 Thomas Renné
Thomas Renné Evi X. Stavrou
Evi X. Stavrou- 1Institute of Clinical Chemistry and Laboratory Medicine, University Medical Center Hamburg-Eppendorf, Hamburg, Germany
- 2Section of Hematology-Oncology, Department of Medicine, Louis Stokes Cleveland Veterans Administration Medical Center, VA Northeast Ohio Healthcare System, Cleveland, OH, United States
- 3Hematology and Oncology Division, Department of Medicine, Case Western Reserve University School of Medicine, Cleveland, OH, United States
Factor XII (FXII) is the zymogen of serine protease, factor XIIa (FXIIa). FXIIa enzymatic activities have been extensively studied and FXIIa inhibition is emerging as a promising target to treat or prevent thrombosis without creating a hemostatic defect. FXII and plasma prekallikrein reciprocally activate each other and result in liberation of bradykinin. Due to its unique structure among coagulation factors, FXII exerts mitogenic activity in endothelial and smooth muscle cells, indicating that zymogen FXII has activities independent of its protease function. A growing body of evidence has revealed that both FXII and FXIIa upregulate neutrophil functions, contribute to macrophage polarization and induce T-cell differentiation. In vivo, these signaling activities contribute to host defense against pathogens, mediate the development of neuroinflammation, influence wound repair and may facilitate cancer maintenance and progression. Here, we review the roles of FXII in innate immunity as they relate to non-sterile and sterile immune responses.
Introduction
Factor XII (FXII) is the zymogen of serine protease, factor XIIa (FXIIa). FXII is converted to its active enzyme (FXIIa) by plasma kallikrein (PKa) or by its unique ability to auto-activate following binding to artificial or biologic surfaces (1). In vivo, FXIIa initiates coagulation via the fibrin-forming “intrinsic” pathway and promotes inflammation via the bradykinin (BK)-producing kallikrein kinin system comprising, high molecular weight kininogen (HK) and plasma prekallikrein (PK). Together, FXII, PK, and HK are termed plasma contact activation system. Furthermore, FXIIa may modulate components of the complement and fibrinolytic systems however to date, these activities have only been demonstrated in vitro. There has been renewed interest in FXII due to the recognition that several substances [e.g., polyphosphate, misfolded protein, vascular collagen, DNA in neutrophil extracellular traps (NETs)] support FXII auto-activation in vivo (2–7) and prior studies showing that FXII deficient (F12−/−) animals are protected from thrombosis without impaired hemostasis (8–11). Similarly, pharmacologic targeting of FXII or FXIIa provided protection from thrombosis without increased incidence of bleeding (7, 12).
Although the enzymatic activities of FXII have been thoroughly studied and the FXIIa protease domain structure is known (13), few zymogen FXII functions have been recognized. Prior reports have shown that FXII deficiency is linked to decreased infiltration of inflammatory cells into skin windows (14). In human plasma, FXII and FXIIa promote neutrophil aggregation and degranulation (15) and FXII with related proteins assembles on the surface of neutrophils (16). FXII has two epidermal growth factor domains that contribute mitogenic activity in endothelial and smooth muscle cells (17, 18), similar to activated Factor X (FXa), protein C and S (19–21). These data indicate that zymogen FXII differentially regulates cell activities independent of its protease function.
Despite its role in pathologic thrombosis, FXII deficiency is rare in humans indicating that it may contribute to homeostatic functions. Here, we review the role of FXII in non-sterile and sterile inflammation. We focus on its differential contribution to host immune responses to infectious pathogens and contrast these to newly appreciated FXII functions in chronic, sterile inflammation.
Role of Factor XII in Infectious Settings
FXIIa Interactions With Bacteria
Experimental and clinical studies have shown a link between FXII and the contact system to infections (22–25). Several mechanisms have been proposed by which contact system components modulate infectious burden and the inflammatory response. First, FXII directly binds to the surface of bacteria, fungi, viruses as well as on neutrophils and neutrophil extracellular traps (NETs) where it autoactivates (26). In a Salmonella-induced pneumonia model in rats, targeting FXIIa using the peptide inhibitor H-D-Pro-Phe-Arg-chloromethylketone ameliorated lung injury and largely prevented bacteria-induced bleeding (27). Another mechanism by which microorganisms can induce FXII zymogen activation, involves contact activation by the inorganic polymer polyphosphate (polyP). Bacterial-derived polyphosphate, similar to polymers exposed by activated platelets, drives contact system activation (2). Microbes contain polyP that serves as their energy storage pool, with a size varying in length from hundreds to thousands of phosphate units (28). PolyP forms calcium ion-rich nanoparticles that trigger FXII autoactivation independently of polymer length on cell surfaces in vivo (29, 30). Consistent with this finding, polyP from Salmonella and E. coli species shows extreme potency at triggering the contact pathway (2, 30). PolyP not only contributes to FXIIa-mediated fibrin formation, but it also interferes with binding of fibrin to tissue-type plasminogen activator or plasminogen (31), thus affecting fibrinolysis. Waack et al. (32) showed that metalloprotease CpaA secreted from Acinetobacter baumanii species inactivates FXII. The cleavage by CpaA was mapped on two positions, 279–280 and 308–309, within the proline-rich region of FXII and cleavage at the 308–309 site was a requirement for inactivation of FXII. At both sites, cleavage takes place between a proline and an O-linked glycosylated threonine residue and deglycosylation of FXII rescues from CpaA cleavage. Strikingly, mutant FXII (Thr309Lys) from patients with hereditary angioedema type III (HAEIII), where O-linked glycosylation at position 309 is lost and inactivation by C1 esterase inhibitor during activation by plasmin is reduced (33, 34), is protected from CpaA inactivation (32). By inactivating FXII, CpaA attenuates important coagulation and inflammatory mechanisms, thus allowing A. baumannii dissemination (32). To date, the structural details of the interaction(s) between FXII and pathogens remain elusive. An exciting possibility would be that FXII functions as a soluble pattern recognition molecule or damage-associated molecular pattern (DAMP), binding to defined sites on microorganisms. Until such time that the surface-binding domains of FXII are fully characterized, this scenario remains speculative.
