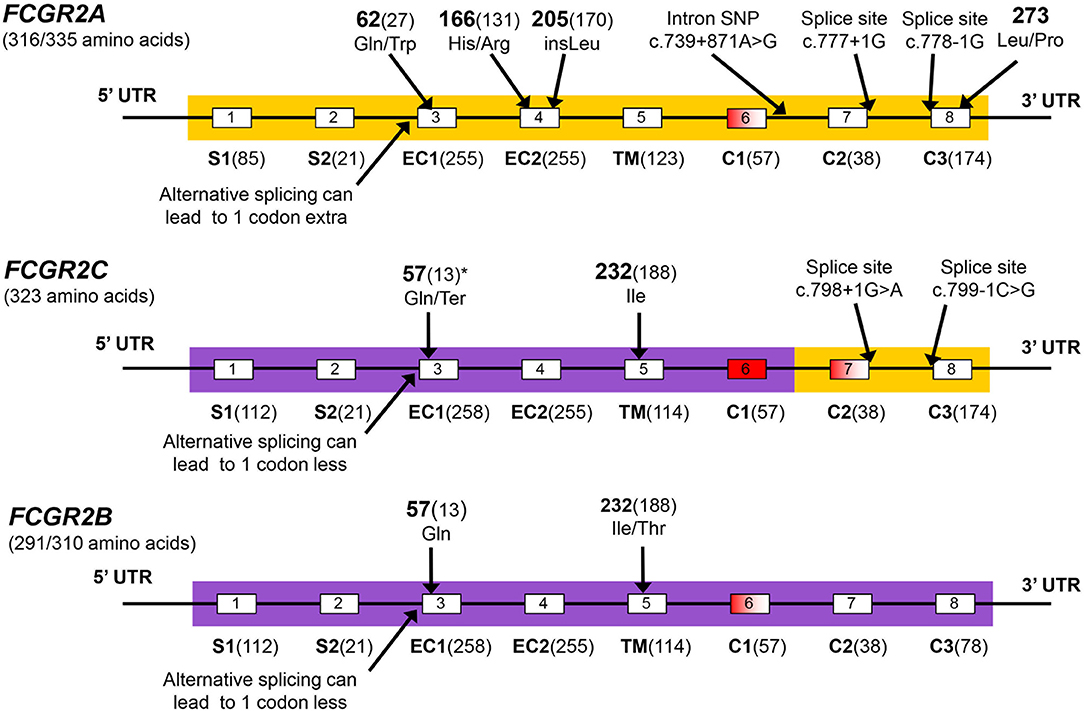
Figure 3. FCGR2 exons. The FCGR2C gene is the crossover product from an unequal crossover between FCGR2A and FCGR2B. Coloring of FCGR2C matches the color of the other FCGR2 genes in the parts where it is highly homologous to that gene. Exons are shown by boxes, white exons are included in all transcripts, red exons are always spliced out, red-shaded exons are spliced out in some transcripts but retained in others. Exon names are below and followed by the number of coding base pairs in that exon. S1, S2, signal peptides; EC1, EC2, extracellular domains; TM, transmembrane domain; C1, C2, C3, cytoplasmic domains. The C3 exons contain an immunoreceptor tyrosine-based activation motif (ITAM) in FCGR2A and FCGR2C, and contains an immunoreceptor tyrosine-based inhibitory motif (ITIM) in FCGR2B. There is a potential confusion with regard to exon numbering in FCGR2 genes: In the FCGR2B gene, transcripts exist that do (FCGR2B1) or do not (FCGR2B2) retain the 57 bp exon6, dependent on the cell type in which the receptor is expressed. In FCGR2A and FCGR2C a very homologous exon6 is present on genomic level, but this exon is always spliced out in FCGR2A and FCGR2C transcripts. Only in some rare cases exon6 is retained in FCGR2A, which results in a gain-of-function FcγRIIA isoform (39). The final two exons of FCGR2A and -2C are often designated exon6 and 7, but to reflect the homology between the 3 FCGR2 genes, we chose to include the potential exon6 in the nomenclature of all FCGR2 genes, designating the final 2 exons exon7 and 8. However, we do not include the base pairs of this exon6 when indicating the nucleotide positions in the FCGR2A and FCGR2C transcripts, because this is not normally done in the literature. In FCGR2A transcripts, further inconsistencies exist as a result of alternative splicing at the beginning of exon3 because of two adjacent splice acceptor sites that can both be used. The most commonly used nucleotide and amino acid numbering is derived from the shorter transcript in which the 3' splice acceptor site is used, so we chose to use this transcript for nucleotide numbering throughout this thesis. Several SNPs are indicated in the figure, all are indicated by their amino-acid position in the full protein, followed by the amino acid position excluding signal peptides between brackets for some of the SNP which are commonly known by that position. *The p.Gln57Ter SNP in FCGR2C is part of a haplotype of 8 SNPs in intron2 and exon3 in FCGR2C, this whole haplotype is identical to FCGR2B in the case of p.57Gln (40). p57Gln is the only non-synonymous coding SNP in this haplotype.
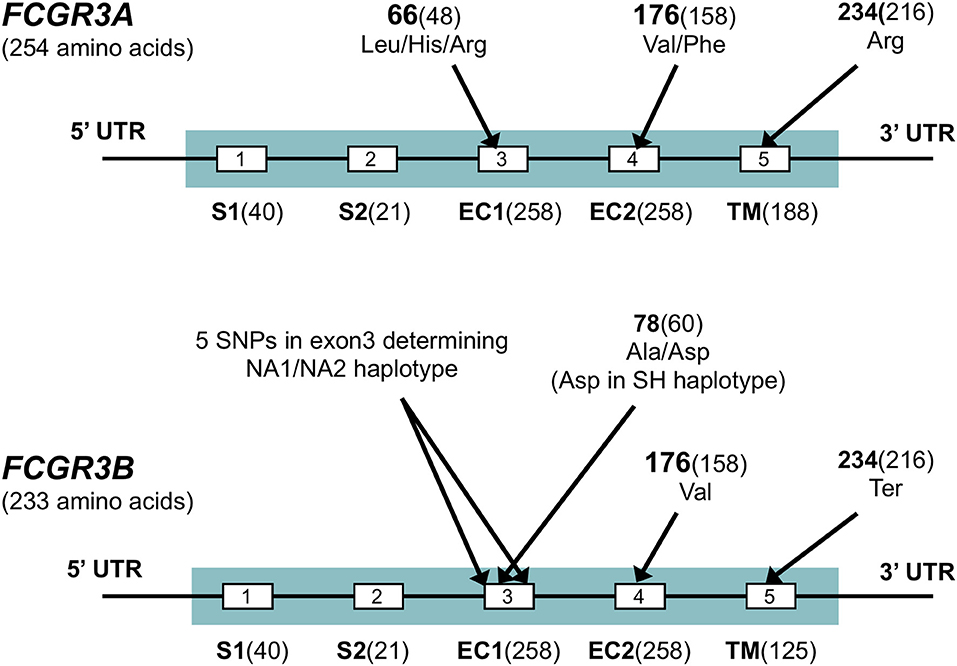
Figure 4. FCGR3 exons. Exons are shown by boxes. Exon names are below and followed by the number of coding base pairs in that exon. S1, S2, signal peptides; EC1, EC2, extracellular domains; TM: transmembrane domain. Several SNPs are indicated in the figure, all are indicated by their amino-acid position in the full protein, followed by the amino acid position excluding signal peptides between brackets for some of the SNP which are commonly known by that position. Sequences for FCGR3A and FCGR3B are very similar, but four non-synonymous differences in the coding sequence exist, most notably the stop codon at p.234 in FCGR3B as indicated in the figure, which truncates the transmembrane domain of this receptor. Other amino acid differences between FCGR3A and FCGR3B include p.147 (Gly in FCGR3A, Asp in FCGR3B), p.158 (Tyr in FCGR3A, His in FCGR3B) and p.203 (Phe in FCGR3A, Ser in FCGR3B) (41). A set of 6 SNPs in exon3 of FCGR3B form three rather well-defined haplotypes, the FCGR3B-NA1, -NA2 and -SH. FCGR3A is identical to NA1 at some sites, but to NA2 at others (41).
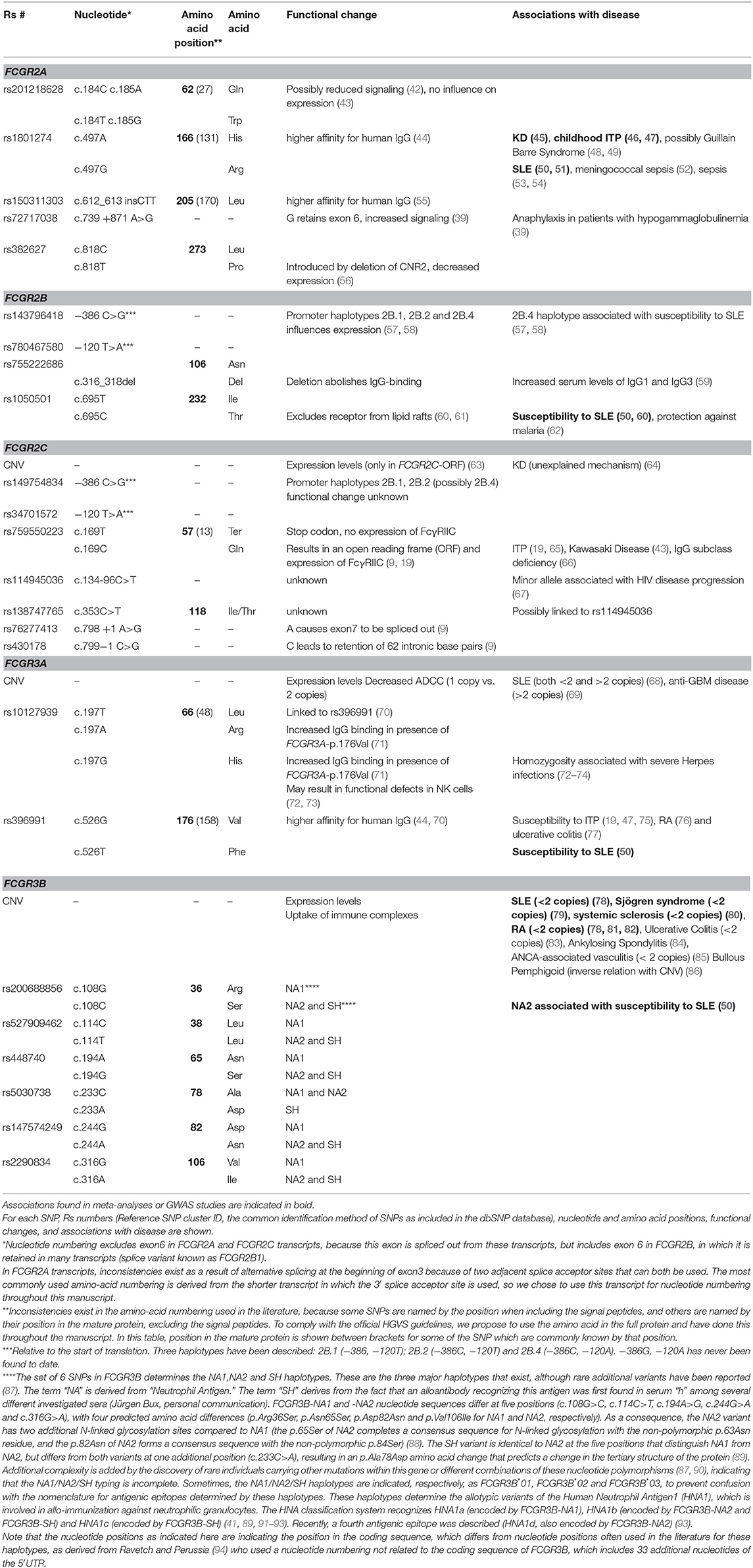
Table 1. Overview of single nucleotide polymorphisms (SNPs) and copy number variation (CNV) at the FCGR2/3 locus.
Besides being polymorphic, some of the low-to-medium-affinity FCGR genes are subject to gene copy number variation (CNV). Although several large-scale studies on CNV have suggested that human FCGR2A and FCGR2B are candidate genes for CNV (95, 96), our group has shown previously that this is not the case. In fact, CNV in the FCGR locus is restricted to FCGR2C, FCGR3A, and FCGR3B (63). CNV at the FCGR2/3 locus always occurs in distinct copy number regions (CNRs) that consist of a complete stretch of 82 kb generated by non-allelic homologous recombination (NAHR) (Figure 2) (56, 97, 98). The clear distinction in CNRs suggests that there must be hotspots for NAHR breakpoints. Breakpoints for the most common CNR1 have been studied in more detail and it appears that these consist of several different breakpoints (81, 97). Exact localization of breakpoints for CNR1 may not always be possible because many potential breakpoints for CNR1 lie within a 24.5 kb block in which no genuine paralogous sequence variants (PSVs) are present (i.e., no clear distinction between paralog A and paralog B can be made and therefore no absolute conclusion on the position of a breakpoint) (99). This block comprises of both the 3′ end of the intergenic region between FCGR3B and FCGR2B and the first exons of FCGR2B. Breakpoints for the rare CNR4 (which have a breakpoint distal of exon3 of FCGR2B) can also lie within this 24.5 kb block. This 24.5 kb “block” may however be a result of a combination of different (smaller) gene conversion events, and Rahbari et al. later showed that it was possible to define breakpoints even within this 24.5 kb block (81).
Nomenclature of Variations at the FCGR2/3 Locus
Many inconsistencies in the nomenclature of SNPs at the FCGR2/3 locus exist, because some SNPs are commonly indicated by the amino acid position in the mature protein (from which the signal peptides have been cleaved off), whereas others are indicated by the amino acid position in the full protein. For some SNPs, both positions are used in the literature, which leads to confusion. We propose to use the amino acid positions in the full protein to avoid possible misunderstanding, following the guidelines of the Human Genome Variation Society (HGVS) nomenclature (100) and have used these positions throughout this review. Table 1 lists also the frequently used positions in the mature protein for some of the SNPs. Further confusion can derive from alternative exon numbering in FCGR2 genes, as explained in Figure 3.
Genetic Analysis of the FCGR2/3 Locus
As a result of the high sequence homology between the genes, genotyping of this locus is very complicated, and it is important to realize that commonly used genome databases such as Ensembl or NCBI BLAST are not in all cases accurate in the distinction between a SNP in one of the FCGR genes and a genuine difference between two homologous FCGR genes (PSV). Detailed knowledge of the organization of the locus and adequate primer design that enables distinction between the paralogs is essential for a proper genetic analysis, and recommendations for analyzing this complex locus have been published before (101). A good source for distinguishing SNPs from PSVs is provided in a Supplementary Table in the article by Mueller et al. (99).
Copy number variation at the locus is commonly determined by multiplex ligation-dependent probe amplification (MLPA) or by paralogue ratio test (PRT) which were concordant in most but not all cases (102). Recently, a method was described that uses data from whole-genome array comparative genomic hybridization (aCGH) to distinguish heterozygous deletion alleles that either include FCGR3A (i.e., CNR2 and CNR3) or include FCGR3B (i.e., CNR1 and CNR4) (81), which allows for a more high throughput method to determine CNV at the FCGR2/3 locus, although only heterozygous deletion alleles could be called with reasonable accuracy. An attempt has also been made to determine FCGR3A and FCGR3B CNV from intensity values derived from an Immunochip platform, allowing the authors to determine CNV in >18,000 individuals (103). This was said to reliably identify cases with 0,1,2 or more copies, although 3 copies could not be reliably be distinguished from higher copy numbers. The authors did however not validate their findings with standard techniques to determine CNV at the FCGR2/3 locus, such as MLPA or the paralogue ratio test (PRT) (104). In our opinion, the fact that in this study the authors could not find any relation of FCGR3B CNV with expression of FcγRIIIB on neutrophils puts serious doubts on the reliability of these data, as expression of FcγRIIIB clearly correlates with CNV of FCGR3B as measured by PRT (105) and MLPA (11, 43, 86).
