 Wai H. Lim
Wai H. Lim Meena Shingde3
Meena Shingde3
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol. , 14 August 2019
Sec. Alloimmunity and Transplantation
Volume 10 - 2019 | https://doi.org/10.3389/fimmu.2019.01944
This article is part of the Research Topic Kidney Transplantation and Immune-Mediated Nephropathies View all 6 articles
The prevalence, pathogenesis, predictors, and natural course of patients with recurrent glomerulonephritis (GN) occurring after kidney transplantation remains incompletely understood, including whether there are differences in the outcomes and advances in the treatment options of specific GN subtypes, including those with de novo GN. Consequently, the treatment options and approaches to recurrent disease are largely extrapolated from the general population, with responses to these treatments in those with recurrent or de novo GN post-transplantation poorly described. Given a greater understanding of the pathogenesis of GN and the development of novel treatment options, it is conceivable that these advances will result in an improved structure in the future management of patients with recurrent or de novo GN. This review focuses on the incidence, genetics, characteristics, clinical course, and risk of allograft failure of patients with recurrent or de novo GN after kidney transplantation, ascertaining potential disparities between “high risk” disease subtypes of IgA nephropathy, idiopathic membranous glomerulonephritis, focal segmental glomerulosclerosis, and membranoproliferative glomerulonephritis. We will examine in detail the management of patients with high risk GN, including the pre-transplant assessment, post-transplant monitoring, and the available treatment options for disease recurrence. Given the relative paucity of data of patients with recurrent and de novo GN after kidney transplantation, a global effort in collecting comprehensive in-depth data of patients with recurrent and de novo GN as well as novel trial design to test the efficacy of specific treatment strategy in large scale multicenter randomized controlled trials are essential to address the knowledge deficiency in this disease.
Primary glomerulonephritis (GN) continues to be one of the leading causes of end-stage kidney disease (ESKD) in the United States (US) and worldwide. According to the 2018 Australia and New Zealand Dialysis and Transplant (ANZDATA) registry report, GN as cause of ESKD accounted for 17% of incident treated ESKD patients (1). In the US, GN as cause of ESKD comprised of 7 and 13% of incident ESKD initiated on dialysis and have received kidney transplants, respectively; with similar proportion reported in the United Kingdom (2, 3). GN is a heterogeneous group of immunological kidney diseases with distinct histological subtypes, causes (primary vs. secondary) and clinical phenotypes, resulting in substantial differences in the prognosis after kidney transplantation in patients with dissimilar GN subtypes, including the risk of disease recurrence post-transplantation (4–7). Consequently, clinicians must provide sufficient information with regards to the risk of disease recurrence post-transplant, when considering the medical suitability of potential kidney transplant candidates with GN for kidney transplantation (4–6, 8–11).
Following kidney transplantation, recurrent or de novo GN in the renal allograft is an important cause of premature allograft failure (12). All GN subtypes can potentially recur after transplantation, with the prevalence of GN recurrence between 3 and 15%, particularly in patients with high risk subtypes of IgA nephropathy, idiopathic membranous GN, focal segmental glomerulosclerosis (FSGS), and membranoproliferative GN (MPGN) (4, 5, 9, 13, 14). Nevertheless, the incidence of de novo or recurrent GN after kidney transplantation is likely to be under-estimated because of the likelihood of selection bias (i.e., systematic differences in the selection and listing of ESKD patients with different GN subtypes), varying biopsy practices, ascertainment of the primary cause of ESKD, differing follow-up period, disparate clinical presentations ranging from asymptomatic urinary abnormalities to rapidly progressive GN, misclassification, and indication bias (where kidney biopsy may be carried out only for specific clinical indication and therefore may fail to identify asymptomatic incidental cases of early disease recurrence) and the competing risk of other causes of allograft failures. The risk of GN recurrence is typically directly related to incremental time post-transplant, with the majority of GN recurrence resulting in allograft failure occurring after 3–5 years post-transplant, although early recurrences can occur in patients with GN subtypes of MPGN and FSGS.
The understanding and characterization of the incidence of GN recurrence occurring after kidney transplantation is largely confined to the availability of data from several large registries and single center reports from the US, Australia, and New Zealand (4, 5, 9, 13). The incidence of GN recurrence post-transplant varies according to GN subtypes and time post-transplantation. Of patients with ESKD secondary to primary GN, particularly FSGS and MPGN, there is a high risk of GN recurrence with a substantial proportion of patients with disease recurrence experiencing premature allograft failure.
Recurrent IgA nephropathy is relatively common, but typically occurs late post-transplant with cumulative incidence of disease recurrence at 15 years of 15% although the risk of disease recurrence may be reducing over time (Table 1) (5, 17). There is substantial variation between studies but likely to reflect disparate follow-up period, differing biopsy practices between centers and the relative benign presentations of the majority of patients (presenting with microscopic hematuria or evidence of IgA deposition in allograft biopsies). Following disease recurrence, up to 40% of patients with recurrent IgA nephropathy have been reported to lose their allografts, predominantly from disease recurrence (up to 60%) (5). Compared with other GN subtypes, the long-term allograft and patient outcomes of patients with IgA nephropathy are substantially better. In the US Renal Data System analysis of 32,131 patients with ESKD secondary to IgA nephropathy, the rates of all-cause mortality, overall allograft failure and death censored allograft failure were 1.2, 3.4, and 2.6 events/100-person-years, respectively; compared with respective 2.8, 6.7, and 5.1 events/100-person-years for patients with MPGN; 2.5, 6.1, and 4.4 events/100-person-years, respectively for patients with FSGS; and 3.1, 6.1, and 4.0 events/100-person-years, respectively for patients with idiopathic membranous GN (6). In this study, the proportion of allograft failure secondary to disease recurrence was 1.6% for those with IgA nephropathy, compared with 5.6, 2.7, and 3.3% for those with MPGN, FSGS, and idiopathic membranous GN, respectively (6). In contrast, data from the ANDATA registry showed that 5-year allograft survival following disease recurrence was similar between IgA nephropathy, idiopathic membranous GN, and FSGS, with the allograft survival of patients with MPGN substantially poorer compared to other GN subtypes (5).
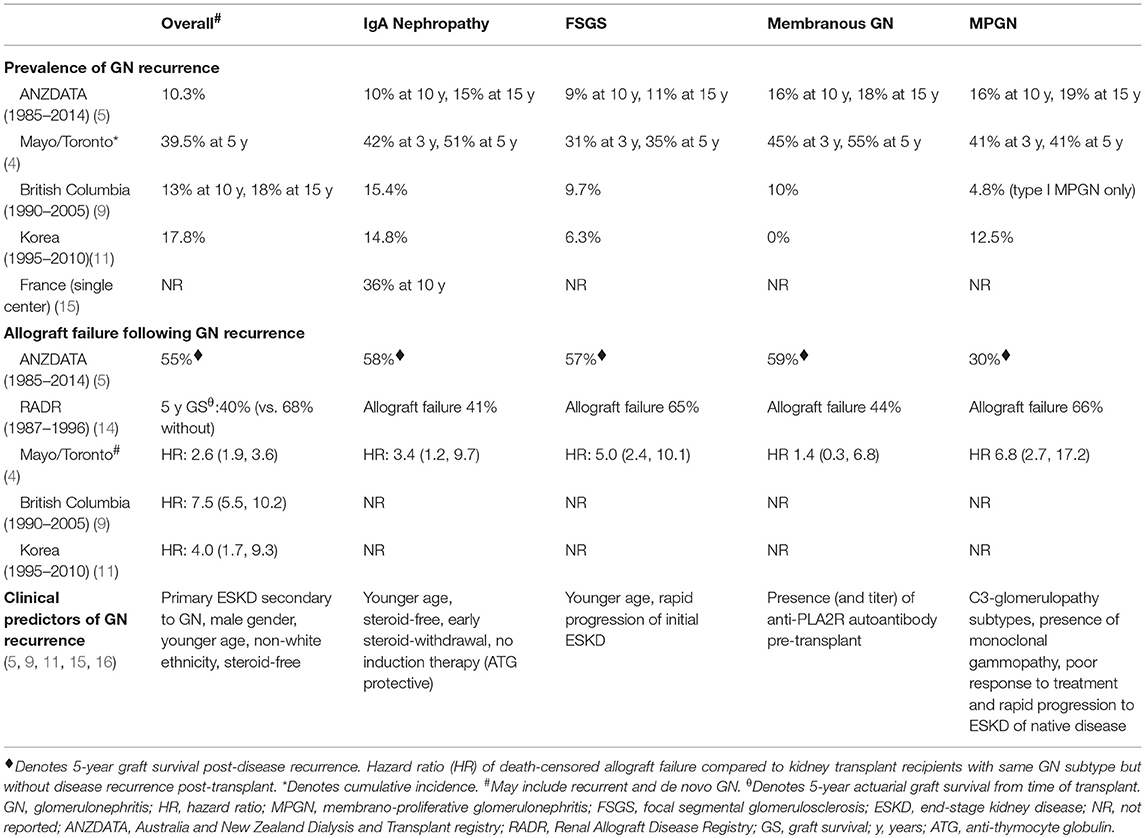
Table 1. Prevalence, risk of allograft failure and clinical predictors of glomerulonephritis recurrence post-kidney transplantation.
Several risk factors for disease recurrence have been described including younger age, recipients of zero-HLA-mismatched live-related donor kidneys, steroid-avoidance or early steroid-withdrawal immunosuppressive regimens, the non-use of induction therapy (whereas anti-thymocyte globulin [ATG] may be associated with a lower risk of recurrence), HLA allelic subtypes, crescentic (and rapidly progressive) IgA nephropathy in the native kidneys and shorter total ischemic time, but given these findings were identified in population cohort studies, it is difficult to ascertain the true causality of these risk factors for disease recurrence (5, 15, 16, 18–25). There are several molecules, including galactose-deficient IgA1 (Gd-IgA1), IgG anti-Gd-IgA1 antibodies, glycan-specific IgG antibodies, and soluble CD89 (an Fc receptor for IgA) that may be implicated in the pathogenesis of IgA nephropathy, the presence of which may portend a greater risk of disease progression and possibly disease recurrence post-transplant (26–32). There are several other non-specific serum and urine biomarkers that may predict the risk of disease recurrence after kidney transplantation, but the prognostic (and predictive) performance of many of these biomarkers have not been truly established or validated in independent population cohorts (Table 2). There are numerous other prognostic markers that have been investigated to predict disease progression of IgA nephropathy affecting the native kidneys, but the clinical relevance of these biomarkers in predicting disease recurrence post-transplant remains undetermined (51).
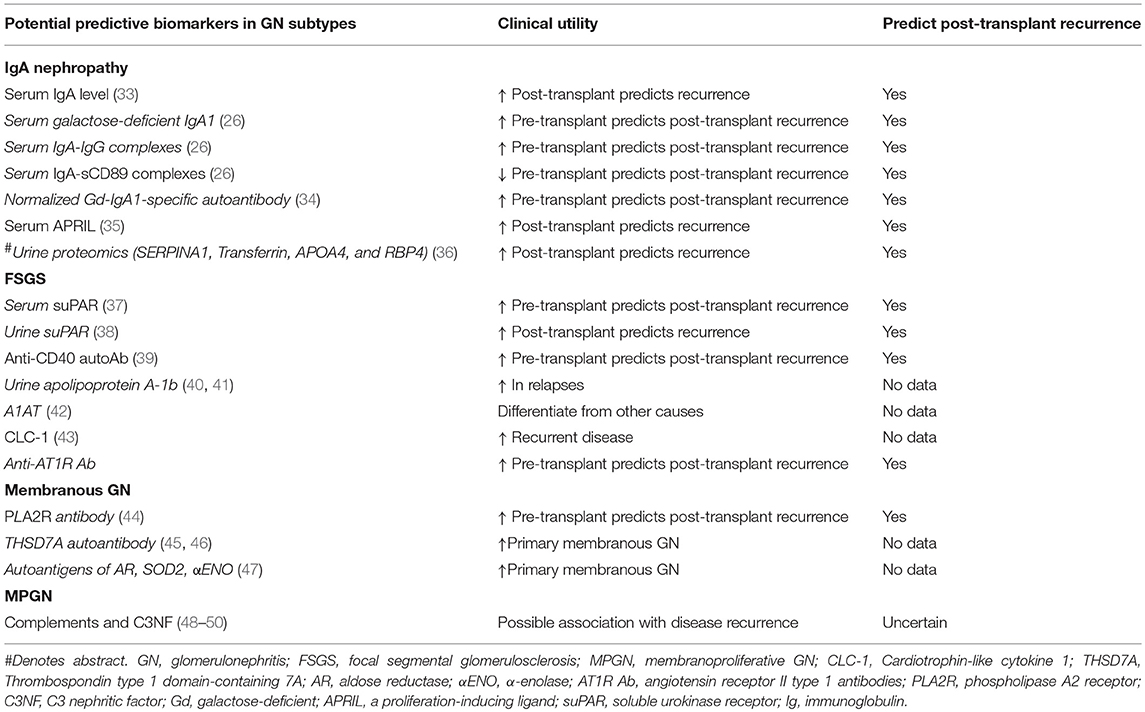
Table 2. Prognostic and predictive biomarkers for glomerulonephritis and recurrence of disease post-kidney transplant.
The optimal treatment of recurrent IgA nephropathy remains unknown and there are no current studies to suggest that alterations in immunosuppression will improve allograft outcomes (52). The current practice is to maintain (or change to) a calcineruin-inhibitor (CNI) and corticosteroids-based immunosuppressive regimen in addition to anti-proteinuric treatments, although the optimal dosing/target therapeutic CNI level or specific CNI type in the treatment of those with recurrent disease remains unknown (Table 3). In cases of crescentic rapidly progressive IgA nephropathy, more aggressive immunosuppression (e.g., cyclophosphamide or rituximab) may be considered but this is largely unproven and unlikely to successfully reverse the disease process (53–56). The potential benefit of tonsillectomy in disease recurrence has been limited to case reports and therefore cannot be recommended as a treatment option for patients with recurrent IgA nephropathy (57, 58).
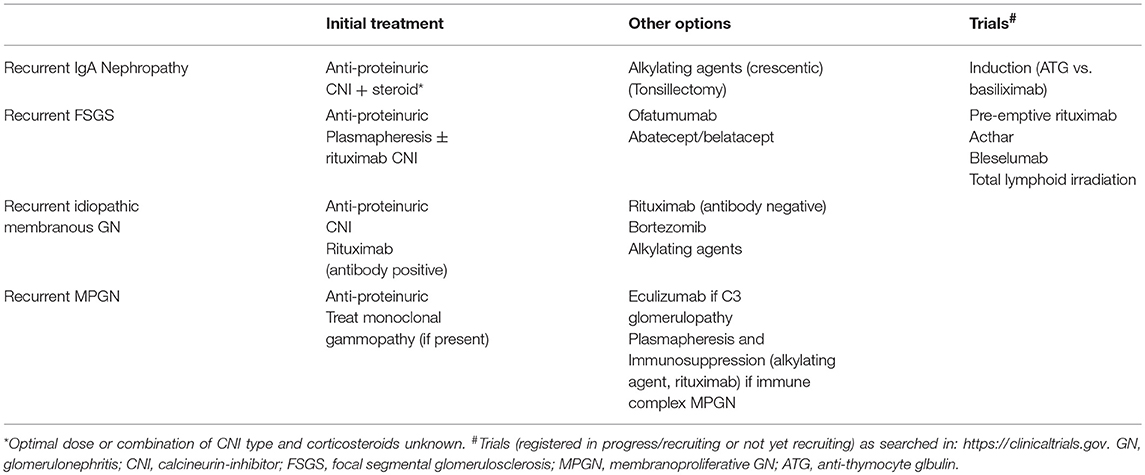
Table 3. Proposed management options for recurrent glomerulonephritis.