FXIIa-Initiated Bradykinin Formation
As discussed above, bacteria can activate the contact system either through direct binding or indirectly through the release of mediators (35). Staphylococcus aureus bacteria release proteinases which directly cleave HK whereas other proteinases such as from Porphyromonas gingivalis, indirectly lead to HK cleavage through proteolytic activation of FXII (36). HK cleavage results in liberation of BK, a potent proinflammatory and vasodilatative peptide (37). BK regulates vascular smooth muscle relaxation, increases vascular permeability and can directly influence leukocyte functions (38, 39). Host infection with hantavirus results in increased enzymatic activities of FXIIa and PKa and subsequent release of BK, leading to enhanced endothelial cell permeability and vascular leak (40). Another virus HSV1 directly binds to FXII and in the presence of PKa, promotes its activation. Inhibiting FXIIa or use of antibodies to FXII, PK, and Factor XI prevented HSV1-initiated clotting (41). Persistent bacteria-mediated contact system activation may result in abnormally high plasma BK levels and consumptive coagulopathy and can induce hypotension and edema, contributing to multi-organ failure. Indeed, in a model of lethal E. coli bacteremia in baboons, FXII activation was related with the hypotension seen in these animals (42). In this primate model, contact activation and FXIIa generation manifested primarily by a significant decrease in HK and a significant increase in PKa, indicative of increased BK formation. Animals treated with C6B7, a monoclonal antibody to FXII, experienced an initial drop in systemic blood pressure that subsequently resolved and extended their overall survival (42). However, all groups including C6B7-treated animals, equally developed disseminated intravascular coagulation (DIC) manifesting with thrombocytopenia, hypofibrinogenemia and decreased factor V levels (42). Interfering with BK activity resulted in attenuation of acute respiratory distress syndrome (ARDS) features in a rat hypothalamic nuclei lesion model (43) and significantly improved outcomes of patients with systemic inflammatory response syndrome caused by Gram-negative bacteria (44). These data indicate that interfering with FXIIa-induced BK formation, alone can have beneficial effects in host defense.
The protective effects of functional FXII deficiency were shown to be pathogen-specific (45). Stroo et al. showed that genetic ablation of FXII conferred a survival advantage from Klebsiella pneumoniae sepsis but not from Streptococcus pneumoniae sepsis (45). The authors postulate that the contact system becomes operative in late disease stages, where damage due to overwhelming inflammation accounts for FXII activation and BK release (45). It is important to note however that in this study, Klebsiella-infected FXII deficient mice also had consistently decreased bacterial loads. This finding raises the intriguing possibility that FXII may preferentially interact with specific pathogens or their soluble mediators to facilitate bacterial maintenance and invasion. Further studies are warranted to mechanistically determine if under certain infectious settings, FXII serves as an in vivo bacterial “fitness factor.”
FXII Responses Independent of Plasma Kallikrein
Although BK is the main inflammatory mediator of the contact system, FXIIa also initiates the classical complement cascade (46) and exerts direct proinflammatory properties in vitro. In the presence of lipopolysaccharide (LPS), FXII exposure of peripheral blood monocytes resulted in enhanced interleukin 1 (IL-1) activity and LPS was also essential for FXII- and FXIIa-triggered expression of IL-6 and IL-23 by splenic dendritic cells (47). Moreover, zymogen FXII and FXIIa were shown to promote neutrophil degranulation (15) and FXII content in bronchoalveolar lavage fluid from ARDS patients, was significantly higher in non-survivors than in survivors (48). In this setting, FXII but not FXIIa, upregulated the expression of inflammatory cytokines interleukin 8 (IL-8), IL-1β, interleukin 6 (IL-6), CXC chemokine ligand 5 (CXCL5), leukemia inhibitory factor (LIF), and tumor necrosis factor alpha (TNF-α), independently of plasma kallikrein (48).
In sum, the aforementioned studies highlight that FXII has a complex role in sepsis eliciting distinct pro-coagulant and pro-inflammatory responses that differentially contribute to infectious outcomes (Figure 1).
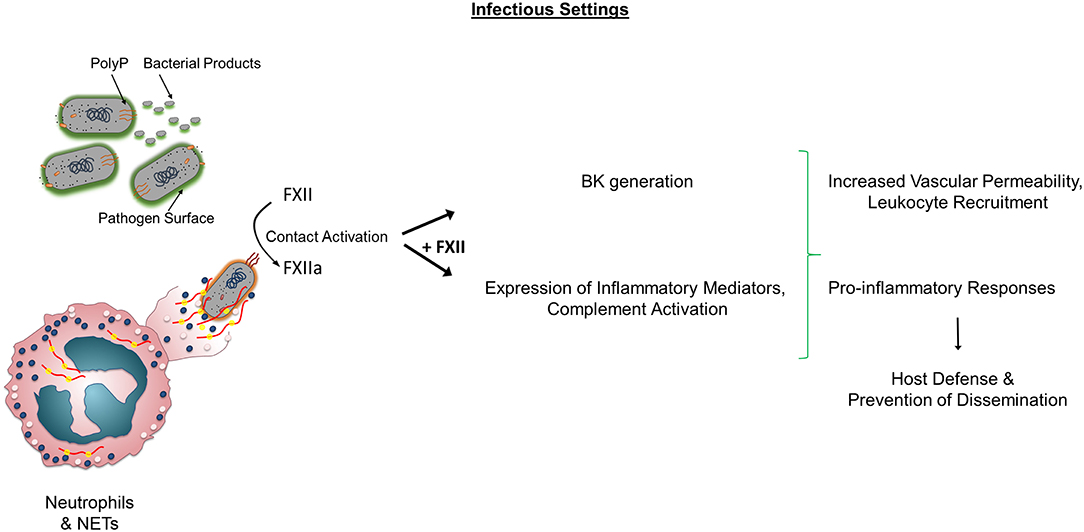
Figure 1. Role of FXII in non-sterile inflammation. In infectious conditions, Factor XII (FXII) activation occurs through various mechanisms including pathogen surfaces, bacterial products, polyP, neutrophils, and NETs. Activated FXII (FXIIa) leads to BK generation and activates components of the complement system. Both FXII and FXIIa increase the expression of pro-inflammatory cytokines and influence leukocyte functions. In certain settings, the sum of these activities contributes to prevention of dissemination. Alternatively, the presence of FXII and subsequent FXIIa and BK generation, lead to adverse outcomes and increased mortality independently of Factor XI activation. PolyP, polyphosphate; BK, Bradykinin; NETs, Neutrophil Extracellular Traps.
Role of FXII in Sterile Inflammation
Discoveries over the past few years have increased our understanding of the diverse FXII functions. Here, we summarize the signaling events for FXII and FXIIa in innate immune cells with a focus on sterile inflammatory conditions and discuss the downstream physiologic and pathologic consequences.
