
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol. , 13 September 2019
Sec. Inflammation
Volume 10 - 2019 | https://doi.org/10.3389/fimmu.2019.02169
This article is part of the Research Topic Ozone as a Driver of Lung Inflammation and Innate Immunity, and as a Model for Lung Disease View all 10 articles
 Milena Sokolowska1,2
Milena Sokolowska1,2 Valerie F. J. Quesniaux3Cezmi A. Akdis1,2
Valerie F. J. Quesniaux3Cezmi A. Akdis1,2 Kian Fan Chung4
Kian Fan Chung4 Bernhard Ryffel3*Dieudonnée Togbe3,5*
Bernhard Ryffel3*Dieudonnée Togbe3,5*Ozone exposure causes irritation, airway hyperreactivity (AHR), inflammation of the airways, and destruction of alveoli (emphysema), the gas exchange area of the lung in human and mice. This review focuses on the acute disruption of the respiratory epithelial barrier in mice. A single high dose ozone exposure (1 ppm for 1 h) causes first a break of the bronchiolar epithelium within 2 h with leak of serum proteins in the broncho-alveolar space, disruption of epithelial tight junctions and cell death, which is followed at 6 h by ROS activation, AHR, myeloid cell recruitment, and remodeling. High ROS levels activate a novel PGAM5 phosphatase dependent cell-death pathway, called oxeiptosis. Bronchiolar cell wall damage and inflammation upon a single ozone exposure are reversible. However, chronic ozone exposure leads to progressive and irreversible loss of alveolar epithelial cells and alveoli with reduced gas exchange space known as emphysema. It is further associated with chronic inflammation and fibrosis of the lung, resembling other environmental pollutants and cigarette smoke in pathogenesis of asthma, and chronic obstructive pulmonary disease (COPD). Here, we review recent data on the mechanisms of ozone induced injury on the different cell types and pathways with a focus on the role of the IL-1 family cytokines and the related IL-33. The relation of chronic ozone exposure induced lung disease with asthma and COPD and the fact that ozone exacerbates asthma and COPD is emphasized.
Human ozone (O3) exposure represents a major health issue (1, 2) playing an important role in the pathogenesis of chronic respiratory diseases such as asthma and chronic obstructive pulmonary disease (COPD). Ozone causes acute epithelial airway wall injury, inflammation, and airway hyperreactivity (AHR). Ozone elicits irritation of the airways with cough, bronchoconstriction, and inflammatory cell infiltration with loss of respiratory function. AHR represents a complex response of the airways to the release of bronchoconstrictive mediators and cholinergic stimulation, and is a hallmark of ozone exposure which is shared with allergic asthma. Furthermore, increased ozone exposure, especially occurring during thunderstorms, provokes severe exacerbations of asthma and may even contribute to the asthma-related deaths (3–7). A recent epidemiologic study revealed that even a short-term exposure to ambient air pollution such as PM2.5, O3, and NO2 significantly increased the risk of asthma mortality (8). Chronic ozone exposure leads to a progressive loss of the gas exchanging alveoli, a phenomenon known as emphysema, usually associated with chronic inflammation, fibrosis, and terminal respiratory failure, observed in patients with chronic obstructive pulmonary disease (COPD) and severe asthma (9). Of note, the pathogenesis of chronic lung diseases is complex and comprises the effects of various environmental particulates, toxins, chemical sand pollutants, detergents, respiratory viruses, microbial dysbiosis as well as allergen exposure, and is influenced by diverse genetic and epigenetic factors (10–15).
The respiratory airway epithelium forms a physical barrier and first line of defense of mucosal immunity (16, 17). Tight junctions (TJ) and adherens junctions (AJ), fluid, mucus, surfactant proteins, and motility of cilia are critical for the barrier control and innate response (18). Ozone impairs the function of critical proteins of the epithelial barrier (19), which will be discussed later. In addition, there is increased proliferation of the airway epithelial cells following exposure to ozone, likely as a result of direct oxidative epithelial damage (20).
Inflammatory cytokines such as members of the IL-1 family, including IL-1α, IL-1β, IL-18, IL-33, and IL-36 (21–23) as well others and several chemokines are upregulated upon ozone exposure and play major roles in the inflammatory and pathogenic response. IL-1 is involved in the inflammatory response (24), while IL-33 may have protective effects in ozone-induced inflammation as discussed below. We review here the most recent findings on ozone involvement in bronchiolar epithelial barrier dysfunction, acute lung injury, inflammation, resolution, and defective repair (20).
The integrity of the epithelial barrier depends on tight junctions (TJ) and adherens junctions (AJ), which insure apicobasal cell polarity, but also mucus, fluid, and function of the cilia (18, 25–27). Tight junctions comprise the claudin family, occluding, and tricellulin. In addition, several scaffolding proteins, such as zonulae occludens (ZO)-1, ZO-2, ZO-3, multi-PDZ domain protein 1, and others have been identified in the tight junctions (28, 29). E-cadherin, as well as TJs were reduced in patients with asthma (30–32). Common respiratory viruses, such as human rhinovirus (HRV) (33, 34) or respiratory syncytial virus (RSV) (35) disrupt and impair airway epithelial barrier and delay healing of infected epithelium (36), through NADPH oxidase-1 and ROS-dependent mechanisms (33, 37, 38). Disruption of tight junctions with leak of the epithelium allows systemic access of irritants, pathogen, and allergens (15, 39), as well as the drainage of host proteins, lipid mediators, or cells into the airway lumen, where they may perpetuate inflammatory response, acting back on epithelium. Depending on the dose of allergen and airway inflammation, the Zo-1 and Cld-18 proteins expression are decreased in eosinophilic asthma, but it is even more pronounced in mixed and neutrophilic asthma phenotype (27). Epithelial barrier is impaired not only in the lower airways of patients with asthma (32), but also in the nasal mucosa of allergic rhinitis due to house dust mite (HDM) (40) displaying reduced occludin and ZO-1 levels. Ozone exposure disrupts tight junction proteins and hence the function of the respiratory barrier as reported recently (27, 41, 42).
Ozone causes immediate damage of the bronchiolar epithelial cell barrier with cell stress, desquamation, and death with leak of protein and DNA into the airspace within 1–2 h. Since several mediators of inflammation are not yet detectable at this time, we postulate that ozone induced ROS has a direct effect on essential components of the bronchiolar cell integrity with reduced cilia function, tight junctions, mucus, and surfactant protein production (43). This first phase of ozone induced damage of the airway epithelium is followed by a second phase at of bronchiolar epithelial injury and cell death with protein leak and influx of neutrophils, ROS expressing myeloid cells, IL-1α and IL-33 production by epithelial and myeloid cells. Thus, the data suggest that ozone causes a biphasic response, an immediate direct injury via ROS and a second damage by myeloid cells, moving to and exacerbating epithelial cell damage. Indeed, neutrophil depletion by antibody against granulocytes attenuates the second phase. Similar disruptions of the respiratory barrier as shown for ozone has been described before by other air pollutants such nitrogen dioxide, disulfide, particle, chemicals, and cigarette smoke- all of which can cause chronic pulmonary diseases, but usually require longer exposure time and at higher concentrations than ozone (38, 44). A schematic view of the initial events at the respiratory barrier and early repair process is given in Figure 1.
