 Lorenza Cannarile
Lorenza Cannarile Domenico V. Delfino
Domenico V. Delfino Sabrina Adorisio
Sabrina Adorisio Carlo Riccardi
Carlo Riccardi Emira Ayroldi
Emira Ayroldi- Section of Pharmacology, Department of Medicine, Medical School, University of Perugia, Perugia, Italy
Glucocorticoid-induced leucine zipper (GILZ) is a protein with multiple biological roles that is upregulated by glucocorticoids (GCs) in both immune and non-immune cells. Importantly, GCs are immunosuppressive primarily due to their regulation of cell signaling pathways that are crucial for immune system activity. GILZ, which is transcriptionally induced by the glucocorticoid receptor (GR), mediates part of these immunosuppressive, and anti-inflammatory effects, thereby controlling immune cell proliferation, survival, and differentiation. The primary immune cells targeted by the immunosuppressive activity of GCs are T cells. Importantly, the effects of GCs on T cells are partially mediated by GILZ. In fact, GILZ regulates T-cell activation, and differentiation by binding and inhibiting factors essential for T-cell function. For example, GILZ associates with nuclear factor-κB (NF-κB), c-Fos, and c-Jun and inhibits NF-κB-, and AP-1-dependent transcription. GILZ also binds Raf and Ras, inhibits activation of Ras/Raf downstream targets, including mitogen-activated protein kinase 1 (MAPK1). In addition GILZ inhibits forkhead box O3 (FoxO3) without physical interaction. GILZ also promotes the activity of regulatory T cells (Tregs) by activating transforming growth factor-β (TGF-β) signaling. Ultimately, these actions inhibit T-cell activation and modulate the differentiation of T helper (Th)-1, Th-2, Th-17 cells, thereby mediating the immunosuppressive effects of GCs on T cells. In this mini-review, we discuss how GILZ mediates GC activity on T cells, focusing mainly on the therapeutic potential of this protein as a more targeted anti-inflammatory/immunosuppressive GC therapy.
Introduction
Glucocorticoids (GCs) are the mainstay of current immunosuppressive and anti-inflammatory therapies (1). Decades of study have revealed that their primary mechanism of action involves GC binding to GC receptors (GRs) to modulate gene transcription (2–5). However, the biological effects of GCs are diverse and are likely controlled by several mechanisms. Given this functional diversity, identifying molecules that are transcriptionally induced by GCs, and can mediate specific GC effects presents a significant challenge.
One potential molecule is glucocorticoid-induced leucine zipper (GILZ), a ubiquitously expressed protein that is primarily under GR transcriptional control. GILZ was originally identified in 1997 when searching for genes that mediate GC-induced apoptosis (6). However, since that time, the roles of GILZ have expanded to include most of the anti-inflammatory, and immunosuppressive effects of GCs (7). Indeed, GILZ is now known to regulate cell apoptosis, proliferation, and differentiation by modulating transcription factors, and signaling pathways associated with host immunity, and inflammation (8–12).
GILZ has a high degree of homology with other members of the TSC22D family. The TSC22D family includes leucine zipper proteins that are differentially expressed and involved in the regulation of multiple biological processes (13). TSC22D isoform heterodimers regulate cell cycle entry and exit (14).
One mechanism by which GCs induce immunosuppression is through regulation of the T-cell response (15, 16). In this review, we discuss the literature concerning how GILZ mediates the effects of GCs on T cells. Regardless of the specific role of GILZ, we highlight information about GC-dependent, and GC-independent GILZ functions to expand the current understanding of the GC mechanism of action. Ultimately, such understanding is critical to improving GC clinical use.
GCs and the T-Cell Response
T-cell activation is an essential part of the adaptive, cell-mediated immune response. GCs modulate T-cell differentiation and activation regulating: (1) antigen-presenting cells (APCs); (2) T helper (Th) cell differentiation; and (3) T-cell receptor (TCR) signaling (Figure 1) (15). The GR acts through genomic and non-genomic mechanisms, regulating adhesion molecules, co-accessory molecules, and cytokines implicated in T-cell activation (17–19).
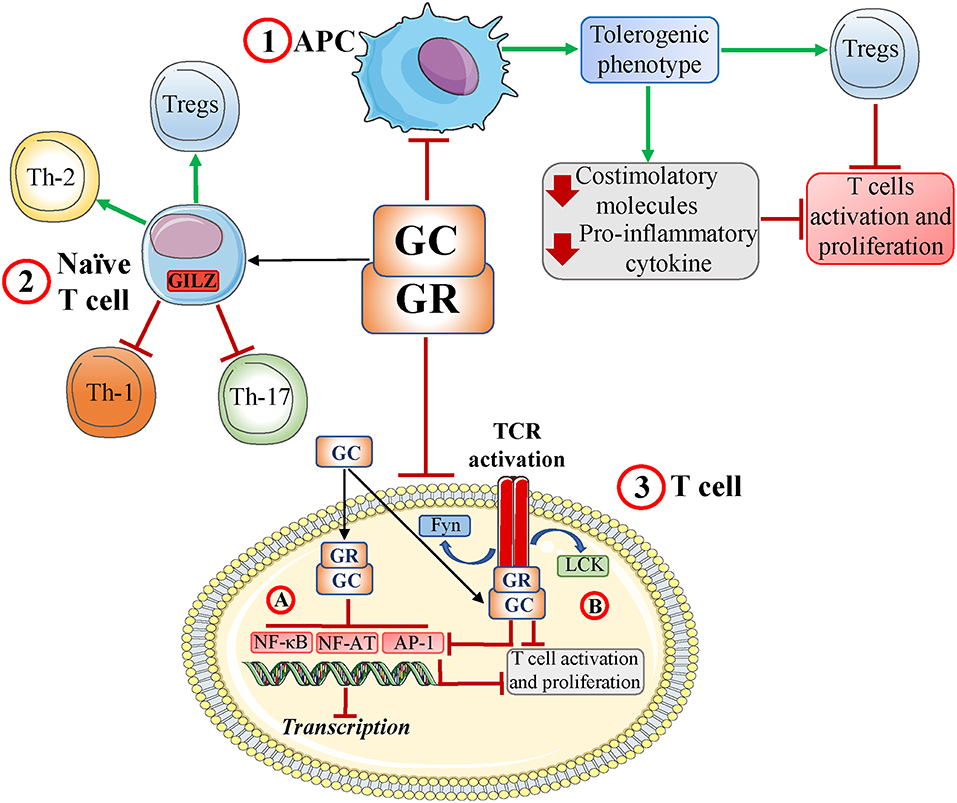
Figure 1. Glucocorticoids and the T-cell response. Glucocorticoid (GCs) and glucocorticoid receptor (GR) interactions induce: (1) a tolerogenic antigen-presenting cell (APC) phenotype with decreased production of both proinflammatory chemokines and costimulatory molecules and development of regulatory T cells (Tregs). This subsequently inhibits T-cell activation; (2) a modulation of naïve T-cell differentiation, including inhibition of Th-1 and Th-17 cell development and induction of Th-2 cells and Tregs; and (3) inhibition of T-cell receptor (TCR) signaling by inhibiting (genomic effects) key transcription factors such as NF-AT, AP-1, and NF-κB (3A) and disruption of TCR-associated multiprotein complexes containing GR, LCK, and FYN (rapid, non-genomic effects) with inhibition of NF-AT, AP-1, and NF-κB (3B). Ultimately, these interactions impair TCR signaling and T-cell activation/proliferation. Red T-headed leaders indicate inhibition; green arrow-headed leaders indicate activation.
