 Marialbert Acosta-Herrera
Marialbert Acosta-Herrera Miguel A. González-Gay
Miguel A. González-Gay Javier Martín
Javier Martín Ana Márquez
Ana Márquez- 1Instituto de Parasitología y Biomedicina “López-Neyra,” CSIC, Granada, Spain
- 2Division of Rheumatology and Epidemiology, Genetics and Atherosclerosis Research Group on Systemic Inflammatory Diseases, Hospital Universitario Marqués de Valdecilla, IDIVAL, University of Cantabria, Santander, Spain
- 3Systemic Autoimmune Disease Unit, Hospital Clínico San Cecilio, Instituto de Investigación Biosanitaria ibs.GRANADA, Granada, Spain
Vasculitides are a heterogeneous group of low frequent disorders, mainly characterized by the inflammation of blood vessels that narrows or occlude the lumen and limits the blood flow, leading eventually to significant tissue and organ damage. These disorders are classified depending on the size of the affected blood vessels in large, medium, and small vessel vasculitis. Currently, it is known that these syndromes show a complex etiology in which both environmental and genetic factors play a major role in their development. So far, these conditions are not curable and the therapeutic approaches are mainly symptomatic. Moreover, a percentage of the patients do not adequately respond to standard treatments. Over the last years, numerous genetic studies have been carried out to identify susceptibility loci and biological pathways involved in vasculitis pathogenesis as well as potential genetic predictors of treatment response. The ultimate goal of these studies is to identify new therapeutic targets and to improve the use of existing drugs to achieve more effective treatments. This review will focus on the main advances made in the field of genetics and pharmacogenetics of vasculitis and their potential application for ameliorating long-term outcomes in patient management and in the development of precision medicine.
Introduction
Systemic vasculitides represent a heterogeneous group of chronic diseases characterized by the inflammation of the blood vessels. These disorders are classified according to the diameter of the affected vessels in large, medium and small vessel vasculitis, and may affect one or several organs and tissues of the body, resulting in different clinical presentations. In the past years, considerable therapeutic advances have been made in the treatment of vasculitis; however, the lack of appropriate therapeutic response and the appearance of side effects remain a major concern (1).
Although the specific mechanisms underlying vasculitis are not fully understood, it is currently known that these conditions show a complex etiology in which both genetic and environmental factors appear to contribute to their pathogenesis (2). In recent years, our knowledge of the genetic landscape of vasculitis has experienced a significant increase, mainly due to the development of large-scale genetic scans, including genome-wide association studies (GWASs) and Immunochip studies, focused on analyzing single-nucleotide polymorphisms (SNPs) in cases and controls (Figure 1). In addition to the human leukocyte antigen (HLA) region, which represents the strongest association in vasculitis, multiple loci located outside the HLA have been shown to play a role in the genetic predisposition to these disorders (Table 1).
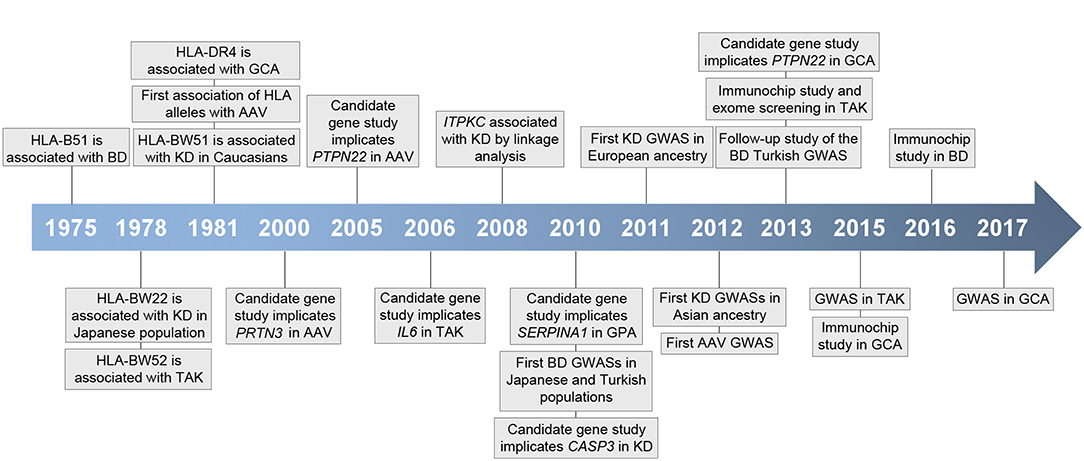
Figure 1. Timeline representing key events in vasculitis genetic research. HLA, human histocompatibility complex; BD, Behçet's disease; GCA, giant cell arteritis; AAV, ANCA-associated vasculitis; KD, Kawasaki's disease; TAK, Takayasu arteritis; GPA, granulomatosis with polyangiitis; GWAS, genome-wide association study.
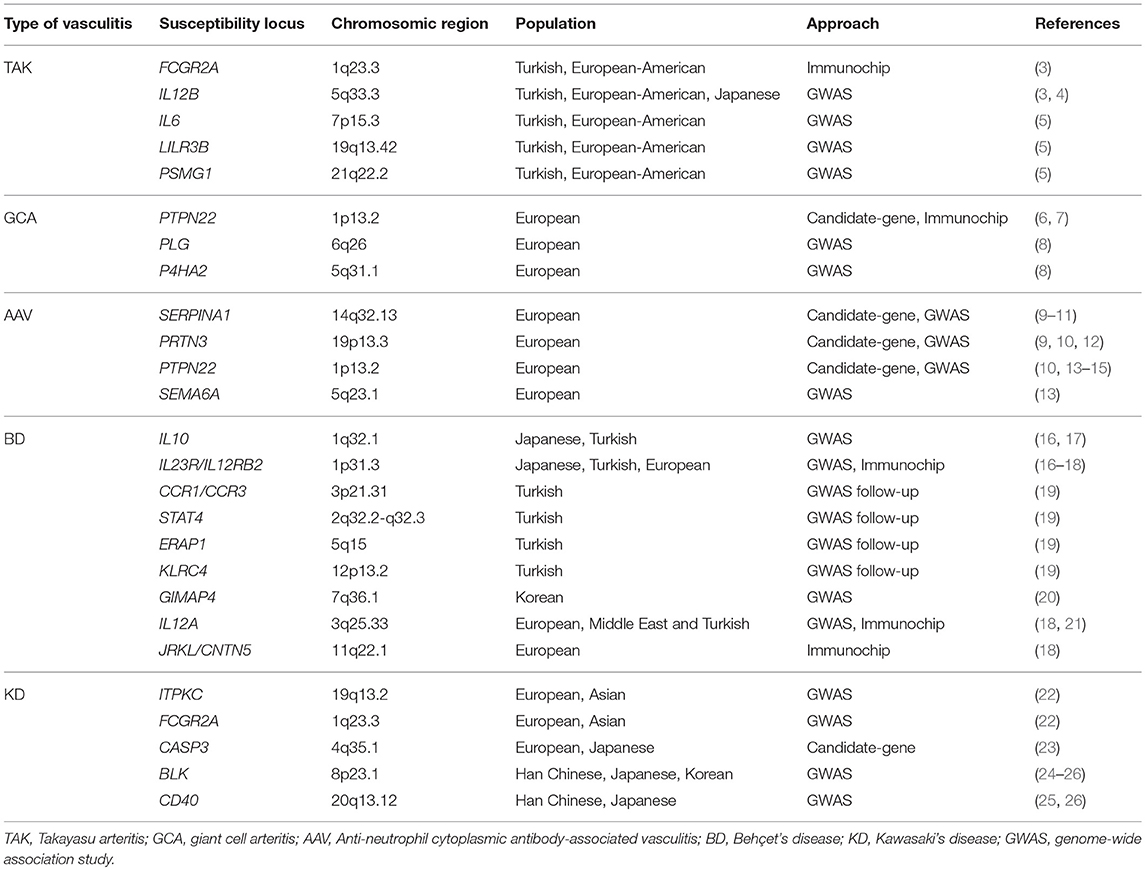
Table 1. Non-HLA loci associated with vasculitis at genome-wide significance level.
Identification of genes and molecular pathways deregulated in vasculitis is crucial to better understand disease pathogenesis and for the development of more effective therapeutic approaches. In this sense, Nelson et al. (27) demonstrated that a drug with genetic support has twice the possibilities of going from phase I to approval in the different phases of drug development, than those drugs without genetic support. The authors found that genes associated with a broad spectrum of human diseases were significantly enriched in target genes for drugs approved in the United States or the European Union, highlighting the importance of the provided genetic knowledge in different drug mechanisms. In addition, genetic studies not only have the potential to identify molecular targets for new therapies, but they also allow us to determine the best way to administer current treatments. In this regard, several pharmacogenetic studies based on candidate genes have identified a number of genetic variants influencing treatment response in different vasculitides (Table 2).
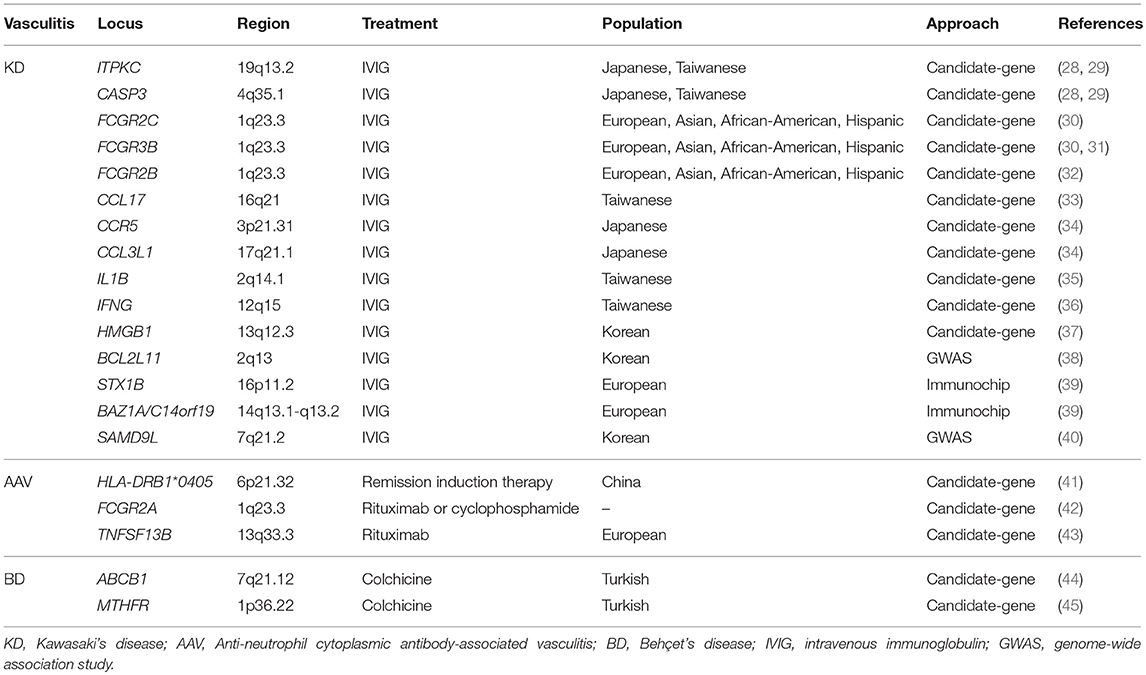
Table 2. Genes associated with treatment response in vasculitis.
This review aims to provide an update of the main findings obtained from genetic and pharmacogenetic studies as well as their potential application to precision medicine in vasculitis.
Contribution of Genetics to New Therapeutic Approaches in Vasculitis
Takayasu Arteritis
Takayasu arteritis (TAK) is a chronic vasculitis characterized by granulomatous inflammation of large vessels, predominantly the aortic arch and its branches, which results in non-specific constitutional symptoms, such as fever and weight loss, and serious complications, including arterial stenosis, occlusion and aneurysm. This disease affects mainly young females with a higher incidence in Asia and Latin America (46).