Similarly, next generation sequencing techniques are currently insufficient to determine FCGCR2/3 SNPs, as multiple variants were mistyped in subjects that were genotyped by whole-exome sequencing (56). Thus, it appears that next generation sequencing techniques will have to be improved greatly before such methods can be used to adequately genotype the FCGR2/3 locus (as well as other complicated loci with duplications and high homology), and it is not sure whether such methods will be present for high-throughput analysis in the near future.
Functional Consequences of SNPs in the FCGR2/3 Genes
In FCGR2A, encoding for FcγRIIA, a well-known SNP rs1801724 is present which results in either a histidine or an arginine at position 166 in the full protein: p.His166Arg (formerly known as p.His131Arg). p.His166Arg is in the IgG binding domain (EC2) (106); FcγRIIA-p.166His has a higher binding affinity for IgG1 and especially IgG2, as compared to FcγRIIA-p.166Arg, but binding to IgG3 and IgG4 is similar for both variants (44). Functionally, mononuclear cells from homozygous FcγRIIA-p.166His individuals produce more IL-1beta when stimulated with IgG2 than heterozygous and homozygous FcγRIIA-p.166Arg individuals (107). Similarly, neutrophils from homozygous FcγRIIA-p.166His individuals have been shown to have increased phagocytosis and degranulation in response to serum-opsonized bacteria and increased rosette formation and phagocytosis in presence of IgG3 anti-RhD sensitized erythrocytes when compared to homozygous FcγRIIA-p.166Arg individuals (108, 109).
In addition to the well-studied FCGR2A-p.His166Arg, several other functional SNPs have been described in FCGR2A: FCGR2A-p.Gln62Trp (formerly known as p.Gln27Trp), a combined SNP of two adjacent nucleotides (known separately as rs9427397 and rs 9427398, and combined as rs201218628) is in linkage disequilibrium with FCGR2C-ORF and the FCGR2B promoter haplotype 2B.4 (43). Compared to p.62Gln, the p.62Trp allele shows similar FcγRII expression amongst neutrophils and monocytes and, even though slightly reduced calcium signaling has been observed in overexpressed cell lines, does not affect neutrophil ADCC in vitro (42). Because of the linkage disequilibrium and given these functional data, it is more likely that 2B.4 variant or FCGR2C-ORF confer the increased risk for autoimmune disease than FCGR2A-p.62Trp.
At position p.273, which normally is a Leucine in FCGR2A, a Proline can be introduced into FCGR2A by deletion of CNR2 which causes the fusion of the proximal part of FCGR2A and the distal part of FCGR2C causing a chimeric FCGR2A/2C gene (56). The Leu/Pro difference is the only amino acid different in this region of FCGR2A and FCGR2C, although additional variants exist in the 3'UTR (56). The chimeric FCGR2A/2C gene shows lower expression levels and lower generation of reactive oxygen species in comparison with the wild-type FCGR2A. This variant could either be seen as a SNP or as the result of the fusion of two genes. FCGR2A-P.Leu273Pro has also been described as a SNP (rs382627, currently flagged as suspect in dbSNP) in a Japanese individual, although it has not been tested whether this individual also had a deletion of CNR2 (110). Compared to leucine, the proline variant was shown in this report to have an increased signaling capacity when expressed in cell lines (110).
Another SNP that may influence the function of FcγRIIA is the intronic rs72717038, which could cause retention of exon6 which is associated with increased signaling capacity (39, 111).
Finally, FCGR2A-p.Val204_Gln205insLeu (rs150311303) is a SNP observed with a minor allele frequency of 8.3% in an African population and confers a higher affinity for human IgG (55).
FCGR2B, encoding for FcγRIIB, also exists in two allelic variants, containing either an isoleucine or a threonine at position 232 in the TM domain (112). As this SNP (p.Ile232Thr) does not affect the IgG-binding EC domains, it has no influence on the binding affinity. However, its localization at the TM domain results in differences in downstream signaling and subsequent inhibition of FcγRI signaling in macrophages and BCR signaling in B cells. In particular, p.Ile232 provides stronger inhibitory signaling than p.Thr232, and this is caused by the exclusion from lipid rafts of FcγRIIB-p.Thr232 (60, 61) (Figure 5). Dendritic cells (DC) also express FcγRIIB, affecting DC maturation end T cell stimulation (113–115), hence FcγRIIB-p.Thr232 may also influence the function of these cells. Other genetic variations influence the expression of FcγRIIB. For instance, in individuals with a CNR1 deletion in the FCGR locus, FcγRIIB can surprisingly also be expressed on the surface of NK cells (9, 11, 99). Expression of FcγRIIB in neutrophils, monocytes or B cells is hardly affected by this deletion (9). Furthermore, two SNPs in the promoter of FCGR2B and FCGR2C, a guanine or cytosine at position −386 and a thymine or adenine at position −120 relative to the start codon, form four haplotypes of which one (−386G, −120A; 2B.3) has never been found in any individual thus far. In case of FCGR2B, the rare 2B.4 promoter haplotype (−386C, −120A) appeared to have higher transcriptional activity than the wild-type promoter 2B.1 (−386G, −120T) (57), resulting in increased expression on neutrophils (10, 11), monocytes (11) whereas the result of this 2B.4 promoter on the expression of FcγRIIB on B cells is less clear and conclusions from various reports range from increased expression (57), no effect (11) to a decreased expression of FcγRIIB on B cells (58); which may differ among different B cell subsets. Functionally, 2B.4 led to a stronger inhibition of B cell receptor signaling without affecting surface expression levels as such (116).
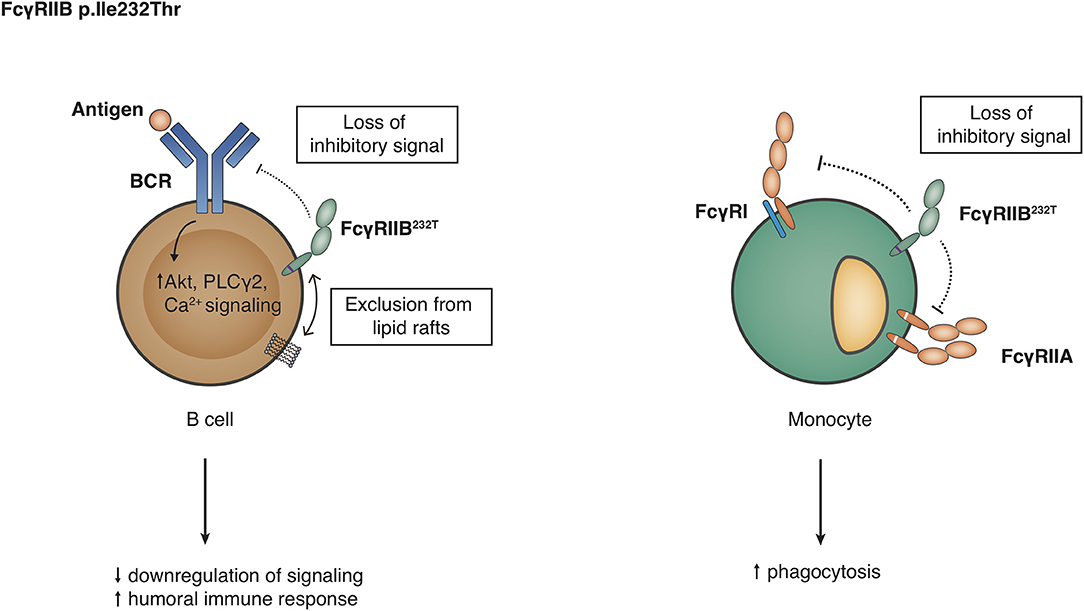
Figure 5. Functional consequences of the FCGR2B-p.Ile232Thr variant. Detailed explanations are given in the main text.
Recently, a rare in-frame deletion c.316_318del, p.Asn106del, rs755222686, was described in the Icelandic population that abolishes IgG binding to FcγRIIb (59). The asparagine residue at position 106 is part of an N-linked glycosylation site, but the absent binding of IgG was not a result of the removal of the glycan, because the same glycan was found at the adjacent asparagine at position 105 in protein encoded by the deletion allele. p.Asn106del was associated with increased levels of IgG1 and IgG3.
In FCGR2C, the previously mentioned p.Gln57Ter SNP (sometimes known as p.Gln13Ter determines whether or not individuals can express FcγRIIC at all. This mutation results in either an open reading frame (classic FCGR2C-ORF, allele frequency ~10–15% in Caucasians) or a stop codon (FCGR2C-Stop) (19). Classically, ORF/Stop genotyping of individuals is done based on this SNP alone. However, we have recently found that some individuals carry splice site mutations in intron7, which leads to alternative transcripts, causing a frameshift in exon8 and the introduction of novel stop codons, leading to an almost complete loss of FcγRIIC expression (9, 43). Genotyping of FCGR2C should therefore include these novel mutations to provide an accurate prediction for FcγRIIC expression.
FcyRIIC on NK cells offers an antibody-mediated, FcyRIIIA-independent pathway to trigger Ca2+ signaling and ADCC (16, 117) (Figure 6). Because of the identity of the intracellular signaling elements of FcyRIIC and FcyRIIA, it can be speculated that FcyRIIC acts as an independent phagocytic receptor on myeloid cells, but this has not been shown experimentally. Transgenic mice expressing FcyRIIC in B cells show enhanced humoral immune responses after T cell dependent and T-independent vaccination (18). Altogether, classic FCGR2C-ORF may thus predispose to autoimmune disease either by providing attenuated innate immune responses or by enhancing the humoral immune response.
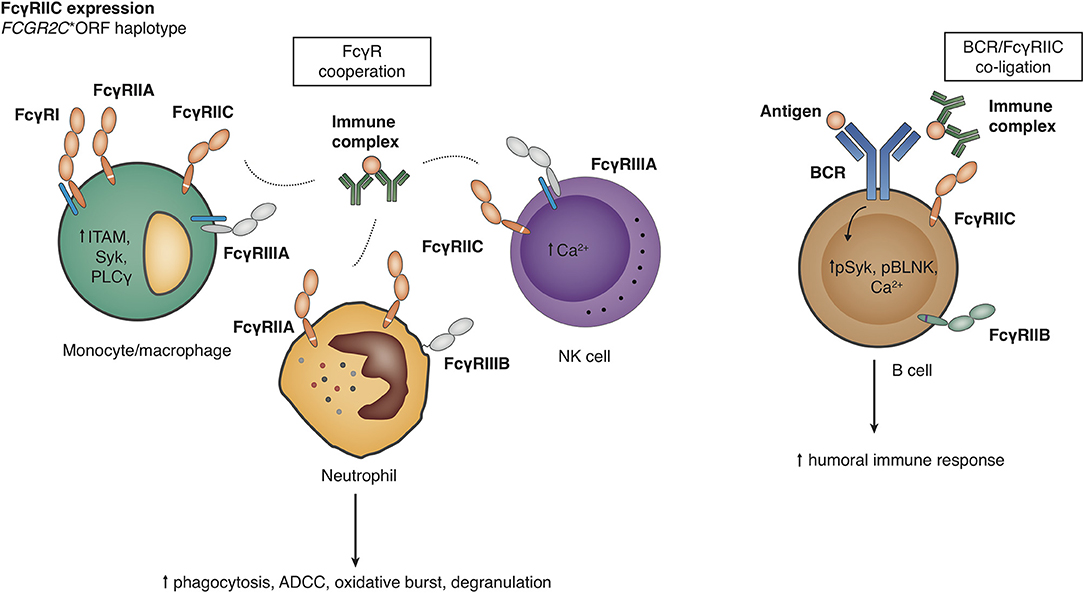
Figure 6. Functional consequences of the FCGR2C-open reading frame (ORF) haplotype. Detailed explanations are given in the main text. Right panel, Effects on the level of B cells have been described by Li et al. (18), but other analyses have not shown FcγRIIC expression on B cell surface in FCGR2C-ORF individuals (17).
In FCGR2C, the promoter haplotypes of FCGR2B as mentioned above, can also be found. In general, only the wildtype and one other promoter haplotype (−386C, −120T; 2B.2) are found. The 2B.2 haplotype is linked to p.57Gln (19, 43).
Recently, a haplotype of several SNPs in and around exon3 has gained a lot of attention because of a possible association with HIV vaccine efficacy. This haplotype consists of c.134-96C>T (rs114945036), p.(Thr118Ile) (rs138747765) and c.391+111G>A (rs78603008) and was associated with vaccine efficacy despite the fact that all but one study participants had a p.57Ter allele. The function of the SNPs in the haplotype is unknown. However, the results of this study must be taken with great caution because the primers used to distinguish FCGR2C from FCGR2B have used specific sites that, according to Mueller et al. (99), are all polymorphic in FCGR2C and can completely resemble FCGR2B. The authors may therefore have missed some FCGR2C alleles in their analysis which may have skewed the results. Later, the haplotype has been reanalyzed in a South African cohort (118) using a specific long range PCR to sequence FCGR2C; these authors have indeed found the c.134-96C>T SNP but not the other SNPs of the haplotype in their cohort. The minor allele of the c.134-96C>T SNP was later found by the same group to be associated with increased odds of HIV-1 disease progression (67).
The FcγRIIIA-encoding FCGR3A gene contains a SNP that results in either a valine or a phenylalanine at position 176 (p.Val176Phe), formerly known as p.Val158Phe, located in the EC2 domain (119). FcγRIIIA-p.176Val has a higher binding affinity for all human IgG classes compared to FcγRIIIA-p.176Phe (44). In ADCC assays, NK cells from FcγRIIIA-p.176Val donors show increased killing of target cells that are opsonized with sub-saturating levels of the human anti-CD20 MoAb Rituximab (23). Another SNP in the FCGR3A gene is a triallelic SNP at position 66; p.66Leu/Arg/His, also formerly known as p.48Leu/Arg/His which is located in the EC1 domain which is not directly involved in binding IgG. Rare homozygosity of p.66His was first described in a patient with recurrent Herpes infections (72) and was later found in two other patients with decreased clearance of Herpes infections (73, 74) and suggested to be a congenital immunodeficiency (73). However, homozygosity for p.66His was also found in a cohort of healthy individuals of European descent (genotype frequency 0.6%) and African descent (genotype frequency 0.1%) (71). Apparently, the clinical phenotype of homozygity for p.66His differs between individuals and recurrent Herpes infections may be associated with but are not directly caused by the mutation.
The FcγRIIIB-encoding FCGR3B gene exists in three polymorphic variant proteins, best known as the NA1, NA2, and SH haplotypes. These haplotypes consist of a set of 6 SNPs in exon3 of FCGR3B (Table 1 and Figure 4). The FCGR3B variants encoded by these haplotypes determine the allotypic variants of the Human Neutrophil Antigen1 (HNA1) (91), which is involved in allo-immunization against neutrophilic granulocytes. The NA1, NA2 and SH haplotypes are sometimes referred to in the literature as HNA1a, HNA1b and HNA1c, respectively, although the latter nomenclature in strict sense determines antigenic epitopes and not genetic haplotypes (see Table 1 for a detailed description). Apart from determining allo-immunization against neutrophils, these haplotypes are known to have functional differences. Compared to NA1, the NA2 and SH variants have two additional N-linked glycosylation sites. The SH variant differs from NA1 and NA2 by a p.Ala78Asp amino acid change that predicts a change in the tertiary structure of the protein (89), although the actual functional consequences of this SNP are not well-characterized. While the binding affinities for IgG1 and IgG3 appear similar between NA1, NA2 and SH (44), neutrophils from FcγRIIIB-NA1NA1 individuals bind and phagocytize IgG-opsonized bacteria and red blood cells more efficiently than those from FcγRIIIB-NA1NA2 and -NA2NA2 individuals (108, 120). It is not known whether the SH is functionally different from the otherwise similar NA2 variant.
