 Patricia Gnieslaw de Oliveira
Patricia Gnieslaw de Oliveira Mirian Farinon
Mirian Farinon Elsa Sanchez-Lopez
Elsa Sanchez-Lopez Shigeki Miyamoto2
Shigeki Miyamoto2 Monica Guma
Monica Guma
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Immunol. , 02 August 2019
Sec. Autoimmune and Autoinflammatory Disorders
Volume 10 - 2019 | https://doi.org/10.3389/fimmu.2019.01743
This article is part of the Research Topic Immunomodulatory Functions of Fibroblast-like Synoviocytes in Joint Inflammation and Destruction during Rheumatoid Arthritis View all 8 articles
Metabolomic studies show that rheumatoid arthritis (RA) is associated with metabolic disruption that may be therapeutically targetable. Among them, glucose metabolism and glycolytic intermediaries seem to have an important role in fibroblast-like synoviocytes (FLS) phenotype and might contribute to early stage disease pathogenesis. RA FLS are transformed from quiescent to aggressive and metabolically active cells and several works have shown that glucose metabolism is increased in activated FLS. Glycolytic inhibitors reduce not only FLS aggressive phenotype in vitro but also decrease bone and cartilage damage in several murine models of arthritis. Essential glycolytic enzymes, including hexokinase 2 (HK2) and 6-phosphofructo-2-kinase/fructose-2,6-biphosphatase (PFKFB) enzymes, have important roles in FLS behavior. Of interest, HK2 is an inducible enzyme present only in the inflamed rheumatic tissues compared to osteoarthritis synovium. It is a contributor to glucose metabolism that could be selectively targeted without compromising systemic homeostasis as a novel approach for combination therapy independent of systemic immunosuppression. More information about metabolic targets that do not compromise global glucose metabolism in normal cells is needed.
Rheumatoid arthritis (RA) pathogenesis includes synovial hyperplasia or pannus, which consists of accumulation of macrophages and fibroblast like synoviocytes (FLS) (1–4), resulting in enhanced invasiveness and destruction of adjacent cartilage and bone (3, 5). FLS are the major component of rheumatoid pannus and have a key role in its formation (4). In healthy individuals, these cells ensure the structural integrity of a normally organized synovial lining (6) and secretes hyaluronic acid and lubricin, two important constituents of synovial fluid that are responsible for lubricating the joint (7, 8). However, after acquiring an aggressive phenotype, FLS have reduced contact inhibition, resistance to apoptosis, increased migration, and increased ability to invade periarticular tissues including bone and cartilage (4, 9). These activated cells produce several mediators that induce angiogenesis, cell growth, and recruitment and activation of immune cells (4). In addition to contributing to the inflammatory environment, FLS also produce matrix metalloproteases (MMPs) that degrade the extracellular matrix and contributes to cartilage destruction (10).
Recently, an increasing number of studies have shown that FLS activation and the subsequent joint damage are associated with an altered metabolism which may be therapeutically targetable. The metabolism of all four major classes of macromolecules (carbohydrates, proteins, lipids, and nucleic acids) will change after cell activation. Carbohydrate metabolism is a fundamental biochemical process that ensures a constant supply of energy to living cells. The most important carbohydrate is glucose, which is first transported into the cell through glucose transporter 1 (GLUT1), then broken down via glycolysis by sequential metabolic enzymes [including hexokinase (HK), aldolase, phosphoglycerate kinase (PGK1), and pyruvate kinase] to generate pyruvate, and afterwards will either enter into the tricarboxylic acid (TCA) cycle and oxidative phosphorylation to generate ATP, or will be converted to lactate via lactate dehydrogenase (LDH).
Activation of FLS by hypoxia, platelet-derived growth factor (PDGF), tumor necrosis factor (TNF), and other inflammatory mediators increases glucose metabolism and transforms the FLS from quiescent to aggressive and metabolically active cells. Specifically, prior work—ours and others—has shown that glucose metabolism is increased in activated FLS, and glycolytic inhibition reduces not only FLS aggressive phenotype in vitro but also decreases bone and cartilage damage in several murine models of arthritis [(11–13); Table 1]. These works have suggested potential metabolic targets to reprogram metabolic disruptions and complement current therapies (11–13). In this brief review, we will summarize what is known about glucose metabolism in FLS and about potential metabolic therapies for RA that could modulate the aggressive behavior of FLS.
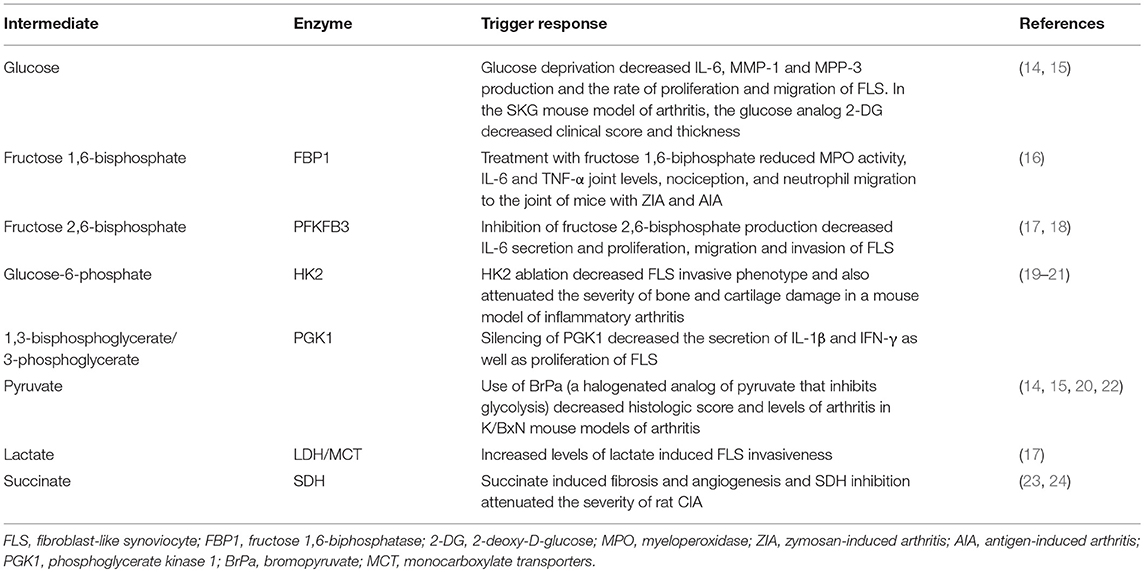
Table 1. Glycolyitic intermediate metabolites and their effect on RA FLS and animal models of arthritis.
