 Krista D. DiSano
Krista D. DiSano Darlene B. Royce
Darlene B. Royce- Department of Neurology, Dartmouth Hitchcock Medical Center and Geisel School of Medicine, Lebanon, NH, United States
Persistent central nervous system (CNS) inflammation, as seen in chronic infections or inflammatory demyelinating diseases such as Multiple Sclerosis (MS), results in the accumulation of various B cell subsets in the CNS, including naïve, activated, memory B cells (Bmem), and antibody secreting cells (ASC). However, factors driving heterogeneous B cell subset accumulation and antibody (Ab) production in the CNS compartment, including the contribution of ectopic lymphoid follicles (ELF), during chronic CNS inflammation remain unclear and is a major gap in our understanding of neuroinflammation. We sought to address this gap using the Theiler's murine encephalomyelitis virus-induced demyelinating disease (TMEV-IDD) model of progressive MS. In this model, injection of the virus into susceptible mouse strains results in a persistent infection associated with demyelination and progressive disability. During chronic infection, the predominant B cell phenotypes accumulating in the CNS were isotype-switched B cells, including Bmem and ASC with naïve/early activated and transitional B cells present at low frequencies. B cell accumulation in the CNS during chronic TMEV-IDD coincided with intrathecal Ab synthesis in the cerebrospinal fluid (CSF). Mature and isotype-switched B cells predominately localized to the meninges and perivascular space, with IgG isotype-switched B cells frequently accumulating in the parenchymal space. Both mature and isotype-switched B cells and T cells occupied meningeal and perivascular spaces, with minimal evidence for spatial organization typical of ELF mimicking secondary lymphoid organs (SLO). Moreover, immunohistological analysis of immune cell aggregates revealed a lack of SLO-like ELF features, such as cell proliferation, cell death, and germinal center B cell markers. Nonetheless, flow cytometric assessment of B cells within the CNS showed enhanced expression of activation markers, including moderate upregulation of GL7 and expression of the costimulatory molecule CD80. B cell-related chemokines and trophic factors, including APRIL, BAFF, CXCL9, CXCL10, CCL19, and CXCL13, were elevated in the CNS. These results indicate that localization of heterogeneous B cell populations, including activated and isotype-switched B cell phenotypes, to the CNS and intrathecal Ab (ItAb) synthesis can occur independently of SLO-like follicles during chronic inflammatory demyelinating disease.
Introduction
The presence of antibody (Ab), antibody-secreting cells (ASC), and multiple other B cell subsets in the CNS compartment are hallmarks of chronic inflammatory processes during persistent infection or demyelinating diseases. In the inflammatory demyelinating disease Multiple Sclerosis (MS), CNS-infiltrating B cells are associated with the presence of oligoclonal IgG bands in the cerebrospinal fluid (CSF), a diagnostic hallmark in 95% of patients resulting from intrathecal antibody (ItAb) production in the CNS compartment (1–3). The exact role of B cells and Ab in MS remains controversial, but nevertheless multiple B cell phenotypes are implicated in MS pathogenesis. IgG and complement deposition are frequently observed in active demyelinating lesions in MS (4–7), indicating local Ab and complement-mediated mechanisms may contribute to demyelination. The success of B cell depletion therapies (BCDT) targeting CD20+ B cells, including mature B cells to plasmablasts, have further implicated B cells in MS pathogenesis, as BCDT reduces the formation of new inflammatory lesions and relapses in relapsing-remitting MS (RRMS) patients and time to confirmed disease progression in young, inflammatory primary progressive MS (PPMS) patients with minimal effects on ItAb (8–12). Collectively, these findings implicate a critical, yet unclear role for multiple B cell phenotypes in MS pathogenesis. However, the mechanisms underlying the recruitment and survival of diverse B cell populations in the CNS as well as factors responsible for fostering Ab production within the CNS compartment in MS and other chronic neuroinflammatory diseases are still largely undetermined.
The exact mechanisms regulating B cell accumulation and persistence in tissues outside of secondary lymphoid organs (SLO) remain unclear. Classically, infiltration of immune cells into non-lymphoid tissue sites, including the CNS, was seen as a random and diffuse process. However, during chronic inflammation lymphoid neogenesis may occur in certain conditions whereby tissue-infiltrating immune cells form highly organized structures resembling SLOs. These structures, known as tertiary lymphoid organs or ectopic lymphoid follicles (ELF), are locally inducible within chronically inflamed sites and contain the organizational, cellular, and molecular features found in SLO essential for propagating immune cell activation and antigen-driven selection, ultimately sustaining tissue-specific immune responses (13–16). Furthermore, similar to SLO, ELF contain B cell-rich infiltrates and are implicated in promoting focal B cell organization to aid activation, affinity maturation, differentiation, class-switch recombination, and local Ab production (13, 16, 17). ELF can develop in nearly every organ in the body during infection, autoimmunity, and tumorigenesis, although the degree to which ELF recapitulate lymphoid tissue-like organization varies (13, 16, 18). These structures can provide a permissive environment for cellular and humoral immune responses independent of primary or secondary lymphoid tissue.
ELFs are particularly prominent during chronic inflammation, yet mouse models of neuroinflammation examining CNS compartmentalized immune responses, including ELF formation, have focused on acute rather than chronic disease. Studies in murine models of CNS demyelinating diseases including mouse hepatitis virus (MHV) (19–23), Sindbis virus infection (24, 25), and experimental autoimmune encephalomyelitis (EAE) (18, 26–29) have highlighted the ability for the CNS to serve as a niche for persistent B cell accumulation. Critical B cell chemokines and trophic factors are constitutively expressed or upregulated during neuroinflammation, including trafficking chemokines, CXCL9/10 and CCL19/21, SLO-organizing chemokines CXCL13 and CXCL12, and survival factors and differentiation factors, including BAFF, APRIL, and IL-21 (19, 20, 22, 26, 27, 30–34). Both infiltrating immune cells and CNS-resident cells, including astrocytes, microglia, and endothelial cells, produce B cell-related chemoattractants and trophic factors in the CNS (19). However, the relative contribution of ELF in fostering CNS compartmentalized B cell responses during acute models of MS remains controversial. In adoptive transfer EAE models, ELFs have been noted within the meninges and express some characteristic lymphoid factors including IL-17, LTα, and chemokines involved in leukocyte homing to and within the lymph nodes such as CXCL13, CXCL9, and CCL19 (28, 35–38).
Conversely, MHV infection, as well as spontaneous B cell-dependent 2D2 IgHMOG MOG EAE, have noted the presence of multiple B cell differentiation phenotypes, including activated and proliferating B cells and lymphoid chemokines with minimal evidence for SLO-like ELF (23, 29). Collectively, studies in viral and autoimmune models emulating the acute phase of MS support the notion that prolonged B cell responses within the CNS can occur irrespective of ELF formation. Nonetheless, ELFs are classically induced during chronic inflammation and therefore may play a more critical role in chronic and progressive phases of the disease. Studies documenting meningeal inflammatory aggregates resembling some features of ELF were found primarily in secondary progressive MS (SPMS) post-mortem brain tissue (36, 37, 39), yet, mouse models of chronic and progressive demyelinating disease course have not evaluated contributions of ELF in promoting CNS compartmentalized immune responses. Thus, the contribution of ELF in promoting persistent B cell activity in the CNS during chronic and progressive phases of MS requires further evaluation.
The pathology of progressive MS is complex and recapitulating all facets of progressive disease in an individual mouse model has proven to be challenging. Nonetheless, Theiler's murine encephalomyelitis virus (TMEV)-induced demyelinating disease (TMEV-IDD) has been established as an accepted model of chronic MS, especially its progressive forms. Chronic TMEV infection of the CNS in susceptible strains of mice produces a progressive clinical disease course exemplified by accrued disability, including spastic paralysis. The model further parallels several hallmarks of progressive MS exhibiting both spinal cord and brain pathology, including demyelination, remyelination, atrophy, neuronal death, axonal injury, and persistent CNS inflammation (40–43). Furthermore, several studies document ItAb synthesis in the CNS during TMEV-IDD, similar to MS (44, 45). The phenotype of accumulating B cells and the role of ELF in supporting prolonged CNS compartmentalized inflammation and Ab production in TMEV-IDD has yet to be determined. Thus far, persistent accumulation of T cells and B cells (46) in the CNS during chronic TMEV-IDD and elevations in CSF Ab despite restored blood-brain barrier (BBB) integrity (46, 47) suggest CNS compartmentalized cellular and humoral immune responses may be fostered during chronic TMEV-IDD.
The present study sought to elucidate B cell phenotypes accumulating in the CNS, and the relative contribution of CNS compartmentalized immune responses, including SLO-like ELF formation, in sustaining B cell accumulation and Ab production in the CNS in TMEV-IDD. Overall, our results demonstrate that ongoing B cell responses and ItAb synthesis in the CNS during chronic TMEV occur independent of SLO-like ELF formation. Our findings broaden our view of the diverse factors involved in supporting CNS compartmentalized B cell responses during CNS inflammatory diseases, a crucial finding for identifying future therapeutic interventions targeting inflammatory aggregates.
Methods
Mice and Infection
Four to five-week-old female SJL mice were purchased from Jackson Laboratories (Bar Harbor, ME) and were intracranially infected with 10 ×106 plaque forming units (PFU) of TMEV (BeAn strain) in 30 μl final volume. All mice were housed under pathogen-free conditions at an accredited facility at Dartmouth College. The control sham-treated group received an intracranial injection with 30 μl of saline solution and were age-matched to TMEV-IDD mice for all experiments. All procedures were conducted under protocols approved by the Institutional Animal Care and Use Committee.
Sample Isolation
Whole blood, cervical lymph nodes (CLN), spinal cord, and CSF were obtained from each mouse at necropsy (n = 32 TMEV-IDD and n = 12 sham from 4 independent experiments). Blood (average 500 μl) was collected by intracardiac puncture and serum was isolated and stored at −80°C. CLN were either paraffin-embedded for immunofluorescence studies (n = 11 TMEV-IDD; n = 3 sham) or processed for flow cytometry (n = 11 TMEV-IDD; n = 3 sham). Spinal cords were divided, with 1/3 of tissue snap frozen and stored at −80°C for gene expression analyses and 2/3 of tissue either left in the spinal column, with vertebrae intact for paraffin-embedding (n = 8 TMEV-IDD; n = 6 sham) or flushed from the spinal column to process for flow cytometry (n = 24 TMEV-IDD; n = 6) as noted below. CSF was collected by cisternal tap as previously described (48). Briefly, the meninges overlaying the cisterna magna were exposed, the surrounding area was gently cleaned to remove any contaminating blood, and a 30 gauge needle was used to puncture the arachnoid membrane. CSF was collected using a glass capillary tube (average 8–10 μl), centrifuged to remove cells, diluted 1:3 in PBS, and stored at −80°C.
RNA Preparation and Real-Time Quantitative Reverse Transcription (RT-PCR)
RNA was extracted from spinal cords using TRIzol (Invitrogen, Foster City, CA). RNA was reverse transcribed using the qScript cDNA Supermix kit (Quanta-Biosciences, Gaithersburg, MD). cDNA was then utilized as the template for real-time RT-PCR based on the 5′ nuclease assay, using the PerfeCTa qPCR FastMix II ROX (Quanta-Biosciences, Gaithersburg, MD). Custom primers and probes were used to detect TMEV mRNA (45), and TaqMan real-time PCR assays (Life Technologies, Grand Island, NY) were used as the primers and probes for all other target genes, including mouse glyceraldehyde phosphate dehydrogenase (GAPDH), the reference gene. TMEV mRNA was assessed by absolute quantification using a standard curve of TMEV plasmids amplified at known concentrations. For the present study, only TMEV positive mice were included in our data analysis. All other targets were analyzed as relative mRNA expression levels calculated by using both the 2−ΔCt method where ΔCt = Cttarget−CtGAPDH, and the 2−ΔΔCt methods where ΔΔCt = ΔCtTMEV−IDD−ΔCtSham (49).
Luminex® Bead-Based Multiplex Assay
Immunoglobulins (Ig) and cytokine levels in the CSF and serum were measured using Luminex® Bead-based Multiplex Assay. The MilliPlex MAP Mouse Immunoglobulin Isotyping Magnetic Bead Panel (EMD Millipore, Burlington, MA) was used to quantify levels of IgA, IgG1, IgG2a, IgG2b, IgG3, and IgM. Likewise, the Bio-Plex Pro Mouse Chemokine Panel 33-plex assay (Bio-Rad, Cambridge, MA) was used to quantify 33 chemokines, including B cell-related chemokines CXCL13, CCL19, CXCL10, IL-6, and CXCL12. Mouse CXCL9 and IL-21 were measured using Luminex® singleplex assays available from Bio-Rad. In order to compare intrathecal production of Igs and chemokines, CSF analytes were normalized to serum analytes by calculating a CSF/serum ratio, which accounts for individual variability in serum concentrations (50, 51).