Genetic studies have shown that the HLA region represents the main genetic risk factor in TAK. Specifically, an association at the genome-wide significance level between the classical allele HLA-B*52:01 and this vasculitis has been reported in TAK patients from Japanese, Turkish and European-American origin (3, 47), and confirmed in Greek, Mexican Mestizo, India, Thai, and Korean populations (48–52). Moreover, independent associations within the HLA class II region, specifically with the DRB1*07 classical allele and DRB1/DQB1 polymorphisms, have also been reported (3, 53, 54); however, additional studies in well-powered populations are required to confirm these findings.
Outside the HLA region, five loci have been consistently associated with TAK through three large-scale genetic analyses (3–5), IL6 (interleukin 6), encoding a cytokine that plays a crucial role in the immune response by regulating the balance between Th17 cells and regulatory T cells (Treg) (55); LILRB3 (leukocyte immunoglobulin like receptor B3), which encodes a protein that binds to HLA class I molecules to inhibit immune cell stimulation (56); IL12B, encoding the p40 subunit of IL-12 and IL-23, two cytokines with a key role in the inflammatory responses mediated by Th1 and Th17 cells, respectively (57); FCGR2A (Fc fragment of IgG receptor IIa) that encodes an immunoglobulin Fcg receptor (FcgR), which has a relevant role in humoral immunity by participating in modulation of antibody production by B cells, phagocytosis, and clearing of immune complexes (58); and an intergenic locus on chromosome 21q22 near PSMG1 (proteasome assembly chaperone 1).
Interestingly, some genes associated with TAK are being explored as therapeutic targets for this vasculitis. On one hand, tocilizumab, a humanized monoclonal antibody against IL-6 receptor (IL-6R), has shown clinical efficacy in TAK patients in several case series studies (59). This efficiency was confirmed in a prospective clinical trial evaluating tocilizumab in refractory TAK, although the primary end-point was not met, probably due to the low number of individuals included in this study (60). A phase III clinical trial evaluating this biological agent as a first-line therapy in TAK patients is currently underway (NCT02101333).
On the other hand, administration of ustekinumab, a monoclonal antibody to the p40 subunit common to IL-12 and IL-23, to patients with active TAK achieved decrease of inflammatory markers but did not improve vascular lesions in a pilot clinical trial (61). In a more recent study, this drug was used to treat a patient with refractory TAK and psoriasis (for which this drug is approved), two diseases that share the genetic risk locus IL12B, with satisfactory results (62). Ustekinumab allowed a significant reduction in glucocorticoid dose and full reduction of vessel wall thickness, thus demonstrating the usefulness of drug repositioning based on the existence of a common genetic component.
Moreover, several evidences, including the association observed between FCGR2A and TAK, indicate that, in addition to T lymphocytes, B cells are also involved in the pathogenesis of this vasculitis. In this regard, depletion of B cells using rituximab, a chimeric anti-CD20 monoclonal antibody, has been shown to be effective in a case series study, achieving clinical and laboratory remission (63). Nevertheless, a randomized control trial is needed in order to confirm the efficacy of rituximab in patients with TAK.
Giant Cell Arteritis
Giant cell arteritis (GCA) is a vasculitis characterized by chronic inflammation of medium- and large-sized blood vessels, mainly the aorta and external carotid arteries and their branches. A severe complication of this disorder is the occlusion of the ophthalmic artery, which leads to acute and irreversible blindness. GCA represents the most frequent vasculitis in elderly individuals from Western countries affecting predominantly women and people over 50 years of age (64).
In the last years, a high number of candidate gene association studies have been performed in GCA, most of them focused on analyzing genes encoding inflammatory cytokines (65). These studies identified the HLA class II region, specifically the classical allele DRB1*04, as the main genetic risk factor in GCA. However, both the low sample size and the lack of replication cohorts of these studies have been limiting factors in the identification of robust genetic associations outside the HLA region. Nevertheless, some of the non-HLA loci associated with this vasculitis using candidate-gene approaches were replicated in different populations (66–71) and, therefore, they represent potential genetic risk factors in GCA, including IL33, which encodes a member of the IL-1 family involved in pro-inflammatory cytokines production, angiogenesis and vascular permeability (72, 73); IL17A, encoding a pro-inflammatory cytokine with a relevant role in the differentiation of Th17 lymphocytes (74); VEGF (vascular endothelial growth factor), encoding a proangiogenic mediator (75); and NLRP1 (NLR family pyrin domain containing 1), encoding a protein implicated in the formation of the inflammasome, which activates caspases 1 and 5 leading to the activation of pro-inflammatory cytokines such as IL-1β and IL-18 (76).
More recently, the emergence of massive genotyping platforms and the formation of a large consortium focused on the study of the genetic basis of GCA have allowed a significant progress in the identification of this genetic component. Until now, two large-scale genetic studies, a GWAS and an Immunochip, have been performed in GCA (6, 8). Both of them have confirmed the classical allele HLA-DRB1*04 as the most consistent association with this vasculitis. In addition, several non-HLA loci have been also found to play a role in the GCA genetic predisposition, including PTPN22 (protein tyrosine phosphatase non-receptor type 22), PLG (plasminogen), and P4HA2 (prolyl 4-hydroxylase subunit alpha 2).
The association between PTPN22 and GCA was initially identified in a candidate-gene association study (7) and subsequently confirmed by using the Immunochip strategy (6). This gene encodes LYP, a tyrosine phosphatase involved in several immune signaling pathways, such as the T cell receptor (TCR) pathway and the humoral activity of B cells. The strongest signal within this locus corresponds to a functional variant (rs2476601), previously associated with multiple immune-mediated disorders, that results in a non-synonymous arginine to tryptophan amino acid change (R620W). It has been described that carrying the rs2476601 risk allele results in enhanced B lymphocyte autoreactivity, deregulated TCR signaling, and reduced capacity for TLR-induced type 1 interferon (IFN) production (77). On the other hand, PLG, encoding plasminogen, is involved in different processes relevant for GCA, such as angiogenesis, lymphocyte recruitment, and production of inflammatory mediators, including tumor necrosis factor alpha (TNF-α) and IL-6 (78), and P4HA2, encoding an isoform of the alpha subunit of the collagen prolyl 4-hydroxylase, is an important hypoxia response gene whose expression is induced by hypoxia-inducible factor-1 (HIF-1), which also induces the expression of other genes involved in GCA such as IL6, MMP9 (matrix metallopeptidase 9), and VEGF (79).
These genetic findings, together with other lines of evidence, have contributed to the identification of several molecular pathways implicated in the GCA pathogenesis. Currently, it is known that both Th1 and Th17 cells are relevant player in GCA with two main cytokine clusters contributing to the local inflammation, the IL-6/IL-17 and the IL-12/IFN-γ axes (74). Interestingly, whereas the inflammatory activity of the IL-6/IL-17 cytokine cluster seems to be affected by glucocorticoid treatment, the IL-12/IFN-γ cytokine cluster is resistant to this therapy. This, together with the adverse events associated with long-term glucocorticoids use, has led to the search for new therapeutical agents.
Considering the major role of IL-6 in the pathogenesis of GCA, the potential use of tocilizumab in the treatment of this vasculitis has been explored. IL-6 inhibition has shown clinical efficacy in several randomized controlled trials (80, 81), thus representing a promising therapeutic strategy for this type of vasculitis. Indeed, tocilizumab has been recently approved to treat GCA by the United States Food and Drug Administration (FDA). Furthermore, other biological agents, such as ustekinumab and abatacept, a fusion protein comprising the Fc region of IgG1 and the extracellular domain of cytotoxic T lymphocyte antigen 4 (CTLA4) that inhibits the co-stimulatory signal required for T cell activation, have also shown encouraging but more moderate results (82, 83). Better powered studies are required in order to evaluate the efficacy of these drugs in GCA.
Finally, the potential therapeutic application of two monoclonal antibodies, anakinra and secukinumab, targeted against IL-1β receptor and IL-17A (one of the genes associated with GCA), respectively, is currently under investigation (NCT02902731 and NCT03765788). Both cytokines are crucial for the differentiation of Th17 cells and, therefore, their inhibition could be a therapeutic option in patients with GCA.
ANCA-Associated Vasculitis
Anti-neutrophil cytoplasmic antibody (ANCA)-associated vasculitis (AAV) is a group of disorders characterized by necrosing inflammation of small vessels, including arterioles, capillaries and venules, that comprises three separate conditions, granulomatosis with polyangiitis (GPA), microscopic polyangiitis (MPA), and eosinophilic granulomatosis with polyangiitis (EGPA). AAV frequently affects small vessels in the respiratory tract and kidneys and is characterized by the presence of antibodies directed against two proteins, proteinase 3 (PR3) and myeloperoxidase (MPO), located on the membrane of monocytes and neutrophils (84).
Both candidate gene association studies and GWASs published in last years have identified several loci associated with these forms of vasculitis (2). Specifically, three GWASs on AAV have been performed so far (9, 10, 13), one in European patients with GPA and MPA and two in North American patients of European descent (one including patients with GPA and the other one including patients with GPA and MPA). Interestingly, these studies have shown that the genetic background of AAV depends on auto-antibody specificity rather than clinically defined disorders. In this regard, different HLA genes have been associated with the different ANCA subgroups; whereas polymorphisms within the DPB1 and DPA1 genes appeared to be associated with PR3-ANCA-positive patients, DQB1 showed a specific effect in the MPO-ANCA subgroup (10).
Additionally, four non-HLA genetic loci, SERPINA1 (serpin family A member 1), PRTN3 (proteinase 3), PTPN22, and SEMA6A (semaphorin 6A) have been associated with AAV at genome-wide significance level (9, 10, 13), the first two showing a specific association with the subgroup of patients positive for PR3-ANCA.
SERPINA1 encodes α1-antitrypsin (α1AT), an inhibitor of the serine proteases, including proteinase 3. The association between this gene and AAV was initially described in a candidate gene study (11), in which a functional genetic variant known to cause a deficient production of α1-AT appeared to be associated with GPA, and subsequently confirmed by GWAS (9, 10). It has been proposed that, since PR3 is a target of α1AT, a decreased production of this inhibitor could result in higher levels of circulating PR3, leading to the synthesis of anti-PR3 ANCA (11).
Regarding PRTN3, the role of this gene in AAV was described in a candidate gene association study, in which a genetic variant affecting a putative transcription factor-binding site was associated with GPA (12). Subsequently, two of the GWASs performed in AAV have confirmed this association (9, 10). Interestingly, the most recent GWAS reported that the lead SNP at this gene (rs62132293), which is in almost complete linkage disequilibrium (LD) with that described in the original study, acts as an expression-quantitative trait locus (eQTL) that results in an increased expression of PRTN3 in neutrophils (10).
The protein encoded by SEMA6A has been characterized as a critical regulator of angiogenesis by modulating VEGF signaling (85). Nevertheless, it should be considered that, although polymorphisms within this locus showed genome-wide significance (13), this association was not subsequently validated in a replication study performed in a well-powered cohort of European AAV patients (including GPA, MPA, and EGPA cases) (86) or in the subsequent GWAS carried out by the same group (10). Therefore, further genetic association studies are needed to confirm the role of SEMA6A in AAV.
Finally, PTPN22 has been consistently associated with both GPA and MPA by candidate gene and genome-wide studies (10, 13–15). The highest signal within the PTPN22 locus lies on the R620W functional variant, the same one associated with GCA, thus pointing to a pleiotropic effect of this polymorphism in both vasculitis.
On the other hand, polymorphisms within the CTLA4 locus, encoding a protein which transmits an inhibitory signal to T cells by blocking the interaction between CD28 on the T cell and CD80 or CD86 on the antigen-presenting cell, have been implicated in AAV by candidate gene analyses in different populations (87–92) and have shown suggestive association in GWASs (9, 13), thus supporting the idea that this locus represents a genetic risk factor for AAV.