Glucose metabolism seems to be especially enhanced in joints with arthritis and involved in RA pathology. The high consumption of glucose by the RA joints can be visualized by PET imaging with 18F-FDG, a probe that detects glycolytic tissues (25). Synovial FLS and macrophages were shown to contribute to FDG-PET accumulation in the RA synovial tissue (26). The synovial tissue of RA patients also presents an enhanced level of lactate compared to non-inflamed synovial tissue (27). The local lower glucose levels and higher ratio of lactate to glucose in the RA synovial tissue suggest an increase in anaerobic cellular metabolism of resident cells, triggered by the inflammation and the hypoxic environment commonly detected in the RA joints (28, 29). This dysregulation is further suggested by the increase of lactate and glucose in the serum of RA patients (30). Moreover, glucose levels are lower in the synovial fluid of RA patients in comparison to non-inflamed synovial fluid (31). This is also supported by metabolic studies using mass spectrometry, which show differential metabolite profile in RA FLS and osteoarthritis (OA) FLS (32). Glycolysis, pentose phosphate pathway (PPP), and amino acid metabolism were different in RA FLS compared to OA FLS (32). In addition, FLS increased its intracellular levels of glucose after TNF stimulation (33).
Shift from oxidative phosphorylation to glycolytic ATP production is a common feature of activated and reactive cells like fibroblasts and macrophages. Micro environmental factors in the RA joint seem to potentiate this metabolic adaptation of FLS and macrophages. The synovial tissue is enriched in hypoxia-inducible factor 1 alpha (HIF1α), a transcription factor induced in hypoxic environments that contributes to RA pathogenesis at multiple steps (29), including supporting enhanced glycolytic activity. Among the HIF1α-transcriptionally regulated glucose metabolism related genes, GLUT1, HK2, and LDH are upregulated in RA FLS (14, 19, 34–36). The effect of HIF1α on glycolysis contributes to FLS survival (37), myeloid recruitment by FLS, angiogenesis (38), and FLS migration and invasion (29). In addition, it promotes the expression in RA FLS of inflammatory mediators that perpetuates interactions with other synovial cells including T and B cells (39, 40).
Other signaling pathways critical for FLS expression of adhesion molecules, pro-inflammatory cytokines, and MMPs, as well as for apoptosis inhibition, and for FLS migration and invasion are mitogen-activated protein kinases (MAPK), nuclear factor kappa B (NF-κB), and phosphoinositide-3-kinase (PI3K)/AKT (41–50). These pathways are activated by both hypoxia and inflammation. They also regulate glucose metabolism through several mechanisms including the upregulation of GLUT1 (51). These pathways are also involved in the phosphorylation of rate-limiting glycolytic enzymes, including 6-phosphofructo-2-kinase/fructose-2,6-bisphosphatases (PFKFB) and HKs (52, 53). JAK/STAT signaling, which also plays a role in FLS activation (54–56), was also shown to mediate glucose uptake and HK2 expression (57). Therefore, the phenotypic changes from FLS at rest to an activated and invasive state are coupled with metabolic alterations like increased GLUT1 and HK2 expression and lactate production (14, 17).
Likewise, the inhibition of glycolysis decreases the aggressive behavior of these cells by decreasing cytokine production, proliferation, migration, and invasion (14, 17). Three targetable glycolytic enzymes were recently shown to be involved in FLS aggressive phenotype. One is the bifunctional PFKFB3 enzyme, which converts fructose-6-phosphate to fructose-2,6-bisP (F2,6BP). F2,6BP is an allosteric activator of 6-phosphofructokinase-1 (PFK-1) and stimulates glycolysis overriding the inhibitory effect of ATP on PFK-1 (58). It was identified as a regulator of insulin/IGF-1 signaling pathway. Suppression of PFKFB3 was found to decrease insulin-stimulated glucose uptake, GLUT4 translocation, Akt signaling, and glycolytic flux (59). In FLS, PFKFB3 inhibition reduced glucose uptake which resulted in decreased lactate production (17, 18). The inhibition of the glycolytic flux by small molecule inhibitors of PFKFB3 significantly reduced FLS migration and invasion, and the production of inflammatory mediators (17). Of interest, FPFK15, a PFKFB3 inhibitor, not only suppressed glucose uptake and lactate secretion but also NF-κB and MAPK activation in RA FLS (18). The second enzyme is the rate-limiting enzyme HK2. Overexpression of HK2 in FLS provides a migratory and invasive advantage that is abolished when HK2 is ablated (19, 20). The last enzyme is phosphoglycerate kinase (PGK)1. Anti-PGK1 siRNA treatment of RA FLS not only decreased cell proliferation and cell migration, but also interleukin (IL)-1β and interferon (IFN)-γ secretion (21).
Since metabolic pathways are highly interconnected, other metabolic pathways described in activated FLS might also affect global glucose metabolism. Glutamine metabolism is increased in FLS, with glutaminase 1 (GLS1) playing a role in regulating the proliferation of these cells. FLS proliferation is reduced under glutamine-deprived conditions, or after GLS1 silencing or inhibition (60). Choline metabolism is also highly activated in FLS. Inhibition of choline kinase (ChoKα) suppressed the RA FLS aggressive phenotype by increasing apoptosis and decreasing cell migration (61). Glycogen synthase 1 (GYS1)-mediated glycogen accumulation was shown to block AMPK activation and to contribute to FLS phenotype as well (62). Finally, RA FLS also overexpresses the neutral amino acid transporter LAT1, and has an increased uptake of leucine after IL-17 stimulation, which potentiate FLS migratory capacity that was eliminated by blocking LAT1 (63). Other amino acids including tryptophan might also play a role in FLS phenotype (64).
Although researchers have suggested a role for metabolic alterations in RA pathology, we are far from understanding which changes are normal responses to cell activation and are transient metabolic responses to acute inflammation, and which are the result of damage and chronic activation that could play a role in driving the pathology of RA. These chronic metabolic changes in FLS can have not only profound effects on the biology of other cells through intermediate metabolites but also can create a new epigenetic landscape that results in a stable FLS activation that is maintained even without continuous stimulation (Figure 1A).