Immune Cell Biology of FXII and FXIIa
Our lab recently reported on the role of FXII in neutrophil proinflammatory responses (49). We showed that autocrine FXII binds to urokinase plasminogen activator receptor (uPAR) with high affinity and their interaction induces Akt2 phosphorylation. In vivo, FXII (which interacts with domain II of uPAR) and HK (which binds to domains II and III), compete for binding and their interaction with uPAR is mutually exclusive (50–52). Moreover, in resting neutrophils uPAR is stored intracellularly secretory vesicles and specific granules (53). Neutrophil activation results in upregulation of uPAR expression and its subsequent translocation to the neutrophil plasma membrane (53). This highly regulated appearance of FXII binding sites on primed neutrophils, partly explains why the FXII-uPAR interaction is not operating continuously. Another rate-limiting step for FXII and uPAR binding is FXII's cell-binding ability. In vivo, FXII binding to cells is augmented when the local free zinc ion concentration rises significantly from baseline plasma levels of ~20 nM (54, 55) to a micromolar (μM) range. The source of extracellular zinc was previously shown to derive from activated cells such as platelets (50, 56). Neutrophils also contain a rich network of zinc transporters and may potentiate the mobilization of zinc toward the extracellular compartment during inflammation. To this end, surface plasmon resonance confirmed that in the absence of zinc, FXII does not interact with uPAR (49). In sum, FXII-uPAR complex formation is a highly regulated process in vivo governed both by uPAR surface expression and the local concentration of zinc ions (49).
Having no intracellular domain, uPAR has to engage other membrane receptors that mediate signals to cells. uPAR has an intimate relationship with integrins regulating their affinity and avidity, but the reverse is also true, integrins are able to modulate the activity of uPAR. In neutrophils and macrophages, this crosstalk permits an indirect connection of uPAR with the integrin interactome (57). In this context, we found that the FXII-uPAR interaction increases the surface expression of neutrophil αMβ2 integrin, leads to intracellular Ca2+ mobilization, and promotes histone citrullination (49). These signaling events, which are independent of FXII enzymatic activity, upregulate neutrophil functions. Specifically, FXII-uPAR promote neutrophil adhesion to extracellular matrices including fibrinogen, increase directional cell migration (chemotaxis) and NET formation (49) (Figure 2). The identification that FXII itself promotes NET formation is novel and therapeutically relevant since the conventional thinking was that their relationship lies purely on activation of circulating FXII on preformed NETs (5). On-going studies in our lab seek to determine if FXII-uPAR-mediated functions in neutrophils are exclusively initiated through αMβ2 or if additional lateral partners are involved in intracellular signal transduction.
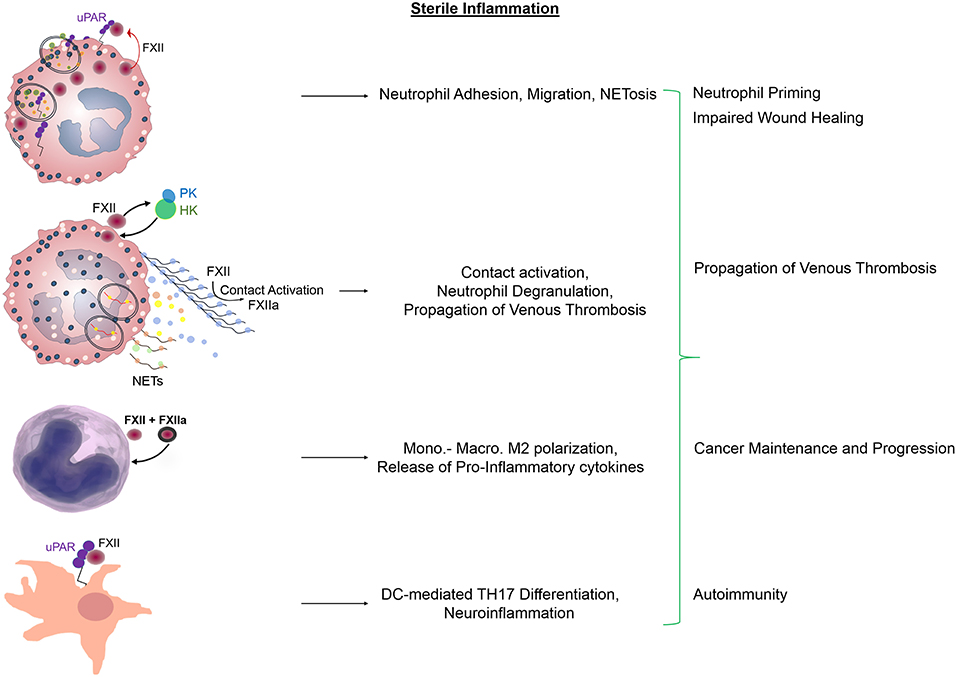
Figure 2. Regulatory functions of FXII and FXIIa in sterile inflammation. Zymogen FXII functions as an autocrine messenger through uPAR to promote Akt2S474 phosphorylation. Propagation of FXII-mediated neutrophil activities includes adhesion, chemotaxis that leads to neutrophil trafficking at sites of inflammation and NET formation. Subsequent contact activation on the surface of preformed NETs leads to FXIIa generation and fibrin formation. In monocytes and macrophages, FXII and FXIIa upregulate the expression of pro-inflammatory mediators and promote cell polarization toward an M2 phenotype. In dendritic cells, FXII signals through uPAR to induce the differentiation of naive T-cells to TH17 cells. These immune cell responses contribute to impaired wound healing, propagation of venous thrombosis, tumor maintenance, and invasion as well as tissue damage during CNS autoimmunity. NETs, neutrophil extracellular traps; Mono, Monocytes; Macro, Macrophages; DC, Dendritic cells; TH17, T-helper cells.
Eventually, neutrophil priming enables circulating FXII to bind onto neutrophils and autoactivate (5, 49). In addition to FXII, HK and PK also assemble on the surface of primed neutrophils (16) via binding to proteoglycans (58), which modulate the liberation of BK (59). This neutrophil-bound contact system generates FXIIa which selectively induces neutrophil aggregation and degranulation (15). FXIIa-mediated activation of PK and/or release of neutrophil-derived tissue kallikrein, may be responsible for the circumscribed formation of BK. The local release of BK in turn may facilitate the escape of neutrophils toward the extracellular space by causing endothelial cell retraction, and permits transudation of plasma content by controlling vascular permeability at sites of inflammation (Figure 2).
In addition to neutrophil pro-inflammatory responses, FXII modulates the functions of an array of innate immune cells (47, 60, 61). Previous studies have shown that macrophages react to FXII and FXIIa, by increasing the production of IL-6, IL-12, and tumor necrosis factor (TNF)-α. Moreover, peripheral blood mononuclear cells treated with FXII acquire an M2-tissue reparative phenotype as seen by increased secretion of IL-4, IL-8, IL-10, and transforming growth factor (TGF)-β. Finally, FXII was shown to promote the differentiation of T helper (Th) naive cells to TH17 cells (62) (Figure 2).
Collectively, these data highlight two key concepts: (i) zymogen FXII is able to directly influence innate immune functions, and (ii) FXII and FXIIa exert distinct and separate cellular effects.
Influence of FXII and FXIIa on Sterile Inflammatory Tissue Responses
Here, we focus on how FXII- and FXIIa-mediated responses in innate and adaptive immune cells are integrated in the activation, regulation and effector mechanisms that lead to diverse pathologies and extend beyond their role in contact activation.