Regarding EGPA, no GWAS has been published in this disease so far and the few associations reported to date have been identified using a candidate-gene strategy. In this regard, an early candidate-gene study reported an association between EGPA and the HLA-DRB4 gene (93), which highlights the existence of different HLA associations for the different AAV subgroups. In addition, polymorphisms within several non-HLA genes, including FCGR3B and IL10, have also been associated with this vasculitis (94, 95). On the other hand, a role of eotaxin-3 in the EGPA pathogenesis was proposed. Interestingly, serum levels of this molecule were found to be increased in active patients and correlated with eosinophil counts, total immunoglobulin E (IgE) levels and acute-phase parameters (96, 97). However, a genetic study failed to identify an association between the gene encoding this molecule, CCL26, and EGPA, maybe due to a lack of statistical power (97). Therefore, the role of this gene as a susceptibility factor for EGPA remains to be clarified.
Although we are still far from fully understanding the pathogenic mechanisms implicated in AAV, genetic studies are contributing to their elucidation. Insights into these pathogenic pathways have opened new strategies for biological treatment. In this line, the central role that ANCA-mediated neutrophil activation plays in these disorders has led to the therapeutic use of B cell depleting drugs for AAV. Rituximab has proved to be highly effective for both remission induction and maintenance treatment (98), representing one of the major breakthroughs of the last decade in the treatment of these vasculitides. Additionally, the therapeutic potential of other B cell-targeting agents, such as belimumab, is being evaluated (NCT01663623). Belimumab is a humanized monoclonal antibody against BAFF, a potent B cell activator, which represents an interesting target for AAV treatment, since it has been reported to increase the production of PR3-ANCA in GPA patients (99).
On the other hand, B cells require T cell help to differentiate into antigen-specific Ig-producing plasma cells. Therefore, blockade of the co-stimulation signal required for full T cell activation using abatacept (CTLA4-Ig) is also an interesting treatment option that has shown clinical efficacy in an open-label clinical trial (100).
Behçet's Disease
Behçet's disease (BD) is an inflammatory disorder that may affect arteries and veins of all sizes. It is characterized by heterogeneous clinical manifestations, including oral and genital ulcers, which are the hallmark lesions of this vasculitis, as well as vascular, gastrointestinal, articular, and central nervous system manifestations. This condition shows a male preponderance and is more frequent in the Middle East and Asia (101).
BD is the vasculitis that has benefited most from the genome-wide era, with five large-scale genetic studies performed on this disorder so far, four GWASs and an Immunochip (16–18, 20, 21). This has lead to the discovery of a significant number of consistent genetic risk loci, including the HLA region that, as in other vasculitis, is the main susceptibility locus for BD. Specifically, an association between the classical allele HLA-B*51 and this disorder has been consistently identified in different ethnic groups during the last years (102). Moreover, dense genotyping and imputation of this region have evidenced additional independent signals. In this regard, a study published in 2013 reported that the association between HLA-B*51 and BD was explained by a SNP located between the HLA-B and MICA genes (103). They also identified three independent signals within the HLA region, located within PSORS1C1 (psoriasis susceptibility 1 candidate 1), upstream HLA-F-AS1 (HLA-F antisense RNA 1), and within HLA-Cw*16:02. In addition, a subsequent Immunochip study also described two signals, HLA-B*57 and HLA-A*03, that showed an independent effect to that conferred by HLA-B*51 (18).
Several loci outside the HLA region have also shown robust associations with this vasculitis. The first GWASs on BD, performed in Japanese and Turkish populations and simultaneously published in 2010, evidenced the role of IL10 and IL23R/IL12RB2 as genetic risk factors in BD (16, 17). The association with the IL23R/IL12RB2 locus was subsequently confirmed in an Immunochip study performed in BD patients from Spain (18). IL10 encodes a cytokine that has an anti-inflammatory role by suppressing the expression of pro-inflammatory cytokines, such as IL-6, IL-12, and IL-1, but also promotes B cell responses by enhancing B cell survival, proliferation, and antibody production (104). The IL23R/IL12RB2 locus contains two genes with a crucial role in the inflammatory response. IL23R encodes a subunit of the IL-23 receptor, whereas IL12RB2 encodes an IL-12 receptor chain. As previously indicated, IL-12 and IL-23 participate in the immune responses mediated by Th1 and Th17 cells, respectively (57).
In addition, a follow-up study, in which data from the Turkish GWAS were imputed, identified four new loci contributing to the BD susceptibility, CCR1/CCR3 (C-C motif chemokine receptor 1/3), STAT4 (signal transducer and activator of transcription 4), KLRC4 (killer cell lectin like receptor C4), and ERAP1 (endoplasmic reticulum aminopeptidase 1) (19). All these loci play relevant roles in the immune response. The CCR1 and CCR3 genes form a chemokine receptor gene cluster, which also includes CCR2, CCRL2, CCR5, and CCXCR1, on the chromosomal region 3p21. These genes encode proteins critical for the recruitment of effector immune cells to the site of inflammation (105). The protein encoded by STAT4 is a member of the STAT family of transcription factors that mediates responses to IL-12, IL-23, and type 1 IFNs, and regulates the differentiation of Th1 and Th17 lymphocytes (106). The signal detected at the KLRC4 region is located within a haplotype block that contains five natural killer (NK) cell receptor genes (KLRK1, KLRC1, KLRC2, KLRC3, and KLRC4), some of which act as co-stimulators for CD4+ and CD8+ T cells (107). Finally, ERAP1 encodes an amino peptidase that is crucial for antigen presentation through HLA class I molecules. Interestingly, ERAP1 variants conferred risk for BD in HLA-B*51 positive individuals preferentially, thus suggesting the existence of an interaction between both proteins (19).
In 2013, a third GWAS performed on BD patients from Korea reported GIMAP4 (guanosine-5′-triphosphatase (GTPase) IMAP family member 4) as a new susceptibility locus (20). This gene encodes a protein belonging to the GTP-binding superfamily and to the immuno-associated nucleotide (IAN) subfamily of nucleotide-binding proteins that seems to play a role in regulating T cell apoptosis (108). Functional studies performed by the authors revealed that the minor allele of the most associated SNP within this region correlated with lower protein activity, and that CD4+ T cells from BD patients have a diminished GIMAP4 expression (20).
A genome-wide association between IL12A, encoding the p35 subunit of IL-12, and BD has also been described in a GWAS performed on an admixed cohort including Western Europeans, Middle Eastern and Turkish cases with BD (21). This association was confirmed in a subsequent Immunochip study, in which the JRKL/CNTN5 (jerky like/contactin 5) locus was also identified as a new genetic risk factor for BD (18). The protein encoded by JRKL has an unknown function, whereas CNTN5 encodes a member of the immunoglobulin superfamily that mediates cell surface interactions during nervous system development (109).
BD treatment has undergone a significant evolution over the years, thanks to the increased knowledge of the pathogenic mechanisms involved in this disease. Genetic findings have evidenced the prominent role of immune responses mediated by Th1 and Th17 cells in BD, with multiple pro-inflammatory molecules contributing to its pathological landscape. This has led to the study of new biological therapies, most of them targeted against cytokines.
In this line, inhibition of IL-1 and IL-6 has shown the most interesting results in both small case series and clinical trials. Specifically, three IL-1 blockers have shown clinical efficacy in BD patients, the IL-1 receptor antagonist anakinra, as well as canakinumab and gevokizumab, targeting the IL-1 molecule directly, which have proved to be effective for all BD manifestations, especially for the most severe ocular involvement (110). In relation to IL-6 inhibition, tocilizumab has proved to be highly effective in treating BD patients with neurological involvement as well as in controlling uveitis, although less promising results were found regarding the treatment of mucocutaneous manifestations (110). Ustekinumab and secukinumab have also shown clinical efficacy in case series studies (111, 112). In addition, the clinical utility of Ustekinumab is currently being evaluated in a phase 2 clinical trial (NCT02648581).
Although there are more clear evidences of T cell involvement in BD, several studies have suggested a possible pathogenic role of B lymphocytes. Indeed, depletion of B cells using rituximab has also emerged as a promising therapy in BD patients (110).
Kawasaki's Disease
Kawasaki's disease (KD) is a systemic vasculitis that affects small and medium size vessels. It mainly affects children younger than 5 years of age, especially of Asian origin. The most serious complication of KD is the development of coronary artery lesions (CALs), representing the main cause of acquired heart disease among children in Japan, Europe and the USA (113).
Although seven GWASs and an Immunochip have been published in both European and Asian KD cohorts in recent years (22, 24–26, 39, 114–116), only a few consistent genetic associations have been described so far, probably due to the lack of statistical power of most of these studies.
As in other vasculitis, the HLA locus seems to be involved in the KD genetic predisposition. However, contradictory results have been found regarding the specific HLA alleles associated with this disease. Whereas early candidate gene studies identified associations with HLA-Bw54 (previously known as Bw22) in Japanese population (117, 118) and with HLA-Bw51 and HLA-B44 in European patients (119–121), a genetic variant located within the HLA class II region (between HLA-DQB2 and HLA-DOB) was identified as the strongest signal in a GWAS performed on KD cases from Japan (26). This association within the HLA class II region was then replicated in an European-American case-parent trio study using the Immunochip platform (39). In addition, a more recent GWAS identified several SNPs within the HLA class I region associated with KD in Korean population, but failed to replicate this association in an independent case/control set from Japan (24). The small sample sizes of these studies and the varying LD patterns observed across different populations are likely explanations of these heterogeneous findings.
Regarding genetic risk factors outside the HLA locus, genome-wide associations within the ITPKC and the FCGR2A loci were identified in a GWAS performed in European KD patients and replicated in independent cohorts of Asian and European descent (22). The KD-associated SNP within FCGR2A is a functional variant encoding a H131R substitution. It has been reported that the presence of arginine instead of histidine at this amino acid position reduces the affinity of the receptor for the IgG2 isotype (122). ITPKC (inositol-trisphosphate 3-kinase C), encoding one of the three isoenzymes of ITPK that phosphorylate inositol 1,4,5-trisphosphate (IP3), was initially implicated in KD by linkage analysis using sib-pairs (123). This same study showed for the first time that ITPKC acts as a negative regulator of T cell activation through the Ca2+/nuclear factor of activated T cells (NFAT) signaling pathway. Interestingly, a subsequent study showed that the genetic variant associated with KD has functional consequences, influencing the ITPKC protein levels, which regulates the production of IL-1β and IL-18 (124). Moreover, using a positional candidate gene study for the 4q35 region, previously linked to KD, Onouchi et al. identified several genome-wide associations within the CASP3 (caspase 3) gene, which encodes a caspase with a crucial role in apoptosis (23). Similarly to the function identified for ITPKC, this study also reported that one of the associated SNPs, located within the 5′ untranslated region of the gene, had functional implications, affecting binding of NFATc2 to the DNA sequence surrounding this polymorphism.
In 2012, two subsequent GWASs, published simultaneously, identified two new susceptibility loci for KD, BLK (BLK proto-oncogene, Src family tyrosine kinase) and CD40 (CD40 molecule) (25, 26). BLK encodes a non-receptor tyrosine-kinase of the src family of proto-oncogenes with a crucial role in B cell receptor signaling, thus participating in B-cell activation and antibody secretion (125). The CD40 gene is a member of the TNF-receptor superfamily that encodes a receptor expressed on antigen-presenting cells involved in inflammation through selection of autoreactive T cells and activation of B and T cells (126).
Additionally, large-scale genetic studies have reported suggestive signals in different loci, including CAMK2D (calcium/calmodulin-dependent protein kinase II delta), CSMD1 (CUB and Sushi multiple domains 1), LNX1 (ligand of numb-protein X1), NAALADL2 (N-acetylated alpha-linked acidic dipeptidase-like 2), TCP1 (t-complex 1), PELI1 (pellino E3 ubiquitin protein ligase 1), DAB1 (Dab reelin signal transducer homolog 1), COPB2 (coatomer protein complex beta-2 subunit), ERAP1, NMNAT2 (nicotinamide nucleotide adenylyltransferase 2), FUT1 [fucosyltransferase 1 (H blood group)], RASIP1 (Ras interacting protein 1), and BRD7P2 (bromodomain containing 7 pseudogene 2) (24, 39, 114–116). However, these associations did not reach genome-wide significance level nor were replicated in later studies and, therefore, they cannot be considered as established susceptibility loci.
