 Matthias Mezger1
Matthias Mezger1 Henry Nording1,2
Henry Nording1,2 Reinhard Sauter1
Reinhard Sauter1 Tobias Graf1
Tobias Graf1 Christian Heim3
Christian Heim3 Nikolas von Bubnoff4Stephan M. Ensminger5
Nikolas von Bubnoff4Stephan M. Ensminger5 Harald F. Langer1,2*
Harald F. Langer1,2*- 1University Hospital, Medical Clinic II, University Heart Center Lübeck, Lübeck, Germany
- 2DZHK (German Research Centre for Cardiovascular Research), Partner Site Hamburg/Lübeck/Kiel, Lübeck, Germany
- 3Department of Cardiac Surgery, University Hospital Erlangen, Erlangen, Germany
- 4Department of Hematology and Oncology, Medical Center, University of Schleswig-Holstein, Lübeck, Germany
- 5Department of Cardiac and Thoracic Vascular Surgery, University Heart Center Lübeck, Lübeck, Germany
Besides mediating hemostatic functions, platelets are increasingly recognized as important players of inflammation. Data from experiments in mice and men revealed various intersection points between thrombosis, hemostasis, and inflammation, which are addressed and discussed in this review in detail. One such example is the intrinsic coagulation cascade that is initiated after platelet activation thereby further propagating and re-enforcing wound healing or thrombus formation but also contributing to the pathophysiology of severe diseases. FXII of the intrinsic pathway connects platelet activation with the coagulation cascade during immune reactions. It can activate the contact system thereby either creating an inflammatory state or accelerating inflammation. Recent insights into platelet biology could show that platelets are equipped with complement receptors. Platelets are important for tissue remodeling after injury has been inflicted to the endothelial barrier and to the subendothelial tissue. Thus, platelets are increasingly recognized as more than just cells relevant for bleeding arrest. Future insights into platelet biology are to be expected. This research will potentially offer novel opportunities for therapeutic intervention in diseases featuring platelet abundance.
Introduction
In recent years, an increasing body of evidence demonstrates that platelets have several functions beyond hemostasis (1, 2). For instance, abundant evidence highlights the role of platelets for atherosclerosis, inflammation, and tissue regeneration. For example, platelets contribute to vascular inflammation of the brain during stroke or experimental autoimmune encephalitis (EAE) (2, 3). As a consequence, the question was raised whether platelets may even be considered as immune cells, despite the fact that they are not equipped with a nucleus (4). Most of the data available today originate from studies performed in small animal disease models and, thus, require validation in the course of the aforementioned diseases in patients. This is especially true in the context of immunology, since there are distinct differences between species (5). In this review, we seek to highlight the role of platelets for immune responses during thromboinflammation. In particular, we will address the relevance of platelet-associated mechanisms directly affecting the course of diseases in patients, as well as translational approaches.
Platelets Originate From Megakaryopoiesis Located in the Bone Marrow
Like other hematopoietic cells, platelets are produced in the bone marrow (4). They are characterized by a small cell size and are missing a nucleus (4). Mean platelet volume in healthy individuals usually is in a range of 7 to 13 femtoliter (6). In the bone marrow, platelets derive from megakaryocytes. So far, one of the most important, but also best investigated factors known to be involved in megakaryopoiesis and platelet development is the cytokine thrombopoietin (TPO) (7). After binding of thrombopoietin to its counterreceptor Myeloproliferative Leukemia Virus Oncogene (c-MPL) on the megakaryocyte surface, intracellular signaling through Janus Kinase 2 (JAK2) is triggered (8, 9). The process of megakaryopoiesis is still incompletely understood, so far. For example, when thrombopoietin or the respective counterreceptor c-MPL is missing, the total amount of platelets is reduced to 10% of the normal platelet count (7). At the same time, platelet function as well as platelet morphology are not altered under these conditions (7) indicating that redundant factors besides thrombopoietin are involved in the process of terminal megakaryocyte maturation (10). Of note in myeloproliferative neoplasias (MPN), megakaryocytes are part of the malignant clone and source of inflammatory cytokines (11, 12), leading to chronic inflammation, constitutional symptoms and induction of fibroblast proliferation and bone marrow fibrosis (13) which are characteristic for primary and secondary myelofibrosis. In addition, this chronic inflammatory state might drive clonal evolution, cardiovascular disease and thrombohemorrhagic complications in these patients (14). The JAK1/2 inhibitor ruxolitinib attenuates inflammatory cytokines in myelofibrosis and clinical responses correlate with cytokine attenuation (15). Furthermore, there are hints pointing toward a role of megakaryocytes in antigen presentation through MHC-I leading to CD8 T-cell activation (16). Additionally, transfer of antigen loaded MHC-I from megakaryocytes to proplatelets has also been described (16). Vice versa, the inflammatory cytokine IL-6 was shown to be linked to increased plasma levels of thrombopoietin and an ultimately increased platelet number in a murine and human setting (17) and TPO was shown to augment platelet P-selectin (CD62P) expression stimulating platelet-leukocyte associations (18).
Platelet Receptors and Interactions
Despite being small particles, platelets are equipped with a multitude of receptors to interact with themselves, with other cells, e.g., endothelial cells and cells of the immune system and, of course, with the extracellular matrix (Figure 1). In general, four types of receptors can be found on platelets: integrins, glycoproteins, selectins and receptors of the immunoglobulin type (Figure 2). First, after injury to the vessel wall has occurred, GPIbα on the platelet surface binds von Willebrand factor (VWF). This is especially important under conditions of high shear stress such as in the arterial branch of the vascular system (19). In addition, platelets bind to exposed subendothelial collagen fibers through glycoprotein VI (GPVI) (20–22), finally leading to a high affinity state of GPIIb/IIIa (23) (Figure 2). Subsequently, fibrinogen can be bound to GPIIb/IIIa on the platelet surface thereby crosslinking platelets with platelets and platelets with endothelial cells (24). Inhibiting GPIIb/IIIa was shown to be beneficial in myocardial infarction (25), however in stroke, clinical studies revealed an increased risk of bleeding (26). Ongoing thrombus formation is further supported by interaction of integrins on the platelet surface with fibrinogen and components of the extracellular matrix such as collagen and laminin. Interaction with fibrinogen is mediated through integrin α5β1 on the platelet surface, binding to laminin is mediated through integrin α6β1 (27). In addition, binding to collagen also involves integrin α2β1, which brings about platelet filopodia and lamellipodia formation (28). After platelet activation has happened, mediators from platelet granules are released further fueling platelet activation (29). Among these are P-selectin, VWF and fibrinogen from α-granules and ADP, calcium and serotonin from dense granules (30–32). Selectins were shown to be involved in platelet-endothelial and platelet-leukocyte interactions as well especially under conditions when the endothelial barrier is not disrupted (Figure 3). For instance, in ischemia-reperfusion injury platelets were shown to be involved in leukocyte recruitment since both adherent leukocytes as well as emigrated leukocytes were significantly reduced when either platelets were depleted through administration of platelet depleting serum or platelet receptors were blocked through administration of blocking antibodies (33). Besides platelets, P-selectin as well as VWF are stored within Weibel-Palade bodies in endothelial cells, too (34, 35). P-selectin on platelets was shown to interact with PSGL-1 expressed on leukocytes (36). The interaction of P-selectin on platelets with PSGL-1 was also demonstrated for platelet-neutrophil interactions (37, 38). In addition, platelets can interact with endothelial cells through binding of GPIbα on platelets to P-selectin expressed on endothelial cells (39). Under conditions of inflammation such as in atherosclerotic lesions, platelet-mediated recruitment of leukocytes through platelet P-selectin has been shown to be followed by platelet binding with CXCR3 to inflamed endothelium expressing CX3CL1 (40).