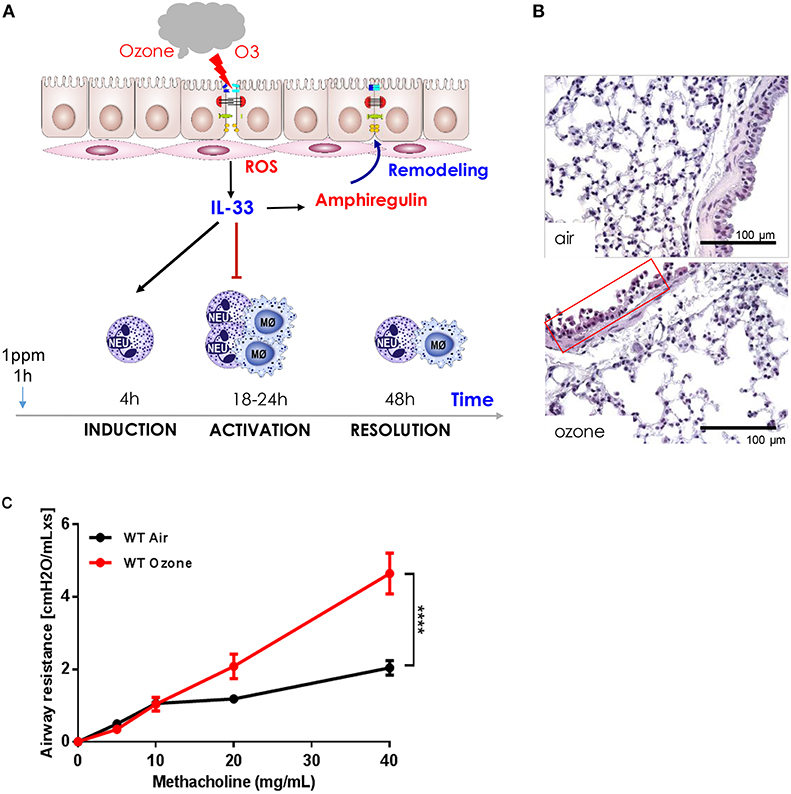
Figure 1. Ozone-induced epithelial barrier dysfunction, AHR, ROS production, cell recruitment, cell desquamation, remodeling, and repair in mice. (A) Scheme showing ozone/ROS-induced disruption of bronchiolar epithelium, cell integrity, tight junctions with protein leak, and cell death with the release of IL-33. Neutrophils and macrophages are recruited within 18 h and macrophages release protective amphiregulin (AREG) with resolution of inflammation and remodeling (43). (B) Micrograph of ozone-induced disruption of bronchiolar epithelium with desquamation, cell death, and focal inflammation at 18 h after exposure (1 h, 1 ppm), scale bar 100 μm. (C) Airways hyperreactivity (AHR): ozone enhances methacholine induced bronchoconstriction measured as resistance (RL) using invasive plethysmography 24 h after exposure (1 h, 1 ppm, unpublished data).
Upon a single ozone exposure, we found enhanced expression of E-cadherin, ZO-1, and claudin-4 using immunofluorescence (43). By contrast, the epithelial E-cadherin, ZO-1, and Cld-4 expression are reduced in the absence of IL-33 (43). Epithelial E-cadherin expression is also reduced in IL-33/ST2 receptor deficient mice, suggesting a protective effect of IL-33. At the transcriptional level, an increased Cld-4 level (4–6 h) or E-cadherin (4–8 h) expression were found. Therefore, the data suggest that IL-33/ST2 signaling has a protective effect. This IL-33-dependant epithelial protection is probably an early defense mechanism in physiological condition. However, in the settings of allergic inflammation, excess of pathogenic Th2 cells and ILC2 in the epithelial wall, reacting to IL-33, lead to increased inflammation, and symptoms of the disease. Interestingly, in the chronic airway inflammation impairment of the barrier consists of decreased expression of several TJ proteins, but also a subsequent increase of TJs, such as CLD-4 (27). Recent reports confirm that ozone exposure impairs the function of epithelial barrier tight junction proteins (27, 41, 42).
Within 24 h after ozone exposure basal cells proliferate and at 48 h the bronchiolar epithelium is restored with firm tight junctions, mucus, and cilia (43). The mechanisms of inflammation including resolution of inflammation, remodeling of airways, and repair mechanisms are an area of intense research (13, 37, 45–47).
Ozone exposure causes bronchoconstriction measured by the pulmonary function tests in humans and airway hyperreactivity (AHR) in rodents and humans as assessed by increased reactivity of the airways to cholinergic stimulation (44, 48). Mice exposed to ozone display a dose-dependent increased of airway resistance (RL) to methacholine aerosol as measured by invasive plethysmograph (Figure 1C). This has also been demonstrated in humans (48).
The underlying mechanisms of AHR are due to the complex response of the airway wall including effect of oxidative stress (49, 50) activation of kinase pathways, the release of chemokines, cytokines (51, 52), and lipid mediators, increased sensitivity of bronchial smooth muscle cells either directly or through an effect on innervation and neuropeptides (53), which is beyond the scope of this review.
The transient receptor potential cation channel subfamily V member1 (TRPV1) is upregulated by ozone in experimental asthma, which is sensitive to a TRPV1 antagonist. The TRPV1 antagonist suppresses the neuropeptide calcitonin gene-related peptide (CGRP) and thymic stromal lymphopoietin (TSLP) (54). Thus, TRPV1 expression may be an important mechanism for asthma exacerbation upon exposure to ozone and other environmental pollutants. Ozone induced AHR and lung inflammation is characterized by increased neutrophils recruitment in the airways and lung (54), although the role of neutrophils in inducing AHR is controversial (55, 56). Interestingly, NKT cell-deficient (CD1d−/− and Jα18−/−) mice are resistant to ozone-induced AHR and have reduced neutrophils. Further, anti-CD1d antibody blockade of NKT cell activation prevented ozone-induced AHR. NKT cells producing IL-17 causes AHR, which was prevented in IL-17A deficient mice or by anti-IL-17 antibody blockade (57, 58). ROS mediated AHR and inflammation further depends on danger activated proteins such as HMGB1, HSPs, RAGE, and others activating the molecular pattern recognition receptors Toll-Like Receptors (TLR) 2 and 4 and the adaptor protein, MyD88 (59). However, the list of endogenous mediators activating AHR includes lipid mediators, prostaglandins and leukotrienes in response to ozone is incomplete (51, 60, 61).
These effects of ozone on bronchoconstriction and AHR raise the possibility that ozone may be involved in underlying a specific type of asthma, namely the neutrophilic inflammatory phenotype of asthma (62, 63). However, the evidence linking neutrophilic asthma to exposure to ozone as a constituent of pollution is unclear, although an increase in exacerbations of asthma in patients with asthma following a peak increase in levels of environmental ozone has been reported (64).
