 Franziska Bayer
Franziska Bayer Stefanie Ascher
Stefanie Ascher Giulia Pontarollo
Giulia Pontarollo Christoph Reinhardt
Christoph Reinhardt- 1Center for Thrombosis and Hemostasis (CTH), University Medical Center Mainz, Johannes Gutenberg University Mainz, Mainz, Germany
- 2German Center for Cardiovascular Research (DZHK), Partner Site RheinMain, Mainz, Germany
The gut microbiota influence host vascular physiology locally in the intestine, but also evoke remote effects that impact distant organ functions. Amongst others, the microbiota affect intestinal vascular remodeling, lymphatic development, cardiac output and vascular function, myelopoiesis, prothrombotic platelet function, and immunovigilance of the host. Experimentally, host-microbiota interactions are investigated by working with animals devoid of symbiotic bacteria, i.e., by the decimation of gut commensals by antibiotic administration, or by taking advantage of germ-free mouse isolator technology. Remarkably, some of the vascular effects that were unraveled following antibiotic treatment were not observed in the germ-free animal models and vice versa. In this review, we will dissect the manifold influences that antibiotics have on the cardiovascular system and their effects on thromboinflammation.
Introduction
During the past decade, microbiome research has started to explore how the densely colonized gut resident ecosystem (microbiota) affects the host's vascular physiology (1, 2). This symbiotic microbial community, whose composition is highly dependent on nutrition, interferes with host metabolism and constitutes a chronic inflammatory stimulus. Nowadays, the various influences of the gut microbiome on vascular inflammatory phenotypes, such as atherosclerosis, myocardial infarction, arterial thrombosis, and stroke, are being increasingly recognized (3–6). Moreover, recent research with germ-free (GF) mouse isolator technology revealed a reduced tendency of arterial thrombus formation in different carotid artery mouse thrombosis models, arguing for a contribution of the gut microbiota to thromboinflammation (7–9).
Indeed, mounting evidence is linking the gut microbiota to the onset of cardiovascular disease and arterial thrombosis (10–12). Among microbial-associated molecular patterns (MAMPs), the microbiota-derived choline-metabolite trimethylamine (TMA) that is produced by gut bacterial TMA-lyases (cutC) and targets the liver, is just emerging as a risk factor for thrombotic manifestations (8, 13–15). Through the action of flavin-containing monoxygenase-3 (FMO3), TMA is oxidized into trimethylamine N-oxide (TMAO) (16), a metabolite that was reported to relieve agonist-induced platelet activation (7). Remarkably, both elevated TMAO plasma levels and the activation of Toll-like receptor (TLR) signaling by MAMPs were shown to accelerate atherogenesis in the apolipoprotein E (Apoe)-deficient mice, which are currently used as an animal model of atherosclerosis (17, 18). Clearly, the influences of the microbiota on atherosclerosis are not limited to the TMAO pathway and pattern-recognition receptors, as additional microbiota-derived factors, such as short chain fatty acids (SCFA), were recently identified (19).
Intriguingly, current research, investigating the impact of the gut microbiota on vascular phenotypes, has revealed discrepancies between broad-spectrum antibiotic treatment protocols and investigations on GF mouse models. To give an example, antibiotic treatment of (Apoe)-deficient mice starting at weaning (4 weeks of age) resulted in a reduced development of aortic root lesions at 20 weeks of age, when the mice were fed with a 1.0% choline-rich chow diet (17). Likewise, Kasahara et al. reported reduced aortic root plaque areas together with reduced macrophage and CD4+ cell infiltration of the aortic sinus, when studying GF Apoe-deficient mice kept on an irradiated chow diet and analyzed at 20 weeks of age (20). To complicate the picture, the aortic root lesion size of GF Apoe-deficient mice kept for 12 weeks on 1.2% choline-rich chow diet starting at the age of 8 weeks was not different compared to conventionally raised (CONV-R) control mice (21). In the study conducted by Wright et al. no differences in aortic root lesion size was reported when GF and CONV-R Apoe-deficient mice were fed with a Western diet at weaning until 22 weeks of age (22). Finally, similar to the study of Lindskog Jonsson et al., Stepankova et al. reported increased atherosclerotic lesion sizes in the thoracic aortas of GF Apoe-deficient mice after feeding with a low cholesterol diet for 3–4 months (3, 21). These seemingly controversial results could be explained by apparently minor differences in the experimental protocols, e.g., the mouse line studied, the various diets used, the feeding time scheme applied, or the normalization of the measured lesion sizes. On the other hand, it is becoming increasingly clear that the interpretation of data collected after microbiota decimation experiments by antibiotics should be considered with caution, given the experimental variables and the several side effects of antibiotic treatment protocols. It should also be kept in mind that antibiotic decimation of the microbiota using broad-spectrum antibiotics represents a selective pressure that favors overgrowth of resistant bacterial taxa (23), which could in principle be causative at least for some of the described outcomes.