Acting directly on T cells, GCs function through different mechanisms, most of which involve GR/transcription factor interaction. GCs affect the activity of transcription factors downstream of TCR activation, including nuclear factor-κB (NF-κB), activator protein-1 (AP-1), and nuclear factor of activated T cells (NF-AT) (15). GCs can also act through non-genomic mechanisms to limit kinase activity downstream of TCR activation, ultimately inhibiting the above-mentioned transcription factors and T-cell activation (20) (Figure 1).
Moreover, GCs can modulate T-cell activation indirectly through other cells such as dendritic cells (DCs), which are professional APCs. DCs have dual functionality, as they both orchestrate adaptive immune responses and also actively maintain peripheral specific tolerance against innocuous antigens (21). The balance between the activating and tolerogenic DC phenotypes is crucial to generating an efficient immune response while also preventing autoimmunity. GCs inhibit DC functions, reducing expression of MHC class II, and costimulatory molecules, decreasing proinflammatory cytokines and increasing anti-inflammatory cytokines such as IL-10 (22). Importantly, GCs can also increase the ability of DCs to capture antigens, suggesting that GCs drive DCs toward a tolerogenic phenotype (23). Tolerogenic DCs induce T-cell suppression and anergy and promote the generation of regulatory T cells (Tregs) (24). Therefore, GC modulation of DCs indirectly inhibits T-cell activation (Figure 1).
GCs can also modulate T cells by targeting tissue macrophages, mast cells, and stromal cells. Myeloid cells modulate T-cell function, acting as APCs and/or secreting inflammatory cytokines in response to stimulation of pattern recognition receptors (PRRs) (15). GCs can attenuate signals downstream of PRR activation, including the transcription factors AP-1, NF-κB, and the mitogen-activated protein kinase 1 (MAPK1) pathway (15, 25, 26). Those signaling changes alter the cytokine network, with important consequences for both inflammation, and T-cell responses. In fact, this mechanism may partially account for both GC inhibition of Th-1 and Th-17 differentiation and GC promotion of Th-2 differentiation and Treg production (27, 28) (Figure 1).
What is the role of GILZ in this context?
GILZ and the T-cell Response
Similar to the GCs, GILZ inhibits innate, and adaptive immune responses, affecting T-cell function (activation, differentiation, and apoptosis) either directly or through APCs (7, 9, 10) (Figure 2).
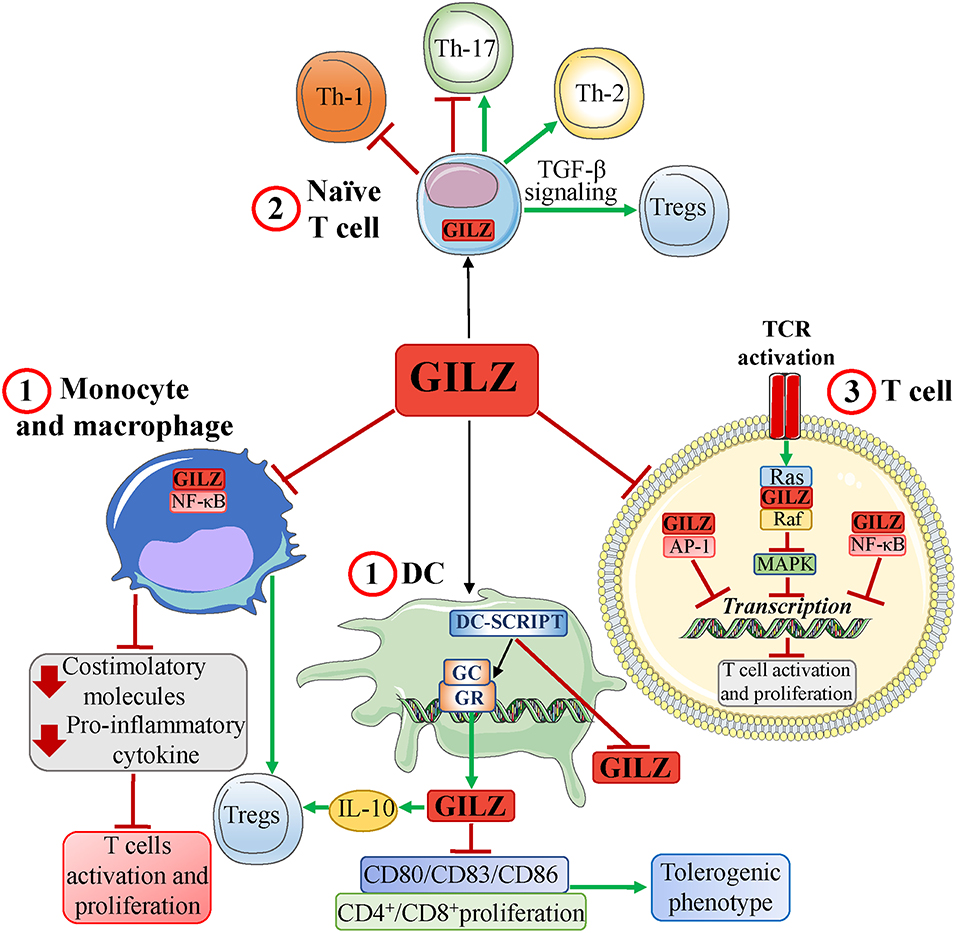
Figure 2. GILZ and the T-cell response. GILZ, expressed basally and/or in response to GCs, induces: (1) a tolerogenic dendritic cell (DC) phenotype and inhibition of human monocyte and mouse macrophage activation via inhibition of NF-κB, thereby limiting production of both proinflammatory chemokines, and costimulatory molecules. In DCs, GILZ expression is regulated by a GR corepressor, DC-specific transcript (DC-SCRIPT), whose recruitment inhibits GILZ expression. GILZ-induced inhibition of APC functions promotes development of Tregs and ultimately inhibits T-cell activation; (2) a modulation of naïve T-cell differentiation (expressing endogenous GILZ) that includes induction of Th-2 cells and Tregs (favoring TGF-β signaling), inhibition of Th-1 cells, and inhibition or development of Th-17 cells; (3) an inhibition of TCR signaling by inhibiting pathways, such as MAPK, and transcription factors, such as AP-1, and NF-κB, through protein-protein interactions. Red T-headed leaders indicate inhibition; green arrow-headed leaders indicate activation.
