 Kwang-Soo Shin1Insu Jeon2Byung-Seok Kim3Il-Kyu Kim1Young-Jun Park3Choong-Hyun Koh1Boyeong Song2Jeong-Mi Lee1Jiyoung Lim2Eun-Ah Bae2Hyungseok Seo2Young Ho Ban4Sang-Jun Ha4Chang-Yuil Kang1,2*
Kwang-Soo Shin1Insu Jeon2Byung-Seok Kim3Il-Kyu Kim1Young-Jun Park3Choong-Hyun Koh1Boyeong Song2Jeong-Mi Lee1Jiyoung Lim2Eun-Ah Bae2Hyungseok Seo2Young Ho Ban4Sang-Jun Ha4Chang-Yuil Kang1,2*- 1Laboratory of Immunology, College of Pharmacy, Seoul National University, Seoul, South Korea
- 2Laboratory of Immunology, Department of Molecular Medicine and Biopharmaceutical Sciences, Graduate School of Convergence Science and Technology, Seoul National University, Seoul, South Korea
- 3Laboratory of Immune Regulation, College of Pharmacy, Seoul National University, Seoul, South Korea
- 4Department of Biochemistry, College of Life Science and Biotechnology, Yonsei University, Seoul, South Korea
Monocyte-derived dendritic cells (moDCs) have been shown to robustly expand during infection; however, their roles in anti-infectious immunity remain unclear. Here, we found that moDCs were dramatically increased in the secondary lymphoid organs during acute LCMV infection in an interferon-γ (IFN-γ)-dependent manner. We also found that priming by moDCs enhanced the differentiation of memory CD8+ T cells compared to differentiation primed by conventional dendritic cells (cDCs) through upregulation of Eomesodermin (Eomes) and T cell factor-1 (TCF-1) expression in CD8+ T cells. Consequently, impaired memory formation of CD8+ T cells in mice that had reduced numbers of moDCs led to defective clearance of pathogens upon rechallenge. Mechanistically, attenuated interleukin-2 (IL-2) signaling in CD8+ T cells primed by moDCs was responsible for the enhanced memory programming of CD8+ T cells. Therefore, our findings unveil a specialization of the antigen-presenting cell subsets in the fate determination of CD8+ T cells during infection and pave the way for the development of a novel therapeutic intervention on infection.
Introduction
CD8+ T cells play a dominant role in the elimination of infectious pathogens. Following their primary activation, CD8+ T cells undergo a fate determination between short-lived effector cells (SLECs) or memory-precursor effector cells (MPECs) (1). It has been suggested that various non-mutually exclusive factors direct the CD8+ T cell fate decision (2). For example, T cell receptor (TCR) signal quantity and quality determine the diversification of progeny from CD8+ T cells. A strong TCR signal licenses CD8+ T cells to undergo effector-prone differentiation, whereas a weak TCR signal diverts the fate of activated CD8+ T cells to memory-like cells (2, 3). Recent studies have proposed that these events result from complex transcriptional regulation (4). Indeed, T-bet and Blimp-1 have been shown to drive the fate of CD8+ T cells into effector cells, while Eomes and TCF1 are involved in differentiation into memory-like cells (1, 5–7). The TCR signal strength is governed by TCR-MHC interaction and co-stimulation delivered by various antigen-presenting cells (APCs) in the secondary lymphoid organs (SLOs); however, the contributions of each APC to the diversification of the CD8+ T cell fate remains poorly characterized.
Upon infection, myeloid cells robustly expand and are recruited to the inflamed sites to clear pathogens or infected cells (8, 9). Myeloid cells are crucial not only for innate immunity but also for initiation of adaptive immune responses. Among them, monocytes have been classically considered to be the major progenitors of macrophages that take part in the clearance of cellular debris and pathogens (10). However, it has recently become evident that adult tissue macrophages mainly develop from the yolk sac (or fetal liver)-derived progenitor cells during embryogenesis (11). Thus, the differentiation and function of monocytes during infection requires further investigation (10). During infection and inflammation, monocytes further differentiate into DC-like cells, which are referred to as monocyte-derived DCs (moDCs) or TNF- and iNOS-producing DCs (Tip-DCs) depending on the context (12, 13). Previous reports have proposed that common monocyte progenitor cells (cMoPs) in the bone marrow (BM) are progenitors of moDCs (14, 15). However, the specific mechanism that drives the differentiation of cMoPs into moDCs needs additional investigation.
Through rapid and robust myelopoiesis upon infection, monocytes and moDCs preferentially populate the antigen-presenting cell (APC) pool in the SLOs. Monocytes and moDCs are known to initiate CD8+ T cell responses by antigen presentation; their roles have been investigated and conflicting results were obtained in various animal models (16). For example, tumor-infiltrating moDCs have been shown to prime CD8+ T cells and induce anti-tumor immunity (15). In contrast, monocytic cells in chronic infections abrogate the induction of anti-infectious CD8+ T cell responses (17). Therefore, specific contribution of monocytes and moDCs to the differentiation of CD8+ T cells remains poorly understood.
In this report, we investigated the role of moDCs in CD8+ T cell fate determination during acute infection. We found that moDCs were expanded during lymphocytic choriomeningitis virus (LCMV) infection; in the case of bone marrow progenitor cells (BMPs), cMoPs differentiated into moDCs in an IFN-γ-dependent manner. In addition, CD8+ T cells that were primed in Ccr2−/− mice, which have reduced numbers of moDCs in SLOs, could not properly develop into memory cells and underwent effector-prone differentiation during the expansion phase of infection. Moreover, Ccr2−/− mice have defects in pathogen clearance upon reinfection due to the defective differentiation and survival of memory CD8+ T cells. Finally, attenuated IL-2 signals provided by moDCs were responsible for the enhanced memory differentiation of CD8+ T cells.
Materials and Methods
Animals
C57BL/6J mice were purchased from the Institute of Medical Science at the University of Tokyo and Balb/c mice from Charles River Laboratory. OT-I [C57BL/6-Tg(TcraTcrb)1100Mjb/J], P14 [B6;D2-Tg(TcrLCMV)327Sdz/JDvsJ], Ccr2−/− (B6.129S4-Ccr2tm1Ifc/J), Ifng−/− [C.129S7(B6)-Ifngtm1Ts/J], and CD45.1 (B6.SJL-PtprcaPepcb/BoyJ) mice were purchased from Jackson Laboratory. CD45.1+ P14 mice were obtained by crossbreeding CD45.1+ mice and C57BL/6J mice. Age (6 to 12 weeks) and sex-matched mice were used for all experiments. All mice were bred and maintained under specific pathogen-free conditions in the Animal Facility of Seoul National University. Experiments with infectious pathogen were performed in the ABL2 vivarium of Seoul National University. All animal protocols were approved by the Institutional Animal Care and Use Committee (IACUC) of Seoul National University.
Infections and IFN-γ Neutralization
For primary infection, mice were injected intraperitoneally (i.p.) with LCMV-Arm [2 × 105 plaque-forming units (PFU)] or intravenously (i.v.) with GP33−41-expressing Listeria monocytogenes [Lm-GP33, 5,000 colony-forming units (CFU)], which were generously donated from Yonsei University. To analyze the host protection capacity of memory cells, mice were infected with Lm-GP33 (5,000 CFU). To neutralize IFN-γ in vivo, mice were treated i.p. with 500 μg anti-IFN-γ mAb (HB170, ATCC) at day 1, 4, and 7 p.i., for LCMV-Arm infection, and at day −1 and 1 p.i., for Lm-GP33 infection. All experimental methods with infectious pathogens were reviewed and approved by the Institutional Biosafety Committee (IBC) of Seoul National University.
Flow Cytometry
Spleen and peripheral lymph nodes were isolated from mice and homogenized using a 70 μm cell strainer (BD Biosciences). Bone marrow cells were isolated by flushing the tibia and femur of mice with a 1 ml syringe. PBMCs in the blood were isolated using Hispaque-1077 (Sigma-Aldrich) following the manufacturer's protocol. Red blood cells (RBC) of single cell suspensions were lysed using RBC Lysis Buffer (BioLegend).
Abs used for flow cytometry were as follows: anti-IA/IE (M5/114.15.2), anti-CD11b (M1/70), anti-CD11c (N418), anti-Ly6G (1A8), anti-Ly6C (HK1.4), anti-CCR2 (475301, R&D Systems), anti-CX3CR1 (SA011F11), anti-F4/80 (BM8, eBioscience), anti-CD64 (X54-5/7.1), anti-CD115 (AFS98), anti-CD135 (A2F10), anti-CD119 (2E2), anti-CD45.1 (A20), anti-CD45.2 (104), anti-H-2Kb (AF6-88.5), anti-H-Db (KH95), anti-CD40 (3/23), anti-CD80 (16-10A1), anti-CD86 (GL-1), anti-PD-L1 (10F.9G2), anti-CD8α (53-6.7), anti-CD3ε (145-2C11), anti-CD25 (PC61), anti-CD62L (MEL-14), anti-KLRG1 (2F1/KLRG1), anti-CD127 (A7R34), anti-IFN-γ (XMG1.2), anti-TNF-α (MP6-XT22), anti-granzyme B (GB11), anti-Eomes (Dan11mag, eBioscience), anti-T-bet (4B10, eBioscience), and anti-TCF1 (C63D9, Cell Signaling). Abs were purchased from BioLegend unless otherwise described. Streptavidin-APC/Cy7 (BioLegend) and Alexa Fluor 647 goat anti-rabbit IgG (Invitrogen) were used for secondary staining.
Single cell suspensions were stained for surface molecules for 30 min at 4°C. Dead cells were excluded from analysis using Fixable Viability Dye (eBioscience). For intracellular cytokine staining, cells were restimulated with GP33−41 peptide (KAVYNFATC, 0.2 μg/ml, Genscript) in the presence of BD GolgiPlug (BD Biosciences) for 4 h, fixed and permeabilized using a Cytofix/Cytoperm kit (BD Biosciences) according to the manufacturer's instructions. To detect transcription factors, a fixation/permeabilization kit purchased from eBioscience was used. For analyzing cellular apoptosis, FITC Annexin V apoptosis Detection Kit I and Propidium Iodide Staining Solution (BD Biosciences) were used. Samples were collected using a FACS LSRFortessa X-20 or FACS Aria III (BD Biosciences), and data were analyzed with FlowJo software (Treestar).
