
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Immunol. , 31 July 2019
Sec. Multiple Sclerosis and Neuroimmunology
Volume 10 - 2019 | https://doi.org/10.3389/fimmu.2019.01811
This article is part of the Research Topic Neuro-Immune Connections to Enable Repair in CNS Disorders View all 20 articles
 Elien Wouters1†
Elien Wouters1† Nienke M. de Wit2†
Nienke M. de Wit2† Jasmine Vanmol1Susanne M. A. van der Pol2Bert van het Hof2
Jasmine Vanmol1Susanne M. A. van der Pol2Bert van het Hof2 Daniela Sommer1
Daniela Sommer1 Melanie Loix1
Melanie Loix1 Dirk Geerts3
Dirk Geerts3 Jan Ake Gustafsson4,5Knut R. Steffensen5
Jan Ake Gustafsson4,5Knut R. Steffensen5 Tim Vanmierlo1,6
Tim Vanmierlo1,6 Jeroen F. J. Bogie1
Jeroen F. J. Bogie1 Jerome J. A. Hendriks1†
Jerome J. A. Hendriks1† Helga E. de Vries2*†
Helga E. de Vries2*†Dysfunction of the blood-brain barrier (BBB) contributes significantly to the pathogenesis of several neuroinflammatory diseases, including multiple sclerosis (MS). Potential players that regulate BBB function are the liver X receptors (LXRs), which are ligand activated transcription factors comprising two isoforms, LXRα, and LXRβ. However, the role of LXRα and LXRβ in regulating BBB (dys)function during neuroinflammation remains unclear, as well as their individual involvement. Therefore, the goal of the present study is to unravel whether LXR isoforms have different roles in regulating BBB function under neuroinflammatory conditions. We demonstrate that LXRα, and not LXRβ, is essential to maintain barrier integrity in vitro. Specific knockout of LXRα in brain endothelial cells resulted in a more permeable barrier with reduced expression of tight junctions. Additionally, the observed dysfunction was accompanied by increased endothelial inflammation, as detected by enhanced expression of vascular cell adhesion molecule (VCAM-1) and increased transendothelial migration of monocytes toward inflammatory stimuli. To unravel the importance of LXRα in BBB function in vivo, we made use of the experimental autoimmune encephalomyelitis (EAE) MS mouse model. Induction of EAE in a constitutive LXRα knockout mouse and in an endothelial specific LXRα knockout mouse resulted in a more severe disease score in these animals. This was accompanied by higher numbers of infiltrating leukocytes, increased endothelial VCAM-1 expression, and decreased expression of the tight junction molecule claudin-5. Together, this study reveals that LXRα is indispensable for maintaining BBB integrity and its immune quiescence. Targeting the LXRα isoform may help in the development of novel therapeutic strategies to prevent BBB dysfunction, and thereby neuroinflammatory disorders.
Liver X receptors (LXRs) belong to a large family of nuclear receptors which upon activation stimulate gene transcription (1). Two LXR isoforms exist in mammals, termed LXRα (NR1H3) and LXRβ (NR1H2), which share over 75% amino acid sequence identity. In the nucleus LXRs form obligate heterodimers with the retinoid X receptor (RXR), together forming the LXR/RXR complex. LXRs play an important role in cholesterol and lipid metabolism. The best described process involving LXR function is reverse cholesterol transport where LXRs facilitate the elimination of excess cholesterol in response to cholesterol precursors or oxysterols (2). However, LXRs appear to be involved in a much broader spectrum of functions.
Recent studies identified LXRs as promising targets to interfere in inflammatory signaling pathways. LXR activation induces anti-inflammatory actions in macrophages by antagonizing NF-κB signaling (3). In the central nervous system (CNS), LXR agonists inhibit the production of proinflammatory cytokines and chemokines in stimulated microglia and reactive astrocytes (4). In several animal models of different CNS disorders, including stroke, Alzheimer's disease (AD), and multiple sclerosis (MS), the activation of LXRs results in a reduction of neuroinflammation, suggesting that LXR targeting may be effective in the treatment of neuroinflammatory disorders (5–8).
The main players in the neuroinflammatory process are proinflammatory cytokines and chemokines. These inflammatory mediators are produced locally within the CNS by glial cells or by leukocytes, which are recruited from the periphery following blood-brain barrier (BBB) breakdown (9). One of the pathological hallmarks of BBB dysfunction seen in neuroinflammatory disorders is increased permeability due to loss of tight junctions and increased leukocyte extravasation (10). During the extravasation process, chemokines presented by the inflamed brain endothelium guide the rolling and firm adhesion of leukocytes on the brain endothelial cell surface. Next, the interaction of integrins on leukocytes with brain endothelial cell adhesion molecules (CAMs) further induces their trans- or paracellular migration into the brain, illustrating the critical role of the BBB in mediating neuroinflammatory disorders (11, 12).
Brain endothelial cells tightly regulate BBB function and are regarded as the gatekeepers of the CNS (13, 14). So far, knowledge on the involvement of LXRs in BBB function is limited and is mostly linked to their function in cholesterol homeostasis. For instance, several studies indicate an upregulation of downstream ATP-binding cassette (ABC) cholesterol transporters after LXR agonism in primary brain endothelial cells (15, 16). Interestingly, LXR activation prevents the downregulation of the tight junctions occludin and zona occludens-1 in ischemic vessels in a mouse model of stroke, indicating that LXRs control BBB integrity (16). To date, it remains unclear whether LXRs regulate BBB function during a neuroinflammatory insult, and whether the LXRα and LXRβ isoforms have a distinct role in controlling BBB integrity. Therefore, the goal of the present study is to unravel whether LXR isoforms have different functions in regulating BBB function under neuroinflammatory conditions.
In this study, we show that LXRα, and not LXRβ, is essential to maintain BBB integrity. Impaired LXRα function in brain endothelial cells resulted in decreased barrier function and increased inflammation as marked by increased endothelial vascular cell adhesion molecule (VCAM-1) expression and enhanced trans-endothelial monocyte migration. Importantly, whole body knockout of LXRα and specific endothelial knockout of LXRα in a neuroinflammatory mouse model, resulted in enhanced extravasation of leukocytes into the brain together with increased VCAM-1 expression and reduced claudin-5 expression in the brain vasculature. Collectively, our findings show that LXRα is essential to maintain BBB function.
The human immortalized cerebral microvascular endothelial cell line hCMEC/D3 (17) was grown in EGM-2 Endothelial Cell Growth Medium-2 BulletKit, including basal medium and supplement components according to the manufacturer's instructions (Lonza, Basel, Switzerland). All cell culture plates were coated with type I collagen (Invitrogen, Thermo Fisher Scientific, Leusden, The Netherlands). Cultures were grown to confluence at 37°C in 5% CO2. hCMEC/D3 cells were detached at 37°C with trypsin/EDTA in PBS (Gibco, Thermo Fisher Scientific).
Selective gene knockdown (KD) was obtained by using a vector-based short hairpin (sh) RNA technique as previously described (18). Recombinant lentiviruses were produced by co-transfecting subconfluent HEK 293T cells with the specific expression plasmids and packaging plasmids (pMDLg/pRRE, pRSV-Rev, and pMD2G) using calcium phosphate as a transfection reagent. HEK 293T cells were cultured in Dulbecco's modified eagle medium (DMEM) supplemented with 10% fetal calf serum (FCS) and 1% penicillin/streptomycin. Cells were cultured at 37°C in 5% CO2. Infectious lentiviruses were collected 48 h after transfection and stored at −80°C. The KD efficiency of all five constructs for each LXR isoform was tested, and the most effective construct used in subsequent experiments for LXRα (NR1H3) was TRC22237, encoding sequence GTGCAGGAGATAGTTGACTTT that target nucleotides 1,043–1,063 of the NM_005693.3 RefSeq. For LXRβ (NR1H2) the most effective construct was TRC275326, encoding sequence GAAGGCATCCACTATCGAGAT that target nucleotides 1,193–1,213 of the NM_007121.5 RefSeq. Subsequently, lentiviruses expressing LXRα- or LXRβ-specific shRNA were used to transduce hCMEC/D3 cells. Control cells were generated by transduction with lentivirus expressing non-targeting shRNA (SHC002, Sigma-Aldrich, St. Louis, MO). Forty-eight hours after infection of hCMEC/D3 cells with the shRNA-expressing lentiviruses, stable cell lines were selected by puromycin treatment (2 μg/ml). The expression knockdown efficiency was determined by quantitative real-time PCR (qRT-PCR).