GC-induced GILZ expression in T cells is involved in multiple GC effects (9); however, its endogenous expression in the naïve T cell suggests a GC-independent function (29).
GCs can modulate T-cell apoptosis, and GILZ can either induce or protect against apoptosis (6). The first studies on apoptosis were performed using the T-cell hybridoma 3DO, which overexpresses GILZ (6, 30). In this cell line, GILZ inhibits both NF-κB (30), and AP-1 (31), behaves as a GC by inhibiting CD3-mediated apoptosis and TCR-driven IL-2 production through Fas/FasL modulation (30). Furthermore, T cells from GILZ-knockout mice (GILZ-KO) show increased antigen-induced T-cell activation (32). These data indicate mutual antagonism between GILZ expression and T-cell activation, suggesting that T cells must inhibit GILZ expression to become activated (29, 33). Moreover, in T cells, GILZ expression mimics the antiproliferative effects of GCs by interacting with Ras and Raf and inhibiting Ras downstream signals, such as MAPK (33, 34) (Figure 2). Notably, IL-2 deprivation in T cells upregulates GILZ (35), whereas IL-2 treatment (35), and T-cell activation (29, 30, 33) decrease GILZ expression. Moreover, IL-2 withdrawal induces cell death and upregulates GILZ by promoting forkhead box O3 (FoxO3) transcriptional activity in GILZ promoter region. In turn, GILZ prevents FoxO3 transcriptional activity, promoting its nuclear exclusion through a mechanism involving the nuclear export receptor Crm1 (36), and inhibiting its own expression and that of the proapoptotic gene Bim. In this case, GILZ protects T cells from IL-2 withdrawal-induced apoptosis by regulating its own expression (35, 37). The role of GILZ in T-cell apoptosis has been further clarified using GILZ transgenic mouse models (GILZ-TG). Thymocytes from GILZ-TG mice undergo apoptosis through caspase-8 activation and Bcl-xL downregulation (38), regulating the thymic repertoire similar to GCs. However, these cells are rescued by TCR-induced apoptosis, suggesting a GC-like mechanism of mutual exclusion (39). In contrast, GILZ does not induce apoptosis in peripheral mature mouse T lymphocytes (40). The ability of GCs to induce the apoptosis of lymphoid cells supports their inclusion in protocols for the treatment of lymphohematopoietic malignancies. GILZ upregulation may underlie these effects of GCs. For example, in multiple myeloma, for which GCs are used, decreasing GILZ levels by siRNA knockdown inhibited GC-induced apoptosis (41).
Constitutive expression of GILZ in naïve T cells (29) plays a major role in their differentiation (Figure 2). GILZ promotes Treg differentiation by activating transforming growth factor-β (TGF-β) signaling (42) and is partly responsible for GC-mediated effects on Tregs (15). In fact, dexamethasone (DEX) treatment augments the frequency of splenic Tregs in WT, but not GILZ-KO, mice (42).
Moreover, GILZ overexpression in CD4+ lymphocytes from GILZ-TG mice promotes Th-2 and inhibits Th-1 differentiation (43); thus, GILZ behaves like GCs (27, 44). As a consequence, GILZ-TG mice are less susceptible to Th-1-mediated diseases, such as experimental dinitrobenzene sulfonic acid- (DNBS-) colitis (45), and spinal cord injury (46). In these models, GILZ-TG mice exhibit an attenuated immune response, which may be explained by GILZ-mediated inhibition of NF-κB, which is crucial for Th-1 cytokine production, in T cells of the intestinal lamina propria, and in spinal cord lesions, respectively (45, 46). Accordingly, injection of mice with either the transactivator of transcription (TAT)-glutathione-S-transferase (GST)-GILZ (TAT-GST–GILZ) fusion protein or high doses of DEX, which upregulates GILZ in mucosal T lymphocytes, rescues mice from Th-1-mediated experimental colitis, again by inhibiting NF-κB (45). GILZ, in this model, is crucial for effects on Tregs cells. In fact, in GILZ-KO mice, the severity of DNBS-colitis is increased compared with WT due to impaired generation of Tregs cells. Transfer of WT Treg cells reverses the augmented vulnerability. DEX ameliorates the symptoms of DNBS-colitis in WT, but not GILZ-KO, through Treg augmentation. Therefore, GC anti-inflammatory activities in this model may be mediated by GILZ expression in T lymphocytes (45), and GILZ-induced Treg generation (42). However, in other murine models of inflammation, GILZ does not appear to be involved in the anti-inflammatory activity of GCs. For example, endogenous GILZ is detectable in the synovia of mice with collagen-induced arthritis (CIA), and in patients with active rheumatoid arthritis, and is upregulated by GC therapeutic doses (47). Moreover, GILZ reduction by RNAi worsens the symptoms of CIA, suggesting a role for GILZ as an endogenous inhibitor (47). However, its deletion does not impair the effects of exogenous GCs in CIA and does not affect the severity of antigen-induced or K/BxN serum–transfer arthritis (32). In fact, no difference in arthritis severity was found between GILZ-KO and WT mice, although antigen-induced T-cell proliferation was higher in GILZ-KO mice. However, injection of adeno-associated virus expressing GILZ (GILZ-rAAV) in CIA mice results in joint GILZ expression and attenuation of joint inflammation without affecting T-cell proliferation (32). These data suggest different roles for GILZ in inflammation, again as a brake for T-cell proliferation, an endogenous natural anti-inflammatory protein, and as a drug.