Moreover, recent research has implicated the intestinal microbiota in arterial thrombosis and it has been proposed that selective inhibition of the TMA-generating gut microbial enzymes could lower thrombotic risk (24). While TMAO has been shown to facilitate agonist-induced platelet activation (7), it was demonstrated that microbiota-derived TLR-ligands promote the activation of hepatic endothelial cells, triggering von Willebrand factor (VWF) synthesis and release (8, 15, 25). In mouse models, both pathways promote carotid artery thrombosis and thrombus growth was reduced in the GF mouse model, linking the gut microbiota mechanistically to arterial thrombosis (7, 8, 26). In the literature, in addition to atherosclerotic phenotypes, there are numerous other examples on microbiota-dependent vascular phenotypes that differ between gnotobiotic mouse models and depletion of the gut microbiota with antibiotics.
Here, we provide an overview on GF mouse isolator technology and antibiotic treatment protocols for microbiota depletion that are widely used to study vascular phenotypes. We will explain the limitations of these mouse models, but also describe the recently gained insights on microbiota-driven influences, affecting vascular physiology, cardiovascular disease development and thromboinflammation.
Differences and Similarities of Germ-Free and Antibiotic-Treated Mice
In 1885, Louis Pasteur claimed that a life without microbial associates is not possible (27). Several years later, in the 1940s, the first colonies of GF rodents were established. Since its beginning, the usage of this technology became a valuable model to understand how the microbiota impacts host physiology and disease processes (28). Gnotobiotic animals such as mice colonized with Altered Schaedler Flora (a defined bacterial community) or germ-free (axenic) mice, lacking all microorganisms, are animal models characterized by a defined colonization status (29). In particular, GF mice are bred and kept for their whole lifetime in sterile isolators to prevent their exposure to microorganisms. These animals are a biological model system to either study the complete absence of microbes, or to investigate the effects of colonization with selected and known microbial species (e.g., in the mono-association experiment) (30). However, the GF mouse model is a labor-intensive technology, which requires permanent controls for the hygiene status of the isolators and special facilities (30, 31). As an alternative and more basic method for the depletion of microbiota, administration of broad-spectrum antibiotics is commonly used. In contrast to GF mice, antibiotic application does not lead to the depletion of all microbes, but can selectively deplete different members of the gut microbiota (32). Furthermore, to prevent dehydration during treatment, Reikvam et al. recommended to gavage mice instead of delivering the antibiotics in the drinking water, or to combine both applications (33).
Importantly, decimation of the microbiota affects the anatomy and function of various organs such as liver and gut (34, 35) (summarized in Figure 1). One of the most evident anatomical changes is the enlarged cecum, observed both in GF and antibiotic-treated mice (30, 33). Furthermore, GF mice present elongated villus structures, a reduced villus width, and poorly developed capillary networks in small intestinal villi (1, 36, 37). In addition to altered organ morphology and physiology, immune cell populations are influenced by antibiotic treatment (38–40). Because the GF animal model and antibiotic decimation of the microbiota may result in different vascular phenotypes with respect to anatomy and function (8, 17, 41, 42), it is important to be aware of the limitations of these experimental models that enable the exploration of microbiota-host interactions.
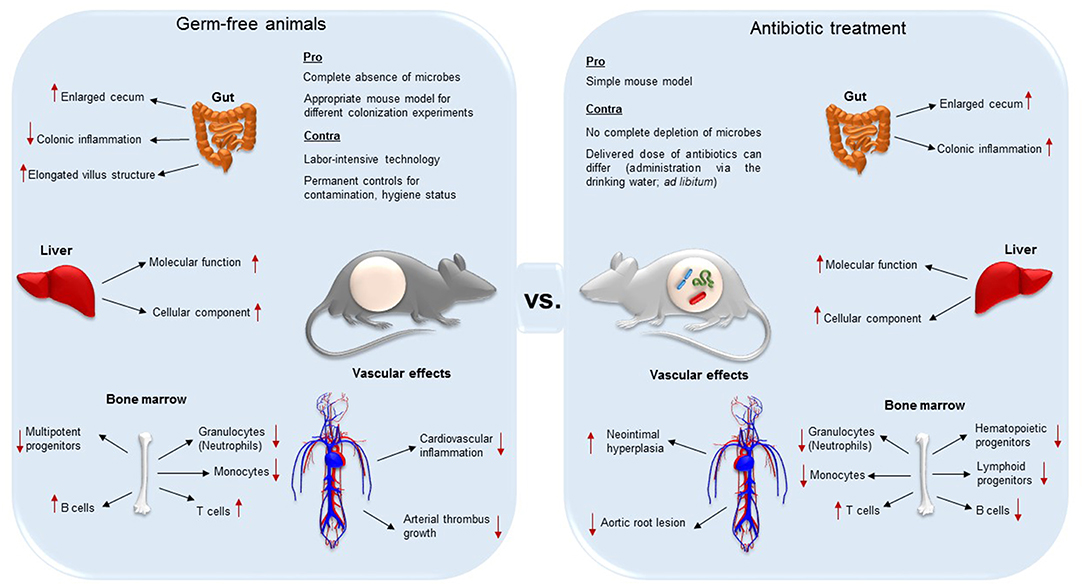
Figure 1. Differences and similarities of germ-free (axenic) and broad-spectrum antibiotic-treated mice (decimation of microbes). While both techniques result in the aberrant enlargement of the cecum, GF mice are less susceptible for colonic inflammation, and present elongated villus structures compared to antibiotic treated animals. In both the animal models, absence of gut microbiota alters protein expression levels in the liver. In the bone marrow, the two mouse models present both reduced granulocytes, monocytes and progenitor cells, but higher T cell levels. On the other hand, while B cells in GF animals are increased, in antibiotic-treated counterparts they were reported to be decreased. Both the complete absence and the decimation of the gut microbiota influences vascular physiology and have an effect on vascular disease. Neointimal hyperplasia, a proliferation and inflammation response to arterial injury, was increased in antibiotic treated rats. Antibiotic treatment leads to a diminished development of aortic root lesion. Additionally, germ-free mice were protected from cardiac inflammation and arterial thrombus growth.