Cell Sorting
For sorting BMPs, BM cells of naïve mice were labeled with biotinylated anti-CD3ε, anti-CD19, anti-CD49b, and anti-Ly6G (all from Biolegend) followed by anti-biotin microbeads (Miltenyi Biotech) and isolated using a MACS LD column (Miltenyi Biotech). The collected cells were further sorted into each BMP subset: total BMPs (lin−c-kit+CD11b−Ly6C−), cMoPs (lin−c-kit+CD115+CD135−CD11b−Ly6C+), MDPs (lin−c-kit+CD115+CD135+CD11b−Ly6C−), CD135+ BMPs (lin−c-kit+CD115−CD135+CD11b−Ly6C−), and CD115−CD135− BMPs (lin−c-kit+CD115−CD135−CD11b−Ly6C−).
To isolate cDCs and moDCs from infected mice, Lin− cells of infected splenocytes (day 4 or 8 p.i.) were isolated using a MACS LD column and further sorted to each subset (cDCs as lin−IA/IE+CD11c+CD11b−CCR2−Ly6C− and moDCs as lin−IA/IE+CD11c−/intCD11b+CCR2+Ly6C+).
CD45.1+ P14 cells used in the in vitro and in vivo experiments were enriched using anti-CD8a microbeads and a MACS LS column (Miltenyi Biotech) and further purified to CD45.1+CD8+ cells by cell sorting.
Cell sorting was conducted using a FACS Aria II or FACS Aria III. The purities of all sorted populations were >95%.
ELISA
The IFN-γ concentration in mouse serum was measured using a mouse IFN-γ ELISA kit (BD Biosciences) according to the manufacturer's protocol. The IL-2 concentrations in the cocultures of T cells and APCs were measured using the following Abs: anti-IL-2 (JES6-1A12) for capture, biotinylated anti-IL-2 (JES6-5H4) and streptavidin-HRP for detection (all from BD Biosciences).
BM Cell Differentiation Assay
To evaluate the differentiation patterns of BMPs in vitro, sorted total BMPs, cMoPs, MDPs, CD135+ BMPs, and CD115−CD135− BMPs (1 × 104 cells/well) were cultured for 4 to 6 days under specific conditions as follows: GM-CSF, GM-CSF plus IL-4, or GM-CSF plus IFN-γ (all from R&D systems). All recombinant cytokines were used at 20 ng/ml, and the culture medium was refreshed every 2 days.
To analyze the differentiation patterns of cMoPs in vivo, sorted cMoPs and non-cMoPs (2 × 105 cells each/mouse) were adoptively transferred into LCMV-Arm-infected recipient mice at day 5 p.i., Donor cells were analyzed at day 3 post transfer (day 8 p.i.).
In vitro APC:T Cell Coculture and in vitro Cytotoxicity Assay
The 1 × 104 APCs (cDCs or moDCs) and 5 × 104 P14 cells were cultured for 3 days in the presence of GP33−41 peptide. To determine the proliferation capacity of P14 cells, the cells were labeled with 5 μM of CellTrace Violet (CTV, Invitrogen) for 15 min prior to incubation. The cocultures in some experiments were treated with recombinant mouse IL-2 (10 ng/ml, Peprotech) or anti-IL-2 mAbs (10 μg/ml, JES6-1A12, eBioscience).
To measure the cytotoxicity of activated CD8+ T cells, equivalent numbers of purified live effector P14 cells from in vitro cocultures or infected mice were cocultured with 51Cr-labeled GP33−41-loaded EL4 cells (ATCC) for 4 h. Target cell specific lysis was measured by a Wallac 1470 Wizard automatic γ-counter (PerkinElmer) and calculated using the following equation; [(sample lysis count per minute (CPM)—spontaneous lysis CPM)/(Triton X-100-mediated lysis CPM—spontaneous lysis CPM)] × 100 (%).
T Cell Adoptive Transfer
To examine the primary immune responses, 1 × 104 purified CD8+ P14 cells from P14 splenocytes were adoptively transferred into WT or Ccr2−/− mice and analyzed at the indicated time points. To establish the memory of P14 cells, 1 × 106 P14 cells isolated from infected mice at day 8 p.i., were adoptively transferred to naïve recipient mice. To evaluate the memory-generation capacity of P14 cells primed by moDCs and cDCs in vitro, P14 cells were activated as indicated in the “in vitro APC:T cell coculture” section and then transferred to infected mice at day 8 p.i., (5 × 105 cells/mouse).
Quantitative Real-Time PCR
Total RNA of sorted P14 cells from infected mice at day 8 p.i., was isolated using TRIzol reagent and reverse-transcribed into cDNA using AmfiRivert II cDNA Synthesis Master Mix (Gendepot). Real-time PCR was performed with a SYBR Green real-time PCR kit (Takara) and LightCycler 1.5 instrument (Roche Diagnostics). Primers were purchased from Cosmo Genetech, and their sequences were as follows: mouse Tbx21 (forward; 5′– ACA AGG GGG CTT CCA ACA AT −3′, reverse; 5′– TGC GTT CTG GTA GGC AGT CA −3′), mouse Eomes (forward; 5′– AGA ACC GTG CCA CAG ACC AA −3′, reverse; 5′– TCG TCA CAG GTT GCT GGA CA −3′), mouse Tcf7 (forward; 5′– GCA CAC TTC GCA GAG ACT TT −3′, reverse; 5′– GTG GAC TGC TGA AAT GTT CG −3′), mouse Prdm1 (forward; 5′– ACT CAG TCG CAT TTG ATG GC −3′, reverse; 5′– GGT CAG TAA GGC TCT TGG GT −3′), mouse Il2 (forward; 5′– CAA CTG TGG TGG ACT TTC TG −3′, reverse; 5′– CCT TGG GGC TTA CAA AAA GAA −3′), and mouse Hprt (forward; 5′– AAG ACT TGC TCG AGA TGT CAT GAA −3′, reverse; 5′– ATC CAG CAG GTC AGC AAA GAA −3′). The value of each gene expression level was normalized to the expression level of mouse Hprt.
Tissue Titration
Tissue titrations were conducted as described previously (18). Spleens of Lm-GP33 infected mice were prepared as a single-cell suspension and then treated with 1% Triton X-100 solution (Sigma-Aldrich). Each diluted suspension was plated on the BHI agar plates (BD Biosciences) and incubated overnight at 37°C. Colonies on the plates were counted next day, and the titers were calculated as CFUs per gram of spleen. Small fragments of the spleens from LCMV-Arm-infected mice were stored in DMEM with 1% FBS (Gibco) and were homogenized completely. Vero cells in 6-well plates were infected with each dilution of the spleen samples for 1 h, subsequently overlaid with Medium-199 (Gibco) agarose gel, and incubated for 4 days. Then, Vero cells were stained with Neutral red solution. Plaques were counted 2 days after the staining and the titers were calculated as PFUs per gram of spleen.
Statistics
Statistical analysis of all data was conducted using GraphPad Prism 5 software (GraphPad Software, Inc.). The unpaired, two-tailed Student's t-test was used to compare two groups, and one-way ANOVA with Bonferroni's multiple comparisons was used to compare more than three groups. Two-way ANOVA with Bonferroni's multiple comparisons was used in Figure 1E, Supplementary Figures 1A,C. P < 0.05 were considered significant.
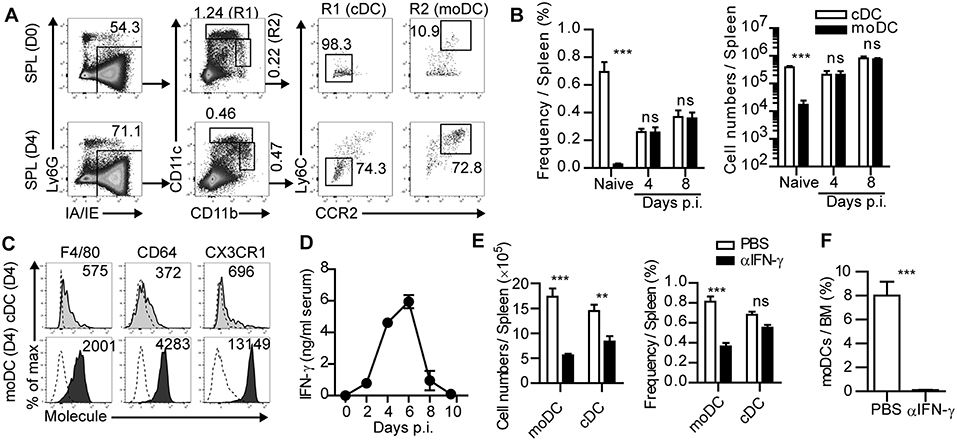
Figure 1. IFN-γ-dependent expansion of monocyte-derived dendritic cells during acute infection. (A) Gating strategies of cDCs and moDCs in the spleen of naïve or LCMV-Arm-infected mice. Numbers indicate the percentages within the gates. (B) Cell numbers and frequencies of cDCs and moDCs in the spleen during LCMV-Arm infection. (C) Expression patterns of indicated surface molecules on cDCs and moDCs in LCMV-Arm-infected mice at day 4 p.i., Numbers indicate the MFI values of each molecule. (D) Kinetics of IFN-γ levels in the serum of LCMV-Arm-infected mice. (E,F) LCMV-Arm-infected mice were treated with IFN-γ-neutralizing Ab. (E) Cell numbers (left) and frequencies (right) of cDCs and moDCs were measured in the indicated organs on day 8 p.i., and are shown as graph plots. (F) Frequency of moDCs in the BM. Data are representative of three independent experiments and are shown as the mean ± SEM. n = 5 per group at each time point. **p < 0.01;***p < 0.001.