Recombinant hCMEC/D3 cell lines (1 × 106 cells/ml) expressing either LXRα shRNA, LXRβ shRNA, or non-targeting shRNA were seeded in 24-well plates in growth medium. Upon confluency, cells were treated with DMSO (VWR, Leuven, Belgium) or with 5 ng/ml TNFα and 5 ng/ml IFNγ (Peprotech, London, UK) for 24 h. EAE animals were sacrificed on day 23 post-adoptive transfer or day 36 post-immunization. Spinal cords were isolated and snap frozen in liquid nitrogen. Total RNA from cultures and tissues was extracted using Qiazol (Qiagen, Venlo, The Netherlands) and the RNeasy mini kit (Qiagen), according to the manufacturer's instructions. RNA concentration and purity were determined with a NanoDrop spectrophotometer (Isogen Life Science, De Meern, The Netherlands). cDNA was synthesized using qScriptTM cDNA SuperMix (Quanta Biosciences, VWR), following manufacturer's guidelines. qRT-PCR was carried out using SYBR green master mix (Applied Biosystems, Waltham, MA) and a Step One Plus detection system (Applied Biosystems). Primers used for qRT-PCR are shown in Table S1. Relative quantitation of gene expression was accomplished using the comparative Ct method. Data were normalized to the most stable reference genes, as previously described (19).
For flow cytometric analysis of VCAM-1, hCMEC/D3 cells (1 × 106 cells/ml) were seeded in 24-well plates. At confluency, cells were treated with DMSO as vehicle control or with 5 ng/ml TNFα and 5 ng/ml IFNγ for 24 h. hCMEC/D3 cells were detached from 24-well plates using 1 mg/ml collagenase type I (Sigma-Aldrich). Washed cells were incubated with mouse anti-human VCAM-1 (AbD Serotec, Kidlington, UK) for 30 min at 4°C. Binding was detected using secondary goat anti-mouse Alexa Fluor 488 (Molecular Probes, Eugene, OR). Omission of primary antibodies served as negative control. Fluorescence intensity was measured using a FACS Calibur flow cytometer (Becton, Dickinson and Company, Franklin Lakes, NJ).
The ECISTM Model 1600R (Applied BioPhysics, Troy, NY) was used to measure the barrier resistance (Rb) of confluent monolayers of hCMEC/D3 cells expressing non-targeting, LXRα or LXRβ shRNA. 100.000 cells were seeded onto each well of an 8W10+ ECIS array (Ibidi, München, Germany). The impedance Z [Ohm's law, potential (V)/current (I)] was measured at multiple frequencies in real-time. ECIS-Wounding was carried out with a current of 5,000 μA at 60 kHz for 20 s to measure proliferation rate of the different cell groups. All ECIS measurements were subjected to a mathematical model to calculate the component of resistance attributed to cell-cell interactions, called barrier resistance (Rb).
Recombinant hCMEC/D3 cells expressing non-targeting shRNA, LXRα shRNA or LXRβ shRNA were seeded at a concentration of 100.000 cells/cm2 onto the upper side of 0.4 μm pore-size collagen-coated Costar Transwell filters (Corning, Corning, NY) in growth medium. Paracellular permeability for FITC-dextran (70 kDa in growth medium, Sigma-Aldrich) from apical to basolateral direction was determined by collecting samples from the lower chambers after 4 h. The fluorescence intensity of the medium in the basolateral compartment was measured using a FLUOstar Galaxy microplate reader (BMG Labtechnologies, Offenburg, Germany, excitation 485 nm and emission 520 nm).
The migrating capacity of isolated monocytes across a monolayer of hCMEC/D3 cells was determined as previously described (20). Briefly, recombinant hCMEC/D3 cells were grown to confluence onto the upper side of 0.4 μm pore-size collagen-coated Costar Transwell filters (Corning) in growth medium and were subsequently exposed to either vehicle or TNFα (5 ng/ml) for 24 h. After washing, 100 μl of primary human monocytes (1 × 106 cells/ml) was added to the upper chamber. The human blood monocytes were isolated from buffy coats of healthy donors (Sanquin, Blood Bank, Amsterdam, The Netherlands) by Ficoll gradient and anti-CD14 beads (21). Following 8 h of migration at 37°C and 5% CO2 in air, the transmigrated monocytes were harvested and quantified using anti-CD14 beads (Flow-CountTM Fluorospheres, Beckman Coulter, Brea, CA) and subsequent FACScan flow cytometer analysis (Becton). The level of migration was calculated as the percentage of migrated monocytes to total monocytes within the field.
Recombinant hCMEC/D3 cells expressing non-targeting shRNA, LXRα shRNA or LXRβ shRNA were grown to confluency in eight-well μ-slides (Ibidi). Cells were washed with ice-cold PBS and fixed in pre-cooled methanol for 10 min at −20°C. Fixed cells were washed and blocked with PBS containing 5% normal goat serum. Subsequently, cells were incubated overnight at 4°C with the primary antibody claudin-5 (Invitrogen). Next, cells were washed and incubated with the secondary antibody goat anti-mouse IgG Alexa Fluor 488 (Invitrogen).
Wild-type C57BL/6JOlaHsd mice were purchased from Envigo (Venray, The Netherlands). LXRα−/− and LXRα loxP/loxP mice on a C57BL/6 background were kindly provided by prof. dr. J.Å. Gustafsson (University of Houston, Houston, USA) (22). Cdh5(PAC)-creERT2 transgenic mice on a C57BL/6 background were kindly provided by prof. dr. Ralf H. Adams (Max Planck Institute, Münster, Germany) (23). All animal experiments were approved by the institutional animal care and use committee of Hasselt University (protocol numbers: 201422 201615 and 201617). The generation of endothelial-specific LXRα-inducible knockout mice was established by crossing LXRα loxP/loxP mice with Cdh5(PAC)-creERT2 transgenic mice to obtain Cdh5(PAC)-creERT2+LXRαLoxP/LoxP mice (LXRαflox/floxCdh5-Cre+/− mice). LXRα loxP/loxP and Cdh5(PAC)-creERT2 littermates were used as controls. Recombination was induced by injecting 10-week-old females intra-peritoneal with 100 μl tamoxifen [Sigma-Aldrich; 20 mg/ml in corn oil (Sigma-Aldrich)] for 5 consecutive days.
At the age of 11 weeks, female C57BL/6 mice were actively immunized subcutaneously with 200 μg myelin oligodendrocyte glycoprotein peptide (MOG35−55) emulsified in 100 μl complete Freund's adjuvant containing 4 mg/ml Mycobacterium tuberculosis (EK-2110 kit; Hooke Laboratories, Massachusetts, USA). Directly after MOG35−55 immunization and after 24 h, mice were intraperitoneally injected with 100 ng (C57BL/6JOlaHsd donor mice adoptive T cell transfer) or 40 ng (endothelial specific knockout mice) pertussis toxin (EK-2110 kit; Hooke Laboratories) to induce a normal or a mild EAE, respectively. Both the control group and experimental group of one experiment received the same amount of PTX. Mice were weighed and clinically evaluated daily for neurological signs of the disease according to manufacturer's mouse EAE scoring guide: 0: no clinical symptoms; 0.5: distal tail paralysis; 1: tail paralysis; 2: mild paraparesis and ataxia; 2.5: moderate paraparesis; 3: complete paralysis of the hind legs; 4: paralysis to the diaphragm; 5: death by EAE.
At day 9 post-immunization, inguinal lymph nodes were isolated from wild-type C57BL/6 donor mice. Next, T cells were collected and cultured at a concentration of 7 × 106 cells/ml in stimulation medium (RPMI medium supplemented with 0.5% Penicillin-Streptomycin, 20 μM β-mercaptoethanol, 10% FCS, 1% Non-Essential Amino Acid, 1% sodium pyruvate and 20 ng/ml IL-23 (Bio-Legend, London, UK) containing 20 μg/ml MOG35−55. After 2 days of incubation, activated cells were intraperitoneally injected into LXRα−/− acceptor mice or wild-type littermates at a density of 15 × 106 cells/ml in PBS.