Functional Consequences of CNV in the FCGR2/3 Genes
CNV (Figure 2) results in differences in expression levels of FcγRIIIB, where an increase in gene copies of FCGR3B very clearly leads to a higher receptor expression at the cell surface (11, 63, 105, 121, 122) and higher mRNA levels (43, 86), and decreased copy number associated with lower soluble serum FcyRIIIB levels released from activated neutrophils (123) (Figure 7). Functionally, increased expression of FcγRIIIB leads to higher binding and uptake of immune complexes by neutrophils (105) and may be associated with increased ROS production (86), which is an intriguing finding because FcγRIIIB has been shown not to contribute to ROS production (27). Interestingly, individuals who are homozygous for a CNR1 deletion, i.e., have no copies of FCGR3B, are generally healthy and apparently not at risk for overwhelming bacterial infections, suggesting that their neutrophil function is sufficient to maintain immune homeostasis (122).
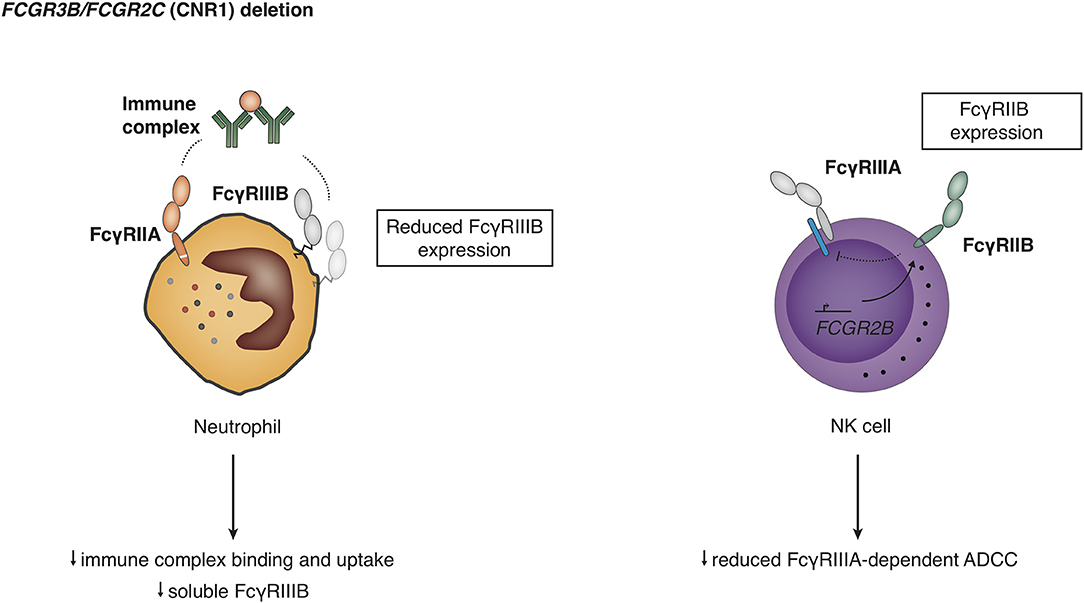
Figure 7. Functional consequences of deletion in the copy number region 1 (CNR1). Detailed explanations are given in the main text.
In addition, a CNR1 deletion leads to expression of FcγRIIB on NK cells (9, 99), which is presumed to result from the molecular effects of the promoter rearrangement, and the ectopic expression of FcγRIIB is either regulated by action of the FCGR2C promoter or by deletion of a negative regulatory elements in the FCGR2B promoter. Humans with CNR1 deletion show a reduced FcyRIIIA-mediated NK cell ADCC, and FcyRIIB expression on NK cells was able to fully inhibit redirected FcyRIIC-mediated ADCC (9).
In case of FcγRIIC, CNV can only play a role in case of classic FCGR2C-ORF alleles, but in these cases the amount of copies of FCGR2C-ORF alleles is clearly linked to expression values (19, 43).
In FcγRIIIA, surface expression of FcγRIIIA on NK cells is also linked to gene copies of FCGR3A (63), although the expression levels are not as clearly associated as in FCGR3B and no significant correlation with mRNA levels could be found (43) suggesting some form of transcriptional regulation. The level of expression on NK cells is, at least for 1 vs. 2 copies, related to the level of killing of target cells in (redirected) ADCC assays (63).
Linkage Disequilibrium at the FCGR2/3 Locus
With all the FCGR2 and FCGR3 genes so closely associated in the FCGR2/3 locus, the different SNPs and CNRs are prone to have a high degree of linkage disequilibrium (LD), and we have recently published a large overview of LD at the locus (43), which showed LD to occur across the locus. The LD found was similar to LD previously found between some selected sets of variants (98, 124, 125). In addition, the FCGR3A-p.66Leu/His/Arg SNP has been shown to be in LD with FCGR3A-p.Val176Phe (70), and this linkage was responsible for an initially observed difference in binding affinity for IgG in FCGR3A-p.66Leu/His/Arg (126), which was later shown to be solely the result of the linkage with the FCGR3A-p.Val176Phe (70), which conferred the difference in binding affinity. However, later binding assays with stratified groups of individuals showed some direct differences in binding affinity resulting from the FCGR3A-66Leu/His/Arg itself (71). Thus, knowledge of LD is very important for a correct interpretation of genotyping results.
Consideration of linkage disequilibrium should have a great impact on all the association studies performed for variants at this locus, which often genotype one or two variants only (mostly the FCGR2A-p.His166Arg and FCGR3A-p.Val176Phe SNPs). Associated variants found in these studies may be the mere reflection of an even stronger association for a variant that is in LD with the identified variant, but was not genotyped in the study (i.e., an increased prevalence of FCGR3A-p.176Val in a certain patient group may simply be the result of the classic FCGR2C-ORF haplotype being even more increased in the same patient group, because these two variants are in LD with each other). If only the FCGR3A-p.Val176Phe SNP is genotyped, this could lead to the wrong conclusion that FCGR3A-p.176Val causes an increased susceptibility to the disease. To be able to most accurately identify a potentially causative variant for an increased disease susceptibility, all the known functional genetic variants at the FCGR2/3 locus have to be genotyped, as we have done with the MLPA technique in our studies. A multiple logistic regression analysis can then identify independent risk markers. Even then, for variants that are in strong LD with other variants, it may be hard to identify independently associated variants, and large groups are needed. For instance, in single logistic regression analyses we observed four variants that are in strong LD with each other, to be all associated with an increased susceptibility to Kawasaki Disease (KD) (43) as well as Immune thrombocytopenic purpura (ITP) (65). It seems likely that only one of these variants (the classic FCGR2C-ORF haplotype) actually causes the increased risk, but to prove this as an independent marker in a multiple logistic regression analysis, many more patients would need to be included in the association study, which is not easily done for rare diseases. On the other hand, for SNPs that are in less strong LD, multiple logistic regression analysis can still identify independent risk markers also in smaller patient groups.
Considering the strong LD and well-defined structure of CNRs at the locus, it may be better to analyze -and report on- some of the genetic variations at the locus as haplotypes, instead of analyzing single variations. This has already been the standard for the SNPs in FCGR3B, which are usually reported as the haplotypes NA1, NA2 and SH. Now, since we know that CNV at the FCGR2/3 locus always occurs in CNRs, we suggest that CNV should be analyzed in the form of these CNRs, which basically form haplotypes. For instance, CNV in FCGR3B never occurs alone, but is always accompanied by CNV of FCGR2C and HSPA7 (in essence, CNV in FCGR3B is in perfect LD with CNV in FCGR2C and HSPA7). Thus, an independent association of FCGR3B CNV with disease is impossible to prove, although in this case seems very likely because both the FCGR2C and HSPA7 genes are pseudogenes in the majority of individuals. However, in case of the most frequent deletion allele of CNR1, the ectopic expression of FcyRIIB on NK cells (9, 99) may also play a role in disease susceptibility, and these effects are impossible to distinguish with genetic association studies.
Similarly, a decreased copy number of FCGR3A, which most often occurs as part of the copy number region CNR2, is in these cases always accompanied by the newly described FCGR2A/2C chimeric gene (56) (in essence, decreased copy number of FCGR3A is in very strong LD with the presence of an FCGR2A/2C chimeric gene). Concluding, the presence of CNV at the FCGR2/3 locus cannot be analyzed separately for single genes, and therefore we think it is better to report the CNV as haplotypes in the form of the different CNRs at the locus.
From a functional point of view, FCGR2C variations should also be reported as haplotypes (classic FCGR2C-ORF, non-classic FCGR2C-ORF and FCGR2C-Stop). These consist of two (possibly three) SNPs that together determine expression of FcγRIIC (43). Analyzing the FCGR2C-p.Gln57Ter alone would include individuals with the non-expressed non-classic FCGR2C-ORF haplotype within the group of the classic FCGR2C-ORF haplotype, whereas we show that on a phenotypic and functional level, that the non-classic FCGR2C-ORF haplotype is similar to FCGR2C-Stop. Therefore, these FCGR2C variations should always be genotyped together and reported as haplotypes. FCGR2C-p.Gln57Ter should not be genotyped alone.
In view of the LD at the locus, and because the FcγR proteins encoded by the genes are functionally related and could thus act together in pathophysiologic mechanisms, it could even be attempted to report extended haplotypes, including all the different SNPs across the whole locus. However, the LD is not absolute for any of the SNPs, and thus the list of possible haplotypes is very long. Therefore, such an approach seems unpractical and confusing. When analyzing the locus as extended haplotypes, disease associations of a single gene encoding one of the FcγRs will not be obvious anymore, which will obscure valuable information on pathophysiology of the disease that is studied.
Ethnic Variation at the FCGR2/3 Locus
Several reports have shown that extensive ethnic variation exists at the FCGR2/3 locus (43, 97, 98), especially for the FCGR2C haplotypes (43, 118), which is relevant for genetic association studies, as it emphasizes the importance of carefully selecting ethnicity-matched control groups.
Evolution of the FCGR2/3 Locus
The FCGR2A gene, defined as a gene that contains an ITAM within its sequence, appears to be specific to primates (127), and has evolved from its ortholog fcgr3 in non-primate animals by NAHR and integration of a retroviral element that included the ITAM (127). The FCGR3(A) gene in primates is an ortholog of non-primate fcgr4 (127), in fact, in some primate species this gene is also indicated as FCGR4. Finally, FCGR2B in primates is an ortholog of fcgr2b (127). Taken together, the basic structure of the FCGR2/3 locus in primates is FCGR2A-HSPA6-FCGR3A(in some cases known as FCGR4)-FCGR2B, with FCGR2A being by far the most distinct from non-primate genes in this locus (127).
The FCGR2C and FCGR3B (and pseudogene HSPA7) genes appear to have evolved more recently. They were formed in a segmental duplication of the FCGR2/3 locus that forms the structure FCGR2A-HSPA6-FCGR3A-FCGR2C-HSPA7-FCGR3B-FCGR2B (15).
Several studies have tried to determine the presence of this segmental duplication in other primates, with sometimes discordant results. It appears that within the great apes (Hominidae family), only the members that are closest to humans (the Homininae subfamily) have the segmental duplication, as shown in humans, chimpanzees (Pan troglodytes) (97, 127) and gorillas (Gorilla gorilla) (127), although one study suggested that even chimpanzees did not have the FCGR2C and FCGR3B genes (128). The other members of the Hominidae family, the orangutans, do not appear to have duplicated the locus (97, 127, 128). A little more distant, the gibbons may however have separate FCGR3A and FCGR3B genes (97) although this could not be replicated in the lar gibbon (Hylobates lar) with gene-specific primers for FCGR2C in a recent study (127). Regarding non-hominoid primates, evidence from all studies suggests that the FCGR3B gene is not present in macaque species (97, 127–130), nor in baboons (97, 130), with the rhesus macaque (Macaca mulatta) (97, 128–130), crab-eating macaque (Macaca fascicularis) (97, 127, 129) and hamadryas baboon (Papio hamadryas) (130) studied in detail. Confirming these genetic findings, no CD16 expression could be detected on neutrophils from macaques or baboons (130). On the other hand, neutrophils of sooty mangabey (Cercocebus atys) did express CD16 (130), which may suggest the presence of an FCGR3B gene, although direct genetic evidence for divergence of the FCGR3 gene could not be found for mangabey (unknown species) (97). With all these studies, it must be taken into account that the complexity of the FCGR2/3 locus may have precluded authors from finding FCGR2C and FCGR3B genes in genomes that are not so well characterized.
In any case, the segmental duplication that created the FCGR2C and FCGR3B genes appears to have occurred relatively recently in evolution and the most thorough and recent study by Lejeune et al. (127) restricts the segmental duplication to Homininae and estimates the event to have occurred <9.2 million years ago. Therefore, it is not so surprising that null variants of these genes are compatible with life in humans; FCGR2C is a pseudogene in >80% of the population and healthy human individuals lacking the FCGR3B gene have been described (122). Indeed, a complete lack of the FCGR3B gene was found in about 0.2% of individuals in a large cohort of >4,000 individuals (56). However, FCGR3B clearly has an important role in the immune system, given the fact that low copy number increases the risk of developing SLE. Apparently, the emergence of FCGR3B was beneficial to the species, and this resulted in the fact that the segmental duplication was maintained, and the FCGR3B gene has evolved to be very different from the FCGR3A gene in expression pattern and function. Evolutionary pressure from helminth infection may have driven this evolution, as an association between large helminth burden and variant frequency in the FCGR3B gene was found in human populations (97).
On the other hand, FCGR2C seems to be not so beneficial, and this result of the segmental duplication has since been modified by evolution to reduce its function. We assume that with the formation of FCGR2C by the segmental duplication of the locus (15), this gene must have been created as a bona fide receptor with an open reading frame. Subsequently, evolution seems to have selected variants that cause reduced function in this receptor through multiple ways: i.e., as a stop codon in exon3, a splice variant in intron7 that abrogates expression (9), and a 3′UTR that does not favor expression when compared with the similar 3′UTR of FCGR2A (56). All these changes indicate that having an active FcγRIIC has been selected against, and the classic FCGR2C-ORF haplotype may be the last functional remnant of the original FCGR2C formed in the segmental duplication. No clear benefits for having an active FcγRIIC are known except for a possible protective effect in helminth infections (97), but it does certainly predispose for certain autoimmune diseases (19, 43). The fact that it does occur at higher frequencies in the European population is intriguing, since it is extremely rare to absent in African populations (43, 118), which constitute the ancestors of the human race. One possibility is that the few classic FCGR2C-ORF alleles that may have been present in the European population after the migration out of Africa, were positively selected (or at least selection against this variant was less strong) and now represent the increased prevalence when compared to African populations. Other possibilities could be a Neanderthal origin of the classic FCGR2C-ORF allele or that it has been newly created in the European population by subsequent recombinations of CNR1 and CNR4, a theory which is supported by the fact that the classic FCGR2C-ORF alleles are actually a haplotype of multiple SNPs in intron2 and exon3 that completely resemble FCGR2B (40). Considering this possibility, one could predict that FCGR2B-Stop alleles (56) were also formed in this way, but the rarity of FCGR2B-Stop alleles suggests great evolutionary pressure on this variant, which may be associated with severe autoimmunity.
Genetic Variation in FCGR2/3 Genes: Associations with Disease
Both SNPs and CNV in FCGR genes have been associated with susceptibility to several auto-immune and infectious diseases. Importantly, despite the extensive linkage disequilibrium at the FCGR2/3 locus, most studies have assessed polymorphisms in relative isolation without a broader consideration of connected variants at the locus. Table 1 provides an overview for a selection of these associations, concentrating on meta-analyses and novel associations that have not been reviewed before by Gillis et al. (3) and Bournazos et al. (131). In general, most of the studies focus on only one or two SNPs; the FCGR2A-p.His166Arg and FCGR3A-p.Val176Phe are the most studied.
FCGR2A-p.His166Arg
Individuals with the variant FCGR2A-p.166Arg have increased susceptibility to SLE, compared to p.166His (50, 132–134), an association which remained also in a GWAS study (135). Although several studies suggest an increased risk of p.166Arg with the development of lupus nephritis (134, 136–138), conflicting evidence exists and a previous meta-analysis did not confirm an association (132).