Up to 1 in 3 patients with primary FSGS will experience disease recurrence after kidney transplantation, with the risk of allograft failure (predominantly from GN recurrence) 5-times the risk compared to those without disease recurrence (Table 1) (4, 5). In an ANZDATA registry analysis comprising of 736 first kidney transplant recipients with biopsy-proven primary FSGS, 10% of patients experienced disease recurrence, with disease recurrence associated with substantially poorer 5-year allograft survival of 52% (95% confidence interval [95%CI], 40%, 63%), compared with 83% (95%CI 79%, 86%) in those without recurrent disease (p < 0.001) (59). However, the true incidence of recurrent disease remains unknown as secondary forms of FSGS can occur late post-transplant, resulting in difficulties in differentiating primary from secondary forms.
In contrast to patients with primary FSGS, familial FSGS in adults, comprising of those with mutations of podocin or structural podocyte proteins [e.g., NPHS2, including those with homozygous or compound heterozygous mutations in podocin or the p.R229Q variant; slit diaphragm-associated transient receptor potential channel C6 [TRPC6] gain-of-function mutation] and apolipoprotein L-1 genotype have low to no risk (<3%) of disease recurrence post-transplant suggesting the relative importance of genetic testing in the evaluation of a subset of patients with adult-onset FSGS for transplantation (60–67). Other than a known family history of chronic kidney disease (which may suggest an autosomal dominant inheritance), many of the genetic mutations associated with adult-onset FSGS have an incomplete penetrance and therefore, the identification of patients with familial FSGS remains challenging (68). Clinicians should consider undertaking genetic screening for patients with adult-onset FSGS when there is uncertainty regarding the likelihood of primary (atypical clinical/pathological features or poor response to immunosuppressive treatment) and secondary (no obvious causes identified) FSGS or when there is a clear family history of FSGS (69, 70). Taking into consideration the cost associated with genetic screening in all patients with adult-onset FSGS, it may not be cost-effective to screen all patients (even with the above criteria) and therefore, it may be more practical to consider screening for patients with a clear family history of FSGS or those with a potential live-related donor for transplantation, including undertaking genetic screening of the donors for the same genetic mutations (if present in the potential recipients). In the absence of genetic mutation in the potential recipients, genetic screening of live-related donors is not currently recommended, although cases of live donors developing FSGS post-donation have been reported (66, 71). A shared-decision approach between clinicians and patients regarding the clinical rationale for genetic screening for patients and potential live-related donors should be considered, balancing between the cost, the clinical utility of the information in the current/future medical management of these patients pre- and post-transplant and the implications for prognostication (post-transplant) and appropriateness of genetic counseling. A similar practical approach to genetic testing may be considered for pediatric patients with FSGS, including those with idiopathic steroid-resistant nephrotic syndrome with FSGS pathology. In a Spanish cohort of 98 children or adolescent patients with FSGS (<18 years at presentation), none of the 7 patients (presented with steroid-resistant nephrotic syndrome and documented NPHS2 mutation) who had received a kidney transplant experienced disease recurrence, suggesting that similar to the adult population, there is a low risk of FSGS recurrence in pediatric patients with genetic FSGS post-transplant (72, 73). There are several other risk factors for disease recurrence in patient with primary FSGS that have been identified, although these are primarily non-specific clinical parameters including younger age at presentation, recipients of live-donor kidneys, non-white ethnicity, severe manifestations of disease at presentation, rapid progression to ESKD, and prior allograft failure from disease recurrence (5, 59, 74).
The pathogenesis of disease recurrence in patients with primary FSGS remains unclear, with no studies confirming the presence of circulating permeability factor(s) causing podocyte injury instigating early disease recurrence. A pathogenic role of the circulating serum soluble urokinase receptor (suPAR) has been proposed, by activating podocyte β(3) integrin resulting in effacement of foot processes and proteinuria, which may contribute to the development of primary FSGS. In two cohorts of pediatric and adult patients with primary FSGS from North America [n = 70, North America–based FSGS clinical trial [FSGS-CT] cohort] (75) and Europe (n = 94, PodoNet Registry cohort) (76), serum suPAR level (threshold of 3,000 pg/ml) was elevated in over 50% of patients (84% FSGS-CT and 55% PoDoNet cohorts), compared with 6% of age- and sex-matched healthy controls (n = 150) (77, 78). Interestingly, in the European cohort, serum suPAR level was higher in patients with familial FSGS compared to those with non-genetic primary FSGS, but this finding will need to be validated in other cohorts. In addition, reduction of serum suPAR level with immunosuppressive treatment was associated with a greater likelihood of achieving clinical remission, raising the possibility that suPAR may be involved in the pathogenesis of this disease (78). However, this patho-physiological link remains debatable (37, 79–81). Furthermore, serum suPAR level can also be non-specifically elevated in other pathological processes including inflammation and infection and has been shown to be an independent prognostic biomarker in predicting future risk of CVD and mortality in the general population (82, 83).
Nevertheless, the diagnostic test accuracy of suPAR in differentiating primary FSGS from other proteinuric diseases or to predict disease recurrence after transplantation remains suboptimal (84, 85) and consequently, the clinical utility of routinely monitoring suPAR levels post-transplant to predict those at risk of disease recurrence remains poorly defined. Other potential biomarkers including urine suPAR, Anti-CD40 autoantibody, and angiotensin receptor II type 1 (AT1R) antibody appear promising but further studies are required to determine the accuracy of these prognostic biomarkers in predicting disease recurrence (Table 2). Close monitoring for proteinuria in high risk patients, with regular checks of urine protein/creatinine ratio or self-check urine dip-stick in the first 3 months post-transplant are recommended, and proceeding to a kidney biopsy (tissues should be sent for electron microscopy to detect early effacement of foot processes) if there is persistent or increasing proteinuria (86).
The management of patients with recurrent primary FSGS remains challenging, with treatment strategies informed predominantly by small case series (Table 3). Plasmapheresis is often preferred and recommended (in the American Society for Apheresis guidelines) in the treatment of primary FSGS recurrence in the allograft of both pediatric and adult patients (87–89). The efficacy of adjunctive therapy including rituximab and CNI such as cyclosporine remains uncertain. The case report of the efficacy of rituximab in ameliorating early primary FSGS recurrence post-transplant in a child with post-transplant lymphoma had generated considerable interest and suggested that B cells may have a role in the pathogenesis of disease recurrence in a subgroup of patients with primary FSGS (90). Recent research showed that rituximab binds sphingomyelin phosphodiesterase acid-like 3b (SMPDL-3b) protein, resulting in preservation of podocyte SMPDL-3b expression, preventing podocyte apoptosis, and maintaining the integrity of podocyte actin cytoskeleton, therefore highlighting the biological rationale of this therapy in patients with FSGS independent of its effect on B cells (91). Given the lack of convincing evidence to suggest B cells is directly implicated in the pathogenesis of primary and recurrent FSGS (despite one small biopsy series showing higher number of glomerular B cells in patients with FSGS) (92), the mechanism by which rituximab may be effective in reducing proteinuria in patients with recurrent FSGS may be through its effect on podocyte function (91, 93). CNI such as cyclosporine, through the inhibitory effect on T cell function and stabilization effect on actin cytoskeleton in kidney podocytes, has been shown to induce clinical remission in patients with recurrent FSGS. In patients already maintained on CNI and have experienced disease recurrence post-transplant, there is no data to suggest that changing to an alternative CNI (e.g., from cyclosporine to tacrolimus or from tacrolimus to cyclosporine) will be effective. However, a switch to CNI if patients were on mammalian target of rapamycin [mTOR]-inhibitor is advocated, given that mTOR-inhibitor, especially at higher doses have been associated with the development of de novo FSGS (94–96). What remains unknown is whether plasmapheresis is always necessary and whether rituximab should be considered as first line therapy for disease recurrence or reserved only for cases of recurrence refractory to plasmapheresis (97).
The roles of pre-emptive plasmapheresis (pre and/or post-transplantation), immunoadsorption therapy, and other novel options such as ofatumumab or B7-1 blockers (abatacept and belatacept) to prevent disease recurrence or to treat recurrence appear promising but the efficacy of these treatments remain debatable and not always consistent or supported in subsequent studies (98–105). Ofatumumab, an anti-CD20 monoclonal antibody that induces profound B cell depletion appears promising in the prevention (n = 1) and treatment (n = 2, achieved partial remission) of recurrent FSGS occurring post-kidney transplant but this needs to be confirmed in large studies (103, 106, 107). The initial treatment success surrounding the efficacy of abatacept, an inhibitor of B7-1 co-stimulatory molecule in achieving clinical remission in 5 patients with FSGS (4 patients with rituximab-resistant recurrent FSGS post-kidney transplant and 1 patient with steroid-resistant primary FSGS; all with positive B7-1 immunostaining of podocytes) has not been corroborated in other cohorts (100). Five subsequent studies showed that abatacept or belatacept was not effective in the treatment of 23 patients with FSGS recurrence post-kidney transplant, although the majority of the patients did not have positive podocyte B7-1 expression (101, 102, 105, 108, 109).
In a retrospective historical-control study of 26 pediatric patients with FSGS, prophylactic pre- and post-transplant plasmapheresis did not prevent FSGS recurrence compared to those who did not undergo similar prophylactic treatment (110). Several pilot studies evaluating the efficacy of an intensive and prolonged plasmapheresis with and without high-dose corticosteroids and maintenance CNI therapy or rituximab (2 studies, n = 22 adult patients with primary FSGS recurrence [2 in native kidneys], up to 15 months of treatment) or immunoadsorption (n = 12 pediatric patients with early FSGS recurrence) may be effective in achieving partial or complete remission but given the retrospective nature of these studies in a small number of patients, these and other treatment regimens will need further appraisal in randomized controlled trials to ascertain the optimal management in preventing or treatment of FSGS recurrence (104, 111, 112). A practical approach in the management of FSGS recurrence post-transplantation should initially include plasmapheresis, maximizing anti-proteinuric therapy, and converting to a CNI-based immunosuppressive regimen (where possible), with B cell depletion (rituximab) considered as adjunctive treatment or in resistant cases (Table 3). There is currently insufficient data to suggest that the pre-emptive use of plasmapheresis ± rituximab will reduce the risk of disease recurrence post-transplant.
Disease recurrence post-transplant from primary MPGN is relatively common, with over 50% of recurrence occurring within the first 24 months post-transplant (Table 1) (5, 13, 48, 49, 113, 114). In patients who had experienced disease recurrence, the risk of allograft failure is relatively high with 5-year allograft survival post-disease recurrence of only 30% (5). The introduction of a new classification of MPGN, which considers the differences in the pathogenesis and histological findings of MPGN subtypes (i.e., immune complex-mediated MPGN and complement-mediated MPGN), has enabled a more accurate assessment of the nature and course of the disease, including the risk of disease recurrence after kidney transplantation (115–117). Immune-complex mediated MPGN is characterized by the glomerular deposition of polyclonal or monoclonal immunoglobulins (Ig), whereas C3 glomerulopathy [comprising of C3GN and dense deposit disease (DDD)] is characterized by the glomerular deposition of C3 in the absence of Ig deposition (115, 117, 118). The pattern of glomerular Ig and complement product deposition may help to differentiate between the MPGN subtypes and has also provided much needed insights and information on the pathogenesis of the different disease processes (4, 116). Even though DDD (previously known as MPGN type II) and C3GN are distinctive diseases, the clinical course, pathogenesis and histological features of these two diseases may be similar. There is a high rate of post-transplant recurrence for both C3GN and DDD, with over 50% of patients with disease recurrence reported to experience allograft failure, although the number of patients in these studies was relatively small (119, 120). The timing and clinical presentations of patients with C3GN and DDD may be dissimilar, with DDD more likely to recur later post-transplant and often associated with no clinical manifestations other than allograft dysfunction. C3GN and DDD are characterized by the presence of strong glomerular staining for C3 and electron deposits on electron microscopy, but these diseases are potentially morphologically distinguishable by the nature and ultrastructural characteristics of these electron dense deposits (115, 121–123). The predominance of C3 deposition suggest the presence of dysregulated alternate complement cascade, resulting in the amplification and subsequent overproduction of C3 and related products of the terminal complement cascade (124). The exact cause of the complement dysregulation remains uncertain, although genetic mutations (e.g., presence of H402 and V62 alleles of Factor H, mutations of Factor H, and I genes) or autoantibodies (e.g., C3 or C4 nephritic factor directed against C3 convertase or Factor H autoantibodies) resulting in dysfunctional complement regulatory proteins and therefore uncontrolled amplification of the C3 protein have been implicated in the pathogenesis of this disease (125–129). In a case series of 21 kidney transplant recipients with C3GN as the cause of ESKD, monoclonal gammopathy was present in 3 of 14 (21%) patients who had experienced disease recurrence (suggesting the involvement of classical complement pathway), which was associated with a more rapid rate of disease recurrence (median time to recurrence 4 vs. 43 months, respectively) and allograft failure compared to those without monoclonal gammopathy (119).
For immune-mediated MPGN, the patterns and types of Ig deposits may have diagnostic and prognostic significance. The presence of serum monoclonal proteins (48, 49), with and without low complement levels (±glomerular C4d deposition) (130), implying activation of the complement cascade was associated with a less favorable clinical course post-transplant, with higher risk of recurrence and disease progression following recurrence. Similarly, the presence of glomerular monoclonal Ig deposits (typically IgG3κ and IgG3λ, but IgG2λ has been reported) was associated with poorer prognosis, characterized by early disease recurrence and substantially greater risk of premature allograft failure following disease recurrence (131–134). Up to 70% of patients with immune-mediated GN and monoclonal deposits have no evidence of plasma cell dyscrasia (i.e., absence of serum and urine monoclonal Ig proteins or evidence of plasma cell dyscrasia in the bone marrow), whereas in the remaining 30% of patients, patients often have detectable elevated monoclonal proteins without fulfilling the criteria for multiple myeloma (often termed “monoclonal gammopathies of renal significance”) (135–137). It is important to note that these disease processes on occasions have overlapping clinical and histological features (e.g., monoclonal gammopathy may be present in both immune complex-mediated and complement-mediated MPGN) and clinicians should consider undertaking a panel of investigations for all patients with MPGN being assessed for transplantation (Figure 1). Despite the advances in the current understanding of the pathogenesis and risk of disease recurrence in patients with MPGN, there continues to be residual uncertainties as to the relationships between patient and disease characteristics and the risk of disease recurrence and longer-term prognosis. Clinicians should be alerted of the need to undertake pre-transplant screening for monoclonal gammopathy ± hematology review in patients with MPGN, while ensuring close monitoring post-transplant for signs of disease recurrence. Open discussion to ensure patients with MPGN are counseled appropriately such that they are cognizant of the risk of disease recurrence post-transplant, weighing between this risk of premature allograft failure (if disease recurs) against that associated with remaining on dialysis (138). Figures 2A–D show the light microscopy and ultrastructural features of a kidney transplant recipient who had developed asymptomatic MPGN recurrence 12-months post-transplant, with similar patterns of electron dense deposits as the primary disease.
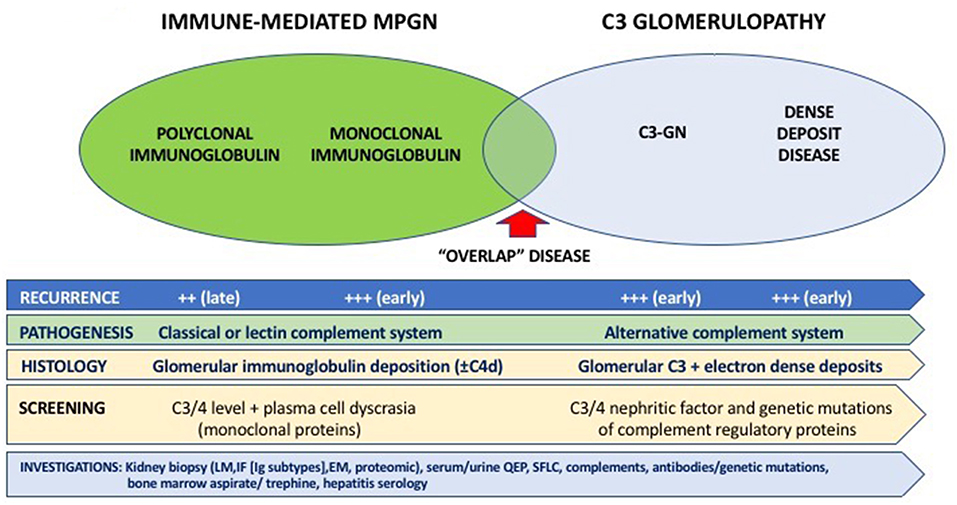
Figure 1. Overview of the classification, pathogenesis, characteristics, and diagnostic assessment of membranoproliferative glomerulonephritis (MPGN).