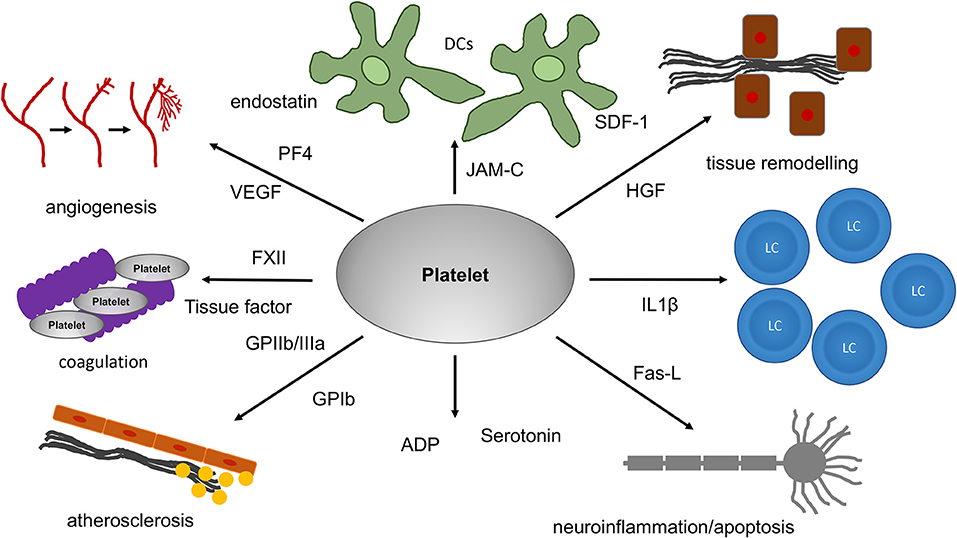
Figure 1. The role of platelets for tissue remodeling, apoptosis and angiogenesis. Platelets can have an influence on tissue remodeling under different pathological and physiological conditions. For tissue remodeling as well as apoptosis and angiogenesis, platelets are equipped with a multitude of receptors. HGF and SDF-1, for example, are of relevance in tissue fibrosis. In addition, platelets can release a multitude of proteins. Among these are proteins with strong pro- or antiangiogenic effects. PF4 and endostatin are known to inhibit angiogenesis. In contrast, VEGF, which platelets can release as well, strongly increases angiogenesis. Since platelets are transported within the blood-stream they can reach almost all organs and tissues thereby even influencing processes associated with inflammation in the brain and systemic diseases like atherosclerosis. In addition, platelet mediated apoptosis through FasL on the platelet surface has been reported in the brain. VEGF, vascular endothelial growth factor; PF4, platelet factor 4; SDF-1, stromal derived factor; HGF, hepatocyte growth factor; JAM-C, junctional adhesion molecule C; IL1β, Interleukin 1β; FasL, Fas-ligand; ADP, adenosine diphosphate; FXII, coagulation factor XII.
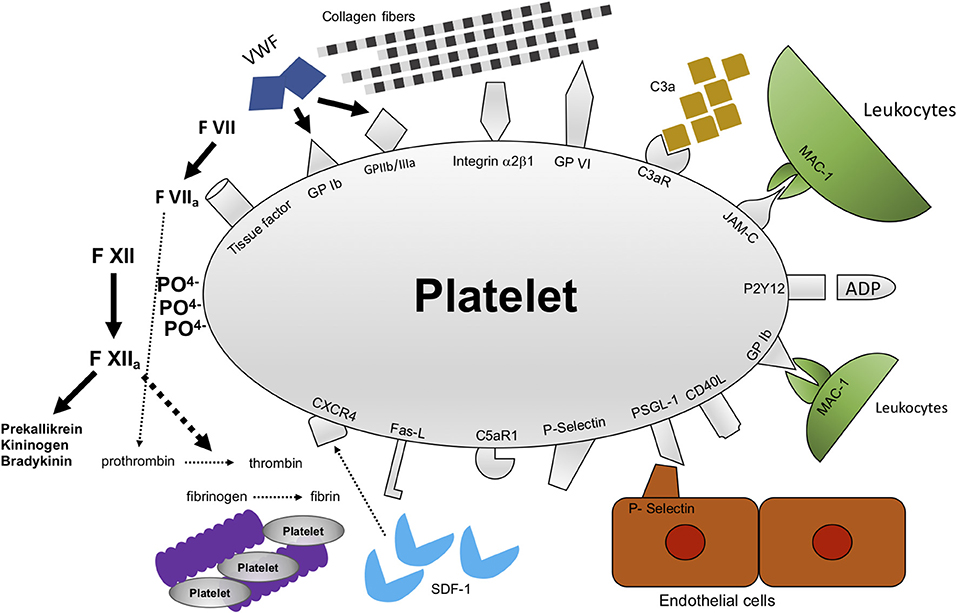
Figure 2. Mechanisms of interactions between platelets and their microenvironment. Platelets were first recognized to be important for thrombosis and hemostasis after vessel injury has happened. In addition, there is accumulating evidence that platelet function goes beyond thrombosis and hemostasis. Platelets interact with a multitude of cells and proteins. Among these are receptors and proteins modulating thrombosis/hemostasis, e.g., GPIbα, GPIIb/IIIa, GPVI, and polyphosphates. In addition, platelets modulate inflammation, e.g., through C3aR, JAM-C, PSGL-1, P-selectin, CXCR4. GPIbα, glycoprotein Ibα; GPIIb/IIIa, glycoprotein IIb/IIIa; GPVI, glycoprotein VI; C3AR, receptor for complement factor 3; JAM-C, junctional adhesion molecule C; CXCR4, C-X-C-chemokine receptor type 4.