Cell Isolation and Flow Cytometry
For B cell analysis via flow cytometry, CLN cells or spinal cord-derived mononuclear cells were isolated from individual TMEV infected or sham mice as previously described (23). Briefly, spinal cords were minced and digested in RPMI supplemented with 10% fetal calf serum (FCS), collagenase type I (1 mg/ml; Worthington Biochemical Corporation, Lakewood, NJ) and DNase I (100 U/ml) (Worthington Biochemical Corporation, Lakewood, NJ). Incubation was followed by the addition of 0.1M EDTA to terminate collagenase activity. Cell pellets were resuspended in RPMI medium, adjusted to 30% Percoll (Pharmacia, Piscataway, NJ), and underlaid with 1 ml of 70% Percoll. Cells were recovered from the 30/70% Percoll interface. Prior to staining, cells were incubated with fixable viability stain 780 APC-Cy7 (BD Biosciences, San Jose, CA). Cells were then incubated with FACS buffer supplemented with 1% mouse serum and rat anti-mouse FcγIII/II mAb (2.4G2; BD Bioscience, San Jose, CA) to prevent non-specific staining. Expression of B cell phenotype and activation surface markers was assessed by staining CLN or CNS cells with Ab specific for CD45 (30-F11; PerCP-Cy5.5), CD19 (1D3; PE-CF594), GL7 (FITC), IgD (11-26; APC), IgM (eB131-15F9; PE), CD138 (281-2; PE), IgG2a/2b (R2-40; FITC) IgG1 (A85-1; FITC), MHCII IAq (KH116; BV421), CD80 (16-10A1; PE) (BD Biosciences, San Jose, CA) (Table 1). Cells were analyzed using a Beckmann Coulter Gallios flow cytometer (BD Biosciences, San Jose, CA) and FlowJo (version 9.7.6) software (Tree Star, Ashland, OR). Voltages for the cytometer's detectors were set based on B cell surface marker expression on B cells isolated from CLN and the same voltages were utilized for analyzing CNS-infiltrating B cells. Cell numbers were calculated based on live cell yields and percentages of gated live cells. Dead cells were excluded using fixable viability stain and comprised <10% of isolated cells. Doublets were excluded based on FSC-Area and FSC-Height.
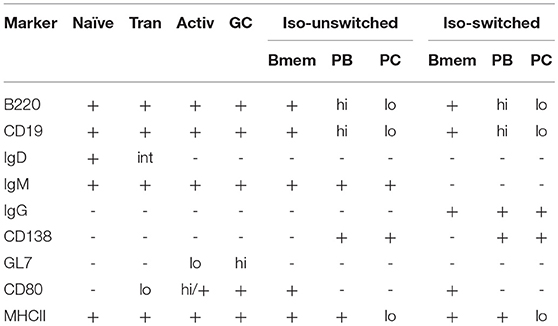
Table 1. Expression of phenotypic and activation markers on B cells, including naïve, transitional (Tran), activated (Activ), isotype-unswitched (Iso-unswitched), and isotype-switched (Iso-switched) subpopulations (52–60).
Immunohistochemistry
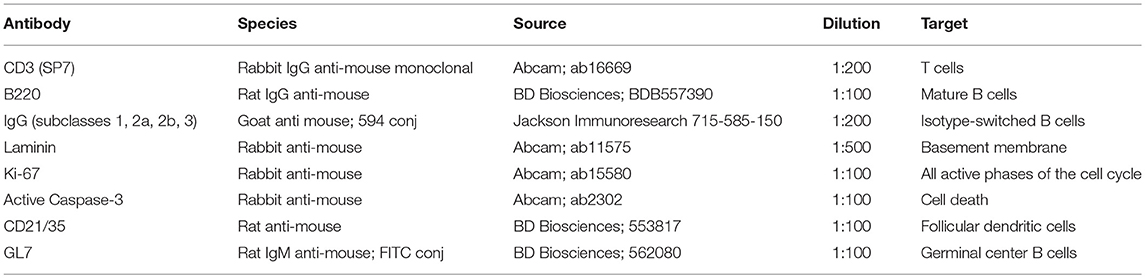
Spinal cords and CLN from PBS-perfused, TMEV-IDD and sham mice were fixed in 10% neutral buffered formalin for 24 h. To retain the meninges, the vertebral column was isolated, trimmed, and spinal cords with intact vertebrae were segmented into cervical, upper thoracic, and lower thoracic regions and embedded in paraffin. Tissue blocks were then surface decalcified using Nitrical nitric acid bone decalcifier (StatLab medical products, McKinney, TX) for 5-10 min, washed, and cut in sections of 4 μm thickness. Deparaffinization and rehydration steps were performed using two xylene 10-min washes followed by two 10-min 100% ethanol washes, and sequential 5-min washes of 95% ethanol, 70% ethanol, and 50% ethanol. Following PBS washes, antigen retrieval was performed at 95°C for 20 min in Tris-EDTA 0.1% Tween-20 buffer followed by cooling. Sections were washed in PBS and blocked with 5% bovine serum albumin and 10% goat serum for 1 h. After blocking, spinal cord sections were incubated with primary antibodies (Table 2). B220 and CD21/35 primary Abs were detected using secondary Ab Alexa Fluor 594 goat anti-rat IgG (Abcam), and caspase-3, Ki-67, CD3, and laminin primary Abs were detected using secondary Ab Alexa Flour 488 goat anti-rabbit IgG (Life Technologies, Grand Island, NY). Sections were mounted with Vectashield Hardset reagent with 4′,6-diamidino-2-phenylindole (DAPI) (Vector Labs, Burlingame, Ca) and examined using a Zeiss LSM 800 confocal microscope with Airyscan (Zeiss, Oberkochen, Germany). Z-series images were collected every 0.2 μm covering a tissue depth of 2-3 μm. Projected images were compiled using Image J software (NIH, http://rsbweb.nih.gov/ij) supplemented with the FIJI plugin set (https://imagej.net/Fiji). Images were assembled for publication and scale bars added using Adobe Photoshop 7.0 software (Adobe Systems, San Jose CA).
All TMEV-IDD mice (n = 8) in our current immunofluorescence studies exhibited CNS inflammatory aggregates, albeit to varying degrees, with no detectable immune cell infiltration in sham-treated mice (n = 6).
Statistical Analyses
Data generated in the present study were analyzed via Prism (version 6.0) software (GraphPad, San Diego CA). Data sets were assessed using a Pearson normality test to determine significant deviations from a normal distribution. Based on the normality results, the parametric Student's t-test or non-parametric Mann-Whitney U test was utilized to compare groups. The exact test used to perform analyses for each data set are denoted within the corresponding figure legend. In all cases, a P < 0.05 was considered significant.
Results
Isotype-Switched B Cells Predominate in the Spinal Cord and Associate With Intrathecal Antibody Synthesis During Chronic TMEV-IDD
To assess B cell phenotypes within the CNS of TMEV-IDD mice, flow cytometry was utilized to examine infiltrating B cells in spinal cords, i.e., a prominent site of inflammation, demyelination, and viral persistence during chronic TMEV infection (41, 46). CD45, a pan immune cell marker, was used to identify infiltrating immune cells in sham and TMEV-IDD spinal cords (Figure 1A; P1). Results revealed prominent CD45hi immune cell infiltration in TMEV-IDD spinal cords (mean number = 50,841; SEM ± 5390), while sham-treated mice showed minimal presence of CD45hi-expressing cells (mean number = 887; SEM ± 257) (Figure 1B). Further analysis of infiltrating CD19+ B cells within total CD45hi cells (Figures 1C,D; P2) revealed 15% (mean = 7,481 cells) were CD45hiCD19+ in TMEV-IDD spinal cords, while CD45hiCD19+ B cells were scarce in sham-treated mice (~15–20 cells; data not shown).
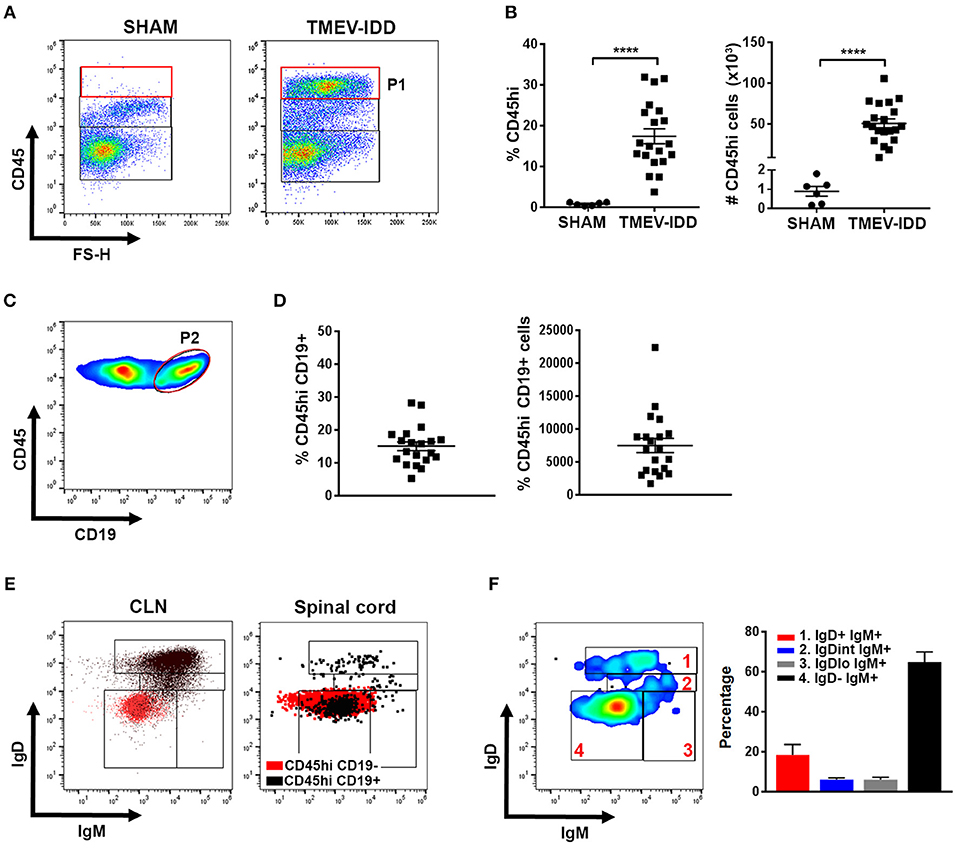
Figure 1. Isotype-switched B cells comprise the majority of spinal cord-infiltrating B cells during chronic TMEV-IDD. (A) Representative gating strategy for CD45hi (P1) cells in the spinal cords of sham-treated or TMEV-infected mice at day 130 post-injection showing minimal CD45hi cells in sham mice. (B) Scatter plots display CD45hi percentage (left) and numbers (right) among total live cells in individual sham or TMEV-IDD mice with mean ± SEM for the group. (C) Representative gating strategy for CD45hi CD19+ B cells (P2) and (D) scatter plots displaying CD19+ B cell percentages (left) and numbers (right) within CD45hi cells for individual TMEV-IDD mice and mean ± SEM for the group. (E) Dot plots show IgD and IgM subpopulations in the CLN (left) and TMEV-IDD spinal cord (right). Gating strategy for B cell subpopulations was initially established in the CLN and then applied to spinal cord-infiltrating B cells. CD45hi CD19− non-B cell immune cells (red) were used as a negative control in both tissues. CD45hi CD19+ B cells (black) were overlaid to confirm gating of IgD−IgM− isotype-switched B cells. (F) Smoothed representative gating strategy (left) and mean percentages ± SEM (right) of B cell phenotypes among CD45hi CD19+ spinal cord-infiltrating B cells identifying IgD and IgM subsets including (1) IgD+IgM+ naïve/early activated, (2) IgDintIgM+ activated, (3) IgD−IgM+ transitional/isotype-unswitched memory/ASC, and (4) IgD−IgM− isotype-switched memory/ASC B cells. Data are representative of three independent experiments with individual TMEV-IDD (n = 6 to 8) or sham (n = 2) mice per experiment. Significant differences between sham and TMEV mice are indicated by ****(p < 0.0001) as determined by an unpaired Student's t-test.
To define the differentiation state of spinal-cord infiltrating B cells, we identified naïve/early activated, activated, transitional or isotype-unswitched ASC/Bmem, isotype-switched ASC/Bmem utilizing IgD and IgM surface expression (Table 1; Figure 1E) (21, 23, 24). B cell immunophenotyping in chronic TMEV-IDD identified the majority of B cells (~60%) as being IgD−IgM− isotype-switched ASC/Bmem phenotype (Figure 1F).
These data, combined with previous results showing pronounced ItAb in the CNS during chronic TMEV-IDD (45), led us to further characterize differentiated B cells accumulating during chronic disease. We examined the canonical surface marker of ASC, CD138 (Table 1). Initial analysis revealed that CD138+ cells expressed moderate levels of CD19 and 20% of total CD19+ cells expressed CD138 (Figure 2A). Among CD19+ CD138+ ASC, an average of 40% were class-switched IgG+ (Figure 2B) and the majority of CD19+ CD138+ ASC expressed MHCII (~90%; Figure 2C), suggesting a plasmablast phenotype.