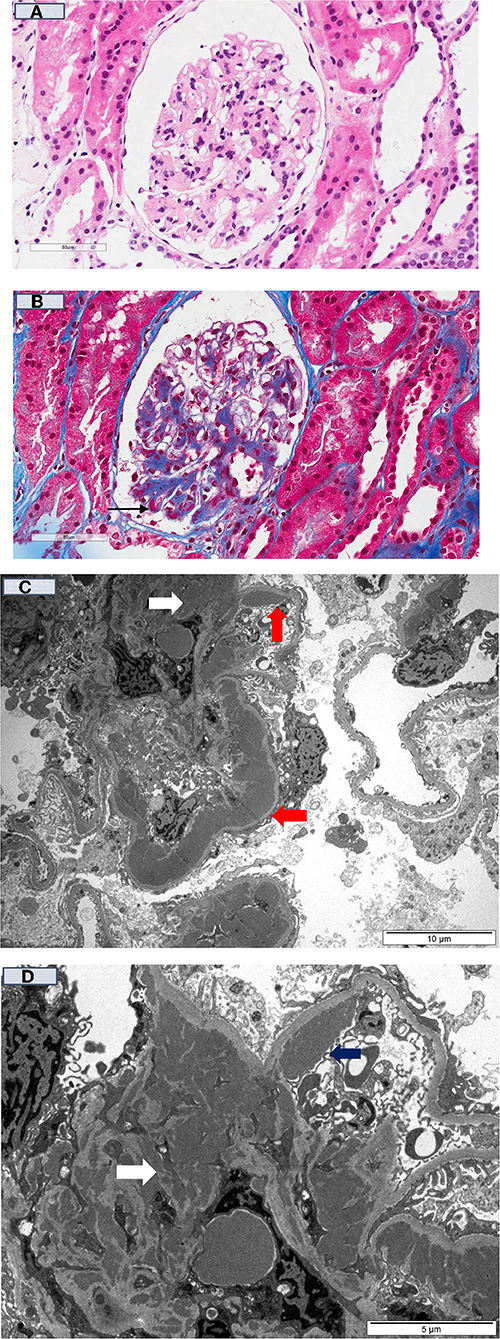
Figure 2. Kidney transplant recipient who had developed recurrent MPGN 12-months post-transplant. (A) [H & E (40x)]: Kidney allograft biopsy showed mesangial matrix expansion and a few thick peripheral capillary loops; (B) [Gomori trichrome stain (40X)] showed the thick peripheral capillary loops containing dense eosinophilic deposits (black arrow); and (C) (10 μm magnification), and (D) (5 μm magnification) showed numerous electron-dense deposits within the mesangium (white arrow) and large band-like intramembranous (red arrow) and subendothelial (blue arrow) electron-dense deposits within the thick peripheral capillary loops (Electron Microscopy).
The approach to treatment for recurrent disease is not well-established, limited to case series of successful treatment with the use of plasmapheresis and other immunosuppressive agents including cyclophosphamide, eculizumab, and rituximab (139–142). However, the optimal treatment strategy remains unknown (100, 101, 143, 144). In a case series of 7 patients with C3 glomerulopathy treated with eculizumab for a duration between 3 and 28 months (n = 5 with C3GN and n = 2 with DDD), 3 (all with C3GN) of the 7 patients had received a kidney transplant. Of these 3 patients, 2 had experienced disease recurrence between 3 and 48 months post-transplant, and 1 patient had developed de novo C3GN at 135 months post-transplant (original ESKD attributed to FSGS). The initiation of eculizumab in these 3 patients resulted in stabilization of kidney function and proteinuria in 2 patients, with no response in 1 patient and treatment with eculizumab was subsequently discontinued (139). Other reports comprising of 6 pediatric patients with MPGN I, C3GN, or DDD (n = 5 native kidneys and n = 1 with recurrence of C3 glomerulopathy post-kidney transplant) showed a modest benefit of eculizumab in reducing proteinuria and stabilization of kidney function suggesting a potential therapeutic role of eculizumab in a subgroup of patients (145, 146). Nevertheless, given the heterogeneity in the use and varying treatment responses to eculizumab in these cases (and small number of cases of post-transplant recurrence), larger studies are required to ascertain the true benefits of eculizumab in the treatment of disease recurrence post-transplant. The use of plasmapheresis and/or rituximab in the treatment of primary MPGN recurrence post-transplant has been limited to a small number of cases (n = 9), with modest response observed in half the cases. In one patient who had developed recurrent crescentic DDD and had failed treatment with rituximab and plasmapheresis, the introduction of eculizumab therapy led to discernible clinical and biochemical responses (119, 140, 141, 147, 148). In a retrospective Spanish cohort study of 60 patients with C3GN (affecting native kidneys), patients who were maintained on immunosuppressive treatment, particularly corticosteroids and mycophenolate were more likely to achieve clinical remission compared to untreated patients or those on other immunosuppressive regimens (149). However, the relevance of this observation in the treatment of disease recurrence post-transplantation is unknown with no current data to support a dose increase of mycophenolate or a change from an alternative regimen to a mycophenolate-based immunosuppressive regimen.
The new classification and current knowledge of MPGN may assist clinicians to adopt a personalized treatment strategy according to the most likely pathogenesis of the disease process (e.g., consideration of plasmapheresis and anti-B cell therapy for immune complex-mediated GN or those with monoclonal gammopathy or to consider eculizumab for C3 glomerulopathy) and the availability of more cases with longer-term follow-up data will be essential in determining the most appropriate treatment options for patients who have development recurrent disease post-transplant (Table 3).
The discoveries of major podocyte antigens and the pathogenic autoantibody against the podocyte antigen phospholipase A2 receptor (PLA2R) have led to breakthroughs in the understanding, management, and treatment of patients with idiopathic membranous GN (150, 151). The ability to test for the presence of anti-PLA2R autoantibody has resulted in improved recognition and differentiating primary vs. secondary membranous GN, as well as assisting clinicians in the management of patients pre and post-transplantation, identifying those patients at high risk of post-transplant disease recurrence that may benefit from more intensive monitoring post-transplant as well as monitoring response to treatment (44, 152, 153). Nevertheless, the absence of PLA2R autoantibody does not definitively exclude cases of idiopathic membranous GN (45, 154). The diagnostic test accuracy of circulating anti-PLA2R autoantibody in differentiating primary from secondary membranous GN is acceptable, with reported test sensitivity of 65% (63–67%), specificity of 97% (97–98%), positive likelihood ratio of 15.65 (9.95–24.62), and negative likelihood ratio of 0.37 (0.32–0.42) (155). Similar test performance accuracy is shown for the positive glomerular staining of PLA2R antigen (155, 156). A second antibody specific for the autoantigen thrombospondin type 1 domain–containing 7A (THSD7A) has been detected in up to 5% of patients with idiopathic membranous GN, typically in those who were seronegative for the PLA2R autoantibody (<15% cases) (45). Nevertheless, up to 20% of patients with idiopathic membranous GN do not have detectable autoantibodies to PLA2R or THSD7A, suggesting the possibility that unidentified autoantibodies targeting other auto-antigens may be contributing (47, 157).
The rate of disease recurrence in patients with idiopathic membranous GN following kidney transplantation is between 30 and 50%, with the disparate detection rates reported in the studies influenced by the characteristics of the cohort (e.g., those with high titres of circulating anti-PLA2R autoantibody have a greater risk of recurrent disease), follow-up period and dissimilar biopsy practices (Table 1) (4, 158). Even though the circulating levels of anti-PLA2R autoantibody tend to decline post-transplant (adsorption into the allograft or the effect of immunosuppression), there is a direct relationship between the titer level and risk of disease recurrence post-transplant. The positive predictive value of pre-transplant anti-PLA2R antibodies for disease recurrence is 83%, but the risk of recurrence in those with idiopathic membranous GN not attributed to anti-PLA2R antibody remains unknown (45, 154). Nevertheless, the diagnostic threshold of anti-PLA2R antibody in defining the risk of disease recurrence remains poorly defined. The utility of monitoring anti-PLA2R antibody post-transplant remains unclear, but should be considered in those with high pre-transplant circulating levels of anti-PLA2R antibody, in those with early disease recurrence (to predict disease progression), to determine response to treatment and to differentiate disease recurrence from de novo membranous GN or other causes of proteinuria (4, 159). The prognostic significance of other biomarkers (antibody to other auto-antigens) shown in Table 2 remains unknown. Figures 3A–C show the light microscopy and ultrastructural features of a kidney transplant recipient who had developed early recurrence of idiopathic membranous GN within a month post-transplant. The transplant allograft biopsy showed features of early membranous GN with a few small subepithelial deposits.
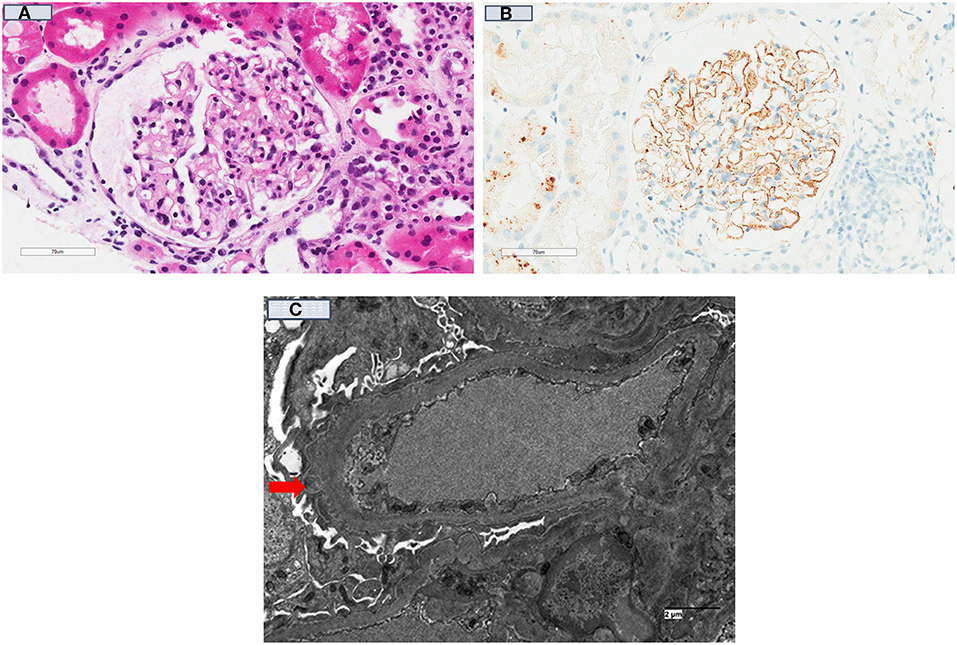
Figure 3. Kidney transplant recipient who had developed recurrent idiopathic membranous glomerulonephritis 3-months post-transplant. (A) [H & E (40x)]: Kidney allograft biopsy showed a glomerulus with no significant changes and no spikes were seen on silver stains; (B) showed diffuse positive granular capillary loop staining with C4d immuno-peroxidase stain; and (C) (Electron Microscopy) showed a capillary loop containing a few small subepithelial electron-dense deposits (red arrow) in keeping with the diagnosis of early recurrent membranous GN.
The treatment of disease recurrence is largely extrapolated from treatment in the general population and typically includes a combination of anti-proteinuric agents, corticosteroids, alkylating agents, CNI, and rituximab. Given that kidney transplant recipients are likely to be maintained on corticosteroids and CNI, most clinicians would advocate continuing (or changing from mTOR inhibitor-based regimen to) CNI and consider the addition of anti-CD20 antibodies rather than introducing alkylating agents to avoid over-immunosuppression resulting in severe infective complications. Rituximab is an effective treatment for native and recurrent membranous GN in the allograft, with up to 80% achieving complete or partial remission with the use of rituximab for early disease recurrence post-transplant (160–163). A personalized approach of prescribing rituximab only to patients with positive anti-PLA2R antibody-associated idiopathic membranous GN in the native kidneys has been suggested (164, 165), but a similar approach has not been advocated for disease recurrence. The decision and timing of initiating specific treatment, in addition to anti-proteinuric treatment for patients with recurrent idiopathic membranous GN remains unknown, as many patients may have subclinical histological recurrence (particularly those with pre-transplant circulating anti-PLA2R antibody) or the proteinuria may be attributed to other concurrent diseases (e.g., transplant glomerulopathy). A single case of complete clinical remission with bortezomib has been described for rituximab-resistant recurrent membranous GN post-transplant, suggesting that depletion of plasma cells (in addition to B cells) may be needed in refractory cases (166). There is currently insufficient information as to the pre-emptive use of rituximab for patients with idiopathic membranous GN and high pre-transplant levels of anti-PLA2R antibody, but this (or early initiation post-transplant) can be considered in those with detectable high levels of anti-PLA2R antibody with prior allograft failure from recurrent membranous GN or have persistent high or increasing levels of circulating anti-PLA2R antibody post-transplant with early histological recurrence. The higher relative risk of allograft failure (up to 50% at 10-years of follow-up) following disease recurrence is a point of concern but must be taken into context the varying rates of disease recurrence, potential ascertainment bias of attributing chronic allograft failure from recurrent disease and the competing risk of other causes of allograft failure and death with a functioning graft (5, 167); and more detailed analysis of these cases to identify potentially modifiable factors that may explain those at risk of allograft failure following disease recurrence are required (e.g., ineffective treatment or delayed institution of treatment). A practical approach post-transplant in patients with idiopathic membranous GN is shown in Figure 4 and Table 3.
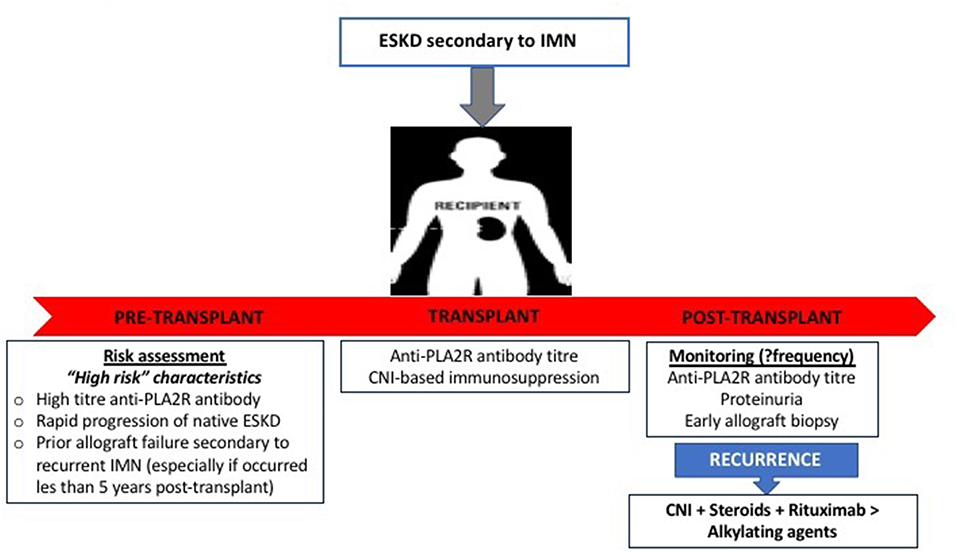
Figure 4. Practical approach to the pre- and post-kidney transplant risk assessment and management of patients with end-stage kidney disease secondary to idiopathic membranous glomerulonephritis.