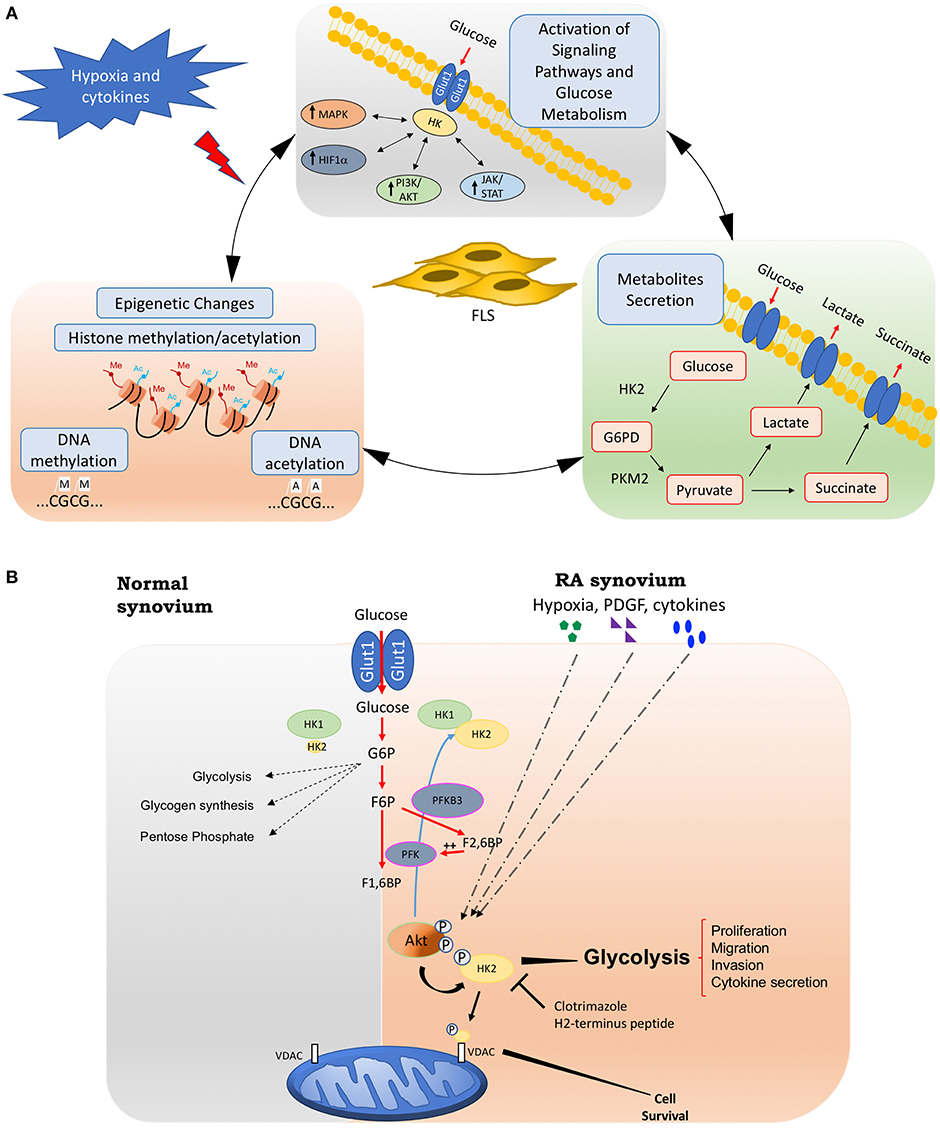
Figure 1. Fibroblasts-like synoviocytes (FLS) glucose metabolism and chronic activation in RA. (A) Chronic glucose metabolic changes induced by hypoxia and inflammatory mediators in FLS will activate many signaling pathways, including HIF, MAPK, PI3K/Akt, and JAK/STAT pathways, which also increases the expression of key glucose metabolism related genes such as GLUT1, HK2, or LDH. Intermediate glucose metabolites including pyruvate, lactate, succinate, a-ketoglutarate, fumarate, and acetyl-coenzyme will create a chronic and sustained FLS activation, either by being secreted extracellularly and triggering profound effects on the biology of other cells, or by inducing a new epigenetic landscape that results in a stable FLS activation that is maintained even without continuous stimulation. (B) Hypoxia, growth factors, and cytokines in arthritis synovium stimulate Akt phosphorylation, which will up-regulate HK2 expression and HK2 phosphorylation. The phosphorylation of HK2 by Akt is accompanied by an increased binding of the enzyme to mitochondrial outer membrane voltage-dependent anion channel (VDAC). Binding to VDAC enhances the affinity of hexokinases. Therefore, HK2 mitochondrial binding might promote glucose metabolism and FLS invasive phenotype. Mitochondrial HK2 might also inhibit apoptosis. Thus, mitochondrial association of HK2 might promote resistance to growth, invasion, and apoptosis of RA FLS, which contribute to joint destruction in RA. Selective HK2 mitochondrial dissociation might be an attractive potential selective target for arthritis therapy and safer than global glycolysis inhibition. HK2, hexokinase 2; G6PD, glucose 6 phosphate dehydrogenase; PKM2, pyruvate kinase muscle isozyme M2; PFK, phosphofructokinase; PFKB3, 6-phosphofructo-2-kinase/fructose-2,6-biphosphatase 3; G6P, glucose-6-phosphate; F6P, fructose-6-phosphate; F1,6BP, Fructose 1,6-bisphosphate; F2,6FB, Fructose 2,6-bisphosphate; VDAC: voltage-dependent anion channel.
Some pro-inflammatory mediators increase several TCA cycle intermediates, and emerging evidence show that these intermediates classically associated with metabolic functions also possess signaling functions as inflammatory mediators and drive chronic activation. For instance, metabolic profiling has revealed itaconic acid as a potential marker of RA, and TNF increased its concentration in the K4IM human fibroblast cell line (65). Importantly, the increased levels in itaconic acid can be attenuated by treatment with infliximab, a biologic drug targeting TNF (65). Succinate is another TCA cycle intermediate that is abundant in RA synovial fluids. Synovial succinate correlates with enhanced release of IL-1β by macrophages in a mechanism that involves the overexpression of succinate receptor Sucnr1/GPR91 (66). In addition, Sucnr1/GPR91 functions as a chemotactic signal for recruitment of dendritic cells into lymph nodes which leads to Th17 cells expansion in a murine model of inflammatory arthritis (67). In FLS, succinate has a myofibroblast effect on FLS (23). A recent paper also evaluated Sucnr1/GPR91 in FLS and concluded that both intra- and extracellular succinate play a role in synovial angiogenesis. Intracellular succinate induced angiogenesis through HIF1α induction, while extracellular succinate increased vascular endothelial growth factor (VEGF) through GPR91 receptor (24).