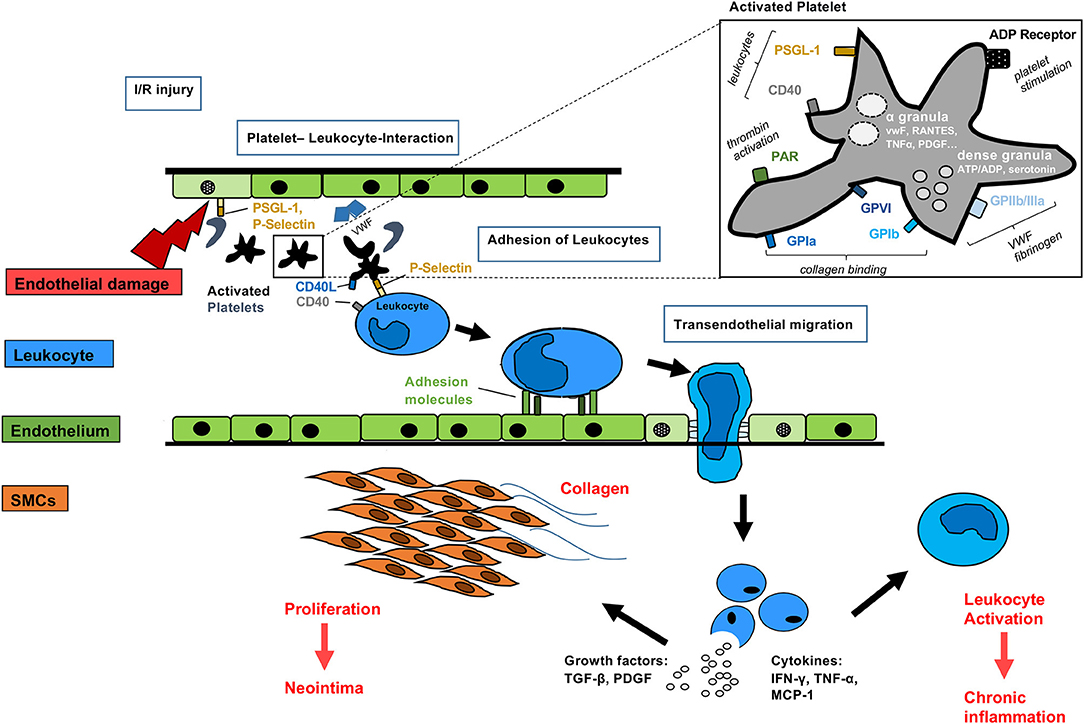
Figure 3. Mechanisms of activated platelets after organ transplantation Platelets may be activated before, during and after allogeneic organ transplantation. Direct interaction with the immune system may occur via simultaneous P-selectin and CD40 binding to leukocytes. Non-nucleated platelets contain dense granules with serotonin, ADP and α-granules with pro-inflammatory cytokines, such as RANTES, TNFα, PDGF, and VWF. Surface receptors including glycoprotein receptors lead to collagen binding, VWF and fibrogen activation. Activated leukocytes adhere to the endothelium of transplanted grafts, migrate and release growth factors and pro-inflammatory cytokines finally leading to smooth muscle cell proliferation, collagen deposition and further leukocyte activation.
Another mechanism especially relevant for the recruitment of dendritic cells to the vascular wall was shown to be mediated by JAM-C (Figure 2), a member of the immunoglobulin family of receptors expressed on platelets, and MAC-1 on dendritic cells (41). Interaction of MAC-1 with platelets bound to endothelial cells has been shown for endothelial transmigration of neutrophils, too (42). Both the interaction of platelets with neutrophils as well as the interaction of platelets with DCs initially requires binding of platelet P-selectin to PSGL-1 on leukocytes (41, 42). Interestingly, MAC-1 was shown to be a binding partner of platelet GPIbα, too (43). Fascinatingly, MAC-1 deficiency goes along with delayed thrombosis although hemostasis seems not to be affected (43). This raises the question whether targeting MAC-1 could offer a way to efficiently inhibit thrombosis without hampering with hemostasis.
Finally, platelets were shown to be involved in the recruitment of CD34+ bone marrow cells and bone marrow progenitor cells to sites of vascular injury (44), linking platelets to tissue remodeling and neointima formation. In this context, platelet P-selectin as well as GPIIb/IIIa were shown to be involved (44). Blockade of platelet receptors with monoclonal antibodies abrogated recruitment of CD34+ cells to sites of vascular injury, further underlining the relevance of platelets for recruitment of bone marrow cells to the vascular wall (44).
Platelets are Closely Linked to the Plasmatic Coagulation System, Thereby Linking Hemostasis and Thrombosis to Inflammation
Platelets mediate thrombosis and hemostasis through the different receptors expressed on the platelet surface but also through soluble mediators released immediately after platelet activation. With respect to hemostasis, platelets operate in parallel to the plasmatic coagulation cascade. In a recent review, the ongoing debate on whether platelets can release tissue factor or not is delineated in a concise fashion (45). Some studies show that platelets can release tissue factor by themselves after activation (46), for instance on the surface of platelet microparticles (47). Tissue factor initiates the extrinsic coagulation cascade (48, 49). On top of that, procoagulant activity was significantly reduced after either tissue factor or F VII were missing in thrombin-activated platelets adhering to fibrinogen (46). Accordingly, in the presence of anti-tissue factor antibody an increased time was observed for clot formation (46). In contrast, other studies were not able to show a role of platelets for tissue-factor mediated coagulation. One study could only show enhanced tissue factor expression when monocytes were present besides platelets, too (48). In another study, no relevant tissue factor secretion could be detected after prolonged stimulation of platelets with lipopolysaccharides (LPS) (50). In the same study, stimulation of a monocyte cell line with LPS yielded large amounts of tissue factor leading to clot formation through activation of the extrinsic coagulation cascade (50), indicating that monocytes in contrast to platelets are responsible for tissue factor production.