Mice were sacrificed on day 23 post-adoptive transfer or day 36 post-immunization. Brains and spinals cords were isolated and snap frozen in optimal cutting temperature (OCT) compound. Material was sectioned using a Leica CM3050 S cryostat (Leica Microsystems, Wetzlar, Germany) to obtain 10 μm slices. Staining was performed on brain and spinal cord sections mounted on coated glass slides (Menzel Gläser Superfrost PLUS, Thermo Scientific, Braunschweig Germany). For colocalization studies, brain sections were air-dried, fixed in ice-cold methanol for 10 min at −20°C, and blocked for 30 min in 10% normal swine serum in PBS. Subsequently, sections were incubated overnight at 4°C with primary antibodies (claudin-5, VCAM-1 and rhodamine-lectin) as indicated in Table S2. Biotin labeled swine anti-rabbit (1:500, Dako, Agilent, Amstelveen, The Netherlands) followed by Alexa 488 labeled streptavidin (1:400, Molecular Probes) was used to detect claudin-5. Sections were incubated for 1 h with their specific secondary antibody. Finally, sections were stained with Hoechst (dilution 1:1000, Molecular Probes) to visualize cellular nuclei and mounted with Mowiol mounting medium. Representative images were taken using a Leica DM6000 microscope (20x objective, Leica Microsystems).
To study immune cell infiltration, spinal cord sections were air-dried and fixed in ice-cold acetone for 10 min at −20°C. Non-specific staining was blocked using Dako protein block (Agilent, Santa Clara, CA) for 30 min. Afterwards, sections were incubated overnight at 4°C with primary antibodies (CD3 and F4/80, Table S2). Secondary goat anti-rat IgG Alexa Fluor 555 (1:400, Thermo Scientific) was used to detect CD3 and F4/80. Representative images were taken using a Nikon eclipse 80i microscope (10× objective) and NIS Elements BR 3.10 software (Nikon, Tokyo, Japan).
Image J version 1.52c (https://imagej.nih.gov/ij/index.html) was used for quantitative analysis of the expression of claudin-5 by recombinant hCMEC/D3 cells expressing non-targeting shRNA, LXRα shRNA, or LXRβ shRNA. For the quantification of the area fraction of the double fluorescent staining of claudin-5 and VCAM-1 overlapping with lectin in control mice, LXRα−/− mice and LXRαflox/floxCdh5-Cre+/− mice, four pictures spanning the hippocampus were taken per animal. The amount of infiltrated immune cells, positive for CD3 and F4/80, was determined by quantitative analysis of six pictures per animal spanning the whole spinal cord.
Data were statistically analyzed using GraphPad Prism v6 (GraphPad Software, La Jolla, CA, USA) and are reported as mean ± standard error of the mean (SEM). D'Agostino-Pearson omnibus normality test was used to test normal distribution. One-way ANOVA (three groups) with Tukey's multiple comparison correction, two-way ANOVA (four groups) with Sidak's multiple comparison correction, or two-tailed unpaired student T-test (two groups) were used for normally distributed data sets. The Mann-Whitney (two groups) analysis was used for non-parametric data sets. No correction, i.e., Bonferroni for multiple statistical comparisons was performed. *P < 0.05, **P < 0.01, ***P < 0.001, and ****P < 0.0001.
To unravel the function of the LXRα and LXRβ isoforms in BBB integrity, we generated a brain endothelial cell line (hCMEC/D3) with a reduced expression of either LXRα or LXRβ. Transduction of brain endothelial cells with lentiviruses expressing LXRα- or LXRβ-specific shRNAs resulted in a reduced expression of LXRα or LXRβ, as determined by qRT-PCR. Only cells with at least 70% knockdown of expression were used in our study (Figure S1A). LXRα and LXRβ knockdown did not affect the proliferation rate of the endothelial cells (Figure S1B). ECIS analysis, used to measure transendothelial electrical resistance, was performed to determine the involvement of LXRα and LXRβ in brain endothelial cell barrier formation. The results of the one-way ANOVA showed a significant difference between the three cell types (p < 0.0001) of all in Figure 1 presented variables. Furthermore, post-hoc analysis revealed that knockdown of LXRα resulted in a significantly reduced barrier resistance compared to LXRβ knockdown cells (p < 0.0001) and the non-targeting control cells (p < 0.0001; Figure 1A). In accordance with a lower barrier resistance, the leakage of FITC-dextran was significantly (p < 0.0001) enhanced in LXRα deficient cells compared to cells lacking LXRβ and non-targeted control cells at 4 h (Figure 1B). Data on barrier formation or stability over the course of 4 h are not shown. Finally, immunocytochemical analysis revealed a decreased expression of the tight junction protein claudin-5 in LXRα-deficient cells (Figure 1C). In addition, quantification of the expression level of claudin-5 in the LXRα knockdown cells showed a significant decrease compared to LXRβ knockdown cells (p < 0.01) and non-targeting control cells (p < 0.01; Figure 1D), further strengthening the importance of LXRα in BBB function. Collectively, these findings demonstrate that LXRα, but not LXRβ, contributes to the formation of endothelial cell-to-cell junctions, thereby controlling BBB integrity.
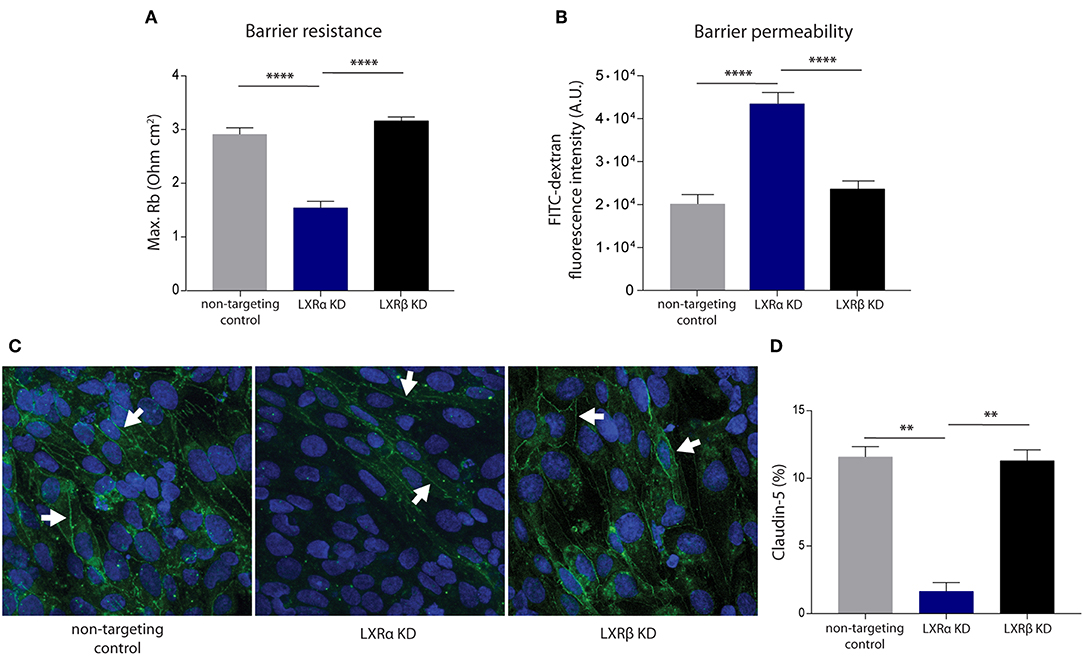
Figure 1. LXRα knockdown in the endothelial cell line hCMEC/D3 decreases BBB integrity. (A) LXRα knockdown cells show a significantly lower intercellular adhesion (Rb) compared to LXRβ knockdown and non-targeted control cells. Data are calculated from impedance measurements (Ohm cm2) ± SEM of two independent experiments performed in 4-fold. (B) Paracellular permeability of 70 kDa FITC-dextran was studied in time. LXRα knockdown cells were more permeable to FITC-dextran compared to control and LXRβ knockdown cells. Data are expressed as mean fluorescence intensity ± SEM after 4 h of three independent experiments performed in 3-fold. (C) Claudin-5 expression was studied using immunocytochemistry. Reduced claudin-5 expression (white arrows) was observed in LXRα knockdown cells compared to control and LXRβ knockdown cells (claudin-5; green, nuclei; blue). (D) Quantitative analysis of claudin-5 expression in non-targeting control, LXRα knockdown, and LXRβ knockdown cells. Statistical significance (one-way ANOVA, with Tukey's multiple comparison correction) is indicated with asterisks: **p < 0.01, ****p < 0.0001.