In contrast to ESKD attributed to primary GN, patients with secondary GN subtypes attributed to systemic diseases [such as atypical hemolytic uremic syndrome (aHUS), systemic lupus erythematosus (SLE), anti-glomerular basement membrane (GBM) disease, and crescentic GN (e.g., from systemic vasculitis)] may experience GN recurrence after kidney transplantation, but these relapses often occur later post-transplant and infrequently lead to allograft failure (8). In patients with anti-neutrophil cytoplasmic antibody (ANCA)-associated vasculitis, the relapse rate (often extra-renal complications) has been reported at 0.02 per patient-years, with no consistent association demonstrated between ANCA subtypes or disease severity prior to transplantation and disease recurrence occurring post-transplant (168–170). The incidence and outcome of patients with recurrent lupus nephritis post-kidney transplantation are unclear, with the majority of the reports from single-center and registry studies. The reported incidence of recurrent lupus nephritis varies between 0 and 44% (171), with patients who had experienced recurrent lupus nephritis at a greater risk of allograft failure and mortality (172–175). However, this association remains inconsistent (176). In patients with anti-GBM disease, disease recurrence after kidney transplant is <5% in the era of modern immunosuppression, and allograft failure from disease recurrence exceedingly rare (177, 178). Prior to the availability of the C5-inhibitor eculizumab (Soliris®), the risk of disease recurrence following transplantation of patients with aHUS was up to 80%, with a substantial proportion of patients losing their allografts from recurrent disease within the first year post-transplant and therefore, kidney transplantation was considered a contraindication for patients with aHUS (179). With prophylactic use of eculizumab, kidney transplantation appeared safe with excellent allograft outcome, even in those with pathogenic mutations known to be associated with a high risk of disease recurrence (180–182). There have been reports of successful live donor kidney transplantation without the use of prophylactic eculizumab but this is generally not recommended or undertaken with extreme caution in patients with certain genetic mutations (e.g., membrane-associated complement regulator membrane cofactor protein mutation) and frequent post-transplant monitoring for disease recurrence (183, 184).
In patients with ESKD where the underlying etiology of the cause of ESKD is uncertain because a biopsy was not undertaken (but with clinical suspicion) or were non-diagnostic with non-specific histological features of interstitial fibrosis and glomerulosclerosis, the incidence of allograft failure attributed to GN is extremely low. Data from the ANZDATA registry showed that 5% of death censored allograft failure of kidney transplant recipients with presumed/advanced GN as cause of ESKD was attributed to GN, compared to over 30% in those with high-risk primary GN subtypes. In this study, almost 90% of the allograft failures from GN were attributed to FSGS, membranous GN and IgA nephropathy, but it is unknown whether these GNs represent recurrent or de novo GN (185). Similar findings have been corroborated in a large Canadian population cohort study where the incidence of post-transplant biopsy-proven GN was diagnosed in 5.7% (9 of 159 patients) of patients with presumed GN (cumulative probability of 11.8% at 15 years post-transplant), with 8 of 9 post-transplant GN cases attributed to FSGS and IgA nephropathy (9).
The true prevalence of de novo GN in kidney transplant recipients remains unknown, but is associated with significantly reduced allograft survival compared to those without de novo glomerular disease. The incidence of de novo GN after kidney transplant varies between 4 and 20%, with FSGS, IgA nephropathy, membranous GN, and MPGN being the most common de novo GN subtypes. There are difficulties in identifying and confirming the presence of de novo GN post-transplant because the cause of native ESKD is often uncertain (kidney biopsies were often not undertaken or were non-diagnostic), the presence of GN in the donor kidney may not be known (particularly in the absence of pre-implantation biopsy), variations in allograft biopsy practices and differences in the histopathological evaluation of allograft biopsies where immunofluorescence and electron microscopy of biopsies may not be routinely performed (8, 186). In a Canadian cohort study, the incidence of de novo GN occurred in 3.4 and 3.6% of those with primary ESKD from GN and those with ESKD from non-GN causes, corresponding to cumulative incidences of 9.6 and 10.5% at 15 years, respectively. In patients whose ESKD was attributed to non-GN causes and had developed de novo GN, over 95% of cases were FSGS (the most common form of de novo GN, 11 of 26 [42%] cases), IgA nephropathy (7 of 26 [27%]), membranous GN (4 of 26 [15%]), and MPGN (3 of 26 [12%]) (9). In this study, the risk of allograft failure was over 7-times greater among recipients who have developed de novo GN, compared to those without disease.
Table 4 shows the subtypes of de novo GN that have been reported, outlining the differences in clinical and histological characteristics of de novo vs. native (or recurrent) GN, as well as potential treatment options and outcomes. The presentations of those with de novo GN are similar to those with primary GN subtypes, ranging from asymptomatic urinary or biochemical abnormalities to overt symptoms and signs of GN with nephrotic syndrome and renal dysfunction. As way of an example, there are several risk factors that may predispose to the development of de novo FSGS, particularly those associated with a reduction in nephron mass (resulting in compensatory hyperfiltration of remaining nephrons such as diabetes, hypertension, BK viral infection, CNI therapy, and rejection) or the introduction of sirolimus (through the effects on podocyte integrity) (96, 186, 225–227). Consequently, it is often difficult to differentiate secondary forms of FSGS (developing post-transplant) from “actual” de novo non-collapsing GN and may to some extent explain why most cases of de novo FSGS tend to occur later post-transplant (compared to the recurrence of primary FSGS). Irrespective of the nature of de novo FSGS, the long-term allograft outcome is relatively poor, particularly in the presence of interstitial fibrosis/tubular atrophy with 5-year allograft survival of <50% after diagnosis (191, 228). Given the heterogeneity of the disease process, the treatment of de novo FSGS predominantly revolves around adequate anti-proteinuric treatment and the removal of the offending agents/factors where possible, but more aggressive therapy (similar to disease recurrence) may be considered, although there are no data to support this approach. Re-transplantation following allograft failure from de novo FSGS can be considered, but clinicians should attempt to establish and exclude or avoid potential causative factors that had resulted in the development of de novo FSGS.
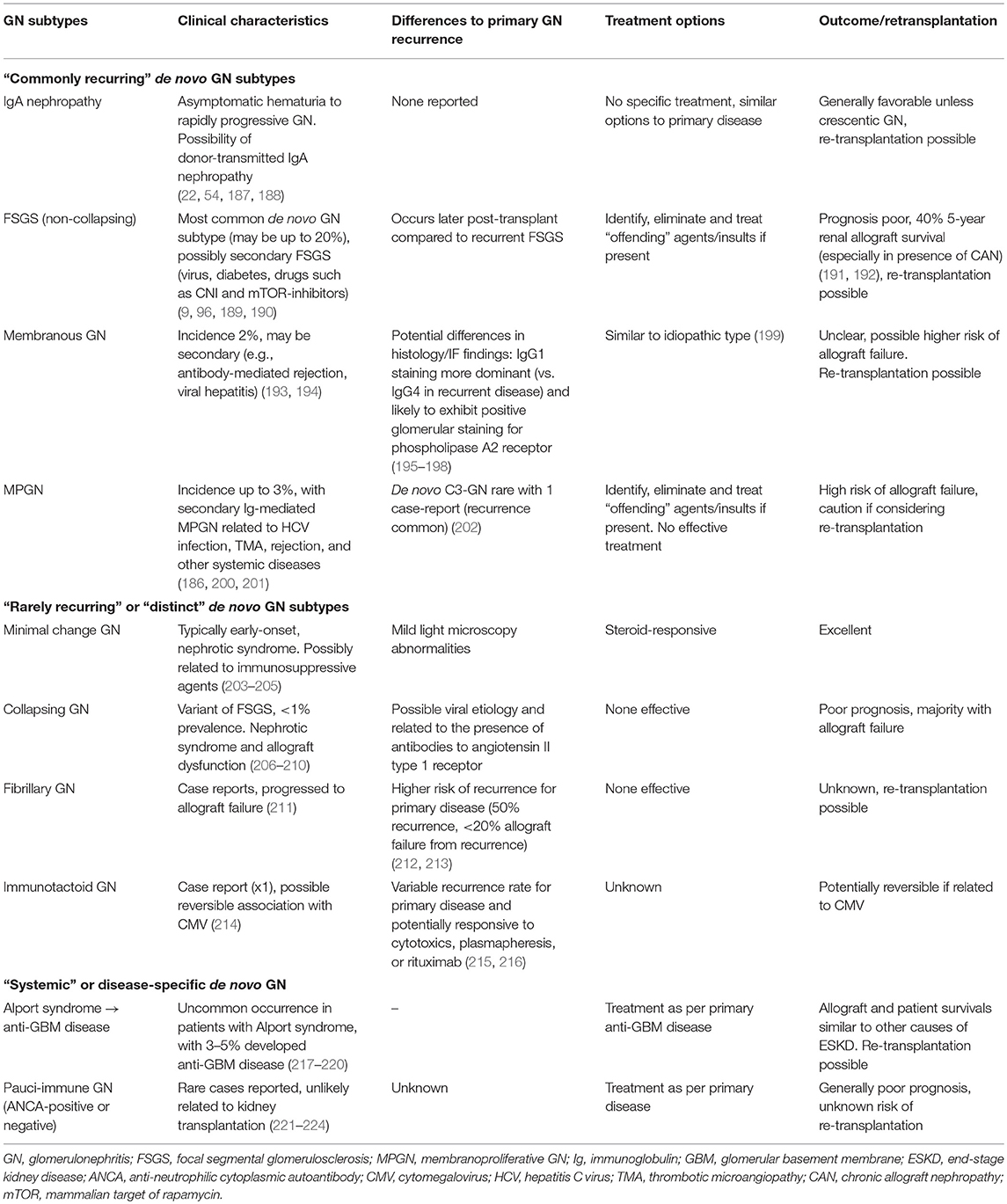
Table 4. Characteristics and differences between de novo glomerulonephritis compared to recurrence of primary glomerulonephritis after kidney transplantation.
The incidence of de novo IgA nephropathy is likely to be underestimated, with asymptomatic IgA deposition not infrequently found (detected incidentally in protocol or indication biopsies or potentially donor-derived asymptomatic disease detected on pre-implantation biopsies) (186, 187). However, presentation with macroscopic hematuria is unusual. The prognosis of those with de novo IgA nephropathy is usually relatively benign with treatment predominantly focuses on anti-proteinuric and/or anti-hypertensive therapy (188). Patients who have developed crescentic de novo IgA nephropathy tend to have a poorer prognosis (22), but the efficacy of more aggressive treatment with steroids and alkylating agent remains unknown. There is insufficient data to determine whether the clinical course of those with recurrent or de novo IgA nephropathy is dissimilar, but re-transplantation of patients with either disease is possible. De novo membranous GN and MPGN are much less common, and the reported cases primarily related to secondary causes including viral infections, rejection, autoimmune disease, CNI and thrombotic microangiopathy (115, 193, 229). The onset of de novo membranous GN or MPGN tend to occur later post-transplant (compared to recurrent disease), with symptoms ranging from asymptomatic detection of mild proteinuria and incidental biopsy findings to nephrotic range proteinuria and rapidly progressive GN (with acute allograft deterioration), but these are often indistinguishable compared to the timing, presentations, and clinical course of those with recurrent diseases (50). For membranous GN, the pattern of Ig staining or glomerular staining for PLA2R may help to differentiate de novo from recurrent disease. In patients with recurrent disease, IgG1 staining in capillary loop deposits was dominant/co-dominant (n = 7 cases), whereas, IgG4 staining in capillary loop deposits was dominant/co-dominant (n = 2 cases) in de novo disease (195). In another small study of 24 patients with recurrent (n = 12) or de novo membranous GN (n = 12), glomerular staining for PLA2R was more common in those with recurrent disease (83% cases, sensitivity and specificity of 83% [95%CI 51–97%] and 92% [95%CI 60–100%], respectively) compared to de novo disease (17%), and suggest that injury to the podocytes occurring post-transplantation triggering the release of podocyte antigens may induce the formation of auto-antibodies and deposition of subepithelial immune complexes (193, 196). These findings will need to be validated in larger cohorts. Nevertheless, in all patients with de novo membranous GN and MPGN, a careful histological examination for potential secondary causes or contributing factors should be undertaken, which will have clinical implication when considering treatment options and future re-transplantation potential for these patients.
Clinicians should be aware of the need to exclude secondary causes in recipients who have developed de novo GN, which is critical when considering treatment options and re-transplant potential (following allograft failure). Nonetheless, the systematic approach to the investigations and subsequent management of kidney transplant recipients with de novo GN is similar to that of patients presenting with GN without a kidney transplant. However, the data informing the clinical and pathological differences between recurrent compared to de novo GN remains limited and therefore the current understanding of the epidemiology, pathogenesis and outcomes of de novo GN is likely to evolve with the availability of future studies.
Despite the advances in the understanding of the epidemiology, pathogenesis, and classification of primary and recurrent GN after kidney transplantation, considerable uncertainty remains in the approach and treatment of post-transplant GN recurrence (230). It is therefore imperative to consider the establishment of a global GN registry, focusing on data collection on the clinical characteristics, histological features, treatment, and outcome of patients who have developed post-transplant GN, particularly those with high risk GN subtypes. The Post-TrANsplant GlOmerular Disease (TANGO) study, an observational, multicenter cohort study was initiated in 2017 by researchers across Europe, North American, and South America, with the goal of collecting patient-level data regarding the risk factor, trajectory of disease activity, and responses to treatment; to develop a bio-repository of human specimens including blood, cell, and tissue samples; and the opportunity to undertake collaborative studies and clinical intervention trials to improve the management and clinical outcomes of these patients (231). Even though the clinical care of patients with recurrent or de novo GN remains challenging, it is an exciting time for the integration of translational GN-related research to novel developments in the understanding and treatment of GN, with the ultimate goal of establishing personalized treatment by effectively tailoring specific treatment strategies to patients with the various types of recurrent or de novo GN.
WL was supported by a Clinical Research Fellowship from the Raine Foundation (University of Western Australia and Health Department of Western Australia) and Jacquot Research Foundation (Royal Australasian College of Physicians). GW was supported by a National Health and Medical Research Council Career Development Fellowship.
WL designed the outline of the manuscript and prepared the tables/figures. MS provided pathology slides. All authors wrote the manuscript.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
1. Australia and New Zealand Dialysis and Transplant Registry. 40th Report, Chapter 1: Incidence of End Stage Kidney Disease. Adelaide, SA: Australia and New Zealand Dialysis and Transplant Registry (2018).
2. United States Renal Data System. USRDS annual data report: Epidemiology of kidney disease in the United States. Chapter 1: Incidence, Prevalence, Patient Characteristics, and Treatment Modalities. Bethesda, MD: National Institutes of Health, National Institute of Diabetes and Digestive and Kidney Diseases (2018).