As shown by genetic studies, both T and B cells participate in the pathogenic mechanisms implicated in KD. The involvement of the FCGR2A gene evidences the relevant role of IgG receptors in the pathogenesis of this vasculitis, providing a biological basis for the use of intravenous immunoglobulin (IVIG), the standard treatment for KD patients. However, approximately up to 20% of patients do not fully respond to this therapy, presenting an increased risk for the development of coronary aneurysms (127). Therefore, several therapeutic options are being tested for treatment of refractory cases.
Considering the role of TNF-α in the pathogenesis of KD, the clinical efficacy of TNF-α inhibitors, such as infliximab and etanercept, has been evaluated. Although treatment with infliximab has shown clinical efficacy in different studies, including reduction of fever duration, markers of inflammation and immunoglobulin reaction rates, its role on the prevention of CALs is still to be determined (128). A phase III trial comparing the efficacy of a second dose of IVIG with infliximab treatment is currently recruiting participants (NCT03065244). In addition, a phase II clinical trial to determine the safety and efficacy of etanercept in reducing the incidence of persistent or recurrent fever in KD patients is currently ongoing (NCT00841789).
On the other hand, studies in mice have demonstrated that both IL-1α and IL-1β are involved in the development of CALs in KD (129, 130). Interestingly, PELI1, encoding a protein that acts an intermediate component in the signaling cascade initiated by the IL-1 receptor (131), showed a suggestive association with CALs development in a KD GWAS (115). Thus, genetic findings also support the role of this pathway as a potential drug target in this vasculitis. Considering this, the potential clinical efficacy of blocking IL-1β receptor using anakinra is currently being explored. Three case reports have reported the beneficial clinical use of this biological agent (128). Moreover, two phase II clinical trials exploring the efficacy of anakinra are currently underway (NCT02179853 and NCT02390596).
Polyarteritis Nodosa
Polyarteritis nodosa (PAN) is a systemic, necrotizing medium-sized vessel vasculitis, mainly affecting adults between the ages of 40–60 years, although it can also appear in children. The clinical features of PAN depend on the affected organs and include systemic symptoms and involvement of the gastrointestinal, renal and peripheral nervous systems.
In 2014, two independent studies identified loss-of-function mutations in the CECR1 (cat eye syndrome chromosome region candidate 1) gene, which encodes the extracellular adenosine deaminase 2 (ADA2), using whole-exome sequencing (132, 133). Interestingly, in many cases, both the clinical manifestations and the histological findings of the deficiency of ADA2 (DADA2) were consistent with the diagnosis of PAN, which suggests that DADA2 contributes to the clinical phenotype of this vasculitis. The ADA2 protein is mainly expressed by myeloid cells and plays a role in the proliferation and differentiation of macrophages. In this regard, its deficiency has been linked to an imbalance in monocytes differentiation toward proinflammatory M1 macrophages (133, 134).
Clinical management of patients with DADA2 is challenging. None of the commonly used immunosuppressive drugs have resulted particularly effective. Anti-TNF agents have shown promise in the management of the inflammatory syndrome and vasculitis; however, this therapy is not able to completely control the disease manifestations in all treated patients. Considering that bone marrow–derived monocytes and macrophages are the main source of secreted ADA2, it was hypothesized that hematopoietic stem cell transplantation (HSCT) could be an effective treatment for this condition. In this regard, two studies have reported that HSCT was able to normalize the plasmatic levels of ADA2 and to control the disease manifestations (135–137), thus suggesting that this therapy could represent a definitive treatment of DADA2. In addition, enzyme-replacement therapies have also been considered as a potential treatment for these conditions. However, the results obtained using this strategy have not been entirely satisfactory (138).
Genetics Determinants of Treatment Response in Vasculitis
The genetic basis of treatment response has only been evaluated in three types of vasculitis so far, KD, AAV, and BD, mainly by means of candidate-gene association studies. This has led to the identification of several potential genetic predictors of treatment efficacy.
Most of the pharmacogenetic studies performed in vasculitis have analyzed genetic variants involved in the resistance to IVIG therapy in KD. As it was already mentioned, this treatment is highly effective, but around 10–20% of patients are resistant and have a higher risk for CALs. Therefore, it is essential to elucidate the causes of this resistance in order to predict the responsiveness of patients during the early stages of the disease.
Polymorphisms previously associated with KD have been evaluated in relation to the IVIG response. A study published in 2013 showed that the risk alleles of the ITPKC and CASP3 susceptibility variants (rs28493229 and rs72689236, respectively) were overrepresented in IVIG resistant patients with respect to responding patients (28). These associations were replicated in a subsequent study (29), thus supporting the role of these genes in the response to IVIG treatment. Interestingly, a functional study demonstrated that the poor response observed in patients homozygous for the risk allele of the ITPKC locus correlated with increased cellular production of IL-1β and IL-18 (124).
On the other hand, additional studies have evaluated the implication of candidate genes in the clinical efficacy of IVIG based on their functional role. Given that the anti-inflammatory activity of IVIG is partly mediated through FcgR (139), the role of several genes encoding these proteins have been explored. In this regard, polymorphisms within FCGR2B, FCGR2C, and FCGR3B have been involved in the response to this drug in different studies performed by the same group (30–32). In addition, genetic variants located within genes encoding chemokine receptors and their ligands, including CCR5 (C-C motif chemokine receptor 5), CCL3L1 (C-C motif chemokine ligand 3 like 1), and CCL17 (C-C motif chemokine ligand 17), as well as genes encoding pro-inflammatory cytokines, such as IL1B and IFNG, have also been implicated in the IVIG treatment resistance (33–36). Moreover, an association between a polymorphism of the HMGB1 (high mobility group box 1) locus, involved in inflammation and cell differentiation, and the clinical efficacy of this treatment has been recently reported (37). However, all these associations need to be confirmed in independent studies.
The genetic basis of IVIG response in KD has also been explored through comprehensive large-scale genetic analyses (38–40). Both GWAS and Immunochip data have been used to identify genetic variants associated with IVIG resistance by stratifying KD patients according to treatment response. A polymorphism within the BCL2L11 (BCL2 like 11) gene showed a specific association at the genome-wide significance level with the subgroup of responder patients in an IVIG response-stratified genome-wide association study (38). The protein encoded by this gene, known as Bim, is an important regulator of the negative selection of B lymphocytes in the bone marrow and of T lymphocytes both in the thymus and the periphery (140). In addition, a very recent GWAS performed in Korean KD patients identified the SAMD9L (sterile alpha-motif domain-containing 9-like) gene as a susceptibility factor for IVIG resistance (40). This gene encodes a cytoplasmic protein involved in multiple cellular processes, such as cell proliferation and innate immune responses to viral infections (141). Several suggestive signals that could be involved in the response to IVIG therapy were identified using the Immunochip platform, including an intronic SNP of the STX1B (syntaxin 1B) gene and a genetic variant located in the intergenic region of BAZ1A (bromodomain adjacent to zinc finger domain 1A) and C14orf19 (39).
A study published in 2017 developed a genetic model to predict IVIG resistance in KD patients (142). In this study, the additive effect of 11 SNPs associated with IVIG response (p < 1 × 10−05) was used to calculate a GWAS-based weighted genetic risk score (wGRS). A significant association between wGRS and the response was found, suggesting that this scoring system can significantly increase the sensitivity and specificity of prediction of IVIG responsiveness.
Regarding AAV, the advances achieved in its therapeutic management in recent years have allowed these forms of vasculitis to go from presenting high mortality to becoming chronic diseases. Currently, the standard treatment for AAV consists of glucocorticoids together with cyclophosphamide or rituximab. However, despite the success of this therapy, a high percentage of patients do not reach complete remission.
Only three pharmacogenetic studies have evaluated the role of genetic variants as predictors of treatment response in AAV so far. One of them, performed in 152 AAV patients from China, was focused on analyzing the possible implication of the HLA locus in the response to remission induction therapy after 6 months (41). Among the 56 HLA-DRB1, HLA-DPB1, HLA-DQB1, and HLA-DQA1 alleles analyzed, HLA-DRB1*0405 appeared to be associated with the clinical efficacy of this treatment; specifically, the proportion of patients showing treatment failure was higher in the subgroup of patients carrying this allele (41.7%) than in the subgroup of patients negative for HLA-DRB1*0405 (12.9%).
On the other hand, the main mechanism through which rituximab achieves B cell depletion is antibody-dependent cell mediated cytotoxicity (ADCC), which is mediated through FcgR. Regarding cyclophosphamide, it requires activation by the hepatic cytochrome P450 (CYP) enzymes. Considering this, a more recent study has explored the role of several polymorphisms, located within three genes encoding FcgR (FCGR2A, FCGR2B, FCGR3A) and two genes encoding different CYP isoforms (CYP2B6 and CYP2C19), in the response to the treatment with rituximab and cyclophosphamide, respectively (42). When both subgroups of patients (96 treated with rituximab and 93 with cyclophosphamide) were individually analyzed, the authors did not find any potential predictor of treatment response among the genetic variants selected. However, when AAV patients were considered as a global cohort, the FCGR2A 519AA genotype was found to predict complete response independently of the induction treatment used.
In addition, a study published in 2017 evaluated the role of several candidate genes in the rituximab response in two independent cohorts of patients with AAV, including MPA and GPA (43). Only one (rs3759467) of the 18 analyzed SNPs showed a consistent association with treatment efficacy. Interestingly, this association was specific for the subgroup of patients PR3-ANCA positive. The associated SNP is located in the 5′ regulatory region of the TNFSF13B gene, encoding the B-cell activating factor BAFF, which has been reported to increase the production of PR3-ANCA in GPA patients (99), as previously mentioned.
Finally, pharmacogenetic studies performed in BD were focused on analyzing genetic factors implicated in the response to colchicine. This drug is the most frequently and widely used for oral and genital ulcers, papulopustular lesions, and arthralgias; however, some patients do not respond to this therapy.
Until now, two genes have been associated with colchicine response in BD. A study published in 2012 identified an association between two SNPs, C3435T and G2677T/A, of the ABCB1 (ATP binding cassette subfamily B member 1) gene and the efficacy of this treatment in a candidate-gene study including a cohort of 68 responder and 37 non-responder patients (44). ABCB1, also known as MDR1 (multidrug resistance), is implicated in drug metabolism by encoding an ATP-dependent drug efflux pump for different xenobiotic compounds, including colchicine (143).
A second pharmacogenetic study, in which 165 responder and 215 non-responder patients were analyzed, reported a role of the MTHFR (methylenetetrahydrofolate reductase) locus in the response to colchicine treatment (45). This gene encodes an enzyme that catalyzes the conversion of 5,10-methylenetetrahydrofolate to 5-methyltetrahydrofolate, a co-substrate for homocysteine remethylation to methionine. In this case, the polymorphism associated with the response, C677T, causes an amino acid substitution from alanine to valine leading to reduced activity and increased thermolability of the enzyme, which in turn results in increased levels of homocysteine (144). It has been described the existence of hyperhomocysteinemia in BD patients, which correlates with thrombosis and ocular involvement (145).
Shared Genetic Component in Vasculitis
Nowadays, it is widely accepted that autoimmune disorders in general and vasculitides in particular share susceptibility genes and molecular pathways influencing their development (146, 147). Indeed, a large number of susceptibility loci described here are common to different vasculitides. The combination of different diseases as a single phenotype in large-scale studies, such as GWAS and Immunochip, has proven to be very useful in the evaluation of this shared genetic component and in the identification of potential drug targets that could be repurposed in related conditions (148–151).
To date, two studies have been conducted combining different forms of vasculitides. In the first one, Carmona et al. (152) combined data from large-vessel vasculitis, namely GCA and TAK, and found a significant genetic correlation within the IL12B locus. Considering this, ustekinumab, which has been successfully used to treat refractory TAK, could be of potential clinical use in GCA. Similarly, Ortiz-Fernandez et al. (153) combined data of different vasculitides (GCA, TAK, AAV, IgA vasculitis, and BD) and identified a common signal within the lysine demethylase 4C (KDM4C) gene, which encodes a histone demethylase involved in epigenetic mechanisms and that could be of potential use in the treatment of these conditions.