There is also evidence that platelets interact with the intrinsic pathway of the coagulation system (Figure 2). Platelets contain polyphosphates that can be externalized onto the platelet membrane, thereby creating a large surface with negative charge triggering activation of coagulation factor XII (contact system of the coagulation cascade) (51). After activation, FXII activates FXI thereby further accelerating coagulation. Additional investigations regarding the role of FXII for coagulation revealed that FXII is necessary for stable thrombi in different models of arterial injury in mice (52). Coagulation Factor XII, besides mediating coagulation, seems to have a role for driving inflammation, as well. After activation, FXII drives activation of the prekallikrein-kininogen-bradykinin-cascade whereby inflammation is triggered (53). Strikingly, in humans FXII deficiency is not associated with an increased bleeding risk. In contrast, in a study population of 74 patients suffering from FXII deficiency, two subjects had already suffered venous thromboembolism at an age <40 years (54). In an experimental design where FXII knockout mice as well as wildtype mice were subjected to transient middle cerebral artery occlusion (tMCAO), FXII knockout mice showed similar reduction in cerebral blood flow in MRI measurements 2 h after tMCAO (55). However, 24 h after tMCAO cerebral blood flow was markedly improved in FXII knockout mice compared to their wildtype counterparts (55). In addition, in tMCAO intravascular fibrin deposits leading to vessel occlusion were reduced in FXII knockout mice. In wildtype animals however, occluding thrombi contain both platelets and fibrin suggesting synergism between FXII mediated fibrin formation and platelets (56). Indeed, knockout mice for FXII showed impaired platelet rich occlusive thrombi in distinct arterial beds (57). This finding was also reported to be of particular relevance in the context of neurovascular inflammation since mice lacking FXII showed protection from ischemic brain damage (55).
Platelets, Despite Lacking a Nucleus, Exhibit Translational Activity and Can Release a Multitude of Active Factors
Early experiments in the 1960 by Warshaw et al. produced first evidence on protein synthesis by platelets since they could inhibit translational activity in platelets with puromycin (58). However, it took several decades and the power of PCR technology and proteomics to demonstrate that platelets are capable of protein synthesis since they incorporate RNA as well as a transcriptional and translational machinery (59). Interestingly, platelets can splice intronic interleukin-1β pre-mRNA generating fully mature interleukin-1β mRNA and interleukin-1β protein (60). Interleukin 1 comprises a group of 11 cytokines with both pro- and anti-inflammatory effects and is a good example demonstrating the role of platelets at an intersection between inflammation and thrombosis (61) (Table 1). The release of a multitude of factors from platelets makes them powerful tools for interaction with various cells and tissues (Figure 1, Table 1). Platelet proteomics could reveal that more than 300 proteins are secreted from human platelets after they become activated by thrombin (62). Among these were proteins responsible for coagulation including the factors FV or FXIII (62), but also proteins such as albumin, platelet factor 4 (PF4, CXCL4) or matrix metalloproteinase inhibitor 1 (62). Finally, factors responsible for tissue remodeling such as CXCL12 (SDF-1) and growth factors, for example hepatocyte growth factor (HGF), can be released by platelets (63, 66). In addition, a specialized way of how translational activity in platelets is modulated through altered ribosome function has been uncovered. It was observed that the platelet lifespan was linked to mRNA and ribosomal RNA content, both of which were reduced with increasing platelet age (78). A recent study could show that loss of the RNA surveillance factor Pelota is a decisive mechanism by which platelets stop decay of their limited pool of mRNA (79). This finding seems to be of relevance in a clinical context, too, since an association of platelet protein synthesis and cardiovascular disease could be demonstrated. Platelet WDR-1 (WD-40 repeat domain 1) mRNA and protein level was found to be significantly lower in patients suffering from cardiovascular disease compared to matched controls (80). WDR-1 is known to enhance actin depolymerizing factor activity (81), thereby altering the platelet cytoskeleton (82). In addition, WDR-1 mutations in mice were linked to defects in neutrophil cytoskeleton and defective platelet production and, finally, brought about macrothrombocytopenia and autoinflammation (83). Whether the autoinflammation observed in this model is attributable only to defective neutrophil function or if platelets are involved as well, e.g., through altered platelet-neutrophil interactions, still remains to be investigated in the future.
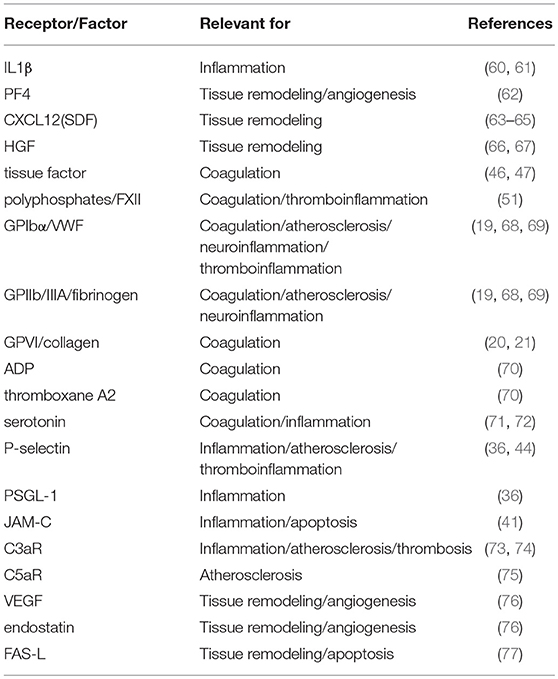
Table 1. Receptors and factors associated with platelets relevant for thromboinflammation.
Platelets Interact With Cells of the Innate Immune System and Endothelial Cells Making Them Important Players Involved in Sepsis and Inflammation-Induced Bleeding
When inflammation takes place, endothelial cells lining the inner surface of blood vessels become activated and change their receptor expression profile and their phenotype. Platelets as well as VWF were shown to be closely linked to inflammation in a neurovascular setting (84). For instance, antibody-mediated blockade of platelet GPIbα, the receptor for VWF, after induction of tMCAO was associated with improved cerebral blood flow in magnetic resonance imaging (85) leading to improved outcome (86). In addition, deficiency in VWF was paralleled with a protective phenotype in tMCAO, as well (87). In contrast, when VWF was reconstituted in VWR knockout mice, protection from cerebral ischemia was diminished (87). A common finding in severe generalized infections and sepsis is that platelet count is markedly reduced (88). In addition, a prolonged time span of thrombocytopenia is correlated with increased mortality in intensive care patients (88). Clinical data, although derived from retrospective studies, indicate that treatment with antiplatelet drugs is beneficial under conditions of severe inflammation/sepsis raising the question of how platelets might be involved in sepsis (89, 90).