Antibiotics and Their Influence on Myeloid Cell Function
In addition to the resulting selection of resistant bacterial taxa (23), antibiotics severely influence the development of the myeloid cell lineage (40), which may greatly hamper the interpretation of results on vascular physiology. It is firmly established that long-term treatment with beta-lactam antibiotics results in an inhibition of granulopoiesis (43). In this clinical study, the presence of granulocyte precursors with the lack of well-differentiated myeloid cells was observed in human bone marrow aspirates, discussed by the authors as a myelotoxic effect of beta-lactam antibiotics. Most important, in bone marrow cultures, Neftel et al. described a direct inhibitory effect of beta-lactam antibiotics on in vitro granulopoiesis. In addition, thrombocytopenia caused by beta-lactam antibiotic treatment was remarked. Likewise, reduced neutrophil counts were reported in the bone marrow of long-term antibiotics treated mice, which was explained by neutrophil aging promoted by MAMPs derived from the gut microbiota (40).
In contrast, Hergott et al. demonstrated that antibiotic treatment accelerates the turnover of circulating neutrophils and inflammatory monocytes, which showed decreased cell counts in the blood, the bone marrow and the spleen (44). In this work, it was described that long-term antibiotic treatment (i.e. 3–4 weeks) with the frequently used cocktail of neomycin, vancomycin, metronidazole, and ampicillin for microbiota decimation was associated with reduced survival of neutrophils and inflammatory monocytes, which was confirmed in the GF mouse model. This phenotype was explained by microbiota-induced sensing of peptidoglycan of nucleotide-binding oligomerization domain-containing protein-1 (NOD1), and by the production of interleukin-17A (IL-17A) in the ileum (44). The requirement of NOD1 sensing in mesenchymal stromal cells in the reduction of immune cells survival was confirmed in a subsequent study with GF mice, showing that these models presented an overall reduced numbers of hematopoietic stem and multipotent progenitor cells (39). By using the same antibiotic cocktail over 14 days and an alternative antibiotic cocktail containing ciprofloxacin and metronidazole, Josefsdottir et al. demonstrated that not only myeloid cell numbers were dramatically reduced, but also the peripheral blood counts of various lymphocyte subsets along with the counts of hematopoietic progenitors were diminished (B-cells, CD4+ T-cells, CD8+ T-cells) (38). Interestingly, it was demonstrated that the cell cycle activity in hematopoietic stem cells and myeloid progenitors was increased in the antibiotic treated group, excluding a direct myelotoxic effect of the antibiotics used by murine bone marrow culture experiments. Of note, there was no antibiotic effect on the hematopoietic progenitor population and on granulocytes in signal transducer and activator of transcription 1 (Stat1)-deficient mice, indicating that upstream pathways of this transcription factor may play a critical role.
While the obvious question for the fundamental differences between antibiotic treatment and GF housing state with regard to hematopoietic functions is apparent, these important studies clearly demonstrate that antibiotic intervention has manifold effects on innate immune cell types, which may lead to inconclusive mechanistic data in hematologic research. Therefore, gnotobiotic models based on GF knock-out mouse strains are essential to control for the validity of results obtained by antibiotic treatment protocols.
Antibiotics and Their Influence on Clotting, Platelet Function and the Vasculature
Antibiotic Treatment Interferes With Vitamin K Metabolism and Clotting Factors
The lipophilic essential vitamin K has two sources: it can be found in green leafed plants as phylloquinone (vitamin K1) and as the microbial metabolite menaquinone (vitamin K2), which is produced by several bacterial species (45–48). Synthesis of clotting factors II, VII, IX, and X and their post-translational modifications in the liver are vitamin K-dependent (45, 46). The correlation between antibiotic treatment and increased bleeding risk due to vitamin K deficiency was already described in 1952 by Dam et al. (47). Over the years, more case reports of hypoprothrombinemic bleeding were published, involving several classes of antibiotics (46, 48–50).
In this context, since 1980s, N-methylthiotetrazole (NMTT) cephalosporins (for example cefoperazone, cefotetan, moxalactam, and cefamandole) administration has been frequently reported to result in impaired hemostasis (45, 46, 50–52). Here, several mechanisms and speculations are discussed in the literature, including: (a), interference of NMTT-side chain with vitamin K metabolism (45), (b), indirect inhibition of vitamin K-dependent blood coagulation (50, 53), and (c), reduction of menaquinone producing bacteria (50, 53). A more recent publication by Fotouhie et al. supports in part hypothesis (c), stating that several risk factors have to concur for the appearance of clinical symptoms (52). The following risk factors were identified: insufficient dietary intake of phylloquinone, modification of normal gut microbial communities via antibiotics, malabsorption of vitamin K, or chronic liver disease (48). Hence, antibiotic treatment protocols may interfere with the synthesis of vitamin K-dependent blood clotting factors.