FXII-Neutrophil Crosstalk in Wound Healing and Tissue Disrepair
Wound healing is an intricate process that consists of multiple phases, each of which is indispensable for proper tissue repair. Prompt initiation and resolution of each wound healing phase namely, hemostasis, inflammation, proliferation, and tissue remodeling, is critical for promoting repair and avoiding scar formation. While neutrophils play a central role in the inflammatory phase, recent evidence suggests that their prompt removal is equally important for orderly wound progression through subsequent phases of healing (63). Animal models show that excess neutrophil entry into wound sites interferes with keratinocyte proliferation and migration (64). In addition to these effects, lengthy neutrophil presence in wounds leads to unrestricted proteolytic activity by neutrophil granular enzymes that eventually leads to a pathologic chain of events leading to matrix degradation and proteolytic inactivation of growth factors and their receptors (65). Indeed, proteomic studies showed that levels of neutrophil granular enzymes, among them neutrophil elastase, are increased in the exudate of non-healing human wounds and is thought to reflect a chronic, inflammatory, tissue-destructive microenvironment (66). In contrast, levels of alpha1-antitrypsin, the physiologic inhibitor of neutrophil elastase, were increased in well-healing wounds (65). Neutrophil elastase is a critical component of NETs and contributes to their function (67). Recent studies show that circulating neutrophils from diabetic individuals are primed constitutively to produce NETs (68), and NETosis delayed diabetic wound healing in mice and humans (69). Therefore, continued recruitment, or buildup of active neutrophils, inevitably prolongs inflammation and contributes to the development of chronic wounds.
In two models of sterile inflammation, we show that neutrophil recruitment is decreased in FXII deficient (F12−/−) mice and this is a bone marrow-endowed function (49). Decreased neutrophil migration at sites of inflammation was associated with reduced NETs into the wound microenvironment and improved wound healing (49). Altogether, these data support that limiting the activity of neutrophils may be beneficial for the treatment of recalcitrant wounds and provide the rationale for our on-going studies, harnessing the FXII-uPAR axis in neutrophils as a therapeutic strategy to promote wound healing. Given that loss of uPA but not uPAR delays wound healing (70), our findings confirm prior studies that the influence on wound healing is ligand dependent, not uPAR dependent.
The FXII-uPAR Axis in Cancer Maintenance and Progression
We previously published our findings on the contribution of FXII and polyP in prostate cancer-associated thrombosis (6). As FXII and FXIIa each exhibit growth factor properties, the question arises as to their potential involvement in regulating cancer cell behavior. uPAR is emerging as a cell surface-associated receptor that contributes to the development, progression, maintenance and metastasis of several cancers including epithelial ovarian cancer (EOC) (71–73). In EOC cells, uPAR has been reported to be overexpressed in more than 90% of ovarian cancer patients whereas, it is absent or minimally expressed in normal ovarian surface epithelium (74). uPAR overexpression in human ovarian cancer is associated with decreased overall survival (71–73). Moreover, global gene expression analysis revealed increased levels of F12 mRNA in EOC tumors but not in normal ovarian epithelium or fallopian tubes (75), and stimulation of ovarian cancer cells with FXII induced cell invasion (76). Interestingly, neutrophils have also recently been recognized to be important players in the ovarian tumor microenvironment, inducing epithelial-to-mesenchymal transition and promoting tumor invasion (53, 77, 78) (Figure 2). In this framework, ascertaining the role of host vs. tumor FXII-uPAR in EOC progression is likely to be a promising line of investigation.
FXII and Thromboinflammation
In a traditional view, the pathogenesis of DVT can be captured by Virchow's triad, which postulates that three main factors contribute to VTE development: reduced blood flow (stasis), vascular endothelial damage, and a hypercoagulable state. This conventional paradigm dramatically shifted with the observation in recent years that neutrophils significantly contribute to thrombosis, termed thromboinflammation (5, 79, 80). The cooperation of platelets with neutrophils was identified using a murine model of DVT in which flow restriction induces thrombosis in the inferior vena cava (IVC) (5). In this model, platelets and neutrophils are promptly recruited to the vessel wall within hours of reduced blood flow and engage in heterotypic cell-cell interactions (5). These interactions facilitate DVT growth and propagation by: (1) supporting additional neutrophil recruitment; and (2) stimulating neutrophils to release NETs which are indispensable for subsequent DVT propagation through FXII autoactivation, coagulation factor assembly and fibrin formation (5) (Figure 2). Exposure of endogenous polyP on activated platelets or intravenous administration of polyP resulted in FXII activation and lethal pulmonary embolism in wild type mice. In contrast, genetic FXII deficiency or a FXIIa inhibitor (2, 81), prevented the development of PE. Recombinant Ixodes ricinus contact phase inhibitor (Ir-CPI), a Kunitz-type protein expressed by the tick Ixodes ricinus, specifically interacts with human FXIIa, FXIa, and PKa and results in prolongation of the aPTT in vitro (81). Intravenous administration of IrCPI in rats and mice caused rescued animals from venous thrombus formation in a dose-dependent fashion (81).
While multiple models have demonstrated the critical role of both FXII and neutrophils in thrombosis, some exceptions seem to exist. FXII and neutrophils were recently shown to be dispensable for vascular occlusion in the large veins of the head following combined “knockdown” of natural anticoagulants, protein C and antithrombin (82). However, unlike the IVC stenosis model which mimics human DVT by reducing venous return, the study by Heestermans et al. involved large alterations in the natural plasma protease inhibitor balance. This setting appears to be more reflective of a state of disseminated intravascular coagulation (DIC) with consumptive drop in platelet count, spontaneous intravascular thrombosis and tissue fibrin deposition, rather than DVT forming as a result of venous stasis in the absence of a severe hypercoagulable state. Although additional studies are required to demonstrate the clinical utility of FXIIa inhibitors in the management of DVT, it will be interesting to also examine whether there is potential for FXII inhibitors that interfere with zymogen FXII activation of cells, as a strategy to mitigate DVT development.
Influence of FXII in Autoimmunity
It is widely accepted that multiple sclerosis develops due to peripheral autoreactive T-cells which are able to pass through the blood–brain barrier, induce diffuse inflammatory lesions in the brain, thus leading to demyelination. Prior studies have shown that effector T-helper cells (TH1 and TH17) play a key role in the development of inflammation and tissue damage during CNS autoimmunity (83–85). Interaction of T cells with dendritic cells (DCs), antigen-presenting cells (APCs), is crucial for proper T-cell differentiation (86, 87). Excess presence of effector T-cells in brain lesions and increased levels of T-cell-derived cytokines in peripheral blood mononuclear cells (PBMCs) of patients also highlight the causal role of autoreactive T-cells in human MS (88, 89). Gobel et al. investigated the role of FXII in autoimmunity using an experimental model of MS (62). They show that F12−/− mice are less prone to developing CNS inflammation and FXII worsened features of experimental autoimmune encephalomyelitis (EAE). Pharmacologic blockade of FXIIa was protective against EAE, independently of Factor XI and kallikrein-kinin generation (62). Mechanistically, FXII stimulated DC-induced TH17-cell generation in a uPAR-dependent manner. These results increased our understanding on the role of FXII in adaptive immunity and its potential as a therapeutic target in autoimmune disease states (Figure 2).