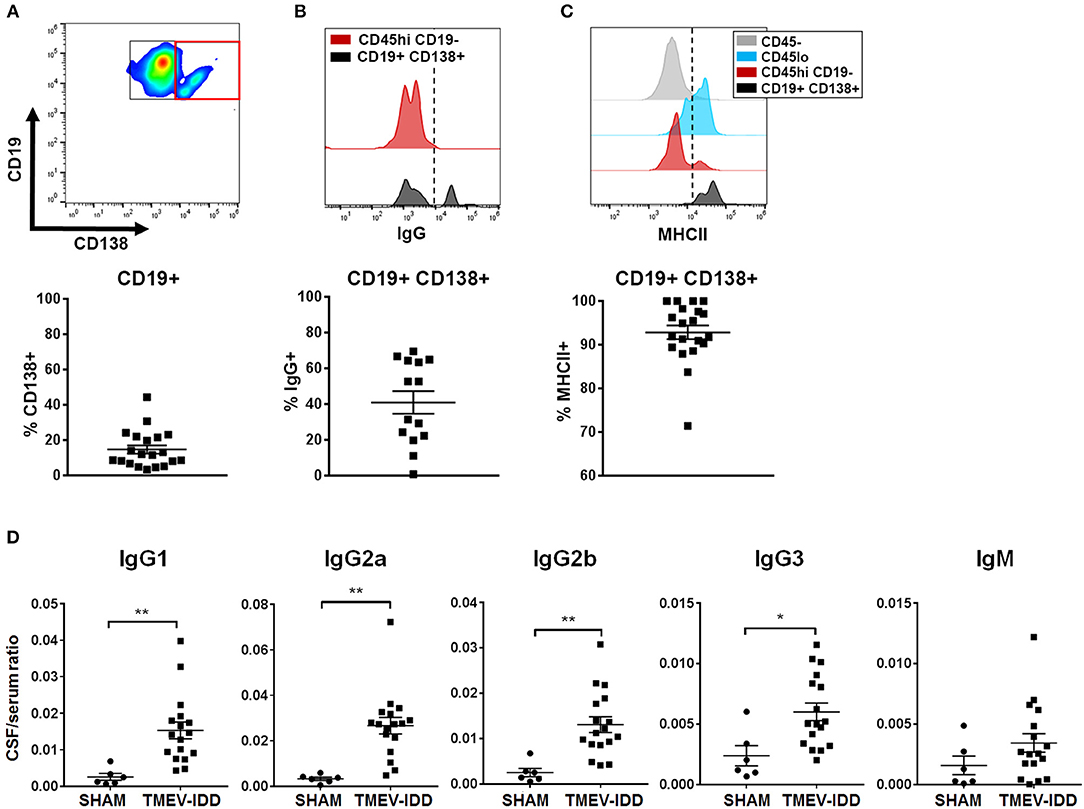
Figure 2. Accumulation of isotype-switched B cells, including ASC, coincides with intrathecal Ig synthesis. (A) Gating strategy for CD138+ ASC (red box) among CD45hi CD19+ cells (top). Scatter plots showing total percentage of CD19+ CD138+ cells (bottom) among CD45hi CD19+ B cells for individual TMEV-IDD spinal cords at day 130 p.i. with mean ± SEM for the group. (B) Histogram (top) depicts IgG expression among CD45hi CD19− non-B cell immune cells as a negative control (red) and IgG expression among CD19+ CD138+ ASC (black). Dotted line denotes cutoff for positive gating of IgG+ cells. Total percentage of IgG+ cells (bottom) among CD45hi CD19+ CD138+ B cells for individual mice and mean ± SEM for the group. (C) Histogram (top) shows MHCII expression among CD45− (astrocytes/neurons/oligodendrocytes; gray) as a negative control, with elevated expression among CD45lo microglia (blue) and CD45hi CD19− non-B cell infiltrating immune cells (red) serving as positive controls. Histogram highlights elevated MHCII expression among CD19+ CD138+ cells (black). Dotted line denotes cutoff for positive gating of MHCII+ cells. Total percentage of MHCII+ (bottom) among CD45hi CD19+ CD138+ B cells for individual mice and mean ± SEM for the group. (D) Immunoglobulin (Ig) protein levels were measured in individual CSF and serum samples using Luminex® technology to measure 6 Ig isotypes simultaneously. Scatter plots depict individual CSF/serum ratios of IgG1, IgG2a, IgG2b, IgG3, or IgM and mean ± SEM for the group. IgA showed no significant differences in CSF/serum ratio compared to sham. Significant differences between sham and TMEV mice are indicated by *(p < 0.05) or **(p < 0.01) as determined by an unpaired Student's t-test.
Flow cytometric analysis revealed the majority of CNS-infiltrating B cells were comprised of an isotype-switched B cell phenotype and previous studies have documented elevated CSF Ab in TMEV-IDD (45). These findings led us to investigate if the presence of differentiated B cells associated with ItAb production in the CSF. Immunoglobulin protein levels were measured in the serum and CSF of sham-treated and chronic TMEV-IDD mice. IgG1 (p = 0.004), IgG2a (p = 0.001), IgG2b (p = 0.002), and IgG3 (p = 0.01) all displayed significantly elevated CSF/serum ratios during chronic TMEV-IDD (Figure 2D) relative to sham-treated mice. IgM CSF/serum ratio was also moderately elevated in TMEV-IDD compared to sham-treated mice but did not reach statistical significance. Elevated CSF/serum IgG isotype ratios indicated CSF IgG was intrathecally produced as BBB integrity is restored during early disease onset (41, 47), indicating that passive transfer of Ab from the serum was unlikely.
Aggregation of Activated and Isotype-Switched B Cells Occurs in Meningeal and Perivascular Spaces
Diverse B cell differentiation phenotypes in TMEV-IDD spinal cords, including extensive isotype-switched ASC/Bmem accumulation and ItAb synthesis in chronic TMEV-IDD, provided evidence for CNS-compartmentalized B cell responses, a phenomenon suggested to occur during the chronic and progressive phases of MS (66).
To determine B cell localization and possible ELF involvement in fostering immune responses during chronic TMEV-IDD, we examined B cells in spinal cords with intact meninges by immunofluorescent staining. We utilized the basement membrane component laminin to delineate vasculature and the meninges, common sites for immune cell aggregation in the CNS (18, 29, 36, 39). B cells were identified via B220, a surface marker present on immature and mature B cells but downregulated on terminally differentiated plasma cells, whereas IgG was utilized to identify isotype-switched B cells, including IgG expressing ASC and Bmem (Table 1). Analysis of B220+ (Figure 3A) or IgG+ (Figure 3B) cell localization relative to laminin in cervical and thoracic spinal cord sections showed prominent B cell infiltration in parenchymal, meningeal, and perivascular spaces. Both B220+ and IgG+ B cells accumulated proximal to the central canal, within the ventral midline vasculature, with evident perivascular cuffing, and frequent aggregation in the ventral lateral meninges, analogous to leptomeningeal immune cell aggregations present in MS patients.
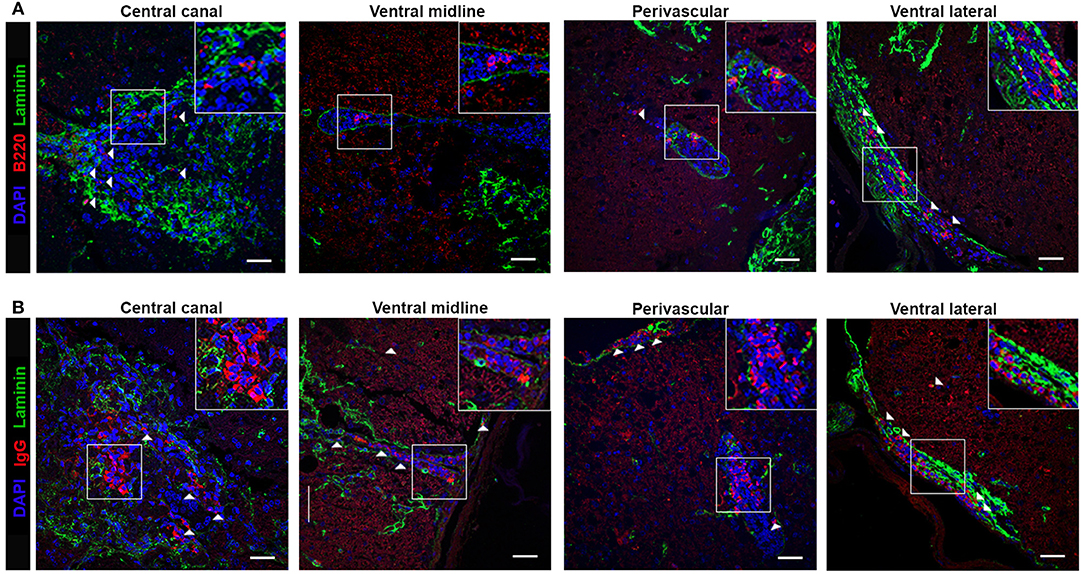
Figure 3. B cell localize to the meninges, perivascular, and parenchymal space in the Theiler's infected spinal cord. TMEV-infected decalcified spinal cords at day 130 p.i. were stained with DAPI (blue), laminin (green) and (A) B220 or (B) IgG (red) as indicated. Representative z stack projected compilations revealing B cell aggregation in the central canal, ventral midline, perivascular space, and ventral lateral meninges. White boxes indicate region selected for cropped insets in each image. White arrows highlight positive B220 or IgG staining as indicated. Z stack compilations represented 6-8 TMEV-infected mice. Scale bar = 50μm.
CNS Inflammatory Aggregates Accumulate in the Absence of Ectopic Lymphoid Follicle Markers
B220+ and IgG+ B cell accumulation within perivascular and meningeal regions led us to examine if these cellular aggregations displayed hallmark SLO-like ELF characteristics, including evidence for spatial organization, differentiation, and antigen-driven selection. Spatial organization was examined by identifying the localization of T cells, key immune cells involved in initiating and organizing ELF formation and in promoting antigen-driven B cell activation, differentiation, and survival (16, 35, 67). Immunofluorescent staining of CD3+ T cells and IgG+ B cells demonstrated T cells aggregated with isotype-switched IgG+ B cells in the ventral lateral meninges and ventral midline, although there was no evidence for a discrete organization of T and B cell regions, a characteristic feature of SLO and SLO-like ELF (Figure 4A). To evaluate B cell differentiation within the CNS compartment, we simultaneously assessed B220 and IgG B cell localization and aggregation within perivascular and meningeal cellular infiltrates. Co-staining of B220 and IgG within spinal cord sections revealed frequent localization of B220+, IgG+, and occasional B220+ IgG+ double-positive cells within cell aggregations in the ventral lateral meninges and ventral midline vasculature with minimal evidence of organized clusters containing IgG and B220-expressing B cells (Figure 4A).