A pharmacological use of the GILZ protein was also shown in experimental autoimmune encephalomyelitis (EAE), an inflammatory model for human multiple sclerosis. GILZ peptide (GILZ-P) binds to and inhibits NF-κB, suppresses T-cell activation, and shows therapeutic efficacy when administered in EAE mice. Specifically, GILZ-P inhibits NF-κB, Th-1 cytokines, and T-Bet transcription but increases expression of GATA-3 and Th-2 cytokines, mimicking GILZ (48), and GC activity (17, 27).
GILZ negatively modulates Th-17 development by binding to IL-21 and Irf4 sites, as demonstrated via ChIP-seq analysis of Th-17 cells. These sites overlap the binding sites of major transcription factors involved in Th-17 polarization. Therefore, GILZ may act as a transcriptional repressor, inducing displacement of Th-17 transcription factors from their sites with inhibitory effects on Th-17 development (49). Consistently, GILZ downregulation in naïve CD4+ T cells is required for development of Th-17 (29). GILZ expression in T cells is protective against several pathologies, including psoriasis, a disease commonly treated with GCs, and myocardial infarction, in which Th-17 lymphokines are pathogenetic (29, 50). However, conflicting data were obtained in vivo with imiquimod (IMQ), a murine model for IL23-, and IL17-dependent psoriasis. Some researchers demonstrate that IMQ-induced psoriasis is more serious in GILZ-deficient mice, with upregulation of Th-17 cytokines and Th-17 proliferation (29). In contrast, other researchers show that IMQ-induced psoriasis is more severe in GILZ-TG mice, with increased Th-17 cytokines (51) (Figure 2). Thus, based on this model, GILZ can be proinflammatory, similar to the effects of prolonged GC treatment (51), or anti-inflammatory (29).
As mentioned, GILZ can modulate T-cell activity indirectly through its actions on APCs. Its effects on myeloid cells have a broad spectrum of action on all cells of the immune system (9). DC subsets constitutively express GILZ at different levels depending on DC functional status (52). Endogenous GCs appear to regulate constitutive DC GILZ expression, whereas exogenous GCs upregulate DC GILZ in vivo and in vitro. Thus, by mediating the effects of GCs, GILZ can regulate the balance between activating and tolerogenic DCs (53, 54). GILZ expression is transcriptionally regulated by the GR, which can either induce or inhibit GILZ by recruiting its corepressor, DC-specific transcript (DC-SCRIPT) (Figure 2). Importantly, neutralizing DC-SCRIPT augments GR-induced GILZ expression (55). This suggests that the tolerogenic-promoting effects of GILZ in DCs are so crucial that a biological brake on its expression is required. Indeed, GILZ overexpression induces a DC tolerogenic phenotype comparable to that induced by GCs (56), downregulating the costimulatory molecules CD86, CD83, and CD80 (57, 58), and reducing CD4+ T-cell proliferation (53) (Figure 2). Knocking down GILZ in activated monocyte-derived DCs (Mo-DCs) promotes more efficient CD8+ T-cell secondary responses (59). In vitro GC treatment of human Mo-DCs induces GILZ expression, driving a DC tolerogenic phenotype that prevents efficient antigen presentation (57), and induces IL-10-promoting Tregs. Together, these changes inhibit the T-cell response (58). This effect is reproduced by GILZ overexpression (60) and abolished by GILZ silencing (57, 59). Finally, GILZ expression in tumor-infiltrating DCs drives a tolerogenic DC phenotype, and T-cell tolerance against the tumor (54). This suggests that tumor cells may “learn” to secrete GCs to induce GILZ as an escape mechanism against the immune system. These results may explain how GCs, both endogenous and exogenously administered, can either block or worsen tumor progression (especially epithelial tumors) through GILZ expression (61).
Similar to DCs, human monocytes and mouse macrophages constitutively express GILZ. GCs further upregulate GILZ expression, which, via inhibition of NF-κB, mediates GC activity in these cells (62–64) (Figure 2). In fact, transfecting GILZ into THP-1 macrophages mimics the effects of GCs and inhibits the production of chemokines, and costimulatory molecules (62). Correspondingly, GILZ is downregulated by Toll-Like agonists, leading to macrophage activation (65). Moreover, GILZ expression is decreased during neuroinflammation, inversely correlating with the development of innate immune responses (66), and in white blood cells from patients with sepsis (67). These findings confirm the immunosuppressive role of GILZ in myeloid cells and the biological necessity of GILZ downregulation for efficient natural or adaptive immune responses.
Furthermore, expression of GILZ, as with GC (15), limits Th-17 differentiation, and induced Treg cell activity by modulating cytokine production by DCs and mesenchymal cells (68, 69). In a mouse model of rheumatoid arthritis, GILZ expression in mesenchymal stem cells (MSCs) is required for therapeutic effectiveness of MSCs in arthritis (68) and inhibition of transferred- Th-1, and Th-17 cells in immunized mice (70). In a model of acute kidney injury, TAT-GST-GILZ fusion protein conferred renoprotection by regulating cross-talk between T cells and neutrophils, reducing proinflammatory type 1 neutrophils and Th-17 cells, and increasing anti-inflammatory type 2 neutrophils and Tregs (71).
The role of GILZ in T-cell activation is even more complex if we consider the effects on its expression following accessory molecule triggering. Indeed, blocking the co-accessory molecule, CD80, enhances GILZ expression in activated CD4+ T cells (72). However, this field of investigation remains unexplored.
Perspective and Expectations
Based on our critical review of the literature, we suggest that GILZ has at least three different functions in T cells: (1) endogenous; (2) mediator of GC activity; and (3) as a drug.
As discussed above, basal endogenous GILZ expression in immune cells has a predominant role in T-cell activation, the development of CD4+ naïve T cells, and the physiological control of inflammation. The latter is demonstrated by the many murine models of inflammation, in which the absence of GILZ aggravates inflammatory pathologies (12, 29, 32, 42, 47). However, GILZ expression in T cells underlies many of the effects of GCs established in experimental in vivo and in vitro models. These models demonstrate that a lack of GILZ inhibits the activity of GCs, and overexpression may mimic GC effects (8–10, 43).