In contrast to the association of p.166Arg variant with SLE, FCGR2A-p.166His is associated with development of ulcerative colitis (77, 139). Furthermore, a genome-wide association study (GWAS) has revealed FCGR2A-p.166His to be strongly associated with susceptibility to Kawasaki disease, a pediatric vasculitis affecting the coronary vasculature in particular (140). This association was confirmed in a GWAS (45). Finally, concerning immune thrombocytopenia, a meta-analysis ascertained an association between FCGR2A-p.166His and susceptibility to childhood ITP, but not adult ITP (46, 75, 141, 142). Taking the genetic associations into account, it may be speculated that a reduced function of FcγRIIA with the p.166Arg variant is associated with a failure to clear circulating immune complexes, which is a hallmark of SLE. On the other end, a relatively more active immune response, conferred by FCGR2A-p.166His, is associated with the development of ITP, Kawasaki disease and inflammatory bowel disease, emphasizing the intricate role of FCGR2A at the interface of multiple pathways leading to autoimmunity.
FCGR3A-p.Val176Phe
A diallelic SNP is responsible for p.Val176Phe polymorphism in FCGR3A. For SLE, a recent meta-analysis established a link with disease susceptibility and p.176Phe (50), and the p.176Phe/Phe genotype is associated with development of lupus nephritis (143). In rheumatoid arthritis, FCGR3A-p.176Val/Val is associated with susceptibility to the disease amongst Europeans, but not Asians (76). Considering ulcerative colitis, there is a small increased susceptibility to develop the disease with the p.176Val allele (77). Finally, several studies have associated the FCGR3A-p.176Val/Val genotype with increased susceptibility for ITP (19, 75, 141, 142).
FCGR2B-p.Ile232Thr
FcγRIIB holds a diallelic SNP that determines the p.Ile232Thr amino acid. The p.232Thr/Thr genotype has been strongly associated with SLE in numerous studies (50, 60, 62, 98, 144–146). Interestingly, the p.232Thr allele is in linkage disequilibrium with deletion of CNR1, but when assessed together both variations are independently associated with susceptibility to SLE (98).
In RA, FCGR2B-p.232Thr is not associated with disease susceptibility, but strongly associated with joint damage (147).
Although studies indicated that the p.232Thr allele is increased in adult ITP (148) and in chronic childhood ITP (149), a recent meta-analysis questioned this association (6 studies) (142). However, in a recent study three childhood ITP patients that were treated with IVIg and had the p.232Thr/Thr genotype failed to respond to IVIg, whereas 17 patients who carried the p.232Ile/Ile genotype and were only observed, all had a remarkable complete recovery from ITP during follow-up without any treatment (150). Such spontaneous recovery during observation at that timepoint is observed in ~20% of individuals overall. These data collectively indicate that the FCGR2B-p.Ile232Thr genotype may be associated with prognosis in childhood ITP.
Classic FCGR2C-ORF and FCGR2B Promoter Haplotype 2B.4
The classic FCGR2C-ORF is in strong linkage disequilibrium with the FCGR2B promoter polymorphism 2B.4 and a third allele, FCGR2A-p.62Trp (43). Only few studies investigated all these variants in the included individuals, and identified relationships between variants can therefore not be distinguished between them.
In adult autoimmune diseases, there is an association of classic FCGR2C-ORF with ITP (19). In rheumatoid arthritis, CD32 expression on NK cells correlated with mild disease, as compared to aggressive disease (151). However, besides classic FCGR2C-ORF that induces FcyRIIC expression, FcyRIIB expression on NK cells from CNR1 deletions must have contributed to this picture, as not all patients with CD32 expression had an ORF allele. Unfortunately, this was not assessed. Presence of classic FCGR2C-ORF was also associated with susceptibility to SLE in one study (18) although this was not found in another study (11). Similarly to RA, the FCGR2B promoter polymorphism 2B.4 also correlated with susceptibility to SLE, and patients with this variant showed reduced autoantibody development and development of lupus nephritis (11, 57, 58). Finally, FCGR2C has been identified as a candidate susceptibility gene for systemic sclerosis (152).
Regarding childhood autoimmune diseases, classic FCGR2C-ORF confers susceptibility to childhood ITP (19, 65) as well as Kawasaki disease (43). Classic FCGR2C-ORF and 2B.4 correlate positively to immunomodulatory treatment with response to intravenous immunoglobulins (IVIg) in childhood ITP (65) and, for 2B.4, in Kawasaki disease (153). Moreover, in ITP, the variants are associated with a transient disease course, and negatively associate with chronic thrombocytopenia (65).
When these observations are combined, the linked variants classic FCGR2C-ORF and 2B.4 are associated with susceptibility to multiple autoimmune diseases. However, where assessed, they conferred a relatively mild disease phenotype and beneficial association with treatment response to IVIg. This suggests that patients without these variants have other contributing determinants to autoimmunity that confer a relatively negative impact on disease severity.
Deletions in Copy Number Region 1 (CNR1)
Copy number variation in FCGR3B can arise from insertions or deletions of CNRs in the FCGR2/3 locus, namely CNR1 and CNR4. Deletions in CNR4 are extremely rare at ~ 0.1% of the population, whereas a deletion in CNR1 is present in 8.6% of the population (56). Almost all studies investigating CNR1 have only determined FCGR3B CNV, but since virtually all deletions or duplications of FCGR3B found by these studies will result from CNV of CNR1 we have used the terms interchangeably in the next paragraphs. As said above, whether the effect driving the association reflects expression levels of FcγRIIIB or the ectopic expression of FcγRIIB on NK cells cannot be determined.
Associations between deletions of FCGR3B and susceptibility to adult autoimmune diseases have been found for SLE (78, 85, 98, 105, 154), ulcerative colitis (83), rheumatoid arthritis (RA) (81, 82, 155–157), ankylosing spondylitis (84), systemic sclerosis (80), primary Sjögren syndrome (SS) (79, 155), microscopic polyangiitis and Wegener's granulomatosis (85). In SLE, deletion of CNR1 is associated with a higher frequency of lupus nephritis (158). We also recently established that CNR1 deletion is associated with chronic and IVIg-resistant immune thrombocytopenia, but not with the transient form of the disease (65). Interestingly, also duplications of the FCGR3B gene were found to be associated with SLE and SS (155) as well as antineutrophil cytoplasmic antibody-associated systemic vasculitis (105), although this was not evident—or conflicted—by other studies (85, 154). In RA, there seemed to be an association with more rheumatoid factor (RF)-positive disease (157), which was not picked up in a smaller study (82). These effects may be modified by other susceptibility genes such as CCL3L1 (155) or deletions, such as ADAM3A (154). In contrast to these systemic autoimmune disease, no association with a deletion of FCGR3B has been found for Graves' disease or Addison's disease (85), which suggests that these organ-specific autoimmune diseases are not influenced by FCGR3B copy numbers, although one study found an association of increased copies of FCGR3B with the a protection against the skin blistering disease Bullous Pemphigoid (86).
Recent meta-analyses confirmed the association between FCGR3B copy numbers and susceptibility with autoimmune diseases for low FCGR3B copies for SLE, Sjogren's syndrome and Wegener's granulomatosis (78, 159). These association were similar amongst Caucasians and Asians (159). For RA, evidence may be less clear (160), as previous meta-analyses had disparate results (82, 156), although the most recent meta-analysis from 2012 (82) did find an association and furthermore a recent study describing a large cohort confirmed the association (81).
Overall, deletions of FCGR3B as part of CNR1 are strongly associated with development of autoimmune diseases. The notion that this association is particularly pronounced for systemic, but not organ-specific autoimmune reactions, are suggestive of divergent pathomechanisms that may be triggered by CNR1 deletions and subsequently predispose to autoimmunity.
The use of Genetic Association Studies at the FCGR2/3 Locus
FCGR2/3 polymorphisms are useful when investigating the role of FcγRs in human disease by means of genetic association studies. In general, such studies can suggest that FcγRs are involved in the pathophysiology of a certain disease. More specifically, association with specific FcγR genetic variations can give a more precise clue, as they may incriminate a certain cell type in the pathophysiology. The best example is the association of FcγRIIIB CNV with SLE (11, 34, 105, 161). Since FcγRIIIB is expressed almost exclusively in neutrophils, this is a strong suggestion that neutrophils are involved in the pathophysiology of SLE. All the other FcγRs are expressed on multiple cell types, and thus, associations of genetic variation in other genes than FCGR3B are less indicative of a specific cellular involvement in a disease. In some cases, it may be only possible to determine whether, in very general terms, the more activating or the less activating FCGR variants are associated with the disease studied, and thus gain insight on the general role of FcγRs in the pathophysiology of the disease. An overview of the more and less activating variants at the FCGR2/3 locus is given in Table 2. Interestingly, several variants that are more activating are in LD with each other: FCGR2A-p.166His, FCGR3A-p.176Val, and the classic FCGR2C-ORF, although this may be ‘balanced' by the LD of the classic FCGR2C-ORF with the 2B.4 promoter haplotype in FCGR2B (43).
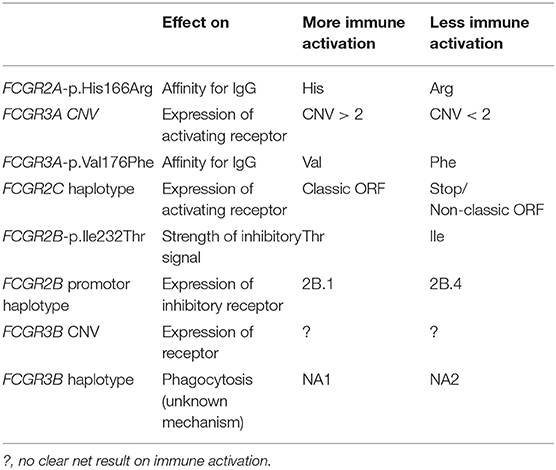
Table 2. Effects of genetic variants at the FCGR2/3 locus on immune function.
Traditionally, it has been thought that activating and inhibitory FcγRs constitute an immunological balance that ensures adequate protection against pathogens, but on the other hand does not result in auto-immunity (7). Simply speaking, FCGR2/3 genetic variation may tip this balance to either side, leading to auto-immunity when the balance is tipped toward the activating side, or leading to decreased immunity against pathogens or cancer cells when the balance is tipped toward the inhibitory side. However, this may be an over-simplification of the matter, and marked differences in FCGR2/3 genetic variations occur between several autoimmune and autoinflammatory diseases. In KD and in immune thrombocytopenia (ITP), it is indeed the case that the more activating variants (FCGR2C-ORF, FCGR3A-p.176Val, FCGR2A-p.166His) are associated with disease susceptibility. However, in other autoinflammatory diseases, the less activating variants, which would be expected to tip the balance toward the inhibitory side, actually predispose to disease. This is for instance the case in SLE, which is associated with the less activating variants FCGR2A-p.166Arg, and the FCGR2B promoter haplotype 2B.4, which causes increased expression of the inhibitory FcγRIIB (10, 11). Another difference between SLE on the one hand and KD and ITP on the other hand is that SLE is associated with low copy number of FCGR3B (as is Sjögren syndrome, systemic sclerosis and possibly RA). Low copy number of FCGR3B is not associated with KD or ITP.
Clearly, autoimmunity is not necessarily associated with more activating FCGR2/3 genetic variations, and FCGR2/3 variants have different, sometimes opposite, effects on different autoimmune and inflammatory diseases, suggesting different pathophysiologic contributions of IgG and FcγRs between the diseases. Possibly, activating FcγRs actually protect against SLE by enabling “waste disposal” of pathogenic immune complexes involved in the disease. On the other hand, in the diseases in which activating variants are associated (KD and ITP), damage done by IgG may be exerted directly by cellular activation via FcγRs, which is enhanced in individuals with more activating variants. In the other diseases (SLE and RA), IgG does not seem to cause harm via cellular activation via FcγRs. Interestingly, the diseases in which the more activating variants are associated with susceptibility, are also the diseases in which IVIg is an effective therapy (KD and ITP), whereas IVIg is of no value in RA, Sjögren syndrome, systemic sclerosis and is possibly beneficial in SLE but this has not been well studied (162, 163), also suggesting a difference in the pathophysiological contribution of FcγRs in these diseases. An explanation for this finding may be that IVIg blocks activating FcγRs, which is beneficial in diseases in which these activating FcγRs are directly involved in pathophysiology, whereas in diseases in which activation of FcγRs does not play a role, blockade of FcγRs is not important.
Altogether, FCGR2/3 association studies suggest that the pathophysiological mechanisms leading to SLE, RA, and Sjögren syndrome may be fundamentally different from the mechanisms leading to KD and ITP, a fact that is supported by the observation that SLE, RA and Sjögren syndrome occur much more frequently in women than in men, whereas in KD and ITP there is a slight predisposition in males.
Concluding, knowledge of FCGR2/3 genetic variation in autoinflammatory and autoimmune diseases may increase our knowledge on the pathophysiology of these complicated and multifactorial diseases, and may be related to effectiveness of IVIg therapy.
FCGR2/3 Genetic Variation and Personalized Medicine
Autoimmunity and Transplant
Perhaps the most clinically useful application of genotyping FCGR2/3 genetic variation could be predicting response to therapy, and FCGR2/3 polymorphisms could be of potential value in personalized medicine. There is clinical data for Kawasaki disease, childhood ITP and SLE that correlated disease outcomes as well as response to treatment are associated with FCGR2/3 variants as well as copy number variation of CNR1 (Table 3). This suggests that it may be possible to use genetic variants to determine prognosis and potentially guide treatment decisions. A key step toward their use would be external validation as well as an investigation of their integration with existing clinical prognostic scores.
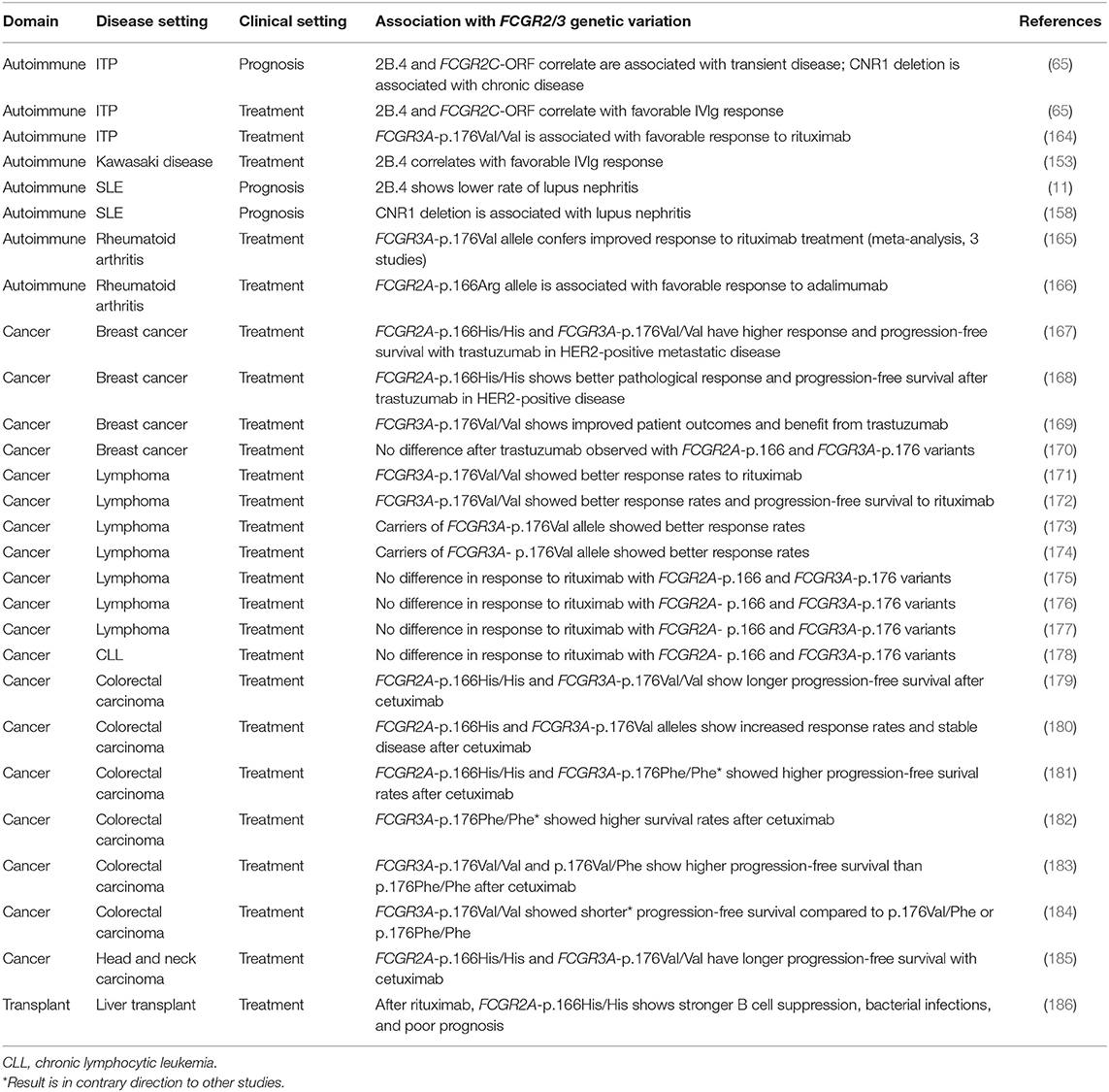
Table 3. Opportunities to use FCGR2/3 locus genotyping in personalized medicine: polymorphisms and copy number variation.