Ozone generates reactive oxygen species (ROS), mitochondrial damage with oxidant stress activating the NLRP3 inflammasome and contributes to AHR, airflow obstruction, and emphysema (2, 65). ROS has a dual effect on cell integrity: at low concentration, it has a beneficial, cytoprotective effect, while at high concentration it is cytotoxic causing cell death. KEAP1 acts as a ROS sensor, which triggers at high ROS levels as found upon ozone exposure a novel caspase-independent cell-death pathway known as oxeiptosis (66, 67) (Figure 2). We found that ROS-induced cell-death depends on interactions of cellular ROS sensor KEAP1 and the phosphatase PGAM5 with antioxidant function and AIFM1, a pro-apoptotic factor. At high ROS concentration PGAM5 is released from the complex and activates the pro-apoptotic factor AIFM1 inducing apoptotic pathway. PGAM5 deficient mice have enhanced lung inflammation with proinflammatory cytokines upon ozone-exposure. Furthermore, in Influenza A virus infection, viral load and lung inflammation were increased in PGAM5 deficient mice, which succumbed to infection (68, 69). Oxeiptosis represents a different, non-canonical cell-death pathway, which is novel, ROS-sensitive, caspase independent, cell-death pathway important for protection against inflammation induced by ROS or viral pathogens (69). The molecular events of oxeiptosis are depicted in Figure 2. Other cell-death pathways include inflammasome-mediated apoptosis and pyroptosis, but also necroptosis and ferroptosis. Their relative roles of these pathways need to be further investigated in the ozone model.
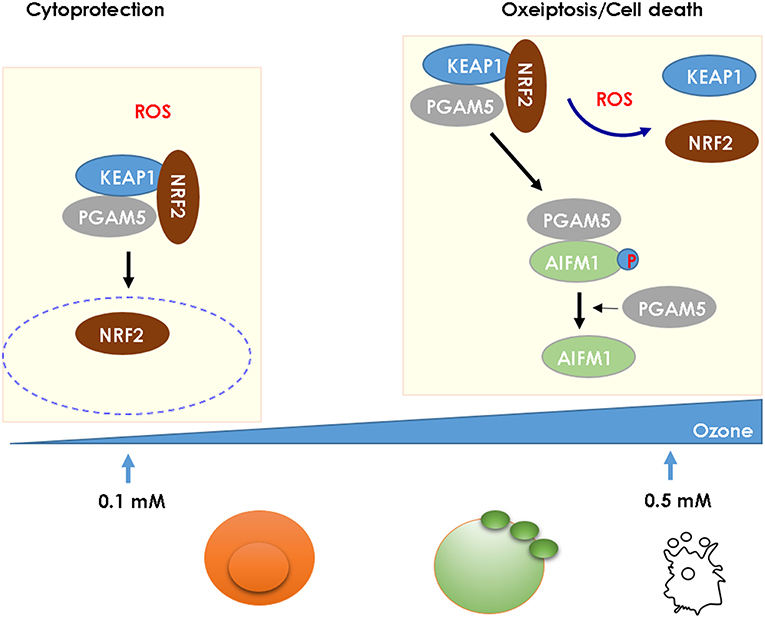
Figure 2. High ROS levels induce oxeiptosis, a novel cell death via the phosphatase PGAM5 activating the death effector AIFM1 in mice. While low concentration of ROS (0.1 mM) release the cytoprotective NRF2 from the KEAP1/PGAM5 complex in the cytosol, at high concentration of ROS (1 mM), the phosphatase PGAM5 is released from the mitochondrial KEAP1/NRF2/PGAM5 complex and PGAM5 dephosphorylates AIFM1, which is the effector protein defining the oxeiptosis death pathway (68).
Cell and tissue damage induce inflammatory cell recruitment including neutrophils, macrophages, innate lymphoid cells, and more. Neutrophils play a critical role in acute and chronic inflammation (70). Neutrophils migrate within and through the vessels depending polarization within activated venules activate platelets present in the bloodstream (71). Neutrophils scan for activated platelets resulting in the redistribution of receptors via the selectin ligand PSGL-1 that drive neutrophil migration allowing the interaction with the endothelium and the circulation before inflammation proceeds. Thus, a close interaction between neutrophils and platelets drives neutrophil migration to sites of injury or cell death. We have described the inflammatory response upon ozone exposure (24). Upon a single ozone exposure at 1 ppm for 1 h the analysis at 4 h after ozone exposure revealed desquamation of epithelial cells with increased CXCL1, CCL2/MIP-2, and IL-6 production, which was followed by increased macrophages and neutrophils in BALF at 6 h. At 24 h, the inflammatory response was further enhanced in the absence of ST2 or IL-33 with predominant neutrophils and interstitial macrophages in the lung. We investigated the role in the second inflammatory phase by GR-1 antibody depletion of neutrophils and other myeloid cells. GR-1 cell depletion with anti-GR1 antibody reduced protein leak, myeloid and epithelial cell in BALF and reduced parenchymal injury and inflammation with reduced MMP9 and increased amphiregulin expression as sign of tissue repair. Therefore, neutrophils contribute to tissue injury and repair as reported in other inflammatory models (72).
Besides T lymphocytes not discussed here, innate lymphocytes (ILC) are involved in the early immune response (54, 73, 74). Tissue-resident ILC-2 is involved in both physiologic and pathologic responses, yet their physical tissue niches are poorly described (75). ILC2 are recruited upon ozone exposure, but there is little information available on their roles in inflammation and airway hyperreactivity (6, 43, 76, 77).
Natural killer T cells expressing IL-17 may induce airway hyperreactivity, which depended on IL-17 expression as mentioned before (57). Platelets are likely involved in ozone induced neutrophilic inflammation, but no studies address their role in ozone induced tissues injury. The role of platelets has been recently studied in allergic inflammation (78).
Fibroblast, stromal cells, pericytes, and endothelial cells are potential targets of ozone (79, 80), which is an area for further investigations. A recent study defined a perivascular fibroblast-like stromal cells producing IL-33 and TSLP which regulate ILC2s and type 2 immunity (75).
Two major previous morphological investigations on the effects of a 4 h exposure to 3 ppm ozone on rat lungs are worth mentioning (81, 82). The epithelial damage was located centrally showing cell death of bronchiolar epithelium and alveolar pneumonocytes. Lesions in capillary endothelium with endothelial swelling were widespread with ring-like formations of endothelial membranes peripherally with alveolar and interstitial edema. These results show that ozone damage is in the centro-acinar regions and affects both endothelial and epithelial cells.
Upon ozone exposure, several interleukins (IL) are increased and may regulate inflammation. Here we focus on two IL-1 family members, IL-1a and IL-33 (23, 83–85), which are known as alarmins, released upon cell stress and death. An overview of IL1 members and IL-33 and their receptors is shown in Figure 3.
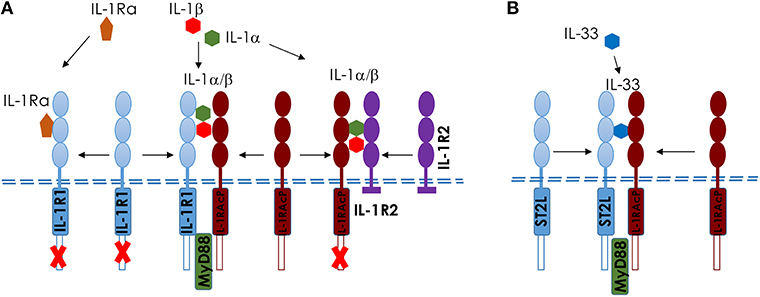
Figure 3. Comparison of IL-1 and IL-33 and their membrane receptors in mice. Scheme of IL-1 (A) and IL-33 (B), showing the different ligands and receptors. (A) IL-1αβ binds to IL-1R1 and induces a conformational change in the extracellular domain of IL-1R1, enabling its interaction with IL-1RAcP, which is required for intracellular signaling, including MyD88-dependent activation. IL-1Ra competes with IL-1αβ for the binding to IL-1R1; the binding of IL-1Ra to IL-1R1 prevents IL-1αβ binding, the recruitment of IL-1RAcP and the activation of intracellular signaling pathways. IL-1R2 acts as a decoy receptor on the cell surface to IL-1RAcP. (B) Interleukin−33 (IL−33) binds to its transmembrane receptor suppression of tumorigenicity 2 (ST2) and induces a conformational change that allows ST2 to interact with IL−1 receptor accessory protein (IL−1RAcP). Activation of ST2 and IL−1RAcP leads to the Toll/IL−1 receptor (TIR) domains clustering and the recruitment of signaling adaptor myeloid differentiation primary response protein 88 (MYD88) (23).