Conclusion
In summary, new perspectives have emerged that call for a reappraisal of the role of FXII in vivo. Far beyond its fundamental property to autoactivate, FXII engages in complex receptor interactions that cooperatively influence immune cell behavior and contribute to physiologic and pathophysiologic responses. The details of the signaling pathways downstream of FXII are still incomplete. The challenge now is to elucidate the subtle molecular details of this versatile orchestrator and its impact on in vivo processes that determine health and disease.
Author Contributions
TR and ES wrote and edited all versions of the manuscript.
Funding
Work in ES's laboratory was supported by a grant from the National Institutes of Health (HL137695) and the Oscar D. Ratnoff Endowed Professorship. TR acknowledges the Deutsche Forschungsgemeinschaft (DFG)—A11/SFB 877 and B8/SFB 841 and a European Research Council grant (ERC-StG-2012-311575_F-12). The contents do not represent the views of the U.S. Department of Veterans Affairs or the United States Government.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Samuel M, Pixley RA, Villanueva MA, Colman RW, Villanueva GB. Human factor XII (Hageman factor) autoactivation by dextran sulfate. Circular dichroism, fluorescence, and ultraviolet difference spectroscopic studies. J Biol Chem. (1992) 267:19691–7.
2. Muller F, Mutch NJ, Schenk WA, Smith SA, Esterl L, Spronk HM, et al. Platelet polyphosphates are proinflammatory and procoagulant mediators in vivo. Cell. (2009) 139:1143–56. doi: 10.1016/j.cell.2009.11.001
3. Maas C, Govers-Riemslag JW, Bouma B, Schiks B, Hazenberg BP, Lokhorst HM, et al. Misfolded proteins activate factor XII in humans, leading to kallikrein formation without initiating coagulation. J Clin Investig. (2008) 118:3208–18. doi: 10.1172/JCI35424
4. van der Meijden PE, Munnix IC, Auger JM, Govers-Riemslag JW, Cosemans JM, Kuijpers MJ, et al. Dual role of collagen in factor XII-dependent thrombus formation. Blood. (2009) 114:881–90. doi: 10.1182/blood-2008-07-171066
5. von Bruhl ML, Stark K, Steinhart A, Chandraratne S, Konrad I, Lorenz M, et al. Monocytes, neutrophils, and platelets cooperate to initiate and propagate venous thrombosis in mice in vivo. J Exp Med. (2012) 209:819–35. doi: 10.1084/jem.20112322
6. Nickel KF, Ronquist G, Langer F, Labberton L, Fuchs TA, Bokemeyer C, et al. The polyphosphate-factor XII pathway drives coagulation in prostate cancer-associated thrombosis. Blood. (2015) 126:1379–89. doi: 10.1182/blood-2015-01-622811
7. Larsson M, Rayzman V, Nolte MW, Nickel KF, Bjorkqvist J, Jamsa A, et al. A factor XIIa inhibitory antibody provides thromboprotection in extracorporeal circulation without increasing bleeding risk. Sci Transl Med. (2014) 6:222ra17. doi: 10.1126/scitranslmed.3006804
8. Renne T, Pozgajova M, Gruner S, Schuh K, Pauer HU, Burfeind P, et al. Defective thrombus formation in mice lacking coagulation factor XII. J Exp Med. (2005) 202:271–81. doi: 10.1084/jem.20050664
9. Kleinschnitz C, Stoll G, Bendszus M, Schuh K, Pauer HU, Burfeind P, et al. Targeting coagulation factor XII provides protection from pathological thrombosis in cerebral ischemia without interfering with hemostasis. J Exp Med. (2006) 203:513–8. doi: 10.1084/jem.20052458
10. Pham M, Stoll G, Nieswandt B, Bendszus M, Kleinschnitz C. Blood coagulation factor XII–a neglected player in stroke pathophysiology. J Mol Med. (2012) 90:119–26. doi: 10.1007/s00109-011-0812-9
11. Matafonov A, Leung PY, Gailani AE, Grach SL, Puy C, Cheng Q, et al. Factor XII inhibition reduces thrombus formation in a primate thrombosis model. Blood. (2014) 123:1739–46. doi: 10.1182/blood-2013-04-499111
12. Revenko AS, Gao D, Crosby JR, Bhattacharjee G, Zhao C, May C, et al. Selective depletion of plasma prekallikrein or coagulation factor XII inhibits thrombosis in mice without increased risk of bleeding. Blood. (2011) 118:5302–11. doi: 10.1182/blood-2011-05-355248
13. Pathak M, Wilmann P, Awford J, Li C, Hamad BK, Fischer PM, et al. Coagulation factor XII protease domain crystal structure. J Thromb Haemost. (2015) 13:580–91. doi: 10.1111/jth.12849
14. Rebuck JW. The skin window as a monitor of leukocytic functions in contact activation factor deficiencies in man. Am J Clin Pathol. (1983) 79:405–13. doi: 10.1093/ajcp/79.4.405
15. Wachtfogel YT, Pixley RA, Kucich U, Abrams W, Weinbaum G, Schapira M, et al. Purified plasma factor XIIa aggregates human neutrophils and causes degranulation. Blood. (1986) 67:1731–7.
16. Henderson LM, Figueroa CD, Muller-Esterl W, Bhoola KD. Assembly of contact-phase factors on the surface of the human neutrophil membrane. Blood. (1994) 84:474–82.