During neuroinflammation, immune activation of the BBB facilitates the migration of leukocytes into the brain. To determine the involvement of LXRα and LXRβ in BBB function under neuroinflammatory conditions, we studied the expression levels of cytokines, chemokines, and adhesion molecules known to be involved in neuroinflammation. Although endothelial cells are capable of transrepression upon LXR activation (Figure S2A), without activation we found no isoform-specific increase or decrease in cytokine or chemokine expression under basal or inflammatory conditions (Figure S2B), using qRT-PCR.
The results of the two-way ANOVA of VCAM-1 mRNA expression showed no main effect of cell type (p = 0.06). However, a significant main effect of inflammation (p < 0.0001) was present. In addition, there was no significant interaction effect between the cell type and inflammation (p = 0.08; Figure 2A). However, the results of the two-way ANOVA of the protein expression of VCAM-1 showed a significant main effect of cell type (p < 0.0001) and a significant main effect of inflammation (p < 0.0001), yet there was no significant interaction effect between the two (p = 0.83). Post-hoc analysis revealed that VCAM-1 protein expression levels were significantly increased in LXRα knockdown cells under basal (p < 0.0001) as well as inflammatory conditions (p < 0.0001) compared to LXRβ knockdown and non-targeted control cells (Figure 2B).
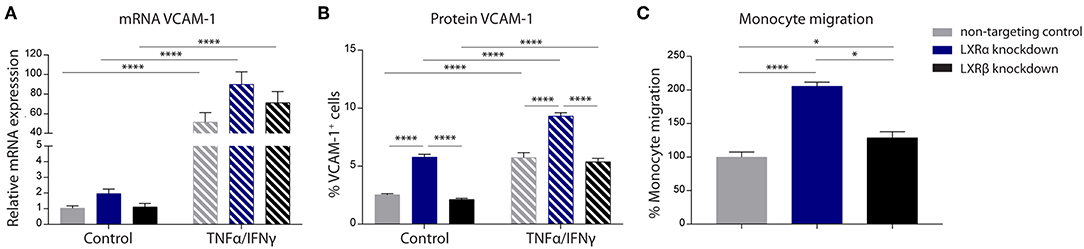
Figure 2. LXRα mediates immune activation of the endothelial cell line hCMEC/D3 and transendothelial migration of monocytes. (A) qRT-PCR of VCAM-1 mRNA expression and (B) FACS analysis of VCAM-1 protein expression levels in LXRα knockdown, LXRβ knockdown, and non-targeted control cells under basal and inflammatory conditions resulted in an increase of VCAM-1 in LXRα knockdown cells on protein level. Data are expressed as mean ± SEM of three independent experiments performed in 3-fold. (C) Primary monocyte migration across confluent monolayers of recombinant hCMEC/D3 cells expressing non-targeting shRNA, LXRα shRNA, or LXRβ shRNA. Knockdown of LXRα increased transmigration of monocytes across the barrier. Data are expressed as mean ± SEM of % migrated cells of total monocytes of two independent experiments performed in 4-fold. Statistical significance (A, B; two-way ANOVA, with Sidak's multiple comparison correction, and C; one-way ANOVA, with Tukey's multiple comparison correction) is indicated with asterisks: *p < 0.05, ****p < 0.0001.
The one-way ANOVA revealed a significant difference (p < 0.0001) between the three cell types regarding monocyte migration. Consistent with increased VCAM-1 expression, endothelial LXRα knockdown resulted in a significantly increased migration of primary human monocytes across the endothelial barrier compared to LXRβ knockdown (p < 0.05) and non-targeting control (p < 0.0001; Figure 2C). Taken together, these results show that LXRα knockdown increases VCAM-1 expression on brain endothelial cells, stimulating transmigration of monocytes across the BBB.
Given the importance of LXRα in maintaining a functional BBB in vitro, we next sought to determine whether LXRα is involved in BBB function during neuroinflammation in vivo. For this purpose, we made use of the experimental autoimmune encephalomyelitis (EAE) MS mouse model. Because LXRs impact T cell proliferation and differentiation (5, 24, 25), we chose a T cell adoptive transfer model in which wild-type T cells are transferred to whole-body LXRα−/− mice. A two-way repeated measures ANOVA showed a significant main effect of the two groups of mice (p < 0.0001), a significant main effect of time (p < 0.0001), and most importantly a significant interaction effect (p < 0.0001). Daily evaluation of the disease severity demonstrated an increase in EAE score in LXRα−/− mice compared to wild-type mice. No differences were observed in disease onset (Figure 3A). The increase in mean clinical score was accompanied by increased inflammatory cytokine and chemokine mRNA expression in the spinal cord of LXRα-deficient animals. In line with our in vitro findings, lack of LXRα resulted in increased VCAM-1 mRNA expression (Figure 3B). Similar, LXRα deficiency increased the mRNA expression of F4/80, suggesting elevated infiltration of peripheral myeloid cells. Immunohistochemical analysis of the spinal cord confirmed the increased infiltration of macrophages (p < 0.05; Figure 3C). In the brain, no significant difference in the expression of VCAM-1 by endothelial cells was observed (Figure 3D). However, LXRα−/− mice did show a significant decrease in claudin-5 expression (p < 0.05) compared to wild-type mice (Figure 3E). Collectively, these findings show that LXRα has a protective function during neuroinflammation.
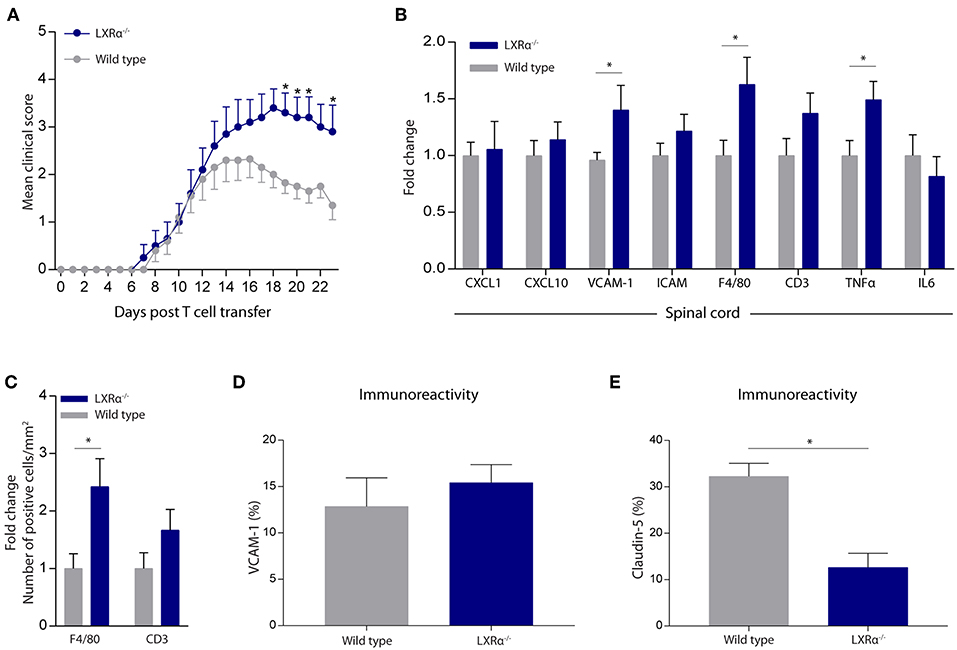
Figure 3. LXRα−/− exacerbates the disease score in a neuroinflammatory mouse model. (A) Adoptive transfer EAE was induced in wild type (WT) and LXRα−/− acceptor mice by immunization with MOG35−55 activated T-cells from WT donor mice. LXRα−/− mice showed a higher mean clinical score during EAE compared to WT mice (n = 10 per group). (B) qRT-PCR analysis of neuroinflammatory marker expression in the spinal cord of WT (n = 7) and LXRα−/− mice (n = 9). (C) Quantitative analysis of the number of infiltrated macrophages (F4/80) and T cells (CD3) into the spinal cord of WT (n = 8) and LXRα−/− mice (n = 7). (D) Quantitative analysis of the immunoreactive area for VCAM-1 and (E) claudin-5 in WT (n = 5) and LXRα−/− mice (n = 5). The values represent the mean ± S.E.M. Statistical significance (A; two-way repeated measures ANOVA, with Sidak's multiple comparison correction, B–E; Mann-Whitney U-test) is indicated with asterisks: *p < 0.05.