Similar to autoimmunity, some data have suggested that patients with the higher-affinity FCGR3A-p.176Val allele show enhanced B cell deletion after rituximab treatment during liver transplant setting (Table 3). On the other hand, these patients may require additional immune protection with IVIg because of increased susceptibility to bacterial infections. These data suggest that FCGR3A genotyping could be used to potentially offer an alternative transplant immunosuppression regimenboth during and following transplantation to intervene early to prevent immunosuppression-related complications.
Cancer Immunotherapy
Association of FCGR2/3 genetic variation has also been extensively evaluated in monoclonal antibody therapy in cancer patients. Antibodies directed against specific tumor antigens may help in eradicating cancer cells, and this takes place in part by cellular effector mechanisms mediated by FcγRs, such as antibody-dependent cellular cytotoxicity (ADCC) and antibody-dependent cellular phagocytosis (ADCP). Thus, the efficacy of the antibody may be related to FCGR2/3 genetic variation. MoAb therapy is costly, and since not all patients seem to benefit from it, predicting an individual patients' response could help to identify the patients likely to respond to this therapy. Extensive data points toward the fact that patients with higher-affinity FCGR2A-p.166His or FCGR3A-p.176Val alleles have an enhanced response to monoclonal antibody-mediated anti-cancer therapy amongst solid tumors of the breast, head and neck, colorectal carcinomas as well as lymphomas (Table 3). These data are supported by extensive molecular evidence. For example, the trastuzumab mediated ADCC of anti-HER2 breast cancer cells is enhanced with FCGR2A-p.166His and FCGR3A-p.176Val, compared to the other alleles (167), and in case of neutrophil-mediated ADCC for FCGR2A-p.166His (187). The same has been observed with NK cell rituximab-mediated ADCC to Daudi (lymphoma) cells in FCGR3A-p.176Val individuals (23). Collectively, these data show on a laboratory and clinical level that individuals with FCGR2A-p.166His and FCGR3A-p.176Val have enhanced responses to antibody-mediated cancer immunotherapy and genotyping may be useful to stratify treatment regimens and potentially adjust dosing to prevent side effects. Notably, some studies have shown discordant results that are in contrast with these findings (Table 3).
Currently, the variation in results do not yet allow a strategy to justify individualized treatments to be on the basis of FCGR2/3 polymorphisms (188). When clinical use of afucosylated IgG antibodies with increased affinity for FcγRIIIA could be combined with FCGR2/3 genotyping, the correlation with efficacy of cancer therapy may be enhanced. A major drawback of most current genotyping studies is the fact that only two SNPs are analyzed, whereas other SNPs at the locus also potentially influence treatment response rates, and the SNPs are in LD with each other. Analysis of all the SNPs and CNV, and analysis as extended haplotypes across the locus may be more useful; the complexity of the locus requires a more comprehensive assessment that includes determination of gene copy numbers, as well as classic FCGR2C-ORF haplotype.
Conclusion
The FCGR2/3 locus is a complex genetic locus with many functional genetic variants in intricate linkage. It holds many disease associations which are different, sometimes with opposite effects, between various autoimmune and autoinflammatory diseases, which may inform us on fundamental differences in pathophysiologic mechanisms. Furthermore, the locus is promising in view of genetic prediction of efficacy of therapy, especially immunotherapy in cancer, although this is currently not yet feasible. Given the complexity of the locus and inaccuracies in the current databases holding reference sequences, research on the locus could benefit from a thorough genetic analysis that sequences through the entire region and can help to establish a correct and proper reference. Such an approach has recently been explored for FCGR3A using long-range sequencing with Nanopore MinION technology, and allowed a complete investigation of polymorphic sites within the gene (189). In any case, to use the full potential of genetic variation at the FCGR2/3 locus, a comprehensive analysis of all SNPs and CNVs together is warranted.
Author Contributions
SN and DS wrote the manuscript. TK and MH edited and critically reviewed the manuscript.
Funding
This work was supported by a research grant from the Landsteiner Foundation for Blood Transfusion Research (LSBR 0916) awarded to TK, a post doctoral stipend awarded to SN by the Stichting Joghem van Loghem and a doctoral stipend to DS by the Studienstiftung des Deutschen Volkes.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
Parts of this review are adapted from the introduction and discussion of the thesis of SN (190), available online at the digital academic repository of the University of Amsterdam: https://pure.uva.nl/ws/files/9872895/Nagelkerke_Thesis_complete_pdf.
References
1. Mocsai A, Ruland J, Tybulewicz VL. The SYK tyrosine kinase: a crucial player in diverse biological functions. Nat Rev Immunol. (2010) 10:387–402. doi: 10.1038/nri2765
2. Smith KG, Clatworthy MR. FcγRIIB in autoimmunity and infection: evolutionary and therapeutic implications. Nat Rev Immunol. (2010) 10:328–43. doi: 10.1038/nri2762
3. Gillis C, Gouel-Cheron A, Jonsson F, Bruhns P. Contribution of human fcgammars to disease with evidence from human polymorphisms and transgenic animal studies. Front Immunol. (2014) 5:254. doi: 10.3389/fimmu.2014.00254
4. Ernst LK, Duchemin AM, Miller KL, Anderson CL. Molecular characterization of six variant Fcgamma receptor class I (CD64) transcripts. Mol Immunol. (1998) 35:943–54. doi: 10.1016/S0161-5890(98)00079-0
5. Brandsma AM, Ten Broeke T, van Dueren den Hollander E, Caniels TG, Kardol-Hoefnagel T, Kuball J, et al. Single nucleotide polymorphisms of the high affinity IgG receptor FcγRI reduce immune complex binding and downstream effector functions. J Immunol. (2017) 199:2432–9. doi: 10.4049/jimmunol.1601929
6. van der Poel CE, Spaapen RM, van de Winkel JG, Leusen JH. Functional characteristics of the high affinity IgG receptor, FcγRI. J Immunol. (2011) 186:2699–704. doi: 10.4049/jimmunol.1003526
7. Nimmerjahn F, Ravetch JV. Fcgamma receptors as regulators of immune responses. Nat Rev Immunol. (2008) 8:34–47. doi: 10.1038/nri2206
8. Vidarsson G, Dekkers G, Rispens T. IgG subclasses and allotypes: from structure to effector functions. Front Immunol. (2014) 5:520. doi: 10.3389/fimmu.2014.00520
9. van der Heijden J, Breunis WB, Geissler J, de Boer M, van den Berg TK, Kuijpers TW. Phenotypic variation in IgG receptors by nonclassical FCGR2C alleles. J Immunol. (2012) 188:1318–24. doi: 10.4049/jimmunol.1003945
10. Su K, Yang H, Li X, Li X, Gibson AW, Cafardi JM, et al. Expression profile of FcγRIIb on leukocytes and its dysregulation in systemic lupus erythematosus. J Immunol. (2007) 178:3272–80. doi: 10.4049/jimmunol.178.5.3272
11. Tsang-A-Sjoe MW, Nagelkerke SQ, Bultink IE, Geissler J, Tanck MW, Tacke CE, et al. Fc-gamma receptor polymorphisms differentially influence susceptibility to systemic lupus erythematosus and lupus nephritis. Rheumatology. (2016) 55:939–48. doi: 10.1093/rheumatology/kev433
12. Hunter S, Indik ZK, Kim MK, Cauley MD, Park JG, Schreiber AD. Inhibition of Fcgamma receptor-mediated phagocytosis by a nonphagocytic Fcgamma receptor. Blood. (1998) 91:1762–68.
13. Syam S, Mero P, Pham T, McIntosh CA, Bruhns P, Booth JW. Differential recruitment of activating and inhibitory Fc gamma RII during phagocytosis. J Immunol. (2010) 184:2966–73. doi: 10.4049/jimmunol.0900016
14. Brooks DG, Qiu WQ, Luster AD, Ravetch JV. Structure and expression of human IgG FcRII(CD32). Functional heterogeneity is encoded by the alternatively spliced products of multiple genes. J Exp Med. (1989) 170:1369–85. doi: 10.1084/jem.170.4.1369
15. Warmerdam PA, Nabben NM, van de Graaf SA, van de Winkel JG, Capel PJ. The human low affinity immunoglobulin G Fc receptor IIC gene is a result of an unequal crossover event. J Biol Chem. (1993) 268:7346–9.
16. Metes D, Ernst LK, Chambers WH, Sulica A, Herberman RB, Morel PA. Expression of functional CD32 molecules on human NK cells is determined by an allelic polymorphism of the FcγRIIC gene. Blood. (1998) 91:2369–80.
17. Nagelkerke SQ, Kuijpers TW. Immunomodulation by IVIg and the Role of Fc-Gamma receptors: classic mechanisms of action after all? Front Immunol. (2015) 5:674. doi: 10.3389/fimmu.2014.00674
18. Li X, Wu J, Ptacek T, Redden DT, Brown EE, Alarcón GS, et al. Allelic-dependent expression of an activating Fc receptor on B cells enhances humoral immune responses. Sci Transl Med. (2013) 5:216ra175. doi: 10.1126/scitranslmed.3007097
19. Breunis WB, van Mirre E, Bruin M, Geissler J, de Boer M, Peters M, et al. Copy number variation of the activating FCGR2C gene predisposes to idiopathic thrombocytopenic purpura. Blood. (2008) 111:1029–38. doi: 10.1182/blood-2007-03-079913
20. Masuda M, Roos D. Association of all three types of Fc gamma R (CD64, CD32, and CD16) with a gamma-chain homodimer in cultured human monocytes. J Immunol. (1993) 151:7188–95.
21. Trinchieri G, Valiante N. Receptors for the Fc fragment of IgG on natural killer cells. Nat Immun. (1993) 12:218–34.
22. Park JG, Isaacs RE, Chien P, Schreiber AD. In the absence of other Fc receptors, Fc gamma RIIIA transmits a phagocytic signal that requires the cytoplasmic domain of its gamma subunit. J Clin Invest. (1993) 92:1967–73. doi: 10.1172/JCI116790
23. Dall'Ozzo S, Tartas S, Paintaud G, Cartron G, Colombat P, Bardos P, et al. Rituximab-dependent cytotoxicity by natural killer cells: influence of FCGR3A polymorphism on the concentration-effect relationship. Cancer Res. (2004) 64:4664–9. doi: 10.1158/0008-5472.CAN-03-2862
24. Anderson CL, Shen L, Eicher DM, Wewers MD, Gill JK. Phagocytosis mediated by three distinct Fc gamma receptor classes on human leukocytes. J Exp Med. (1990) 171:1333–45. doi: 10.1084/jem.171.4.1333
25. Golay J, Valgardsdottir R, Musaraj G, Giupponi D, Spinelli O, Introna M. Human neutrophils express low levels of FcγRIIIA, which plays a role in PMN activation. Blood. (2019) 133:1395–405. doi: 10.1182/blood-2018-07-864538
26. Huizinga TW, van Kemenade F, Koenderman L, Dolman KM, von dem Borne AE, Tetteroo PA, et al. The 40-kDa Fc gamma receptor (FcRII) on human neutrophils is essential for the IgG-induced respiratory burst and IgG-induced phagocytosis. J Immunol. (1989) 142:2365–9.
27. Huizinga TW, Dolman KM, van der Linden NJ, Kleijer M, Nuijens JH, von dem Borne AE, et al. Phosphatidylinositol-linked FcRIII mediates exocytosis of neutrophil granule proteins, but does not mediate initiation of the respiratory burst. J Immunol. (1990) 144:1432–7.
28. Vossebeld PJ, Kessler J, von dem Borne AE, Roos D, Verhoeven AJ. Heterotypic Fc gamma R clusters evoke a synergistic Ca2+ response in human neutrophils. J Biol Chem. (1995) 270:10671–9. doi: 10.1074/jbc.270.18.10671
29. Vossebeld PJ, Homburg CH, Roos D, Verhoeven AJ. The anti-Fc gamma RIII mAb 3G8 induces neutrophil activation via a cooperative actin of Fc gamma RIIIb and Fc gamma RIIa. Int J Biochem Cell Biol. (1997) 29:465–73. doi: 10.1016/S1357-2725(96)00160-4
30. Edberg JC, Moon JJ, Chang DJ, Kimberly RP. Differential regulation of human neutrophil FcγRIIa (CD32) and FcγRIIIb (CD16)-induced Ca2+ transients. J Biol Chem. (1998) 273:8071–9. doi: 10.1074/jbc.273.14.8071
31. Fernandes MJ, Lachance G, Pare G, Rollet-Labelle E, Naccache PH. Signaling through CD16b in human neutrophils involves the Tec family of tyrosine kinases. J Leukoc Biol. (2005) 78:524–32. doi: 10.1189/jlb.0804479
32. Derer S, Glorius P, Schlaeth M, Lohse S, Klausz K, Muchhal U, et al. Increasing FcγRIIa affinity of an FcγRIII-optimized anti-EGFR antibody restores neutrophil-mediated cytotoxicity. MAbs. (2014) 6:409–21. doi: 10.4161/mabs.27457
33. Brandsma AM, Bondza S, Evers M, Koutstaal R, Nederend M, Jansen JHM, et al. Potent Fc receptor signaling by IgA leads to superior killing of cancer cells by neutrophils compared to IgG. Front Immunol. (2019) 10:704. doi: 10.3389/fimmu.2019.00704
34. Chen K, Nishi H, Travers R, Tsuboi N, Martinod K, Wagner DD, et al. Endocytosis of soluble immune complexes leads to their clearance by FcγRIIIB but induces neutrophil extracellular traps via FcγRIIA in vivo. Blood. (2012) 120:4421–31. doi: 10.1182/blood-2011-12-401133
35. Krauss JC, PooH, Xue W, Mayo-Bond L, Todd RF III, Petty HR. Reconstitution of antibody-dependent phagocytosis in fibroblasts expressing Fc gamma receptor IIIB and the complement receptor type 3. J Immunol. (1994) 153:1769–77.