Antibiotic Treatment and Platelets
Other reasons for impaired hemostasis and thrombus formation following antibiotics intake are thrombocytopenia or impaired platelet function. In the early 1970s, the antibiotic ristocetin got withdrawn from clinical use because it induced platelet aggregation (54). As a result, many research groups then replaced ristocetin administration with vancomycin, as the two molecules share many chemical properties. However, even if vancomycin itself is not able to directly trigger platelet aggregation, it was reported to induce precipitates in which platelets are incorporated (55, 56). A more recent publication revealed the underlying mechanism for vancomycin-induced thrombocytopenia. Towhid et al. described platelet apoptosis paralleled by mitochondria depolarization, activation of caspase-3, cell membrane scrambling and ceramide formation. Further tests revealed that Ca2+ is necessary for vancomycin to cause these effects. They hypothesized that the cell scrambling induces an accelerated clearance of platelets from the blood, resulting in the observed thrombocytopenia. Importantly, all the described side effects are induced by exploiting vancomycin concentrations normally reached during standard protocols of antibiotic treatments (57).
Penicillin antibiotics may also have an effect on platelets. Clinically relevant concentrations of penicillin G and carbenicillin seem to have a global effect on platelet membrane receptors, as platelets become less responsive to physiologic agonists and fail to aggregate with bovine factor VIIIa stimulation (45). Subsequently, Pastakia et al. have shown that penicillin G inhibits thrombin-induced upregulation of GPIb-IX levels on the platelet surface (58, 59). In contrast, other beta-lactam antibiotics (ceftriaxone, ceftazidime, and aztreonam) did not inhibit platelet aggregation (60). On the contrary, ceftriaxone (and to a lesser extent aztreonam) was shown to enhance platelet aggregation, but the underlying molecular mechanisms remain unclear. Another antibiotic presenting inhibitory effect on in vitro platelet aggregation, even after oral treatment, is metronidazole (61).
The underlying mechanisms for thrombocytopenia (i.e., a circulating platelet count inferior to 150,000/mm3) are increased platelet consumption/destruction or reduced platelet production. Linezolid, an antibiotic commonly used to treat vancomycin-resistant enterococci and methicillin-resistant Staphylococcus aureus infections, is known to induce myelosuppression, but is more frequently associated with thrombo- than pancytopenia (62). It was described that linezolid has no direct toxic effects on platelets and it does not affect the differentiation of hematopoietic stem cells, but via phosphorylation of an enzyme relevant for platelet release, it induces thrombocytopenia (62). Although some influences of antibiotics on platelet functions were identified, the information on effects of antibiotic treatment on the coagulation system remains sparse.
Antibiotic Treatment and Vasomodulatory Effects
Since the 1950s, aminoglycoside antibiotics are known to have hypotensive effects and a negative inotropic effect on the heart (63). Gentamicin was reported to cause hypotension resulting from vasodilatation and relaxation of smooth muscle cells, thus yielding reduced vascular resistance, together with decreased cardiac contraction force and bradycardia. The proposed mechanism is an impaired Ca2+ influx. Neomycin, gentamicin and, to a lesser extent, streptomycin, and kanamycin were demonstrated to have vasorelaxant effects on the cerebral arteries of dogs (64). All four antibiotics inhibited vasoconstriction after administering depolarizing concentrations of potassium chloride (64). Two mechanisms were proposed: (a), the inhibition of phospholipase C (an enzyme that catalyzes the production of the second messenger molecule inositol trisphosphate, which drives the release of Ca2+ from the sarcoplasmatic reticulum) and (b), direct interference with Ca2+ influx by blocking L-type voltage-dependent Ca2+ channels (64). Belus et al. revealed the mechanism for the aminoglycoside antibiotics neomycin, gentamicin, and streptomycin negative inotropic effect. The resting and transient intracellular Ca2+ levels of rat ventricular myocytes are decreased, leading to reduced contractility (65). Furthermore, high dosages of vancomycin (glycopeptide antibiotic) and tobramycin (aminoglycoside antibiotic) have relaxing effects on the vascular smooth muscles (66). Not only aminoglycoside antibiotics, but also beta-lactam antibiotics seem to have an influence on Ca2+-influx in the vascular endothelium (60). This indicates that vascular function is significantly affected by the administration of several different antibiotics.