A role of interleukin-1 (IL-1)-associated cytokines has been reviewed recently (24). We investigated inflammation after a single ozone exposure (1 ppm for 1 h) using IL-1α-, IL-1β-, and IL-18-deficient mice or neutralizing anti-IL-1α antibody to investigate their role in epithelial cell death. IL-1α was increased within 1 h after ozone exposure. Epithelial injury, inflammation and AHR were IL-1α-dependent. Further, we found that IL-1α signaling via IL-1R1/MyD88 was type I alveolar epithelial cell dependent as demonstrated by cell specific MyD88 deletion in mice (86). By contrast, inflammation and epithelial injury were less reduced in absence of IL-1β and IL-18. In conclusion, the IL-1α induced tissue damage and inflammation is mediated by IL-1R/MyD88 signaling in epithelial cells. Interestingly, in more chronic disease settings, IL-1β is increased in the lungs of mice with neutrophilic phenotype of asthma and in patients with neutrophilic asthma (27). Additionally, in humans IL-1β may lead to impairment of epithelial barrier function and increase of mucus production (27). Therefore, IL-1α or IL-1 receptor may represent a therapeutic target to attenuate ozone-induced lung inflammation and airway hyperreactivity. The use of neutralizing IL-1β antibody and IL1-RA antagonist is in fact used for different experimental conditions.
IL-33 is another alarmin released rapidly upon ozone and may have a protective role. IL-33 has homeostatic functions and is involved in injury and repair. The alarmin IL-33 is expressed at steady state in tissue cells and released upon airway epithelial injury and repair during inflammatory process (87, 88). IL-33 has functions in both innate and adaptive immune response (85). IL-33 binds to ST2 chain known as IL-33R or IL-1RL1 (89) which associates with IL-1RAcP (23). The full length, 35 kDa IL-33 is cleaved by different proteases and is produced by neutrophils, macrophages, or mast cells that full length IL-33 to several active moieties that are about 30-fold more active than the full length form (84, 90, 91). IL-33 receptor ST2 is was first detected in high endothelial venules in lymphoid tissues in mice (92), but is found in most innate immune cells including mast cells (85), innate lymphoid cells (85), myeloid and dendritic cells (93), and to a lesser extent on Th2 cells (85). Further, nuclear IL-33 may associate with chromatin in vivo, but the function of nuclear IL-33 is still unresolved.
IL-33 expression was increased in epithelial and macrophages and other myeloid cells in mice upon ozone exposure. In the absence of IL-33 or IL-33R/ST2, epithelial cell injury with protein leak and myeloid cell recruitment and inflammation was further increased, while tight junction proteins E-cadherin and ZO-1, ROS expression in neutrophils, and AHR were diminished (43). ST2 antibody neutralization recapitulated the enhanced ozone induced neutrophilic inflammation, while the administration of rmIL-33 reduced neutrophil recruitment in IL-33 deficient mice (69). These data demonstrate that ozone causes an immediate barrier injury, which precedes myeloid cell mediated inflammatory injury under the control of the IL-33/ST2 axis. Thus, IL-33/ST2 signaling appears to be critical for the maintenance of intact epithelial barrier and inflammation.
Surfactant proteins mucus are the first line protection protecting the respiratory epithelium. Surfactant protein-D (SP-D), produced by the airway epithelium, is multimeric protein sensitive to oxidative stress. SP-D directly inhibits extracellular DNA trap formation by eosinophils. Allergic airway sensitization and ozone exposure augmented eosinophilia and nos2 mRNA (iNOS) activation in the lung tissue with modification of SP-D in the airways. Thus, the regulatory feedback between SP-D and eosinophils appears to be destroyed by the NO-rich oxidative lung tissue environment in asthma exacerbations (94).
The protective role of antioxidants against ROS/RNS induced injury and inflammation environmental pollutants has been reviewed (38, 95). A comprehensive analysis of oxidative stress in ozone induced lung injury and mitochondrial dysfunction allowed to identify potential druggable pathways (2, 38, 95, 96). Indeed, several interventions attenuate ozone induced inflammation such N-acetylcysteine (97), hydrogen disulfide (98), MIF antagonist (99) as well as blockade of IL-1α (86) or IL-17A (100), recombinant IL-33 (69), L-arginine promoting DNA repair (101), Taurine (102). Also, blocking ROS-induced airway inflammation by apocynin, an NADPH inhibitor has been targeted in asthma and COPD in human clinical trials (47, 103). Furthermore, DNA released upon cell death and is highly inflammatory. In particle-induced lung injury, enzymatic degradation of DNA by DNase I reduced inflammation (10, 11). The list therapeutic targets are not exhaustive, but new mechanistic insights may lead to antagonists that are more efficacious. However, reduction of airborne pollution, especially high levels of ozone and smog, would be the most efficacious measure to prevent chronic respiratory disease.
Chronic ozone exposure causes chronic lung inflammation, emphysema, and interstitial fibrosis with progressive loss of lung function in man and rodents. Furthermore, ozone enhances the development of chronic lung diseases such as allergic and non-allergic asthma, COPD, and emphysema. Recent studies demonstrated the importance of IL-1α, IL-33, and IL-17A axis in ozone induced lung injury and inflammation, and a role of PGAM5 emerged, which defines a novel cell death pathway known as oxeiptosis. Understanding the fundamental mechanisms of injury and defective repair likely related to DNA damage, and defining critical targets require further investigations including clinical and epidemiological studies. Chronic progressive lung diseases could be prevented to a great extent by reducing environmental air pollution, tobacco, and wood fire smoke and smog containing ozone exposure.
MS, BR, and DT wrote the review. KC, VQ, and CA inspired the work and corrected. All authors read the contribution.
This work was supported by le Centre National de la Recherche Scientifique (CNRS), European Regional Development Fund (FEDER) (N° 2016-00110366 and EX005756), and Respir Ozone (n°2014-00091905).
DT is employee at ArtImmune.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
AHR, airways hyperreactivity; AIFM1, mitochondrial apoptosis inducing factor 1; AJ, adherens junctions; AREG, amphiregulin; BAL, broncho alveolar lavage to quantify cell counts; BALF, broncho alveolar lavage fluid to detect protein levels; GR1, Ly-6G antibody depleting neutrophils; IL-33, interleukin-33; IL-33R/ST2, interleukin-33 receptor/ suppression of tumorigenicity 2; CXCL1/KC, keratinocyte chemoattractant; COPD, chronic obstructive pulmonary disease; LCN-2, lipocalin 2; CCL1/MCP-1, chemokine ligand 2; CCL2/MIP-2, chemokine (C-X-C motif) ligand 2; MMP-9, matrix metalloproteinase 9; MPO, myeloperoxidase; PGAM5, mitochondrial serine/threonine-protein phosphatase; rmIL-33, recombinant mouse IL-33; ROS, reactive oxygen species; RNS, reactive nitrogen species; TIMP-1, tissue inhibitor metalloproteinase 1; TJ, tight junctions; WT, wild type mice.