Results
IFN-γ-Dependent Expansion of Monocyte-Derived Dendritic Cells During Acute Infection
Various types of APCs in inflamed tissues and lymphoid organs are known to initiate adaptive immune responses during infection. Among those APCs, we sought to determine the role of moDCs in anti-infectious immune responses. Initially, we investigated the frequencies and numbers of moDCs and cDCs in lymphoid organs after acute LCMV (LCMV-Arm) infection. Based on a previous report (19), cDCs and moDCs in the spleen were defined by their cell surface marker expression patterns including Ly6G−IA/IE+CD11chiCD11blow/hiLy6C−CCR2− cells and Ly6G−IA/IE+CD11clow/intCD11bhiLy6C+CCR2+ cells, respectively (Figure 1A). moDCs were sparsely present in uninfected mice, while cDCs represented a dominant APC subset at steady state. However, moDCs rapidly accumulated in the spleen after LCMV-Arm infection and became an abundant APC population during the expansion phase of infection (Figures 1A,B). In addition, high expression levels of F4/80, CD64, and CX3CR1 in moDCs compared to those in cDCs indicated that moDCs were distinct from cDCs (Figure 1C).
We further investigated the underlying mechanism of the accumulation of moDCs during infection. Recent reports have demonstrated that IFN-γ induces the differentiation, migration and expansion of inflammatory monocyte lineage cells during infection (20–22). In this regard, the level of serum IFN-γ was elevated in LCMV-Arm-infected mice ~2 days prior to the accumulation of moDCs (Figure 1D). To determine whether the accumulation of moDCs was regulated by IFN-γ, we treated infected mice with IFN-γ-neutralizing Ab (Supplementary Figure 1A). IFN-γ neutralization led the slight increase in the viral loads in mice (Supplementary Figure 1B) (23). Although the absolute numbers of cDCs and moDCs were decreased along with reductions in the numbers of splenocytes, we found that the frequency of moDCs was markedly reduced in the spleen of mice that received IFN-γ-neutralizing Ab, while the frequency of cDCs did not decrease significantly (Figure 1E and Supplementary Figure 1C). Importantly, moDCs were almost completely absent in the BM of LCMV-Arm-infected mice with neutralized IFN-γ (Figure 1F). IFN-γ neutralization did not lead to the reductions of BMPs, but it rather induced the enhanced BMP frequencies (Supplementary Figure 1D). Furthermore, IFN-γ neutralization did not induce apoptotic death of moDCs (Supplementary Figure 1E). Thus, we concluded that the IFN-γ-dependent accumulation of moDCs in the periphery is due to the increased generation of the cells rather than the enhanced migration from the BM, their enhanced survival, or the effects of IFN-γ on the frequencies of BMPs. We also confirmed that IFN-γ is required for moDC generation in the spleen and BM during LCMV-Arm infection using IFN-γ-deficient mice (Supplementary Figure 2A). Additionally, we found that the frequency of moDCs was also increased in GP33−41-expressing Listeria monocytogenes (Lm-GP33)-infected mice (Supplementary Figures 2B,C). Consistent with the result on LCMV-Arm infection, accumulation of moDCs was dependent on IFN-γ during Lm-GP33 infection (Supplementary Figures 2B,C). Altogether, these results indicate that moDCs robustly expand in an IFN-γ-dependent manner during an acute viral and bacterial infection.
IFN-γ Acts Directly on Common Monocyte Progenitor Cells and Promotes the Differentiation of moDCs
The observed role of IFN-γ in the surge of moDCs in the periphery after acute infection prompted us to investigate whether IFN-γ can influence moDC generation from specific BMPs. To achieve this goal, we sorted lineage−c-kit+ BMPs from naïve mice and differentiated them with GM-CSF, GM-CSF plus IL-4, or GM-CSF plus IFN-γ. In line with a previous report, BMPs cultured with GM-CSF differentiated into two distinct populations including GM-Macs (CD11c+IA/IEint) and GM-DCs (CD11c+IA/IEhi), which represent monocyte-derived macrophages and dendritic cells, respectively (24). While the addition of IL-4 favors differentiation of GM-DCs, IFN-γ promoted the differentiation of Ly6C+CD11b+ cells instead of GM-Macs or GM-DCs (Figure 2A). Collectively, these results demonstrate that IFN-γ diverts the fate of BMPs from GM-Macs or GM-DCs to moDCs.
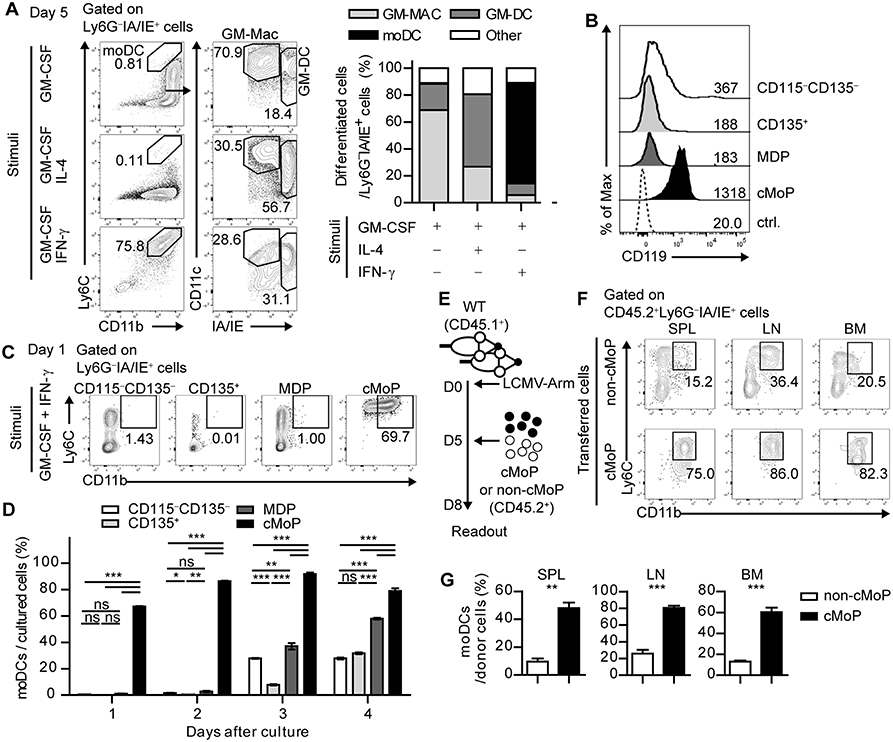
Figure 2. IFN-γ acts directly on common monocyte progenitor cells and promotes the differentiation of moDCs. (A) Differentiation patterns of total BMPs at day 5 under different stimuli (GM-CSF alone, GM-CSF + IL-4, and GM-CSF + IFN-γ are shown as flow cytometry plots (left) and graphs (right). Numbers in the plots indicate the percentages within the gates. (B) IFN-γR (CD119) expression levels of the BMP subsets. Numbers indicate the MFI values of each subset. (C,D) Daily differentiation of the subdivided BMP subsets into moDCs under GM-CSF and IFN-γ stimulation. (C) Representative flow cytometry plots of sorted BMP subsets after a day of culture (D) Graph shows the proportions of cells that differentiate into moDCs in each BMP subset. (E–G) Sorted cMoPs or non-cMoPs were transferred into LCMV-Arm-infected mice on day 5 p.i., and their differentiation into moDCs was analyzed 3 days later. (E) Experimental schedule. (F–G) Differentiation patterns of donor cells into moDCs in the indicated organs are shown as flow cytometry plots (F) and graphs (G). Numbers in the plots indicate the percentages within the gates. Data are representative of two or three independent experiments and are shown as the mean ± SEM. *p < 0.05; **p < 0.01; ***p < 0.001.
Next, we examined the subpopulation of BMPs that directly responds to IFN-γ. BMPs can be subdivided into four subpopulations based on cell surface expression of CD115 (M-CSFR) and CD135 (Flt3): CD115−CD135+ cells as CD135+ BMPs, CD115+CD135+ cells as monocyte-DC progenitors (MDPs), CD115+CD135− cells as cMoPs, and CD115−CD135− cells as resting CD115−CD135− BMPs. Each BMP subpopulation expressed different levels of Ly6C and CD11b (Supplementary Figure 3A). Interestingly, cMoPs displayed a high expression level of CD119 (IFN-γR) compared with that in the other populations (Figure 2B). We isolated these cells and differentiated each subpopulation in the presence of GM-CSF and IFN-γ. In line with the high IFN-γR expression, only cMoPs immediately differentiated into CD11b+IA/IE+Ly6C+Ly6G− moDC phenotype cells in response to IFN-γ in agreement with the earlier findings that suggested that cMoPs is a progenitor population of moDCs (Figures 2C,D and Supplementary Figure 3B) (14, 15). The differentiations of other populations into moDCs beginning on day 3 of the culture were likely due to a developmental hierarchy of BMPs. To directly investigate whether cMoPs can differentiate into moDCs, we sorted cMoPs and other resting BMPs (non-cMoPs) from naive mice and transferred them into LCMV-Arm-infected congenically marked (CD45.1+) recipient mice on day 5 post infection (p.i.) when IFN-γ was abundant. The frequencies of moDCs among donor cells were analyzed 3 days later (Figure 2E). Consistent with the in vitro experiments, the majority of cMoPs differentiated into moDCs, while non-cMoPs showed limited differentiation into moDCs (Figures 2F,G). Taken together, these data suggest that IFN-γ acts directly on cMoPs to promote their differentiation into moDCs in LCMV Arm-infected mice. The accumulation of cMoPs in mice that received IFN-γ-neutralizing Ab (Supplementary Figure 1B) could be explained by these results; cMoPs rarely received IFN-γ signaling, could not develop into moDCs, and consequently accumulated in the BM.