Pyruvate is another metabolite generated during glycolysis and is converted to acetyl-CoA to fuel the TCA cycle. Acetyl-CoA is an important cofactor that catalyzes the transfer of an acetyl group. Histone acetyltransferases (HATs) are enzymes that use this co-factor and regulate histone acetylation and therefore link metabolism and epigenetics in cells (68, 69). Other metabolites elevated after glucose metabolism activation such as fumarate, succinate, and lactate also modify chromatin and nucleic acid-modifying enzymes activity by competitively inhibiting substrate utilization (68). The relationship between metabolic intermediates and chromatin-modifying enzymes implies that metabolic changes could directly affect gene expression by modulating chromatin-modifying enzymes and triggering epigenetic dysfunction (68, 69). Of interest, several reports have shown that epigenetic alterations, such as histone modification, might contribute to RA pathogenesis (70). In fact, a comprehensive epigenomic characterization of RA FLS has recently been described (71), suggesting that synovial fibroblasts stimulation results in a stable activation that is maintained even without continuous stimulation through epigenetic changes. Further FLS studies are needed to better understand the epigenetic modifications affecting metabolic gene expression and glucose metabolism that can drive chronic RA FLS activation and may help to identify novel metabolic targets.
The concept of metabolic reprogramming to improve immunotherapy and to complement current therapies is being slowly translated into the autoimmune disease field (11, 72–74). In fact, glycolytic inhibitors not only reduce FLS aggressive phenotype in vitro but also decrease bone and cartilage damage in several murine models of arthritis. More specifically, ablation of glycolytic genes or treatment with 3-bromopyruvate, which antagonizes HK2, significantly reduced the severity of several murine arthritis models (14, 15, 19, 20, 22). Although HK2 specific inhibitors are not available, HK2 can be inhibited by the use of 2-deoxyglucose (2-DG), which is a derivative of glucose that can be phosphorylated by HK2 but not mobilized through succeeding steps of glycolysis. Murine studies have shown that 2-DG reduces cell proliferation and the severity of a spontaneous model of RA arthritis (15). HK2 ablation has also attenuated the severity of bone and cartilage damage in a murine model of inflammatory arthritis (19). Interestingly, the administration of fructose 1,6-bisphosphate (FBP), a glycolytic intermediate, decreased arthritis scores in two different animal models. Mechanistic studies showed that this metabolic intermediate activated the anti-inflammatory adenosinergic pathway instead of enhancing FLS glycolysis (16). Treatment with a saponin that inhibits succinate dehydrogenase (SDH) activity ameliorated the clinical symptoms of the arthritis as well as histopathologic features of synovial hyperplasia, infiltration of inflammatory cells, and fibrosis (23). In addition, treatment with dimethylmalonate, another inhibitor of SDH, decreased succinate content in the synovial tissue of rats with collagen-induced arthritis (CIA) in addition to amelioration of the disease (24). Finally, inhibition of the enzyme ChoKα (61) and GLS1 (62) also ameliorated the severity of experimental autoimmune arthritis.
Yet, although all these works have demonstrated a role of glucose metabolism in RA, inhibiting global glucose metabolism is not desirable. In addition, inhibition of some of the above pathways can have other detrimental effects. For instance, a recent report demonstrated a key anti-inflammatory function of HIF1α by driving the expression of IL-10 in B cells (75). PFKFB3 activity is also defective in CD4 T cells in RA patients which results in energy deprivation that prone cells to undergo apoptosis (76). Thus, there is a need of finding specific metabolic targets that are induced in activated FLS.
Out of all the glycolytic enzymes described to play a role in RA pathogenesis, HK2 could function as a selective metabolic target (Figure 1B). HKs catalyze the phosphorylation of glucose to glucose-6-phosphate (G6P) that facilitates glucose entry into cells. G6P initiates several metabolic pathways that need glucose, including glycolysis, the hexosamine pathway, glycogen synthesis, and the PPP (77). HK2 also plays important roles in angiogenesis (78). HKs has four different isoforms: HK1 is the ubiquitous isoform in all adult issues. However, HK2 is an inducible isoform that is only highly expressed in skeletal and cardiac muscles, and adipose tissue (77). HK2 is also highly upregulated in tumor cells and HK2 inhibition synergies with anti-tumor treatment and improves response to therapy (79). In addition to its canonical metabolic roles in tumor or cardiac tissues, HK2 translocates to the nucleus or mitochondria and triggers an autophagic and anti-apoptotic responses through its interaction with the voltage-dependent anion channel (VDAC) (80, 81). Of interest, HK2 plays a small role in inflammation driven by T cells, so HK2 inhibition should have limited immunosuppressive effects (82). Importantly, we and others have shown that the synovial expression of HK2 is elevated only in RA compared to OA samples (19, 20). Given HK2 selective overexpression in inflamed RA synovium, its small role in T cells, and its expression in a very limited number of adult tissues, HK2 is an attractive selective target for arthritis therapy that is safer than global glucose metabolism inhibition (19). In addition to its expression profile, its diverse effects at various cellular compartments could offer another level of specificity since targeting a specific intracellular compartment of HK2 (i.e., cytosol, nucleus, or mitochondria) would also provide a selective means to block deleterious effects of this enzyme in RA without affecting glucose metabolism in normal cells. Therefore, HK2 could be selectively targeted offering a safer and novel additional approach for combination therapy in RA joint disease independent of systemic immunosuppression.
Of interest, rheumatologists already have antimetabolites in the current RA armamentarium, such as methotrexate and leflunomide. Although they were thought to inhibit the proliferation of synovial and immune cells, methotrexate and other disease-modifying antirheumatic drugs (DMARDs) also have effect on glucose metabolism. For instance, methotrexate treatment significantly reduced HK2 expression and glucose/fructose carriers (SLC2A5, a member of the solute carrier family 2) in human FLS, suggesting that FLS glycolytic activity can be modulated by methotrexate (83). Anti-TNF treatment decreases the synovial expression of GLUT1 and of the glycolytic enzymes pyruvate kinase muscle isozyme M2 (PKM2) and glyceraldehyde 3-phosphate dehydrogenase (GAPDH) in patients that responded to TNF inhibition compared to non-responders (17). Anti-IL-6 receptor therapy inhibited oxidative stress and improved endothelial function in RA leucocytes, although whether or not this therapy also has a metabolic effect on the synovial tissue is not known yet (84). Finally, inhibition of JAK/STAT3 signaling with tofacinib, a drug approved for severe RA and active psoriasis, induces oxidative phosphorylation and maximal respiratory capacity of FLS while shutting down key glycolytic enzymes including HK2 and LDH. This effect correlated with the reduction of inflammatory mediators and FLS activation (85).