3. Byrne C, Caskey F, Castledine C, Davenport A, Dawnay A, Fraser S, et al. UK renal registry: 20th annual report of the renal association. Nephron. (2018) 139(Suppl. 1):490958. doi: 10.1159/000490958
4. Cosio FG, Cattran DC. Recent advances in our understanding of recurrent primary glomerulonephritis after kidney transplantation. Kidney Int. (2017) 91:304–14. doi: 10.1016/j.kint.2016.08.030
5. Allen PJ, Chadban SJ, Craig JC, Lim WH, Allen RDM, Clayton PA, et al. Recurrent glomerulonephritis after kidney transplantation: risk factors and allograft outcomes. Kidney Int. (2017) 92:461–9. doi: 10.1016/j.kint.2017.03.015
6. O'Shaughnessy MM, Liu S, Montez-Rath ME, Lenihan CR, Lafayette RA, Winkelmayer WC. Kidney transplantation outcomes across GN Subtypes in the United States. J Am Soc Nephrol. (2017) 28:632–44. doi: 10.1681/ASN.2016020126
7. O'Shaughnessy MM, Montez-Rath ME, Lafayette RA, Winkelmayer WC. Patient characteristics and outcomes by GN subtype in ESRD. Clin J Am Soc Nephrol. (2015) 10:1170–8. doi: 10.2215/CJN.11261114
8. Golgert WA, Appel GB, Hariharan S. Recurrent glomerulonephritis after renal transplantation: an unsolved problem. Clin J Am Soc Nephrol. (2008) 3:800–7. doi: 10.2215/CJN.04050907
9. Chailimpamontree W, Dmitrienko S, Li G, Balshaw R, Magil A, Shapiro RJ, et al. Probability, predictors, and prognosis of posttransplantation glomerulonephritis. J Am Soc Nephrol. (2009) 20:843–51. doi: 10.1681/ASN.2008050454
10. Choy BY, Chan TM, Lai KN. Recurrent glomerulonephritis after kidney transplantation. Am J Transplant. (2006) 6:2535–42. doi: 10.1111/j.1600-6143.2006.01502.x
11. An JN, Lee JP, Oh YJ, Oh YK, Ha JW, Chae DW, et al. Incidence of post-transplant glomerulonephritis and its impact on graft outcome. Kidney Res Clin Pract. (2012) 31:219–26. doi: 10.1016/j.krcp.2012.09.004
12. El-Zoghby ZM, Stegall MD, Lager DJ, Kremers WK, Amer H, Gloor JM, et al. Identifying specific causes of kidney allograft loss. Am J Transplant. (2009) 9:527–35. doi: 10.1111/j.1600-6143.2008.02519.x
13. Briganti EM, Russ GR, McNeil JJ, Atkins RC, Chadban SJ. Risk of renal allograft loss from recurrent glomerulonephritis. N Engl J Med. (2002) 347:103–9. doi: 10.1056/NEJMoa013036
14. Hariharan S, Adams MB, Brennan DC, Davis CL, First MR, Johnson CP, et al. Recurrent and de novo glomerular disease after renal transplantation: a report from Renal Allograft Disease Registry (RADR). Transplantation. (1999) 68:635–41. doi: 10.1097/00007890-199909150-00007
15. Berthoux F, El Deeb S, Mariat C, Diconne E, Laurent B, Thibaudin L. Antithymocyte globulin (ATG) induction therapy and disease recurrence in renal transplant recipients with primary IgA nephropathy. Transplantation. (2008) 85:1505–7. doi: 10.1097/TP.0b013e3181705ad4
16. Leeaphorn N, Garg N, Khankin EV, Cardarelli F, Pavlakis M. Recurrence of IgA nephropathy after kidney transplantation in steroid continuation versus early steroid-withdrawal regimens: a retrospective analysis of the UNOS/OPTN database. Transpl Int. (2018) 31:175–86. doi: 10.1111/tri.13075
17. Moroni G, Longhi S, Quaglini S, Gallelli B, Banfi G, Montagnino G, et al. The long-term outcome of renal transplantation of IgA nephropathy and the impact of recurrence on graft survival. Nephrol Dial Transpl. (2013) 28:1305–14. doi: 10.1093/ndt/gfs472
18. Pippias M, Stel VS, Aresté-Fosalba N, Couchoud C, Fernandez-Fresnedo G, Finne P, et al. Long-term kidney transplant outcomes in primary glomerulonephritis: analysis from the ERA-EDTA registry. Transplantation. (2016) 100:1955–62. doi: 10.1097/TP.0000000000000962
19. McDonald SP, Russ GR. Recurrence of IgA nephropathy among renal allograft recipients from living donors is greater among those with zero HLA mismatches. Transplantation. (2006) 82:759–62. doi: 10.1097/01.tp.0000230131.66971.45
20. Clayton P, McDonald S, Chadban S. Steroids and recurrent IgA nephropathy after kidney transplantation. Am J Transpl. (2011) 11:1645–9. doi: 10.1111/j.1600-6143.2011.03667.x
21. Andresdottir MB, Haasnoot GW, Persijn GG, Claas FH. HLA-B8, DR3: a new risk factor for graft failure after renal transplantation in patients with underlying immunoglobulin A nephropathy. Clin Transplant. (2009) 23:660–5. doi: 10.1111/j.1399-0012.2009.01059.x
22. Kowalewska J, Yuan S, Sustento-Reodica N, Nicosia RF, Smith KD, Davis CL, et al. IgA nephropathy with crescents in kidney transplant recipients. Am J Kidney Dis. (2005) 45:167–75. doi: 10.1053/j.ajkd.2004.09.030
23. Avasare RS, Rosenstiel PE, Zaky ZS, Tsapepas DS, Appel GB, Markowitz GS, et al. Predicting post-transplant recurrence of iga nephropathy: the importance of crescents. Am J Nephrol. (2017) 45:99–106. doi: 10.1159/000453081
24. Wang AY, Lai FM, Yu AW, Lam PK, Chow KM, Choi PC, et al. Recurrent IgA nephropathy in renal transplant allografts. Am J Kidney Dis. (2001) 38:588–96. doi: 10.1053/ajkd.2001.26885
25. Nijim S, Vujjini V, Alasfar S, Luo X, Orandi B, Delp C, et al. Recurrent IgA nephropathy after kidney transplantation. Transplant Proc. (2016) 48:2689–94. doi: 10.1016/j.transproceed.2016.08.011
26. Berthelot L, Robert T, Vuiblet V, Tabary T, Braconnier A, Dramé M, et al. Recurrent IgA nephropathy is predicted by altered glycosylated IgA, autoantibodies and soluble CD89 complexes. Kidney Int. (2015) 88:815–22. doi: 10.1038/ki.2015.158
27. Robert T, Berthelot L, Cambier A, Rondeau E, Monteiro RC. Molecular insights into the pathogenesis of IgA nephropathy. Trends Mol Med. (2015) 21:762–75. doi: 10.1016/j.molmed.2015.10.003
28. Placzek WJ, Yanagawa H, Makita Y, Renfrow MB, Julian BA, Rizk DV, et al. Serum galactose-deficient-IgA1 and IgG autoantibodies correlate in patients with IgA nephropathy. PLoS ONE. (2018) 13:e0190967. doi: 10.1371/journal.pone.0190967
29. Zhao YF, Zhu L, Liu LJ, Shi SF, Lv JC, Zhang H. Pathogenic role of glycan-specific IgG antibodies in IgA nephropathy. BMC Nephrol. (2017) 18:301. doi: 10.1186/s12882-017-0722-3
30. Berthoux F, Suzuki H, Thibaudin L, Yanagawa H, Maillard N, Mariat C, et al. Autoantibodies targeting galactose-deficient IgA1 associate with progression of IgA nephropathy. J Am Soc Nephrol. (2012) 23:1579–87. doi: 10.1681/ASN.2012010053
31. Suzuki H, Fan R, Zhang Z, Brown R, Hall S, Julian BA, et al. Aberrantly glycosylated IgA1 in IgA nephropathy patients is recognized by IgG antibodies with restricted heterogeneity. J Clin Invest. (2009) 119:1668–77. doi: 10.1172/JCI38468
32. Wyld ML, Chadban SJ. Recurrent IgA nephropathy after kidney transplantation. Transplantation. (2016) 100:1827–32. doi: 10.1097/TP.0000000000001093
33. Garnier AS, Duveau A, Demiselle J, Croué A, Subra JF, Sayegh J, et al. Early post-transplant serum IgA level is associated with IgA nephropathy recurrence after kidney transplantation. PLoS ONE. (2018) 13:e0196101. doi: 10.1371/journal.pone.0196101
34. Berthoux F, Suzuki H, Mohey H, Maillard N, Mariat C, Novak J, et al. Prognostic value of serum biomarkers of autoimmunity for recurrence of IgA nephropathy after kidney transplantation. J Am Soc Nephrol. (2017) 28:1943–50. doi: 10.1681/ASN.2016060670
35. Martin-Penagos L, Benito-Hernández A, Martín J, Gómez-Román J, San Segundo D, López-Hoyos M, et al. APRIL serum levels relate to recurrence of IgA nephropathy. Transplantation. (2018) 102:S10. doi: 10.1097/01.tp.0000542547.90273.0b
36. Lee S, Kin D, Kim J, Jeon H. Identification of urinary biomarkers for recurrence of IgA nephropathy after kidney transplantation using proteomic methods. Transplantation. (2018) 102:S580. doi: 10.1097/01.tp.0000543458.26810.ce
37. Wei C, El Hindi S, Li J, Fornoni A, Goes N, Sageshima J, et al. Circulating urokinase receptor as a cause of focal segmental glomerulosclerosis. Nat Med. (2011) 17:952–60. doi: 10.1038/nm.2411
38. Huang J, Liu G, Zhang YM, Cui Z, Wang F, Liu XJ, et al. Urinary soluble urokinase receptor levels are elevated and pathogenic in patients with primary focal segmental glomerulosclerosis. BMC Med. (2014) 12:81. doi: 10.1186/1741-7015-12-81
39. Delville M, Sigdel TK, Wei C, Li J, Hsieh SC, Fornoni A, et al. A circulating antibody panel for pretransplant prediction of FSGS recurrence after kidney transplantation. Sci Transl Med. (2014) 6:256ra136. doi: 10.1126/scitranslmed.3008538
40. Lopez-Hellin J, Cantarell C, Jimeno L, Sanchez-Fructuoso A, Puig-Gay N, Guirado L, et al. A form of apolipoprotein a-I is found specifically in relapses of focal segmental glomerulosclerosis following transplantation. Am J Transplant. (2013) 13:493–500. doi: 10.1111/j.1600-6143.2012.04338.x
41. Puig-Gay N, Jacobs-Cacha C, Sellarès J, Guirado L, González Roncero F, Jiménez C, et al. Apolipoprotein A-Ib as a biomarker of focal segmental glomerulosclerosis recurrence after kidney transplantation: diagnostic performance and assessment of its prognostic value - a multi-centre cohort study. Transpl Int. (2019) 32:313–22. doi: 10.1111/tri.13372
42. Pérez V, Ibernón M, López D, Pastor MC, Navarro M, Navarro-Muñoz M, et al. Urinary peptide profiling to differentiate between minimal change disease and focal segmental glomerulosclerosis. PLoS ONE. (2014) 9:e87731. doi: 10.1371/journal.pone.0087731
43. McCarthy ET, Sharma M, Savin VJ. Circulating permeability factors in idiopathic nephrotic syndrome and focal segmental glomerulosclerosis. Clin J Am Soc Nephrol. (2010) 5:2115–21. doi: 10.2215/CJN.03800609
44. Quintana LF, Blasco M, Seras M, Pérez NS, López-Hoyos M, Villarroel P, et al. Antiphospholipase A2 receptor antibody levels predict the risk of posttransplantation recurrence of membranous nephropathy. Transplantation. (2015) 99:1709–14. doi: 10.1097/TP.0000000000000630
45. Tomas NM, Beck LH Jr, Meyer-Schwesinger C, Seitz-Polski B, Ma H, Zahner G, et al. Thrombospondin type-1 domain-containing 7A in idiopathic membranous nephropathy. N Engl J Med. (2014) 371:2277–87. doi: 10.1056/NEJMoa1409354
46. Herwig J, Skuza S, Sachs W, Sachs M, Failla AV, Rune G, et al. Thrombospondin type 1 domain-containing 7A localizes to the slit diaphragm and stabilizes membrane dynamics of fully differentiated podocytes. J Am Soc Nephrol. (2019) 30:824–39. doi: 10.1681/ASN.2018090941
47. Murtas C, Bruschi M, Candiano G, Moroni G, Magistroni R, Magnano A, et al. Coexistence of different circulating anti-podocyte antibodies in membranous nephropathy. Clin J Am Soc Nephrol. (2012) 7:1394–400. doi: 10.2215/CJN.02170312
48. Alasfar S, Carter-Monroe N, Rosenberg AZ, Montgomery RA, Alachkar N. Membranoproliferative glomerulonephritis recurrence after kidney transplantation: using the new classification. BMC Nephrol. (2016) 17:7. doi: 10.1186/s12882-015-0219-x
49. Lorenz EC, Sethi S, Leung N, Dispenzieri A, Fervenza FC, Cosio FG. Recurrent membranoproliferative glomerulonephritis after kidney transplantation. Kidney Int. (2010) 77:721–8. doi: 10.1038/ki.2010.1
50. Ponticelli C, Glassock RJ. Posttransplant recurrence of primary glomerulonephritis. Clin J Am Soc Nephrol. (2010) 5:2363–72. doi: 10.2215/CJN.06720810
51. Nafar M, Samavat S. Biomarkers in IgA nephropathy. In: Patel VB, Preedy VR editors. Biomarkers in Kidney Disease. Dordrecht: Springer Science+Business Media (2015). p. 1–29.