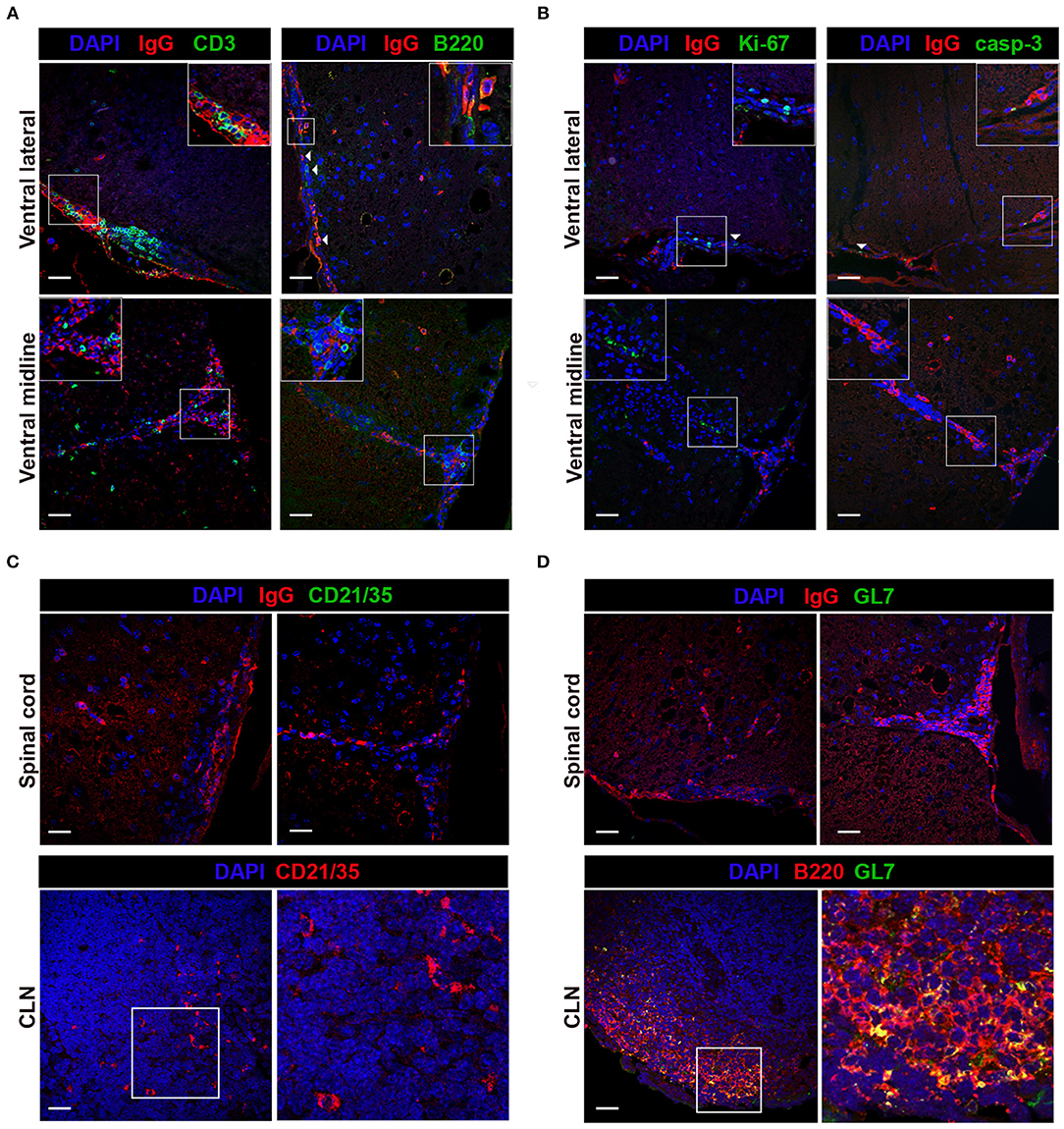
Figure 4. Immune cells aggregate in TMEV-IDD spinal cords in absence of conventional SLO-like ELF features. TMEV-infected decalcified spinal cords from day 130 p.i. were stained and assessed for markers of SLO-like ELF. Spinal cord representative images depict the ventral meninges and the ventral midline, regions with frequent cellular aggregation. White boxes indicate the region cropped for insets within each image. (A) CD3 or B220 (green), IgG (red), and DAPI (blue) were co-stained to assess co-aggregation and organization of infiltrating immune cells. Inset within IgG B220 costained representative shows B220+IgG+ double-positive B cell. (B) Ki-67 or caspase-3 (green), IgG (red) and DAPI (blue) were used to determine cell proliferation and cell death in B cell-containing aggregates. White arrows indicate presence of Ki-67 or caspase-3 immunoreactivity. (C) FDC presence in cellular aggregates was assessed in TMEV-IDD spinal cords using CD21/35 (green) with DAPI (blue) and IgG (red). As a positive control, CD21/35 immunostaining was confirmed at day 27 p.i. in CLN tissue using CD21/35 (red) and DAPI (blue). White box within CLN indicates the region cropped for the representative image (right) highlighting CD21/35+ cells. (D) GC B cells in TMEV-IDD spinal cords were identified using GL7 (green), with IgG (red), and DAPI (blue). CLN tissue (day 27 p.i.) was used to verify GL7 (green) immunostaining with B220 (red) and DAPI (blue). White box with CLN indicates the region cropped for the representative image (right) highlighting GL7+ B220+ (yellow) GC B cells. Representative z stack compilations from 6-8 mice. Scale bar = 50μm.
Although cellular organization akin to SLO was not evident, simultaneous accumulation of T cells with immature, mature, and isotype-switched B cells in meningeal and perivascular spaces of TMEV-IDD spinal cords was apparent. To further evaluate whether immune cell aggregates expressed common features of SLO-like ELF, we utilized markers of proliferation and cell death, events indicative of ongoing antigen-driven selection within germinal center (GC) reactions in SLO and ELF (13, 16). Proliferation was assessed using Ki-67, and active caspase-3 was used as an indicator of apoptosis (Table 2). Immunohistochemical analysis of Ki-67 and caspase-3 expression revealed sparse immunoreactivity within meningeal and perivascular cellular aggregates containing IgG+ cells (Figure 4B). Further evaluation of perivascular and meningeal aggregates also showed the absence of the follicular dendritic cell (FDC) marker CD21/35, indicating an absence of FDCs, a specialized stromal cell in SLO and ELF involved in trapping and presenting antigen in B cell follicles and in secretion of B cell supporting chemokines, adhesion molecules, and trophic factors (Table 2; Figure 4C) (14). To evaluate GC formation, structures essential for antigen-driven selection, somatic hypermutation, and isotype switching, we utilized GL7, a surface antigen present at high levels on GC B cells during GC reactions in SLO and ELF (Table 2) (52, 68). Immunostaining of TMEV-IDD spinal cords showed no evidence of GL7 immunopositivity within immune cell aggregates containing IgG+ B cells (Figure 4D). Thus, histological assessment revealed minimal evidence for SLO-like ELF formation.
CNS Infiltrating B Cells Display Elevations in Activation Markers During Chronic TMEV-IDD
The absence of detectable GL7 expression in spinal cords by IHC was further confirmed by flow cytometry, a more sensitive method of detection. Flow cytometry showed minimal evidence for spinal cord-infiltrating B cells expressing a conventional GL7hi GC phenotype as depicted in the TMEV-IDD CLN (Figures 5A,B; red box), though modest GL7 expression was observed on a small proportion of spinal cord-infiltrating B cells (Figures 5B,C). Among GL7-expressing B cells in the spinal cord, the mean fluorescence intensity (MFI) was comparable to GL7int “activated” B cells (Table 1) in the CLN (Figures 5A,B) (23, 52, 68). GL7 expression analysis identified 10% or approximately 800 infiltrating B cells as expressing an “activated” GL7int phenotype, with minimal detection of GL7hi or “GC” phenotype infiltrating cells (Figure 5D). In TMEV-IDD spinal cords, GL7int B cells were mostly IgD−, confirming that the GL7 expression was elevated on activated/transitional B cells (data not shown). Observed GL7int expression among a proportion of B cells within the spinal cord implied these B cells had acquired an activated phenotype.
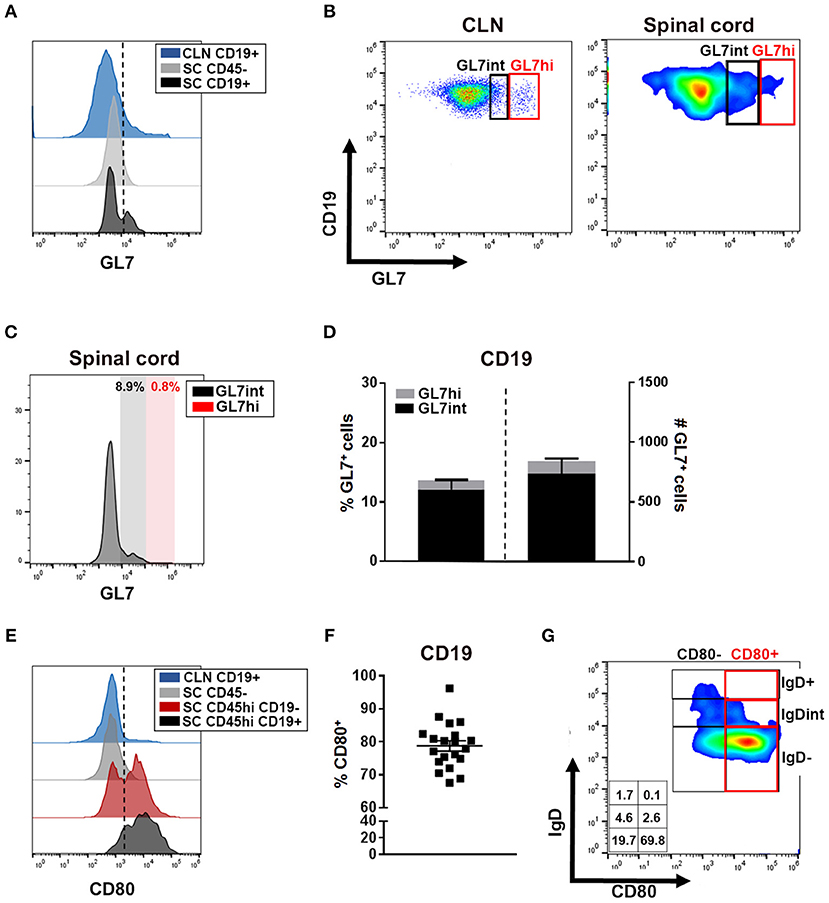
Figure 5. CNS-infiltrating B cells express markers of activation. (A) Histogram depicts GL7 expression on CLN CD19+ cells (blue) as a positive control, spinal cord CD45− (astrocytes/neurons/oligodendrocytes; gray) as a negative control, and spinal cord-infiltrating CD19+ B cells (black). Dotted line delineates cutoff for positive gating of GL7. (B) Representative dot plot of GL7 expression with CLN (day 27 post infection -p.i.-) demarcating activated/pre-GC GL7int (black box) and germinal center (GC) phenotype GL7hi (red box) with the same gating applied to the representative smoothed dot plot of B cells isolated from the chronic TMEV-IDD spinal cord. (C) Histograms of the mean fluorescence intensity (MFI) of GL7 expressing cells among spinal cord CD45hi CD19+ cells identifying minimal GL7int (black) activated B cells and GL7hi (red) GC phenotype B cells. (D) Stacked bar graphs display the mean ± SEM percentage (left bar and axis) and mean ± SEM number (right bar and axis) of GL7int and GL7hi B cells within total CD45hi CD19+ GL7+ spinal cord infiltrating B cells. (E) Histogram depicts CD80 expression among CLN CD19+ B cells (blue) at day 27 p.i. showing restricted CD80+ cells, CD45− (astrocyte/neurons/oligodendrocyte; gray) expression as a negative control, and CD45hi CD19− non-B cell immune cells (red) expression as a positive control. CD80 expression on CD45hi CD19+ spinal cord infiltrating B cells is depicted in black. Dotted line delineates cutoff for gating CD80 positive cells. (F) CD45hi CD19+ B cells and scatter plot showing individual percentages and group mean ± SEM of CD19+ CD80+ B cells within the spinal cord at day 130 p.i. (G) Representative dot plot showing expression of CD80+ predominately on IgD− B cells in the spinal cord with corresponding percentages for each population indicated within the table inset. Results represent three independent experiments with 6 to 8 mice per experiment.
Following stimulation, including CD4 T cell-dependent help, GL7 surface expression is moderately upregulated, increasing B cell activation and functional capabilities, including antigen presentation (52, 53). To further assess B cell activation, we quantified the expression of CD80, a costimulatory molecule upregulated following stimulation by factors including BCR or CD40 engagement which, upon signaling, enhances B cell survival, proliferation, and differentiation (54, 69). Analysis of CD80 surface expression among CD45hi CD19+ B cells revealed expression on approximately 80% of infiltrating B cells (Figures 5E,F). Among CD80+ B cells, the majority were an IgD− phenotype, indicating CD80+ B cells include highly-activated, transitional, and/or Bmem phenotypes (Figure 5G). Extensive CD80 expression on CNS-infiltrating B cells suggested most B cells accumulating during chronic TMEV-IDD display an activated phenotype possibly driven by antigen and CD4 T cell-mediated help (54, 70, 71), or perhaps, by polyclonal activation (72).
Elevated Transcript and Protein Levels of B Cell Trafficking Chemokines and Factors Supporting Differentiation and Survival
Heterogeneous B cell differentiation phenotypes, including B cells displaying an activated phenotype, accumulating in parallel with intrathecal Ab synthesis led us to examine if cellular aggregations were still associated with factors involved in fostering B cell activation, survival, and accumulation in the absence of SLO-like ELF. Therefore, we evaluated whether factors promoting B cell trafficking, differentiation, and survival were present in chronic TMEV-IDD. IL-21 is a cytokine produced predominately by CD4 T cells and is essential for promoting B cell survival and differentiation (73, 74). Additionally, IL-6, BAFF, and APRIL are cytokines which play crucial roles in B cell survival and differentiation and are primarily produced by astrocytes within the CNS during viral infection and autoimmunity (19, 75–77). Gene expression analysis of these factors revealed constitutive expression of IL-21, IL-6, BAFF, and APRIL within spinal cords (Figure 6A), consistent with previous studies highlighting basal expression within CNS tissue (21, 25).