17. Gordon EM, Venkatesan N, Salazar R, Tang H, Schmeidler-Sapiro K, Buckley S, et al. Factor XII-induced mitogenesis is mediated via a distinct signal transduction pathway that activates a mitogen-activated protein kinase. Proc Natl Acad Sci USA. (1996) 93:2174–9. doi: 10.1073/pnas.93.5.2174
18. LaRusch GA, Mahdi F, Shariat-Madar Z, Adams G, Sitrin RG, Zhang WM, et al. Factor XII stimulates ERK1/2 and Akt through uPAR, integrins, and the EGFR to initiate angiogenesis. Blood. (2010) 115:5111–20. doi: 10.1182/blood-2009-08-236430
19. Gasic GP, Arenas CP, Gasic TB, Gasic GJ. Coagulation factors X, Xa, and protein S as potent mitogens of cultured aortic smooth muscle cells. Proc Natl Acad Sci USA. (1992) 89:2317–20. doi: 10.1073/pnas.89.6.2317
20. Fernandez JA, Heeb MJ, Xu X, Singh I, Zlokovic BV, Griffin JH. Species-specific anticoagulant and mitogenic activities of murine protein S. Haematologica. (2009) 94:1721–31. doi: 10.3324/haematol.2009.009233
21. Bohm A, Flosser A, Ermler S, Fender AC, Luth A, Kleuser B, et al. Factor-Xa-induced mitogenesis and migration require sphingosine kinase activity and S1P formation in human vascular smooth muscle cells. Cardiovasc Res. (2013) 99:505–13. doi: 10.1093/cvr/cvt112
22. Ben Nasr A, Herwald H, Sjobring U, Renne T, Muller-Esterl W, Bjorck L. Absorption of kininogen from human plasma by Streptococcus pyogenes is followed by the release of bradykinin. Biochem J. (1997) 326(Pt. 3):657–60. doi: 10.1042/bj3260657
23. Ben Nasr A, Olsen A, Sjobring U, Muller-Esterl W, Bjorck L. Assembly of human contact phase proteins and release of bradykinin at the surface of curli-expressing Escherichia coli. Mol Microbiol. (1996) 20:927–35. doi: 10.1111/j.1365-2958.1996.tb02534.x
24. Herwald H, Morgelin M, Olsen A, Rhen M, Dahlback B, Muller-Esterl W, et al. Activation of the contact-phase system on bacterial surfaces–a clue to serious complications in infectious diseases. Nat Med. (1998) 4:298–302. doi: 10.1038/nm0398-298
25. Isenring J, Kohler J, Nakata M, Frank M, Jans C, Renault P, et al. Streptococcus gallolyticus subsp. gallolyticus endocarditis isolate interferes with coagulation and activates the contact system. Virulence. (2018) 9:248–61. doi: 10.1080/21505594.2017.1393600
26. Maas C, Oschatz C, Renne T The plasma contact system 2.0. Semin Thromb Hemost. (2011) 37:375–81. doi: 10.1055/s-0031-1276586
27. Persson K, Morgelin M, Lindbom L, Alm P, Bjorck L, Herwald H. Severe lung lesions caused by Salmonella are prevented by inhibition of the contact system. J Exp Med. (2000) 192:1415–24. doi: 10.1084/jem.192.10.1415
28. Kornberg A, Rao NN, Ault-Riche D. Inorganic polyphosphate: a molecule of many functions. Annu Rev Biochem. (1999) 68:89–125. doi: 10.1146/annurev.biochem.68.1.89
29. Verhoef JJ, Barendrecht AD, Nickel KF, Dijkxhoorn K, Kenne E, Labberton L, et al. Polyphosphate nanoparticles on the platelet surface trigger contact system activation. Blood. (2017) 129:1707–17. doi: 10.1182/blood-2016-08-734988
30. Smith SA, Choi SH, Davis-Harrison R, Huyck J, Boettcher J, Rienstra CM, et al. Polyphosphate exerts differential effects on blood clotting, depending on polymer size. Blood. (2010) 116:4353–9. doi: 10.1182/blood-2010-01-266791
31. Mutch NJ, Engel R, Uitte de Willige S, Philippou H, Ariens RA. Polyphosphate modifies the fibrin network and down-regulates fibrinolysis by attenuating binding of tPA and plasminogen to fibrin. Blood. (2010) 115:3980–8. doi: 10.1182/blood-2009-11-254029
32. Waack U, Warnock M, Yee A, Huttinger Z, Smith S, Kumar A, et al. CpaA is a glycan-specific adamalysin-like protease secreted by Acinetobacter baumannii that inactivates coagulation factor XII. MBio. (2018) 9:e01606-18. doi: 10.1128/mBio.01606-18
33. de Maat S, Bjorkqvist J, Suffritti C, Wiesenekker CP, Nagtegaal W, Koekman A, et al. Plasmin is a natural trigger for bradykinin production in patients with hereditary angioedema with factor XII mutations. J Allergy Clin Immunol. (2016) 138:1414–23 e9. doi: 10.1016/j.jaci.2016.02.021
34. Bjorkqvist J, de Maat S, Lewandrowski U, Di Gennaro A, Oschatz C, Schonig K, et al. Defective glycosylation of coagulation factor XII underlies hereditary angioedema type III. J Clin Invest. (2015) 125:3132–46. doi: 10.1172/JCI77139
35. Frick IM, Akesson P, Herwald H, Morgelin M, Malmsten M, Nagler DK, et al. The contact system–a novel branch of innate immunity generating antibacterial peptides. EMBO J. (2006) 25:5569–78. doi: 10.1038/sj.emboj.7601422
36. Imamura T, Potempa J, Pike RN, Travis J. Dependence of vascular permeability enhancement on cysteine proteinases in vesicles of Porphyromonas gingivalis. Infect Immun. (1995) 63:1999–2003.