Growing evidence suggests that the study of activated metabolism not only of immune cells but also of stroma cells including FLS can provide critical pathways for therapeutic intervention. Pre-clinical studies in mouse models of inflammatory arthritis strongly suggest that agents that interfere with certain steps of glycolysis can be therapeutic in RA and have identified potential targetable glycolytic enzymes such as HK2, and glycolytic intermediate metabolites (Table 1). In addition, therapeutic effects of DMARDs could be due, at least partially, to the inhibition of glucose metabolism, highlighting the pathogenic role of this metabolic pathway. As global inhibition of glucose metabolism is not desirable, more information about inducible glycolytic genes, the specific distribution of these targets, their effect in different cellular compartments, and their additional non-metabolic functions, may help us to identify new targets that do not compromise global glucose metabolism in normal cells.
All authors listed have made a substantial, direct and intellectual contribution to the work, and approved it for publication.
MG was supported by the National Institutes of Health under Award Numbers R01AR073324 and by Rheumatology Research Foundation.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
1. Smolen JS, Aletaha D, McInnes IB. Rheumatoid arthritis. Lancet. (2016) 388:2023–38. doi: 10.1016/S0140-6736(16)30173-8
2. Wegner N, Lundberg K, Kinloch A, Fisher B, Malmström V, Feldmann M, et al. Autoimmunity to specific citrullinated proteins gives the first clues to the etiology of rheumatoid arthritis. Immunol Rev. (2010) 233:34–54. doi: 10.1111/j.0105-2896.2009.00850.x
3. McInnes IB, Schett G. The pathogenesis of rheumatoid arthritis. N Engl J Med. (2011) 365:2205–19. doi: 10.1056/NEJMra1004965
4. Bottini N, Firestein G. Duality of fibroblast-like synoviocytes in RA: passive responders and imprinted aggressors. Nat Rev Rheumatol. (2013) 9:10. doi: 10.1038/nrrheum.2012.190
5. Smolen JS, Aletaha D, Barton A, Burmester GR, Emery P, Firestein GS, et al. Rheumatoid arthritis. Nat Rev Dis Primers. (2018) 4:18001. doi: 10.1038/nrdp.2018.1
6. Kiener HP, Watts GF, Cui Y, Wright J, Thornhill TS, Sköld M, et al. Synovial fibroblasts self-direct multicellular lining architecture and synthetic function in three-dimensional organ culture. Arthritis Rheum. (2010) 62:742–52. doi: 10.1002/art.27285
7. Filer A. The fibroblast as a therapeutic target in rheumatoid arthritis. Curr Opin Pharmacol. (2013) 13:413–9. doi: 10.1016/j.coph.2013.02.006
8. Bartok B, Firestein GS. Fibroblast-like synoviocytes: key effector cells in rheumatoid arthritis. Immunol Rev. (2010) 233:233–55. doi: 10.1111/j.0105-2896.2009.00859.x
9. Turner JD, Filer A. The role of the synovial fibroblast in rheumatoid arthritis pathogenesis. Curr Opin Rheumatol. (2015) 27:175–82. doi: 10.1097/BOR.0000000000000148
10. Tolboom TC, van der Helm-Van Mil AH, Nelissen RG, Breedveld FC, Toes RE, Huizinga TW. Invasiveness of fibroblast-like synoviocytes is an individual patient characteristic associated with the rate of joint destruction in patients with rheumatoid arthritis. Arthritis Rheum. (2005) 52:1999–2002. doi: 10.1002/art.21118
11. Guma M, Tiziani S, Firestein GS. Metabolomics in rheumatic diseases: desperately seeking biomarkers. Nat Rev Rheumatol. (2016) 12:269–81. doi: 10.1038/nrrheum.2016.1
12. Falconer J, Murphy AN, Young S, Clark AR, Tiziani S, Guma M, et al. Synovial cell metabolism and chronic inflammation in rheumatoid arthritis. Arthritis Rheumatol. (2018) 70:984–99. doi: 10.1002/art.40504
13. Fearon U, Canavan M, Biniecka M, Veale DJ. Hypoxia, mitochondrial dysfunction and synovial invasiveness in rheumatoid arthritis. Nat Rev Rheumatol. (2016) 12:385–97. doi: 10.1038/nrrheum.2016.69
14. Garcia-Carbonell R, Divakaruni AS, Lodi A, Vicente-Suarez I, Saha A, Cheroutre H, et al. Critical role of glucose metabolism in rheumatoid arthritis fibroblast-like synoviocytes. Arthritis Rheumatol. (2016) 68:1614–26. doi: 10.1002/art.39608
15. Abboud G, Choi SC, Kanda N, Zeumer-Spataro L, Roopenian DC, Morel L. Inhibition of glycolysis reduces disease severity in an autoimmune model of rheumatoid arthritis. Front Immunol. (2018) 9:1973. doi: 10.3389/fimmu.2018.01973
16. Veras FP, Peres RS, Saraiva AL, Pinto LG, Louzada-Junior P, Cunha TM, et al. Fructose 1,6-bisphosphate, a high-energy intermediate of glycolysis, attenuates experimental arthritis by activating anti-inflammatory adenosinergic pathway. Sci Rep. (2015) 5:15171. doi: 10.1038/srep15171
17. Biniecka M, Canavan M, McGarry T, Gao W, McCormick J, Cregan S, et al. Dysregulated bioenergetics: a key regulator of joint inflammation. Ann Rheum Dis. (2016) 75:2192–200. doi: 10.1136/annrheumdis-2015-208476
18. Zou Y, Zeng S, Huang M, Qiu Q, Xiao Y, Shi M, et al. Inhibition of 6-phosphofructo-2-kinase suppresses fibroblast-like synoviocytes-mediated synovial inflammation and joint destruction in rheumatoid arthritis. Br J Pharmacol. (2017) 174:893–908. doi: 10.1111/bph.13762
19. Bustamante MF, Oliveira PG, Garcia-Carbonell R, Croft AP, Smith JM, Serrano RL, et al. Hexokinase 2 as a novel selective metabolic target for rheumatoid arthritis. Ann Rheum Dis. (2018) 77:1636–43. doi: 10.1136/annrheumdis-2018-213103
20. Song G, Lu Q, Fan H, Zhang X, Ge L, Tian R, et al. Inhibition of hexokinases holds potential as treatment strategy for rheumatoid arthritis. Arthritis Res Ther. (2019) 21:87. doi: 10.1186/s13075-019-1865-3