36. Treffers LW, van Houdt M, Bruggeman CW, Heineke MH, Zhao XW, van der Heijden J, et al. FcγRIIIb restricts antibody-dependent destruction of cancer cells by human neutrophils. Front Immunol. (2018) 9:3124. doi: 10.3389/fimmu.2018.03124
37. Tsuboi N, Asano K, Lauterbach M, Mayadas TN. Human neutrophil Fcgamma receptors initiate and play specialized nonredundant roles in antibody-mediated inflammatory diseases. Immunity. (2008) 28:833–46. doi: 10.1016/j.immuni.2008.04.013
38. Skilbeck CA, Lu X, Sheikh S, Savage CO, Nash GB. Capture of flowing human neutrophils by immobilised immunoglobulin: roles of Fc-receptors CD16 and CD32. Cell Immunol. (2006) 241:26–31. doi: 10.1016/j.cellimm.2006.07.007
39. van der Heijden J, Geissler J, van Mirre E, van Deuren M, van der Meer JW, Salama A, et al. A novel splice variant of FcγRIIa: A risk factor for anaphylaxis in patients with hypogammaglobulinemia. J Allergy Clin Immunol. (2013) 131:1408–16. doi: 10.1016/j.jaci.2013.02.009
40. Su K, Wu J, Edberg JC, McKenzie SE, Kimberly RP. Genomic organization of classical human low-affinity Fcgamma receptor genes. Genes Immun. (2002) 3 (Suppl. 1):S51–56. doi: 10.1038/sj.gene.6363879
41. Matsuo K, Procter J, Stroncek D. Variations in genes encoding neutrophil antigens NA1 and NA2. Transfusion. (2000) 40:645–53. doi: 10.1046/j.1537-2995.2000.40060645.x
42. Flinsenberg TW, Janssen WJ, Herczenik E, Boross P, Nederend M, Jongeneel LH, et al. A novel FcγRIIa Q27W gene variant is associated with common variable immune deficiency through defective FcγRIIa downstream signaling. Clin Immunol. (2014) 155:108–17. doi: 10.1016/j.clim.2014.09.006
43. Nagelkerke SQ, Tacke CE, Breunis WB, Tanck MWT, Geissler J, Png E, et al. Extensive ethnic variation and linkage disequilibrium at the FCGR2/3 locus: different genetic associations revealed in kawasaki disease. Front Immunol. (2019) 10:185. doi: 10.3389/fimmu.2019.00185
44. Bruhns P, Iannascoli B, England P, Mancardi DA, Fernandez N, Jorieux S, et al. Specificity and affinity of human Fcgamma receptors and their polymorphic variants for human IgG subclasses. Blood. (2009) 113:3716–25. doi: 10.1182/blood-2008-09-179754
45. Duan J, Lou J, Zhang Q, Ke J, Qi Y, Shen N, et al. A genetic variant rs1801274 in FCGR2A as a potential risk marker for Kawasaki disease: a case-control study and meta-analysis. PLoS ONE. (2014) 9:e103329. doi: 10.1371/journal.pone.0103329
46. Wang D, Hu SL, Cheng XL, Yang JY. FCGR2A rs1801274 polymorphism is associated with risk of childhood-onset idiopathic (immune) thrombocytopenic purpura: evidence from a meta-analysis. Thromb Res. (2014) 134:1323–7. doi: 10.1016/j.thromres.2014.10.003
47. Xu J, Zhao L, Zhang Y, Guo Q, Chen H. CD16 and CD32 gene polymorphisms may contribute to risk of idiopathic thrombocytopenic purpura. Med Sci Monit. (2016) 22:2086–96. doi: 10.12659/MSM.895390
48. Wu LY, Zhou Y, Qin C, Hu BL. The effect of TNF-alpha, FcγR and CD1 polymorphisms on Guillain-Barre syndrome risk: evidences from a meta-analysis. J Neuroimmunol. (2012) 243:18–24. doi: 10.1016/j.jneuroim.2011.12.003
49. Mansour LA, Girgis MY, Abdulhay M, ElEinein EI, ElHawary R, Hanna MO. Polymorphisms of immunoglobulin G Fc receptors in pediatric guillain-barre syndrome. Neuropediatrics. (2016) 47:151–6. doi: 10.1055/s-0036-1579633
50. Zhu XW, Wang Y, Wei YH, Zhao PP, Wang XB, Rong JJ, et al. Comprehensive assessment of the association between FCGRs polymorphisms and the risk of systemic lupus erythematosus: evidence from a meta-analysis. Sci Rep. (2016) 6:31617. doi: 10.1038/srep31617
51. Yuan H, Pan HF, Li LH, Feng JB, Li WX, Li XP, et al. Meta analysis on the association between FγRIIa-R/H131 polymorphisms and systemic lupus erythematosus. Mol Biol Rep. (2009) 36:1053–8. doi: 10.1007/s11033-008-9280-x
52. van Sorge NM, van der Pol WL, van de Winkel JG. FcγR polymorphisms: Implications for function, disease susceptibility and immunotherapy. Tissue Antigens. (2003) 61:189–202. doi: 10.1034/j.1399-0039.2003.00037.x
53. West SD, Ziegler A, Brooks T, Krencicki M, Myers O, Mold C. An FcγRIIa polymorphism with decreased C-reactive protein binding is associated with sepsis and decreased monocyte HLA-DR expression in trauma patients. J Trauma Acute Care Surg. (2015) 79:773–81. doi: 10.1097/TA.0000000000000837
54. Beppler J, Koehler-Santos P, Pasqualim G, Matte U, Alho CS, Dias FS, et al. Fc Gamma Receptor IIA (CD32A) R131 polymorphism as a marker of genetic susceptibility to sepsis. Inflammation. (2015) 39:518–25. doi: 10.1007/s10753-015-0275-1
55. Omar AH, Shibata H, Yasunami M, Yamazaki A, Ofori MF, Akanmori BD, et al. The rs150311303 polymorphism in FcγRIIa enhances IgG binding capacity. Scand J Immunol. (2012) 76:167–74. doi: 10.1111/j.1365-3083.2012.02715.x
56. Nagelkerke SQ, Tacke CE, Breunis WB, Geissler J, Sins JW, Appelhof B, et al. Nonallelic homologous recombination of the FCGR2/3 locus results in copy number variation and novel chimeric FCGR2 genes with aberrant functional expression. Genes Immun. (2015) 16:422–9. doi: 10.1038/gene.2015.25
57. Su K, Wu J, Edberg JC, Li X, Ferguson P, Cooper GS, et al. A promoter haplotype of the immunoreceptor tyrosine-based inhibitory motif-bearing FcγRIIb alters receptor expression and associates with autoimmunity. I. Regulatory FCGR2B polymorphisms and their association with systemic lupus erythematosus. J Immunol. (2004) 172:7186–91. doi: 10.4049/jimmunol.172.11.7186
58. Blank MC, Stefanescu RN, Masuda E, Marti F, King PD, Redecha PB, et al. Decreased transcription of the human FCGR2B gene mediated by the−343 G/C promoter polymorphism and association with systemic lupus erythematosus. Hum Genet. (2005) 117:220–27. doi: 10.1007/s00439-005-1302-3
59. Jonsson S, Sveinbjornsson G, de Lapuente Portilla AL, Swaminathan B, Plomp R, Dekkers G, et al. Identification of sequence variants influencing immunoglobulin levels. Nat Genet. (2017) 49:1182–91. doi: 10.1038/ng.3897
60. Floto RA, Clatworthy MR, Heilbronn KR, Rosner DR, MacAry PA, Rankin A, et al. Loss of function of a lupus-associated FcγRIIb polymorphism through exclusion from lipid rafts. Nat Med. (2005) 11:1056–8. doi: 10.1038/nm1288
61. Kono H, Kyogoku C, Suzuki T, Tsuchiya N, Honda H, Yamamoto K, et al. FcγRIIB Ile232Thr transmembrane polymorphism associated with human systemic lupus erythematosus decreases affinity to lipid rafts and attenuates inhibitory effects on B cell receptor signaling. Hum Mol Genet. (2005) 14:2881–92. doi: 10.1093/hmg/ddi320
62. Willcocks LC, Carr EJ, Niederer HA, Rayner TF, Williams TN, Yang W, et al. A defunctioning polymorphism in FCGR2B is associated with protection against malaria but susceptibility to systemic lupus erythematosus. Proc Natl Acad Sci USA. (2010) 107:7881–5. doi: 10.1073/pnas.0915133107
63. Breunis WB, van Mirre E, Geissler J, Laddach N, Wolbink G, van der Schoot E, et al. Copy number variation at the FCGR locus includes FCGR3A, FCGR2C and FCGR3B but not FCGR2A and FCGR2B. Hum Mutat. (2009) 30:E640–50. doi: 10.1002/humu.20997
64. Makowsky R, Wiener HW, Ptacek TS, Silva M, Shendre A, Edberg JC, et al. FcγR gene copy number in Kawasaki disease and intravenous immunoglobulin treatment response. Pharmacogenet Genomics. (2013) 23:455–62. doi: 10.1097/FPC.0b013e328363686e
65. Schmidt DE, Heitink-Pollé KMJ, Laarhoven AG, Bruin MCA, Veldhuisen B, Nagelkerke SQ, et al. Transient and chronic childhood immune thrombocytopenia are distinctly affected by Fc-γ receptor polymorphisms. Blood Adv. (2019) 3:2003–12. doi: 10.1182/bloodadvances.2019000068
66. Wågström P, Yamada-Fowler N, Dahle C, Nilsdotter-Augustinsson Å, Bengnér M, Söderkvist P, et al. Fcgamma-receptor polymorphisms associated with clinical symptoms in patients with immunoglobulin G subclass deficiency. Infect Dis. (2018) 50:853–8. doi: 10.1080/23744235.2018.1510183
67. Lassauniere R, Paximadis M, Ebrahim O, Chaisson RE, Martinson NA, Tiemessen CT. The FCGR2C allele that modulated the risk of HIV-1 infection in the Thai RV144 vaccine trial is implicated in HIV-1 disease progression. Genes Immun. (2018). doi: 10.1038/s41435-018-0053-9
68. Chen JY, Wang CM, Chang SW, Cheng CH, Wu YJ, Lin JC, et al. Association of FCGR3A and FCGR3B copy number variations with systemic lupus erythematosus and rheumatoid arthritis in Taiwanese patients. Arthritis Rheumatol. (2014) 66:3113–21. doi: 10.1002/art.38813
69. Zhou XJ, Lv JC, Bu DF, Yu L, Yang YR, Zhao J, et al. Copy number variation of FCGR3A rather than FCGR3B and FCGR2B is associated with susceptibility to anti-GBM disease. Int Immunol. (2010) 22:45–51. doi: 10.1093/intimm/dxp113
70. Koene HR, Kleijer M, Algra J, Roos D, von dem Borne AE, de Haas M. Fc gammaRIIIa-158V/F polymorphism influences the binding of IgG by natural killer cell Fc gammaRIIIa, independently of the Fc gammaRIIIa-48L/R/H phenotype. Blood. (1997) 90:1109–14. doi: 10.1016/S0165-2478(97)85823-3
71. Dong C, Ptacek TS, Redden DT, Zhang K, Brown EE, Edberg JC, et al. Fcgamma receptor IIIa single-nucleotide polymorphisms and haplotypes affect human IgG binding and are associated with lupus nephritis in african americans. Arthritis Rheumatol. (2014) 66:1291–9. doi: 10.1002/art.38337
72. Jawahar S, Moody C, Chan M, Finberg R, Geha R, Chatila T. Natural Killer (NK) cell deficiency associated with an epitope-deficient Fc receptor type IIIA (CD16-II). Clin Exp Immunol. (1996) 103:408–13. doi: 10.1111/j.1365-2249.1996.tb08295.x
73. Grier JT, Forbes LR, Monaco-Shawver L, Oshinsky J, Atkinson TP, Moody C, et al. Human immunodeficiency-causing mutation defines CD16 in spontaneous NK cell cytotoxicity. J Clin Invest. (2012) 122:3769–80. doi: 10.1172/JCI64837
74. de Vries E, Koene HR, Vossen JM, Gratama JW, von dem Borne AE, Waaijer JL, et al. Identification of an unusual Fc gamma receptor IIIa (CD16) on natural killer cells in a patient with recurrent infections. Blood. (1996) 88:3022–7.
75. Carcao MD, Blanchette VS, Wakefield CD, Stephens D, Ellis J, Matheson K, et al. Fcgamma receptor IIa and IIIa polymorphisms in childhood immune thrombocytopenic purpura. Br J Haematol. (2003) 120:135–41. doi: 10.1046/j.1365-2141.2003.04033.x
76. Lee YH, Ji JD, Song GG. Associations between FCGR3A polymorphisms and susceptibility to rheumatoid arthritis: a metaanalysis. J Rheumatol. (2008) 35:2129–35. doi: 10.3899/jrheum.080186
77. Asano K, Matsushita T, Umeno J, Hosono N, Takahashi A, Kawaguchi T, et al. A genome-wide association study identifies three new susceptibility loci for ulcerative colitis in the Japanese population. Nat Genet. (2009) 41:1325–9. doi: 10.1038/ng.482
78. McKinney C, Merriman TR. Meta-analysis confirms a role for deletion in FCGR3B in autoimmune phenotypes. Hum Mol Genet. (2012) 21:2370–76. doi: 10.1093/hmg/dds039
79. Nossent JC, Rischmueller M, Lester S. Low copy number of the Fc-gamma receptor 3B gene FCGR3B is a risk factor for primary Sjogren's syndrome. J Rheumatol. (2012) 39:2142–7. doi: 10.3899/jrheum.120294
80. McKinney C, Broen JC, Vonk MC, Beretta L, Hesselstrand R, Hunzelmann N, et al. Evidence that deletion at FCGR3B is a risk factor for systemic sclerosis. Genes Immun. (2012) 13:458–60. doi: 10.1038/gene.2012.15
81. Rahbari R, Zuccherato LW, Tischler G, Chihota B, Ozturk H, Saleem S, et al. Understanding the genomic structure of copy-number variation of the low-affinity fcgamma receptor region allows confirmation of the association of FCGR3B deletion with rheumatoid arthritis. Hum Mutat. (2017) 38:390–9. doi: 10.1002/humu.23159
82. Graf SW, Lester S, Nossent JC, Hill CL, Proudman SM, Lee A, et al. Low copy number of the FCGR3B gene and rheumatoid arthritis: a case-control study and meta-analysis. Arthritis Res Ther. (2012) 14:R28. doi: 10.1186/ar3731
83. Asano K, Matsumoto T, Umeno J, Hirano A, Esaki M, Hosono N, et al. Impact of allele copy number of polymorphisms in FCGR3A and FCGR3B genes on susceptibility to ulcerative colitis. Inflamm Bowel Dis. (2013) 19:2061–8. doi: 10.1097/MIB.0b013e318298118e
84. Wang L, Yang X, Cai G, Xin L, Xia Q, Zhang X, et al. Association study of copy number variants in FCGR3A and FCGR3B gene with risk of ankylosing spondylitis in a Chinese population. Rheumatol Int. (2016) 36:437–42. doi: 10.1007/s00296-015-3384-0
85. Fanciulli M, Norsworthy PJ, Petretto E, Dong R, Harper L, Kamesh L, et al. FCGR3B copy number variation is associated with susceptibility to systemic, but not organ-specific, autoimmunity. Nat Genet. (2007) 39:721–3. doi: 10.1038/ng2046
86. Recke A, Vidarsson G, Ludwig RJ, Freitag M, Möller S, Vonthein R, et al. Allelic and copy-number variations of FcγRs affect granulocyte function and susceptibility for autoimmune blistering diseases. J Autoimmun. (2015) 61:36–44. doi: 10.1016/j.jaut.2015.05.004
87. Adu B, Dodoo D, Adukpo S, Hedley PL, Arthur FK, Gerds TA, et al. Fc Gamma Receptor IIIB (FcγRIIIB) polymorphisms are associated with clinical malaria in ghanaian children. PLoS ONE. (2012) 7:e46197. doi: 10.1371/journal.pone.0046197
88. Ory PA, Clark MR, Kwoh EE, Clarkson SB, Goldstein IM. Sequences of complementary DNAs that encode the NA1 and NA2 forms of Fc receptor III on human neutrophils. J Clin Invest. (1989) 84:1688–91. doi: 10.1172/JCI114350
89. Bux J, Stein EL, Bierling P, Fromont P, Clay M, Stroncek D, et al. Characterization of a new alloantigen (SH) on the human neutrophil Fc gamma receptor IIIb. Blood. (1997) 89:1027–34.