The use of GILZ as a drug is a great challenge given the potential side effects on metabolism. However, many experimental models support and encourage this possibility. Experiments with fusion proteins TAT-GST-GILZ, and HHph-GILZ, viral constructs GILZ-rAAV expressing GILZ, and GILZ-peptide GILZ-P provide examples of achieving pharmacokinetic, pharmacodynamic, and therapeutic efficacy using GILZ in vivo as a drug (29, 32, 45, 47, 73). Many of the experimental models discussed above involve pathologies due to an imbalance of the development of naïve CD4+ cells, demonstrating how the therapeutic activity of GILZ is related to actions on T cells (11, 12, 45, 47).
GCs inhibit T-cell activation through genomic and non-genomic mechanisms. GR-mediated genomic regulation induces immunosuppressive molecules, including GILZ (8, 15, 74–76). GCs also modulate T-cell activity through non-genomic mechanisms that occur immediately after drug exposure (77, 78). In T cells, the GR physically associates with the TCR in a multiprotein complex with LCK, and FYN. Short-term treatment with DEX induces the non-genomic destruction of this complex, thereby limiting TCR activation (20) (Figure 1). Is it possible to hypothesize that GCs regulate GILZ function and/or expression through both genomic and non-genomic mechanisms? The regulation of GILZ by GC non-genomic effects would lay the groundwork for several future lines of study. In particular, because GC-induced GILZ transcription in T cells interacts with and inhibits TCR-triggered signaling pathways and transcription factors, it is likely that there is a GC-induced non-genomic effect on constitutive GILZ expression. This would reveal another mechanism by which GCs regulate the T-cell response. Such a mechanism might provide further explanation for the basal level of GILZ in immune cells (63, 79). Therefore, it would be interesting to investigate whether GC/GR interactions induce rapid changes in the cytoplasmic basal pool of GILZ, as such GILZ expression may have alternative functions compared to those of peak GILZ activation induced by GR-mediated transcription. Ultimately, building on our understanding of the molecular mechanisms involving GCs and GILZ may improve the use of GCs as clinical therapeutics and limit treatment-related side effects.
Author Contributions
EA wrote the review and suggested the general topic. SA prepared the figures. LC and DD discussed and reviewed the manuscript. CR suggested the general topic and discussed and reviewed the manuscript.
Funding
This research was funded by the Progetti di Ricerca di Rilevante Interesse Nazionale (PRIN) 2015 (2015ZT9HXY) and the Fondazione Cassa di Risparmio di Perugia, Ricerca Scientifica e Tecnologica (2016.0123.021).
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Rhen T, Cidlowski JA. Antiinflammatory action of glucocorticoids–new mechanisms for old drugs. N Engl J Med. (2005) 353:1711–23. doi: 10.1056/NEJMra050541
2. Barnes PJ. Anti-inflammatory actions of glucocorticoids: molecular mechanisms. Clin Sci. (1998) 94:557–72 doi: 10.1042/cs0940557
3. Baschant U, Tuckermann J. The role of the glucocorticoid receptor in inflammation and immunity. J Steroid Biochem Mol Biol. (2010) 120:69–75. doi: 10.1016/j.jsbmb.2010.03.058
4. Biddie SC, Conway-Campbell BL, Lightman SL. Dynamic regulation of glucocorticoid signalling in health and disease. Rheumatology. (2012) 51:403–12. doi: 10.1093/rheumatology/ker215
5. Hubner S, Dejager L, Libert C, Tuckermann JP. The glucocorticoid receptor in inflammatory processes: transrepression is not enough. Biol Chem. (2015) 396:1223–31. doi: 10.1515/hsz-2015-0106
6. D'Adamio F, Zollo O, Moraca R, Ayroldi E, Bruscoli S, Bartoli A, et al. A new dexamethasone-induced gene of the leucine zipper family protects T lymphocytes from TCR/CD3-activated cell death. Immunity. (1997) 7:803–12 doi: 10.1016/S1074-7613(00)80398-2
7. Ayroldi E, Riccardi C. Glucocorticoid-induced leucine zipper (GILZ): a new important mediator of glucocorticoid action. Faseb J. (2009) 23:3649–58. doi: 10.1096/fj.09-134684
8. Ayroldi E, Macchiarulo A, Riccardi C. Targeting glucocorticoid side effects: selective glucocorticoid receptor modulator or glucocorticoid-induced leucine zipper? a perspective. Faseb J. (2014) 28:5055–70. doi: 10.1096/fj.14-254755
9. Ronchetti S, Migliorati G, Riccardi C. GILZ as a mediator of the anti-inflammatory effects of glucocorticoids. Front Endocrinol. (2015) 6:170. doi: 10.3389/fendo.2015.00170
10. Beaulieu E, Morand EF. Role of GILZ in immune regulation, glucocorticoid actions and rheumatoid arthritis. Nat Rev Rheumatol. (2011) 7:340–8. doi: 10.1038/nrrheum.2011.59
11. Cheng Q, Morand E, Yang YH. Development of novel treatment strategies for inflammatory diseases-similarities and divergence between glucocorticoids and GILZ. Front Pharmacol. (2014) 5:169. doi: 10.3389/fphar.2014.00169
12. Bereshchenko O, Migliorati G, Bruscoli S, Riccardi C Glucocorticoid-induced leucine zipper: a novel anti-inflammatory molecule. Front Pharmacol. (2019) 10:308. doi: 10.3389/fphar.2019.00308
13. Soundararajan R, Wang J, Melters D, Pearce D. Differential activities of glucocorticoid-induced leucine zipper protein isoforms. J Biol Chem. (2007) 282:36303–13. doi: 10.1074/jbc.M707287200