21. Zhao Y, Yan X, Li X, Zheng Y, Li S, Chang X. PGK1, a glucose metabolism enzyme, may play an important role in rheumatoid arthritis. Inflamm Res. (2016) 65:815–25. doi: 10.1007/s00011-016-0965-7
22. Okano T, Saegusa J, Nishimura K, Takahashi S, Sendo S, Ueda Y, et al. 3-bromopyruvate ameliorate autoimmune arthritis by modulating Th17/Treg cell differentiation and suppressing dendritic cell activation. Sci Rep. (2017) 7:42412. doi: 10.1038/srep42412
23. Li Y, Zheng JY, Liu JQ, Yang J, Liu Y, Wang C, et al. Succinate/NLRP3 inflammasome induces synovial fibroblast activation: therapeutical effects of clematichinenoside AR on arthritis. Front Immunol. (2016) 7:532. doi: 10.3389/fimmu.2016.00532
24. Li Y, Liu Y, Wang C, Xia WR, Zheng JY, Yang J, et al. Succinate induces synovial angiogenesis in rheumatoid arthritis through metabolic remodeling and HIF-1alpha/VEGF axis. Free Radic Biol Med. (2018) 126:1–14. doi: 10.1016/j.freeradbiomed.2018.07.009
25. Lee SJ, Jeong JH, Lee CH, Ahn BC, Eun JS, Kim NR, et al. Development and validation of an (18) F-FDG PET/CT-based tool for the evaluation of joint counts and disease activity in patients with rheumatoid arthritis. Arthritis Rheumatol. (2019) 2019:40860. doi: 10.1002/art.40860
26. Matsui T, Nakata N, Nagai S, Nakatani A, Takahashi M, Momose T, et al. Inflammatory cytokines and hypoxia contribute to 18F-FDG uptake by cells involved in pannus formation in rheumatoid arthritis. J Nucl Med. (2009) 50:920–6. doi: 10.2967/jnumed.108.060103
27. Volchenkov R, Dung Cao M, Elgstoen KB, Goll GL, Eikvar K, Bjorneboe O, et al. Metabolic profiling of synovial tissue shows altered glucose and choline metabolism in rheumatoid arthritis samples. Scand J Rheumatol. (2017) 46:160–1. doi: 10.3109/03009742.2016.1164242
28. Quinonez-Flores CM, Gonzalez-Chavez SA, Pacheco-Tena C. Hypoxia and its implications in rheumatoid arthritis. J Biomed Sci. (2016) 23:62. doi: 10.1186/s12929-016-0281-0
29. Hua S, Dias TH. Hypoxia-inducible factor (HIF) as a target for novel therapies in rheumatoid arthritis. Front Pharmacol. (2016) 7:184. doi: 10.3389/fphar.2016.00184
30. Young SP, Kapoor SR, Viant MR, Byrne JJ, Filer A, Buckley CD, et al. The impact of inflammation on metabolomic profiles in patients with arthritis. Arthritis Rheum. (2013) 65:2015–23. doi: 10.1002/art.38021
31. Anderson JR, Chokesuwattanaskul S, Phelan MM, Welting TJM, Lian LY, Peffers MJ, et al. (1)H NMR metabolomics identifies underlying inflammatory pathology in osteoarthritis and rheumatoid arthritis synovial joints. J Proteome Res. (2018) 17:3780–90. doi: 10.1021/acs.jproteome.8b00455
32. Ahn JK, Kim S, Hwang J, Kim J, Kim KH, Cha HS. GC/TOF-MS-based metabolomic profiling in cultured fibroblast-like synoviocytes from rheumatoid arthritis. Joint Bone Spine. (2016) 83:707–13. doi: 10.1016/j.jbspin.2015.11.009
33. Ahn JK, Kim S, Hwang J, Kim J, Lee YS, Koh EM, et al. Metabolomic elucidation of the effects of curcumin on fibroblast-like synoviocytes in rheumatoid arthritis. PLoS ONE. (2015) 10:e0145539. doi: 10.1371/journal.pone.0145539
34. Hurter K, Spreng D, Rytz U, Schawalder P, Ott-Knusel F, Schmokel H. Measurements of C-reactive protein in serum and lactate dehydrogenase in serum and synovial fluid of patients with osteoarthritis. Vet J. (2005) 169:281–5. doi: 10.1016/j.tvjl.2004.01.027
35. Wright AJ, Husson ZMA, Hu DE, Callejo G, Brindle KM, Smith ESJ. Increased hyperpolarized [1-(13) C] lactate production in a model of joint inflammation is not accompanied by tissue acidosis as assessed using hyperpolarized (13) C-labelled bicarbonate. NMR Biomed. (2018) 31:e3892. doi: 10.1002/nbm.3892
36. Bustamante MF, Garcia-Carbonell R, Whisenant KD, Guma M. Fibroblast-like synoviocyte metabolism in the pathogenesis of rheumatoid arthritis. Arthritis Res Ther. (2017) 19:110. doi: 10.1186/s13075-017-1303-3
37. Del Rey MJ, Valin A, Usategui A, Garcia-Herrero CM, Sanchez-Arago M, Cuezva JM, et al. Hif-1alpha knockdown reduces glycolytic metabolism and induces cell death of human synovial fibroblasts under normoxic conditions. Sci Rep. (2017) 7:3644. doi: 10.1038/s41598-017-03921-4
38. del Rey MJ, Izquierdo E, Caja S, Usategui A, Santiago B, Galindo M, et al. Human inflammatory synovial fibroblasts induce enhanced myeloid cell recruitment and angiogenesis through a hypoxia-inducible transcription factor 1alpha/vascular endothelial growth factor-mediated pathway in immunodeficient mice. Arthritis Rheum. (2009) 60:2926–34. doi: 10.1002/art.24844
39. Hu F, Liu H, Xu L, Li Y, Liu X, Shi L, et al. Hypoxia-inducible factor-1alpha perpetuates synovial fibroblast interactions with T cells and B cells in rheumatoid arthritis. Eur J Immunol. (2016) 46:742–51. doi: 10.1002/eji.201545784
40. Hu F, Mu R, Zhu J, Shi L, Li Y, Liu X, et al. Hypoxia and hypoxia-inducible factor-1alpha provoke toll-like receptor signalling-induced inflammation in rheumatoid arthritis. Ann Rheum Dis. (2014) 73:928–36. doi: 10.1136/annrheumdis-2012-202444
41. Vincenti MP, Coon CI, Brinckerhoff CE. Nuclear factor kappaB/p50 activates an element in the distal matrix metalloproteinase 1 promoter in interleukin-1beta-stimulated synovial fibroblasts. Arthritis Rheum. (1998) 41:1987–94.
42. Miagkov AV, Kovalenko DV, Brown CE, Didsbury JR, Cogswell JP, Stimpson SA, et al. NF-kappaB activation provides the potential link between inflammation and hyperplasia in the arthritic joint. Proc Natl Acad Sci U S A. (1998) 95:13859–64.