CD8+ T Cells Primed by moDCs Have Reduced Effector Function Than Those Primed by cDCs
Given that moDCs become the dominant population of APCs during the expansion phase of acute viral infection, moDCs may be involved in modulating antiviral T cell responses. Thus, we determined whether infection-induced moDCs play an essential role in the virus-specific CD8+ T cell responses. To compare the ability of each DC subset to stimulate CD8+ T cell responses, we isolated moDCs and cDCs from the LCMV-Arm-infected mice on day 4 p.i., and cocultured the cells in the presence of various doses of cognate peptide (GP33−41) with Cell trace violet (CTV)-labeled P14 cells, which express TCRs that recognize the epitope peptide of the LCMV glycoprotein. moDCs were shown to have lower priming capacity than that of cDCs under low antigenic peptide stimulation (20 ng/ml), suggesting that moDCs deliver weak signals to CD8+ T cells (Figures 3A,B). Then, we compared the surface phenotypes of the P14 cells that were primed by each DC subset. Analysis of surface marker expression levels revealed that, while CD44 expressions were comparable between the two cell types, cDC-stimulated P14 cells (P14cDC) were more activated than moDC-stimulated P14 cells (P14moDC) as shown by elevated CD25 (IL-2Rα) and CD69 levels. Instead, P14moDC showed upregulated expressions of CD122 (IL-2/15 Rβ) and CD132 (common γ chain) compared to P14cDC.CD127 (IL-7R) levels were similar between P14cDC and P14moDC, and IL-15Rα was rarely detectable in both cell types (Figure 3C). Interestingly, when P14 cells were plotted by their coexpressions of CD25 and CD62L, P14moDC displayed a predominantly memory-like phenotype (CD25lowCD62Lhi), whereas P14cDC had a higher fraction of effector-like cells (CD25hiCD62Llow) (Figure 3D) (25, 26). Moreover, P14moDC showed reduced expressions of effector molecules [IFN-γ, TNF-α, and granzyme B (GzmB)] compared with those of P14cDC (Figure 3E). Similar results were obtained in the experiments using APCs isolated from infected mice on day 8 p.i., Day 8 moDCs were indistinguishable from day 4 moDCs in terms of T cell-stimulating capacity (Supplementary Figures 4A–D).
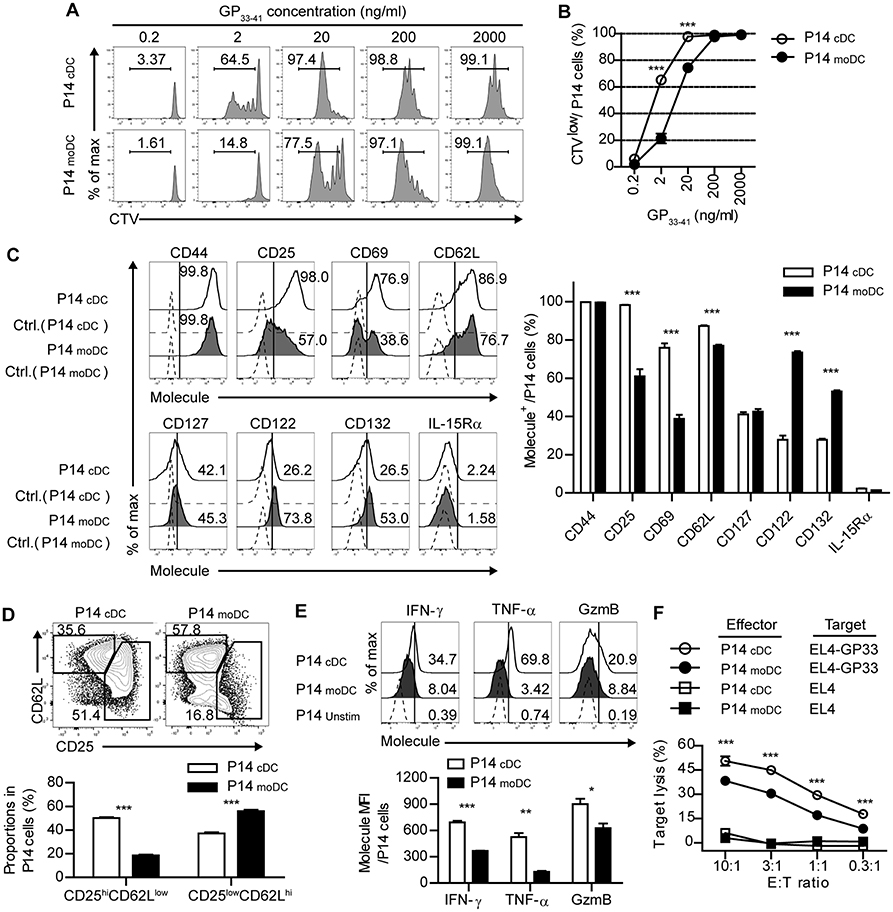
Figure 3. CD8+ T cells primed by moDCs have reduced effector function than those primed by cDCs. (A,B) Representative histograms (A) and graph (B) of CTV dilutions in P14 cells primed by cDCs or moDCs in the presence of different doses of GP33−41 peptide for 3 days. Numbers in the histograms indicate the percentage of cells that were divided at least once. (C–F) P14 cells were activated by cDCs or moDCs in the presence of 0.2 μg/ml GP33−41 peptide for 3 days. (C) Expression levels of indicated surface molecules are shown as histograms (right) and a graph plot (left). Numbers in the histograms indicate the percentages of positive cells for each molecule. (D) Coexpression of CD25 and CD62L on P14 cells primed by cDCs or moDCs are shown as flow cytometry plots (upper) and graph (lower). Numbers in the plots indicate the percentages within each gate. (E) Secretion levels of the indicated effector molecules in P14 cells that were primed by cDCs or moDCs are shown as histograms (upper) and graph (lower). Numbers in the histograms indicate the percentages of positive cells for each molecule. (F) in vitro target killing ability of P14 cells primed by cDCs or moDCs. Cr51-labeled GP33−41-loaded EL4 tumor cells were used as the target cells. Data are representative of three independent experiments and are shown as the mean ± SEM. *p < 0.05; **p < 0.01; ***p < 0.001.
To directly compare antigen-specific cytolytic function of P14moDC with that of P14cDC, we cocultured each type of CD8+ effector T cells with GP33−41-loaded EL4 tumor cells. Consistent with the expression levels of effector molecules, P14moDC exhibited lower target killing ability than that of P14cDC (Figure 3F). These data suggest that moDCs were not efficient in generation of effector CD8+ T cells but had an ability to induce memory precursor CD8+ T cells.
Stimulation by moDCs Dictates the Developmental Program of Memory CD8+ T Cells by Transcriptional Regulation
It has been shown that the fate determination of CD8+ T cells is regulated by the expression of several transcription factors (4). To further identify whether the signal delivered by moDCs directs CD8+ T cells to memory cells, we analyzed the transcriptional changes of P14cDC and P14moDC. Interestingly, P14moDC expressed low levels of T-bet and partly differentiated into Eomes+ cells while P14cDC differentiated into T-bet+ effector cells (Figures 4A,B). Moreover, TCF1, a transcription factor associated with memory T cell differentiation (7), was maintained at relatively higher levels in P14moDC compared to that in P14cDC (Figure 4A). The TCF1 expression level in P14moDC remained constant regardless of the number of cell divisions, whereas that in P14cDC was inversely correlated with the number of cell divisions (Figure 4C). To evaluate the memory formation ability of CD8+ T cells activated by moDCs, equivalent numbers of the viable P14cDC or P14moDC were transferred into LCMV-infected recipient mice on day 8 p.i., and analyzed on day 28 post transfer (day 36 p.i.) (Figure 4D). The memory phenotype of P14cDC and P14moDC showed no difference at the memory time point; however, recipient mice that received P14moDC had a higher number of donor cells than recipient mice that received P14cDC, suggesting that moDCs induced long-term survival of P14moDC (Figures 4E–G). Overall, these results suggest that moDC stimulation diverts the fate of CD8+ T cells to memory cells.
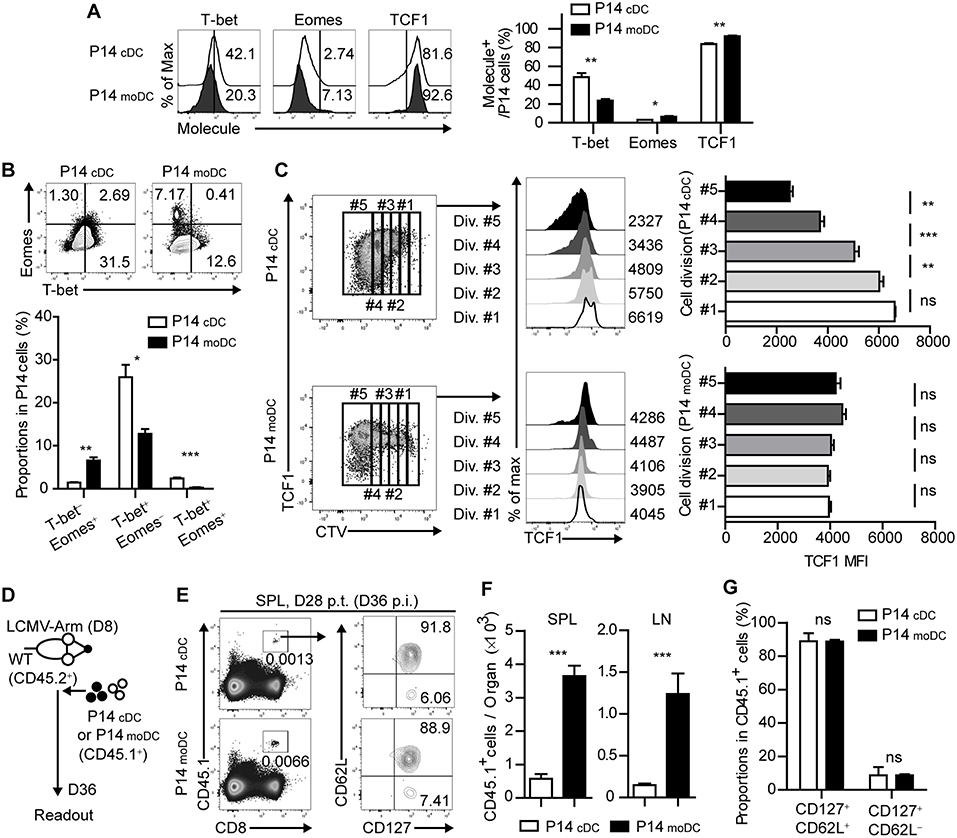
Figure 4. Stimulation by moDCs dictates the developmental program of memory CD8+ T cells by transcriptional regulation. (A) Expression levels of the indicated transcription factors in P14 cells primed by cDCs or moDCs are shown as histograms (upper) and graph (lower). Numbers in the histograms indicate the percentages of positive cells for each molecule. (B) Coexpressions of T-bet and Eomes in P14 cells primed by cDCs or moDCs are shown as flow cytometry plots (upper) and graph (lower). Numbers in the plots indicate the percentages of the cells in each quadrant. (C) P14 cells primed by cDCs or moDCs were gated by their cell division (left). TCF1 expression levels of each gate are shown as histograms (center) and graphs (right). Numbers in the histograms indicate the MFI values of TCF1 expression in each gate. (D–G) CD45.1+ P14 cells were primed in vitro by cDCs or moDCs, transferred to infected recipient mice on day 8 p.i., and analyzed on day 28 post transfer. (D) Experimental schedule. (E) Representative flow cytometry plots of donor cells in the spleens of recipient mice. (F) Graphs show the number of donor P14 cells in the indicated organs. (G) Graphs show the coexpressions of CD127 and CD62L of the donor cells in the spleen of the recipient mice. Data are representative of two or three independent experiments and are shown as the mean ± SEM. *p < 0.05; **p < 0.01; ***p < 0.001.