52. Chandrakantan A, Ratanapanichkich P, Said M, Barker CV, Julian BA. Recurrent IgA nephropathy after renal transplantation despite immunosuppressive regimens with mycophenolate mofetil. Nephrol Dial Transplant. (2005) 20:1214–21. doi: 10.1093/ndt/gfh773
53. Zagkotsis G, Vourlakou C, Paraskevopoulos A, Apostolou T. Recurrence of crescentic IgA nephropathy after renal transplantation. CEN Case Rep. (2018) 7:268–73. doi: 10.1007/s13730-018-0341-2
54. Tang Z, Ji SM, Chen DR, Wen JQ, Chen JS, Liu ZH, et al. Recurrent or de novo IgA nephropathy with crescent formation after renal transplantation. Ren Fail. (2008) 30:611–6. doi: 10.1080/08860220802134516
55. Lafayette RA, Canetta PA, Rovin BH, Appel GB, Novak J, Nath KA, et al. A Randomized, controlled trial of rituximab in IgA nephropathy with proteinuria and renal dysfunction. J Am Soc Nephrol. (2017) 28:1306–13. doi: 10.1681/ASN.2016060640
56. Courtney AE, McNamee PT, Nelson WE, Maxwell AP. Does angiotensin blockade influence graft outcome in renal transplant recipients with IgA nephropathy? Nephrol Dial Transplant. (2006) 21:3550–4. doi: 10.1093/ndt/gfl506
57. Hotta K, Fukasawa Y, Akimoto M, Tanabe T, Sasaki H, Fukuzawa N, et al. Tonsillectomy ameliorates histological damage of recurrent immunoglobulin A nephropathy after kidney transplantation. Nephrology. (2013) 18:808–12. doi: 10.1111/nep.12151
58. Kennoki T, Ishida H, Yamaguchi Y, Tanabe K. Proteinuria-reducing effects of tonsillectomy alone in IgA nephropathy recurring after kidney transplantation. Transplantation. (2009) 88:935–41. doi: 10.1097/TP.0b013e3181b75374
59. Francis A, Trnka P, McTaggart SJ. Long-term outcome of kidney transplantation in recipients with focal segmental glomerulosclerosis. Clin J Am Soc Nephrol. (2016) 11:2041–6. doi: 10.2215/CJN.03060316
60. Bertelli R, Ginevri F, Caridi G, Dagnino M, Sandrini S, Di Duca M, et al. Recurrence of focal segmental glomerulosclerosis after renal transplantation in patients with mutations of podocin. Am J Kidney Dis. (2003) 41:1314–21. doi: 10.1016/S0272-6386(03)00364-0
61. Freedman BI, Pastan SO, Israni AK, Schladt D, Julian BA, Gautreaux MD, et al. APOL1 genotype and kidney transplantation outcomes from deceased african american donors. Transplantation. (2016) 100:194–202. doi: 10.1097/TP.0000000000000969
62. Lee BT, Kumar V, Williams TA, Abdi R, Bernhardy A, Dyer C, et al. The APOL1 genotype of African American kidney transplant recipients does not impact 5-year allograft survival. Am J Transplant. (2012) 12:1924–8. doi: 10.1111/j.1600-6143.2012.04033.x
63. Jungraithmayr TC, Hofer K, Cochat P, Chernin G, Cortina G, Fargue S, et al. Screening for NPHS2 mutations may help predict FSGS recurrence after transplantation. J Am Soc Nephrol. (2011) 22:579–85. doi: 10.1681/ASN.2010010029
64. Machuca E, Hummel A, Nevo F, Dantal J, Martinez F, Al-Sabban E, et al. Clinical and epidemiological assessment of steroid-resistant nephrotic syndrome associated with the NPHS2 R229Q variant. Kidney Int. (2009) 75:727–35. doi: 10.1038/ki.2008.650
65. Hofstra JM, Lainez S, van Kuijk WH, Schoots J, Baltissen MP, Hoefsloot LH, et al. New TRPC6 gain-of-function mutation in a non-consanguineous Dutch family with late-onset focal segmental glomerulosclerosis. Nephrol Dial Transplant. (2013) 28:1830–8. doi: 10.1093/ndt/gfs572
66. Conlon PJ, Lynn K, Winn MP, Quarles LD, Bembe ML, Pericak-Vance M, et al. Spectrum of disease in familial focal and segmental glomerulosclerosis. Kidney Int. (1999) 56:1863–71. doi: 10.1046/j.1523-1755.1999.00727.x
67. Weber S, Gribouval O, Esquivel EL, Morinière V, Tête MJ, Legendre C, et al. NPHS2 mutation analysis shows genetic heterogeneity of steroid-resistant nephrotic syndrome and low post-transplant recurrence. Kidney Int. (2004) 66:571–9. doi: 10.1111/j.1523-1755.2004.00776.x
68. Gbadegesin RA, Lavin PJ, Hall G, Bartkowiak B, Homstad A, Jiang R, et al. Inverted formin 2 mutations with variable expression in patients with sporadic and hereditary focal and segmental glomerulosclerosis. Kidney Int. (2012) 81:94–9. doi: 10.1038/ki.2011.297
69. Büscher AK, Beck BB, Melk A, Hoefele J, Kranz B, Bamborschke D, et al. Rapid response to cyclosporin A and favorable renal outcome in nongenetic versus genetic steroid-resistant nephrotic syndrome. Clin J Am Soc Nephrol. (2016) 11:245–53. doi: 10.2215/CJN.07370715
70. Lepori N, Zand L, Sethi S, Fernandez-Juarez G, Fervenza FC. Clinical and pathological phenotype of genetic causes of focal segmental glomerulosclerosis in adults. Clin Kidney J. (2018) 11:179–90. doi: 10.1093/ckj/sfx143
71. Rood IM, Deegens JK, Wetzels JF. Genetic causes of focal segmental glomerulosclerosis: implications for clinical practice. Nephrol Dial Transplant. (2012) 27:882–90. doi: 10.1093/ndt/gfr771
72. Santín S, Bullich G, Tazón-Vega B, García-Maset R, Giménez I, Silva I, et al. Clinical utility of genetic testing in children and adults with steroid-resistant nephrotic syndrome. Clin J Am Soc Nephrol. (2011) 6:1139–48. doi: 10.2215/CJN.05260610
73. Santín S, Tazón-Vega B, Silva I, Cobo MÁ, Giménez I, Ruíz P, et al. Clinical value of NPHS2 analysis in early- and adult-onset steroid-resistant nephrotic syndrome. Clin J Am Soc Nephrol. (2011) 6:344–54. doi: 10.2215/CJN.03770410
74. Pinto J, Lacerda G, Cameron JS, Turner DR, Bewick M, Ogg CS. Recurrence of focal segmental glomerulosclerosis in renal allografts. Transplantation. (1981) 32:83–9. doi: 10.1097/00007890-198108000-00001
75. Gipson DS, Trachtman H, Kaskel FJ, Greene TH, Radeva MK, Gassman JJ, et al. Clinical trial of focal segmental glomerulosclerosis in children and young adults. Kidney Int. (2011) 80:868–78. doi: 10.1038/ki.2011.195
76. Trautmann A, Bodria M, Ozaltin F, Gheisari A, Melk A, Azocar M, et al. Spectrum of steroid-resistant and congenital nephrotic syndrome in children: the PodoNet registry cohort. Clin J Am Soc Nephrol. (2015) 10:592–600. doi: 10.2215/CJN.06260614
77. Wigger M, Schaible J, Muscheites J, Kundt G, Haffner D, Fischer DC. Fetuin-A serum concentrations in healthy children. Ann Clin Biochem. (2009) 46(Pt 6):511–3. doi: 10.1258/acb.2009.009037
78. Wei C, Trachtman H, Li J, Dong C, Friedman AL, Gassman JJ, et al. Circulating suPAR in two cohorts of primary FSGS. J Am Soc Nephrol. (2012) 23:2051–9. doi: 10.1681/ASN.2012030302
79. Franco Palacios CR, Lieske JC, Wadei HM, Rule AD, Fervenza FC, Voskoboev N, et al. Urine but not serum soluble urokinase receptor (suPAR) may identify cases of recurrent FSGS in kidney transplant candidates. Transplantation. (2013) 96:394–9. doi: 10.1097/TP.0b013e3182977ab1
80. Maas RJH, Wetzels JFM, Deegens JKJ. Serum-soluble urokinase receptor concentration in primary FSGS. Kidney Int. (2012) 81:1043–4. doi: 10.1038/ki.2012.32
81. Bock ME, Price HE, Gallon L, Langman CB. Serum soluble urokinase-type plasminogen activator receptor levels and idiopathic FSGS in children: a single-center report. Clin J Am Soc Nephrol. (2013) 8:1304–11. doi: 10.2215/CJN.07680712
82. Backes Y, van der Sluijs KF, Mackie DP, Tacke F, Koch A, Tenhunen JJ, et al. Usefulness of suPAR as a biological marker in patients with systemic inflammation or infection: a systematic review. Intensive Care Med. (2012) 38:1418–28. doi: 10.1007/s00134-012-2613-1
83. Eugen-Olsen J, Andersen O, Linneberg A, Ladelund S, Hansen TW, Langkilde A, et al. Circulating soluble urokinase plasminogen activator receptor predicts cancer, cardiovascular disease, diabetes and mortality in the general population. J Intern Med. (2010) 268:296–308. doi: 10.1111/j.1365-2796.2010.02252.
84. Sinha A, Bajpai J, Saini S, Bhatia D, Gupta A, Puraswani M, et al. Serum-soluble urokinase receptor levels do not distinguish focal segmental glomerulosclerosis from other causes of nephrotic syndrome in children. Kidney Int. (2014) 85:649–58. doi: 10.1038/ki.2013.546
85. Halleck F, Staeck O, Neumayer H, Budde K, Dmytro K. suPAR in transplant patients with FSGS recurrence. Abstract# 1407. Transplantation. (2014) 98:97. doi: 10.1097/00007890-201407151-00299
86. Cravedi P, Kopp JB, Remuzzi G. Recent progress in the pathophysiology and treatment of FSGS recurrence. Am J Transplant. (2013) 13:266–74. doi: 10.1111/ajt.12045
87. Straatmann C, Kallash M, Killackey M, Iorember F, Aviles D, Bamgbola O, et al. Success with plasmapheresis treatment for recurrent focal segmental glomerulosclerosis in pediatric renal transplant recipients. Pediatr Transplant. (2014) 18:29–34. doi: 10.1111/petr.12185
88. Tran MH, Chan C, Pasch W, Carpenter P, Ichii H, Foster C. Treatment of focal segmental glomerulosclerosis recurrence in the renal allograft: a report of two cases. Case Rep Nephrol Dial. (2016) 6:53–60. doi: 10.1159/000445428
89. Schwartz J, Winters JL, Padmanabhan A, Balogun RA, Delaney M, Linenberger ML, et al. Guidelines on the use of therapeutic apheresis in clinical practice-evidence-based approach from the Writing Committee of the American Society for Apheresis: the sixth special issue. J Clin Apher. (2013) 28:145–284. doi: 10.1002/jca.21276
90. Pescovitz MD, Book BK, Sidner RA. Resolution of recurrent focal segmental glomerulosclerosis proteinuria after rituximab treatment. N Engl J Med. (2006) 354:1961–3. doi: 10.1056/NEJMc055495
91. Fornoni A, Sageshima J, Wei C, Merscher-Gomez S, Aguillon-Prada R, Jauregui AN, et al. Rituximab targets podocytes in recurrent focal segmental glomerulosclerosis. Sci Transl Med. (2011) 3:85ra46. doi: 10.1126/scitranslmed.3002231
92. Benz K, Büttner M, Dittrich K, Campean V, Dötsch J, Amann K. Characterisation of renal immune cell infiltrates in children with nephrotic syndrome. Pediatr Nephrol. (2010) 25:1291–8. doi: 10.1007/s00467-010-1507-0
93. Yoo TH, Pedigo CE, Guzman J, Correa-Medina M, Wei C, Villarreal R, et al. Sphingomyelinase-like phosphodiesterase 3b expression levels determine podocyte injury phenotypes in glomerular disease. J Am Soc Nephrol. (2015) 26:133–47. doi: 10.1681/ASN.2013111213
94. Kang HG, Ha IS, Cheong HI. Recurrence and treatment after renal transplantation in children with FSGS. Biomed Res Int. (2016) 2016:6832971. doi: 10.1155/2016/6832971
95. Tejani AT, Butt K, Trachtman H, Suthanthiran M, Rosenthal CJ, Khawar MR. Cyclosporine A induced remission of relapsing nephrotic syndrome in children. Kidney Int. (1988) 33:729–34. doi: 10.1038/ki.1988.59
96. Letavernier E, Bruneval P, Mandet C, Duong Van Huyen JP, Péraldi MN, Helal I, et al. High sirolimus levels may induce focal segmental glomerulosclerosis de novo. Clin J Am Soc Nephrol. (2007) 2:326–33. doi: 10.2215/CJN.03751106
97. Hickson LJ, Gera M, Amer H, Iqbal CW, Moore TB, Milliner DS, et al. Kidney transplantation for primary focal segmental glomerulosclerosis: outcomes and response to therapy for recurrence. Transplantation. (2009) 87:1232–9. doi: 10.1097/TP.0b013e31819f12be
98. Gonzalez E, Ettenger R, Rianthavorn P, Tsai E, Malekzadeh M. Preemptive plasmapheresis and recurrence of focal segmental glomerulosclerosis in pediatric renal transplantation. Pediatr Transplant. (2011) 15:495–501. doi: 10.1111/j.1399-3046.2011.01478.x
99. Gohh RY, Yango AF, Morrissey PE, Monaco AP, Gautam A, Sharma M, et al. Preemptive plasmapheresis and recurrence of FSGS in high-risk renal transplant recipients. Am J Transplant. (2005) 5:2907–12. doi: 10.1111/j.1600-6143.2005.01112.x
100. Yu CC, Fornoni A, Weins A, Hakroush S, Maiguel D, Sageshima J, et al. Abatacept in B7–1-positive proteinuric kidney disease. N Engl J Med. (2013) 369:2416–23. doi: 10.1056/NEJMoa1304572
101. Kristensen T, Ivarsen P, Povlsen JV. Unsuccessful treatment with abatacept in recurrent focal segmental glomerulosclerosis after kidney transplantation. Case Rep Nephrol Dial. (2017) 7:1–5. doi: 10.1159/000454947
102. Delville M, Baye E, Durrbach A, Audard V, Kofman T, Braun L, et al. B7–1 Blockade Does not improve post-transplant nephrotic syndrome caused by recurrent FSGS. J Am Soc Nephrol. (2016) 27:2520–7. doi: 10.1681/ASN.2015091002
103. Kienzl-Wagner K, Rosales A, Scheidl S, Giner T, Bösmüller C, Rudnicki M, et al. Successful management of recurrent focal segmental glomerulosclerosis. Am J Transplant. (2018) 18:2818–22. doi: 10.1111/ajt.14998
104. Allard L, Kwon T, Krid S, Bacchetta J, Garnier A, Novo R, et al. Treatment by immunoadsorption for recurrent focal segmental glomerulosclerosis after paediatric kidney transplantation: a multicentre French cohort. Nephrol Dial Transplant. (2018) 33:954–63. doi: 10.1093/ndt/gfx214
105. Grellier J, Del Bello A, Milongo D, Guilbeau-Frugier C, Rostaing L, Kamar N. Belatacept in recurrent focal segmental glomerulosclerosis after kidney transplantation. Transpl Int. (2015) 28:1109–10. doi: 10.1111/tri.12574
106. Bernard J, Bruel A, Allain-Launay E, Dantal J, Roussey G. Ofatumumab in post-transplantation recurrence of a pediatric steroid-resistant idiopathic nephrotic syndrome. Pediatr Transplant. (2018) 22:e13175. doi: 10.1111/petr.13175
107. Solomon S, Zolotnitskaya A, Del Rio M. Ofatumumab in post-transplantation recurrence of focal segmental glomerulosclerosis in a child. Pediatr Transplant. (2019) 23:e13413. doi: 10.1111/petr.13413
108. Alachkar N, Carter-Monroe N, Reiser J. Abatacept in B7–1-positive proteinuric kidney disease. N Engl J Med. (2014) 370:1263–4. doi: 10.1056/NEJMc1400502
109. Garin EH, Reiser J, Cara-Fuentes G, Wei C, Matar D, Wang H, et al. Case series: CTLA4-IgG1 therapy in minimal change disease and focal segmental glomerulosclerosis. Pediatr Nephrol. (2015) 30:469–77. doi: 10.1007/s00467-014-2957-6
110. Verghese PS, Rheault MN, Jackson S, Matas AJ, Chinnakotla S, Chavers B. The effect of peri-transplant plasmapheresis in the prevention of recurrent FSGS. Pediatr Transplant. (2018) 22:e13154. doi: 10.1111/petr.13154
111. Canaud G, Zuber J, Sberro R, Royale V, Anglicheau D, Snanoudj R, et al. Intensive and prolonged treatment of focal and segmental glomerulosclerosis recurrence in adult kidney transplant recipients: a pilot study. Am J Transplant. (2009) 9:1081–6. doi: 10.1111/j.1600-6143.2009.02580.x