Precision Medicine in Vasculitis: From Genetic Findings to Clinical Application
The goal of precision medicine is to maximize treatment efficacy by developing more targeted drugs directed against biological pathways with a pathogenic role in the disease, as well as by optimizing the use of existing drugs, through the a priori selection of those patients who will benefit from a certain treatment.
As described in this review, it is now clear that genetic studies offer great potential for understanding the molecular mechanisms involved in vasculitis. Thus, insight into disease pathogenesis is progressively leading to new ways for targeted biologic treatment. Moreover, based on the moderate effects provided by the thousands of genome-wide SNPs identified by GWAS, nowadays it is possible to predict each individual susceptibility by means of the polygenic risk score (PRS) analysis, which have been recently performed in other diseases with remarkable results (154). Currently, PRSs are being calculated for different phenotypes separately and, as a potential next step, parallel calculation and disease comparisons of PRS could reflect shared and opposite mechanisms in different vasculitides. However, although these diseases have benefited from the genome-wide era, genetic studies conducted to date still lack enough statistical power to detect variants with moderate effects and, consequently, only a few consistent genetic risk loci have been identified so far. Therefore, further genetic studies in larger cohorts are crucial to obtain information on the missing heritability of these disorders.
Moreover, in recent years, the translation of GWAS/Immunochip findings into biological insights has been challenging, mainly due to the difficulty of identifying causal variants, as well as by the fact that many of the disease-associated SNPs are located in non-coding regions of the genome. Therefore, substantial effort is needed to move from association signals to understanding the functional implication of the genes. In this sense, integration of genomic data with other–omic information, such as epigenomic and transcriptomic data, has become a useful approach to unravel the mechanisms underlying complex diseases. Thus, a better understanding of the interaction between these factors will allow us to obtain a clearer picture of the molecular network involved in the pathogenesis of vasculitis, so that we may turn basic biological knowledge into targets for new therapeutic approaches.
On the other hand, it is likely that a better use of existing drugs will improve the clinical management of vasculitis. In this regard, prediction of those patients that will respond to a specific drug based on their molecular profiles results essential. Although several genetic variants have been described as potential predictor of drug efficacy, mainly in KD but also in AAV and BD, at present, no validated biological biomarker exists to predict treatment response in vasculitis. Again, large-scale genetic studies including well-powered cohorts will be essential to identify genetic profiles that help to classify vasculitis patients and to guide the selection of the most appropriate therapeutic intervention.
It is, therefore, expected that genetic findings in vasculitis continue to open new ways for targeted biologic therapies and improve the use of existing drugs, which will lead to a more personalized application of treatment in the future. However, multiple issues must be overcome before precision medicine can be effectively implemented, which will necessarily require great collaborative efforts among vasculitis expertise research groups.
Author Contributions
MA-H and AM wrote this review. MG-G and JM critically read and edited the manuscript.
Funding
This work was supported by the following grants: PI18/00040 from Instituto de Salud Carlos III (ISCIII, Spanish Ministry of Economy, Industry, and Competitiveness) and the Cooperative Research Thematic Network (RETICS) programme (RD16/0012/0013 and RD16/0012/0009) (RIER), from ISCIII (Spanish Ministry of Economy, Industry and Competitiveness). AM is recipient of a Miguel Servet fellowship (CP17/00008) from ISCIII (Spanish Ministry of Economy, Industry and Competitiveness).
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The handling editor declared a past co-authorship with one of the authors JM.
The reviewer DM declared a past co-authorship with one of the authors with the authors JM and AM to the handling editor.
References
1. Katsuyama T, Sada KE, Makino H. Current concept and epidemiology of systemic vasculitides. Allergol Int. (2014) 63:505–13. doi: 10.2332/allergolint.14-RAI-0778
2. Carmona FD, Martin J, Gonzalez-Gay MA. Genetics of vasculitis. Curr Opin Rheumatol. (2015) 27:10–7. doi: 10.1097/BOR.0000000000000124
3. Saruhan-Direskeneli G, Hughes T, Aksu K, Keser G, Coit P, Aydin SZ, et al. Identification of multiple genetic susceptibility loci in Takayasu arteritis. Am J Hum Genet. (2013) 93:298–305. doi: 10.1016/j.ajhg.2013.05.026
4. Terao C, Yoshifuji H, Kimura A, Matsumura T, Ohmura K, Takahashi M, et al. Two susceptibility loci to Takayasu arteritis reveal a synergistic role of the IL12B and HLA-B regions in a Japanese population. Am J Hum Genet. (2013) 93:289–97. doi: 10.1016/j.ajhg.2013.05.024
5. Renauer PA, Saruhan-Direskeneli G, Coit P, Adler A, Aksu K, Keser G, et al. Identification of susceptibility loci in IL6, RPS9/LILRB3, and an intergenic locus on chromosome 21q22 in Takayasu arteritis in a genome-wide association study. Arthritis Rheumatol. (2015) 67:1361–8. doi: 10.1002/art.39035
6. Carmona FD, Mackie SL, Martin JE, Taylor JC, Vaglio A, Eyre S, et al. A large-scale genetic analysis reveals a strong contribution of the HLA class II region to giant cell arteritis susceptibility. Am J Hum Genet. (2015) 96:565–80. doi: 10.1016/j.ajhg.2015.02.009
7. Serrano A, Marquez A, Mackie SL, Carmona FD, Solans R, Miranda-Filloy JA, et al. Identification of the PTPN22 functional variant R620W as susceptibility genetic factor for giant cell arteritis. Ann Rheum Dis. (2013) 72:1882–6. doi: 10.1136/annrheumdis-2013-203641
8. Carmona FD, Vaglio A, Mackie SL, Hernandez-Rodriguez J, Monach PA, Castaneda S, et al. A genome-wide association study identifies risk alleles in plasminogen and P4HA2 associated with giant cell arteritis. Am J Hum Genet. (2017) 100:64–74. doi: 10.1016/j.ajhg.2016.11.013
9. Lyons PA, Rayner TF, Trivedi S, Holle JU, Watts RA, Jayne DR, et al. Genetically distinct subsets within ANCA-associated vasculitis. N Engl J Med. (2012) 367:214–23. doi: 10.1056/NEJMoa1108735
10. Merkel PA, Xie G, Monach PA, Ji X, Ciavatta DJ, Byun J, et al. Identification of functional and expression polymorphisms associated with risk for antineutrophil cytoplasmic autoantibody-associated vasculitis. Arthritis Rheumatol. (2017) 69:1054–66. doi: 10.1002/art.40034
11. Mahr AD, Edberg JC, Stone JH, Hoffman GS St, Clair EW, Specks U, et al. Alpha1-antitrypsin deficiency-related alleles Z and S and the risk of Wegener's granulomatosis. Arthritis Rheum. (2010) 62:3760–7. doi: 10.1002/art.27742
12. Gencik M, Meller S, Borgmann S, Fricke H. Proteinase 3 gene polymorphisms and Wegener's granulomatosis. Kidney Int. (2000) 58:2473–7. doi: 10.1046/j.1523-1755.2000.00430.x
13. Xie G, Roshandel D, Sherva R, Monach PA, Lu EY, Kung T, et al. Association of granulomatosis with polyangiitis (Wegener's) with HLA-DPB1*04 and SEMA6A gene variants: evidence from genome-wide analysis. Arthritis Rheum. (2013) 65:2457–68. doi: 10.1002/art.38036
14. Carr EJ, Niederer HA, Williams J, Harper L, Watts RA, Lyons PA, et al. Confirmation of the genetic association of CTLA4 and PTPN22 with ANCA-associated vasculitis. BMC Med Genet. (2009) 10:121. doi: 10.1186/1471-2350-10-121
15. Jagiello P, Aries P, Arning L, Wagenleiter SE, Csernok E, Hellmich B, et al. The PTPN22 620W allele is a risk factor for Wegener's granulomatosis. Arthritis Rheum. (2005) 52:4039–43. doi: 10.1002/art.21487
16. Mizuki N, Meguro A, Ota M, Ohno S, Shiota T, Kawagoe T, et al. Genome-wide association studies identify IL23R-IL12RB2 and IL10 as Behcet's disease susceptibility loci. Nat Genet. (2010) 42:703–6. doi: 10.1038/ng.624
17. Remmers EF, Cosan F, Kirino Y, Ombrello MJ, Abaci N, Satorius C, et al. Genome-wide association study identifies variants in the MHC class I, IL10, and IL23R-IL12RB2 regions associated with Behcet's disease. Nat Genet. (2010) 42:698–702. doi: 10.1038/ng.625
18. Ortiz-Fernandez L, Carmona FD, Montes-Cano MA, Garcia-Lozano JR, Conde-Jaldon M, Ortego-Centeno N, et al. Genetic analysis with the immunochip platform in behcet disease. Identification of residues associated in the HLA class I region and new susceptibility loci. PLoS ONE. (2016) 11:e0161305. doi: 10.1371/journal.pone.0161305
19. Kirino Y, Bertsias G, Ishigatsubo Y, Mizuki N, Tugal-Tutkun I, Seyahi E, et al. Genome-wide association analysis identifies new susceptibility loci for Behcet's disease and epistasis between HLA-B*51 and ERAP1. Nat Genet. (2013) 45:202–7. doi: 10.1038/ng.2520
20. Lee YJ, Horie Y, Wallace GR, Choi YS, Park JA, Choi JY, et al. Genome-wide association study identifies GIMAP as a novel susceptibility locus for Behcet's disease. Ann Rheum Dis. (2013) 72:1510–6. doi: 10.1136/annrheumdis-2011-200288
21. Kappen JH, Medina-Gomez C, van Hagen PM, Stolk L, Estrada K, Rivadeneira F, et al. Genome-wide association study in an admixed case series reveals IL12A as a new candidate in Behcet disease. PLoS ONE. (2015) 10:e0119085. doi: 10.1371/journal.pone.0119085
22. Khor CC, Davila S, Breunis WB, Lee YC, Shimizu C, Wright VJ, et al. Genome-wide association study identifies FCGR2A as a susceptibility locus for Kawasaki disease. Nat Genet. (2011) 43:1241–6. doi: 10.1038/ng.981
23. Onouchi Y, Ozaki K, Buns JC, Shimizu C, Hamada H, Honda T, et al. Common variants in CASP3 confer susceptibility to Kawasaki disease. Hum Mol Genet. (2010) 19:2898–906. doi: 10.1093/hmg/ddq176
24. Kim JJ, Yun SW, Yu JJ, Yoon KL, Lee KY, Kil HR, et al. A genome-wide association analysis identifies NMNAT2 and HCP5 as susceptibility loci for Kawasaki disease. J Hum Genet. (2017) 62:1023–9. doi: 10.1038/jhg.2017.87
25. Lee YC, Kuo HC, Chang JS, Chang LY, Huang LM, Chen MR, et al. Two new susceptibility loci for Kawasaki disease identified through genome-wide association analysis. Nat Genet. (2012) 44:522–5. doi: 10.1038/ng.2227
26. Onouchi Y, Ozaki K, Burns JC, Shimizu C, Terai M, Hamada H, et al. A genome-wide association study identifies three new risk loci for Kawasaki disease. Nat Genet. (2012) 44:517–21. doi: 10.1038/ng.2220