CD8+ T Cells Fail to Differentiate Into MPECs in CCR2-Deficient Mice
To determine the role of moDCs in the fate determination of CD8+ T cells during acute viral infection, we used CCR2-deficient (Ccr2−/−) mice. While the frequency and composition of cDCs was comparable to that in the WT mice, moDCs in SLOs were dramatically reduced in the Ccr2−/− mice compared to WT mice during infection (Supplementary Figures 5A,B). The virus titers of Ccr2−/− mice and WT mice showed no differences during the acute phase of infection (Supplementary Figure 5C). We transferred CD45.1+ P14 cells into WT and Ccr2−/− mice, which were subsequently infected with LCMV-Arm, and analyzed the transferred P14 cells on day 8 p.i., To dissect the fate of CD8+ T cells, we subdivided the donor T cells by the distinct expression pattern of KLRG1 and CD127. When WT and Ccr2−/− mice bearing CD45.1+ P14 cells were infected, the frequency of KLRG1+CD127− cells, identified as short-lived effector cells (SLECs), was significantly increased in P14 cells in Ccr2−/− mice compared with that in WT mice. In addition, the frequency of KLRG1−CD127+ cells, which represent memory precursor cells (MPECs), was significantly reduced in Ccr2−/− hosts, suggesting that CCR2+ cells contributed to the efficient generation of memory CD8+ T cells during infection (Figures 5A,B). We also examined the effector function of P14 cells in WT and Ccr2−/− mice. The intensity, but not the frequency, of IFN-γ or TNF-α-producing T cells among P14 cells was elevated in Ccr2−/− mice compared with that in WT mice (Figure 5C). Furthermore, P14 cells from Ccr2−/− mice showed more potent target cell killing activity than those from WT mice (Figure 5D).
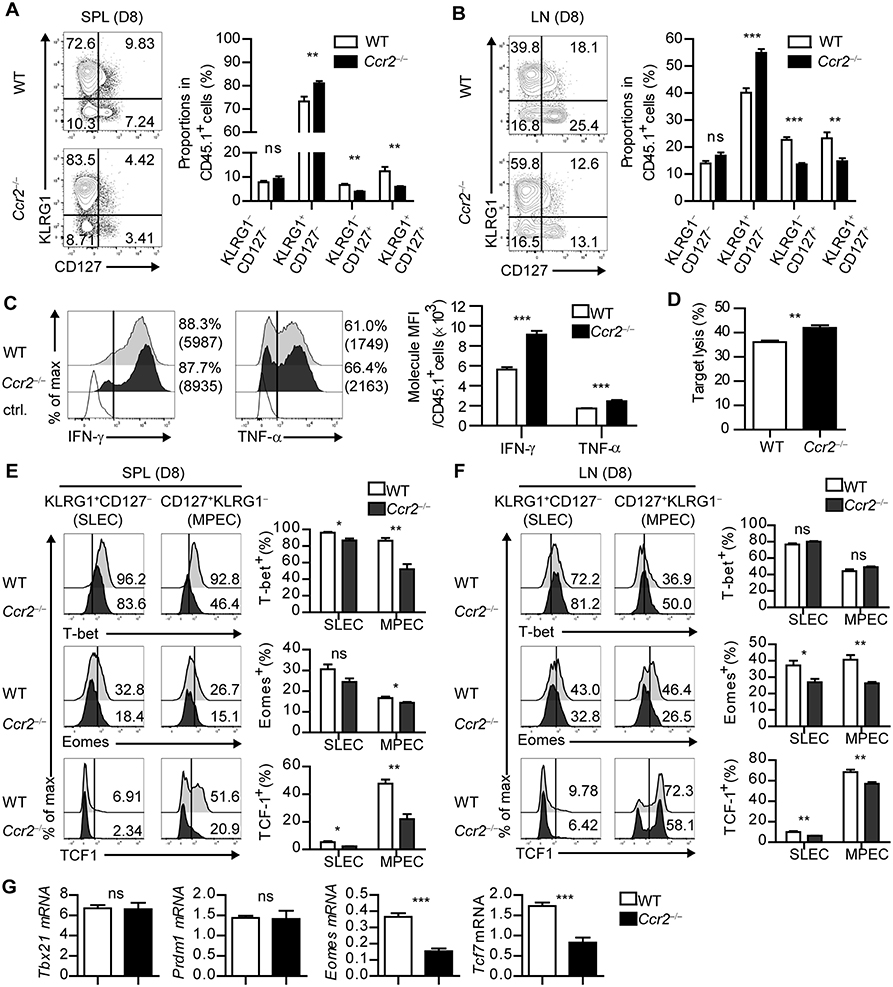
Figure 5. CD8+ T cells fail to differentiate into MPECs in CCR2-deficient mice. (A,B) Coexpressions of KLRG1 and CD127 of CD45.1+ P14 cells in the spleen and LN of LCMV-Arm-infected WT and Ccr2−/− mice on day 8 p.i., are shown as flow cytometry plots (left) and graph (right). Numbers in the plots indicate the percentage of cells within each quadrant. (C) Secretion levels of IFN-γ and TNF-α in CD45.1+ P14 cells in the spleen of WT and Ccr2−/− mice on day 8 p.i., are shown as histograms (left) and graph (right). (D) In vitro target killing ability of CD45.1+ P14 cells from WT and Ccr2−/− splenocytes on day 8 p.i., Cr51-labeled GP33−41-loaded EL4 tumor cells were used as the target cells. (E,F) Expression levels of T-bet, Eomes and TCF1 in SLECs and MPECs of CD45.1+ P14 cells from WT and Ccr2−/− splenocytes and LN cells on day 8 p.i., are shown as histograms (upper) and graph (lower). Numbers in the histograms indicate the percentage of positive cells for each molecule. (G) Gene expression levels of Tbx21, Prdm1, Eomes, and Tcf7 in CD45.1+ P14 cells from WT and Ccr2−/− splenocytes on day 8 p.i., were measured by real-time PCR. Expression levels were normalized to mHprt. Data are representative of three independent experiments and are shown as the mean ± SEM. n = 5 per group. *p < 0.05; **p < 0.01; ***p < 0.001.
To determine whether the altered effector/memory differentiation of virus-specific CD8+ T cells in Ccr2−/− mice reflects molecular changes in CD8+ T cells, we compared the transcriptional profile of SLECs and MPECs in transferred P14 cells of WT and Ccr2−/− mice. Consistent with the results of the in vitro studies (Figure 4), the expressions of Eomes and TCF1 in each P14 cell subset were decreased in Ccr2−/− mice compared with those in WT mice and the effect was more prominent in MPECs (Figures 5E,F). Despite the reduced numbers of moDCs, SLECs, and MPECs of P14 cells in Ccr2−/− mice had low levels of T-bet expression, suggesting that other factors could have contributed to the transcriptional changes in CD8+ T cells. In addition, the expressions of Tcf7 and Eomes in P14 cells were significantly reduced in Ccr2−/− recipient mice compared to WT mice, while the expressions of Tbx21 and Prdm1 were similar (Figure 5G). Collectively, these results suggest that moDCs can induce memory precursor cells from naïve CD8+ T cells by regulation of transcription factor expression including Eomes and TCF1.
CD8+ T Cells Primed in CCR2-Deficient Mice Cannot Respond to Reinfection
Then, we analyzed transferred P14 cells at the memory phase of infection. On day 35 p.i., P14 cells were barely detectable in the spleen and LN of Ccr2−/− mice (Supplementary Figures 6A,C), which may be due to reduced frequencies of MPECs in Ccr2−/− mice at the early phase of infection. However, the phenotype of the remaining P14 cells in WT and Ccr2−/− mice was similar except that fraction of CD127−CD62L− cells was slightly increased in splenic P14 cells of Ccr2−/− mice (Supplementary Figures 6B,D). Additionally, P14 memory cells in Ccr2−/− mice had similar inflammatory potency compared with those in WT mice (Supplementary Figure 6E). These results suggest that moDCs play a major role in maintaining the memory CD8+ T cell pool without affecting their functions.
To examine whether virus-specific CD8+ T cell priming by moDCs leads to enhanced protection against rechallenge, we harvested CD45.1+ effector P14 cells from WT or Ccr2−/− mice on day 8 post LCMV-Arm infection. Then, we transferred an equivalent number of each type of P14 cells into naive WT recipient mice to establish LCMV-specific memory CD8+ T cells in the hosts (Figure 6A). As expected, P14 cells primed in WT mice preferentially survived in the hosts compared to those primed in Ccr2−/− mice, as shown by reduced numbers of CD45.1+ P14 cells from Ccr2−/− mice in the blood, spleen, and liver in recipient mice over 20 days post transfer, with a minor alteration in the phenotype of these cells (Figures 6B–D). We infected recipient mice with Lm-GP33 on day 26 after transfer. Consistent with the reduced memory P14 cell numbers before reinfection, mice that received effector P14 cells from Ccr2−/− mice could not control bacterial burden while mice that received P14 cells from WT mice showed reduced bacterial burden compared with the untransferred control (Figure 6E). Taken together, our data suggest that moDCs induced by infection are crucial in the long-term survival of CD8+ T cells, thus enabling efficient clearance of pathogens by the hosts upon reinfection.