112. Staeck O, Slowinski T, Lieker I, Wu K, Rudolph B, Schmidt D, et al. Recurrent primary focal segmental glomerulosclerosis managed with intensified plasma exchange and concomitant monitoring of soluble urokinase-type plasminogen activator receptor-mediated podocyte beta3-integrin activation. Transplantation. (2015) 99:2593–7. doi: 10.1097/TP.0000000000000914
113. Green H, Rahamimov R, Rozen-Zvi B, Pertzov B, Tobar A, Lichtenberg S, et al. Recurrent membranoproliferative glomerulonephritis type I after kidney transplantation: a 17-year single-center experience. Transplantation. (2015) 99:1172–7. doi: 10.1097/TP.0000000000000459
114. Andresdottir MB, Assmann KJ, Hoitsma AJ, Koene RA, Wetzels JF. Recurrence of type I membranoproliferative glomerulonephritis after renal transplantation: analysis of the incidence, risk factors, and impact on graft survival. Transplantation. (1997) 63:1628–33. doi: 10.1097/00007890-199706150-00016
115. Sethi S, Fervenza FC. Membranoproliferative glomerulonephritis–a new look at an old entity. N Engl J Med. (2012) 366:1119–31. doi: 10.1056/NEJMra1108178
116. Sethi S, Fervenza FC. Standardized classification and reporting of glomerulonephritis. Nephrol Dial Transplant. (2019) 34:193–9. doi: 10.1093/ndt/gfy220
117. Sethi S, Haas M, Markowitz GS, D'Agati VD, Rennke HG, Jennette JC, et al. Mayo clinic/renal pathology society consensus report on pathologic classification, diagnosis, and reporting of GN. J Am Soc Nephrol. (2016) 27:1278–87. doi: 10.1681/ASN.2015060612
118. Sethi S, Fervenza FC. Membranoproliferative glomerulonephritis: pathogenetic heterogeneity and proposal for a new classification. Semin Nephrol. (2011) 31:341–8. doi: 10.1016/j.semnephrol.2011.06.005
119. Zand L, Lorenz EC, Cosio FG, Fervenza FC, Nasr SH, Gandhi MJ, et al. Clinical findings, pathology, and outcomes of C3GN after kidney transplantation. J Am Soc Nephrol. (2014) 25:1110–7. doi: 10.1681/ASN.2013070715
120. Medjeral-Thomas NR, O'Shaughnessy MM, O'Regan JA, Traynor C, Flanagan M, Wong L, et al. C3 glomerulopathy: clinicopathologic features and predictors of outcome. Clin J Am Soc Nephrol. (2014) 9:46–53. doi: 10.2215/CJN.04700513
121. Smith RJ, Alexander J, Barlow PN, Botto M, Cassavant TL, Cook HT, et al. New approaches to the treatment of dense deposit disease. J Am Soc Nephrol. (2007) 18:2447–56. doi: 10.1681/ASN.2007030356
122. Fakhouri F, Frémeaux-Bacchi V, Noël LH, Cook HT, Pickering MC. C3 glomerulopathy: a new classification. Nat Rev Nephrol. (2010) 6:494–9. doi: 10.1038/nrneph.2010.85
123. Pickering MC, D'Agati VD, Nester CM, Smith RJ, Haas M, Appel GB, et al. C3 glomerulopathy: consensus report. Kidney Int. (2013) 84:1079–89. doi: 10.1038/ki.2013.377
124. Sethi S, Fervenza FC, Zhang Y, Nasr SH, Leung N, Vrana J, et al. Proliferative glomerulonephritis secondary to dysfunction of the alternative pathway of complement. Clin J Am Soc Nephrol. (2011) 6:1009–17. doi: 10.2215/CJN.07110810
125. Sethi S, Fervenza FC, Zhang Y, Zand L, Vrana JA, Nasr SH, et al. C3 glomerulonephritis: clinicopathological findings, complement abnormalities, glomerular proteomic profile, treatment, and follow-up. Kidney Int. (2012) 82:465–73. doi: 10.1038/ki.2012.212
126. Sethi S, Nester CM, Smith RJ. Membranoproliferative glomerulonephritis and C3 glomerulopathy: resolving the confusion. Kidney Int. (2012) 81:434–41. doi: 10.1038/ki.2011.399
127. Servais A, Noël LH, Roumenina LT, Le Quintrec M, Ngo S, Dragon-Durey MA, et al. Acquired and genetic complement abnormalities play a critical role in dense deposit disease and other C3 glomerulopathies. Kidney Int. (2012) 82:454–64. doi: 10.1038/ki.2012.63
128. Servais A, Frémeaux-Bacchi V, Lequintrec M, Salomon R, Blouin J, Knebelmann B, et al. Primary glomerulonephritis with isolated C3 deposits: a new entity which shares common genetic risk factors with haemolytic uraemic syndrome. J Med Genet. (2007) 44:193–9. doi: 10.1136/jmg.2006.045328
129. Ohi H, Yasugi T. Occurrence of C3 nephritic factor and C4 nephritic factor in membranoproliferative glomerulonephritis (MPGN). Clin Exp Immunol. (1994) 95:316–21. doi: 10.1111/j.1365-2249.1994.tb06530.x
130. Sethi S, Nasr SH, De Vriese AS, Fervenza FC. C4d as a diagnostic tool in proliferative GN. J Am Soc Nephrol. (2015) 26:2852–9. doi: 10.1681/ASN.2014040406
131. Nasr SH, Sethi S, Cornell LD, Fidler ME, Boelkins M, Fervenza FC, et al. Proliferative glomerulonephritis with monoclonal IgG deposits recurs in the allograft. Clin J Am Soc Nephrol. (2011) 6:122–32. doi: 10.2215/CJN.05750710
132. Nasr SH, Markowitz GS, Stokes MB, Seshan SV, Valderrama E, Appel GB, et al. Proliferative glomerulonephritis with monoclonal IgG deposits: a distinct entity mimicking immune-complex glomerulonephritis. Kidney Int. (2004) 65:85–96. doi: 10.1111/j.1523-1755.2004.00365.x
133. Sumida K, Ubara Y, Marui Y, Nakamura M, Takaichi K, Tomikawa S, et al. Recurrent proliferative glomerulonephritis with monoclonal IgG deposits of IgG2lambda subtype in a transplanted kidney: a case report. Am J Kidney Dis. (2013) 62:587–90. doi: 10.1053/j.ajkd.2013.01.013
134. Czarnecki PG, Lager DJ, Leung N, Dispenzieri A, Cosio FG, Fervenza FC. Long-term outcome of kidney transplantation in patients with fibrillary glomerulonephritis or monoclonal gammopathy with fibrillary deposits. Kidney Int. (2009) 75:420–7. doi: 10.1038/ki.2008.577
135. Nasr SH, Valeri AM, Cornell LD, Fidler ME, Sethi S, D'Agati VD, et al. Renal monoclonal immunoglobulin deposition disease: a report of 64 patients from a single institution. Clin J Am Soc Nephrol. (2012) 7:231–9. doi: 10.2215/CJN.08640811
136. Leung N, Bridoux F, Hutchison CA, Nasr SH, Cockwell P, Fermand JP, et al. Monoclonal gammopathy of renal significance: when MGUS is no longer undetermined or insignificant. Blood. (2012) 120:4292–5. doi: 10.1182/blood-2012-07-445304
137. Leung N, Gertz MA. The spectrum of monoclonal gammopathies affecting the kidney. Leuk Lymphoma. (2012) 53:1656–7. doi: 10.3109/10428194.2012.673229
138. Bridoux F, Leung N, Hutchison CA, Touchard G, Sethi S, Fermand JP, et al. Diagnosis of monoclonal gammopathy of renal significance. Kidney Int. (2015) 87:698–711. doi: 10.1038/ki.2014.408
139. Welte T, Arnold F, Kappes J, Seidl M, Häffner K, Bergmann C, et al. Treating C3 glomerulopathy with eculizumab. BMC Nephrol. (2018) 19:7. doi: 10.1186/s12882-017-0802-4
140. Muczynski KA. Plasmapheresis maintained renal function in an allograft with recurrent membranoproliferative glomerulonephritis type I. Am J Nephrol. (1995) 15:446–9. doi: 10.1159/000168882
141. Farooqui M, Alsaad K, Aloudah N, Alhamdan H. Treatment-resistant recurrent membranoproliferative glomerulonephritis in renal allograft responding to rituximab: case report. Transpl Proc. (2015) 47:823–6. doi: 10.1016/j.transproceed.2015.02.003
142. Lien YH, Scott K. Long-term cyclophosphamide treatment for recurrent type I membranoproliferative glomerulonephritis after transplantation. Am J Kidney Dis. (2000) 35:539–43. doi: 10.1016/S0272-6386(00)70211-3
143. Dantal J, Baatard R, Hourmant M, Cantarovich D, Buzelin F, Soulillou JP. Recurrent nephrotic syndrome following renal transplantation in patients with focal glomerulosclerosis. A one-center study of plasma exchange effects. Transplantation. (1991) 52:827–31. doi: 10.1097/00007890-199111000-00014
144. Cochat P, Kassir A, Colon S, Glastre C, Tourniaire B, Parchoux B, et al. Recurrent nephrotic syndrome after transplantation: early treatment with plasmaphaeresis and cyclophosphamide. Pediatr Nephrol. (1993) 7:50–4. doi: 10.1007/BF00861567
145. Lebreton C, Bacchetta J, Dijoud F, Bessenay L, Fremeaux-Bacchi V, Sellier-Leclerc AL. C3 glomerulopathy and eculizumab: a report on four paediatric cases. Pediatr Nephrol. (2017) 32:1023–8. doi: 10.1007/s00467-017-3619-2
146. Spartà G, Gaspert A, Neuhaus TJ, Weitz M, Mohebbi N, Odermatt U, et al. Membranoproliferative glomerulonephritis and C3 glomerulopathy in children: change in treatment modality? A report of a case series. Clin Kidney J. (2018) 11:479–90. doi: 10.1093/ckj/sfy006
147. McCaughan JA, O'Rourke DM, Courtney AE. Recurrent dense deposit disease after renal transplantation: an emerging role for complementary therapies. Am J Transplant. (2012) 12:1046–51. doi: 10.1111/j.1600-6143.2011.03923.x
148. Pérez-Sáez MJ, Toledo K, Navarro MD, Lopez-Andreu M, Redondo MD, Ortega R, et al. Recurrent membranoproliferative glomerulonephritis after second renal graft treated with plasmapheresis and rituximab. Transpl Proc. (2011) 43:4005–9. doi: 10.1016/j.transproceed.2011.09.079
149. Rabasco C, Cavero T, Román E, Rojas-Rivera J, Olea T, Espinosa M, et al. Effectiveness of mycophenolate mofetil in C3 glomerulonephritis. Kidney Int. (2015) 88:1153–60. doi: 10.1038/ki.2015.227
150. Hoxha E, Thiele I, Zahner G, Panzer U, Harendza S, Stahl RA. Phospholipase A2 receptor autoantibodies and clinical outcome in patients with primary membranous nephropathy. J Am Soc Nephrol. (2014) 25:1357–66. doi: 10.1681/ASN.2013040430
151. Hofstra JM, Wetzels JF. Phospholipase A2 receptor antibodies in membranous nephropathy: unresolved issues. J Am Soc Nephrol. (2014) 25:1137–9. doi: 10.1681/ASN.2014010091
152. Gupta G, Fattah H, Ayalon R, Kidd J, Gehr T, Quintana LF, et al. Pre-transplant phospholipase A2 receptor autoantibody concentration is associated with clinically significant recurrence of membranous nephropathy post-kidney transplantation. Clin Transplant. (2016) 30:461–9. doi: 10.1111/ctr.12711
153. Leon J, Pérez-Sáez MJ, Batal I, Beck LH, Rennke HG, Canaud G, et al. Membranous nephropathy post-transplantation: an update of the pathophysiology and management. Transplantation. (2019). doi: 10.1097/TP.0000000000002758. [Epub ahead of print].
154. Debiec H, Lefeu F, Kemper MJ, Niaudet P, Deschênes G, Remuzzi G, et al. Early-childhood membranous nephropathy due to cationic bovine serum albumin. N Engl J Med. (2011) 364:2101–10. doi: 10.1056/NEJMoa1013792
155. Li W, Zhao Y, Fu P. Diagnostic test accuracy of serum anti-PLA2R autoantibodies and glomerular PLA2R antigen for diagnosing idiopathic membranous nephropathy: an updated meta-analysis. Front Med. (2018) 5:1. doi: 10.3389/fmed.2018.00101
156. Debiec H, Ronco P. PLA2R autoantibodies and PLA2R glomerular deposits in membranous nephropathy. N Engl J Med. (2011) 364:689–90. doi: 10.1056/NEJMc1011678
157. Cattran DC, Brenchley PE. Membranous nephropathy: integrating basic science into improved clinical management. Kidney Int. (2017) 91:566–74. doi: 10.1016/j.kint.2016.09.048
158. Passerini P, Malvica S, Tripodi F, Cerutti R, Messa P. Membranous nephropathy (MN) recurrence after renal transplantation. Front Immunol. (2019) 10:1326. doi: 10.3389/fimmu.2019.01326
159. Debiec H, Martin L, Jouanneau C, Dautin G, Mesnard L, Rondeau E, et al. Autoantibodies specific for the phospholipase A2 receptor in recurrent and de novo membranous nephropathy. Am J Transplant. (2011) 11:2144–52. doi: 10.1111/j.1600-6143.2011.03643.x
160. El-Zoghby ZM, Grande JP, Fraile MG, Norby SM, Fervenza FC, Cosio FG. Recurrent idiopathic membranous nephropathy: early diagnosis by protocol biopsies and treatment with anti-CD20 monoclonal antibodies. Am J Transplant. (2009) 9:2800–7. doi: 10.1111/j.1600-6143.2009.02851.x
161. Ruggenenti P, Chiurchiu C, Brusegan V, Abbate M, Perna A, Filippi C, et al. Rituximab in idiopathic membranous nephropathy: a one-year prospective study. J Am Soc Nephrol. (2003) 14:1851–7. doi: 10.1097/01.ASN.0000071511.35221.B3
162. Fervenza FC, Abraham RS, Erickson SB, Irazabal MV, Eirin A, Specks U, et al. Rituximab therapy in idiopathic membranous nephropathy: a 2-year study. Clin J Am Soc Nephrol. (2010) 5:2188–98. doi: 10.2215/CJN.05080610
163. Dahan K, Debiec H, Plaisier E, Cachanado M, Rousseau A, Wakselman L, et al. Rituximab for severe membranous nephropathy: a 6-month trial with extended follow-up. J Am Soc Nephrol. (2017) 28:348–58. doi: 10.1681/ASN.2016040449
164. Ruggenenti P, Debiec H, Ruggiero B, Chianca A, Pellé T, Gaspari F, et al. Anti-phospholipase A2 receptor antibody titer predicts post-rituximab outcome of membranous nephropathy. J Am Soc Nephrol. (2015) 26:2545–58. doi: 10.1681/ASN.2014070640
165. Dahan K, Gillion V, Johanet C, Debiec H, Ronco P. The role of PLA2R antibody in treatment of membranous nephropathy. Kidney Int Rep. (2018) 3:498–501. doi: 10.1016/j.ekir.2017.10.013
166. Barbari A, Chehadi R, Kfoury Assouf H, Kamel G, Jaafar M, Abdallah A, et al. Bortezomib as a novel approach to early recurrent membranous glomerulonephritis after kidney transplant refractory to combined conventional rituximab therapy. Exp Clin Transplant. (2017) 15:350–4. doi: 10.6002/ect.2016.0350
167. Grupper A, Cornell LD, Fervenza FC, Beck LH Jr, Lorenz E, Cosio FG. Recurrent membranous nephropathy after kidney transplantation: treatment and long-term implications. Transplantation. (2016) 100:2710–6. doi: 10.1097/TP.0000000000001056
168. Geetha D, Eirin A, True K, Valentina Irazabal M, Specks U, Seo P, et al. Renal transplantation in antineutrophil cytoplasmic antibody-associated vasculitis: a multicenter experience. Transplantation. (2011) 91:1370–5. doi: 10.1097/TP.0b013e31821ab9aa
169. Gera M, Griffin MD, Specks U, Leung N, Stegall MD, Fervenza FC. Recurrence of ANCA-associated vasculitis following renal transplantation in the modern era of immunosupression. Kidney Int. (2007) 71:1296–301. doi: 10.1038/sj.ki.5002244
170. Elmedhem A, Adu D, Savage CO. Relapse rate and outcome of ANCA-associated small vessel vasculitis after transplantation. Nephrol Dial Transplant. (2003) 18:1001–4. doi: 10.1093/ndt/gfg087
171. Nyberg G, Blohmé I, Persson H, Olausson M, Svalander C. Recurrence of SLE in transplanted kidneys: a follow-up transplant biopsy study. Nephrol Dial Transplant. (1992) 7:1116–23.