1. Al-Hegelan M, Tighe RM, Castillo C, Hollingsworth JW. Ambient ozone and pulmonary innate immunity. Immunol Res. (2011) 49:173–91. doi: 10.1007/s12026-010-8180-z
2. Wiegman CH, Michaeloudes C, Haji G, Narang P, Clarke CJ, Russell KE, et al. Oxidative stress-induced mitochondrial dysfunction drives inflammation and airway smooth muscle remodeling in patients with chronic obstructive pulmonary disease. J Allergy Clin Immunol. (2015) 136:769–80. doi: 10.1016/j.jaci.2015.01.046
3. D'Amato G, Annesi-Maesano I, Cecchi L, D'Amato M. Latest news on relationship between thunderstorms and respiratory allergy, severe asthma, and deaths for asthma. Allergy. (2019) 74:9–11. doi: 10.1111/all.13616
4. Hew M, Lee J, Susanto NH, Prasad S, Bardin PG, Barnes S, et al. The 2016 Melbourne thunderstorm asthma epidemic: Risk factors for severe attacks requiring hospital admission. Allergy. (2019) 74:122–30. doi: 10.1111/all.13609
5. Ji M, Cohan DS, Bell ML. Meta-analysis of the association between short-term exposure to ambient ozone and respiratory hospital admissions. Environ Res Lett. (2011) 6:024006. doi: 10.1088/1748-9326/6/2/024006
6. Mathews JA, Krishnamoorthy N, Kasahara DI, Cho Y, Wurmbrand AP, Ribeiro L, et al. IL-33 drives augmented responses to ozone in obese mice. Environ Health Perspect. (2017) 125:246–53. doi: 10.1289/EHP272
7. Miller RL, Peden DB. Environmental effects on immune responses in patients with atopy and asthma. J Allergy Clin Immunol. (2014) 134:1001–8. doi: 10.1016/j.jaci.2014.07.064
8. Liu Y, Pan J, Zhang H, Shi C, Li G, Peng Z, et al. Short-term exposure to ambient air pollution and asthma mortality. Am J Respir Crit Care Med. (2019) 200:24–32. doi: 10.1164/rccm.201810-1823OC
9. Shah PL, Herth FJ, van Geffen WH, Deslee G, Slebos DJ. Lung volume reduction for emphysema. Lancet Respir Med. (2017) 5:147–56. doi: 10.1016/S2213-2600(16)30221-1
10. Benmerzoug S, Rose S, Bounab B, Gosset D, Duneau L, Chenuet P, et al. STING-dependent sensing of self-DNA drives silica-induced lung inflammation. Nat Commun. (2018) 9:5226. doi: 10.1038/s41467-018-07425-1
11. Benmerzoug S, Ryffel B, Togbe D, Quesniaux VFJ. Self-DNA sensing in lung inflammatory diseases. Trends Immunol. (2019) 40:719–34. doi: 10.1016/j.it.2019.06.001
12. Kurai D, Saraya T, Ishii H, Takizawa H. Virus-induced exacerbations in asthma and COPD. Front Microbiol. (2013) 4:293. doi: 10.3389/fmicb.2013.00293
13. Sokolowska M, Chen LY, Liu Y, Martinez-Anton A, Logun C, Alsaaty S, et al. Dysregulation of lipidomic profile and antiviral immunity in response to hyaluronan in patients with severe asthma. J Allergy Clin Immunol. (2017) 139:1379–83. doi: 10.1016/j.jaci.2016.09.031
14. Sokolowska M, Frei R, Lunjani N, Akdis CA, O'Mahony L. Microbiome and asthma. Asthma Res Pract. (2018) 4:1. doi: 10.1186/s40733-017-0037-y
15. Wang M, Tan G, Eljaszewicz A, Meng Y, Wawrzyniak P, Acharya S, et al. Laundry detergents and detergent residue after rinsing directly disrupt tight junction barrier integrity in human bronchial epithelial cells. J Allergy Clin Immunol. (2019) 143:1892–903. doi: 10.1016/j.jaci.2018.11.016
16. Georas SN, Rezaee F. Epithelial barrier function: at the front line of asthma immunology and allergic airway inflammation. J Allergy Clin Immunol. (2014) 134:509–20. doi: 10.1016/j.jaci.2014.05.049
17. Lambrecht BN, Hammad H. Allergens and the airway epithelium response: gateway to allergic sensitization. J Allergy Clin Immunol. (2014) 134:499–507. doi: 10.1016/j.jaci.2014.06.036
18. Price ME, Sisson JH. Redox regulation of motile cilia in airway disease. Redox Biol. (2019) 2019:101146. doi: 10.1016/j.redox.2019.101146
19. Rothenberg ME, Saito H, Peebles RS Jr. Advances in mechanisms of allergic disease in 2016. J Allergy Clin Immunol. (2017) 140:1622–31. doi: 10.1016/j.jaci.2017.08.029
20. Salmon M, Koto H, Lynch OT, Haddad EB, Lamb NJ, Quinlan GJ, et al. Proliferation of airway epithelium after ozone exposure: effect of apocynin and dexamethasone. Am J Respir Crit Care Med. (1998) 157:970–7. doi: 10.1164/ajrccm.157.3.9704067
21. Akdis M, Aab A, Altunbulakli C, Azkur K, Costa RA, Crameri R, et al. Interleukins (from IL-1 to IL-38), interferons, transforming growth factor β, and TNF-α: Receptors, functions, and roles in diseases. J Allergy Clin Immunol. (2016) 138:984–1010. doi: 10.1016/j.jaci.2016.06.033
22. Borthwick LA. The IL-1 cytokine family and its role in inflammation and fibrosis in the lung. Semin Immunopathol. (2016) 38:517–34. doi: 10.1007/s00281-016-0559-z
23. Palomo J, Dietrich D, Martin P, Palmer G, Gabay C. The interleukin (IL)-1 cytokine family–balance between agonists and antagonists in inflammatory diseases. Cytokine. (2015) 76:25–37. doi: 10.1016/j.cyto.2015.06.017
24. Michaudel C, Couturier-Maillard A, Chenuet P, Maillet I, Mura C, Couillin I, et al. Inflammasome, IL-1 and inflammation in ozone-induced lung injury. Am J Clin Exp Immunol. (2016) 5:33–40.