To elucidate whether increased CNS infiltration of inflammatory cells in LXRα−/− EAE mice is due to the absence of LXRα in the endothelium of the BBB, endothelial-specific knockouts of LXRα were generated by crossing LXRα loxP/loxP and Cdh5(PAC)-creERT2 transgenic mice (LXRαflox/floxCdh5-Cre+/−). No significant difference (repeated measures two-way ANOVA, Sidak's multiple comparison correction) was observed in disease score between LXRα loxP/loxP and Cdh5(PAC)-creERT2 transgenic mice over time (data not shown). Therefore, both control groups were combined for further analyses.
Endothelial knockout of LXRα resulted in a significant reduction in LXRα expression in endothelial cell isolates (Supplementary Materials and Methods, Figure S3). A two-way repeated measures ANOVA showed a significant main effect of the two groups of mice (p < 0.0001), a significant main effect of time (p < 0.0001), and most importantly a significant interaction effect (p < 0.0001). During a mild EAE (mean clinical score 1–2 in control animals), endothelial-specific deletion of LXRα resulted in a more severe mean clinical score compared to control animals with no difference in disease onset (Figure 4A). This increase in mean clinical score was associated with increased cytokine and chemokine expression in the spinal cord of endothelial-specific LXRα-deficient animals (Figure 4B). In addition, a significant increase in VCAM-1 mRNA expression together with enhanced F4/80 macrophage marker mRNA expression was found (p < 0.05; Figure 4B). Immunohistochemical analysis of the spinal cord showed a significantly increased migration of peripheral leukocytes (CD3, p < 0.05; Figures 5A,B). Moreover, immunohistochemical analysis of the brain tissue showed an enhanced expression of VCAM-1 (p < 0.05) and a decrease in claudin-5 expression (p < 0.05) in LXRα−/− mice (Figures 6A,B). The expression levels of VCAM-1 were similar to the expression levels of VCAM-1 in the whole-body LXRα−/− mice (Figure 3D vs. Figure 6B). Comparing claudin-5 expression levels between whole-body and endothelial specific LXRα−/− mice revealed a significant higher expression of claudin-5 (p < 0.01) in the latter group (Figure 3E vs. Figure 6B). These results demonstrate that LXRα deficiency in endothelial cells aggravates the disease course in a mouse model of neuroinflammation.
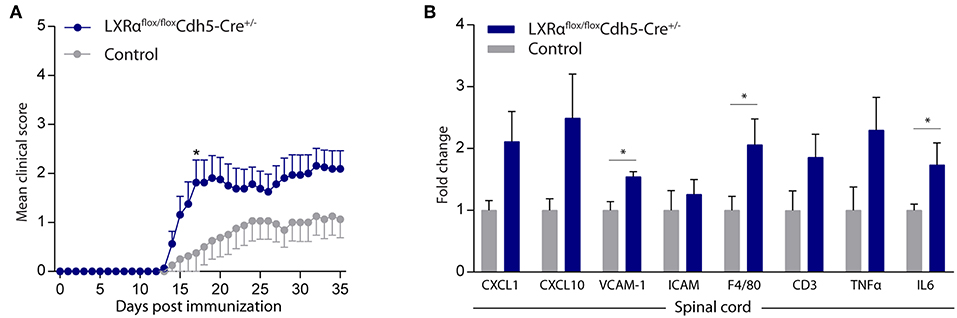
Figure 4. Endothelial specific knockout of LXRα worsens disease progression in a neuroinflammatory mouse model. (A) Mild EAE (mean clinical score 1–2 in control animals) was initiated in a cohort of 11-week-old Cdh5(PAC)-creERT2+LXRαLoxP/LoxP mice (LXRαflox/floxCdh5-Cre+/−). LXRα loxP/loxP and Cdh5(PAC)-creERT2 transgenic mice were used as controls. LXRαflox/floxCdh5-Cre+/−mice show a higher clinical score during the disease course compared with control mice (n = 8 per group). (B) qRT-PCR analysis of neuroinflammatory marker expression in the spinal cord of control (n = 8) and LXRαflox/floxCdh5-Cre+/− mice (n = 8). The values represent the mean ± S.E.M. Statistical significance (A; two-way repeated measures ANOVA, with Sidak's multiple comparison correction, B; Mann-Whitney U-test) is indicated with asterisks: *p < 0.05.
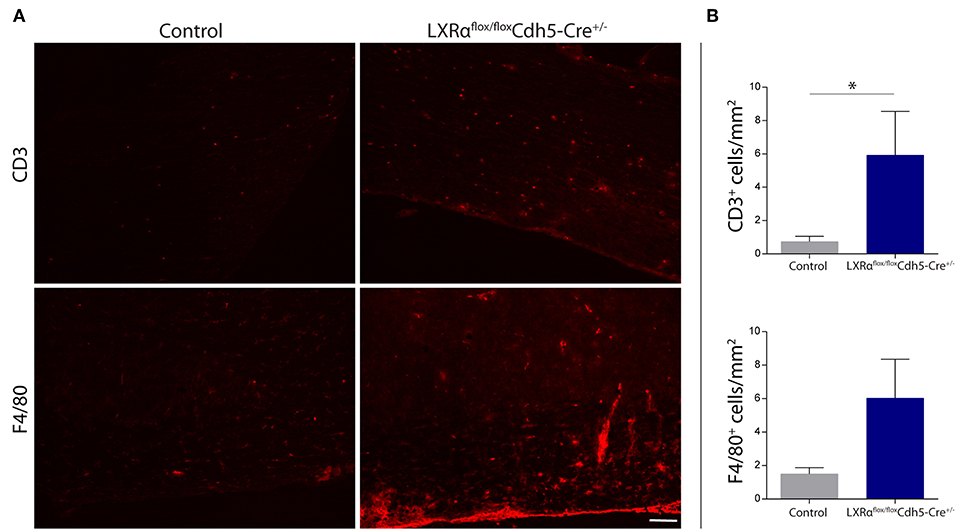
Figure 5. Increased leukocyte infiltration in the spinal cord of LXRαflox/floxCdh5-Cre+/− mice. (A) Immunofluorescent labeling of leukocytes (CD3, upper panel) or macrophages (F4/80, lower panel) in the spinal cord of control and LXRαflox/floxCdh5-Cre+/− mice. Bar: 100 μm. (B) Quantitative analysis of the number of infiltrated macrophages (F4/80) and T cells (CD3) into the spinal cord of control (n = 8) and LXRαflox/floxCdh5-Cre+/− mice (n = 8 per group). The values represent the mean ± S.E.M. Statistical significance (Mann-Whitney U-test) is indicated with asterisks: *p < 0.05.