27. Nelson MR, Tipney H, Painter JL, Shen J, Nicoletti P, Shen Y, et al. The support of human genetic evidence for approved drug indications. Nat Genet. (2015) 47:856–60. doi: 10.1038/ng.3314
28. Onouchi Y, Suzuki Y, Suzuki H, Terai M, Yasukawa K, Hamada H, et al. ITPKC and CASP3 polymorphisms and risks for IVIG unresponsiveness and coronary artery lesion formation in Kawasaki disease. Pharmacogenomics J. (2013) 13:52–9. doi: 10.1038/tpj.2011.45
29. Kuo HC, Hsu YW, Wu CM, Chen SH, Hung KS, Chang WP, et al. A replication study for association of ITPKC and CASP3 two-locus analysis in IVIG unresponsiveness and coronary artery lesion in Kawasaki disease. PLoS ONE. (2013) 8:e69685. doi: 10.1371/journal.pone.0069685
30. Makowsky R, Wiener HW, Ptacek TS, Silva M, Shendre A, Edberg JC, et al. FcgammaR gene copy number in Kawasaki disease and intravenous immunoglobulin treatment response. Pharmacogenet Genomics. (2013) 23:455–62. doi: 10.1097/FPC.0b013e328363686e
31. Shrestha S, Wiener H, Shendre A, Kaslow RA, Wu J, Olson A, et al. Role of activating FcgammaR gene polymorphisms in Kawasaki disease susceptibility and intravenous immunoglobulin response. Circ Cardiovasc Genet. (2012) 5:309–16. doi: 10.1161/CIRCGENETICS.111.962464
32. Shrestha S, Wiener HW, Olson AK, Edberg JC, Bowles NE, Patel H, et al. Functional FCGR2B gene variants influence intravenous immunoglobulin response in patients with Kawasaki disease. J Allergy Clin Immunol. (2011) 128:677–80. doi: 10.1016/j.jaci.2011.04.027
33. Lee CP, Huang YH, Hsu YW, Yang KD, Chien HC, Yu HR, et al. TARC/CCL17 gene polymorphisms and expression associated with susceptibility and coronary artery aneurysm formation in Kawasaki disease. Pediatr Res. (2013) 74:545–51. doi: 10.1038/pr.2013.134
34. Mamtani M, Matsubara T, Shimizu C, Furukawa S, Akagi T, Onouchi Y, et al. Association of CCR2-CCR5 haplotypes and CCL3L1 copy number with Kawasaki Disease, coronary artery lesions, and IVIG responses in Japanese children. PLoS ONE. (2010) 5:e11458. doi: 10.1371/journal.pone.0011458
35. Weng KP, Hsieh KS, Ho TY, Huang SH, Lai CR, Chiu YT, et al. IL-1B polymorphism in association with initial intravenous immunoglobulin treatment failure in Taiwanese children with Kawasaki disease. Circ J. (2010) 74:544–51. doi: 10.1253/circj.CJ-09-0664
36. Huang YH, Hsu YW, Lu HF, Wong HS, Yu HR, Kuo HC, et al. Interferon-gamma Genetic Polymorphism and Expression in Kawasaki Disease. Medicine (Baltimore). (2016) 95:e3501. doi: 10.1097/MD.0000000000003501
37. Ahn JG, Bae Y, Shin D, Nam J, Kim KY, Kim DS. HMGB1 gene polymorphism is associated with coronary artery lesions and intravenous immunoglobulin resistance in Kawasaki disease. Rheumatology (Oxford). (2018) 2018:356. doi: 10.1093/rheumatology/key356
38. Kwon YC, Kim JJ, Yun SW, Yu JJ, Yoon KL, Lee KY, et al. BCL2L11 is associated with kawasaki disease in intravenous immunoglobulin responder patients. Circ Genom Precis Med. (2018) 11:e002020. doi: 10.1161/CIRCGEN.117.002020
39. Shendre A, Wiener HW, Zhi D, Vazquez AI, Portman MA, Shrestha S. High-density genotyping of immune loci in Kawasaki disease and IVIG treatment response in European-American case-parent trio study. Genes Immun. (2014) 15:534–42. doi: 10.1038/gene.2014.47
40. Kim JJ, Yun SW, Yu JJ, Yoon KL, Lee KY, Kil HR, et al. Identification of SAMD9L as a susceptibility locus for intravenous immunoglobulin resistance in Kawasaki disease by genome-wide association analysis. Pharmacogenomics J. (2019) 2019:85. doi: 10.1038/s41397-019-0085-1
41. Chang DY, Luo H, Zhou XJ, Chen M, Zhao MH. Association of HLA genes with clinical outcomes of ANCA-associated vasculitis. Clin J Am Soc Nephrol. (2012) 7:1293–9. doi: 10.2215/CJN.13071211
42. Cartin-Ceba R, Indrakanti D, Specks U, Stone JH, Hoffman GS, Kallenberg CG, et al. The pharmacogenomic association of Fcgamma receptors and cytochrome P450 enzymes with response to rituximab or cyclophosphamide treatment in antineutrophil cytoplasmic antibody-associated vasculitis. Arthritis Rheumatol. (2017) 69:169–75. doi: 10.1002/art.39822
43. Alberici F, Smith RM, Fonseca M, Willcocks LC, Jones RB, Holle JU, et al. Association of a TNFSF13B (BAFF) regulatory region single nucleotide polymorphism with response to rituximab in antineutrophil cytoplasmic antibody-associated vasculitis. J Allergy Clin Immunol. (2017) 139:1684–7 e10. doi: 10.1016/j.jaci.2016.08.051
44. Rustemoglu A, Gul U, Gumus-Akay G, Gonul M, Yigit S, Bozkurt N, et al. MDR1 gene polymorphisms may be associated with Behcet's disease and its colchicum treatment response. Gene. (2012) 505:333–9. doi: 10.1016/j.gene.2012.05.040
45. Karakus N, Yigit S, Kalkan G, Rustemoglu A, Inanir A, Gul U, et al. Association between the methylene tetrahydrofolate reductase gene C677T mutation and colchicine unresponsiveness in Behcet's disease. Mol Vis. (2012) 18:1696–700.
46. Keser G, Direskeneli H, Aksu K. Management of Takayasu arteritis: a systematic review. Rheumatology (Oxford). (2014) 53:793–801. doi: 10.1093/rheumatology/ket320
47. Terao C, Yoshifuji H, Ohmura K, Murakami K, Kawabata D, Yurugi K, et al. Association of Takayasu arteritis with HLA-B 67:01 and two amino acids in HLA-B protein. Rheumatology (Oxford). (2013) 52:1769–74. doi: 10.1093/rheumatology/ket241
48. Karageorgaki ZT, Bertsias GK, Mavragani CP, Kritikos HD, Spyropoulou-Vlachou M, Drosos AA, et al. Takayasu arteritis: epidemiological, clinical, and immunogenetic features in Greece. Clin Exp Rheumatol. (2009) 27:S33–9.
49. Lee SW, Kwon OJ, Park MC, Oh HB, Park YB, Lee SK. HLA alleles in Korean patients with Takayasu arteritis. Clin Exp Rheumatol. (2007) 25:S18–22.
50. Vargas-Alarcon G, Flores-Dominguez C, Hernandez-Pacheco G, Zuniga J, Gamboa R, Soto ME, et al. Immunogenetics and clinical aspects of Takayasu's arteritis patients in a Mexican Mestizo population. Clin Exp Rheumatol. (2001) 19:439–43.
51. Charoenwongse P, Kangwanshiratada O, Boonnam R, Hoomsindhu U. The association between the HLA antigens and Takayasu's arteritis in Thai patients. Int J Cardiol. (1998) 66(Suppl 1):S117–20.
52. Mehra NK, Jaini R, Balamurugan A, Kanga U, Prabhakaran D, Jain S, et al. Immunogenetic analysis of Takayasu arteritis in Indian patients. Int J Cardiol. (1998) 66(Suppl 1):S127–32; discussion S33.
53. Lv N, Wang Z, Dang A, Zhu X, Liu Y, Zheng D, et al. HLA-DQA1, DQB1 and DRB1 alleles associated with Takayasu arteritis in the Chinese Han population. Hum Immunol. (2015) 76:241–4. doi: 10.1016/j.humimm.2015.01.008
54. Dang A, Wang B, Zhang Y, Zhang P, Huang J, Liu G, et al. Association of the HLA-DRB1 gene with susceptibility to aortoarteritis in a Chinese Han population. Hypertens Res. (2002) 25:631–4. doi: 10.1291/hypres.25.631
55. Kimura A, Kishimoto T. IL-6: regulator of Treg/Th17 balance. Eur J Immunol. (2010) 40:1830–5. doi: 10.1002/eji.201040391
56. Sloane DE, Tedla N, Awoniyi M, Macglashan DW Jr, Borges L, Austen KF, et al. Leukocyte immunoglobulin-like receptors: novel innate receptors for human basophil activation and inhibition. Blood. (2004) 104:2832–9. doi: 10.1182/blood-2004-01-0268
57. Teng MW, Bowman EP, McElwee JJ, Smyth MJ, Casanova JL, Cooper AM, et al. IL-12 and IL-23 cytokines: from discovery to targeted therapies for immune-mediated inflammatory diseases. Nat Med. (2015) 21:719–29. doi: 10.1038/nm.3895
58. Tan Sardjono C, Mottram PL, Hogarth PM. The role of FcgammaRIIa as an inflammatory mediator in rheumatoid arthritis and systemic lupus erythematosus. Immunol Cell Biol. (2003) 81:374–81. doi: 10.1046/j.1440-1711.2003.01182.x
59. Samson M, Espigol-Frigole G, Terrades-Garcia N, Prieto-Gonzalez S, Corbera-Bellalta M, Alba-Rovira R, et al. Biological treatments in giant cell arteritis & Takayasu arteritis. Eur J Intern Med. (2018) 50:12–9. doi: 10.1016/j.ejim.2017.11.003
60. Nakaoka Y, Isobe M, Takei S, Tanaka Y, Ishii T, Yokota S, et al. Efficacy and safety of tocilizumab in patients with refractory Takayasu arteritis: results from a randomised, double-blind, placebo-controlled, phase 3 trial in Japan (the TAKT study). Ann Rheum Dis. (2018) 77:348–54. doi: 10.1136/annrheumdis-2017-211878
61. Terao C, Yoshifuji H, Nakajima T, Yukawa N, Matsuda F, Mimori T. Ustekinumab as a therapeutic option for Takayasu arteritis: from genetic findings to clinical application. Scand J Rheumatol. (2016) 45:80–2. doi: 10.3109/03009742.2015.1060521
62. Yachoui R, Kreidy M, Siorek M, Sehgal R. Successful treatment with ustekinumab for corticosteroid- and immunosuppressant-resistant Takayasu's arteritis. Scand J Rheumatol. (2018) 47:246–7. doi: 10.1080/03009742.2017.1278788
63. Pazzola G, Muratore F, Pipitone N, Crescentini F, Cacoub P, Boiardi L, et al. Rituximab therapy for Takayasu arteritis: a seven patients experience and a review of the literature. Rheumatology (Oxford). (2017) 2017:249. doi: 10.1093/rheumatology/kex249
64. Salvarani C, Cantini F, Hunder GG. Polymyalgia rheumatica and giant-cell arteritis. Lancet. (2008) 372:234–45. doi: 10.1016/S0140-6736(08)61077-6
65. Carmona FD, Gonzalez-Gay MA, Martin J. Genetic component of giant cell arteritis. Rheumatology (Oxford). (2014) 53:6–18. doi: 10.1093/rheumatology/ket231
66. Boiardi L, Casali B, Nicoli D, Farnetti E, Chen Q, Macchioni P, et al. Vascular endothelial growth factor gene polymorphisms in giant cell arteritis. J Rheumatol. (2003) 30:2160–4.
67. Enjuanes A, Benavente Y, Hernandez-Rodriguez J, Queralt C, Yague J, Jares P, et al. Association of NOS2 and potential effect of VEGF, IL6, CCL2 and IL1RN polymorphisms and haplotypes on susceptibility to GCA–a simultaneous study of 130 potentially functional SNPs in 14 candidate genes. Rheumatology (Oxford). (2012) 51:841–51. doi: 10.1093/rheumatology/ker429
68. Marquez A, Hernandez-Rodriguez J, Cid MC, Solans R, Castaneda S, Fernandez-Contreras ME, et al. Influence of the IL17A locus in giant cell arteritis susceptibility. Ann Rheum Dis. (2014) 73:1742–5. doi: 10.1136/annrheumdis-2014-205261
69. Marquez A, Solans R, Hernandez-Rodriguez J, Cid MC, Castaneda S, Ramentol M, et al. A candidate gene approach identifies an IL33 genetic variant as a novel genetic risk factor for GCA. PLoS ONE. (2014) 9:e113476. doi: 10.1371/journal.pone.0113476
70. Rueda B, Lopez-Nevot MA, Lopez-Diaz MJ, Garcia-Porrua C, Martin J, Gonzalez-Gay MA. A functional variant of vascular endothelial growth factor is associated with severe ischemic complications in giant cell arteritis. J Rheumatol. (2005) 32:1737–41.