37. Renne T. The procoagulant and proinflammatory plasma contact system. Semin Immunopathol. (2012) 34:31–41. doi: 10.1007/s00281-011-0288-2
38. Araujo RC, Kettritz R, Fichtner I, Paiva AC, Pesquero JB, Bader M. Altered neutrophil homeostasis in kinin B1 receptor-deficient mice. Biol Chem. (2001) 382:91–5. doi: 10.1515/BC.2001.014
39. Austinat M, Braeuninger S, Pesquero JB, Brede M, Bader M, Stoll G, et al. Blockade of bradykinin receptor B1 but not bradykinin receptor B2 provides protection from cerebral infarction and brain edema. Stroke. (2009) 40:285–93. doi: 10.1161/STROKEAHA.108.526673
40. Taylor SL, Wahl-Jensen V, Copeland AM, Jahrling PB, Schmaljohn CS. Endothelial cell permeability during hantavirus infection involves factor XII-dependent increased activation of the kallikrein-kinin system. PLoS Pathog. (2013) 9:e1003470. doi: 10.1371/journal.ppat.1003470
41. Gershom ES, Sutherland MR, Lollar P, Pryzdial EL. Involvement of the contact phase and intrinsic pathway in herpes simplex virus-initiated plasma coagulation. J Thromb Haemost. (2010) 8:1037–43. doi: 10.1111/j.1538-7836.2010.03789.x
42. Pixley RA, De La Cadena R, Page JD, Kaufman N, Wyshock EG, Chang A, et al. The contact system contributes to hypotension but not disseminated intravascular coagulation in lethal bacteremia. In vivo use of a monoclonal anti-factor XII antibody to block contact activation in baboons. J Clin Invest. (1993) 91:61–8. doi: 10.1172/JCI116201
43. de Oliveira GG, Antonio MP. Adult respiratory distress syndrome (ARDS): the pathophysiologic role of catecholamine-kinin interactions. J Trauma. (1988) 28:246–53. doi: 10.1097/00005373-198802000-00023
44. Fein AM, Bernard GR, Criner GJ, Fletcher EC, Good JT Jr, Knaus WA, et al. Treatment of severe systemic inflammatory response syndrome and sepsis with a novel bradykinin antagonist, deltibant (CP-0127). Results of a randomized, double-blind, placebo-controlled trial. CP-0127 SIRS and Sepsis Study Group. JAMA. (1997) 277:482–7. doi: 10.1001/jama.277.6.482
45. Stroo I, Zeerleder S, Ding C, Luken BM, Roelofs J, de Boer OJ, et al. Coagulation factor XI improves host defence during murine pneumonia-derived sepsis independent of factor XII activation. Thromb Haemost. (2017) 117:1601–14. doi: 10.1160/TH16-12-0920
46. Ghebrehiwet B, Randazzo BP, Dunn JT, Silverberg M, Kaplan AP. Mechanisms of activation of the classical pathway of complement by Hageman factor fragment. J Clin Invest. (1983) 71:1450–6. doi: 10.1172/JCI110898
47. Toossi Z, Sedor JR, Mettler MA, Everson B, Young T, Ratnoff OD. Induction of expression of monocyte interleukin 1 by Hageman factor (factor XII). Proc Natl Acad Sci USA. (1992) 89:11969–72. doi: 10.1073/pnas.89.24.11969
48. Hess R, Wujak L, Hesse C, Sewald K, Jonigk D, Warnecke G, et al. Coagulation factor XII regulates inflammatory responses in human lungs. Thromb Haemost. (2017) 117:1896–907. doi: 10.1160/TH16-12-0904
49. Stavrou EX, Fang C, Bane KL, Long AT, Naudin C, Kucukal E, et al. Factor XII and uPAR upregulate neutrophil functions to influence wound healing. J Clin Invest. (2018) 128:944–59. doi: 10.1172/JCI92880
50. Mahdi F, Madar ZS, Figueroa CD, Schmaier AH. Factor XII interacts with the multiprotein assembly of urokinase plasminogen activator receptor, gC1qR, and cytokeratin 1 on endothelial cell membranes. Blood. (2002) 99:3585–96. doi: 10.1182/blood.V99.10.3585
51. Mahdi F, Shariat-Madar Z, Kuo A, Carinato M, Cines DB, Schmaier AH. Mapping the interaction between high molecular mass kininogen and the urokinase plasminogen activator receptor. J Biol Chem. (2004) 279:16621–8. doi: 10.1074/jbc.M313850200
52. Reddigari SR, Shibayama Y, Brunnee T, Kaplan AP. Human hageman factor (factor XII) and high molecular weight kininogen compete for the same binding site on human umbilical vein endothelial cells. J Biol Chem. (1993) 268:11982–7.
53. Plesner T, Ploug M, Ellis V, Ronne E, Hoyer-Hansen G, Wittrup M, et al. The receptor for urokinase-type plasminogen activator and urokinase is translocated from two distinct intracellular compartments to the plasma membrane on stimulation of human neutrophils. Blood. (1994) 83:808–15.
54. Bal W, Sokolowska M, Kurowska E, Faller P. Binding of transition metal ions to albumin: sites, affinities and rates. Biochim Biophys Acta. (2013) 1830:5444–55. doi: 10.1016/j.bbagen.2013.06.018
55. Haase H, Hebel S, Engelhardt G, Rink L. The biochemical effects of extracellular Zn(2+) and other metal ions are severely affected by their speciation in cell culture media. Metallomics. (2015) 7:102–11. doi: 10.1039/C4MT00206G
56. Zhu D, Su Y, Zheng Y, Fu B, Tang L, Qin YX. Zinc regulates vascular endothelial cell activity through zinc-sensing receptor ZnR/GPR39. Am J Physiol Cell Physiol. (2018) 314:C404–14. doi: 10.1152/ajpcell.00279.2017
57. Zaidel-Bar R, Itzkovitz S, Ma'ayan A, Iyengar R, Geiger B. Functional atlas of the integrin adhesome. Nat Cell Biol. (2007) 9:858–67. doi: 10.1038/ncb0807-858
58. Renne T, Dedio J, David G, Muller-Esterl W. High molecular weight kininogen utilizes heparan sulfate proteoglycans for accumulation on endothelial cells. J Biol Chem. (2000) 275:33688–96. doi: 10.1074/jbc.M000313200
59. Renne T, Schuh K, Muller-Esterl W. Local bradykinin formation is controlled by glycosaminoglycans. J Immunol. (2005) 175:3377–85. doi: 10.4049/jimmunol.175.5.3377
60. Barbasz A, Kozik A. The assembly and activation of kinin-forming systems on the surface of human U-937 macrophage-like cells. Biol Chem. (2009) 390:269–75. doi: 10.1515/BC.2009.032
61. Vorlova S, Koch M, Manthey HD, Cochain C, Busch M, Chaudhari SM, et al. Coagulation factor XII induces pro-inflammatory cytokine responses in macrophages and promotes atherosclerosis in mice. Thromb Haemost. (2017) 117:176–87. doi: 10.1160/TH16-06-0466
62. Gobel K, Pankratz S, Asaridou CM, Herrmann AM, Bittner S, Merker M, et al. Blood coagulation factor XII drives adaptive immunity during neuroinflammation via CD87-mediated modulation of dendritic cells. Nat Commun. (2016) 7:11626. doi: 10.1038/ncomms11626
63. Widgerow AD. Cellular resolution of inflammation–catabasis. Wound Repair Regen. (2012) 20:2–7. doi: 10.1111/j.1524-475X.2011.00754.x