14. Dragotto J, Canterini S, Del Porto P, Bevilacqua A, Fiorenza MT. The interplay between TGF-beta-stimulated TSC22 domain family proteins regulates cell-cycle dynamics in medulloblastoma cells. J Cell Physiol. (2019) 234:18349–60. doi: 10.1002/jcp.28468
15. Cain DW, Cidlowski JA. Immune regulation by glucocorticoids. Nat Rev Immunol. (2017) 17:233–47. doi: 10.1038/nri.2017.1
16. Libert C, Dejager L. How steroids steer T cells. Cell Rep. (2014) 7:938–9. doi: 10.1016/j.celrep.2014.04.041
17. De Bosscher K, Vanden Berghe W, Haegeman G. The interplay between the glucocorticoid receptor and nuclear factor-kappaB or activator protein-1: molecular mechanisms for gene repression. Endocr Rev. (2003) 24:488–522. doi: 10.1210/er.2002-0006
18. Ratman D, Vanden Berghe W, Dejager L, Libert C, Tavernier J, Beck IM, et al. How glucocorticoid receptors modulate the activity of other transcription factors: a scope beyond tethering. Mol Cell Endocrinol. (2013) 380:41–54. doi: 10.1016/j.mce.2012.12.014
19. Langlais D, Couture C, Balsalobre A, Drouin J, The Stat3/GR interaction code: predictive value of direct/indirect DNA recruitment for transcription outcome. Mol Cell. (2012) 47:38–49. doi: 10.1016/j.molcel.2012.04.021
20. Lowenberg M, Verhaar AP, van den Brink GR, Hommes DW Glucocorticoid signaling: a nongenomic mechanism for T-cell immunosuppression. Trends Mol Med. (2007) 13:158–63. doi: 10.1016/j.molmed.2007.02.001
21. Steinman RM, Hawiger D, Nussenzweig MC. Tolerogenic dendritic cells. Annu Rev Immunol. (2003) 21:685–711. doi: 10.1146/annurev.immunol.21.120601.141040
22. van den Heuvel MM, van Beek NM, Broug-Holub E, Postmus PE, Hoefsmit EC, Beelen RH, Kraal G. Glucocorticoids modulate the development of dendritic cells from blood precursors. Clin Exp Immunol. (1999) 115:577–83 doi: 10.1046/j.1365-2249.1999.00811.x
23. Chamorro S, Garcia-Vallejo JJ, Unger WW, Fernandes RJ, Bruijns SC, Laban S, et al. TLR triggering on tolerogenic dendritic cells results in TLR2 up-regulation and a reduced proinflammatory immune program. J Immunol. (2009) 183:2984–94. doi: 10.4049/jimmunol.0801155
24. Iberg CA, Jones A, Hawiger D. Dendritic cells as inducers of peripheral tolerance. Trends Immunol. (2017) 38:793–804. doi: 10.1016/j.it.2017.07.007
25. Scheinman RI, Cogswell PC, Lofquist AK, Baldwin AS Jr. Role of transcriptional activation of I kappa B alpha in mediation of immunosuppression by glucocorticoids. Science. (1995) 270:283–6 doi: 10.1126/science.270.5234.283
26. Ayroldi E, Cannarile L, Migliorati G, Nocentini G, Delfino DV, Riccardi C. Mechanisms of the anti-inflammatory effects of glucocorticoids: genomic and nongenomic interference with MAPK signaling pathways. Faseb J. (2012) 26:4805–20. doi: 10.1096/fj.12-216382
27. Elenkov IJ. Glucocorticoids and the Th1/Th2 balance. Ann N Y Acad Sci. (2004) 1024:138–46. doi: 10.1196/annals.1321.010
28. Cari L, De Rosa F, Nocentini G, Riccardi C. Context-dependent effect of glucocorticoids on the proliferation, differentiation, and apoptosis of regulatory T cells: a review of the empirical evidence and clinical applications. Int J Mol Sci. (2019) 20:e1142. doi: 10.3390/ijms20051142
29. Jones SA, Perera DN, Fan H, Russ BE, Harris J, Morand EF. GILZ regulates Th17 responses and restrains IL-17-mediated skin inflammation. J Autoimmun. (2015) 61:73–80. doi: 10.1016/j.jaut.2015.05.010
30. Ayroldi E, Migliorati G, Bruscoli S, Marchetti C, Zollo O, Cannarile L, et al. Modulation of T-cell activation by the glucocorticoid-induced leucine zipper factor via inhibition of nuclear factor kappaB. Blood. (2001) 98:743–53 doi: 10.1182/blood.V98.3.743
31. Mittelstadt PR, Ashwell JD. Inhibition of AP-1 by the glucocorticoid-inducible protein GILZ. J Biol Chem. (2001) 276:29603–10. doi: 10.1074/jbc.M101522200
32. Ngo D, Beaulieu E, Gu R, Leaney A, Santos L, Fan H, et al. Divergent effects of endogenous and exogenous glucocorticoid-induced leucine zipper in animal models of inflammation and arthritis. Arthritis Rheum. (2013) 65:1203–12. doi: 10.1002/art.37858
33. Ayroldi E, Zollo O, Bastianelli A, Marchetti C, Agostini M, Di Virgilio R, et al. GILZ mediates the antiproliferative activity of glucocorticoids by negative regulation of Ras signaling. J Clin Invest. (2007) 117:1605–15. doi: 10.1172/JCI30724
34. Ayroldi E, Zollo O, Macchiarulo A, Di Marco B, Marchetti C, Riccardi C Glucocorticoid-induced leucine zipper inhibits the raf-extracellular signal-regulated kinase pathway by binding to Raf-1. Mol Cell Biol. (2002) 22:7929–41. doi: 10.1128/MCB.22.22.7929-7941.2002
35. Asselin-Labat ML, David M, Biola-Vidamment A, Lecoeuche D, Zennaro MC, Bertoglio J, et al. GILZ, a new target for the transcription factor FoxO3, protects T lymphocytes from interleukin-2 withdrawal-induced apoptosis. Blood. (2004) 104:215–23. doi: 10.1182/blood-2003-12-4295
36. Latre de Late P, Pepin A, Assaf-Vandecasteele H, Espinasse C, Nicolas V, Asselin-Labat ML, et al. Glucocorticoid-induced leucine zipper (GILZ) promotes the nuclear exclusion of FOXO3 in a Crm1-dependent manner. J Biol Chem. (2010) 285:5594–605. doi: 10.1074/jbc.M109.068346
37. Asselin-Labat ML, Biola-Vidamment A, Kerbrat S, Lombes M, Bertoglio J, Pallardy M. FoxO3 mediates antagonistic effects of glucocorticoids and interleukin-2 on glucocorticoid-induced leucine zipper expression. Mol Endocrinol. (2005) 19:1752–64. doi: 10.1210/me.2004-0206