Conclusions
GF mouse models and antibiotic treatment protocols are frequently used as comparable methods to investigate the effect of gut microbiota in triggering inflammatory vascular phenotypes. In times of spreading antimicrobial resistance, every unnecessary use of antibiotics should be carefully weighed up with the expected findings (67). As pointed out in this review, antibiotic treatment results in significant changes of host physiology, but the underlying mechanisms remain often unclear. Since no standardized antibiotic treatment regimen exists (68) and residual surviving microbiota differs dependent on the animal facility and housing conditions, the reproducibility of this experimental procedure is at stake (69, 70). Therefore, all the functional studies on the gut microbiota exploiting antibiotic treatment should at least mention the limitations of this technique and point out what kind of controls were included. In contrast, the GF mouse model remains the state-of-the-art approach for studying host-microbe and microbe-microbe interactions, since mono-colonization, minimal microbial consortia or humanized microbial consortia are standardized experimental approaches (71). Therefore, if GF mouse technology or gnotobiotic animal models are available and applicable, the use of antibiotic treatment should certainly be reconsidered.
Author Contributions
All authors listed have made a substantial, direct and intellectual contribution to the work, and approved it for publication.
Funding
This work was supported by the BMBF (01EO1503), by a DFG Individual Grant to CR (RE 3450/5-2), a project grant from the Boehringer Ingelheim Stiftung (Novel and neglected cardiovascular risk factors) to CR, and by an intramural Stufe 1 project grant (Inneruniversitäre Forschungsförderung).
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Reinhardt C, Bergentall M, Greiner TU, Schaffner F, Ostergren-Lundén G, Petersen LC, et al. Tissue factor and PAR1 promote microbiota-induced intestinal vascular remodelling. Nature. (2012) 483:627–31. doi: 10.1038/nature10893
2. Howitt MR, Garrett WS. A complex microworld in the gut: gut microbiota and cardiovascular disease connectivity. Nat Med. (2012) 18:1188–9. doi: 10.1038/nm.2895
3. Stepankova R, Tonar Z, Bartova J, Nedorost L, Rossman P, Ploedne R, et al. Absence of microbiota (germ-free conditions) accelerates the atherosclerosis in ApoE-deficient mice fed standard low cholesterol diet. J Atheroscler Thromb. (2010) 17:796–804. doi: 10.5551/jat.3285
4. Martínez I, Wallace G, Zhang C, Legge R, Benson AK, Carr TP, et al. Diet-induced metabolic improvements in a hamster model of hypercholesterolemia are strongly linked to alterations of the gut microbiota. Appl Environ Microbiol. (2009) 75:4175–84. doi: 10.1128/AEM.00380-09
5. Caesar R, Fåk F, Bäckhed F. Effects of gut microbiota on obesity and atherosclerosis via modulation of inflammation and lipid metabolism. J Intern Med. (2010) 268:320–8. doi: 10.1111/j.1365-2796.2010.02270.x
6. Benakis C, Brea D, Caballero S, Faraco G, Moore J, Murphy M, et al. Commensal microbiota affects ischemic stroke outcome by regulating intestinal γδ T cells. Nat Med. (2016) 22:516–23. doi: 10.1038/nm.4068
7. Zhu W, Gregory JC, Org E, Buffa JA, Gupta N, Wang Z, et al. Gut microbial metabolite TMAO enhances platelet hyperreactivity and thrombosis risk. Cell. (2016) 165:111–24. doi: 10.1016/j.cell.2016.02.011
8. Jäckel S, Kiouptsi K, Lillich M, Hendrikx T, Khandagale A, Kollar B, et al. Gut microbiota regulate hepatic von Willebrand factor synthesis and arterial thrombus formation via Toll-like receptor-2. Blood. (2017) 130:542–53. doi: 10.1182/blood-2016-11-754416
9. Skye SM, Zhu W, Romano KA, Guo CJ, Wang Z, Jia X, et al. Microbial transplantation with human gut commensals containing cutC is sufficient to transmit enhanced platelet reactivity and thrombosis potential. Circ Res. (2018) 123:1164–76. doi: 10.1161/CIRCRESAHA.118.313142
10. Ascher S, Reinhardt C. The gut microbiota: an emerging risk factor for cardiovascular and cerebrovascular disease. Eur J Immunol. (2018) 48:564–75. doi: 10.1002/eji.201646879
11. Kiouptsi K, Reinhardt C. Contribution of the commensal microbiota to atherosclerosis and arterial thrombosis. Br J Pharmacol. (2018) 175:4439–49. doi: 10.1111/bph.14483
12. Reinhardt C. The gut microbiota as an influencing factor of arterial thrombosis. Hämostaseologie. (2018) 39: 173–9. doi: 10.1055/s-0038-1675357
13. Rath S, Heidrich B, Pieper DH, Vital M. Uncovering the trimethylamine-producing bacteria oft he human gut microbiota. Microbiome. (2017) 5:54. doi: 10.1186/s40168-017-0271-9
14. Smith JL, Wishnok JS, Deen WM. Metabolism and excretion of methylamines in rats. Toxicol Appl Pharmacol. (1994) 125:296–308. doi: 10.1006/taap.1994.1076
15. Carnevale R, Raparelli V, Nocella C, Bartimoccia S, Novo M, Severino A, et al. Gut-derived endotoxin stimulates factor VIII secretion from endothelial cells. Implications for hypercoagulability in cirrhosis. J Hepatol. (2017) 67:950–6. doi: 10.1016/j.jhep.2017.07.002