Interestingly, in thrombocytopenia inflammatory mediators like LPS increased the risk of bleeding (91), underlining the importance of platelets not only for thrombosis and hemostasis but also for inflammation. This finding was demonstrated in various models of inflammation in mice. For instance, when mice were suffering from thrombocytopenia, induction of contact dermatitis resulted in increased bleeding compared to WT animals (91). Investigations using the dorsal skinfold chamber model, which enables in vivo observations of the vascular system, further confirmed the relevance of inflammation for bleeding in cases of thrombocytopenia (91). Mice suffering from severe thrombocytopenia as well as inflammation induced by application of LPS showed spontaneous intraalveolar hemorrhage (91). In addition, inflammatory bleedings under conditions of thrombocytopenia in the skin as well as in the lungs, were shown to be strongly dependent on neutrophil-endothelial interactions, thereby involving endothelial VE-Cadherin (92). In addition, platelet GPVI was shown to attenuate inflammation-induced bleeding mediated through neutrophils by binding to exposed subendothelial collagen (93). Recently, the maintenance of inflammatory hemostasis was shown to be organ- and stimulus-dependent, i.e., GPVI and GPIIb/IIIa were not required for prevention of intraalveolar bleeding after LPS challenge, while GPIbα attenuated inflammation-induced bleeding in the lung (94).
However, in sepsis adverse outcome is not only due to inflammation-induced bleeding. Instead, increased mortality could be observed irrespective of bleeding in cases when thrombocytopenia was present (95). Here, platelet transfusion was shown to be beneficial with respect to sepsis (95). The mechanisms behind the protective role of platelets observed, involved prostaglandin E2 biosynthesis within platelets through COX-1, and activation of prostaglandin receptors on macrophages (95). Furthermore, platelets have been demonstrated to protect from extracorporeal-circulation induced inflammatory lung injury. Here, platelet transfusion was associated with milder lung injury which also went along with decreased levels of TNFα and neutrophil elastase which were measured in the blood plasma (96). Blocking platelet GPIIb/IIIa through administration of Tirofiban reversed the observed effects (96). Finally, platelets were shown to limit neutrophil-induced endothelial damage by interaction with neutrophil elastase, as well (93).
Furthermore, the interplay of DCs and platelets was shown to be of paramount importance with respect to bacterial infections. Listeria monocytogenes is a bacterial infection threatening immunocompromised individuals as well as creating fetal infections that finally can lead to abortion (97, 98). Recently, new insights on the role of platelets for infection with Listeria monocytogenes were published. Platelets, with the aid of complement C3 and GPIbα, were shown to be directly involved in the transport of Listeria to splenic CD8α+ DCs, which then cross-present antigenic peptides via MHC-I to T-helper cells thereby creating an adaptive immune response (99). Besides Listeria, C3-mediated platelet association could be demonstrated for other gram-positive bacteria, e.g., Staphylococcus aureus (99). This further underlines the importance of platelets for any defense against bacteria and the development of adaptive immunity. In addition, blockade of GPIIb/IIIa had protective effects when sepsis was induced in mice (100). Furthermore, when GPIIb/IIIa was blocked, reduced levels of apoptosis in splenocytes in an in vitro approach were observed (100).
Another link of platelets to inflammation could be demonstrated in vivo in a mouse model of immune complex-mediated systemic shock, since interaction of platelets with immune complexes led to serotonin release from platelets (71) (Table 1). After platelet depletion, injection of immune complexes did not create a systemic shock-reaction according to the clinical scores (71). Further investigations of the mechanisms underlying platelet response to immune complex-mediated shock revealed that GPIIb/IIIa is necessary in this context since dysfunctional receptor ligand interaction due to mutated fibrinogen abrogated immune complex-mediated systemic shock (71). In contrast, neither P-selectin nor GPIbα were required for immune complex-mediated shock (71).
Platelets Interact With the Complement System
The complement system is a very old and well-conserved cascade of proteases produced in the liver, which are involved in clearance of dead cells as well as pathogens that have passed the natural barrier between the body and the environment (101, 102). The complement system is involved in various human pathologies associated with dysregulated platelet function and disseminated thrombosis in microvessels, e.g., hemolytic uremic syndrome (HUS) (103). In atypical HUS (aHUS), deposition of complement factors C3 and C9 could be verified on the platelet membrane (104). Furthermore, CD40L expression was increased on platelets, indicating platelet activation (104). Complement receptors for C3a and C5a are expressed on platelets, too (73, 75) (Table 1). Interestingly, in HUS renal dysfunction is caused through microthrombosis in the renal vascular system and mutations in complement C3 were shown to predispose to development of aHUS (105). Investigating the role of platelets for cardiovascular disease, we were able to show in a clinical study with patients suffering from coronary artery disease that expression of complement receptor for C5a on platelets (C5aR) could be correlated to markers of platelet activation (75). Interestingly, in vitro investigations of platelets with flow cytometry performed after platelets had been stimulated with oxidized low-density lipoprotein (oxLDL) revealed that expression of C5aR and P-selectin increased after platelet incubation with oxLDL (75). In the same study, an inverse correlation between platelet bound oxLDL and plasma C5a could also be observed (75). Another study performed in this field uncovered a strong correlation between expression of C3aR and GPIIb/IIIa on human platelets with known coronary artery disease (73), further highlighting the intimate connection between the complement system and platelets. When thrombi of cardiovascular patients were analyzed, coexpression of C3aR and GPIIb/IIIa was evident (73). Additional investigations in vivo in a mouse model deficient for either C3aR or C3 revealed that C3a affects not only bleeding time but also tissue injury after stroke, myocardial infarction and thrombosis (73). Reconstitution of mice deficient for C3aR with WT platelets, could reverse the observed protective effects of C3aR deficiency with respect to thrombosis-related ischemic injury (73). Bleeding was aggravated in the C3aR knockout mice, which could be reversed after transfusion of WT platelets (74). In addition there is increasing evidence that platelet P-selectin could be a receptor for C3b, underlining the close intersection between coagulation and inflammation (106). The crosstalk between platelet activation and the complement cascade is a good example of how closely platelets link inflammation to thrombosis and vice versa.