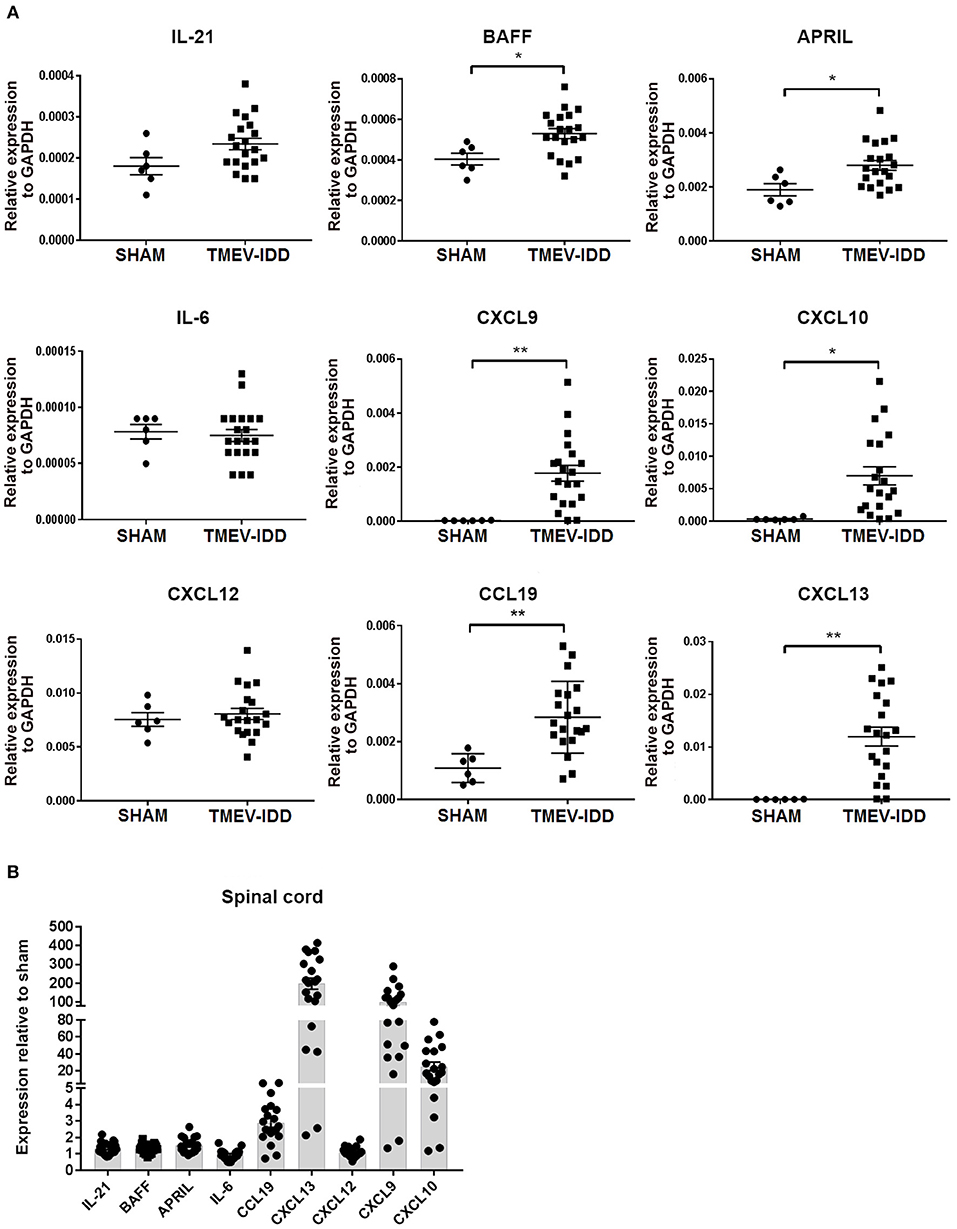
Figure 6. B cell accumulation in the central nervous system coincides with enhanced transcript expression of B-cell supportive chemokines. (A) Relative gene expression levels for IL-21, BAFF, APRIL, IL-6, CXCL9, CXCL10, CXCL12, CCL19, CXCL13 in the spinal cords of individual sham and chronic TMEV-infected mice (day 130 post-infection -p.i-), normalized by the housekeeping gene glyceraldehyde phosphate dehydrogenase (GAPDH) with mean ± SEM plotted for each group. (B) Scatter plot showing fold-changes in cytokine and chemokine expression compared to sham mice. The fold-change was obtained by normalizing the gene expression number to those of GAPDH, then comparing the samples to the average GAPDH-normalized gene expression in sham mice. Data depict the means ± SEM for at least 6 to 8 individual infected mice and 2 sham mice derived from three independent experiments. Significant differences between sham and infected are indicated (*P < 0.05; **P < 0.005). In all cases, a P value of <0.05 was considered significant, as determined by an unpaired Student's t-test or Mann-Whitney U-test.
Further comparisons among sham-treated and TMEV-IDD mice showed modest upregulation of IL-21 (p = 0.06), with significant upregulation of APRIL (p = 0.01) and BAFF (p = 0.01) transcripts. IL-6 transcript levels were comparable to sham-treated mice (Figure 6A). Overall, IL-21, APRIL, and BAFF mRNA levels in TMEV-IDD spinal cords were twice as high as shams (Figure 6B).
CXCL9, CXCL10, CCL19, CXCL12, and CXCL13 are important for B-cell related trafficking and organization and are known to be produced in the CNS (21, 27, 32, 78). Analysis of chemokine transcript levels revealed constitutive expression of CCL19 and CXCL12 in spinal cords (Figure 6A), confirming previous results of basal expression in the CNS (21, 25). In TMEV-IDD mice, CXCL9 (p = 0.003), CXCL10 (p = 0.01), CCL19 (p = 0.002), and CXCL13 (p = 0.001) mRNA levels were all significantly increased compared to shams. Overall, CXCL13 showed a ~200-fold increase in expression, CXCL9 a ~100-fold increase in expression and CXCL10 a ~20-fold increase in expression, when TMEV-IDD spinal cords were compared to sham spinal cords (Figure 6B). Conversely, CXCL12 mRNA levels were similar in the spinal cords of TMEV-IDD and sham-treated mice.
To confirm that elevated transcript levels resulted in enhanced protein levels in the CNS compartment, we also evaluated protein levels in CSF from sham-treated and TMEV-IDD mice for all analytes previously tested by gene expression available for quantification by Luminex magnetic bead assay. Assessment of B cell differentiation and supporting factors, IL-21 and IL-6, confirmed previous gene expression results with minimal alterations in protein levels for both analytes in TMEV-IDD CSF samples compared to controls and overall low expression ranging from 10-30 pg/ml (Figure 7A). Further analysis of B cell trafficking and organizing chemokine protein levels revealed a modest increase in CXCL9 production in TMEV-IDD CSF (mean = 227 pg/ml) compared to sham (mean = 143 pg/ml), although results did not approach significance. Similarly, CXCL12 showed elevated production in TMEV-IDD (mean = 352 pg/ml) compared to sham (mean = 193 pg/ml) although results were only near to significance (p = 0.09). CCL19 and CXCL13 both displayed significantly increased expression, with CCL19 reaching an average of 689 pg/ml in TMEV-IDD CSF compared to sham levels of approximately 319 pg/ml (p = 0.02) and CXCL13 exhibiting the highest protein levels of CXCL13 at an average of 7820 pg/ml compared to sham levels of 2249 pg/ml (p = 0.01), coinciding with high upregulation in transcript levels detected by gene expression analysis in TMEV-IDD spinal cords. Conversely, CXCL10 protein levels in sham and TMEV-IDD spinal cords diverged from transcript levels, with comparable CXCL10 protein levels in sham and TMEV-IDD CSF (~1600 pg/mL), though CXCL10 protein levels in the CSF showed the second highest expression among all analytes assessed.
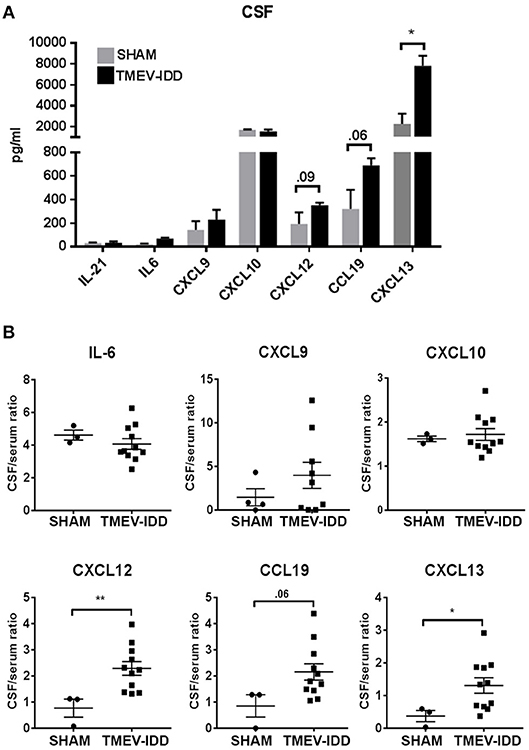
Figure 7. Selective B cell supportive chemokines are elevated in the cerebrospinal fluid during chronic TMEV-IDD. (A) Cerebrospinal fluid (CSF) levels (in pg/mL) and (B) CSF/serum ratios of B cell supporting cytokines in sham (n = 3–4) and chronic TMEV-infected mice (n = 11–12) at day 130 post-infection (p.i.) collected from three independent experiments. CSF levels are expressed as mean ± SEM for each group and CSF/serum ratios are expressed as dot plots showing individual values and the mean ± SEM for each group. Significant differences between sham and TMEV-infected mice are indicated (*P < 0.05; **P< 0.005) as determined by an unpaired Student's t-test.
B Cell Trafficking and Organizing Chemokines Are Intrathecally Produced During Chronic TMEV-IDD
The above data showing elevated transcript and protein levels of B cell trafficking and organizing chemokines led us to examine if these factors were also intrathecally produced. To assess if elevation in intrathecal production of B cell-related chemo-attractants and trophic factors was evident in the CSF of TMEV-IDD mice, we evaluated protein levels of B cell chemokines in the CSF and serum to establish a CSF/serum ratio. Results revealed moderately increased CXCL9 and CCL19 (p = 0.06) CSF/serum ratios, with significant increases in CSF/serum ratios among CXCL13 (p = 0.03) and CXCL12 (p = 0.005) in TMEV-IDD compared to sham-treated mice (Figure 7B). IL-6 and CXCL10 CSF/serum ratios revealed minimal alterations in chronic TMEV-IDD compared to sham-treated CSF samples. Elevated CSF/serum ratios in the absence of detectable increased BBB permeability [(47); manuscript submitted] suggest intrathecal production is responsible for the observed elevations in B cell trafficking chemokines in the CNS compartment.
Discussion
Our experiments focused on elucidating B cell phenotypes and factors promoting persistent accumulation in the CNS compartment during chronic TMEV-IDD. Previous studies have shown a prominent involvement of the B cells in the CNS compartment during TMEV-IDD, including robust ItAb production and IgG+ and CD138+ B cells localizing to the CNS (47). Further studies highlighting upregulated B-cell immune response pathway genes in the deep CLN during the early phase of the disease and a relative downregulation during chronic TMEV-IDD also suggested a shift in B cell inflammation from the peripheral to CNS compartment during chronic TMEV-IDD (79). The spectrum of B cells involved in the CNS and their localization and organization within the CNS has not been extensively investigated and served as the foci of this work.
Our studies demonstrate that within the CNS during the progressive phase of TMEV-IDD, activated and isotype-switched B cells, including Bmem and ASC preferentially accumulate relative to other B cell phenotypes. These cells likely underlie the observed elevations in CSF Ab production, including ItAb of IgG1, IgG2a, IgG2b, IgG3. Present results revealed that the majority of CD138+ ASC express MHCII, indicating a plasmablast phenotype. Our data, as well as previous studies in TMEV-IDD (47), have shown that few CD138+ ASCs display Ki-67 immunopositivity, suggesting their proliferative capacity is decreased (47). It is possible that our flow cytometry results are skewed toward detecting a plasmablast phenotype B cell due to downregulation of CD19 on terminally differentiated plasma cells (80). However, other viral-induced encephalitic models such as MHV and Sindbis have confirmed high percentages of CNS-infiltrating CD138+ MHCII “plasmablast” phenotype B cells in the inflamed CNS (24). Since most classical plasmablasts continue to proliferate and thus are Ki-67+, the isotype-switched B cells exhibiting a CD19hi, CD138+, MHCII+ Ki-67− phenotype in our experiments may represent a B cell phase intermediate between a plasmablast and terminally differentiated plasma cells, such as the early differentiated CD138hi MHCII+ plasma cell phenotype previously documented in kidney inflammation and observed following sheep red blood cell immunization (80, 81). MHCII-expressing ASCs have essential implications during neuroinflammation as MHCII+ ASC can not only sustain Ab production but can also engage in antigen presentation to potentiate local immune responses.
B cell phenotyping in chronic TMEV-IDD spinal cords also highlighted extensive CD80+ B cell infiltration, suggesting the majority of accumulating B cells exhibit an activated phenotype associated with an enhanced capacity for antigen presentation, cytokine production, survival, and differentiation (54, 82). CD80 is also upregulated on Bmem (55, 56, 83). During chronic TMEV-IDD a high proportion of spinal cord-infiltrating B cells expressed an isotype-switched, differentiated phenotype (~70%), yet only 20% of total CD19+ were CD138+ ASC. These data, in combination with the observed high number of CD80+ B cells, suggest CD138− isotype-switched B cells are Bmem in chronic TMEV-IDD. Future studies validating the presence of Bmem during chronic TMEV-IDD are required to assess conventional surface molecules present on murine Bmem, including co-expression of CD80 with PD-L2 and CD73 (84, 85). Investigating Bmem during chronic TMEV-IDD will be essential for our understanding of B cells in chronic demyelinating disease, including MS, as Bmem are antigen-experienced cells with potent antigen presentation capabilities, producing large amounts of pro- and anti-inflammatory cytokines, and have the potential to rapidly convert into antigen-specific ASC to sustain CNS-compartmentalized Ab synthesis (86, 87).