16. Bennett BJ, de Aguiar Vallim TQ, Wang Z, Shih DM, Meng Y, Gregory J, et al. Trimethylamine-N-oxide, a metabolite associated with atherosclerosis, exhibits complex genetic and dietary regulation. Cell Metab. (2013) 17:49–60. doi: 10.1016/j.cmet.2012.12.011
17. Wang Z, Klipfell E, Bennett BJ, Koeth R, Levison BS, Dugar B, et al. Gut flora metabolism of phosphatidylcholine promotes cardiovascular disease. Nature. (2011) 472:57–63. doi: 10.1038/nature09922
18. Michelsen KS, Wong MH, Shah PK, Zhang W, Yano J, Doherty TM, et al. Lack of Toll-like receptor 4 or myeloid differentiation factor 88 reduces atherosclerosis and alters plaque phenotype in mice deficient in apolipoprotein E. Proc Natl Acad Sci USA. (2004) 101:10679–84. doi: 10.1073/pnas.0403249101
19. Brandsma E, Kloosterhuis NJ, Koster M, Dekker DC, Gijbels MJJ, van der Velden S, et al. A proinflammatory gut microbiota increases systemic inflammation and accelerates atherosclerosis. Circ Res. (2019) 124:94–100. doi: 10.1161/CIRCRESAHA.118.313234
20. Kasahara K, Tanoue T, Yamashita T, Yodoi K, Matsumoto T, Emoto T, et al. Commensal bacteria at the crossroad between cholesterol homeostasis and chronic inflammation in atherosclerosis. J Lipid Res. (2017) 58:519–28. doi: 10.1194/jlr.M072165
21. Lindskog Jonsson A, Caesar R, Akrami R, Reinhardt C, Fåk Hållenius F, Borén J, et al. Impact of gut microbiota and diet on the development of atherosclerosis in Apoe−/− mice. Arterioscler Thromb Vasc Biol. (2018) 38:2318–26. doi: 10.1161/ATVBAHA.118.311233
22. Wright SD, Burton C, Hernandez M, Hassing H, Montenegro J, Mundt S, et al. Infectious agents are not necessary for murine atherosclerosis. J Exp Med. (2000) 191:1437–42. doi: 10.1084/jem.191.8.1437
23. Ubeda C, Taur Y, Jenq RR, Equinda MJ, Son T, Samstein M, et al. Vancomycin-resistant Enterococcus domination in intestinal microibota is enabled by antibiotic treatment in mice and precedes bloodstream invasion in humans. J Clin Invest. (2010) 120:4332–41. doi: 10.1172/JCI43918
24. Roberts AB, Gu X, Buffa JA, Hurd AG, Wang Z, Zhu W, et al. Development of a gut microbe-targeted nonlethal therapeutic to inhibit thrombosis potential. Nat Med. (2018) 24:1407–17. doi: 10.1038/s41591-018-0128-1
25. Into T, Kanno Y, Dohkan J, Nakashima M, Inomata M, Shibata K, et al. Pathogen recognition by Toll-like receptor 2 activates Weibel-Palade body exocytosis in human aortic endothelial cells. J Biol Chem. (2007) 282:8134–41. doi: 10.1074/jbc.M609962200
26. Zhu W, Buffa JA, Wang Z, Warrier M, Schugar R, Shih DM, et al. Flavin monooxygenase 3, the host hepatic enzyme in the metaorganismal trimethylamine N-oxide-generating pathway, modulates platelet responsiveness and thrombosis risk. J Thormb Haemost. (2018) 16:1857–72. doi: 10.1111/jth.14234
27. Gordon HA, Pesti L. The gnotobiotic animal as a tool in the study of host microbial relationships. Bacteriol Rev. (1971) 35:390–429.
28. Yi P, Li L. The germfree murine animal: an important animal model for research on the relationship between gut microbiota and the host. Vet Microbiol. (2012) 157:1–7. doi: 10.1016/j.vetmic.2011.10.024
29. Brand MW, Wannemuehler MJ, Phillips GJ, Proctor A, Overstreet A-M, Jergens AE, et al. The altered Schaedler Flora: continued application of a defined murine microbial community. ILAR J. (2015) 56:169–78. doi: 10.1093/ilar/ilv012
30. Nicklas W, Keubler L, Bleich A. Maintaining and monitoring the defined microbiota status of gnotobiotic rodents. ILAR J. (2015) 56:241–9. doi: 10.1093/ilar/ilv029
31. Fontaine CA, Skorupski AM, Vowles CJ, Anderson NE, Poe AS, Eaton KA. How free of germs is germ-free? Detection of bacterial contamination in a germ free mouse unit. Gut Microbes. (2015) 6:225–33. doi: 10.1080/19490976.2015.1054596
32. Schubert AM, Sinani H, Schloss PD. Antibiotic-induced alteration of the gut murine microbiota and subsequent effects on colonization resistance against clostridium difficile. MBio. (2015) 6:e00974–15. doi: 10.1128/mBio.00974-15
33. Reikvam DH, Erofeev A, Sandvik A, Grcic V, Jahnsen FL, Gaustad P, et al. Depletion of murine intestinal microbiota: effects on gut mucosa and epithelial gene expression. PLoS ONE. (2011) 6:e17996. doi: 10.1371/journal.pone.0017996
34. Hernández-Chirlaque C, Aranda CJ, Ocón B, Capitan-Cañadas F, Ortega-González M, Carrero JJ, et al. Germ-free and antibiotic-treated mice are highly susceptible to epithelial injury in DSS colitis. J Crohns Colitis. (2016) 10:1324–35. doi: 10.1093/ecco-jcc/jjw096
35. Kuno T, Hirayama-Kurogi M, Ito S, Ohtsuki S. Effect of the intestinal flora on protein expression of drug-metabolizing enzymes and transporters in the liver and kidney of germ-free and antibiotics-treated mice. Mol Pharm. (2016) 13:2691–701. doi: 10.1021/acs.molpharmaceut.6b00259
36. Sommer F, Bäckhed F. The gut microbiota–masters of host development and physiology. Nat Rev Microbiol. (2013) 11:227–38. doi: 10.1038/nrmicro2974
37. Abrams GD, Bauer H, Sprinz H. Influence of the normal flora on mucosal morphology and cellular renewal in the ileum. A comparison of germ-free and conventional mice. Lab Invest. (1963) 12:355–64.