The Role of Platelets for Tissue Remodeling, Apoptosis and Angiogenesis
The Role of Platelets for Tissue Remodeling
Besides the established function of platelets for coagulation, there is also increasing evidence for a function of platelets in tissue remodeling and angiogenesis (Figure 1, Table 1). For example, platelets are directly involved in the process of atherosclerosis, even before any thrombotic event. Massberg et al. could demonstrate in a mouse model of ApoE deficient mice suffering from severe atherosclerosis, that the development of atherosclerotic lesions was preceded by platelet adhesion to the endothelium through interaction of GP Ibα and GPIIb/IIIa with the arterial wall (68). Platelet adhesion was the first event preceding atherosclerotic plaque formation, followed by leukocyte adhesion to the vascular wall (68). There is conclusive evidence supporting a role of platelets not only for acute atherothrombosis, for instance in myocardial infarction, but also in the process of chronic vascular inflammation. For example, individuals suffering from familial hypercholesterolemia display elevated levels of platelet microparticles in the blood (107). These microparticles exhibit markers of platelet activation such as P-selectin or GPIIb/IIIa and tissue factor (107). Furthermore, MRI-imaging performed in these patients revealed increased atherosclerotic plaque burden reflecting dangerous lipid-rich cores prone to rupture, particularly in the case when tissue factor bearing microparticles were present (107). Increased levels of platelet microparticles were also reported in patients suffering from severe heart failure requiring cardiac assist device therapy (108). These patients often suffer both from bleeding disorders as well as thromboembolic complications (109) and management of coagulation is a major concern.
As already mentioned, platelets can release various factors already known from tissue remodeling processes (Figure 1). Among those factors released from platelets are CXCL12 and hepatocyte growth factor (HGF), respectively (44, 63, 66) (Table 1). HGF is highly relevant in tissue fibrosis and remodeling as investigated in an in vivo model, in which Syrian hamsters suffering from hereditary cardiomyopathy were treated with HGF (67). The animals showed severe cardiac dysfunction and fibrosis. After treatment with recombinant HGF for 3 weeks, cardiac fibrosis was ameliorated (67). This was accompanied by reduction of transforming growth factor β 1 (TGFβ1) and type I collagen (67). Interestingly, platelets themselves were shown to interfere with HGF, as well, since they could inhibit migration of mesenchymal stem cells to apoptotic cardiomyocytes. HMGB-1 released by platelets was directly involved in this process (110, 111). However, the inhibitory role of platelets for recruitment of stem cells has also been questioned since an inhibitory effect on recruitment of vascular endothelium has been reported, too (112).
CXCL 12 also known as SDF-1, which can be released by platelets, is involved in neointima formation after vascular injury through recruitment of vascular smooth muscle cell progenitors (64, 65). First, SDF-1 is released by media smooth muscle cells undergoing apoptosis after injury has happened (65). Subsequently, SDF-1 binds to platelets which then are attached to the vessel wall at sites of injury (65). This is followed by attachment of smooth muscle cell progenitor cells to the platelets mediated by P-selectin and CXCR4, the SDF-1 counterreceptor (65). With respect to cardiovascular disease it has been demonstrated that platelet expression levels of SDF-1 correlate with adverse outcomes (113).
Platelets were shown to be involved in experimental autoimmune encephalitis (EAE) as well, linking them to another field of neuroinflammation and tissue remodeling besides stroke (69, 114). Experimental autoimmune encephalitis is a preclinical model for the human disease multiple sclerosis (MS), during the course of which inflammation of the brain is induced through administration of central nervous tissue or myelin peptides (115). Both in the human as well as in the murine disease, platelet specific CD41 was shown to be upregulated in brain tissue (69, 116). In addition, the course of the disease was affected as a result of platelet depletion. When platelets were depleted through administration of platelet depleting serum in the effector phase of the disease, reduced microgliosis within inflamed brain tissue could be observed. Both interfering with platelet GPIbα as well as platelet GPIIb and the GPIbα counterreceptor on leukocytes, MAC-1, through administration of blocking antibodies was able to ameliorate EAE (69). Recently, the importance of the timing of platelet depletion for the course of the disease was further supported by another study (114). Platelet depletion in the immunization phase of EAE did not have an impact on the course of the disease (69, 114). Microarray analysis of the spinal cords after induction of EAE revealed several factors relevant for inflammation such as CCL2, CCL5, CXCR4, and IL1β which were downregulated significantly after platelet depletion (69). The question whether platelets themselves, are drivers of neuroinflammation in the context of experimental autoimmune encephalitis, or whether they contribute to experimental autoimmune encephalitis through recruitment of inflammatory leukocytes either by receptor-ligand interaction with leukocytes or by releasing inflammatory mediators is still open. Hopefully, future research will be able to resolve this question thereby significantly improving therapies for patients suffering from multiple sclerosis.
The Role of Platelets for Angiogenesis
Angiogenesis in general is a tightly regulated process that is modulated by a multitude of cells and soluble factors (117, 118). Angiogenesis can be beneficial, for instance with respect to wound healing and tissue regeneration (117, 118). However, angiogenesis can also be harmful since tumors need a so-called angiogenic switch to grow beyond a certain size (117, 118). Angiogenesis is tightly associated with inflammation (119). Some of the proteins released from platelets possess angiogenic potential either exerting pro- or antiangiogenic responses on endothelial cells (Table 1). Experimental approaches using Matrigel, an extracellular matrix from the Engelbrecht-Holm-Swarm (EHS) sarcoma, together with endothelial cells is an established way to investigate angiogenesis in vitro (120). In a Matrigel model, a proangiogenic effect of platelets could be demonstrated after endothelial cells and platelets had been added (121). Interestingly, platelets directly adhere to endothelial cells (121). Adding platelet supernatant to the Matrigel showed significantly reduced tube formation compared to adding platelets (121), further supporting that direct platelet-endothelial interaction is necessary for the observed proangiogenic effect in vitro. Regarding platelet physiology, differential release of pro- or antiangiogenic factors happens depending on the stimulus (122). There are hints that ADP as well as GPVI favor a proangiogenic phenotype of platelets (122). In addition, platelets can release VEGF after stimulation with ADP (76). In contrast, PAR-4 favors an antiangiogenic phenotype of platelets (122). This was also observed, when platelets were stimulated with thromboxane A2 since this triggered release of the antiangiogenic agent endostatin (76). Platelets have been recognized as a major source of vascular endothelial growth factor (VEGF) (123), one of the most important growth factors involved in angiogenesis (124). When VEGF is released, it can bind to a variety of growth factor receptors thereby directing proangiogenic effects (124). After VEGF has bound to endothelial cells, they start to proliferate and to form tubes, which results in the formation of new vessels with recruitment of pericytes as well as smooth muscle cells (124).