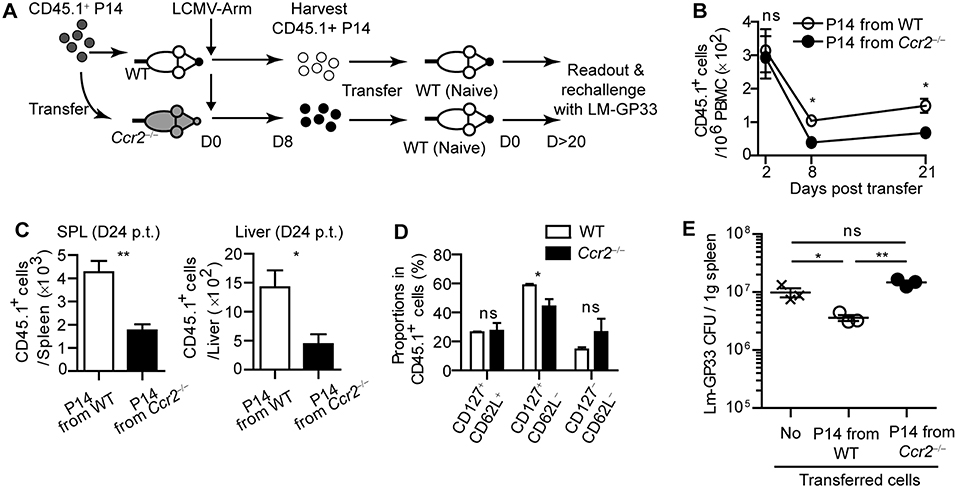
Figure 6. CD8+ T cells primed in CCR2-deficient mice cannot respond to reinfection. Effector P14 cells of WT and Ccr2−/− mice at day 8 p.i., were sorted and equivalent numbers of the cells were transferred into naïve mice. Recipient mice were analyzed at least 20 days after transfer. (A) Experimental schedule. (B) Temporally enumerated CD45.1+ P14 cells in blood PBMCs of recipient mice after the transfer of effector P14 cells from WT and Ccr2−/− mice. (C) The frequencies of CD45.1+ P14 cells in the spleen (left) and liver (right) of recipient mice on day 24 post transfer of P14 cells from WT and Ccr2−/− mice. (D) The memory phenotypes of CD45.1+ P14 cells in the spleen of recipient mice on day 24 post transfer of P14 cells from WT and Ccr2−/− mice. (E) Recipient mice were challenged with Lm-GP33 at day 26 post transfer. Graph shows the bacterial titers in the spleen of recipient mice at day 3 after rechallenge. Data are representative of two independent experiments and are shown as the mean ± SEM. n = 3–4 per group. *p < 0.05; **p < 0.01.
Defective IL-2 Signaling Grants moDCs an Ability to Induce Memory CD8+ T Cells
Finally, we sought to investigate the underlying mechanisms that mediate the differentiation of memory-precursor cells during the interaction between CD8+ T cells and moDCs. Three signals have been known to be responsible for the initial activation of CD8+ T cells: TCR-MHC interaction, co-stimulation and cytokine signaling (27, 28). moDCs showed no defect in the expression of MHCI and co-stimulatory molecules (CD40, CD80, and CD86) compared to those of cDCs (Supplementary Figure 7). Thus, we hypothesized that defective cytokine signaling caused moDCs to deliver relatively weak signals to CD8+ T cells. Interestingly, the differentiation patterns of CD8+ T cells primed in the presence of a high dose of IL-2 and in the presence of a low dose of IL-2 resembled P14cDC and P14moDC, respectively (29, 30). Accordingly, the levels of IL-2 in the supernatants of P14 cells cocultured with cDCs were higher than in those cocultured with moDCs (Figure 7A). Moreover, only cDCs expressed IL-2 at the transcript level (Figure 7B). In addition, an analysis of IL-2 secretion levels in the cocultures 12 h after culture revealed that cDCs produced more IL-2 than moDCs and P14cDC expressed high levels of IL-2 compared to P14moDC (Supplementary Figure 8). Therefore, we hypothesized that defective IL-2 signaling is responsible for the differentiation of CD8+ T cells into CD25lowCD62Lhi memory precursor cells.
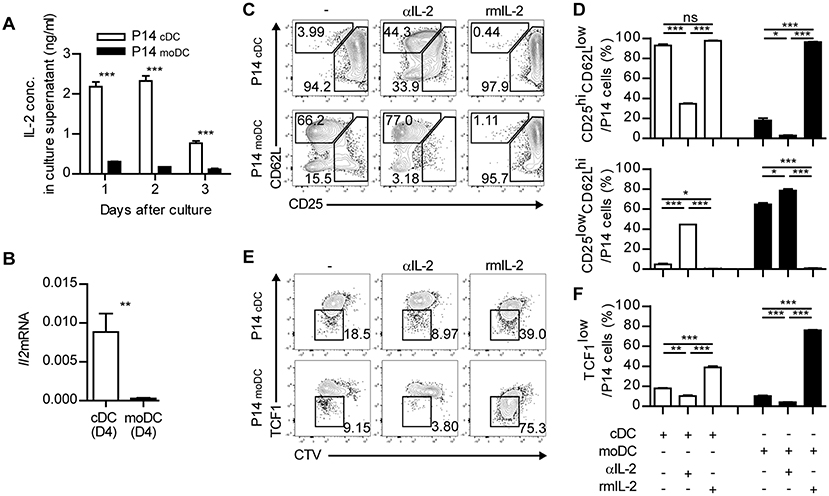
Figure 7. Defective IL-2 signaling grants moDCs an ability to induce memory CD8+ T cells. (A) IL-2 concentrations in the supernatant from the cultures of P14 cells with cDCs or moDCs. (B) Gene expression levels of Il2 in cDCs and moDCs isolated from LCMV-Arm-infected splenocytes. (C–F) Recombinant IL-2 or anti-IL-2 mAbs were added to the cultures of P14 cells with moDCs or cDCs. (C,D) Coexpressions of CD25 and CD62L under each condition are shown as flow cytometry plots (C) and graphs (D). (E,F) Expression levels of TCF1 are shown as flow cytometry plots (E) and graph (F). Numbers in the plots indicate the percentage within each gate. Data are representative of two independent experiments and are shown as the mean ± SEM. *p < 0.05; **p < 0.01; ***p < 0.001.
To test this possibility, we cultured CD8+ T cells with cDCs or moDCs in the presence of anti-IL-2 mAbs (αIL-2) or recombinant IL-2 (rmIL-2). In agreement with our hypothesis, P14cDC cells cultured with αIL-2 predominantly differentiated into CD25lowCD62Lhi memory phenotype cells suggesting that IL-2 was required for the full effector cell differentiation of P14 cells (Figures 7C,D). Due to the endogenous IL-2 secretion of P14cDC, adding rmIL-2 to cocultures showed no effect on the surface phenotypes of P14cDC. P14moDC, which were mainly differentiated into CD25lowCD62Lhi memory precursor cells, were converted into CD25hiCD62Llow effector cells by adding rmIL-2 to the cocultures. We also found that the culture of P14cDC with αIL-2 abrogated the downregulation of TCF1, whereas the culture of P14moDC with rmIL-2 showed dramatic suppression of TCF1 expression (Figures 7E,F). Collectively, these results suggest that IL-2 signaling is important for modulation of the fate of CD8+ T cells during priming and that moDCs promote memory-prone differentiation of CD8+ T cells by delivering defective signaling such as IL-2.
Discussion
Understanding the mechanism of the influence of myeloid cells on the development of T cell immunity during infection is important for establishing strategies for development of vaccines against infections. Here, we demonstrate that moDCs dramatically expand in an IFN-γ dependent manner during acute infection. More importantly, CD8+ T cells activated by moDCs preferentially differentiate to memory cells by inducing Eomes expression and maintaining TCF1 expression, while those cells that are primed by cDCs undergo effector-prone differentiation through upregulation of T-bet and downregulation of TCF1. Moreover, we found that the difference in IL-2 production between the two APC subsets results in the different outcomes of the primed CD8+ T cells.
APC subsets with distinct properties have been proposed to be crucial for the differentiation of T cells (31, 32). Our study suggests that the interaction between T cells and distinct APC subsets during priming regulates the fate of CD8+ T cells. Since cDCs are enriched in the SLOs during the initial phase of acute viral infection, they may serve as the primary professional APCs that prime CD8+ T cells resulting in the predominant generation of effector T cells. As the increased effector CD8+ T cell populations primed by cDCs clear the pathogen by producing effector molecules including IFN-γ, the elevated IFN-γ also contributes to the accumulation of moDCs in the SLOs. As a result, moDCs become abundant in the SLOs during the expansion phases of infection. It has been suggested that decreased TCR stimulation strength due to the reduced antigen burden after pathogen clearance results in the differentiation of memory CD8+ T cells (2, 4); however, our data suggest another possibility that changes in the APC subset composition also contribute to the effector/memory fate determination of CD8+ T cells. Our data also revealed that, in addition to its direct effect on effector CD8+ T cell expansion (33), IFN-γ can also influence memory CD8+ T cell generation indirectly by inducing moDCs. In line with this notion, generation of memory CD8+ T cell was abrogated in IFN-γR-deficient mice (34, 35). Therefore, we suggest that the changes in APC composition as well as the environment, including antigen abundance and CD4 T cell help, contribute to memory CD8+ T cell formation (36).
Previous studies have shown that IFN-γ induces the differentiation of moDCs by influencing cMoPs (14, 15, 37). We confirmed this finding and found that IFN-γ potentiated moDC differentiation by directly acting on cMoPs. Although granulocyte-macrophage progenitor cells (GMPs) are known to express IFN-γR (38), we extended this finding by showing that IFN-γRhi cMoPs were the major precursor for moDC. A previous study has demonstrated that IFN-γ provokes the expansion of myeloid cells through an indirect pathway (39). We found an additional pathway by which IFN-γ induces the expansion of monocyte-derived cells, such as moDCs, via direct action on cMoPs. It is tempting to investigate why and how cMoPs selectively upregulate CD119 expression during monopoiesis.