90. Yamamoto K, Sugita N, Kobayashi T, Okuda K, van de Winkel JG, Yoshie H. Evidence for a novel polymorphism affecting both N-linked glycosylation and ligand binding of the IgG receptor IIIB (CD16). Tissue Antigens. (2001) 57:363–6. doi: 10.1034/j.1399-0039.2001.057004363.x
91. Flesch BK, Curtis BR, de Haas M, Lucas G, Sachs UJ. Update on the nomenclature of human neutrophil antigens and alleles. Transfusion. (2016) 56:1477–9. doi: 10.1111/trf.13575
92. Aan de Kerk DJ, Jansen MH, Ten Berge IJ, van Leeuwen EM, Kuijpers TW. Identification of B cell defects using age-defined reference ranges for in vivo and in vitro B cell differentiation. J Immunol. (2013) 190:5012–9. doi: 10.4049/jimmunol.1201807
93. Reil A, Sachs UJ, Siahanidou T, Flesch BK, Bux J. HNA-1d: a new human neutrophil antigen located on Fcgamma receptor IIIb associated with neonatal immune neutropenia. Transfusion. (2013) 53:2145–51. doi: 10.1111/trf.12086
94. Ravetch JV, Perussia B. Alternative membrane forms of Fc gamma RIII(CD16) on human natural killer cells and neutrophils. Cell type-specific expression of two genes that differ in single nucleotide substitutions. J Exp Med. (1989) 170:481–97. doi: 10.1084/jem.170.2.481
95. de Smith AJ, Tsalenko A, Sampas N, Scheffer A, Yamada NA, Tsang P, et al. Array CGH analysis of copy number variation identifies 1284 new genes variant in healthy white males: implications for association studies of complex diseases. Hum Mol Genet. (2007) 16:2783–94. doi: 10.1093/hmg/ddm208
96. Redon R, Ishikawa S, Fitch KR, Feuk L, Perry GH, Andrews TD, et al. Global variation in copy number in the human genome. Nature. (2006) 444:444–54. doi: 10.1038/nature05329
97. Machado LR, Hardwick RJ, Bowdrey J, Bogle H, Knowles TJ, Sironi M, et al. Evolutionary history of copy-number-variable locus for the low-affinity Fcgamma receptor: mutation rate, autoimmune disease, and the legacy of helminth infection. Am J Hum Genet. (2012) 90:973–85. doi: 10.1016/j.ajhg.2012.04.018
98. Niederer HA, Willcocks LC, Rayner TF, Yang W, Lau YL, Williams TN, et al. Copy number, linkage disequilibrium and disease association in the FCGR locus. Hum Mol Genet. (2010) 19:3282–94. doi: 10.1093/hmg/ddq216
99. Mueller M, Barros P, Witherden AS, Roberts AL, Zhang Z, Schaschl H, et al. Genomic pathology of SLE-associated copy-number variation at the FCGR2C/FCGR3B/FCGR2B Locus. Am J Hum Genet. (2013) 92:28–40. doi: 10.1016/j.ajhg.2012.11.013
100. den Dunnen JT, Dalgleish R, Maglott DR, Hart RK, Greenblatt MS, McGowan-Jordan J, et al. HGVS recommendations for the description of sequence variants: 2016 Update. Hum Mutat. (2016) 37:564–9. doi: 10.1002/humu.22981
101. Hargreaves CE, Rose-Zerilli MJ, Machado LR, Iriyama C, Hollox EJ, Cragg MS, et al. Fcgamma receptors: genetic variation, function, and disease. Immunol Rev. (2015) 268:6–24. doi: 10.1111/imr.12341
102. Hargreaves CE, Iriyama C, Rose-Zerilli MJ, Nagelkerke SQ, Hussain K, Ganderton R, et al. Evaluation of high-throughput genomic assays for the Fc Gamma receptor locus. PLoS ONE. (2015) 10:e0142379. doi: 10.1371/journal.pone.0142379
103. Franke L, el Bannoudi H, Jansen DT, Kok K, Trynka G, Diogo D, et al. Association analysis of copy numbers of FC-gamma receptor genes for rheumatoid arthritis and other immune-mediated phenotypes. Eur J Hum Genet. (2015) 24:263–70. doi: 10.1038/ejhg.2015.95
104. Haridan US, Mokhtar U, Machado LR, Abdul Aziz AT, Shueb RH, Zaid M, et al. A comparison of assays for accurate copy number measurement of the low-affinity Fc gamma receptor genes FCGR3A and FCGR3B. PLoS ONE. (2015) 10:e0116791. doi: 10.1371/journal.pone.0116791
105. Willcocks LC, Lyons PA, Clatworthy MR, Robinson JI, Yang W, Newland SA, et al. Copy number of FCGR3B, which is associated with systemic lupus erythematosus, correlates with protein expression and immune complex uptake. J Exp Med. (2008) 205:1573–82. doi: 10.1084/jem.20072413
106. Warmerdam PA, van de Winkel JG, Gosselin EJ, Capel PJ. Molecular basis for a polymorphism of human Fc gamma receptor II (CD32). J Exp Med. (1990) 172:19–25. doi: 10.1084/jem.172.1.19
107. Yamamoto K, Kobayashi T, Sugita N, Tai H, Yoshie H. The FcγRIIa polymorphism influences production of interleukin-1 by mononuclear cells. Int J Immunogenet. (2007) 34:369–72. doi: 10.1111/j.1744-313X.2007.00701.x
108. Bredius RG, Fijen CA, De Haas M, Kuijper EJ, Weening RS, Van de Winkel JG, et al. Role of neutrophil Fc gamma RIIa (CD32) and Fc gamma RIIIb (CD16) polymorphic forms in phagocytosis of human IgG1- and IgG3-opsonized bacteria and erythrocytes. Immunology. (1994) 83:624–30.
109. Nicu EA, Van der Velden U, Everts V, Van Winkelhoff AJ, Roos D, Loos BG. Hyper-reactive PMNs in FcγRIIa 131 H/H genotype periodontitis patients. J Clin Periodontol. (2007) 34:938–45. doi: 10.1111/j.1600-051X.2007.01136.x
110. Tada M, Ishii-Watabe A, Maekawa K, Fukushima-Uesaka H, Kurose K, Suzuki T, et al. Genetic polymorphisms of FCGR2A encoding Fcgamma receptor IIa in a Japanese population and functional analysis of the L273P variant. Immunogenetics. (2012) 64:869–77. doi: 10.1007/s00251-012-0646-9
111. Anania JC, Trist HM, Palmer CS, Tan PS, Kouskousis BP, Chenoweth AM, et al. The rare anaphylaxis-associated FcγRIIa3 exhibits distinct characteristics from the canonical FcγRIIa1. Front Immunol. (2018) 9:1809. doi: 10.3389/fimmu.2018.01809
112. Li X, Wu J, Carter RH, Edberg JC, Su K, Cooper GS, et al. A novel polymorphism in the Fcgamma receptor IIB (CD32B) transmembrane region alters receptor signaling. Arthritis Rheum. (2003) 48:3242–52. doi: 10.1002/art.11313
113. den Dunnen J, Vogelpoel LT, Wypych T, Muller FJ, de Boer L, Kuijpers TW, et al. IgG opsonization of bacteria promotes Th17 responses via synergy between TLRs and FcγRIIa in human dendritic cells. Blood. (2012) 120:112–21. doi: 10.1182/blood-2011-12-399931
114. Boruchov AM, Heller G, Veri MC, Bonvini E, Ravetch JV, Young JW. Activating and inhibitory IgG Fc receptors on human DCs mediate opposing functions. J Clin Invest. (2005) 115:2914–23. doi: 10.1172/JCI24772
115. Pellerin A, Otero K, Czerkowicz JM, Kerns HM, Shapiro RI, Ranger AM, et al. Anti-BDCA2 monoclonal antibody inhibits plasmacytoid dendritic cell activation through Fc-dependent and Fc-independent mechanisms. EMBO Mol Med. (2015) 7:464–76. doi: 10.15252/emmm.201404719
116. Su K, Li X, Edberg JC, Wu J, Ferguson P, Kimberly RP. A promoter haplotype of the immunoreceptor tyrosine-based inhibitory motif-bearing FcγRIIb alters receptor expression and associates with autoimmunity. II. Differential binding of GATA4 and Yin-Yang1 transcription factors and correlated receptor expression and function. J Immunol. (2004) 172:7192–9. doi: 10.4049/jimmunol.172.11.7192
117. Metes D, Manciulea M, Pretrusca D, Rabinowich H, Ernst LK, Popescu I, et al. Ligand binding specificities and signal transduction pathways of Fc gamma receptor IIc isoforms: the CD32 isoforms expressed by human NK cells. Eur J Immunol. (1999) 29:2842–52.
118. Lassauniere R, Tiemessen CT. Variability at the FCGR locus: characterization in Black South Africans and evidence for ethnic variation in and out of Africa. Genes Immun. (2016) 17:93–104. doi: 10.1038/gene.2015.60
119. Wu J, Edberg JC, Redecha PB, Bansal V, Guyre PM, Coleman K, et al. A novel polymorphism of FcγRIIIa (CD16) alters receptor function and predisposes to autoimmune disease. J Clin Invest. (1997) 100:1059–70. doi: 10.1172/JCI119616
120. Salmon JE, Edberg JC, Kimberly RP. Fc gamma receptor III on human neutrophils. Allelic variants have functionally distinct capacities. J Clin Invest. (1990) 85:1287–95. doi: 10.1172/JCI114566
121. Koene HR, Kleijer M, Roos D, de Haas M, von dem Borne AE. Fc gamma RIIIB gene duplication: evidence for presence and expression of three distinct Fc gamma RIIIB genes in NA(1+,2+)SH(+) individuals. Blood. (1998) 91:673–79.
122. de Haas M, Kleijer M, van Zwieten R, Roos D, von dem Borne AE. Neutrophil Fc gamma RIIIb deficiency, nature, and clinical consequences: a study of 21 individuals from 14 families. Blood. (1995) 86:2403–13.
123. Huizinga TW, de Haas M, Kleijer M, Nuijens JH, Roos D, von dem Borne AE. Soluble Fc gamma receptor III in human plasma originates from release by neutrophils. J Clin Invest. (1990) 86:416–23. doi: 10.1172/JCI114727
124. Lejeune J, Thibault G, Ternant D, Cartron G, Watier H, Ohresser M. Evidence for linkage disequilibrium between Fcgamma RIIIa-V158F and Fcgamma RIIa-H131R polymorphisms in white patients, and for an Fcgamma RIIIa-restricted influence on the response to therapeutic antibodies. J Clin Oncol. (2008) 26:5489–91. doi: 10.1200/JCO.2008.19.4118
125. Lejeune J, Piegu B, Gouilleux-Gruart V, Ohresser M, Watier H, Thibault G. FCGR2C genotyping by pyrosequencing reveals linkage disequilibrium with FCGR3A V158F and FCGR2A H131R polymorphisms in a Caucasian population. MAbs. (2012) 4:784–7. doi: 10.4161/mabs.22287
126. de Haas M, Koene HR, Kleijer M, de Vries E, Simsek S, van Tol MJ, et al. A triallelic Fc gamma receptor type IIIA polymorphism influences the binding of human IgG by NK cell Fc gamma RIIIa. J Immunol. (1996) 156:2948–55.
127. Lejeune J, Brachet G, Watier H. Evolutionary story of the low/medium-affinity IgG Fc receptor gene cluster. Front Immunol. (2019) 10:1297. doi: 10.3389/fimmu.2019.01297
128. Akula S, Mohammadamin S, Hellman L. Fc Receptors for immunoglobulins and their appearance during vertebrate evolution. PLoS ONE. (2014) 9:e96903. doi: 10.1371/journal.pone.0096903
129. Hogarth PM, Anania JC, Wines BD. The FcγR of humans and non-human primates and their interaction with IgG: implications for induction of inflammation, resistance to infection and the use of therapeutic monoclonal antibodies. Curr Top Microbiol Immunol. (2014) 382:321–52. doi: 10.1007/978-3-319-07911-0_15
130. Rogers KA, Scinicariello F, Attanasio R. IgG Fc receptor III homologues in nonhuman primate species: genetic characterization and ligand interactions. J Immunol. (2006) 177:3848–56. doi: 10.4049/jimmunol.177.6.3848
131. Bournazos S, Woof JM, Hart SP, Dransfield I. Functional and clinical consequences of Fc receptor polymorphic and copy number variants. Clin Exp Immunol. (2009) 157:244–54. doi: 10.1111/j.1365-2249.2009.03980.x
132. Karassa FB, Trikalinos TA, Ioannidis JP. Role of the Fcgamma receptor IIa polymorphism in susceptibility to systemic lupus erythematosus and lupus nephritis: a meta-analysis. Arthritis Rheum. (2002) 46:1563–71. doi: 10.1002/art.10306
133. Duits AJ, Bootsma H, Derksen RH, Spronk PE, Kater L, Kallenberg CG, et al. Skewed distribution of IgG Fc receptor IIa (CD32) polymorphism is associated with renal disease in systemic lupus erythematosus patients. Arthritis Rheum. (1995) 38:1832–6. doi: 10.1002/art.1780381217
134. Vigato-Ferreira IC, Toller-Kawahisa JE, Pancoto JA, Mendes-Junior CT, Martinez EZ, Donadi EA, et al. FcγRIIa and FcγRIIIb polymorphisms and associations with clinical manifestations in systemic lupus erythematosus patients. Autoimmunity. (2014) 47:451–8. doi: 10.3109/08916934.2014.921809
135. Harley JB, Alarcón-Riquelme ME, Criswell LA, Jacob CO, Kimberly RP, Tsao BP, et al. Genome-wide association scan in women with systemic lupus erythematosus identifies susceptibility variants in ITGAM, PXK, KIAA1542 and other loci. Nat Genet. (2008) 40:204–10. doi: 10.1038/ng.81
136. Norsworthy P, Theodoridis E, Botto M, Athanassiou P, Beynon H, Gordon C, et al. Overrepresentation of the Fcgamma receptor type IIA R131/R131 genotype in caucasoid systemic lupus erythematosus patients with autoantibodies to C1q and glomerulonephritis. Arthritis Rheum. (1999) 42:1828–32.