The maintenance of diverse B cell phenotypes within the CNS during neuroinflammatory diseases, including MS, is suggested to occur by two different mechanisms: (1) continuous recruitment from the peripheral compartment (88, 89) and (2) CNS compartmentalized inflammation, such as ELF formation (36, 37, 39, 90–92). In chronic and progressive MS, evidence for CNS compartmentalized B cell activity is supported by the relative absence of new gadolinium-enhancing lesions in the CNS caused by inflammatory cells derived from the periphery, and relatively reduced effectiveness for peripherally administered disease-modifying therapies, including anti-CD20, in eliminating CSF B cells (12, 93, 94). Furthermore, CNS compartmentalized inflammation including meningeal leukocyte aggregates, CSF B cells, and ItAb is associated with disease progression in MS (39). However, there is a significant gap in our understanding of the factors required for supporting B cell localization and functional activity within the CNS, including ELF formation, in MS and its animal models. During chronic TMEV-IDD as shown above, ItAb synthesis coincided with the accumulation of heterogeneous B cell subsets, including mature and isotype-switched B cells, frequently aggregating within meningeal and perivascular regions of the spinal cord, often localizing with T cells. Meningeal inflammatory aggregates lacked several conventional features of SLO-like ELF necessary for driving local B cell differentiation into antigen-specific Bmem or ASC. Spatial organization, heightened proliferation and cell death indicative of antigen-driven selection, FDC stromal networks, and detectable GL7 expression were all absent in chronic TMEV-IDD, a finding similar to previous studies in viral-induced demyelination models of MS and B cell-dependent EAE models (23, 29).
Use of the term ELF in the CNS encapsulates a diverse array of lymphoid tissue-like organization, and it remains to be determined if meningeal or perivascular aggregations can exhibit all the features of SLO-like ELF. The potential for the CNS to create an immune-competent environment resembling an SLO-like ELF requires a supportive stromal cell network to provide conventional trafficking and organizational chemokines such as CXCL12, CXCL13, CCL19/CCL21 and pro-inflammatory cytokines, including IL-17 and LTα, which can induce and perpetuate lymphocyte accumulation and organization (18). In our studies, conventional B cell trafficking, organizing, and trophic factors were expressed in the CNS and may aid heterogeneous B cell phenotype accumulation and ItAb synthesis. These common features of meningeal aggregations during neuroinflammation occurred in the absence of SLO-like ELF formation and GC markers essential for fostering antigen-driven selection of immune cells and inducing somatic hypermutation, class switch recombination, and local differentiation. Specifically, discretely organized T and B cell follicles, supportive FDC stromal cells, Ki-67+ proliferating and caspase-3+ cells indicating ongoing antigen-driven selection, and germinal center GL7hi B cells were searched for, but not found, in our studies in the chronic phase of TMEV-IDD.
The characteristics of the meningeal and perivascular clusters of inflammatory cells in TMEV-IDD may present insights into the controversial issues surrounding the existence and role of ELFs in MS. Patients with RRMS and progressive forms of MS exhibit meningeal inflammatory aggregates (18, 36, 37, 39, 95–99), yet the presence of leukocyte aggregations does not confirm the meninges provide a niche for SLO-like ELF formation. Moreover, the pathology and molecules associated with meningeal inflammation have not been definitively determined. For instance, in MS there is no evidence thus far that the leukocyte aggregations in the CNS involve somatic hypermutation, including activation-induced deaminase (AID) expression, an enzyme essential for hypermutation of variable Ig genes and increased B cell receptor affinity for the antigen. In the viral-induced mouse model of demyelination, MHV, CNS inflammatory aggregates were observed yet AID transcript expression was absent among sorted B cells isolated from the CNS (21). Thus, leukocyte aggregations described in the CNS in MS may more closely resemble the “immature,” non-SLO-like ELFs described in our experiments in TMEV-IDD, in contrast to ELFs outside of the CNS in chronic inflammatory diseases which are more SLO-like such as inducible forms of lymphoid tissue, i.e., iBALT (13, 16). This difference between CNS lymphoid aggregates and ELFs in inflammatory conditions outside of the CNS may be due to an inherent inability of the CNS to develop and organize SLO-like ELFs, even with chronic inflammation.
Our studies did not document SLO-like ELF in driving B cell persistence in the CNS compartment but revealed frequent immune cell aggregation within meningeal spaces. Moreover, upregulation of markers, including GL7 and CD80, on B cells in chronic TMEV-IDD suggests the majority are highly activated and may be antigen-experienced (52–54, 68, 69). It remains unclear whether B cells acquire an activated phenotype in the peripheral or CNS compartment or the exact mechanisms driving activation (polyclonal activators vs. CD4 T cell help). Frequent immune cell aggregation in both meningeal and perivascular spaces and CNS-infiltrating B cells displaying an activated/helped phenotype suggest CD4 T cell-dependent activation may occur in the CNS compartment. The meninges are known to be a niche for harboring cells essential for promoting ongoing T cell activation and effector responses, suggesting B cell activation may be supported by similar mechanisms (100, 101). The abundance of T cells in meningeal and perivascular aggregates in the CNS during chronic TMEV-IDD resembles immune aggregates described in other viral infections of the CNS as well as in SPMS patients (23, 29, 37). Although studies examining B and T cell contact-dependent interactions within the CNS remain limited, following MHV infection, B and T cell interactions were evident within meningeal spaces in the brain which may promote local B cell activation (23). It remains to be determined if CD4 T cells are necessary and/or sufficient for propagating B cell activation, sustained accumulation, and potential differentiation in the CNS in mouse models of MS and MS patients. An important factor derived from T cells which supports B cell survival and differentiation in SLO, IL-21, showed minimal elevations during chronic TMEV-IDD in contrast to reports of elevated IL-21 production in the CNS during the acute phase of viral-induced encephalitis (21, 25). Nonetheless, increased GL7 and CD80 expression suggests most infiltrating B cells during chronic TMEV-IDD exhibited a contact-dependent CD4 T cell “helped” phenotype. Further studies are needed to determine if T cell secreted factors or contact-dependent mechanisms are essential for sustaining B cell accumulation and activation within the CNS compartment.
Our results and previous studies (19–21, 25, 27, 28) highlight that diverse factors may foster persistent B cell accumulation and ItAb in the CNS compartment. These factors may range in complexity from the expression of adhesion molecules such as CEACAM1, which alone can promote meningeal aggregation, to ectopic expression of lymphoid chemokines, cytokines and trophic factors supporting aggregation, to well-defined ELF resembling SLO (16, 18, 35, 102). Our study demonstrates that fully developed ectopic germinal centers are not required to support diverse B cell phenotypes and chronic CNS ItAb production. Irrespective of the level of organization, meningeal aggregates have been extensively associated with underlying cortical demyelination, gray matter pathology, diffuse axonal and neuronal loss (95–99, 103, 104). Thus, it is imperative to further delineate factors supporting meningeal aggregations, as potential mechanisms for dissociating pathogenic inflammatory aggregations in MS and other neuroinflammatory diseases will likely depend on the level of organization and molecular/cellular factors sustaining these structures. A limitation of the current study characterizing CNS compartmentalized inflammation in a chronic model of MS is the absence of mechanistic studies linking inflammatory aggregates with disability and/or neurodegeneration. Nonetheless, our data provide an important platform for investigating the role of inflammatory aggregates in neurodegeneration and for probing targets to disturb aggregates. Previous studies documenting neuronal death, axonal damage, and demyelination within the ventral regions of the spinal cord (40, 46, 105–107) in concert with our findings of frequent inflammatory aggregation in the ventral spinal cord, suggest a possible role of inflammatory aggregates in adjacent neurodegeneration.
Overall, the data show the ability for the CNS to foster isotype-switched B cell accumulation, ItAb synthesis, and production of B cell related chemokines and trophic factors, and meningeal inflammatory aggregates within the CNS compartment during a chronic, progressive mouse model of MS.
Data Availability
All raw data and protocols used to generate datasets for this study will be made available by the authors upon request to any qualified researcher.
Ethics Statement
This study was carried out in accordance with the recommendations of the Institutional Animal Care and Use Committee. The protocol was approved by the Institutional Animal Care and Use Committee.
Author Contributions
KD designed the experiments and conceived the original idea with the supervision of AP and FG. KD and DR carried out all experiments. KD conducted all data analysis and data interpretation. KD wrote the manuscript with support from AP and FG.
Funding
This project has been funded by the Bornstein research fund within the Department of Neurology at Dartmouth-Hitchcock Medical Center.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors thank the staff of the Center for Comparative Medicine and Research (CCMR) at Dartmouth College for their expert care of the mice used for this study. The authors also acknowledge the NCCC light microscopy shared resource (IPIM) supported in part by NCI Cancer Center Support Grant 5P30 CA023108-37 and also NIH S10 SIG award S10OD21616 funding the LSM800 confocal.