43. Han Z, Boyle DL, Chang L, Bennett B, Karin M, Yang L, et al. c-Jun N-terminal kinase is required for metalloproteinase expression and joint destruction in inflammatory arthritis. J Clin Invest. (2001) 108:73–81. doi: 10.1172/JCI12466
44. Han Z, Chang L, Yamanishi Y, Karin M, Firestein GS. Joint damage and inflammation in c-Jun N-terminal kinase 2 knockout mice with passive murine collagen-induced arthritis. Arthritis Rheum. (2002) 46:818–23. doi: 10.1002/art.10104
45. Pillinger MH, Rosenthal PB, Tolani SN, Apsel B, Dinsell V, Greenberg J, et al. Cyclooxygenase-2-derived E prostaglandins down-regulate matrix metalloproteinase-1 expression in fibroblast-like synoviocytes via inhibition of extracellular signal-regulated kinase activation. J Immunol. (2003) 171:6080–9. doi: 10.4049/jimmunol.171.11.6080
46. Bradley K, Scatizzi JC, Fiore S, Shamiyeh E, Koch AE, Firestein GS, et al. Retinoblastoma suppression of matrix metalloproteinase 1, but not interleukin-6, through a p38-dependent pathway in rheumatoid arthritis synovial fibroblasts. Arthritis Rheum. (2004) 50:78–87. doi: 10.1002/art.11482
47. Hwang SY, Kim JY, Kim KW, Park MK, Moon Y, Kim WU, et al. IL-17 induces production of IL-6 and IL-8 in rheumatoid arthritis synovial fibroblasts via NF-kappaB- and PI3-kinase/Akt-dependent pathways. Arthritis Res Ther. (2004) 6:R120–8. doi: 10.1186/ar1038
48. Bartok B, Boyle DL, Liu Y, Ren P, Ball ST, Bugbee WD, et al. PI3 kinase delta is a key regulator of synoviocyte function in rheumatoid arthritis. Am J Pathol. (2012) 180:1906–16. doi: 10.1016/j.ajpath.2012.01.030
49. Li GQ, Zhang Y, Liu D, Qian YY, Zhang H, Guo SY, et al. PI3 kinase/Akt/HIF-1alpha pathway is associated with hypoxia-induced epithelial-mesenchymal transition in fibroblast-like synoviocytes of rheumatoid arthritis. Mol Cell Biochem. (2013) 372:221–31. doi: 10.1007/s11010-012-1463-z
50. Duvel K, Yecies JL, Menon S, Raman P, Lipovsky AI, Souza AL, et al. Activation of a metabolic gene regulatory network downstream of mTOR complex 1. Mol Cell. (2010) 39:171–83. doi: 10.1016/j.molcel.2010.06.022
51. Barthel A, Okino ST, Liao J, Nakatani K, Li J, Whitlock JP Jr, et al. Regulation of GLUT1 gene transcription by the serine/threonine kinase Akt1. J Biol Chem. (1999) 274:20281–6.
52. Miyamoto S, Murphy AN, Brown JH. Akt mediates mitochondrial protection in cardiomyocytes through phosphorylation of mitochondrial hexokinase-II. Cell Death Differ. (2008) 15:521–9. doi: 10.1038/sj.cdd.4402285
53. Deprez J, Vertommen D, Alessi DR, Hue L, Rider MH. Phosphorylation and activation of heart 6-phosphofructo-2-kinase by protein kinase B and other protein kinases of the insulin signaling cascades. J Biol Chem. (1997) 272:17269–75.
54. Gao W, McCormick J, Connolly M, Balogh E, Veale DJ, Fearon U. Hypoxia and STAT3 signalling interactions regulate pro-inflammatory pathways in rheumatoid arthritis. Ann Rheum Dis. (2015) 74:1275–83. doi: 10.1136/annrheumdis-2013-204105
55. Migita K, Komori A, Torigoshi T, Maeda Y, Izumi Y, Jiuchi Y, et al. CP690,550 inhibits oncostatin M-induced JAK/STAT signaling pathway in rheumatoid synoviocytes. Arthritis Res Ther. (2011) 13:R72. doi: 10.1186/ar3333
56. Rosengren S, Corr M, Firestein GS, Boyle DL. The JAK inhibitor CP-690,550 (tofacitinib) inhibits TNF-induced chemokine expression in fibroblast-like synoviocytes: autocrine role of type I interferon. Ann Rheum Dis. (2012) 71:440–7. doi: 10.1136/ard.2011.150284
57. Heiss EH, Schachner D, Donati M, Grojer CS, Dirsch VM. Increased aerobic glycolysis is important for the motility of activated VSMC and inhibited by indirubin-3'-monoxime. Vascular Pharmacol. (2016) 83:47–56. doi: 10.1016/j.vph.2016.05.002
58. Clem BF, O'Neal J, Tapolsky G, Clem AL, Imbert-Fernandez Y, Kerr DA II, et al. Targeting 6-phosphofructo-2-kinase (PFKFB3) as a therapeutic strategy against cancer. Mol Cancer Ther. (2013) 12:1461–70. doi: 10.1158/1535-7163.MCT-13-0097
59. Trefely S, Khoo PS, Krycer JR, Chaudhuri R, Fazakerley DJ, Parker BL, et al. Kinome screen identifies PFKFB3 and glucose metabolism as important regulators of the insulin/insulin-like growth factor (IGF)-1 signaling pathway. J Biol Chem. (2015) 290:25834–46. doi: 10.1074/jbc.M115.658815
60. Takahashi S, Saegusa J, Sendo S, Okano T, Akashi K, Irino Y, et al. Glutaminase 1 plays a key role in the cell growth of fibroblast-like synoviocytes in rheumatoid arthritis. Arthritis Res Ther. (2017) 19:76. doi: 10.1186/s13075-017-1283-3
61. Guma M, Sanchez-Lopez E, Lodi A, Garcia-Carbonell R, Tiziani S, Karin M, et al. Choline kinase inhibition in rheumatoid arthritis. Ann Rheum Dis. (2015) 74:1399–407. doi: 10.1136/annrheumdis-2014-205696
62. Shi M, Wang J, Xiao Y, Wang C, Qiu Q, Lao M, et al. Glycogen metabolism and rheumatoid arthritis: the role of glycogen synthase 1 in regulation of synovial inflammation via blocking AMP-activated protein kinase activation. Front Immunol. (2018) 9:1714. doi: 10.3389/fimmu.2018.01714
63. Yu Z, Lin W, Rui Z, Jihong P. Fibroblast-like synoviocyte migration is enhanced by IL-17-mediated overexpression of L-type amino acid transporter 1 (LAT1) via the mTOR/4E-BP1 pathway. Amino Acids. (2018) 50:331–40. doi: 10.1007/s00726-017-2520-4