137. Dijstelbloem HM, Bijl M, Fijnheer R, Scheepers RH, Oost WW, Jansen MD, et al. Fcgamma receptor polymorphisms in systemic lupus erythematosus: association with disease and in vivo clearance of immune complexes. Arthritis Rheum. (2000) 43:2793–800. doi: 10.1002/1529-0131(200012)43:12<2793::AID-ANR20>3.0.CO;2-6
138. Salmon JE, Millard S, Schachter LA, Arnett FC, Ginzler EM, Gourley MF, et al. Fc gamma RIIA alleles are heritable risk factors for lupus nephritis in African Americans. J Clin Invest. (1996) 97:1348–54. doi: 10.1172/JCI118552
139. Castro-Dopico T, Dennison TW, Ferdinand JR, Mathews RJ, Fleming A, Clift D, et al. Anti-commensal IgG drives intestinal inflammation and type 17 immunity in ulcerative colitis. Immunity. (2019) 50:1099–114.e1010. doi: 10.1016/j.immuni.2019.02.006
140. Khor CC, Davila S, Breunis WB, Lee YC, Shimizu C, Wright VJ, et al. Genome-wide association study identifies FCGR2A as a susceptibility locus for Kawasaki disease. Nat Genet. (2011) 43:1241–6. doi: 10.1038/ng.981
141. Pavkovic M, Petlichkovski A, Karanfilski O, Cevreska L, Stojanovic A. FC gamma receptor polymorphisms in patients with immune thrombocytopenia. Hematology. (2018) 23:163–8. doi: 10.1080/10245332.2017.1377902
142. Li G, Gao L, Ma R, Tian W, Mingzhi L. Associations between FCGR polymorphisms and immune thrombocytopenia: a meta-analysis. Scand J Immunol. (2019) 89:e12758. doi: 10.1111/sji.12758
143. Karassa FB, Trikalinos TA, Ioannidis JP. The Fc gamma RIIIA-F158 allele is a risk factor for the development of lupus nephritis: a meta-analysis. Kidney Int. (2003) 63:1475–82. doi: 10.1046/j.1523-1755.2003.00873.x
144. Chu ZT, Tsuchiya N, Kyogoku C, Ohashi J, Qian YP, Xu SB, et al. Association of Fcgamma receptor IIb polymorphism with susceptibility to systemic lupus erythematosus in Chinese: a common susceptibility gene in the Asian populations. Tissue Antigens. (2004) 63:21–7. doi: 10.1111/j.1399-0039.2004.00142.x
145. Kyogoku C, Dijstelbloem HM, Tsuchiya N, Hatta Y, Kato H, Yamaguchi A, et al. Fcgamma receptor gene polymorphisms in Japanese patients with systemic lupus erythematosus: contribution of FCGR2B to genetic susceptibility. Arthritis Rheum. (2002) 46:1242–54. doi: 10.1002/art.10257
146. Chen JY, Wang CM, Ma CC, Luo SF, Edberg JC, Kimberly RP, et al. Association of a transmembrane polymorphism of Fcgamma receptor IIb (FCGR2B) with systemic lupus erythematosus in Taiwanese patients. Arthritis Rheum. (2006) 54:3908–17. doi: 10.1002/art.22220
147. Radstake TR, Franke B, Wenink MH, Nabbe KC, Coenen MJ, Welsing P, et al. The functional variant of the inhibitory Fcgamma receptor IIb (CD32B) is associated with the rate of radiologic joint damage and dendritic cell function in rheumatoid arthritis. Arthritis Rheum. (2006) 54:3828–37. doi: 10.1002/art.22275
148. Satoh T, Miyazaki K, Shimohira A, Amano N, Okazaki Y, Nishimoto T, et al. Fcgamma receptor IIB gene polymorphism in adult Japanese patients with primary immune thrombocytopenia. Blood. (2013) 122:1991–2. doi: 10.1182/blood-2013-05-501858
149. Bruin M, Bierings M, Uiterwaal C, Révész T, Bode L, Wiesman ME, et al. Platelet count, previous infection and FCGR2B genotype predict development of chronic disease in newly diagnosed idiopathic thrombocytopenia in childhood: results of a prospective study. Br J Haematol. (2004) 127:561–7. doi: 10.1111/j.1365-2141.2004.05235.x
150. Heitink-Pollé KMJ, Uiterwaal CSPM, Porcelijn L, Tamminga RYJ, Smiers FJ, van Woerden NL, et al. Intravenous immunoglobulin vs observation in childhood immune thrombocytopenia: a randomized controlled trial. Blood. (2018) 132:883–91. doi: 10.1182/blood-2018-02-830844
151. Stewart-Akers AM, Cunningham A, Wasko MC, Morel PA. Fc gamma R expression on NK cells influences disease severity in rheumatoid arthritis. Genes Immun. (2004) 5:521–9. doi: 10.1038/sj.gene.6364121
152. Gorlova OY, Li Y, Gorlov I, Ying J, Chen WV, Assassi S, et al. Gene-level association analysis of systemic sclerosis: a comparison of African-Americans and White populations. PLoS ONE. (2018) 13:e0189498. doi: 10.1371/journal.pone.0189498
153. Shrestha S, Wiener HW, Olson AK, Edberg JC, Bowles NE, Patel H, et al. Functional FCGR2B gene variants influence intravenous immunoglobulin response in patients with Kawasaki disease. J Allergy Clin Immunol. (2011) 128:677–80. doi: 10.1016/j.jaci.2011.04.027
154. Barbosa FB, Simioni M, Wiezel CEV, Torres FR, Molck MC, Bonilla MM, et al. Copy number variation in the susceptibility to systemic lupus erythematosus. PLoS ONE. (2018) 13:e0206683. doi: 10.1371/journal.pone.0206683
155. Mamtani M, Anaya JM, He W, Ahuja SK. Association of copy number variation in the FCGR3B gene with risk of autoimmune diseases. Genes Immun. (2010) 11:155–60. doi: 10.1038/gene.2009.71
156. McKinney C, Fanciulli M, Merriman ME, Phipps-Green A, Alizadeh BZ, Koeleman BP, et al. Association of variation in Fcgamma receptor 3B gene copy number with rheumatoid arthritis in Caucasian samples. Ann Rheum Dis. (2010) 69:1711–6. doi: 10.1136/ard.2009.123588
157. Robinson JI, Carr IM, Cooper DL, Rashid LH, Martin SG, Emery P, et al. Confirmation of association of FCGR3B but not FCGR3A copy number with susceptibility to autoantibody positive rheumatoid arthritis. Hum Mutat. (2012) 33:741–9. doi: 10.1002/humu.22031
158. Aitman TJ, Dong R, Vyse TJ, Norsworthy PJ, Johnson MD, Smith J, et al. Copy number polymorphism in Fcgr3 predisposes to glomerulonephritis in rats and humans. Nature. (2006) 439:851–5. doi: 10.1038/nature04489
159. Lee YH, Bae SC, Seo YH, Kim JH, Choi SJ, Ji JD, et al. Association between FCGR3B copy number variations and susceptibility to autoimmune diseases: a meta-analysis. Inflamm Res. (2015) 64:983–91. doi: 10.1007/s00011-015-0882-1
160. Marques RB, Thabet MM, White SJ, Houwing-Duistermaat JJ, Bakker AM, Hendriks GJ, et al. Genetic variation of the Fc gamma receptor 3B gene and association with rheumatoid arthritis. PLoS ONE. (2010) 5:e13173. doi: 10.1371/journal.pone.0013173
161. Yuan J, Zhao D, Wu L, Xu X, Pang Y, Zhang J, et al. FCGR3B copy number loss rather than gain is a risk factor for systemic lupus erythematous and lupus nephritis: a meta-analysis. Int J Rheum Dis. (2015) 18:392–7. doi: 10.1111/1756-185X.12342
162. Mulhearn B, Bruce IN. Indications for IVIG in rheumatic diseases. Rheumatology. (2015) 54:383–91. doi: 10.1093/rheumatology/keu429
163. Sakthiswary R, D'Cruz D. Intravenous immunoglobulin in the therapeutic armamentarium of systemic lupus erythematosus: a systematic review and meta-analysis. Medicine. (2014) 93:e86. doi: 10.1097/MD.0000000000000086
164. Cooper N, Stasi R, Cunningham-Rundles S, Cesarman E, McFarland JG, Bussel JB. Platelet-associated antibodies, cellular immunity and FCGR3a genotype influence the response to rituximab in immune thrombocytopenia. Br J Haematol. (2012) 158:539–47. doi: 10.1111/j.1365-2141.2012.09184.x
165. Lee YH, Bae SC, Song GG. Functional FCGR3A 158 V/F and IL-6−174 C/G polymorphisms predict response to biologic therapy in patients with rheumatoid arthritis: a meta-analysis. Rheumatol Int. (2014) 34:1409–15. doi: 10.1007/s00296-014-3015-1
166. Dávila-Fajardo CL, van der Straaten T, Baak-Pablo R, Medarde Caballero C, Cabeza Barrera J, Huizinga TW, et al. FcGR genetic polymorphisms and the response to adalimumab in patients with rheumatoid arthritis. Pharmacogenomics. (2015) 16:373–81. doi: 10.2217/pgs.14.178
167. Musolino A, Naldi N, Bortesi B, Pezzuolo D, Capelletti M, Missale G, et al. Immunoglobulin G fragment C receptor polymorphisms and clinical efficacy of trastuzumab-based therapy in patients with HER-2/neu-positive metastatic breast cancer. J Clin Oncol. (2008) 26:1789–96. doi: 10.1200/JCO.2007.14.8957
168. Tamura K, Shimizu C, Hojo T, Akashi-Tanaka S, Kinoshita T, Yonemori K, et al. FcγR2A and 3A polymorphisms predict clinical outcome of trastuzumab in both neoadjuvant and metastatic settings in patients with HER2-positive breast cancer. Ann Oncol. (2011) 22:1302–7. doi: 10.1093/annonc/mdq585
169. Gavin PG, Song N, Kim SR, Lipchik C, Johnson NL, Bandos H, et al. Association of polymorphisms in FCGR2A and FCGR3A with degree of trastuzumab benefit in the adjuvant treatment of ERBB2/HER2-positive breast cancer: analysis of the NSABP B-31 trial. JAMA Oncol. (2017) 3:335–41. doi: 10.1001/jamaoncol.2016.4884
170. Hurvitz SA, Betting DJ, Stern HM, Quinaux E, Stinson J, Seshagiri S, et al. Analysis of Fcgamma receptor IIIa and IIa polymorphisms: lack of correlation with outcome in trastuzumab-treated breast cancer patients. Clin Cancer Res. (2012) 18:3478–86. doi: 10.1158/1078-0432.CCR-11-2294
171. Cartron G, Dacheux L, Salles G, Solal-Celigny P, Bardos P, Colombat P, et al. Therapeutic activity of humanized anti-CD20 monoclonal antibody and polymorphism in IgG Fc receptor FcγRIIIa gene. Blood. (2002) 99:754–8. doi: 10.1182/blood.V99.3.754
172. Weng WK, Levy R. Two immunoglobulin G fragment C receptor polymorphisms independently predict response to rituximab in patients with follicular lymphoma. J Clin Oncol. (2003) 21:3940–7. doi: 10.1200/JCO.2003.05.013
173. Persky DO, Dornan D, Goldman BH, Braziel RM, Fisher RI, Leblanc M, et al. Fc gamma receptor 3a genotype predicts overall survival in follicular lymphoma patients treated on SWOG trials with combined monoclonal antibody plus chemotherapy but not chemotherapy alone. Haematologica. (2012) 97:937–42. doi: 10.3324/haematol.2011.050419
174. Kim DH, Jung HD, Kim JG, Lee JJ, Yang DH, Park YH, et al. FCGR3A gene polymorphisms may correlate with response to frontline R-CHOP therapy for diffuse large B-cell lymphoma. Blood. (2006) 108:2720–25. doi: 10.1182/blood-2006-01-009480
175. Ghesquieres H, Cartron G, Seymour JF, Delfau-Larue MH, Offner F, Soubeyran P, et al. Clinical outcome of patients with follicular lymphoma receiving chemoimmunotherapy in the PRIMA study is not affected by FCGR3A and FCGR2A polymorphisms. Blood. (2012) 120:2650–7. doi: 10.1182/blood-2012-05-431825
176. Carlotti E, Palumbo GA, Oldani E, Tibullo D, Salmoiraghi S, Rossi A, et al. FcγRIIIA and FcγRIIA polymorphisms do not predict clinical outcome of follicular non-Hodgkin's lymphoma patients treated with sequential CHOP and rituximab. Haematologica. (2007) 92:1127–130. doi: 10.3324/haematol.11288
177. Prochazka V, Papajik T, Gazdova J, Divoka M, Rozmanova S, Faber E, et al. FcγRIIIA receptor genotype does not influence an outcome in patients with follicular lymphoma treated with risk-adapted immunochemotherapy. Neoplasma. (2011) 58:263–70. doi: 10.4149/neo_2011_03_263
178. Dornan D, Spleiss O, Yeh RF, Duchateau-Nguyen G, Dufour A, Zhi J, et al. Effect of FCGR2A and FCGR3A variants on CLL outcome. Blood. (2010) 116:4212–22. doi: 10.1182/blood-2010-03-272765
179. Bibeau F, Lopez-Crapez E, Di Fiore F, Thezenas S, Ychou M, Blanchard F, et al. Impact of FcγRIIa-FcγRIIIa polymorphisms and KRAS mutations on the clinical outcome of patients with metastatic colorectal cancer treated with cetuximab plus irinotecan. J Clin Oncol. (2009) 27:1122–29. doi: 10.1200/JCO.2008.18.0463
180. Qiu LX, Mao C, Zhang J, Zhu XD, Liao RY, Xue K, et al. Predictive and prognostic value of KRAS mutations in metastatic colorectal cancer patients treated with cetuximab: a meta-analysis of 22 studies. Eur J Cancer. (2010) 46:2781–7. doi: 10.1016/j.ejca.2010.05.022
181. Zhang W, Azuma M, Lurje G, Gordon MA, Yang D, Pohl A, et al. Molecular predictors of combination targeted therapies (cetuximab, bevacizumab) in irinotecan-refractory colorectal cancer (BOND-2 study). Anticancer Res. (2010) 30:4209–17.
182. Dahan L, Norguet E, Etienne-Grimaldi MC, Formento JL, Gasmi M, Nanni I, et al. Pharmacogenetic profiling and cetuximab outcome in patients with advanced colorectal cancer. BMC Cancer. (2011) 11:496. doi: 10.1186/1471-2407-11-496
183. Calemma R, Ottaiano A, Trotta AM, Nasti G, Romano C, Napolitano M, et al. Fc gamma receptor IIIa polymorphisms in advanced colorectal cancer patients correlated with response to anti-EGFR antibodies and clinical outcome. J Transl Med. (2012) 10:232. doi: 10.1186/1479-5876-10-232
184. Zhang W, Gordon M, Schultheis AM, Yang DY, Nagashima F, Azuma M, et al. FCGR2A and FCGR3A polymorphisms associated with clinical outcome of epidermal growth factor receptor expressing metastatic colorectal cancer patients treated with single-agent cetuximab. J Clin Oncol. (2007) 25:3712–8. doi: 10.1200/JCO.2006.08.8021
185. Magnes T, Melchardt T, Hufnagl C, Weiss L, Mittermair C, Neureiter D, et al. The influence of FCGR2A and FCGR3A polymorphisms on the survival of patients with recurrent or metastatic squamous cell head and neck cancer treated with cetuximab. Pharmacogenomics J. (2018) 18:474–9. doi: 10.1038/tpj.2017.37
186. Sakai H, Tanaka Y, Tazawa H, Shimizu S, Verma S, Ohira M, et al. Effect of Fc-gamma receptor polymorphism on rituximab-mediated B cell depletion in ABO-incompatible adult living donor liver transplantation. Transplant Direct. (2017) 3:e164. doi: 10.1097/TXD.0000000000000683
187. Treffers LW, Zhao XW, van der Heijden J, Nagelkerke SQ, van Rees DJ, Gonzalez P, et al. Genetic variation of human neutrophil Fcgamma receptors and SIRPalpha in antibody-dependent cellular cytotoxicity towards cancer cells. Eur J Immunol. (2017) 8:344–54. doi: 10.1002/eji.201747215
188. Mellor JD, Brown MP, Irving HR, Zalcberg JR, Dobrovic A. A critical review of the role of Fc gamma receptor polymorphisms in the response to monoclonal antibodies in cancer. J Hematol Oncol. (2013) 6:1. doi: 10.1186/1756-8722-6-1
189. Mahaweni NM, Olieslagers TI, Rivas IO, Molenbroeck SJJ, Groeneweg M, Bos GMJ, et al. A comprehensive overview of FCGR3A gene variability by full-length gene sequencing including the identification of V158F polymorphism. Sci Rep. (2018) 8:15983. doi: 10.1038/s41598-018-34258-1
Keywords: Fc gamma receptor (FcγR), genetic variation, autoinflammatory and autoimmune diseases, immunotherapy, mechanisms of disease
Citation: Nagelkerke SQ, Schmidt DE, de Haas M and Kuijpers TW (2019) Genetic Variation in Low-To-Medium-Affinity Fcγ Receptors: Functional Consequences, Disease Associations, and Opportunities for Personalized Medicine. Front. Immunol. 10:2237. doi: 10.3389/fimmu.2019.02237
Received: 19 July 2019; Accepted: 04 September 2019;
Published: 03 October 2019.
Edited by:
Latha P. Ganesan, Wexner Medical Center, The Ohio State University, United StatesReviewed by:
Jeanette Leusen, University Medical Center Utrecht, NetherlandsRalf J. Ludwig, Universität zu Lübeck, Germany
Edward Hollox, University of Leicester, United Kingdom
Copyright © 2019 Nagelkerke, Schmidt, de Haas and Kuijpers. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Sietse Q. Nagelkerke, s.nagelkerke@sanquin.nl