25. Ivanov AI, Naydenov NG. Dynamics and regulation of epithelial adherens junctions: recent discoveries and controversies. Int Rev Cell Mol Biol. (2013) 303:27–99. doi: 10.1016/B978-0-12-407697-6.00002-7
26. Lambrecht BN, Hammad H. The immunology of asthma. Nat Immunol. (2015) 16:45–56. doi: 10.1038/ni.3049
27. Tan HT, Hagner S, Ruchti F, Radzikowska U, Tan G, Altunbulakli C, et al. Tight junction, mucin, and inflammasome-related molecules are differentially expressed in eosinophilic, mixed, and neutrophilic experimental asthma in mice. Allergy. (2019) 74:294–307. doi: 10.1111/all.13619
28. De Benedetto A, Rafaels NM, McGirt LY, Ivanov AI, Georas SN, Cheadle C, et al. Tight junction defects in patients with atopic dermatitis. J Allergy Clin Immunol. (2011) 127:773–86.e1–7. doi: 10.1016/j.jaci.2010.10.018
29. Sugita K, Steer CA, Martinez-Gonzalez I, Altunbulakli C, Morita H, Castro-Giner F, et al. Type 2 innate lymphoid cells disrupt bronchial epithelial barrier integrity by targeting tight junctions through IL-13 in asthmatic patients. J Allergy Clin Immunol. (2018) 141:300–10.e11. doi: 10.1016/j.jaci.2017.02.038
30. Hackett TL, Warner SM, Stefanowicz D, Shaheen F, Pechkovsky DV, Murray LA, et al. Induction of epithelial-mesenchymal transition in primary airway epithelial cells from patients with asthma by transforming growth factor-beta1. Am J Respir Crit Care Med. (2009) 180:122–33. doi: 10.1164/rccm.200811-1730OC
31. Looi K, Buckley AG, Rigby PJ, Garratt LW, Iosifidis T, Zosky GR, et al. Effects of human rhinovirus on epithelial barrier integrity and function in children with asthma. Clin Exp Allergy. (2018) 48:513–24. doi: 10.1111/cea.13097
32. Wawrzyniak P, Wawrzyniak M, Wanke K, Sokolowska M, Bendelja K, Ruckert B, et al. Regulation of bronchial epithelial barrier integrity by type 2 cytokines and histone deacetylases in asthmatic patients. J Allergy Clin Immunol. (2017) 139:93–103. doi: 10.1016/j.jaci.2016.03.050
33. Comstock AT, Ganesan S, Chattoraj A, Faris AN, Margolis BL, Hershenson MB, et al. Rhinovirus-induced barrier dysfunction in polarized airway epithelial cells is mediated by NADPH oxidase 1. J Virol. (2011) 85:6795–808. doi: 10.1128/JVI.02074-10
34. Sajjan U, Wang Q, Zhao Y, Gruenert DC, Hershenson MB. Rhinovirus disrupts the barrier function of polarized airway epithelial cells. Am J Respir Crit Care Med. (2008) 178:1271–81. doi: 10.1164/rccm.200801-136OC
35. Kast JI, McFarlane AJ, Globinska A, Sokolowska M, Wawrzyniak P, Sanak M, et al. Respiratory syncytial virus infection influences tight junction integrity. Clin Exp Immunol. (2017) 190:351–9. doi: 10.1111/cei.13042
36. Faris AN, Ganesan S, Chattoraj A, Chattoraj SS, Comstock AT, Unger BL, et al. Rhinovirus delays cell repolarization in a model of injured/regenerating human airway epithelium. Am J Respir Cell Mol Biol. (2016) 55:487–99. doi: 10.1165/rcmb.2015-0243OC
37. Jakiela B, Gielicz A, Plutecka H, Hubalewska-Mazgaj M, Mastalerz L, Bochenek G, et al. Th2-type cytokine-induced mucus metaplasia decreases susceptibility of human bronchial epithelium to rhinovirus infection. Am J Respir Cell Mol Biol. (2014) 51:229–41. doi: 10.1165/rcmb.2013-0395OC
38. Wieczfinska J, Sokolowska M, Pawliczak R. NOX modifiers-just a step away from application in the therapy of airway inflammation? Antioxid Redox Signal. (2015) 23:428–45. doi: 10.1089/ars.2013.5783
39. Mattila P, Joenvaara S, Renkonen J, Toppila-Salmi S, Renkonen R. Allergy as an epithelial barrier disease. Clin Transl Allergy. (2011) 1:5. doi: 10.1186/2045-7022-1-5
40. Steelant B, Farre R, Wawrzyniak P, Belmans J, Dekimpe E, Vanheel H, et al. Impaired barrier function in patients with house dust mite-induced allergic rhinitis is accompanied by decreased occludin and zonula occludens-1 expression. J Allergy Clin Immunol. (2016) 137:1043–53.e5. doi: 10.1016/j.jaci.2015.10.050
41. Kim BG, Lee PH, Lee SH, Park CS, Jang AS. Impact of ozone on claudins and tight junctions in the lungs. Environ Toxicol. (2018) 33:798–806. doi: 10.1002/tox.22566
42. Van Itallie CM, Anderson JM. Phosphorylation of tight junction transmembrane proteins: many sites, much to do. Tissue Barriers. (2018) 6:e1382671. doi: 10.1080/21688370.2017.1382671
43. Michaudel C, Mackowiak C, Maillet I, Fauconnier L, Akdis C, Sokolowska M, et al. Ozone exposure induces respiratory barrier biphasic injury and inflammation controlled by Interleukin-33. J Allergy Clin Immunol. (2018) 142:942–58. doi: 10.1016/j.jaci.2017.11.044
44. Chitano P, Hosselet JJ, Mapp CE, Fabbri LM. Effect of oxidant air pollutants on the respiratory system: insights from experimental animal research. Eur Respir J. (1995) 8:1357–71. doi: 10.1183/09031936.95.08081357
45. Martinez-Anton A, Sokolowska M, Kern S, Davis AS, Alsaaty S, Taubenberger JK, et al. Changes in microRNA and mRNA expression with differentiation of human bronchial epithelial cells. Am J Respir Cell Mol Biol. (2013) 49:384–95. doi: 10.1165/rcmb.2012-0368OC
46. Sokolowska M, Stefanska J, Wodz-Naskiewicz K, Pawliczak R. Cytosolic phospholipase A2 group IVA influence on GM-CSF expression in human lung cells: a pilot study. Med Sci Monit. (2010) 16:BR300-6.