64. Szanto S, Koreny T, Mikecz K, Glant TT, Szekanecz Z, Varga J. Inhibition of indoleamine 2,3-dioxygenase-mediated tryptophan catabolism accelerates collagen-induced arthritis in mice. Arthritis Res Ther. (2007) 9:R50. doi: 10.1186/ar2205
65. Michopoulos F, Karagianni N, Whalley NM, Firth MA, Nikolaou C, Wilson ID, et al. Targeted metabolic profiling of the Tg197 mouse model reveals itaconic acid as a marker of rheumatoid arthritis. J Proteome Res. (2016) 15:4579–90. doi: 10.1021/acs.jproteome.6b00654
66. Littlewood-Evans A, Sarret S, Apfel V, Loesle P, Dawson J, Zhang J, et al. GPR91 senses extracellular succinate released from inflammatory macrophages and exacerbates rheumatoid arthritis. J Exp Med. (2016) 213:1655–62. doi: 10.1084/jem.20160061
67. Saraiva AL, Veras FP, Peres RS, Talbot J, de Lima KA, Luiz JP, et al. Succinate receptor deficiency attenuates arthritis by reducing dendritic cell traffic and expansion of Th17 cells in the lymph nodes. FASEB J. (2018) 2018:fj201800285. doi: 10.1096/fj.201800285
68. Reid MA, Dai Z, Locasale JW. The impact of cellular metabolism on chromatin dynamics and epigenetics. Nat Cell Biol. (2017) 19:1298–306. doi: 10.1038/ncb3629
69. Etchegaray JP, Mostoslavsky R. Interplay between metabolism and epigenetics: a nuclear adaptation to environmental changes. Mol Cell. (2016) 62:695–711. doi: 10.1016/j.molcel.2016.05.029
70. Doody KM, Bottini N, Firestein GS. Epigenetic alterations in rheumatoid arthritis fibroblast-like synoviocytes. Epigenomics. (2017) 9:479–92. doi: 10.2217/epi-2016-0151
71. Ai R, Laragione T, Hammaker D, Boyle DL, Wildberg A, Maeshima K, et al. Comprehensive epigenetic landscape of rheumatoid arthritis fibroblast-like synoviocytes. Nat Commun. (2018) 9:1921. doi: 10.1038/s41467-018-04310-9
72. McGarry T, Fearon U. Cell metabolism as a potentially targetable pathway in RA. Nat Rev Rheumatol. (2019) 15:70–2. doi: 10.1038/s41584-018-0148-8
73. Rhoads JP, Major AS, Rathmell JC. Fine tuning of immunometabolism for the treatment of rheumatic diseases. Nat Rev Rheumatol. (2017) 13:313–20. doi: 10.1038/nrrheum.2017.54
74. Weyand CM, Goronzy JJ. Immunometabolism in early and late stages of rheumatoid arthritis. Nat Rev Rheumatol. (2017) 13:291–301. doi: 10.1038/nrrheum.2017.49
75. Meng X, Grotsch B, Luo Y, Knaup KX, Wiesener MS, Chen XX, et al. Hypoxia-inducible factor-1alpha is a critical transcription factor for IL-10-producing B cells in autoimmune disease. Nat Commun. (2018) 9:251. doi: 10.1038/s41467-017-02683-x
76. Yang Z, Fujii H, Mohan SV, Goronzy JJ, Weyand CM. Phosphofructokinase deficiency impairs ATP generation, autophagy, and redox balance in rheumatoid arthritis T cells. J Exp Med. (2013) 210:2119–34. doi: 10.1084/jem.20130252
77. Wilson JE. Isozymes of mammalian hexokinase: structure, subcellular localization and metabolic function. J Exp Biol. (2003) 206(Pt 12):2049–57. doi: 10.1242/jeb.00241
78. Yu P, Wilhelm K, Dubrac A, Tung JK, Alves TC, Fang JS, et al. FGF-dependent metabolic control of vascular development. Nature. (2017) 545:224–8. doi: 10.1038/nature22322
79. Jones AT, Narov K, Yang J, Sampson JR, Shen MH. Efficacy of dual inhibition of glycolysis and glutaminolysis for therapy of renal lesions in Tsc2(+/-) mice. Neoplasia. (2019) 21:230–8. doi: 10.1016/j.neo.2018.12.003
80. Chang CH, Curtis JD, Maggi LB Jr, Faubert B, Villarino AV, O'Sullivan D, et al. Posttranscriptional control of T cell effector function by aerobic glycolysis. Cell. (2013) 153:1239–51. doi: 10.1016/j.cell.2013.05.016
81. Yu X, Li S. Non-metabolic functions of glycolytic enzymes in tumorigenesis. Oncogene. (2016) 36:2629–36. doi: 10.1038/onc.2016.410
82. Mehta MM, Weinberg SE, Steinert EM, Chhiba K, Martinez CA, Gao P, et al. Hexokinase 2 is dispensable for T cell-dependent immunity. Cancer Metab. (2018) 6:10. doi: 10.1186/s40170-018-0184-5
83. Shervington L, Darekar A, Shaikh M, Mathews R, Shervington A. Identifying reliable diagnostic/predictive biomarkers for rheumatoid arthritis. Biomark Insights. (2018) 13:1177271918801005. doi: 10.1177/1177271918801005
84. Ruiz-Limon P, Ortega R, Arias de la Rosa I, Abalos-Aguilera MDC, Perez-Sanchez C, Jimenez-Gomez Y, et al. Tocilizumab improves the proatherothrombotic profile of rheumatoid arthritis patients modulating endothelial dysfunction, NETosis, and inflammation. Transl Res. (2017) 183:87–103. doi: 10.1016/j.trsl.2016.12.003
Keywords: rheumatoid arthritis, fibroblast-like synoviocytes, hexokinase-2, glucose metabolism, glycolytic inhibitors
Citation: de Oliveira PG, Farinon M, Sanchez-Lopez E, Miyamoto S and Guma M (2019) Fibroblast-Like Synoviocytes Glucose Metabolism as a Therapeutic Target in Rheumatoid Arthritis. Front. Immunol. 10:1743. doi: 10.3389/fimmu.2019.01743
Received: 15 May 2019; Accepted: 10 July 2019;
Published: 02 August 2019.
Edited by:
Hanshi Xu, First Affiliated Hospital of Sun Yat-sen University, ChinaReviewed by:
Aleksander M. Grabiec, Jagiellonian University, PolandCopyright © 2019 de Oliveira, Farinon, Sanchez-Lopez, Miyamoto and Guma. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Monica Guma, bWd1bWFAdWNzZC5lZHU=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.