Furthermore, platelets have also been implicated in ischemia-induced revascularization after arterial occlusion, which is primarily achieved by arteriogenesis (125). Platelets have been shown to recruit bone marrow-derived cells in response to ischemia in mouse models of hindlimb ischemia or tumor implantation in mice (126). After platelet depletion through administration of an anti GPIbα antibody, levels of bone marrow cells within the tissue were significantly reduced (126). Among the different factors mediating platelet function, α-granules as well as the antiangiogenic protein thrombospondin were shown to be relevant for the observed effects (126). In addition, a role of platelet microparticles for angiogenesis has been reported, too (127).
These effects may be the reason why platelet-rich plasma (PRP) is a potent agent to foster wound healing. Using PRP in a patient collective suffering from dehiscent sternal wounds or necrotic skin ulcers has been shown to be beneficial since duration of hospital stay after administration of PRP was almost half of the time in patients with dehiscent sternal wounds compared to standard care (128). Unfortunately, detailed investigations of the underlying mechanisms were not undertaken, so far, and we are left with speculating on the molecular mechanisms involved. Nevertheless, a COCHRANE review has confirmed the clinical benefit of the administration of platelet-rich plasma in patients with diabetes and chronic wounds (129), offering promise for future platelet-based therapies in this field.
The Role of Platelets for Apoptosis
Recently, there were hints from preclinical studies in a murine model that platelets may be involved in the process of apoptosis (Figure 1, Table 1). Apoptosis in general can be induced both through external signaling as well as through internal pathways (130). The external pathway involves several factors (TNFα, FasL, TRAIL), which after binding to their respective receptor activate an intracellular signaling cascade finally leading to activation of a set of specialized enzymes the so-called caspases (130). Besides the external pathway there is an intrinsic way how apoptosis can be initiated as well. The intrinsic pathway relies on cytochrome c which activates caspases after their release from mitochondria (130). Finally the cells undergo a special program which finally leads to cellular clearance (130). Schleicher et al. could demonstrate that platelets can be found in the brain tissue after experimental stroke in the tMCAO stroke model. Apoptosis in the brain tissue was reduced after platelet depletion (77). Similar observations were made when GPIbα deficient mice were used (77). Further analysis revealed that platelets express FasL in their membrane thereby mediating apoptosis (77). In contrast, Bax/Bak signaling of the internal pathway of apoptosis was not required but additionally contributed to apoptosis (77). A previously unrecognized role of platelets for apoptosis was also identified in the context of platelet-DC interactions. JAM-C was shown to be directly involved in platelet-DC interactions mediating apoptosis of DCs (41). Platelets were directly responsible for the recruitment of DCs to the vessel wall. In vivo, when no vascular lesion was present in a model of carotid artery injury, no DCs were recruited to the vessel wall. In contrast, after vascular injury, the number of DCs adhering to the vessel increased markedly (41).
Finally, patients suffering from human immunodeficiency virus (HIV) were shown to have an increased rate of cardiovascular events (131), despite having achieved stable disease by means of combined antiretroviral therapy. In addition, in vitro investigations could show that markers of platelet activation, e.g., P-selectin were upregulated under this condition (132). Furthermore, activation of the intrinsic pathway of apoptosis was more prevalent in platelets from patients suffering from HIV infection despite being under viral control (132), suggesting dysregulated platelet function as one possible contributing factor to increased numbers of cardiovascular events. This was further underlined in a HIV positive patient collective suffering from acute coronary syndrome where, despite receiving Aspirin and P2Y12 inhibitor therapy, high residual platelet reactivity could be measured (133). However, the mechanism underlying the observed dysregulated platelet function in patients suffering from HIV is incompletely understood, yet. Altogether, the data reported strongly point to an intimate connection of platelets and inflammation and a function of platelets beyond thrombosis and hemostasis.
Platelets and Cancer
A common finding is that cancer is associated with thrombosis and embolism (Trousseau phenomenon). The increased risk of thrombosis in cancer can be attributed to a variety of mechanisms, including increased expression and release of procoagulant factors and microparticles by tumor cells and platelets, abnormal tumor vascularity and increased inflammation [reviewed in (134)]. An increasing body of evidence indicates that, in addition to increasing the risk of thrombosis, platelets can also contribute to tumor progression and metastasis by altering the tumor microenvironment, by expression of growth factors and proangiogenic factors and by assisting neoplastic cells to evade apoptosis (76, 135–137). In addition, platelets can promote metastasis by protecting tumor cells in circulation from immune surveillance and by assisting tumor cell adhesion and transmigation of the vascular endothelium (138, 139). Investigations in the field of platelets and cancer have shown that induction of thrombocytopenia by platelet-depleting antibodies increased the efficacy of paclitaxel therapy in a murine model of breast cancer, likely through increased tumor vascular permeability (140). In addition, platelet depletion caused intratumor hemorrhage in different tumor models in mice (141). Interestingly, no increase in intratumor hemorrhage could be observed when GPIbα was blocked (141). However, as expected, tail bleeding time was markedly increased after blockade of GPIbα (141). In patients suffering from ovarian cancer, the patients' platelet count had a prognostic relevance. Thrombocytosis was associated with reduced overall survival and resistance to chemotherapy (142, 143). Although these results point to a central role of platelets in cancer biology, these insights did not translate to therapeutic strategies exploiting the function of platelets in cancer progression and metastasis, so far. While the use of low-molecular-weight heparin has been demonstrated to reduce the rate of recurrent thrombosis and thus represents the current standard in patients with cancer who suffered venous thromboembolism (144), the use of antiplatelet therapies in patients with cancer remains controversial. Although retrospective analyses suggest protective effects of daily aspirin for some cancers (145), antiplatelet drugs have not entered cancer therapy.