71. Serrano A, Carmona FD, Castaneda S, Solans R, Hernandez-Rodriguez J, Cid MC, et al. Evidence of association of the NLRP1 gene with giant cell arteritis. Ann Rheum Dis. (2013) 72:628–30. doi: 10.1136/annrheumdis-2012-202609
72. Choi YS, Choi HJ, Min JK, Pyun BJ, Maeng YS, Park H, et al. Interleukin-33 induces angiogenesis and vascular permeability through ST2/TRAF6-mediated endothelial nitric oxide production. Blood. (2009) 114:3117–26. doi: 10.1182/blood-2009-02-203372
73. Schmitz J, Owyang A, Oldham E, Song Y, Murphy E, McClanahan TK, et al. IL-33, an interleukin-1-like cytokine that signals via the IL-1 receptor-related protein ST2 and induces T helper type 2-associated cytokines. Immunity. (2005) 23:479–90. doi: 10.1016/j.immuni.2005.09.015
74. Weyand CM, Goronzy JJ. Immune mechanisms in medium and large-vessel vasculitis. Nat Rev Rheumatol. (2013) 9:731–40. doi: 10.1038/nrrheum.2013.161
75. Ferrara N, Gerber HP, LeCouter J. The biology of VEGF and its receptors. Nat Med. (2003) 9:669–76. doi: 10.1038/nm0603-669
76. Tschopp J, Martinon F, Burns K. NALPs: a novel protein family involved in inflammation. Nat Rev Mol Cell Biol. (2003) 4:95–104. doi: 10.1038/nrm1019
77. Bottini N, Peterson EJ. Tyrosine phosphatase PTPN22: multifunctional regulator of immune signaling, development, and disease. Annu Rev Immunol. (2014) 32:83–119. doi: 10.1146/annurev-immunol-032713-120249
78. Miles LA, Parmer RJ. Plasminogen receptors: the first quarter century. Semin Thromb Hemost. (2013) 39:329–37. doi: 10.1055/s-0033-1334483
79. Gilkes DM, Bajpai S, Chaturvedi P, Wirtz D, Semenza GL. Hypoxia-inducible factor 1 (HIF-1) promotes extracellular matrix remodeling under hypoxic conditions by inducing P4HA1, P4HA2, and PLOD2 expression in fibroblasts. J Biol Chem. (2013) 288:10819–29. doi: 10.1074/jbc.M112.442939
80. Stone JH, Klearman M, Collinson N. Trial of tocilizumab in giant-cell arteritis. N Engl J Med. (2017) 377:1494–5. doi: 10.1056/NEJMc1711031
81. Villiger PM, Adler S, Kuchen S, Wermelinger F, Dan D, Fiege V, et al. Tocilizumab for induction and maintenance of remission in giant cell arteritis: a phase 2, randomised, double-blind, placebo-controlled trial. Lancet. (2016) 387:1921–7. doi: 10.1016/S0140-6736(16)00560-2
82. Conway R, O'Neill L, O'Flynn E, Gallagher P, McCarthy GM, Murphy CC, et al. Ustekinumab for the treatment of refractory giant cell arteritis. Ann Rheum Dis. (2016) 75:1578–9. doi: 10.1136/annrheumdis-2016-209351
83. Langford CA, Cuthbertson D, Ytterberg SR, Khalidi N, Monach PA, Carette S, et al. A randomized, double-blind trial of abatacept (CTLA-4Ig) for the treatment of giant cell arteritis. Arthritis Rheumatol. (2017) 69:837–45. doi: 10.1002/art.40044
84. Jarrot PA, Kaplanski G. Pathogenesis of ANCA-associated vasculitis: an update. Autoimmun Rev. (2016) 15:704–13. doi: 10.1016/j.autrev.2016.03.007
85. Segarra M, Ohnuki H, Maric D, Salvucci O, Hou X, Kumar A, et al. Semaphorin 6A regulates angiogenesis by modulating VEGF signaling. Blood. (2012) 120:4104–15. doi: 10.1182/blood-2012-02-410076
86. Wieczorek S, Holle JU, Cohen Tervaert JW, Harper L, Moosig F, Gross WL, et al. The SEM6A6 locus is not associated with granulomatosis with polyangiitis or other forms of antineutrophil cytoplasmic antibody-associated vasculitides in Europeans: comment on the article by Xie et al. Arthritis Rheumatol. (2014) 66:1400–1. doi: 10.1002/art.38367
87. Giscombe R, Wang X, Huang D, Lefvert AK. Coding sequence 1 and promoter single nucleotide polymorphisms in the CTLA-4 gene in Wegener's granulomatosis. J Rheumatol. (2002) 29:950–3.
88. Huang D, Giscombe R, Zhou Y, Lefvert AK. Polymorphisms in CTLA-4 but not tumor necrosis factor-alpha or interleukin 1beta genes are associated with Wegener's granulomatosis. J Rheumatol. (2000) 27:397–401.
89. Kamesh L, Heward JM, Williams JM, Gough SC, Chavele KM, Salama A, et al. CT60 and +49 polymorphisms of CTLA 4 are associated with ANCA-positive small vessel vasculitis. Rheumatology (Oxford). (2009) 48:1502–5. doi: 10.1093/rheumatology/kep280
90. Lee YH, Choi SJ, Ji JD, Song GG. CTLA-4 and TNF-alpha promoter-308 A/G polymorphisms and ANCA-associated vasculitis susceptibility: a meta-analysis. Mol Biol Rep. (2012) 39:319–26. doi: 10.1007/s11033-011-0741-2
91. Slot MC, Sokolowska MG, Savelkouls KG, Janssen RG, Damoiseaux JG, Tervaert JW. Immunoregulatory gene polymorphisms are associated with ANCA-related vasculitis. Clin Immunol. (2008) 128:39–45. doi: 10.1016/j.clim.2008.03.506
92. Zhou Y, Huang D, Paris PL, Sauter CS, Prock KA, Hoffman GS. An analysis of CTLA-4 and proinflammatory cytokine genes in Wegener's granulomatosis. Arthritis Rheum. (2004) 50:2645–50. doi: 10.1002/art.20385
93. Vaglio A, Martorana D, Maggiore U, Grasselli C, Zanetti A, Pesci A, et al. HLA-DRB4 as a genetic risk factor for Churg-Strauss syndrome. Arthritis Rheum. (2007) 56:3159–66. doi: 10.1002/art.22834
94. Martorana D, Bonatti F, Alberici F, Gioffredi A, Reina M, Urban ML, et al. Fcgamma-receptor 3B (FCGR3B) copy number variations in patients with eosinophilic granulomatosis with polyangiitis. J Allergy Clin Immunol. (2016) 137:1597–9 e8. doi: 10.1016/j.jaci.2015.09.053
95. Wieczorek S, Hellmich B, Arning L, Moosig F, Lamprecht P, Gross WL, et al. Functionally relevant variations of the interleukin-10 gene associated with antineutrophil cytoplasmic antibody-negative Churg-Strauss syndrome, but not with Wegener's granulomatosis. Arthritis Rheum. (2008) 58:1839–48. doi: 10.1002/art.23496
96. Polzer K, Karonitsch T, Neumann T, Eger G, Haberler C, Soleiman A, et al. Eotaxin-3 is involved in Churg-Strauss syndrome–a serum marker closely correlating with disease activity. Rheumatology (Oxford). (2008) 47:804–8. doi: 10.1093/rheumatology/ken033
97. Zwerina J, Bach C, Martorana D, Jatzwauk M, Hegasy G, Moosig F, et al. Eotaxin-3 in Churg-Strauss syndrome: a clinical and immunogenetic study. Rheumatology (Oxford). (2011) 50:1823–7. doi: 10.1093/rheumatology/keq445
98. McClure M, Gopaluni S, Jayne D, Jones R. B cell therapy in ANCA-associated vasculitis: current and emerging treatment options. Nat Rev Rheumatol. (2018) 14:580–91. doi: 10.1038/s41584-018-0065-x
99. Lepse N, Land J, Rutgers A, Kallenberg CG, Stegeman CA, Abdulahad WH, et al. Toll-like receptor 9 activation enhances B cell activating factor and interleukin-21 induced anti-proteinase 3 autoantibody production in vitro. Rheumatology (Oxford). (2016) 55:162–72. doi: 10.1093/rheumatology/kev293
100. Langford CA, Monach PA, Specks U, Seo P, Cuthbertson D, McAlear CA, et al. An open-label trial of abatacept (CTLA4-IG) in non-severe relapsing granulomatosis with polyangiitis (Wegener's). Ann Rheum Dis. (2014) 73:1376–9. doi: 10.1136/annrheumdis-2013-204164
101. Zeidan MJ, Saadoun D, Garrido M, Klatzmann D, Six A, Cacoub P. Behcet's disease physiopathology: a contemporary review. Auto Immun Highlights. (2016) 7:4. doi: 10.1007/s13317-016-0074-1
102. de Menthon M, Lavalley MP, Maldini C, Guillevin L, Mahr A. HLA-B51/B5 and the risk of Behcet's disease: a systematic review and meta-analysis of case-control genetic association studies. Arthritis Rheum. (2009) 61:1287–96. doi: 10.1002/art.24642
103. Hughes T, Coit P, Adler A, Yilmaz V, Aksu K, Duzgun N, et al. Identification of multiple independent susceptibility loci in the HLA region in Behcet's disease. Nat Genet. (2013) 45:319–24. doi: 10.1038/ng.2551
104. Iyer SS, Cheng G. Role of interleukin 10 transcriptional regulation in inflammation and autoimmune disease. Crit Rev Immunol. (2012) 32:23–63. doi: 10.1615/CritRevImmunol.v32.i1.30
105. Rossi D, Zlotnik A. The biology of chemokines and their receptors. Annu Rev Immunol. (2000) 18:217–42. doi: 10.1146/annurev.immunol.18.1.217
106. Watford WT, Hissong BD, Bream JH, Kanno Y, Muul L, O'Shea JJ. Signaling by IL-12 and IL-23 and the immunoregulatory roles of STAT4. Immunol Rev. (2004) 202:139–56. doi: 10.1111/j.0105-2896.2004.00211.x
107. Raulet DH. Roles of the NKG2D immunoreceptor and its ligands. Nat Rev Immunol. (2003) 3:781–90. doi: 10.1038/nri1199
108. Schnell S, Demolliere C, van den Berk P, Jacobs H. Gimap4 accelerates T-cell death. Blood. (2006) 108:591–9. doi: 10.1182/blood-2005-11-4616
109. Shimoda Y, Watanabe K. Contactins: emerging key roles in the development and function of the nervous system. Cell Adh Migr. (2009) 3:64–70. doi: 10.4161/cam.3.1.7764
110. Vitale A, Rigante D, Lopalco G, Emmi G, Bianco MT, Galeazzi M, et al. New therapeutic solutions for Behcet's syndrome. Expert Opin Investig Drugs. (2016) 25:827–40. doi: 10.1080/13543784.2016.1181751
111. Di Scala G, Bettiol A, Cojan RD, Finocchi M, Silvestri E, Emmi G. Efficacy of the anti-IL 17 secukinumab in refractory Behcet's syndrome: a preliminary study. J Autoimmun. (2019) 97:108–13. doi: 10.1016/j.jaut.2018.09.002
112. Mirouse A, Barete S, Monfort JB, Resche-Rigon M, Bouyer AS, Comarmond C, et al. Ustekinumab for Behcet's disease. J Autoimmun. (2017) 82:41–6. doi: 10.1016/j.jaut.2017.05.002
113. Shulman ST, Rowley AH. Kawasaki disease: insights into pathogenesis and approaches to treatment. Nat Rev Rheumatol. (2015) 11:475–82. doi: 10.1038/nrrheum.2015.54
114. Burgner D, Davila S, Breunis WB, Ng SB, Li Y, Bonnard C, et al. A genome-wide association study identifies novel and functionally related susceptibility Loci for Kawasaki disease. PLoS Genet. (2009) 5:e1000319. doi: 10.1371/journal.pgen.1000319
115. Kim JJ, Hong YM, Sohn S, Jang GY, Ha KS, Yun SW, et al. A genome-wide association analysis reveals 1p31 and 2p13.3 as susceptibility loci for Kawasaki disease. Hum Genet. (2011) 129:487–95. doi: 10.1007/s00439-010-0937-x
116. Tsai FJ, Lee YC, Chang JS, Huang LM, Huang FY, Chiu NC, et al. Identification of novel susceptibility Loci for kawasaki disease in a Han chinese population by a genome-wide association study. PLoS ONE. (2011) 6:e16853. doi: 10.1371/journal.pone.0016853
117. Kato S, Kimura M, Tsuji K, Kusakawa S, Asai T, Juji T, et al. HLA antigens in Kawasaki disease. Pediatrics. (1978) 61:252–5.