47. Stefanska J, Sarniak A, Wlodarczyk A, Sokolowska M, Pniewska E, Doniec Z, et al. Apocynin reduces reactive oxygen species concentrations in exhaled breath condensate in asthmatics. Exp Lung Res. (2012) 38:90–9. doi: 10.3109/01902148.2011.649823
48. Seltzer J, Bigby BG, Stulbarg M, Holtzman MJ, Nadel JA, Ueki IF, et al. O3-induced change in bronchial reactivity to methacholine and airway inflammation in humans. J Appl Physiol. (1986) 60:1321–6. doi: 10.1152/jappl.1986.60.4.1321
49. Tsukagoshi H, Haddad EB, Sun J, Barnes PJ, Chung KF. Ozone-induced airway hyperresponsiveness: role of superoxide anions, NEP, and BK receptors. J Appl Physiol. (1995) 78:1015–22. doi: 10.1152/jappl.1995.78.3.1015
50. Williams AS, Issa R, Leung SY, Nath P, Ferguson GD, Bennett BL, et al. Attenuation of ozone-induced airway inflammation and hyper-responsiveness by c-Jun NH2 terminal kinase inhibitor SP600125. J Pharmacol Exp Ther. (2007) 322:351–9. doi: 10.1124/jpet.107.121624
51. Aizawa H, Chung KF, Leikauf GD, Ueki I, Bethel RA, O'Byrne PM, et al. Significance of thromboxane generation in ozone-induced airway hyperresponsiveness in dogs. J Appl Physiol. (1985) 59:1918–23. doi: 10.1152/jappl.1985.59.6.1918
52. Koto H, Salmon M, Haddad el B, Huang TJ, Zagorski J, Chung KF. Role of cytokine-induced neutrophil chemoattractant (CINC) in ozone-induced airway inflammation and hyperresponsiveness. Am J Respir Crit Care Med. (1997) 156:234–9. doi: 10.1164/ajrccm.156.1.9606095
53. Li F, Zhang M, Hussain F, Triantaphyllopoulos K, Clark AR, Bhavsar PK, et al. Inhibition of p38 MAPK-dependent bronchial contraction after ozone by corticosteroids. Eur Respir J. (2011) 37:933–42. doi: 10.1183/09031936.00021110
54. Li S, Morita H, Sokolowska M, Tan G, Boonpiyathad T, Opitz L, et al. Gene expression signatures of circulating human type 1, 2, and 3 innate lymphoid cells. J Allergy Clin Immunol. (2019) 143:2321–5. doi: 10.1016/j.jaci.2019.01.047
55. O'Byrne PM, Walters EH, Gold BD, Aizawa HA, Fabbri LM, Alpert SE, et al. Neutrophil depletion inhibits airway hyperresponsiveness induced by ozone exposure. Am Rev Respir Dis. (1984) 130:214–9. doi: 10.1164/arrd.1984.130.2.214
56. Evans TW, Brokaw JJ, Chung KF, Nadel JA, McDonald DM. Ozone-induced bronchial hyperresponsiveness in the rat is not accompanied by neutrophil influx or increased vascular permeability in the trachea. Am Rev Respir Dis. (1988) 138:140–4. doi: 10.1164/ajrccm/138.1.140
57. Pichavant M, Goya S, Meyer EH, Johnston RA, Kim HY, Matangkasombut P, et al. Ozone exposure in a mouse model induces airway hyperreactivity that requires the presence of natural killer T cells and IL-17. J Exp Med. (2008) 205:385–93. doi: 10.1084/jem.20071507
58. Pinart M, Zhang M, Li F, Hussain F, Zhu J, Wiegman C, et al. IL-17A modulates oxidant stress-induced airway hyperresponsiveness but not emphysema. PLoS ONE. (2013) 8:e58452. doi: 10.1371/journal.pone.0058452
59. Williams AS, Leung SY, Nath P, Khorasani NM, Bhavsar P, Issa R, et al. Role of TLR2, TLR4, and MyD88 in murine ozone-induced airway hyperresponsiveness and neutrophilia. J Appl Physiol. (2007) 103:1189–95. doi: 10.1152/japplphysiol.00172.2007
60. Nakano H, Aizawa H, Matsumoto K, Fukuyama S, Inoue H, Hara N. Cyclooxygenase-2 participates in the late phase of airway hyperresponsiveness after ozone exposure in guinea pigs. Eur J Pharmacol. (2000) 403:267–75. doi: 10.1016/S0014-2999(00)00524-0
61. Wagner JG, Jiang Q, Harkema JR, Illek B, Patel DD, Ames BN, et al. Ozone enhancement of lower airway allergic inflammation is prevented by gamma-tocopherol. Free Radic Biol Med. (2007) 43:1176–88. doi: 10.1016/j.freeradbiomed.2007.07.013
62. Chung KF. Neutrophilic asthma: a distinct target for treatment? Lancet Respir Med. (2016) 4:765–7. doi: 10.1016/S2213-2600(16)30232-6
63. Qiu R, Xie J, Chung KF, Li N, Yang Z, He M, et al. Asthma phenotypes defined from parameters obtained during recovery from a hospital-treated exacerbation. J Allergy Clin Immunol Pract. (2018) 6:1960–7. doi: 10.1016/j.jaip.2018.02.012
64. Halonen JI, Lanki T, Yli-Tuomi T, Kulmala M, Tiittanen P, Pekkanen J. Urban air pollution, and asthma and COPD hospital emergency room visits. Thorax. (2008) 63:635–41. doi: 10.1136/thx.2007.091371
65. Li F, Xu M, Wang M, Wang L, Wang H, Zhang H, et al. Roles of mitochondrial ROS and NLRP3 inflammasome in multiple ozone-induced lung inflammation and emphysema. Respir Res. (2018) 19:230. doi: 10.1186/s12931-018-0931-8
66. Scaturro P, Pichlmair A. Oxeiptosis-a cell death pathway to mitigate damage caused by radicals. Cell Death Differ. (2018) 25:1191–3. doi: 10.1038/s41418-018-0134-3
67. Scaturro P, Pichlmair A. Oxeiptosis: a discreet way to respond to radicals. Curr Opin Immunol. (2019) 56:37–43. doi: 10.1016/j.coi.2018.10.006
68. Kesavardhana S, Kanneganti TD. Stressed-out ROS take a silent death route. Nat Immunol. (2018) 19:103–5. doi: 10.1038/s41590-017-0034-6
69. Holze C, Michaudel C, Mackowiak C, Haas DA, Benda C, Hubel P, et al. Oxeiptosis, a ROS-induced caspase-independent apoptosis-like cell-death pathway. Nat Immunol. (2018) 19:130–40. doi: 10.1038/s41590-017-0013-y
70. Soehnlein O, Steffens S, Hidalgo A, Weber C. Neutrophils as protagonists and targets in chronic inflammation. Nat Rev Immunol. (2017) 17:248–61. doi: 10.1038/nri.2017.10
71. Sreeramkumar V, Adrover JM, Ballesteros I, Cuartero MI, Rossaint J, Bilbao I, et al. Neutrophils scan for activated platelets to initiate inflammation. Science. (2014) 346:1234–8. doi: 10.1126/science.1256478
72. Pain M, Bermudez O, Lacoste P, Royer PJ, Botturi K, Tissot A, et al. Tissue remodelling in chronic bronchial diseases: from the epithelial to mesenchymal phenotype. Eur Respir Rev. (2014) 23:118–30. doi: 10.1183/09059180.00004413
73. Cording S, Medvedovic J, Lecuyer E, Aychek T, Dejardin F, Eberl G. Mouse models for the study of fate and function of innate lymphoid cells. Eur J Immunol. (2018) 48:1271–80. doi: 10.1002/eji.201747388
74. Vivier E, Artis D, Colonna M, Diefenbach A, Di Santo JP, Eberl G, et al. Innate lymphoid cells: 10 years on. Cell. (2018) 174:1054–66. doi: 10.1016/j.cell.2018.07.017
75. Dahlgren MW, Jones SW, Cautivo KM, Dubinin A, Ortiz-Carpena JF, Farhat S, et al. Adventitial stromal cells define group 2 innate lymphoid cell tissue niches. Immunity. (2019) 50:707–22.e6. doi: 10.1016/j.immuni.2019.02.002
76. Kumagai K, Lewandowski R, Jackson-Humbles DN, Li N, Van Dyken SJ, Wagner JG, et al. Ozone-induced nasal type 2 immunity in mice is dependent on innate lymphoid cells. Am J Respir Cell Mol Biol. (2016) 54:782–91. doi: 10.1165/rcmb.2015-0118OC
77. Ong CB, Kumagai K, Brooks PT, Brandenberger C, Lewandowski RP, Jackson-Humbles DN, et al. Ozone-induced type 2 immunity in nasal airways. development and lymphoid cell dependence in mice. Am J Respir Cell Mol Biol. (2016) 54:331–40. doi: 10.1165/rcmb.2015-0165OC
78. Obeso D, Mera-Berriatua L, Rodriguez-Coira J, Rosace D, Fernandez P, Martin-Antoniano IA, et al. Multi-omics analysis points to altered platelet functions in severe food-associated respiratory allergy. Allergy. (2018) 73:2137–49. doi: 10.1111/all.13563
79. Gross KB, White HJ, Sargent NE. The effect of ozone inhalation on metabolic functioning of vascular endothelium and on ventilatory function. Toxicol Appl Pharmacol. (1991) 109:336–51. doi: 10.1016/0041-008X(91)90180-M
80. Robertson S, Colombo ES, Lucas SN, Hall PR, Febbraio M, Paffett ML, et al. CD36 mediates endothelial dysfunction downstream of circulating factors induced by O3 exposure. Toxicol Sci. (2013) 134:304–11. doi: 10.1093/toxsci/kft107
81. Plopper CG, Dungworth DL, Tyler WS. Morphometric evaluation of pulmonary lesions in rats exposed to ozone. Am J Pathol. (1973) 71:395–408.