References
1. Imrell K, Landtblom AM, Hillert J, Masterman T. Multiple sclerosis with and without CSF bands: clinically indistinguishable but immunogenetically distinct. Neurology. (2006) 67:1062–4. doi: 10.1212/01.wnl.0000237343.93389.35
2. Cruz M, Olsson T, Ernerudh J, Hojeberg B, Link H. Immunoblot detection of oligoclonal anti-myelin basic protein IgG antibodies in cerebrospinal fluid in multiple sclerosis. Neurology. (1987) 37:1515–9. doi: 10.1212/WNL.37.9.1515
3. Kabat EA, Glusman M, Knaub V. Quantitative estimation of the albumin and gamma globulin in normal and pathologic cerebrospinal fluid by immunochemical methods. Am J Med. (1948) 4:653–62. doi: 10.1016/S0002-9343(48)90389-1
4. Prineas JW, Graham JS. Multiple sclerosis: capping of surface immunoglobulin G on macrophages engaged in myelin breakdown. Ann Neurol. (1981) 10:149–58. doi: 10.1002/ana.410100205
5. Breij EC, Brink BP, Veerhuis R, van den Berg C, Vloet R, Yan R, et al. Homogeneity of active demyelinating lesions in established multiple sclerosis. Ann Neurol. (2008) 63:16–25. doi: 10.1002/ana.21311
6. Genain CP, Cannella B, Hauser SL, Raine CS. Identification of autoantibodies associated with myelin damage in multiple sclerosis. Nat Med. (1999) 5:170–5. doi: 10.1038/5532
7. Lucchinetti C, Bruck W, Parisi J, Scheithauer B, Rodriguez M, Lassmann H. Heterogeneity of multiple sclerosis lesions: implications for the pathogenesis of demyelination. Ann Neurol. (2000) 47:707–17.3. doi: 10.1002/1531-8249(200006)47:6<707::AID-ANA3>3.0.CO;2-Q
8. Hauser SL, Waubant E, Arnold DL, Vollmer T, Antel J, Fox RJ, et al. B-cell depletion with rituximab in relapsing-remitting multiple sclerosis. N Engl J Med. (2008) 358:676–88. doi: 10.1056/NEJMoa0706383
9. Hauser SL, Bar-Or A, Comi G, Giovannoni G, Hartung HP, Hemmer B, et al. Ocrelizumab versus interferon beta-1a in relapsing multiple sclerosis. N Engl J Med. (2016) 376:221–34 doi: 10.1056/NEJMoa1601277
10. Hawker K, O'Connor P, Freedman MS, Calabresi PA, Antel J, Simon J, et al. Rituximab in patients with primary progressive multiple sclerosis: results of a randomized double-blind placebo-controlled multicenter trial. Ann Neurol. (2009) 66:460–71. doi: 10.1002/ana.21867
11. Montalban X, Hauser SL, Kappos L, Arnold DL, Bar-Or A, Comi G, et al. Ocrelizumab versus placebo in primary progressive multiple sclerosis. N Engl J Med. (2016) 376:209–20 doi: 10.1056/NEJMoa1606468
12. Cross AH, Stark JL, Lauber J, Ramsbottom MJ, Lyons JA. Rituximab reduces B cells and T cells in cerebrospinal fluid of multiple sclerosis patients. J Neuroimmunol. (2006) 180:63–70. doi: 10.1016/j.jneuroim.2006.06.029
13. Carragher DM, Rangel-Moreno J, Randall TD. Ectopic lymphoid tissues and local immunity. Semin Immunol. (2008) 20:26–42. doi: 10.1016/j.smim.2007.12.004
14. Buckley CD, Barone F, Nayar S, Bénézech C, Caamaño J. Stromal cells in chronic inflammation and tertiary lymphoid organ formation. Annu Rev Immunol. (2015) 33:715–45. doi: 10.1146/annurev-immunol-032713-120252
15. Drayton DL, Liao S, Mounzer RH, Ruddle NH. Lymphoid organ development: from ontogeny to neogenesis. Nat Immunol. (2006) 7:344–53. doi: 10.1038/ni1330
16. Pitzalis C, Jones GW, Bombardieri M, Jones SA. Ectopic lymphoid-like structures in infection, cancer and autoimmunity. Nat Rev Immunol. (2014) 14:447–62. doi: 10.1038/nri3700
17. Humby F, Bombardieri M, Manzo A, Kelly S, Blades MC, Kirkham B, et al. Ectopic lymphoid structures support ongoing production of class-switched autoantibodies in rheumatoid synovium. PLoS Med. (2009) 6:e1. doi: 10.1371/journal.pmed.0060001
18. Pikor NB, Prat A, Bar-Or A, Gommerman JL. Meningeal tertiary lymphoid tissues and multiple sclerosis: a gathering place for diverse types of immune cells during CNS autoimmunity. Front Immunol. (2015) 6:657. doi: 10.3389/fimmu.2015.00657
19. Phares TW, Marques CP, Stohlman SA, Hinton DR, Bergmann CC. Factors supporting intrathecal humoral responses following viral encephalomyelitis. J Virol. (2011) 85:2589–98. doi: 10.1128/JVI.02260-10
20. Phares TW, Stohlman SA, Hinton DR, Bergmann CC. Astrocyte-derived CXCL10 drives accumulation of antibody-secreting cells in the central nervous system during viral encephalomyelitis. J Virol. (2013) 87:3382–92. doi: 10.1128/JVI.03307-12
21. Phares TW, DiSano KD, Stohlman SA, Bergmann CC. Progression from IgD+ IgM+ to isotype-switched B cells is site specific during coronavirus-induced encephalomyelitis. J Virol. (2014) 88:8853–67. doi: 10.1128/JVI.00861-14
22. Phares TW, DiSano KD, Stohlman SA, Segal BM, Bergmann CC. CXCL13 promotes isotype-switched B cell accumulation to the central nervous system during viral encephalomyelitis. Brain Behav Immun. (2016) 54:128–139. doi: 10.1016/j.bbi.2016.01.016
23. DiSano KD, Stohlman SA, Bergmann CC. Activated GL7+ B cells are maintained within the inflamed CNS in the absence of follicle formation during viral encephalomyelitis. Brain Behav Immun. (2017) 60:71–83. doi: 10.1016/j.bbi.2016.09.022
24. Metcalf TU, Griffin DE. Alphavirus-induced encephalomyelitis: antibody-secreting cells and viral clearance from the nervous system. J Virol. (2011) 85:11490–501. doi: 10.1128/JVI.05379-11
25. Metcalf TU, Baxter VK, Nilaratanakul V, Griffin DE. Recruitment and retention of B cells in the central nervous system in response to alphavirus encephalomyelitis. J Virol. (2013) 87:2420–9. doi: 10.1128/JVI.01769-12
26. Bagaeva LV, Rao P, Powers JM, Segal BM. CXC chemokine ligand 13 plays a role in experimental autoimmune encephalomyelitis. J Immunol. (2006) 176:7676–85. doi: 10.4049/jimmunol.176.12.7676
27. Columba-Cabezas S, Serafini B, Ambrosini E, Aloisi F. Lymphoid chemokines CCL19 and CCL21 are expressed in the central nervous system during experimental autoimmune encephalomyelitis: implications for the maintenance of chronic neuroinflammation. Brain Pathol. (2003) 13:38–51. doi: 10.1111/j.1750-3639.2003.tb00005.x
28. Magliozzi R, Columba-Cabezas S, Serafini B, Aloisi F. Intracerebral expression of CXCL13 and BAFF is accompanied by formation of lymphoid follicle-like structures in the meninges of mice with relapsing experimental autoimmune encephalomyelitis. J Neuroimmunol. (2004) 148:11–23. doi: 10.1016/j.jneuroim.2003.10.056
29. Dang AK, Tesfagiorgis Y, Jain RW, Craig HC, Kerfoot SM. Meningeal infiltration of the spinal cord by non-classically activated b cells is associated with chronic disease course in a spontaneous B cell-dependent model of CNS autoimmune disease. Front Immunol. (2015) 6:470. doi: 10.3389/fimmu.2015.00470
30. Bleul CC, Fuhlbrigge RC, Casasnovas JM, Aiuti A, Springer TA. A highly efficacious lymphocyte chemoattractant, stromal cell-derived factor 1 (SDF-1). J Exp Med. (1996) 184:1101–9. doi: 10.1084/jem.184.3.1101
31. Phares TW, DiSano KD, Hinton DR, Hwang M, Zajac AJ, Stohlman SA, et al. IL-21 optimizes T cell and humoral responses in the central nervous system during viral encephalitis. J Neuroimmunol. (2013) 263:43–54. doi: 10.1016/j.jneuroim.2013.07.019
32. Kowarik MC, Cepok S, Sellner J, Grummel V, Weber MS, Korn T, et al. CXCL13 is the major determinant for B cell recruitment to the CSF during neuroinflammation. J Neuroinflamm. (2012) 9:93. doi: 10.1186/1742-2094-9-93
33. Bajetto A, Bonavia R, Barbero S, Florio T, Schettini G. Chemokines and their receptors in the central nervous system. Front Neuroendocrinol. (2001) 22:147–84. doi: 10.1006/frne.2001.0214
34. Cartier L, Hartley O, Dubois-Dauphin M, Krause KH. Chemokine receptors in the central nervous system: role in brain inflammation and neurodegenerative diseases. Brain Res Brain Res Rev. (2005) 48:16–42. doi: 10.1016/j.brainresrev.2004.07.021
35. Peters A, Pitcher LA, Sullivan JM, Mitsdoerffer M, Acton SE, Franz B, et al. Th17 cells induce ectopic lymphoid follicles in central nervous system tissue inflammation. Immunity. (2011) 35:986–96. doi: 10.1016/j.immuni.2011.10.015
36. Magliozzi R, Howell O, Vora A, Serafini B, Nicholas R, Puopolo M, et al. Meningeal B-cell follicles in secondary progressive multiple sclerosis associate with early onset of disease and severe cortical pathology. Brain. (2007) 130:1089–104. doi: 10.1093/brain/awm038
37. Serafini B, Rosicarelli B, Magliozzi R, Stigliano E, Aloisi F. Detection of ectopic B-cell follicles with germinal centers in the meninges of patients with secondary progressive multiple sclerosis. Brain Pathol. (2004) 14:164–74. doi: 10.1111/j.1750-3639.2004.tb00049.x
38. Kuerten S, Schickel A, Kerkloh C, Recks MS, Addicks K, Ruddle NH, et al. Tertiary lymphoid organ development coincides with determinant spreading of the myelin-specific T cell response. Acta Neuropathol. (2012) 124:861–73. doi: 10.1007/s00401-012-1023-3
39. Howell OW, Reeves CA, Nicholas R, Carassiti D, Radotra B, Gentleman SM, et a. Meningeal inflammation is widespread and linked to cortical pathology in multiple sclerosis. Brain. (2011) 134:2755–71. doi: 10.1093/brain/awr182
40. McGavern DB, Murray PD, Rodriguez M. Quantitation of spinal cord demyelination, remyelination, atrophy, and axonal loss in a model of progressive neurologic injury. J Neurosci Res. (1999) 58:492–504.
41. Oleszak EL, Chang JR, Friedman H, Katsetos CD, Platsoucas CD. Theiler's virus infection: a model for multiple sclerosis. Clin Microbiol Rev. (2004) 17:174–207. doi: 10.1128/CMR.17.1.174-207.2004
42. Tsunoda I, Fujinami RS. Neuropathogenesis of Theiler's murine encephalomyelitis virus infection, an animal model for multiple sclerosis. J Neuroimmune Pharmacol. (2010) 5:355–69. doi: 10.1007/s11481-009-9179-x
43. Lipton HL. Theiler's virus infection in mice: an unusual biphasic disease process leading to demyelination. Infect Immun. (1975) 11:1147–55.
44. Pachner AR, Brady J, Narayan K. Antibody-secreting cells in the central nervous system in an animal model of MS: phenotype, association with disability, and in vitro production of antibody. J Neuroimmunol. (2007) 190:112–20. doi: 10.1016/j.jneuroim.2007.09.001
45. Pachner AR, Li L, Narayan K. Intrathecal antibody production in an animal model of multiple sclerosis. J Neuroimmunol. (2007) 185:57–63. doi: 10.1016/j.jneuroim.2007.01.017
46. Gerhauser I, Hansmann F, Ciurkiewicz M, Löscher W, Beineke A. Facets of Theiler's murine encephalomyelitis virus-induced diseases: an update. Int J Mol Sci. (2019) 20:E448. doi: 10.3390/ijms20020448
47. Pachner AR, Li L, Lagunoff D. Plasma cells in the central nervous system in the Theiler's virus model of multiple sclerosis. J Neuroimmunol. (2011) 232:35–40. doi: 10.1016/j.jneuroim.2010.09.026
48. Li L, Narayan K, Pak E, Pachner AR. Intrathecal antibody production in a mouse model of Lyme neuroborreliosis. J Neuroimmunol. (2006) 173:56–68. doi: 10.1016/j.jneuroim.2005.11.019
49. Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCT Method. Methods. (2001) 25:402–8. doi: 10.1006/meth.2001.1262
50. Andersson M, Alvarez-Cermeño J, Bernardi G, Cogato I, Fredman P, Frederiksen J, et al. Cerebrospinal fluid in the diagnosis of multiple sclerosis: a consensus report. J Neurol Neurosurg Psychiatry. (1994) 57:897–902. doi: 10.1136/jnnp.57.8.897
51. Mehta PD. Diagnostic usefulness of cerebrospinal fluid in multiple sclerosis. Crit Rev Clin Lab Sci. (1991) 28:233–51. doi: 10.3109/10408369109106864
52. Balogh A, Adori M, Torok K, Matko J, Laszlo G. A closer look into the GL7 antigen: its spatio-temporally selective differential expression and localization in lymphoid cells and organs in human. Immunol Lett. (2010) 130:89–96. doi: 10.1016/j.imlet.2009.12.008
53. Cervenak L, Magyar A, Boja R, Laszlo G. Differential expression of GL7 activation antigen on bone marrow B cell subpopulations and peripheral B cells. Immunol Lett. (2001) 78:89–96. doi: 10.1016/S0165-2478(01)00239-5
54. Clatza A, Bonifaz LC, Vignali DA, Moreno J. CD40-induced aggregation of MHC class II and CD80 on the cell surface leads to an early enhancement in antigen presentation. J Immunol. (2003) 171:6478–87. doi: 10.4049/jimmunol.171.12.6478
55. Anderson SM, Tomayko MM, Ahuja A, Haberman AM, Shlomchik MJ. New markers for murine memory B cells that define mutated and unmutated subsets. J Exp Med. (2007) 204:2103–14. doi: 10.1084/jem.20062571
56. Zuccarino-Catania GV, Sadanand S, Weisel FJ, Tomayko MM, Meng H, Kleinstein SH, et al. CD80 and PD-L2 define functionally distinct memory B cell subsets that are independent of antibody isotype. Nat Immunol. (2014) 15:631–7. doi: 10.1038/ni.2914
57. Shinall SM, Gonzalez-Fernandez M, Noelle RJ, Waldschmidt TJ. Identification of murine germinal center B cell subsets defined by the expression of surface isotypes and differentiation antigens. J Immunol. (2000) 164:5729–38. doi: 10.4049/jimmunol.164.11.5729
58. Tellier J, Nutt SL. Standing out from the crowd: How to identify plasma cells. Eur J Immunol. (2017) 47:1276–79. doi: 10.1002/eji.201747168
59. Sanderson RD, Lalor P, Bernfield M. B lymphocytes express and lose syndecan at specific stages of differentiation. Cell Regul. (1989) 1:27–35. doi: 10.1091/mbc.1.1.27
60. Treanor B. B-cell receptor: from resting state to activate. Immunology. (2012) 136:21–7. doi: 10.1111/j.1365-2567.2012.03564.x
61. Sixt M, Engelhardt B, Pausch F, Hallmann R, Wendler O, Sorokin LM. Endothelial cell laminin isoforms, laminins 8 and 10, play decisive roles in T cell recruitment across the blood-brain barrier in experimental autoimmune encephalomyelitis. J Cell Biol. (2001) 153:933–46. doi: 10.1083/jcb.153.5.933
62. Eriksdotter-Nilsson M, Björklund H, Olson L. Laminin immunohistochemistry: a simple method to visualize and quantitate vascular structures in the mammalian brain. J Neurosci Methods. (1986) 17:275–86. doi: 10.1016/0165-0270(86)90128-7
63. Scholzen T, Gerdes J. The Ki-67 protein: from the known and the unknown. J Cell Physiol. (2000) 182:311–22. doi: 10.1002/(SICI)1097-4652(200003)182:3<311::AID-JCP1>3.0.CO;2-9
64. Allen CD, Cyster JG. Follicular dendritic cell networks of primary follicles and germinal centers: phenotype and function. Semin Immunol. (2008) 20:14–25. doi: 10.1016/j.smim.2007.12.001
65. Li J, Yuan J. Caspases in apoptosis and beyond. Oncogene. (2008) 27:6194–206. doi: 10.1038/onc.2008.297
66. Michel L, Touil H, Pikor NB, Gommerman JL, Prat A, Bar-Or A. B cells in the multiple sclerosis central nervous system: trafficking and contribution to CNS-compartmentalized inflammation. Front Immunol. (2015) 6:636. doi: 10.3389/fimmu.2015.00636
67. Grogan JL, Ouyang W. A role for Th17 cells in the regulation of tertiary lymphoid follicles. Eur J Immunol. (2012) 42:2255–62. doi: 10.1002/eji.201242656
68. Naito Y, Takematsu H, Koyama S, Miyake S, Yamamoto H, Fujinawa R, et al. Germinal center marker GL7 probes activation-dependent repression of N-glycolylneuraminic acid, a sialic acid species involved in the negative modulation of B-cell activation. Mol Cell Biol. (2007) 27:3008–22. doi: 10.1128/MCB.02047-06
69. Yellin MJ, Sinning J, Covey LR, Sherman W, Lee JJ, Glickman-Nir E, et al. T lymphocyte T cell-B cell-activating molecule/CD40-L molecules induce normal B cells or chronic lymphocytic leukemia B cells to express CD80 (B7/BB-1) and enhance their costimulatory activity. J Immunol. (1994) 153:666–74.