118. Matsuda I, Hattori S, Nagata N, Fruse A, Nambu H. HLA antigens in mucocutaneous lymph node syndrome. Am J Dis Child. (1977) 131:1417–8.
119. Kaslow RA, Bailowitz A, Lin FY, Koslowe P, Simonis T, Israel E. Association of epidemic Kawasaki syndrome with the HLA-A2, B44, Cw5 antigen combination. Arthritis Rheum. (1985) 28:938–40.
120. Krensky AM, Berenberg W, Shanley K, Yunis EJ. HLA antigens in mucocutaneous lymph node syndrome in New England. Pediatrics. (1981) 67:741–3.
121. Krensky AM, Grady S, Shanley KM, Berenberg W, Yunis EJ. Epidemic and endemic HLA-B and DR associations in mucocutaneous lymph node syndrome. Hum Immunol. (1983) 6:75–7.
122. Warmerdam PA, van de Winkel JG, Vlug A, Westerdaal NA, Capel PJ. A single amino acid in the second Ig-like domain of the human Fc gamma receptor II is critical for human IgG2 binding. J Immunol. (1991) 147:1338–43.
123. Onouchi Y, Gunji T, Burns JC, Shimizu C, Newburger JW, Yashiro M, et al. ITPKC functional polymorphism associated with Kawasaki disease susceptibility and formation of coronary artery aneurysms. Nat Genet. (2008) 40:35–42. doi: 10.1038/ng.2007.59
124. Alphonse MP, Duong TT, Shumitzu C, Hoang TL, McCrindle BW, Franco A, et al. Inositol-triphosphate 3-kinase C mediates inflammasome activation and treatment response in kawasaki disease. J Immunol. (2016) 197:3481–9. doi: 10.4049/jimmunol.1600388
125. Nemazee D, Weigert M. Revising B cell receptors. J Exp Med. (2000) 191:1813–7. doi: 10.1084/jem.191.11.1813
126. Peters AL, Stunz LL, Bishop GA. CD40 and autoimmunity: the dark side of a great activator. Semin Immunol. (2009) 21:293–300. doi: 10.1016/j.smim.2009.05.012
127. Wallace CA, French JW, Kahn SJ, Sherry DD. Initial intravenous gammaglobulin treatment failure in Kawasaki disease. Pediatrics. (2000) 105:E78. doi: 10.1542/peds.105.6.e78
128. Galeotti C, Kaveri SV, Cimaz R, Kone-Paut I, Bayry J. Predisposing factors, pathogenesis and therapeutic intervention of Kawasaki disease. Drug Discov Today. (2016) 21:1850–7. doi: 10.1016/j.drudis.2016.08.004
129. Lee Y, Schulte DJ, Shimada K, Chen S, Crother TR, Chiba N, et al. Interleukin-1beta is crucial for the induction of coronary artery inflammation in a mouse model of Kawasaki disease. Circulation. (2012) 125:1542–50. doi: 10.1161/CIRCULATIONAHA.111.072769
130. Wakita D, Kurashima Y, Crother TR, Noval Rivas M, Lee Y, Chen S, et al. Role of interleukin-1 signaling in a mouse model of kawasaki disease-associated abdominal aortic aneurysm. Arterioscler Thromb Vasc Biol. (2016) 36:886–97. doi: 10.1161/ATVBAHA.115.307072
131. Medvedev AE, Murphy M, Zhou H, Li X. E3 ubiquitin ligases Pellinos as regulators of pattern recognition receptor signaling and immune responses. Immunol Rev. (2015) 266:109–22. doi: 10.1111/imr.12298
132. Navon Elkan P, Pierce SB, Segel R, Walsh T, Barash J, Padeh S, et al. Mutant adenosine deaminase 2 in a polyarteritis nodosa vasculopathy. N Engl J Med. (2014) 370:921–31. doi: 10.1056/NEJMoa1307362
133. Zhou Q, Yang D, Ombrello AK, Zavialov AV, Toro C, Stone DL, et al. Early-onset stroke and vasculopathy associated with mutations in ADA2. N Engl J Med. (2014) 370:911–20. doi: 10.1056/NEJMoa1307361
134. Zavialov AV, Gracia E, Glaichenhaus N, Franco R, Lauvau G. Human adenosine deaminase 2 induces differentiation of monocytes into macrophages and stimulates proliferation of T helper cells and macrophages. J Leukoc Biol. (2010) 88:279–90. doi: 10.1189/jlb.1109764
135. Hashem H, Kumar AR, Muller I, Babor F, Bredius R, Dalal J, et al. Hematopoietic stem cell transplantation rescues the hematological, immunological, and vascular phenotype in DADA2. Blood. (2017) 130:2682–8. doi: 10.1182/blood-2017-07-798660
136. Hsu AP, West RR, Calvo KR, Cuellar-Rodriguez J, Parta M, Kelly SJ, et al. Adenosine deaminase type 2 deficiency masquerading as GATA2 deficiency: successful hematopoietic stem cell transplantation. J Allergy Clin Immunol. (2016) 138:628–30 e2. doi: 10.1016/j.jaci.2016.03.016
137. Van Eyck L Jr, Hershfield MS, Pombal D, Kelly SJ, Ganson NJ, Moens L, et al. Hematopoietic stem cell transplantation rescues the immunologic phenotype and prevents vasculopathy in patients with adenosine deaminase 2 deficiency. J Allergy Clin Immunol. (2015) 135:283–7 e5. doi: 10.1016/j.jaci.2014.10.010
138. Ombrello AK, Qin J, Hoffmann PM, Kumar P, Stone D, Jones A, et al. Treatment Strategies for Deficiency of Adenosine Deaminase 2. N Engl J Med. (2019) 380:1582–4. doi: 10.1056/NEJMc1801927
139. Samuelsson A, Towers tl, Ravetch JV. Anti-inflammatory activity of IVIG mediated through the inhibitory Fc receptor. Science. (2001) 291:484–6. doi: 10.1126/science.291.5503.484
140. Hughes P, Bouillet P, Strasser A. Role of Bim and other Bcl-2 family members in autoimmune and degenerative diseases. Curr Dir Autoimmun. (2006) 9:74–94. doi: 10.1159/000090773
141. Lemos de Matos A, Liu J, McFadden G, Esteves PJ. Evolution and divergence of the mammalian SAMD9/SAMD9L gene family. BMC Evol Biol. (2013) 13:121. doi: 10.1186/1471-2148-13-121
142. Kuo HC, Wong HS, Chang WP, Chen BK, Wu MS, Yang KD, et al. Prediction for intravenous immunoglobulin resistance by using weighted genetic risk score identified from genome-wide association study in kawasaki disease. Circ Cardiovasc Genet. (2017) 10:1625. doi: 10.1161/CIRCGENETICS.116.001625
143. Richaud-Patin Y, Soto-Vega E, Jakez-Ocampo J, Llorente L. P-glycoprotein in autoimmune diseases. Autoimmun Rev. (2004) 3:188–92. doi: 10.1016/j.autrev.2003.08.002
144. Liew SC, Gupta ED. Methylenetetrahydrofolate reductase (MTHFR) C677T polymorphism: epidemiology, metabolism and the associated diseases. Eur J Med Genet. (2015) 58:1–10. doi: 10.1016/j.ejmg.2014.10.004
145. Aksu K, Turgan N, Oksel F, Keser G, Ozmen D, Kitapcioglu G, et al. Hyperhomocysteinaemia in Behçet's disease. Rheumatology (Oxford). (2001) 40:687–90. doi: 10.1093/rheumatology/40.6.687
146. Parkes M, Cortes A, van Heel DA, Brown MA. Genetic insights into common pathways and complex relationships among immune-mediated diseases. Nat Rev Genet. (2013) 14:661–73. doi: 10.1038/nrg3502
147. Zhernakova A, Withoff S, Wijmenga C. Clinical implications of shared genetics and pathogenesis in autoimmune diseases. Nat Rev Endocrinol. (2013) 9:646–59. doi: 10.1038/nrendo.2013.161
148. Acosta-Herrera M, Kerick M, Gonzalez-Serna D, Wijmenga C, Franke A, Gregersen PK, et al. Genome-wide meta-analysis reveals shared new loci in systemic seropositive rheumatic diseases. Ann Rheum Dis. (2019) 78:311–9. doi: 10.1136/annrheumdis-2018-214127
149. Ellinghaus D, Jostins L, Spain SL, Cortes A, Bethune J, Han B, et al. Analysis of five chronic inflammatory diseases identifies 27 new associations and highlights disease-specific patterns at shared loci. Nat Genet. (2016) 48:510–8. doi: 10.1038/ng.3528
150. Li YR, Li J, Zhao SD, Bradfield JP, Mentch FD, Maggadottir SM, et al. Meta-analysis of shared genetic architecture across ten pediatric autoimmune diseases. Nat Med. (2015) 21:1018–27. doi: 10.1038/nm.3933
151. Marquez A, Kerick M, Zhernakova A, Gutierrez-Achury J, Chen WM, Onengut-Gumuscu S, et al. Meta-analysis of Immunochip data of four autoimmune diseases reveals novel single-disease and cross-phenotype associations. Genome Med. (2018) 10:97. doi: 10.1186/s13073-018-0604-8
152. Carmona FD, Coit P, Saruhan-Direskeneli G, Hernandez-Rodriguez J, Cid MC, Solans R, et al. Analysis of the common genetic component of large-vessel vasculitides through a meta-Immunochip strategy. Sci Rep. (2017) 7:43953. doi: 10.1038/srep43953
153. Ortiz-Fernandez L, Carmona FD, Lopez-Mejias R, Gonzalez-Escribano MF, Lyons PA, Morgan AW, et al. Cross-phenotype analysis of Immunochip data identifies KDM4C as a relevant locus for the development of systemic vasculitis. Ann Rheum Dis. (2018) 77:589–95. doi: 10.1136/annrheumdis-2017-212372
Keywords: systemic vasculitis, polymorphism, genome-wide association studies, immunochip, precision medicine
Citation: Acosta-Herrera M, González-Gay MA, Martín J and Márquez A (2019) Leveraging Genetic Findings for Precision Medicine in Vasculitis. Front. Immunol. 10:1796. doi: 10.3389/fimmu.2019.01796
Received: 20 May 2019; Accepted: 16 July 2019;
Published: 02 August 2019.
Edited by:
Giacomo Emmi, University of Florence, ItalyReviewed by:
Augusto Vaglio, University of Parma, ItalyDavide Martorana, University Hospital of Parma, Italy
Copyright © 2019 Acosta-Herrera, González-Gay, Martín and Márquez. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ana Márquez, YW5hbWFvcnRAaXBiLmNzaWMuZXM=