38. Josefsdottir KS, Baldridge MT, Kadmon CS, King KY. Antibiotics impair murine hematopoiesis by depleting the intestinal microbiota. Blood. (2017) 129:729–39. doi: 10.1182/blood-2016-03-708594
39. Iwamura C, Bouladoux N, Belkaid Y, Sher A, Jankovic D. Sensing of the microbiota by NOD1 in mesenchymal stromal cells regulates murine hematopoiesis. Blood. (2017) 129:171–6. doi: 10.1182/blood-2016-06-723742
40. Zhang D, Chen G, Manwani D, Mortha A, Xu C, Faith JJ, et al. Neutrophil ageing is regulated by the microbiome. Nature. (2015) 525:528–32. doi: 10.1038/nature15367
41. Ho KJ, Xiong L, Hubert NJ, Nadimpalli A, Wun K, Chang EB, et al. Vancomycin treatment and butyrate supplementation modulate gut microbe composition and severity of neointimal hyperplasia after arterial injury. Physiol Rep. (2015) 3:e12627. doi: 10.14814/phy2.12627
42. Karbach SH, Schönfelder T, Brandão I, Wilms E, Hörmann N, Jäckel S, et al. Gut microbiota promote angiotensin II-induced arterial hypertension and vascular dysfunction. J Am Heart Assoc. (2016) 5:e003698. doi: 10.1161/JAHA.116.003698
43. Neftel KA, Hauser SP, Müller MR. Inhibition of granulopoiesis in vivo and in vitro by β-lactam antibiotics. J Infect Dis. (1985) 152:90–8. doi: 10.1093/infdis/152.1.90
44. Hergott CB, Roche AM, Tamashiro E, Clarke TB, Bailey AG, Laughlin A, et al. Peptidoglycan from the gut microbiota governs the lifespan of circulating phagocytes at homeostasis. Blood. (2016) 127:2460–71. doi: 10.1182/blood-2015-10-675173
45. Sattler F, Weitekamp MA, Seyegh A, Ballard JO. Impaired hemostasis caused by beta-lactam antibiotics. Am J Surg. (1988) 155:30–9. doi: 10.1016/S0002-9610(88)80209-5
46. Shevchuk YM, Conly JM. Antibiotic-associated hypoprothrombinemia: a review of prospective studies, 1966-1988. Rev Infect Dis. (1990) 12:1109–26. doi: 10.1093/clinids/12.6.1109
47. Dam H, Dyggve H, Larsen H, Plum P. The relation of vitamin K deficiency to hemorrhagic disease of the newborn. Adv Pediatr. (1952) 5:129–53.
48. Goldman HI, Deposito F. Hypoprothrombinemic bleeding in young infants. Association with diarrhea, antibiotics, and milk substitutes. Am J Dis Child. (1966) 111:430–2. doi: 10.1001/archpedi.1966.02090070128021
49. Pineo GF, Gallus AS, Hirsh J. Unexpected vitamin K deficiency in hospitalized patients. Can Med Assoc J. (1973) 109:880–3.