38. Delfino DV, Agostini M, Spinicelli S, Vito P, Riccardi C. Decrease of Bcl-xL and augmentation of thymocyte apoptosis in GILZ overexpressing transgenic mice. Blood. (2004) 104:4134–41. doi: 10.1182/blood-2004-03-0920
39. Delfino DV, Agostini M, Spinicelli S, Vacca C, Riccardi C. Inhibited cell death, NF-kappaB activity and increased IL-10 in TCR-triggered thymocytes of transgenic mice overexpressing the glucocorticoid-induced protein GILZ. Int Immunopharmacol. (2006) 6:1126–34. doi: 10.1016/j.intimp.2006.02.001
40. Pozzesi N, Gizzi S, Gori F, Vacca C, Cannarile L, Riccardi C, et al. IL-2 induces and altered CD4/CD8 ratio of splenic T lymphocytes from transgenic mice overexpressing the glucocorticoid-induced protein GILZ. J Chemother. (2007) 19:562–9. doi: 10.1179/joc.2007.19.5.562
41. Kervoelen C, Menoret E, Gomez-Bougie P, Bataille R, Godon C, Marionneau-Lambot S, et al. Dexamethasone-induced cell death is restricted to specific molecular subgroups of multiple myeloma. Oncotarget. (2015) 6:26922–34. doi: 10.18632/oncotarget.4616
42. Bereshchenko O, Coppo M, Bruscoli S, Biagioli M, Cimino M, Frammartino T, et al. GILZ promotes production of peripherally induced treg cells and mediates the crosstalk between glucocorticoids and TGF-beta signaling. Cell Rep. (2014) 7:464–75. doi: 10.1016/j.celrep.2014.03.004
43. Cannarile L, Fallarino F, Agostini M, Cuzzocrea S, Mazzon E, Vacca C, et al. Increased GILZ expression in transgenic mice up-regulates Th-2 lymphokines. Blood. (2006) 107:1039–47. doi: 10.1182/blood-2005-05-2183
44. Miyaura H, Iwata M. Direct and indirect inhibition of Th1 development by progesterone and glucocorticoids. J Immunol. (2002) 168:1087–94. doi: 10.4049/jimmunol.168.3.1087
45. Cannarile L, Cuzzocrea S, Santucci L, Agostini M, Mazzon E, Esposito E, et al. Glucocorticoid-induced leucine zipper is protective in Th1-mediated models of colitis. Gastroenterology. (2009) 136:530–41. doi: 10.1053/j.gastro.2008.09.024
46. Esposito E, Bruscoli S, Mazzon E, Paterniti I, Coppo M, Velardi E, et al. Glucocorticoid-induced leucine zipper (GILZ) over-expression in T lymphocytes inhibits inflammation and tissue damage in spinal cord injury. Neurotherapeutics. (2012) 9:210–25. doi: 10.1007/s13311-011-0084-7
47. Beaulieu E, Ngo D, Santos L, Yang YH, Smith M, Jorgensen C, et al. Glucocorticoid-induced leucine zipper is an endogenous antiinflammatory mediator in arthritis. Arthritis Rheum. (2010) 62:2651–61. doi: 10.1002/art.27566
48. Srinivasan M, Janardhanam S. Novel p65 binding glucocorticoid-induced leucine zipper peptide suppresses experimental autoimmune encephalomyelitis. J Biol Chem. (2011) 286:44799–810. doi: 10.1074/jbc.M111.279257
49. Yosef N, Shalek AK, Gaublomme JT, Jin H, Lee Y, Awasthi A, et al. Dynamic regulatory network controlling TH17 cell differentiation. Nature. (2013) 496:461–8. doi: 10.1038/nature11981
50. Baban B, Yin L, Qin X, Liu JY, Shi X, Mozaffari MS. The role of GILZ in modulation of adaptive immunity in a murine model of myocardial infarction. Exp Mol Pathol. (2017) 102:408–14. doi: 10.1016/j.yexmp.2017.05.002
51. Carceller E, Ballegeer M, Deckers J, Riccardi C, Bruscoli S, Hochepied T, et al. Overexpression of glucocorticoid-induced leucine zipper (GILZ) increases susceptibility to imiquimod-induced psoriasis and involves cutaneous activation of TGF-beta1. Sci Rep. (2016) 6:38825. doi: 10.1038/srep38825
52. Calmette J, Bertrand M, Vetillard M, Ellouze M, Flint S, Nicolas V, et al. Glucocorticoid-induced leucine zipper protein controls macropinocytosis in dendritic cells. J Immunol. (2016) 197:4247–56. doi: 10.4049/jimmunol.1600561
53. Vetillard M, Schlecht-Louf G Glucocorticoid-induced leucine zipper: fine-tuning of dendritic cells function. Front Immunol. (2018) 9:1232. doi: 10.3389/fimmu.2018.01232
54. Lebson L, Wang T, Jiang Q, Whartenby KA. Induction of the glucocorticoid-induced leucine zipper gene limits the efficacy of dendritic cell vaccines. Cancer Gene Ther. (2011) 18:563–70. doi: 10.1038/cgt.2011.23
55. Hontelez S, Karthaus N, Looman MW, Ansems M, Adema GJ. DC-SCRIPT regulates glucocorticoid receptor function and expression of its target GILZ in dendritic cells. J Immunol. (2013) 190:3172–9. doi: 10.4049/jimmunol.1201776
56. Krzysiek R. Role of glucocorticoid-induced leucine zipper (GILZ) expression by dendritic cells in tolerance induction. Transplant Proc. (2010) 42:3331–2. doi: 10.1016/j.transproceed.2010.07.038
57. Cohen N, Mouly E, Hamdi H, Maillot MC, Pallardy M, Godot V, et al. GILZ expression in human dendritic cells redirects their maturation and prevents antigen-specific T lymphocyte response. Blood. (2006) 107:2037–44. doi: 10.1182/blood-2005-07-2760
58. Hamdi H, Godot V, Maillot MC, Prejean MV, Cohen N, Krzysiek R, et al. Induction of antigen-specific regulatory T lymphocytes by human dendritic cells expressing the glucocorticoid-induced leucine zipper. Blood. (2007) 110:211–9. doi: 10.1182/blood-2006-10-052506
59. Cathelin D, Met O, Svane IM. Silencing of the glucocorticoid-induced leucine zipper improves the immunogenicity of clinical-grade dendritic cells. Cytotherapy. (2013) 15:740–9. doi: 10.1016/j.jcyt.2013.02.005