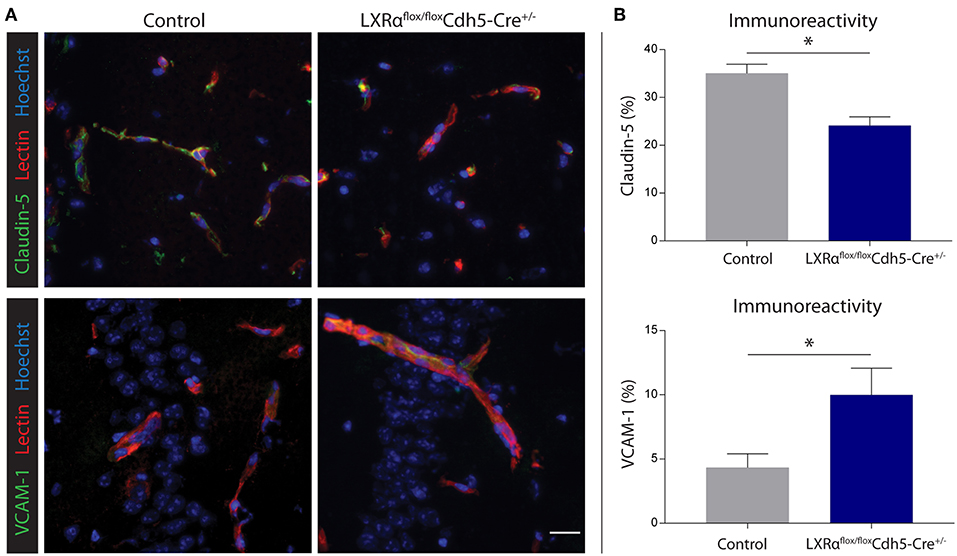
Figure 6. Endothelial specific knockout of LXRα results in decreased barrier integrity. (A) Immunofluorescent double labeling indicated that endothelial cells (red) of LXRαflox/floxCdh5-Cre+/− mice express less claudin-5 (green, upper panel) and more VCAM-1 (green, lower panel) compared to control animals. Nuclei were counterstained with Hoechst (blue). Bar: 20 μm. (B) Quantitative analysis of the immunoreactive area for claudin-5 and VCAM-1 in control (n = 5) and LXRαflox/floxCdh5-Cre+/− mice (n = 7). The values represent the mean ± S.E.M. Statistical significance (Mann-Whitney U-test) is indicated with asterisks: *p < 0.05.
The BBB is a highly specialized structure essential for CNS homeostasis. In this study, we determined the effect of both LXR isoforms on BBB integrity during neuroinflammation. Our experiments performed in vitro show that mainly LXRα, and not LXRβ, is important in maintaining proper barrier function. In addition, under neuroinflammatory conditions LXRα knockdown resulted in increased VCAM-1 expression by the endothelial cells, which was accompanied by an increase in monocyte migration across the barrier. Moreover, our in vitro findings were confirmed in vivo where endothelial specific knockout mice under neuroinflammatory conditions showed a higher disease score, increased peripheral leukocytes extravasation into the spinal cord, together with higher VCAM-1 expression and lower claudin-5 expression in the brain compared to control mice.
Our results demonstrate a difference in function between the LXRα and LXRβ isoform in brain endothelial cells. Even though LXRα is present in lower levels than LXRβ in the endothelial cells in mice and in the hCMEC/D3 cell line used in our experiments, we still observe a significant effect after knockdown of LXRα on BBB integrity, indicating that LXRα is essential in the regulation of BBB function. The two LXR isoforms share high sequence homology, but differ in their tissue distribution and function. LXRα is mainly expressed in the liver, intestine, adipose tissue, and macrophages and regulates for example reverse cholesterol transport in human macrophages and bile acid metabolism in the liver. On the other hand, LXRβ is more ubiquitously expressed and is involved in processes like lipid metabolism in the CNS and water transport in the pancreas (26, 27). A large amount of research has been performed using general LXR agonists that can target both LXR isoforms, thereby neglecting the possibility that both isoforms might exert different functions (16, 28, 29). Our results indicate that it is crucial to study the individual role of the distinct isoforms in different tissues, either by developing specific agonists or by generating a specific knockout in the tissue of interest.
We further show that whole-body and endothelium specific knockout of LXRα results in a decreased barrier integrity and increased inflammatory burden in a mouse model of neuroinflammation. Although the expression levels of VCAM-1 are similar between the two groups, comparing claudin-5 expression levels revealed a significant difference, where the endothelial specific LXRα−/− mice show higher expression. This difference in expression level might be the result of the interaction between astrocytes and pericytes with the endothelial cells. Both cell types are important in maintaining BBB properties, and lacking LXRα might influence the functional interaction. Other studies have used synthetic agonists to investigate LXR function in the vasculature. For instance, the treatment of human umbilical vein endothelial cells (HUVECs) with GW3965, a LXR agonist, inhibited the adhesion of monocytes to endothelial cells (28). Furthermore, LXR activation by T0901317 in a mouse model of ischemic stroke selectively prevented the downregulation of occludin and ZO-1 on ischemic microvessels (16). A more recent paper demonstrated that LXR activation by GW3965 also positively modulated the microvasculature in an Alzheimer mouse model (29). However, these studies did not make a distinction between LXRα and LXRβ. Our results suggest that these beneficial processes at the endothelial level are controlled by the LXRα isoform, and not by LXRβ.
Several other studies showed that LXR activation is able to suppress EAE and reduce CNS inflammation (5, 24, 25). In these papers, the protective impact of the LXR agonists was attributed to their impact on T cell proliferation and differentiation. However, their effect on BBB function has not been described. The results of our study suggest that specifically the activation of LXRα might ameliorate the EAE disease course via regulating BBB integrity and inflammation. By maintaining BBB integrity, i.e., less VCAM-1 expression and maintaining tight junction expression, less immune cells might be able to infiltrate the brain. Interestingly, we only observed an effect on endothelial specific LXRα knockdown during a mild EAE (mean clinical score 1–2 in control animals), and not during a normal EAE (clinical score above 2—data not shown). This could partly be explained by the effect of pertussis toxin (PTX) on BBB permeability and leukocyte recruitment (30, 31). Therefore, by inducing a mild EAE using lower PTX concentration, BBB integrity changes caused by LXRα knockdown could still contribute to disease severity. Of note, Cdh5 is also expressed in some cells of the hematopoietic system, including macrophages (32), which implies that macrophages in our generated LXRαflox/floxCdh5-Cre+/− mice may also lack LXRα expression, which may influence disease to a certain extent. However, since we observe similar effects using our in vitro assays using brain endothelial cells that lack LXRα, we are confident that the majority of observed effects are due to the role of LXRα in the endothelium. It will be very interesting to define the impact of LXR isoform-specific agonists on BBB integrity and inflammation, once these become available. Nevertheless, it is important to take into account that hepatic LXRα activation promotes hepatic steatosis and dyslipidemia (33, 34). Therefore, targeting of LXRα via liposomes or adeno-associated viruses specific for endothelial cells would be useful.
So far, we can only speculate about the underlying pathways. One possible mechanism that could contribute to the observed effects, is ABCA1 induction by LXRs. This transporter is not only important for the efflux of intracellular free cholesterol, but also has an anti-inflammatory effect in both the brain and in the peripheral circulation (35–37). Moreover, ABCA1 is able to suppress metalloproteinase-9 (MMP9) expression in the ischemic brain (38). MMP9 is an important inducer of BBB damage presumably via the degradation of tight junction proteins and basement membrane extracellular matrix proteins (39). In macrophages, the stimulation of LXRs results in decreased MMP9 expression (40). Consequently, the induction of MMP9 expression in LXRα deficient endothelial cells could result in BBB damage. Interestingly, our LXRα knockdown cells showed higher MMP9 mRNA expression (data not shown). However, further studies are needed to determine whether this pathway is responsible for the LXRα mediated effects in endothelial cells.
Another possible mechanism is a process called epithelial to mesenchymal transition (EMT), which is driven by the transcription factor Snail. During EMT, the epithelial phenotype shifts through changes in gene expression, loss of cell polarity and cell-cell adhesion, and reorganization of the cytoskeleton, ultimately leading to a more migratory and invasive phenotype (41). Interestingly, in different cancer cell lines the presence or overexpression of LXRα positively contributes to their migratory abilities and Snail expression, whereas the opposite is observed in epithelial cells, where absence of LXRα results in a higher Snail expression (42–44). EMT has also been described for (brain) endothelial cells (EndoMT) and might underlie the observed changes in the endothelial cells when LXRα is absent (45–48).