50. Nomoto H, Hatta K, Usui C, Ito M, Kita Y, Arai H. Vitamin K deficiency due to prolongation of antibiotic treatment and decrease in food intake in a catatonia patient. Psychosomatics. (2011) 52:486–7. doi: 10.1016/j.psym.2011.01.026
51. Cai Z, Yang W, He Y, Chen Q, Wang S, Luo X, et al. Cefoperazone/sulbactam-induced abdominal wall hematoma and upper gastrointestinal bleeding: a case report and review of the literature. Drug Saf Case Rep. (2016) 3:2. doi: 10.1007/s40800-016-0025-9
52. Fotouhie A, Desai H, Parsa NA, King S. Gastrointestinal bleeding secondary to trimethoprim-sulfamethoxazole-induced vitamin K deficiency. BMJ Case Rep. (2016) 2016:bcr2016214437. doi: 10.1136/bcr-2016-214437
53. Schentag JJ, Welage LS, Grasela TH, Adelman MH. Determinants of antibiotic-associated hypoprothrombinemia. Pharmacotherapy. (1987) 7:80–6. doi: 10.1002/j.1875-9114.1987.tb03522.x
54. Perkins HR. Vancomycin and related antibiotics. Pharmacol Ther. (1982) 16:181–97. doi: 10.1016/0163-7258(82)90053-5
55. Coller BS, Lundberg WB, Gralnick HR. Effects of vancomycin on platelets, plasma proteins and hepatitis B surface antigen. Thromb Diath Haemorrh. (1975) 34:83–93. doi: 10.1055/s-0038-1651448
56. Coller BS, Gralnick HR. Studies on the mechanism of ristocetin-induced platelet agglutination. Effects of structural modification of ristocetin and vancomycin. J Clin Invest. (1977) 60:302–12. doi: 10.1172/JCI108778
57. Towhid ST, Schmidt EM, Tolios A, Münzer P, Schmid E, Borst O, et al. Stimulation of platelet death by vancomycin. Cell Physiol Biochem. (2013) 31:102–12. doi: 10.1159/000343353
58. Pastakia KB, Terle D, Prodouz KN. Penicillin inhibits agonist-induced expression of platelet surface membrane glycoproteins. Ann N Y Acad Sci. (1993) 677:437–9. doi: 10.1111/j.1749-6632.1993.tb38810.x
59. Pastakia KB, Terle D, Prodouz KN. Penicillin-induced dysfunction of platelet membrane glycoproteins. J Lab Clin Med. (1993) 121:546–54.
60. Togna GI, Togna AR, Caprino L. Beta-lactam antibiotic-mediated changes in platelet reactivity and vascular endothelial functions. Pharmacol Toxicol. (2001) 88:277–81. doi: 10.1034/j.1600-0773.2001.d01-117.x
61. Prado-Franceschi J, Antunes E. Effect of metronidazole on platelet aggregation. Braz J Med Biol Res. (1989) 22:1497–500.
62. Tajima M, Kato Y, Matsumoto J, Hirosawa I, Suzuki M, Takashio Y, et al. Linezolid-induced thrombocytopenia is caused by suppression of platelet production via phosphorylation of myosin light chain 2. Biol Pharm Bull. (2016) 39:1846–51. doi: 10.1248/bpb.b16-00427
63. Gotanda K, Yanagisawa T, Satoh K, Taira N. Are the cardiovascular effects of gentamicin similar to tose of calcium antagonists? Jpn J Pharmacol. (1988) 47:217–27. doi: 10.1254/jjp.47.217
64. Gergawy M, Vollrath B, Cook D. The mechanism by which aminoglycoside antibiotics cause vasodilation of canine cerebral arteries. Br J Pharmacol. (1998) 125:1150–7. doi: 10.1038/sj.bjp.0702180
65. Belus A, White E. Effects of antibiotics on the contractility and Ca2+ transients of rat cardiac myocytes. Eur J Pharmacol. (2001) 412:121–6. doi: 10.1016/S0014-2999(01)00717-8
66. Richter J, Zhou J, Pavlovic D, Scheibe R, Bac VH, Blumenthal J, et al. Vancomycin and to lesser extent tobramycin have vasomodulatory effects in experimental endotoxemia in the rat. Clin Hemorheol Microcirc. (2010) 46:37–49. doi: 10.3233/CH-2010-1331
67. ECDC. Surveillance of Antimicrobial Resistance in Europe – Annual Report of the European Antimicrobial Resistance Surveillance Network (EARS-Net) 2017. EU publications (2018).
68. Kennedy EA, King KY, Baldridge MT. Mouse microbiota models: comparing germ-free mice and antibiotics treatment as tools for modifying gut bacteria. Front Physiol. (2018) 9:1534. doi: 10.3389/fphys.2018.01534
69. Macpherson AJ, McCoy KD. Standardised animal models of host microbial mutualism. Mucosal Immunol. (2015) 8:476–86. doi: 10.1038/mi.2014.113
70. Tremaroli V, Bäckhed F. Functional interactions between the gut microbiota and host metabolism. Nature. (2012) 489:242–9. doi: 10.1038/nature11552
Keywords: platelets, germ-free mouse models, antibiotics, thrombosis, microbiota, vascular function
Citation: Bayer F, Ascher S, Pontarollo G and Reinhardt C (2019) Antibiotic Treatment Protocols and Germ-Free Mouse Models in Vascular Research. Front. Immunol. 10:2174. doi: 10.3389/fimmu.2019.02174
Received: 03 June 2019; Accepted: 28 August 2019;
Published: 12 September 2019.
Edited by:
Benoît Ho-Tin-Noé, Institut National de la Santé et de la Recherche Médicale (INSERM), FranceReviewed by:
Karen Ho, Northwestern University, United StatesDalil Hannani, UMR5525 Techniques de l'Ingénierie Médicale et de la Complexité Informatique, Mathématiques et Applications, Grenoble (TIMC-IMAG), France
Copyright © 2019 Bayer, Ascher, Pontarollo and Reinhardt. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Christoph Reinhardt, Y2hyaXN0b3BoLnJlaW5oYXJkdCYjeDAwMDQwO3VuaW1lZGl6aW4tbWFpbnouZGU=
†These authors have contributed equally to this work