60. Calmette J, Ellouze M, Tran T, Karaki S, Ronin E, Capel F, et al. Glucocorticoid-induced leucine zipper enhanced expression in dendritic cells is sufficient to drive regulatory T cells expansion in vivo. J Immunol. (2014) 193:5863–72. doi: 10.4049/jimmunol.1400758
61. Ayroldi E, Cannarile L, Delfino DV, Riccardi C. A dual role for glucocorticoid-induced leucine zipper in glucocorticoid function: tumor growth promotion or suppression? Cell Death Dis. (2018) 9:463. doi: 10.1038/s41419-018-0558-1
62. Berrebi D, Bruscoli S, Cohen N, Foussat A, Migliorati G, Bouchet-Delbos L, et al. Synthesis of glucocorticoid-induced leucine zipper (GILZ) by macrophages: an anti-inflammatory and immunosuppressive mechanism shared by glucocorticoids and IL-10. Blood. (2003) 101:729–38. doi: 10.1182/blood-2002-02-0538
63. Hoppstadter J, Kiemer AK. Glucocorticoid-induced leucine zipper (GILZ) in immuno suppression: master regulator or bystander? Oncotarget. (2015) 6:38446–57. doi: 10.18632/oncotarget.6197
64. Hamdi H, Bigorgne A, Naveau S, Balian A, Bouchet-Delbos L, Cassard-Doulcier AM, et al. Glucocorticoid-induced leucine zipper: a key protein in the sensitization of monocytes to lipopolysaccharide in alcoholic hepatitis. Hepatology. (2007) 46:1986–92. doi: 10.1002/hep.21880
65. Hoppstadter J, Diesel B, Linnenberger R, Hachenthal N, Flamini S, Minet M, et al. Amplified host defense by toll-like receptor-mediated downregulation of the glucocorticoid-induced leucine zipper (GILZ) in macrophages. Front Immunol. (2018) 9:3111. doi: 10.3389/fimmu.2018.03111
66. Witek E, Hickman D, Lahiri DK, Srinivasan M. Glucocorticoid induced leucine zipper in lipopolysaccharide induced neuroinflammation. Front Aging Neurosci. (2018) 10:432. doi: 10.3389/fnagi.2018.00432
67. Ballegeer M, Vandewalle J, Eggermont M, Van Isterdael G, Dejager L, De Bus L, et al. Overexpression of gilz protects mice against lethal septic peritonitis. Shock. (2018) 52:208–214. doi: 10.1097/SHK.0000000000001252
68. Luz-Crawford P, Tejedor G, Mausset-Bonnefont AL, Beaulieu E, Morand EF, Jorgensen C, et al. Glucocorticoid-induced leucine zipper governs the therapeutic potential of mesenchymal stem cells by inducing a switch from pathogenic to regulatory Th17 cells in a mouse model of collagen-induced arthritis. Arthritis Rheumatol. (2015) 67:1514–24. doi: 10.1002/art.39069
69. Yang N, Baban B, Isales CM, Shi XM. Crosstalk between bone marrow-derived mesenchymal stem cells and regulatory T cells through a glucocorticoid-induced leucine zipper/developmental endothelial locus-1-dependent mechanism. FASEB J. (2015) 29:3954–63. doi: 10.1096/fj.15-273664
70. Luz-Crawford P, Espinosa-Carrasco G, Ipseiz N, Contreras R, Tejedor G, Medina DA, et al. Gilz-activin a as a novel signaling axis orchestrating mesenchymal stem cell and Th17 cell interplay. Theranostics. (2018) 8:846–59. doi: 10.7150/thno.21793
71. Baban B, Marchetti C, Khodadadi H, Malik A, Emami G, Lin PC, et al. Glucocorticoid-induced leucine zipper promotes neutrophil and T-cell polarization with protective effects in acute kidney injury. J Pharmacol Exp Ther. (2018) 367:483–93. doi: 10.1124/jpet.118.251371
72. Dudhgaonkar SP, Janardhanam SB, Kodumudi KN, Srinivasan M. CD80 blockade enhance glucocorticoid-induced leucine zipper expression and suppress experimental autoimmune encephalomyelitis. J Immunol. (2009) 183:7505–13. doi: 10.4049/jimmunol.0902056
73. Pinheiro I, Dejager L, Petta I, Vandevyver S, Puimege L, Mahieu T, et al. LPS resistance of SPRET/Ei mice is mediated by Gilz, encoded by the Tsc22d3 gene on the X chromosome. EMBO Mol Med. (2013) 5:456–70. doi: 10.1002/emmm.201201683
74. Franchimont D. Overview of the actions of glucocorticoids on the immune response: a good model to characterize new pathways of immunosuppression for new treatment strategies. Ann N Y Acad Sci. (2004) 1024:124–37. doi: 10.1196/annals.1321.009
75. Barnes PJ. Glucocorticosteroids. Handb Exp Pharmacol. (2017) 237:93–115. doi: 10.1007/164_2016_62
76. Vandewalle J, Luypaert A, De Bosscher K, Libert C. Therapeutic mechanisms of glucocorticoids. Trends Endocrinol Metab. (2018) 29:42–54. doi: 10.1016/j.tem.2017.10.010
77. Boldizsar F, Talaber G, Szabo M, Bartis D, Palinkas L, Nemeth P, et al. Emerging pathways of non-genomic glucocorticoid (GC) signalling in T cells. Immunobiology. (2010) 215:521–6. doi: 10.1016/j.imbio.2009.10.003
78. Panettieri RA, Schaafsma D, Amrani Y, Koziol-White C, Ostrom R, Tliba O Non-genomic effects of glucocorticoids: an updated view. Trends Pharmacol Sci. (2019) 40:38–49. doi: 10.1016/j.tips.2018.11.002
Keywords: glucocorticoids, glucocorticoid-induced leucine zipper (GILZ), T-cell activation, T-cell, immune response, glucocorticoid receptor
Citation: Cannarile L, Delfino DV, Adorisio S, Riccardi C and Ayroldi E (2019) Implicating the Role of GILZ in Glucocorticoid Modulation of T-Cell Activation. Front. Immunol. 10:1823. doi: 10.3389/fimmu.2019.01823
Received: 09 April 2019; Accepted: 18 July 2019;
Published: 07 August 2019.
Edited by:
Massimo Gadina, National Institute of Arthritis and Musculoskeletal and Skin Diseases (NIAMS), United StatesReviewed by:
Marc Pallardy, Université Paris-Sud, FranceGéraldine Schlecht-Louf, Université Paris-Sud, France
Copyright © 2019 Cannarile, Delfino, Adorisio, Riccardi and Ayroldi. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Emira Ayroldi, ZW1pcmEuYXlyb2xkaUB1bmlwZy5pdA==