172. Contreras G, Mattiazzi A, Guerra G, Ortega LM, Tozman EC, Li H, et al. Recurrence of lupus nephritis after kidney transplantation. J Am Soc Nephrol. (2010) 21:1200–7. doi: 10.1681/ASN.2009101093
173. Goral S, Ynares C, Shappell SB, Snyder S, Feurer ID, Kazancioglu R, et al. Recurrent lupus nephritis in renal transplant recipients revisited: it is not rare. Transplantation. (2003) 75:651–6. doi: 10.1097/01.TP.0000053750.59630.83
174. Bunnapradist S, Chung P, Peng A, Hong A, Chung P, Lee B, et al. Outcomes of renal transplantation for recipients with lupus nephritis: analysis of the Organ Procurement and Transplantation Network database. Transplantation. (2006) 82:612–8. doi: 10.1097/01.tp.0000235740.56573.c6
175. Zhang L, Lee G, Liu X, Pascoe EM, Badve SV, Boudville NC, et al. Long-term outcomes of end-stage kidney disease for patients with lupus nephritis. Kidney Int. (2016) 89:1337–45. doi: 10.1016/j.kint.2016.02.014
176. Moroni G, Tantardini F, Gallelli B, Quaglini S, Banfi G, Poli F, et al. The long-term prognosis of renal transplantation in patients with lupus nephritis. Am J Kidney Dis. (2005) 45:903–11. doi: 10.1053/j.ajkd.2005.01.038
177. Sauter M, Schmid H, Anders HJ, Heller F, Weiss M, Sitter T. Loss of a renal graft due to recurrence of anti-GBM disease despite rituximab therapy. Clin Transplant. (2009) 23:132–6. doi: 10.1111/j.1399-0012.2008.00912.x
178. Tang W, McDonald SP, Hawley CM, Badve SV, Boudville NC, Brown FG, et al. Anti-glomerular basement membrane antibody disease is an uncommon cause of end-stage renal disease. Kidney Int. (2013) 83:503–10. doi: 10.1038/ki.2012.375
179. Le Quintrec M, Zuber J, Moulin B, Kamar N, Jablonski M, Lionet A, et al. Complement genes strongly predict recurrence and graft outcome in adult renal transplant recipients with atypical hemolytic and uremic syndrome. Am J Transplant. (2013) 13:663–75. doi: 10.1111/ajt.12077
180. Fakhouri F, Hourmant M, Campistol JM, Cataland SR, Espinosa M, Gaber AO, et al. Terminal complement inhibitor eculizumab in adult patients with atypical hemolytic uremic syndrome: a single-arm, open-label trial. Am J Kidney Dis. (2016) 68:84–93. doi: 10.1053/j.ajkd.2015.12.034
181. Alasfar S, Alachkar N. Atypical hemolytic uremic syndrome post-kidney transplantation: two case reports and review of the literature. Front Med. (2014) 1:52. doi: 10.3389/fmed.2014.00052
182. Sellier-Leclerc AL, Fremeaux-Bacchi V, Dragon-Durey MA, Macher MA, Niaudet P, Guest G, et al. Differential impact of complement mutations on clinical characteristics in atypical hemolytic uremic syndrome. J Am Soc Nephrol. (2007) 18:2392–400. doi: 10.1681/ASN.2006080811
183. Duineveld C, Verhave JC, Berger SP, van de Kar NCAJ, Wetzels JFM. Living donor kidney transplantation in atypical hemolytic uremic syndrome: a case series. Am J Kidney Dis. (2017) 70:770–7. doi: 10.1053/j.ajkd.2017.06.024
184. Cruzado JM, de Córdoba SR, Melilli E, Bestard O, Rama I, Sánchez-Corral P, et al. Successful renal transplantation in a patient with atypical hemolytic uremic syndrome carrying mutations in both factor I and MCP. Am J Transplant. (2009) 9:1477–83. doi: 10.1111/j.1600-6143.2009.02647.x
185. Lim WH, Wong G, McDonald SP, Chakera A, Luxton G, Isbel NM, et al. Long-term outcomes of kidney transplant recipients with end-stage kidney disease attributed to presumed/advanced glomerulonephritis or unknown cause. Sci Rep. (2018) 8:9021. doi: 10.1038/s41598-018-27151-4
186. Ponticelli C, Moroni G, Glassock RJ. de novo glomerular diseases after renal transplantation. Clin J Am Soc Nephrol. (2014) 9:1479–87. doi: 10.2215/CJN.12571213
187. Suzuki K, Honda K, Tanabe K, Toma H, Nihei H, Yamaguchi Y. Incidence of latent mesangial IgA deposition in renal allograft donors in Japan. Kidney Int. (2003) 63:2286–94. doi: 10.1046/j.1523-1755.63.6s.2.x
188. Shabaka A, Perez-Flores I, Cortes JA, Sanchez-Fructuoso AI. de novo IgA nephropathy in a renal allograft. Exp Clin Transplant. (2019) 17:550–3. doi: 10.6002/ect.2016.0266
189. Tanawattanacharoen S, Falk RJ, Jennette JC, Kopp JB. Parvovirus B19 DNA in kidney tissue of patients with focal segmental glomerulosclerosis. Am J Kidney Dis. (2000) 35:1166–74. doi: 10.1016/S0272-6386(00)70055-2
190. Shimizu A, Higo S, Fujita E, Mii A, Kaneko T. Focal segmental glomerulosclerosis after renal transplantation. Clin Transplant. (2011) 25(Suppl. 23):6–14. doi: 10.1111/j.1399-0012.2011.01452.x
191. Cosio FG, Frankel WL, Pelletier RP, Pesavento TE, Henry ML, Ferguson RM. Focal segmental glomerulosclerosis in renal allografts with chronic nephropathy: implications for graft survival. Am J Kidney Dis. (1999) 34:731–8. doi: 10.1016/S0272-6386(99)70400-2
192. Patel RD, Vanikar AV, Nigam LA, Kanodia KV, Suthar KS, Patel HV. de novo focal segmental glomerulosclerosis in renal allograft-histological presentation and clinical correlation: single centre experience. J Clin Diagn Res. (2017) 11:EC39–42. doi: 10.7860/JCDR/2017/25502.9728
193. Ponticelli C, Glassock RJ. de novo membranous nephropathy (MN) in kidney allografts. A peculiar form of alloimmune disease? Transpl Int. (2012) 25:1205–10. doi: 10.1111/j.1432-2277.2012.01548.x
194. Schwarz A, Krause PH, Offermann G, Keller F. Impact of de novo membranous glomerulonephritis on the clinical course after kidney transplantation. Transplantation. (1994) 58:650–4. doi: 10.1097/00007890-199409000-00002
195. Kearney N, Podolak J, Matsumura L, Houghton D, Troxell M. Patterns of IgG subclass deposits in membranous glomerulonephritis in renal allografts. Transpl Proc. (2011) 43:3743–6. doi: 10.1016/j.transproceed.2011.10.042
196. Larsen CP, Walker PD. Phospholipase A2 receptor (PLA2R) staining is useful in the determination of de novo versus recurrent membranous glomerulopathy. Transplantation. (2013) 95:1259–62. doi: 10.1097/TP.0b013e31828a947b
197. Monga G, Mazzucco G, Basolo B, Quaranta S, Motta M, Segoloni G, et al. Membranous glomerulonephritis (MGN) in transplanted kidneys: morphologic investigation on 256 renal allografts. Mod Pathol. (1993) 6:249–58.
198. Doke T, Sato W, Takahashi K, Hayashi H, Koide S, Sasaki H, et al. Post-transplant membranous nephropathy associated with chronic active antibody-mediated rejection and Hepatitis C infection after deceased donor renal transplantation. Intern Med. (2016) 55:375–80. doi: 10.2169/internalmedicine.55.5468
199. Poduval RD, Josephson MA, Javaid B. Treatment of de novo and recurrent membranous nephropathy in renal transplant patients. Semin Nephrol. (2003) 23:392–9. doi: 10.1016/S0270-9295(03)00057-3
200. Roth D, Cirocco R, Zucker K, Ruiz P, Viciana A, Burke G, et al. de novo membranoproliferative glomerulonephritis in hepatitis C virus-infected renal allograft recipients. Transplantation. (1995) 59:1676–82. doi: 10.1097/00007890-199506270-00006
201. Abbas F, El Kossi M, Jin JK, Sharma A, Halawa A. de novo glomerular diseases after renal transplantation: How is it different from recurrent glomerular diseases? World J Transplant. (2017) 7:285–300. doi: 10.5500/wjt.v7.i6.285
202. Nahm JH, Song SH, Kim YS, Cheong HI, Lim BJ, Kim BS, et al. de novo C3 glomerulonephritis in a renal allograft. Ultrastruct Pathol. (2016) 40:112–5. doi: 10.3109/01913123.2016.1154634
203. Zafarmand AA, Baranowska-Daca E, Ly PD, Tsao CC, Choi YJ, Suki WN, et al. de novo minimal change disease associated with reversible post-transplant nephrotic syndrome. A report of five cases and review of literature. Clin Transplant. (2002) 16:350–61. doi: 10.1034/j.1399-0012.2002.02023.x
204. Markowitz GS, Stemmer CL, Croker BP, D'Agati VD. de novo minimal change disease. Am J Kidney Dis. (1998) 32:508–13. doi: 10.1053/ajkd.1998.v32.pm9740171
205. Mainra R, Mulay A, Bell R, Karpinski J, Hoar S, Knoll G, et al. Sirolimus use and de novo minimal change nephropathy following renal transplantation. Transplantation. (2005) 80:1816. doi: 10.1097/01.tp.0000181385.68835.a0
206. Moudgil A, Shidban H, Nast CC, Bagga A, Aswad S, Graham SL, et al. Parvovirus B19 infection-related complications in renal transplant recipients: treatment with intravenous immunoglobulin. Transplantation. (1997) 64:1847–50. doi: 10.1097/00007890-199712270-00037
207. Tomlinson L, Boriskin Y, McPhee I, Holwill S, Rice P. Acute cytomegalovirus infection complicated by collapsing glomerulopathy. Nephrol Dial Transplant. (2003) 18:187–9. doi: 10.1093/ndt/18.1.187
208. Meehan SM, Pascual M, Williams WW, Tolkoff-Rubin N, Delmonico FL, Cosimi AB, et al. de novo collapsing glomerulopathy in renal allografts. Transplantation. (1998) 65:1192–7. doi: 10.1097/00007890-199805150-00009
209. Swaminathan S, Lager DJ, Qian X, Stegall MD, Larson TS, Griffin MD. Collapsing and non-collapsing focal segmental glomerulosclerosis in kidney transplants. Nephrol Dial Transplant. (2006) 21:2607–14. doi: 10.1093/ndt/gfl225
210. Alachkar N, Gupta G, Montgomery RA. Angiotensin antibodies and focal segmental glomerulosclerosis. N Engl J Med. (2013) 368:971–3. doi: 10.1056/NEJMc1207233
211. Isaac J, Herrara GA, Shihab FS. de novo fibrillary glomerulopathy in the renal allograft of a patient with systemic lupus erythematosus. Nephron. (2001) 87:365–8. doi: 10.1159/000045944
212. Samaniego M, Nadasdy GM, Laszik Z, Nadasdy T. Outcome of renal transplantation in fibrillary glomerulonephritis. Clin Nephrol. (2001) 55:159–66.
213. Nasr SH, Valeri AM, Cornell LD, Fidler ME, Sethi S, Leung N, et al. Fibrillary glomerulonephritis: a report of 66 cases from a single institution. Clin J Am Soc Nephrol. (2011) 6:775–84. doi: 10.2215/CJN.08300910
214. Rao KV, Hafner GP, Crary GS, Anderson WR, Crosson JT. de novo immunotactoid glomerulopathy of the renal allograft: possible association with cytomegalovirus infection. Am J Kidney Dis. (1994) 24:97–103. doi: 10.1016/S0272-6386(12)80167-3
215. Sathyan S, Khan FN, Ranga KV. A case of recurrent immunotactoid glomerulopathy in an allograft treated with rituximab. Transpl Proc. (2009) 41:3953–5. doi: 10.1016/j.transproceed.2009.03.100
216. Carles X, Rostaing L, Modesto A, Orfila C, Cisterne JM, Delisle MB, et al. Successful treatment of recurrence of immunotactoid glomerulopathy in a kidney allograft recipient. Nephrol Dial Transplant. (2000) 15:897–900. doi: 10.1093/ndt/15.6.897
217. Byrne MC, Budisavljevic MN, Fan Z, Self SE, Ploth DW. Renal transplant in patients with Alport's syndrome. Am J Kidney Dis. (2002) 39:769–75. doi: 10.1053/ajkd.2002.31997
218. Gumber MR, Kute VB, Goplani KR, Vanikar AV, Shah PR, Patel HV, et al. Outcome of renal transplantation in Alport's syndrome: a single-center experience. Transpl Proc. (2012) 44:261–3. doi: 10.1016/j.transproceed.2011.11.035
219. Temme J, Kramer A, Jager KJ, Lange K, Peters F, Müller GA, et al. Outcomes of male patients with Alport syndrome undergoing renal replacement therapy. Clin J Am Soc Nephrol. (2012) 7:1969–76. doi: 10.2215/CJN.02190312
220. Gillion V, Dahan K, Cosyns JP, Hilbert P, Jadoul M, Goffin E, et al. Genotype and outcome after kidney transplantation in alport syndrome. Kidney Int Rep. (2018) 3:652–60. doi: 10.1016/j.ekir.2018.01.008
221. Sagmeister MS, Weiss M, Eichhorn P, Habicht A, Habersetzer R, Fischereder M, et al. Case report: de novo ANCA-associated vasculitis after kidney transplantation treated with rituximab and plasma exchange. BMC Nephrol. (2018) 19:270. doi: 10.1186/s12882-018-1086-z
222. Tabata H, Honda K, Moriyama T, Itabashi M, Taneda S, Takei T, et al. Two cases of ANCA-associated vasculitis in post-transplant kidney: relapse and de novo. Clin Transplant. (2009) 23(Suppl. 20):49–53. doi: 10.1111/j.1399-0012.2009.01010.x
223. Haruyama N, Tsuchimoto A, Masutani K, Noguchi H, Suehiro T, Kitada H, et al. de novo myeloperoxidase-antineutrophil cytoplasmic antibody (MPO-ANCA)-associated glomerulonephritis 31 years after living-donor kidney transplantation. CEN Case Rep. (2015) 4:14–9. doi: 10.1007/s13730-014-0131-4
224. Asif A, Toral C, Diego J, Miller J, Roth D. de novo ANCA-associated vasculitis occurring 14 years after kidney transplantation. Am J Kidney Dis. (2000) 35:E10. doi: 10.1016/S0272-6386(00)70222-8
225. Li RM, Branton MH, Tanawattanacharoen S, Falk RA, Jennette JC, Kopp JB. Molecular identification of SV40 infection in human subjects and possible association with kidney disease. J Am Soc Nephrol. (2002) 13:2320–30. doi: 10.1097/01.ASN.0000028249.06596.CF
226. Olyaei AJ, de Mattos AM, Bennett WM. Nephrotoxicity of immunosuppressive drugs: new insight and preventive strategies. Curr Opin Crit Care. (2001) 7:384–9. doi: 10.1097/00075198-200112000-00003
227. Letavernier E, Bruneval P, Vandermeersch S, Perez J, Mandet C, Belair MF, et al. Sirolimus interacts with pathways essential for podocyte integrity. Nephrol Dial Transplant. (2009) 24:630–8. doi: 10.1093/ndt/gfn574
228. Gupta R, Sharma A, Mahanta PJ, Agarwal SK, Dinda AK. Focal and segmental glomerulosclerosis in renal allograft recipients: a clinico-pathologic study of 37 cases. Saudi J Kidney Dis Transplant. (2013) 24:8–14. doi: 10.4103/1319-2442.106231
229. Hammoud H, Haem J, Laurent B, Alamartine E, Diab N, Defilippis JP, et al. Glomerular disease during HCV infection in renal transplantation. Nephrol Dial Transplant. (1996) 11(Suppl. 4): 54–5. doi: 10.1093/ndt/11.supp4.54
230. de Fijter JW. Recurrence of glomerulonephritis: an underestimated and unmet medical need. Kidney Int. (2017) 92:294–6. doi: 10.1016/j.kint.2017.04.007
Keywords: recurrent disease, glomerulonephritis, kidney transplantation, recurrent glomerulonephritis, de novo glomerulonephritis, allograft failure
Citation: Lim WH, Shingde M and Wong G (2019) Recurrent and de novo Glomerulonephritis After Kidney Transplantation. Front. Immunol. 10:1944. doi: 10.3389/fimmu.2019.01944
Received: 15 April 2019; Accepted: 01 August 2019;
Published: 14 August 2019.
Edited by:
Josep M. Grinyó, University of Barcelona, SpainReviewed by:
Lionel P. E. Rostaing, Université Grenoble Alpes, FranceCopyright © 2019 Lim, Shingde and Wong. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Wai H. Lim, d2FpLmxpbUBoZWFsdGgud2EuZ292LmF1
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.