Among the different signals that regulate the fate of CD8+ T cells, defective IL-2 signaling in the cocultures of moDCs and CD8+ T cells was shown to lead the memory-prone differentiation of CD8+ T cells. It has been suggested that the fates of CD8+ T cells are regulated by the differential exposures to IL-2 signals (40). Indeed, IL-2 signaling has been known to induce the terminal differentiation of CD8+ T cells by activating STAT-5 and consequently abrogate the memory CD8+ T cell generation (41). IL-2 intracellular cytokine staining showed that P14 T cells expressed higher levels of IL-2 than DCs in the cocultures, suggesting that CD8+ T cells were the main source of IL-2 in the cocultures (Supplementary Figure 8). In addition, we found that P14cDC secreted more IL-2 than P14moDC. It leads to the question of why CD8+ T cells primed by cDCs secrete higher levels of IL-2 than those primed by moDCs. Interestingly, previous studies reported that the IL-2 production by DCs allows them to stimulate T cells. DC-derived IL-2 accumulation at the DC-T contact site has been considered to be important in the stimulation of T cells (42–44). From the viewpoint of these findings, our data suggest that cDCs, potent IL-2 providers, induce robust IL-2 production from CD8+ T cells and dictate terminal differentiation of CD8+ T cells. On the other hand, moDCs stimulate CD8+ T cells suboptimally via attenuated IL-2 signaling and promote the differentiation of memory-phenotype cells. In summary, DCs are likely to contribute to the fate determination of CD8+ T cells by regulating IL-2 signaling, especially at the initial priming phase.
Long-term survival of memory CD8+ T cells is mediated by homeostatic proliferation (45). Our in vitro coculture analysis revealed that P14moDC expressed high levels of CD122 and CD132, which are the subunits of IL-15 receptor, compared to P14cDC. As IL-15 plays an important role in maintaining memory T cells (46, 47), it might have been involved in the long-term survival of P14moDC when P14moDC were adoptively transferred to mice. Although IL-15Rα was not detected in P14moDC, other IL-15Rα-expressing cells could have delivered IL-15 signaling to P14moDC via trans-presentation (48). Thus, we suggest that moDCs direct CD8+ T cells to express IL-15 receptors, enabling CD8+ T cells to survive at the memory phase.
The mechanisms for effector/memory differentiation of CD8+ T cells have been incompletely understood. Asymmetric cell division, which directs proximal and distal daughter cells to differentiate into SLECs and MPECs, respectively, has recently emerged as one of the mechanisms explaining how effector and memory progenies occur from naïve parental T cells (26, 49). A previous report suggested that strong TCR stimulation is required for the initiation of asymmetric cell division of CD8+ T cells. On the contrary, weak TCR stimulation preferentially leads to symmetric cell division of CD8+ T cells, resulting in the increased generation of MPECs (50). These results suggest that different TCR stimulation capacity of each DC subset could determine the fate of CD8+ T cells through differential regulation of the symmetry of cell division. In this regard, it would be interesting to compare the asymmetry in T cell division upon stimulation with different DC subsets.
We used Ccr2−/− mice to define the roles of moDCs in CD8+ T cell differentiation during infection in vivo. The reduced memory CD8+ T cell responses in Ccr2−/− mice correlated with our in vitro studies that demonstrated the specialized role of moDCs in the induction of memory precursor cells. We were unable to rule out the contribution of monocytes to memory CD8+ T cell differentiation because Ccr2−/− mice exhibited a decreased frequency not only of moDCs but also of monocytes (Supplementary Figure 3B). However, monocytes themselves have been considered as less efficient APCs in CD8+ T cell stimulation (51). Monocytes can uptake foreign antigens, but they present antigens to T cells after subsequent differentiation into moDCs (52). Although a recent report have suggested that Ly6C+ monocytes can prime CD8+ T cells efficiently, a majority of Ly6C+CD11b+ cells in that report showed the feature of moDCs that express a certain level of CD11c (16). Thus, we suggest that reduction in moDCs is mainly responsible for defective memory CD8+ T cell formation in Ccr2−/− mice.
It should be noted that the role of monocyte-derived cells in establishing defense mechanisms against pathogens is context-dependent. A previous report demonstrated that Tip-DCs, which share many phenotypic characteristics with moDCs in our experimental setting, mediate innate immune responses but are dispensable for T cell priming in Lm infection (13). We also showed that CD8+ T cells primed in Ccr2−/− mice exhibited no defects in their cytokine production capacity and cytotoxicity during LCMV-Arm infection. However, we identified an unrecognized role of moDCs in triggering memory CD8+ T cell generation under cognate antigenic stimulation. Interestingly, during the late phase of LCMV chronic (CL-13) infection, monocytes have been shown to acquire a myeloid-derived suppressor cell (MDSC)-like feature that abrogates CD8+ T cell proliferation and drives T cell exhaustion (17). The underlying factors that educate monocytic cells to play opposing roles during acute and chronic infections remain to be elucidated.
Taken together, our study demonstrates a crucial role of moDCs in the generation of memory CD8+ T cells during acute antiviral immune responses. Our findings expand the understanding of the link between myelopoiesis and CD8+ T cell differentiation during acute viral infection and have implications for the development of novel vaccine strategies against infection.
Data Availability
The raw data supporting the conclusions of this manuscript will be made available by the authors, without undue reservation, to any qualified researcher.
Ethics Statement
All animal experiments were approved by and carried out in accordance with the approved guidelines of the Institutional Animal Care and Use Committee (IACUC) of Seoul National University.
Author Contributions
K-SS and C-YK designed the study. K-SS, IJ, C-HK, BS, J-ML, JL, E-AB, and HS performed experiments. K-SS, B-SK, Y-JP, and C-YK analyzed and interpreted the data. YB and S-JH provided reagents. K-SS, I-KK, B-SK, and C-YK wrote the manuscript. C-YK supervised the study.
Funding
This research was supported by grants from the Basic Science Research Program (NRF-2018R1A2A1A05077627) and the Bio & Medical Technology Development Program (NRF-2016M3A9B5941426) through the National Research Foundation of Korea funded by the Ministry of Science & ICT.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We appreciate the staff of the National Center for Inter-university Research Facilities (NCIRF, Seoul National University) for assistance with the cell sorting by flow cytometry.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2019.01887/full#supplementary-material
References
1. Joshi NS, Cui W, Chandele A, Lee HK, Urso DR, Hagman J, et al. Inflammation directs memory precursor and short-lived effector CD8(+) T cell fates via the graded expression of T-bet transcription factor. Immunity. (2007) 27:281–95. doi: 10.1016/j.immuni.2007.07.010
2. Chang JT, Wherry EJ, Goldrath AW. Molecular regulation of effector and memory T cell differentiation. Nat Immunol. (2014) 15:1104–5. doi: 10.1038/ni.3031
3. Sallusto F, Geginat J, Lanzavecchia A. Central memory and effector memory T cell subsets: function, generation, and maintenance. Annu Rev Immunol. (2004) 22:745–63. doi: 10.1146/annurev.immunol.22.012703.104702
4. Kaech SM, Cui W. Transcriptional control of effector and memory CD8+ T cell differentiation. Nat Rev Immunol. (2012) 12:749–61. doi: 10.1038/nri3307
5. Kallies A, Xin A, Belz GT, Nutt SL. Blimp-1 transcription factor is required for the differentiation of effector CD8(+) T cells and memory responses. Immunity. (2009) 31:283–95. doi: 10.1016/j.immuni.2009.06.021
6. Banerjee A, Gordon SM, Intlekofer AM, Paley MA, Mooney EC, Lindsten T, et al. Cutting edge: the transcription factor eomesodermin enables CD8+ T cells to compete for the memory cell niche. J Immunol. (2010) 185:4988–92. doi: 10.4049/jimmunol.1002042
7. Zhou X, Yu S, Zhao DM, Harty JT, Badovinac VP, Xue HH. Differentiation and persistence of memory CD8(+) T cells depend on T cell factor 1. Immunity. (2010) 33:229–40. doi: 10.1016/j.immuni.2010.08.002
8. Boettcher S, Manz MG. Regulation of inflammation- and infection-driven hematopoiesis. Trends Immunol. (2017) 38:345–57. doi: 10.1016/j.it.2017.01.004
9. Glatman Zaretsky A, Engiles JB, Hunter CA. Infection-induced changes in hematopoiesis. J Immunol. (2014) 192:27–33. doi: 10.4049/jimmunol.1302061
10. Jakubzick CV, Randolph GJ, Henson PM. Monocyte differentiation and antigen-presenting functions. Nat Rev Immunol. (2017) 17:349–62. doi: 10.1038/nri.2017.28
11. Ginhoux F, Jung S. Monocytes and macrophages: developmental pathways and tissue homeostasis. Nat Rev Immunol. (2014) 14:392–404. doi: 10.1038/nri3671
12. Schlitzer A, McGovern N, Ginhoux F. Dendritic cells and monocyte-derived cells: two complementary and integrated functional systems. Semin Cell Dev Biol. (2015) 41:9–22. doi: 10.1016/j.semcdb.2015.03.011
13. Serbina NV, Salazar-Mather TP, Biron CA, Kuziel WA, Pamer EG. TNF/iNOS-producing dendritic cells mediate innate immune defense against bacterial infection. Immunity. (2003) 19:59–70. doi: 10.1016/S1074-7613(03)00171-7
14. Askenase MH, Han SJ, Byrd AL, Morais da Fonseca D, Bouladoux N, Wilhelm C, et al. Bone-marrow-resident NK cells prime monocytes for regulatory function during infection. Immunity. (2015) 42:1130–42. doi: 10.1016/j.immuni.2015.05.011
15. Sharma MD, Rodriguez PC, Koehn BH, Baban B, Cui Y, Guo G, et al. Activation of p53 in immature myeloid precursor cells controls differentiation into Ly6c(+)CD103(+) monocytic antigen-presenting cells in tumors. Immunity. (2018) 48:91–106 e6. doi: 10.1016/j.immuni.2017.12.014
16. Larson SR, Atif SM, Gibbings SL, Thomas SM, Prabagar MG, Danhorn T, et al. Ly6C(+) monocyte efferocytosis and cross-presentation of cell-associated antigens. Cell Death Differ. (2016) 23:997–1003. doi: 10.1038/cdd.2016.24
17. Norris BA, Uebelhoer LS, Nakaya HI, Price AA, Grakoui A, Pulendran B. Chronic but not acute virus infection induces sustained expansion of myeloid suppressor cell numbers that inhibit viral-specific T cell immunity. Immunity. (2013) 38:309–21. doi: 10.1016/j.immuni.2012.10.022
18. Ban YH, Oh S-C, Seo S-H, Kim S-M, Choi I-P, Greenberg PD, et al. miR-150-Mediated Foxo1 regulation programs CD8+ T cell differentiation. Cell Rep. (2017) 20:2598–611. doi: 10.1016/j.celrep.2017.08.065
19. Chow KV, Lew AM, Sutherland RM, Zhan Y. Monocyte-derived dendritic cells promote Th polarization, whereas conventional dendritic cells promote Th proliferation. J Immunol. (2016) 196:624–36. doi: 10.4049/jimmunol.1501202
20. MacNamara KC, Oduro K, Martin O, Jones DD, McLaughlin M, Choi K, et al. Infection-induced myelopoiesis during intracellular bacterial infection is critically dependent upon IFN-gamma signaling. J Immunol. (2011) 186:1032–43. doi: 10.4049/jimmunol.1001893
21. de Bruin AM, Libregts SF, Valkhof M, Boon L, Touw IP, Nolte MA. IFNgamma induces monopoiesis and inhibits neutrophil development during inflammation. Blood. (2012) 119:1543–54. doi: 10.1182/blood-2011-07-367706
22. Cunningham CR, Champhekar A, Tullius MV, Dillon BJ, Zhen A, de la Fuente JR, et al. Type I and type II interferon coordinately regulate suppressive dendritic cell fate and function during viral persistence. PLoS Pathog. (2016) 12:e1005356. doi: 10.1371/journal.ppat.1005356
23. Moskophidis D, Battegay M, Bruendler MA, Laine E, Gresser I, Zinkernagel RM. Resistance of lymphocytic choriomeningitis virus to alpha/beta interferon and to gamma interferon. J Virol. (1994) 68:1951–5.