In conclusion, we show that LXRs have different roles in regulating BBB function under neuroinflammatory conditions. More specially, we demonstrate that LXRα, and not LXRβ, is needed to maintain barrier integrity. Endothelial specific knockdown of LXRα in vitro and in vivo resulted in a more permeable barrier with less tight junctions, increased expression of adhesion molecule VCAM-1, and in an increased transendothelial migration of peripheral leukocytes across the barrier. Understanding the mechanisms by which BBB permeability is regulated during neuroinflammation may help in the development of therapeutic strategies, i.e., targeted delivery or selective activation of LXRα, to prevent BBB leakage and peripheral leukocyte infiltration during the early stages of neuroinflammatory diseases.
This study was carried out in accordance with the recommendations of the institutional animal care and use committee of Hasselt University. The protocol was approved by the institutional animal care and use committee of Hasselt University (protocol numbers: 201422, 201615, and 201617).
EW, NdW, and JV are responsible for the generation of all data. SvdP, BvhH, DS, and ML gave technical support during experiments. EW and NdW wrote the manuscript. DG provided constructs for the generation of LXR knockdown brain endothelial cells. JG and KS provided the animals. JB and TV performed the adoptive T cell transfer and supervised the research. JH and HdV helped in designing the work and provided feedback on the manuscript. All the authors have read and approved the manuscript.
This work was supported by grants to NdW from ZonMw Memorabel program (project nr: 733050105), to JG a fellowship from Robert A. Welch (E-0004) and a grant from the Swedish Research Council, and to TV from FWO (project nr: 1506916N). The received funding had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript. Research at the Department of Molecular Cell Biology and immunology is part of the neuro-infection and neuroinflammation research program of Amsterdam Neuroscience.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We would like to thank the Advanced Optical Microscopy core facility in O|2 (AO|2M) for their expertise of and help with the microscopy applications (http://www.ao2m.amsterdam).
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2019.01811/full#supplementary-material
1. Zelcer N, Tontonoz P. Liver X receptors as integrators of metabolic and inflammatory signaling. J Clin Invest. (2006) 116:607–14. doi: 10.1172/JCI27883
2. Tontonoz P, Mangelsdorf DJ. Liver X receptor signaling pathways in cardiovascular disease. Mol Endocrinol. (2003) 17:985–93. doi: 10.1210/me.2003-0061
3. Joseph SB, Castrillo A, Laffitte BA, Mangelsdorf DJ, Tontonoz P. Reciprocal regulation of inflammation and lipid metabolism by liver X receptors. Nat Med. (2003) 9:213–9. doi: 10.1038/nm820
4. Zhang-Gandhi CX, Drew PD. Liver X receptor and retinoid X receptor agonists inhibit inflammatory responses of microglia and astrocytes. J Neuroimmunol. (2007) 183:50–9. doi: 10.1016/j.jneuroim.2006.11.007
5. Hindinger C, Hinton DR, Kirwin SJ, Atkinson RD, Burnett ME, Bergmann CC, et al. Liver X receptor activation decreases the severity of experimental autoimmune encephalomyelitis. J Neurosci Res. (2006) 84:1225–34. doi: 10.1002/jnr.21038
6. Cui W, Sun Y, Wang Z, Xu C, Peng Y, Li R. Liver X receptor activation attenuates inflammatory response and protects cholinergic neurons in APP/PS1 transgenic mice. Neuroscience. (2012) 210:200–10. doi: 10.1016/j.neuroscience.2012.02.047
7. Cheng O, Ostrowski RP, Liu W, Zhang JH. Activation of liver X receptor reduces global ischemic brain injury by reduction of nuclear factor-κB. Neuroscience. (2010) 166:1101–9. doi: 10.1016/j.neuroscience.2010.01.024
8. Mailleux J, Vanmierlo T, Bogie JFJ, Wouters E, Lütjohann D, Hendriks JJA, et al. Active liver X receptor signaling in phagocytes in multiple sclerosis lesions. Mult Scler J. (2018) 24:279–89. doi: 10.1177/1352458517696595
9. Shastri A, Bonifati DM, Kishore U. Innate immunity and neuroinflammation. Mediators of Inflammation. (2013) 2013:342931. doi: 10.1155/2013/342931
10. Obermeier B, Daneman R, Ransohoff RM. Development, maintenance and disruption of the blood-brain barrier. Nat Med. (2013) 19:1584–96. doi: 10.1038/nm.3407
11. Zlokovic B V. The blood-brain barrier in health and chronic neurodegenerative disorders. Neuron. (2008) 57:178–201. doi: 10.1016/j.neuron.2008.01.003
12. Man S, Ubogu EE, Ransohoff RM. Inflammatory cell migration into the central nervous system: a few new twists on an old tale. Brain Pathol. (2007) 17:243–50. doi: 10.1111/j.1750-3639.2007.00067.x
13. Greene C, Hanley N, Campbell M. Claudin-5: gatekeeper of neurological function. Fluids Barriers CNS. (2019) 16:3. doi: 10.1186/s12987-019-0123-z
14. Vanmierlo T, Bogie JFJ, Mailleux J, Vanmol J, Lütjohann D, Mulder M, et al. Plant sterols: friend or foe in CNS disorders? Prog Lipid Res. (2015) 58:26–39. doi: 10.1016/j.plipres.2015.01.003
15. Panzenboeck U, Kratzer I, Sovic A, Wintersperger A, Bernhart E, Hammer A, et al. Regulatory effects of synthetic liver X receptor- and peroxisome-proliferator activated receptor agonists on sterol transport pathways in polarized cerebrovascular endothelial cells. Int J Biochem Cell Biol. (2006) 38:1314–29. doi: 10.1016/j.biocel.2006.01.013
16. Elali A, Hermann DM. Liver X receptor activation enhances blood-brain barrier integrity in the ischemic brain and increases the abundance of ATP-binding cassette transporters ABCB1 and ABCC1 on brain capillary cells. Brain Pathol. (2012) 22:175–87. doi: 10.1111/j.1750-3639.2011.00517.x
17. Weksler BB, Subileau EA, Perrière N, Charneau P, Holloway K, Leveque M, et al. Blood-brain barrier-specific properties of a human adult brain endothelial cell line. FASEB J. (2005) 19:1872–4. doi: 10.1096/fj.04-3458fje
18. Reijerkerk A, Lakeman KAM, Drexhage JAR, Van Het Hof B, Van Wijck Y, Van Der Pol SMA, et al. Brain endothelial barrier passage by monocytes is controlled by the endothelin system. J Neurochem. (2012) 121:730–7. doi: 10.1111/j.1471-4159.2011.07393.x
19. Vandesompele J, De Preter K, Pattyn F, Poppe B, Van Roy N, De Paepe A, et al. The multifunctional FUS, EWS and TAF15 proto-oncoproteins show cell type-specific expression patterns and involvement in cell spreading and stress response. Genome Biol. (2002) 3:research0034.1. doi: 10.1186/1471-2121-9-37
20. Viegas P, Chaverot N, Enslen H, Perriere N, Couraud P-O, Cazaubon S. Junctional expression of the prion protein PrPC by brain endothelial cells: a role in trans-endothelial migration of human monocytes. J Cell Sci. (2006) 119:4634–43. doi: 10.1242/jcs.03222
21. Elkord E, Williams PE, Kynaston H, Rowbottom AW. Human monocyte isolation methods influence cytokine production from in vitro generated dendritic cells. Immunology. (2005) 114:204–12. doi: 10.1111/j.1365-2567.2004.02076.x
22. Alberti S, Schuster G, Parini P, Feltkamp D, Diczfalusy U, Rudling M, et al. Hepatic cholesterol metabolism and resistance to dietary cholesterol in LXRβ-deficient mice. J Clin Invest. (2001) 107:565–73. doi: 10.1172/JCI9794