64. Mori R, Power KT, Wang CM, Martin P, Becker DL. Acute downregulation of connexin43 at wound sites leads to a reduced inflammatory response, enhanced keratinocyte proliferation and wound fibroblast migration. J Cell Sci. (2006) 119:5193–203. doi: 10.1242/jcs.03320
65. Buchstein N, Hoffmann D, Smola H, Lang S, Paulsson M, Niemann C, et al. Alternative proteolytic processing of hepatocyte growth factor during wound repair. Am J Pathol. (2009) 174:2116–28. doi: 10.2353/ajpath.2009.080597
66. Eming SA, Koch M, Krieger A, Brachvogel B, Kreft S, Bruckner-Tuderman L, et al. Differential proteomic analysis distinguishes tissue repair biomarker signatures in wound exudates obtained from normal healing and chronic wounds. J Proteome Res. (2010) 9:4758–66. doi: 10.1021/pr100456d
67. Brinkmann V, Reichard U, Goosmann C, Fauler B, Uhlemann Y, Weiss DS, et al. Neutrophil extracellular traps kill bacteria. Science. (2004) 303:1532–5. doi: 10.1126/science.1092385
68. Wong SL, Demers M, Martinod K, Gallant M, Wang Y, Goldfine AB, et al. Diabetes primes neutrophils to undergo NETosis, which impairs wound healing. Nat Med. (2015) 21:815–9. doi: 10.1038/nm.3887
69. Fadini GP, Menegazzo L, Rigato M, Scattolini V, Poncina N, Bruttocao A, et al. NETosis delays diabetic wound healing in mice and humans. Diabetes. (2016) 65:1061–71. doi: 10.2337/db15-0863
70. Connolly BM, Choi EY, Gardsvoll H, Bey AL, Currie BM, Chavakis T, et al. Selective abrogation of the uPA-uPAR interaction in vivo reveals a novel role in suppression of fibrin-associated inflammation. Blood. (2010) 116:1593–603. doi: 10.1182/blood-2010-03-276642
71. Pedersen N, Schmitt M, Ronne E, Nicoletti MI, Hoyer-Hansen G, Conese M, et al. A ligand-free, soluble urokinase receptor is present in the ascitic fluid from patients with ovarian cancer. J Clin Invest. (1993) 92:2160–7. doi: 10.1172/JCI116817
72. Memarzadeh S, Kozak KR, Chang L, Natarajan S, Shintaku P, Reddy ST, et al. Urokinase plasminogen activator receptor: Prognostic biomarker for endometrial cancer. Proc Natl Acad Sci USA. (2002) 99:10647–52. doi: 10.1073/pnas.152127499
73. Davidson B, Trope CG, Reich R. The role of the tumor stroma in ovarian cancer. Front Oncol. (2014) 4:104. doi: 10.3389/fonc.2014.00104
74. Kenny HA, Leonhardt P, Ladanyi A, Yamada SD, Montag A, Im HK, et al. Targeting the urokinase plasminogen activator receptor inhibits ovarian cancer metastasis. Clin Cancer Res. (2011) 17:459–71. doi: 10.1158/1078-0432.CCR-10-2258
75. Wang E, Ngalame Y, Panelli MC, Nguyen-Jackson H, Deavers M, Mueller P, et al. Peritoneal and subperitoneal stroma may facilitate regional spread of ovarian cancer. Clin Cancer Res. (2005) 11:113–22.
76. Wang R, Zhang T, Ma Z, Wang Y, Cheng Z, Xu H, et al. The interaction of coagulation factor XII and monocyte/macrophages mediating peritoneal metastasis of epithelial ovarian cancer. Gynecol Oncol. (2010) 117:460–6. doi: 10.1016/j.ygyno.2010.02.015
77. Bednarska K, Klink M, Wilczynski JR, Szyllo K, Malinowski A, Sulowska Z, et al. Heterogeneity of the Mac-1 expression on peripheral blood neutrophils in patients with different types of epithelial ovarian cancer. Immunobiology. (2016) 221:323–32. doi: 10.1016/j.imbio.2015.10.003
78. Mayer C, Darb-Esfahani S, Meyer AS, Hubner K, Rom J, Sohn C, et al. Neutrophil granulocytes in ovarian cancer - induction of epithelial-to-mesenchymal-transition and tumor cell migration. J Cancer. (2016) 7:546–54. doi: 10.7150/jca.14169
79. Martinod K, Demers M, Fuchs TA, Wong SL, Brill A, Gallant M, et al. Neutrophil histone modification by peptidylarginine deiminase 4 is critical for deep vein thrombosis in mice. Proc Natl Acad Sci USA. (2013) 110:8674–9. doi: 10.1073/pnas.1301059110
80. Fuchs TA, Brill A, Duerschmied D, Schatzberg D, Monestier M, Myers DD Jr, et al. Extracellular DNA traps promote thrombosis. Proc Natl Acad Sci USA. (2010) 107:15880–5. doi: 10.1073/pnas.1005743107
81. Decrem Y, Rath G, Blasioli V, Cauchie P, Robert S, Beaufays J, et al. Ir-CPI, a coagulation contact phase inhibitor from the tick Ixodes ricinus, inhibits thrombus formation without impairing hemostasis. J Exp Med. (2009) 206:2381–95. doi: 10.1084/jem.20091007
82. Heestermans M, Salloum-Asfar S, Streef T, Laghmani EH, Salvatori D, Luken BM, et al. Mouse venous thrombosis upon silencing of anticoagulants depends on tissue factor and platelets, not FXII or neutrophils. Blood. (2019) 133:2090–9. doi: 10.1182/blood-2018-06-853762
83. Steinman L. A brief history of T(H)17, the first major revision in the T(H)1/T(H)2 hypothesis of T cell-mediated tissue damage. Nat Med. (2007) 13:139–45. doi: 10.1038/nm1551
84. Steinman L. A rush to judgment on Th17. J Exp Med. (2008) 205:1517–22. doi: 10.1084/jem.20072066
85. Korn T, Bettelli E, Oukka M, Kuchroo VK. IL-17 and Th17 cells. Annu Rev Immunol. (2009) 27:485–517. doi: 10.1146/annurev.immunol.021908.132710
86. Wu HY, Quintana FJ, da Cunha AP, Dake BT, Koeglsperger T, Starossom SC, et al. In vivo induction of Tr1 cells via mucosal dendritic cells and AHR signaling. PLoS ONE. (2011) 6:e23618. doi: 10.1371/journal.pone.0023618
87. Quintana FJ, Basso AS, Iglesias AH, Korn T, Farez MF, Bettelli E, et al. Control of T(reg) and T(H)17 cell differentiation by the aryl hydrocarbon receptor. Nature. (2008) 453:65–71. doi: 10.1038/nature06880
88. Lock C, Hermans G, Pedotti R, Brendolan A, Schadt E, Garren H, et al. Gene-microarray analysis of multiple sclerosis lesions yields new targets validated in autoimmune encephalomyelitis. Nat Med. (2002) 8:500–8. doi: 10.1038/nm0502-500
Keywords: factor XII, uPAR, contact activation, innate immunity, sepsis, neutrophil extracellular traps, cancer progression, wound healing
Citation: Renné T and Stavrou EX (2019) Roles of Factor XII in Innate Immunity. Front. Immunol. 10:2011. doi: 10.3389/fimmu.2019.02011
Received: 14 June 2019; Accepted: 08 August 2019;
Published: 22 August 2019.
Edited by:
Krasimir Kolev, Semmelweis University, HungaryReviewed by:
Yi Wu, Temple University, United StatesSven Hammerschmidt, University of Greifswald, Germany
Copyright © 2019 Renné and Stavrou. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Evi X. Stavrou, ZXZpLnN0YXZyb3VAY2FzZS5lZHU=