24. Helft J, Bottcher J, Chakravarty P, Zelenay S, Huotari J, Schraml BU, et al. GM-CSF mouse bone marrow cultures comprise a heterogeneous population of CD11c(+)MHCII(+) macrophages and dendritic cells. Immunity. (2015) 42:1197–211. doi: 10.1016/j.immuni.2015.05.018
25. Kalia V, Sarkar S, Subramaniam S, Haining WN, Smith KA, Ahmed R. Prolonged interleukin-2Ralpha expression on virus-specific CD8+ T cells favors terminal-effector differentiation in vivo. Immunity. (2010) 32:91–103. doi: 10.1016/j.immuni.2009.11.010
26. Arsenio J, Kakaradov B, Metz PJ, Kim SH, Yeo GW, Chang JT. Early specification of CD8+ T lymphocyte fates during adaptive immunity revealed by single-cell gene-expression analyses. Nat Immunol. (2014) 15:365–72. doi: 10.1038/ni.2842
27. Mescher MF, Curtsinger JM, Agarwal P, Casey KA, Gerner M, Hammerbeck CD, et al. Signals required for programming effector and memory development by CD8+ T cells. Immunol Rev. (2006) 211:81–92. doi: 10.1111/j.0105-2896.2006.00382.x
28. Haring JS, Badovinac VP, Harty JT. Inflaming the CD8+ T cell response. Immunity. (2006) 25:19–29. doi: 10.1016/j.immuni.2006.07.001
29. Manjunath N, Shankar P, Wan J, Weninger W, Crowley MA, Hieshima K, et al. Effector differentiation is not prerequisite for generation of memory cytotoxic T lymphocytes. J Clin Invest. (2001) 108:871–8. doi: 10.1172/JCI200113296
30. Mueller K, Schweier O, Pircher H. Efficacy of IL-2- versus IL-15-stimulated CD8 T cells in adoptive immunotherapy. Eur J Immunol. (2008) 38:2874–85. doi: 10.1002/eji.200838426
31. Kim TS, Gorski SA, Hahn S, Murphy KM, Braciale TJ. Distinct dendritic cell subsets dictate the fate decision between effector and memory CD8(+) T cell differentiation by a CD24-dependent mechanism. Immunity. (2014) 40:400–13. doi: 10.1016/j.immuni.2014.02.004
32. Belz GT, Heath WR, Carbone FR. The role of dendritic cell subsets in selection between tolerance and immunity. Immunol Cell Biol. (2002) 80:463–8. doi: 10.1046/j.1440-1711.2002.01116.x
33. Whitmire JK, Tan JT, Whitton JL. Interferon-gamma acts directly on CD8+ T cells to increase their abundance during virus infection. J Exp Med. (2005) 201:1053–9. doi: 10.1084/jem.20041463
34. Sercan O, Hammerling GJ, Arnold B, Schuler T. Cutting edge: innate immune cells contribute to the IFN- -dependent regulation of antigen-specific CD8+ T cell homeostasis. J Immunol. (2006) 176:735–9. doi: 10.4049/jimmunol.176.2.735
35. Sercan O, Stoycheva D, Hammerling GJ, Arnold B, Schuler T. IFN-gamma receptor signaling regulates memory CD8+ T cell differentiation. J Immunol. (2010) 184:2855–62. doi: 10.4049/jimmunol.0902708
36. Wherry EJ, Ahmed R. Memory CD8 T-cell differentiation during viral infection. J Virol. (2004) 78:5535–45. doi: 10.1128/JVI.78.11.5535-5545.2004
37. Goldszmid RS, Caspar P, Rivollier A, White S, Dzutsev A, Hieny S, et al. NK cell-derived interferon-gamma orchestrates cellular dynamics and the differentiation of monocytes into dendritic cells at the site of infection. Immunity. (2012) 36:1047–59. doi: 10.1016/j.immuni.2012.03.026
38. de Bruin AM, Buitenhuis M, van der Sluijs KF, van Gisbergen KP, Boon L, Nolte MA. Eosinophil differentiation in the bone marrow is inhibited by T cell-derived IFN-gamma. Blood. (2010) 116:2559–69. doi: 10.1182/blood-2009-12-261339
39. Schurch CM, Riether C, Ochsenbein AF. Cytotoxic CD8+ T cells stimulate hematopoietic progenitors by promoting cytokine release from bone marrow mesenchymal stromal cells. Cell Stem Cell. (2014) 14:460–72. doi: 10.1016/j.stem.2014.01.002
40. Kalia V, Sarkar S. Regulation of effector and memory CD8 T cell differentiation by IL-2-A balancing act. Front Immunol. (2018) 9:2987. doi: 10.3389/fimmu.2018.02987
41. Pipkin ME, Sacks JA, Cruz-Guilloty F, Lichtenheld MG, Bevan MJ, Rao A. Interleukin-2 and inflammation induce distinct transcriptional programs that promote the differentiation of effector cytolytic T cells. Immunity. (2010) 32:79–90. doi: 10.1016/j.immuni.2009.11.012
42. Granucci F, Vizzardelli C, Pavelka N, Feau S, Persico M, Virzi E, et al. Inducible IL-2 production by dendritic cells revealed by global gene expression analysis. Nat Immunol. (2001) 2:882. doi: 10.1038/ni0901-882
43. Boyman O, Sprent J. The role of interleukin-2 during homeostasis and activation of the immune system. Nat Rev Immunol. (2012) 12:180–90. doi: 10.1038/nri3156
44. Granucci F, Feau S, Angeli V, Trottein F, Ricciardi-Castagnoli P. Early IL-2 production by mouse dendritic cells is the result of microbial-induced priming. J Immunol. (2003) 170:5075–81. doi: 10.4049/jimmunol.170.10.5075
45. Surh CD, Sprent J. Homeostasis of naive and memory T cells. Immunity. (2008) 29:848–62. doi: 10.1016/j.immuni.2008.11.002
46. Becker TC, Wherry EJ, Boone D, Murali-Krishna K, Antia R, Ma A, et al. Interleukin 15 is required for proliferative renewal of virus-specific memory CD8 T cells. J Exp Med. (2002) 195:1541–8. doi: 10.1084/jem.20020369
47. Zhang X, Sun S, Hwang I, Tough DF, Sprent J. Potent and selective stimulation of memory-phenotype CD8+ T cells in vivo by IL-15. Immunity. (1998) 8:591–9. doi: 10.1016/S1074-7613(00)80564-6
48. Dubois S, Mariner J, Waldmann TA, Tagaya Y. IL-15Rα Recycles and presents IL-15 in trans to neighboring cells. Immunity. (2002) 17:537–47. doi: 10.1016/S1074-7613(02)00429-6
49. Chang JT, Palanivel VR, Kinjyo I, Schambach F, Intlekofer AM, Banerjee A, et al. Asymmetric T lymphocyte division in the initiation of adaptive immune responses. Science. (2007) 315:1687–91. doi: 10.1126/science.1139393
50. King CG, Koehli S, Hausmann B, Schmaler M, Zehn D, Palmer E. T cell affinity regulates asymmetric division, effector cell differentiation, and tissue pathology. Immunity. (2012) 37:709–20. doi: 10.1016/j.immuni.2012.06.021
51. Hey YY, Quah B, O'Neill HC. Antigen presenting capacity of murine splenic myeloid cells. BMC Immunol. (2017) 18:4. doi: 10.1186/s12865-016-0186-4
Keywords: monocyte-derived dendritic cells, memory CD8+ T cells, IFN-γ, LCMV, acute infection
Citation: Shin K-S, Jeon I, Kim B-S, Kim I-K, Park Y-J, Koh C-H, Song B, Lee J-M, Lim J, Bae E-A, Seo H, Ban YH, Ha S-J and Kang C-Y (2019) Monocyte-Derived Dendritic Cells Dictate the Memory Differentiation of CD8+ T Cells During Acute Infection. Front. Immunol. 10:1887. doi: 10.3389/fimmu.2019.01887
Received: 01 May 2019; Accepted: 25 July 2019;
Published: 16 August 2019.
Edited by:
Surojit Sarkar, University of Washington, United StatesReviewed by:
Jason Kyle Whitmire, University of North Carolina at Chapel Hill, United StatesVandana Kalia, University of Washington, United States
Copyright © 2019 Shin, Jeon, Kim, Kim, Park, Koh, Song, Lee, Lim, Bae, Seo, Ban, Ha and Kang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Chang-Yuil Kang, Y3lrYW5nQHNudS5hYy5rcg==