82. Plopper CG, Dungworth DL, Tyler WS. Pulmonary lesions in rats exposed to ozone. A correlated light and electron microscopic study. Am J Pathol. (1973) 71:375–94.
83. Dinarello CA. Overview of the IL-1 family in innate inflammation and acquired immunity. Immunol Rev. (2018) 281:8–27. doi: 10.1111/imr.12621
84. Liew FY, Girard JP, Turnquist HR. Interleukin-33 in health and disease. Nat Rev Immunol. (2016) 16:676–89. doi: 10.1038/nri.2016.95
85. Molofsky AB, Savage AK, Locksley RM. Interleukin-33 in tissue homeostasis, injury, and inflammation. Immunity. (2015) 42:1005–19. doi: 10.1016/j.immuni.2015.06.006
86. Michaudel C, Maillet I, Fauconnier L, Quesniaux V, Chung KF, Wiegman C, et al. Interleukin-1alpha mediates ozone-induced myeloid differentiation factor-88-dependent epithelial tissue injury and inflammation. Front Immunol. (2018) 9:916. doi: 10.3389/fimmu.2018.00916
87. Cayrol C, Girard JP. IL-33: an alarmin cytokine with crucial roles in innate immunity, inflammation and allergy. Curr Opin Immunol. (2014) 31:31–7. doi: 10.1016/j.coi.2014.09.004
88. Martin NT, Martin MU. Interleukin 33 is a guardian of barriers and a local alarmin. Nat Immunol. (2016) 17:122–31. doi: 10.1038/ni.3370
89. Schmitz J, Owyang A, Oldham E, Song Y, Murphy E, McClanahan TK, et al. IL-33, an interleukin-1-like cytokine that signals via the IL-1 receptor-related protein ST2 and induces T helper type 2-associated cytokines. Immunity. (2005) 23:479–90. doi: 10.1016/j.immuni.2005.09.015
90. Lefrancais E, Duval A, Mirey E, Roga S, Espinosa E, Cayrol C, et al. Central domain of IL-33 is cleaved by mast cell proteases for potent activation of group-2 innate lymphoid cells. Proc Natl Acad Sci USA. (2014) 111:15502–7. doi: 10.1073/pnas.1410700111
91. Lefrancais E, Roga S, Gautier V, Gonzalez-de-Peredo A, Monsarrat B, Girard JP, et al. IL-33 is processed into mature bioactive forms by neutrophil elastase and cathepsin G. Proc Natl Acad Sci USA. (2012) 109:1673–8. doi: 10.1073/pnas.1115884109
92. Carriere V, Roussel L, Ortega N, Lacorre DA, Americh L, Aguilar L, et al. IL-33, the IL-1-like cytokine ligand for ST2 receptor, is a chromatin-associated nuclear factor in vivo. Proc Natl Acad Sci USA. (2007) 104:282–7. doi: 10.1073/pnas.0606854104
93. Besnard AG, Togbe D, Guillou N, Erard F, Quesniaux V, Ryffel B. IL-33-activated dendritic cells are critical for allergic airway inflammation. Eur J Immunol. (2011) 41:1675–86. doi: 10.1002/eji.201041033
94. Yousefi S, Sharma SK, Stojkov D, Germic N, Aeschlimann S, Ge MQ, et al. Oxidative damage of SP-D abolishes control of eosinophil extracellular DNA trap formation. J Leukoc Biol. (2018) 104:205–14. doi: 10.1002/JLB.3AB1117-455R
95. Poljsak B, Fink R. The protective role of antioxidants in the defence against ROS/RNS-mediated environmental pollution. Oxid Med Cell Longev. (2014) 2014:671539. doi: 10.1155/2014/671539
96. Wiegman CH, Li F, Clarke CJ, Jazrawi E, Kirkham P, Barnes PJ, et al. A comprehensive analysis of oxidative stress in the ozone-induced lung inflammation mouse model. Clin Sci. (2014) 126:425–40. doi: 10.1042/CS20130039
97. Li F, Wiegman C, Seiffert JM, Zhu J, Clarke C, Chang Y, et al. Effects of N-acetylcysteine in ozone-induced chronic obstructive pulmonary disease model. PLoS ONE. (2013) 8:e80782. doi: 10.1371/journal.pone.0080782
98. Zhang P, Li F, Wiegman CH, Zhang M, Hong Y, Gong J, et al. Inhibitory effect of hydrogen sulfide on ozone-induced airway inflammation, oxidative stress, and bronchial hyperresponsiveness. Am J Respir Cell Mol Biol. (2015) 52:129–37. doi: 10.1165/rcmb.2013-0415OC
99. Russell KE, Chung KF, Clarke CJ, Durham AL, Mallia P, Footitt J, et al. The MIF antagonist ISO-1 attenuates corticosteroid-insensitive inflammation and airways hyperresponsiveness in an ozone-induced model of COPD. PLoS ONE. (2016) 11:e0146102. doi: 10.1371/journal.pone.0146102
100. Zhang M, Fei X, Zhang GQ, Zhang PY, Li F, Bao WP, et al. Role of neutralizing anti-murine interleukin-17A monoclonal antibody on chronic ozone-induced airway inflammation in mice. Biomed Pharmacother. (2016) 83:247–56. doi: 10.1016/j.biopha.2016.06.041
101. Cui Y, Gao CQ, Sun G, Zhou Y, Qu F, Tang C, et al. L-arginine promotes DNA repair in cultured bronchial epithelial cells exposed to ozone: involvement of the ATM pathway. Cell Biol Int. (2011) 35:273–80. doi: 10.1042/CBI20090252
102. Schuller-Levis GB, Gordon RE, Park E, Pendino KJ, Laskin DL. Taurine protects rat bronchioles from acute ozone-induced lung inflammation and hyperplasia. Exp Lung Res. (1995) 21:877–88. doi: 10.3109/01902149509031768
Keywords: inflammation, cell death, interleukins, mucus, tight junctions, innate immunity
Citation: Sokolowska M, Quesniaux VFJ, Akdis CA, Chung KF, Ryffel B and Togbe D (2019) Acute Respiratory Barrier Disruption by Ozone Exposure in Mice. Front. Immunol. 10:2169. doi: 10.3389/fimmu.2019.02169
Received: 10 May 2019; Accepted: 28 August 2019;
Published: 13 September 2019.
Edited by:
Rudolf Lucas, Augusta University, United StatesCopyright © 2019 Sokolowska, Quesniaux, Akdis, Chung, Ryffel and Togbe. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Bernhard Ryffel, YnJ5ZmZlbEBjbnJzLW9ybGVhbnMuZnI=; Dieudonnée Togbe, ZHRvZ2JlQGNucnMtb3JsZWFucy5mcg==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.