70. Lenschow DJ, Sperling AI, Cooke MP, Freeman G, Rhee L, Decker DC, et al. Differential up-regulation of the B7-1 and B7-2 costimulatory molecules after Ig receptor engagement by antigen. J Immunol. (1994) 153:1990–7.
71. Buisman AM, de Rond CG, Oztürk K, Ten Hulscher HI, van Binnendijk RS. Long-term presence of memory B-cells specific for different vaccine components. Vaccine. (2009) 28:179–86. doi: 10.1016/j.vaccine.2009.09.102
72. Montes CL, Acosta-Rodriguez EV, Merino MC, Bermejo DA, Gruppi A. Polyclonal B cell activation in infections: infectious agents' devilry or defense mechanism of the host? J Leukoc Biol. (2007) 82:1027–32. doi: 10.1189/jlb.0407214
73. Ozaki K, Spolski R, Feng CG, Qi CF, Cheng J, Sher A, et al. A critical role for IL-21 in regulating immunoglobulin production. Science. (2002) 298:1630–4. doi: 10.1126/science.1077002
74. Spolski R, Leonard WJ. Interleukin-21: basic biology and implications for cancer and autoimmunity. Annu Rev Immunol. (2008) 26:57–79. doi: 10.1146/annurev.immunol.26.021607.090316
75. Krumbholz M, Theil D, Derfuss T, Rosenwald A, Schrader F, Monoranu CM, et al. BAFF is produced by astrocytes and up-regulated in multiple sclerosis lesions and primary central nervous system lymphoma. J Exp Med. 201 (2005) 195–200. doi: 10.1084/jem.20041674
76. Thangarajh M, Masterman T, Hillert J, Moerk S, Jonsson R. A proliferation-inducing ligand (APRIL) is expressed by astrocytes and is increased in multiple sclerosis. Scand J Immunol. (2007) 65:92–8. doi: 10.1111/j.1365-3083.2006.01867.x
77. Van Wagoner NJ, Benveniste EN. Interleukin-6 expression and regulation in astrocytes. J Neuroimmunol. (1999) 100:124–39. doi: 10.1016/S0165-5728(99)00187-3
78. Lalor SJ, Segal BM. Lymphoid chemokines in the CNS. J Neuroimmunol. (2010) 224:56–61. doi: 10.1016/j.jneuroim.2010.05.017
79. Navarrete-Talloni MJ, Kalkuhl A, Deschl U, Ulrich R, Kummerfeld M, Rohn K, et al. Transient peripheral immune response and central nervous system leaky compartmentalization in a viral model for multiple sclerosis. Brain Pathol. (2010) 20:890–901. doi: 10.1111/j.1750-3639.2010.00383.x
80. Pracht K, Meinzinger J, Daum P, Schulz SR, Reimer D, Hauke M, et al. A new staining protocol for detection of murine antibody-secreting plasma cell subsets by flow cytometry. Eur J Immunol. (2017) 47:1389–92. doi: 10.1002/eji.201747019
81. Lacotte S, Decossas M, Le Coz C, Brun S, Muller S, Dumortier H. Early differentiated CD138(high) MHCII+ IgG+ plasma cells express CXCR3 and localize into inflamed kidneys of lupus mice. PLoS ONE. (2013) 8:e58140. doi: 10.1371/journal.pone.0058140
82. Banchereau J, Bazan F, Blanchard D, Briere F, Galizzi JP, van Kooten C, et al. The CD40 antigen and its ligand. Annu Rev Immunol. (1994) 12:881–922. doi: 10.1146/annurev.iy.12.040194.004313
83. Küppers R. Human memory B cells: memory B cells of a special kind. Immunol Cell Biol. (2008) 86:635–6. doi: 10.1038/icb.2008.59
84. Tomayko MM, Steinel NC, Anderson SM, Shlomchik MJ. Cutting edge: hierarchy of maturity of murine memory B cell subsets. J Immunol. (2010) 185:7146–50. doi: 10.4049/jimmunol.1002163
85. Conter LJ, Song E, Shlomchik MJ, Tomayko MM. CD73 expression is dynamically regulated in the germinal center and bone marrow plasma cells are diminished in its absence. PLoS ONE. (2014) 9:e92009. doi: 10.1371/journal.pone.0092009
86. Hebeis BJ, Klenovsek K, Rohwer P, Ritter U, Schneider A, Mach M, et al. Activation of virus-specific memory B cells in the absence of T cell help. J Exp Med. (2004) 199:593–602. doi: 10.1084/jem.20030091
87. Kurosaki T, Kometani K, Ise W. Memory B cells. Nat Rev Immunol. (2015) 15:149–59. doi: 10.1038/nri3802
88. Palanichamy A, Apeltsin L, Kuo TC, Sirota M, Wang S, Pitts SJ, et al. Immunoglobulin class-switched B cells form an active immune axis between CNS and periphery in multiple sclerosis. Sci Transl Med. (2014) 6:248ra106. doi: 10.1126/scitranslmed.3008930
89. Stern JN, Yaari G, Vander Heiden JA, Church G, Donahue WF, Hintzen RQ, et al. B cells populating the multiple sclerosis brain mature in the draining cervical lymph nodes. Sci Transl Med. (2014) 6:248ra107. doi: 10.1126/scitranslmed.3008879
90. Colombo M, Dono M, Gazzola P, Roncella S, Valetto A, Chiorazzi N, et al. Accumulation of clonally related B lymphocytes in the cerebrospinal fluid of multiple sclerosis patients. J Immunol. (2000) 164:2782–9. doi: 10.4049/jimmunol.164.5.2782
91. Eggers EL, Michel BA, Wu H, Wang SZ, Bevan CJ, Abounasr A, et al. Clonal relationships of CSF B cells in treatment-naive multiple sclerosis patients. JCI Insight. (2017) 2:92724. doi: 10.1172/jci.insight.92724
92. Corcione A, Aloisi F, Serafini B, Capello E, Mancardi GL, Pistoia V, et al. B-cell differentiation in the CNS of patients with multiple sclerosis. Autoimmun Rev. (2005) 4:549–54. doi: 10.1016/j.autrev.2005.04.012
93. Cepok S, Jacobsen M, Schock S, Omer B, Jaekel S, Böddeker I, et al. Patterns of cerebrospinal fluid pathology correlate with disease progression in multiple sclerosis. Brain. (2001) 124:2169–76. doi: 10.1093/brain/124.11.2169
94. Thompson AJ, Kermode AG, Wicks D, MacManus DG, Kendall BE, Kingsley DP, et al. Major differences in the dynamics of primary and secondary progressive multiple sclerosis. Ann Neurol. (1991) 29:53–62. doi: 10.1002/ana.410290111
95. Choi SR, Howell OW, Carassiti D, Magliozzi R, Gveric D, Muraro PA, et al. Meningeal inflammation plays a role in the pathology of primary progressive multiple sclerosis. Brain. (2012) 135:2925–37. doi: 10.1093/brain/aws189
96. Magliozzi R, Howell OW, Reeves C, Roncaroli F, Nicholas R, Serafini B, et al. A Gradient of neuronal loss and meningeal inflammation in multiple sclerosis. Ann Neurol. (2010) 68:477–93. doi: 10.1002/ana.22230
97. Androdias G, Reynolds R, Chanal M, Ritleng C, Confavreux C, Nataf S. Meningeal T cells associate with diffuse axonal loss in multiple sclerosis spinal cords. Ann Neurol. (2010) 68:465–76. doi: 10.1002/ana.22054
98. Lucchinetti CF, Popescu BF, Bunyan RF, Moll NM, Roemer SF, Lassmann H, et al. Inflammatory cortical demyelination in early multiple sclerosis. N Engl J Med. (2011) 365:2188–97. doi: 10.1056/NEJMoa1100648
99. Kooi EJ, Geurts JJ, van Horssen J, Bo L, van der Valk P. Meningeal inflammation is not associated with cortical demyelination in chronic multiple sclerosis. J Neuropathol Exp Neurol. (2009) 68:1021–8. doi: 10.1097/NEN.0b013e3181b4bf8f
100. Bartholomaus I, Kawakami N, Odoardi F, Schlager C, Miljkovic D, Ellwart JW, et al. Effector T cell interactions with meningeal vascular structures in nascent autoimmune CNS lesions. Nature. (2009) 462:94–8. doi: 10.1038/nature08478
101. Kivisäkk P, Imitola J, Rasmussen S, Elyaman W, Zhu B, Ransohoff RM, et al. Localizing central nervous system immune surveillance: meningeal antigen-presenting cells activate T cells during experimental autoimmune encephalomyelitis. Ann Neurol. (2009) 65:457–69. doi: 10.1002/ana.21379
102. Rovituso DM, Scheffler L, Wunsch M, Kleinschnitz C, Dörck S, Ulzheimer J, et al. CEACAM1 mediates B cell aggregation in central nervous system autoimmunity. Sci Rep. (2016) 6:29847. doi: 10.1038/srep29847
103. Howell OW, Schulz-Trieglaff EK, Carassiti D, Gentleman SM, Nicholas R, Roncaroli F, et al. Extensive grey matter pathology in the cerebellum in multiple sclerosis is linked to inflammation in the subarachnoid space. Neuropathol Appl Neurobiol. (2015) 41:798–813. doi: 10.1111/nan.12199
104. Kutzelnigg A, Lucchinetti CF, Stadelmann C, Brück W, Rauschka H, Bergmann M, et al. Cortical demyelination and diffuse white matter injury in multiple sclerosis. Brain. (2005) 128:2705–12. doi: 10.1093/brain/awh641
105. Leitzen E, Jin W, Herder V, Beineke A, Elmarabet SA, Baumgartner W, et al. Comparison of reported spinal cord lesions in progressive multiple sclerosis with theiler's murine encephalomyelitis virus induced demyelinating disease. Int J Mol Sci. (2019) 20:E989. doi: 10.3390/ijms20040989
106. McGavern DB, Murray PD, Rivera-Quinones C, Schmelzer JD, Low PA, Rodriguez M. Axonal loss results in spinal cord atrophy, electrophysiological abnormalities and neurological deficits following demyelination in a chronic inflammatory model of multiple sclerosis. Brain. (2000) 123 (Pt 3):519–31. doi: 10.1093/brain/123.3.519
107. Sathornsumetee S, McGavern DB, Ure DR, Rodriguez M. Quantitative ultrastructural analysis of a single spinal cord demyelinated lesion predicts total lesion load, axonal loss, and neurological dysfunction in a murine model of multiple sclerosis. Am J Pathol. (2000) 157:1365–76. doi: 10.1016/S0002-9440(10)64650-0
Keywords: multiple sclerois, ectopic lymphoid follicles, B cells, TMEV-IDD, neuroinflammation
Citation: DiSano KD, Royce DB, Gilli F and Pachner AR (2019) Central Nervous System Inflammatory Aggregates in the Theiler's Virus Model of Progressive Multiple Sclerosis. Front. Immunol. 10:1821. doi: 10.3389/fimmu.2019.01821
Received: 01 April 2019; Accepted: 18 July 2019;
Published: 02 August 2019.
Edited by:
Jorge Ivan Alvarez, University of Pennsylvania, United StatesReviewed by:
Steven M. Kerfoot, University of Western Ontario, CanadaJen Gommerman, University of Toronto, Canada
Copyright © 2019 DiSano, Royce, Gilli and Pachner. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Krista D. DiSano, krista.d.disano@hitchcock.org