23. Wang Y, Nakayama M, Pitulescu ME, Schmidt TS, Bochenek ML, Sakakibara A, et al. Ephrin-B2 controls VEGF-induced angiogenesis and lymphangiogenesis. Nature. (2010) 465:483–6. doi: 10.1038/nature09002
24. Xu J, Wagoner G, Douglas JC, Drew PD. Liver X receptor agonist regulation of Th17 lymphocyte function in autoimmunity. J Leukoc Biol. (2009) 86:401–9. doi: 10.1189/jlb.1008600
25. Cui G, Qin X, Wu L, Zhang Y, Sheng X, Yu Q, et al. Liver X receptor (LXR) mediates negative regulation of mouse and human Th17 differentiation. J Clin Invest. (2011) 121:658–70. doi: 10.1172/JCI42974
26. Seol W, Choi HS, Moore DD. Isolation of proteins that interact specifically with the retinoid X receptor: two novel orphan receptors. Mol Endocrinol. (2014) 9:72–85. doi: 10.1210/mend.9.1.7760852
27. Ishibashi M, Filomenko R, Rebe C, Chevriaux A, Varin A, Derangere V, et al. Knock-down of the oxysterol receptor LXRa impairs cholesterol efflux in human primary macrophages: lack of compensation by LXRb activation. Biochem Pharmacol. (2013) 86:122–9. doi: 10.1016/j.bcp.2012.12.024
28. Bi X, Song J, Gao J, Zhao J, Wang M, Scipione CA, et al. Activation of liver X receptor attenuates lysophosphatidylcholine-induced IL-8 expression in endothelial cells via the NF-κB pathway and SUMOylation. J Cell Mol Med. (2016) 20:2249–58. doi: 10.1111/jcmm.12903
29. Sandoval-Hernández AG, Restrepo A, Cardona-Gómez GP, Arboleda G. LXR activation protects hippocampal microvasculature in very old triple transgenic mouse model of Alzheimer's disease. Neurosci Lett. (2016) 621:15–21. doi: 10.1016/j.neulet.2016.04.007
30. Kügler S, Böcker K, Heusipp G, Greune L, Kim KS, Schmidt MA. Pertussis toxin transiently affects barrier integrity, organelle organization and transmigration of monocytes in a human brain microvascular endothelial cell barrier model. Cell Microbiol. (2007) 9:619–32. doi: 10.1111/j.1462-5822.2006.00813.x
31. Kerfoot SM, Long EM, Hickey MJ, Andonegui G, Lapointe BM, Zanardo RCO, et al. TLR4 contributes to disease-inducing mechanisms resulting in central nervous system autoimmune disease. J Immunol. (2014) 173:7070–7. doi: 10.4049/jimmunol.173.11.7070
32. Gentek R, Ghigo C, Hoeffel G, Bulle MJ, Msallam R, Gautier G, et al. Hemogenic endothelial fate mapping reveals dual developmental origin of mast cells. Immunity. (2018) 48:1160–1171.e5. doi: 10.1016/j.immuni.2018.04.025
33. Lund EG, Peterson LB, Adams AD, Lam MHN, Burton CA, Chin J, et al. Different roles of liver X receptor α and β in lipid metabolism: Effects of an α-selective and a dual agonist in mice deficient in each subtype. Biochem Pharmacol. (2006) 71:453–63. doi: 10.1016/j.bcp.2005.11.004
34. Quinet EM, Savio DA, Halpern AR, Chen L, Schuster GU, Gustafsson J-A, et al. Liver X receptor (LXR)-beta regulation in LXR -deficient mice: implications for therapeutic targeting. Mol Pharmacol. (2006) 70:1340–9. doi: 10.1124/mol.106.022608
35. Tang C, Liu Y, Kessler PS, Vaughan AM, Oram JF. The macrophage cholesterol exporter ABCA1 functions as an anti-inflammatory receptor. J Biol Chem. (2009) 284:32336–43. doi: 10.1074/jbc.M109.047472
36. Ito A, Hong C, Rong X, Zhu X, Tarling EJ, Hedde PN, et al. LXRs link metabolism to inflammation through Abca1-dependent regulation of membrane composition and TLR signaling. Elife. (2015) 4:e08009. doi: 10.7554/eLife.08009
37. Karasinska JM, de Haan W, Franciosi S, Ruddle P, Fan J, Kruit JK, et al. ABCA1 influences neuroinflammation and neuronal death. Neurobiol Dis. (2013) 54:445–55. doi: 10.1016/j.nbd.2013.01.018
38. Cui X, Chopp M, Zacharek A, Karasinska JM, Cui Y, Ning R, et al. Deficiency of brain ATP-binding cassette transporter A-1 exacerbates blood-brain barrier and white matter damage after stroke. Stroke. (2015) 46:827–34. doi: 10.1161/STROKEAHA.114.007145
39. Yang Y, Estrada EY, Thompson JF, Liu W, Rosenberg GA. Matrix metalloproteinase-mediated disruption of tight junction proteins in cerebral vessels is reversed by synthetic matrix metalloproteinase inhibitor in focal ischemia in rat. J Cereb Blood Flow Metab. (2007) 27:697–709. doi: 10.1038/sj.jcbfm.9600375
40. Castrillo A, Joseph SB, Marathe C, Mangelsdorf DJ, Tontonoz P. Liver X receptor-dependent repression of matrix metalloproteinase-9 expression in macrophages. J Biol Chem. (2003) 278:10443–9. doi: 10.1074/jbc.M213071200
41. Lamouille S, Xu J, Derynck R. Molecular mechanisms of epithelial-mesenchymal transition. Nat Rev Mol Cell Biol. (2014) 15:178–96. doi: 10.1038/nrm3758
42. Bellomo C, Caja L, Fabregat I, Mikulits W, Kardassis D, Heldin CH, et al. Snail mediates crosstalk between TGFβ and LXRα in hepatocellular carcinoma. Cell Death Differ. (2018) 25:885–903. doi: 10.1038/s41418-017-0021-3
43. Ji L, Zhang B, Zhao G. Liver X receptor α (LXRα) promoted invasion and EMT of gastric cancer cells by regulation of NF-κB activity. Hum Cell. (2017) 30:124–32. doi: 10.1007/s13577-016-0157-3
44. Kim H-J, Andersson LC, Bouton D, Warner M, Gustafsson J-A. Stromal growth and epithelial cell proliferation in ventral prostates of liver X receptor knockout mice. Proc Natl Acad Sci USA. (2009) 106:558–63. doi: 10.1073/pnas.0811295106
45. Zeisberg EM, Tarnavski O, Zeisberg M, Dorfman AL, McMullen JR, Gustafsson E, et al. Endothelial-to-mesenchymal transition contributes to cardiac fibrosis. Nat Med. (2007) 13:952–61. doi: 10.1038/nm1613
46. Medici D, Shore EM, Lounev VY, Kaplan FS, Kalluri R, Olsen BR. Conversion of vascular endothelial cells into multipotent stem-like cells. Nat Med. (2010) 16:1400–8. doi: 10.1038/nm.2252
47. Troletti CD, de Goede P, Kamermans A, de Vries HE. Molecular alterations of the blood-brain barrier under inflammatory conditions: the role of endothelial to mesenchymal transition. Biochim Biophys Acta Mol Basis Dis. (2016) 1862:452–60. doi: 10.1016/j.bbadis.2015.10.010
48. Derada Troletti C, Fontijn RD, Gowing E, Charabati M, van Het Hof B, Didouh I, et al. Inflammation-induced endothelial to mesenchymal transition promotes brain endothelial cell dysfunction and occurs during multiple sclerosis pathophysiology. Cell Death Dis. (2019) 10:45. doi: 10.1038/s41419-018-1294-2
Keywords: blood-brain barrier, permeability, endothelium, liver X receptors, neuroinflammation
Citation: Wouters E, de Wit NM, Vanmol J, van der Pol SMA, van het Hof B, Sommer D, Loix M, Geerts D, Gustafsson JA, Steffensen KR, Vanmierlo T, Bogie JFJ, Hendriks JJA and de Vries HE (2019) Liver X Receptor Alpha Is Important in Maintaining Blood-Brain Barrier Function. Front. Immunol. 10:1811. doi: 10.3389/fimmu.2019.01811
Received: 06 February 2019; Accepted: 17 July 2019;
Published: 31 July 2019.
Edited by:
Fabienne Brilot, University of Sydney, AustraliaReviewed by:
Fred Lühder, University of Göttingen, GermanyCopyright © 2019 Wouters, de Wit, Vanmol, van der Pol, van het Hof, Sommer, Loix, Geerts, Gustafsson, Steffensen, Vanmierlo, Bogie, Hendriks and de Vries. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Helga E. de Vries, aGUuZGV2cmllc0B2dW1jLm5s
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.