Platelets Participate in Vascular Remodeling After Organ Transplantation
During the process of solid organ transplantation, platelets can be activated at multiple points. Activation and subsequent degranulation may already occur during graft procurement in organ donors (146). Brain death boosts a catecholamine storm resulting in organ malperfusion (147). Furthermore, blood or platelet transfusions administered perioperatively can promote platelet activation. Prolonged ischemia during organ procurement or transplantation results in platelet activation via P-selectin and CD40L (148, 149). Platelet activation in organ recipients can occur in patients with preexisting diseases such as atherosclerosis (150) or in contact with bioincompatible surfaces such as in dialysis patients, patients with a ventricular assist device and, of course, through contact with surfaces during extracorporeal circulation at time of transplantation (151, 152). Subsequently, activated platelets may trigger an inflammatory reaction of endothelial cells and interfere with leukocytes resulting in cellular rejection (153, 154) (Figure 3). These processes may lead to the development of cardiac allograft vasculopathy and are the basis for a series of experiments regarding the role of platelets for chronic rejection after heart transplantation (155, 156). The experimental therapeutic approach of platelet inhibition with the P2Y12 ADP receptor blocker clopidogrel especially in combination with an mTOR inhibitor was very effective in a mouse aortic transplantation model, where allograft vasculopathy was almost abolished (157, 158). These findings paved the ground for a multi-center clinical trial called CEDRIC. However, the CEDRIC trial (Clopidogrel add on Certican: Effects on Coronary Diameter Reduction and Intimal Hyperplasia in Long-term follow-up after Cardiac Transplantation) had to be terminated due to recruiting problems and therefore further studies are necessary to ultimately validate this concept for a broad clinical application. Taken together, platelets play an important role in vascular remodeling after organ transplantation through both antithrombotic properties and the above-mentioned immune modulatory effects (Figure 3).
Concluding Remarks
Taken together there is increasing evidence for a role of platelets beyond hemostasis and thrombosis. Platelets are closely connected to inflammation. Contextual examples for this intimate connection between platelets and thromboinflammation are the plasmatic coagulation system as well as the complement system. Nonetheless, a lot of questions are still unanswered. One such question is how the beneficial effect of FXII deficiency on ischemic stroke can be explained. Solving this question might point the way how the outcome of this—sadly—very often disabling disease might be improved for patients. Another promising field of research is the close connection of the complement system and platelets to diseases featuring disseminated thrombosis, e.g., hemolytic uremic syndrome. Effective therapies for HUS are still missing, which raises the question, whether a clinically beneficial resolution of thrombus formation can be achieved by modulation of platelet function. Future research in platelet biology has the potential to show us even more novel, previously unexpected ways how platelets are directly involved in the most fundamental processes of health and disease.
Author Contributions
MM, HN, RS, TG, CH, NvB, SE, and HL wrote parts of the manuscript. HL conceptualized and submitted the manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Gawaz M, Langer H, May AE. Platelets in inflammation and atherogenesis. J Clin Invest. (2005) 115:3378–84. doi: 10.1172/JCI27196
2. Mezger M, Gobel K, Kraft P, Meuth SG, Kleinschnitz C, Langer HF. Platelets and vascular inflammation of the brain. Hamostaseologie. (2015) 35:244–51. doi: 10.5482/HAMO-14-11-0071
3. Langer HF, Chavakis T. Platelets and neurovascular inflammation. Thromb Haemost. (2013) 110:888–93. doi: 10.1160/TH13-02-0096
4. Garraud O, Cognasse F. Are platelets cells? and if yes, are they immune cells? Front Immunol. (2015) 6:70–70. doi: 10.3389/fimmu.2015.00070
5. Mestas J, Hughes CCW. Of mice and not men: differences between mouse and human immunology. J Immunol. (2004) 172:2731 doi: 10.4049/jimmunol.172.5.2731
6. Bessman JD, Williams LJ, Gilmer PRJr. Platelet size in health and hematologic disease. Am J Clin Pathol. (1982) 78:150–3. doi: 10.1093/ajcp/78.2.150
7. Bunting S, Widmer R, Lipari T, Rangell L, Steinmetz H, Carver-Moore K, et al. Normal platelets and megakaryocytes are produced in vivo in the absence of thrombopoietin. Blood. (1997) 90:3423–9.
8. Hitchcock IS, Chen MM, King JR, Kaushansky K. YRRL motifs in the cytoplasmic domain of the thrombopoietin receptor regulate receptor internalization and degradation. Blood. (2008) 112:2222–31. doi: 10.1182/blood-2008-01-134049
9. Patel SR, Hartwig JH, Italiano JEJr. The biogenesis of platelets from megakaryocyte proplatelets. J Clin Invest. (2005) 115:3348–54. doi: 10.1172/JCI26891
10. Shivdasani RA, Rosenblatt MF, Zucker-Franklin D, Jackson CW, Hunt P, Saris CJ, et al. Transcription factor NF-E2 is required for platelet formation independent of the actions of thrombopoietin/MGDF in megakaryocyte development. Cell. (1995) 81:695–704. doi: 10.1016/0092-8674(95)90531-6
11. Ciurea SO, Merchant D, Mahmud N, Ishii T, Zhao Y, Hu W, et al. Pivotal contributions of megakaryocytes to the biology of idiopathic myelofibrosis. Blood. (2007) 110:986–93. doi: 10.1182/blood-2006-12-064626
12. Tefferi A, Vaidya R, Caramazza D, Finke C, Lasho T, Pardanani A. Circulating interleukin (IL)-8, IL-2R, IL-12, and IL-15 levels are independently prognostic in primary myelofibrosis: a comprehensive cytokine profiling study. J Clin Oncol. (2011) 29:1356–63. doi: 10.1200/JCO.2010.32.9490
13. Desterke C, Martinaud C, Ruzehaji N, Bousse-Kerdiles MC. Le Bousse-Kerdiles, inflammation as a keystone of bone marrow stroma alterations in primary myelofibrosis. Mediators Inflamm. (2015) 2015:415024. doi: 10.1155/2015/415024
14. Hasselbalch HC. Perspectives on chronic inflammation in essential thrombocythemia, polycythemia vera, and myelofibrosis: is chronic inflammation a trigger and driver of clonal evolution and development of accelerated atherosclerosis and second cancer? Blood. (2012) 119:3219–25. doi: 10.1182/blood-2011-11-394775
15. Verstovsek S, Kantarjian H, Mesa RA, Pardanani AD, Cortes-Franco J, Thomas DA, et al. Safety and efficacy of INCB018424, a JAK1 and JAK2 inhibitor, in myelofibrosis. N Engl J Med. (2010) 363:1117–27. doi: 10.1056/NEJMoa1002028
16. Zufferey A, Speck ER, Machlus KR, Aslam R, Guo L, McVey MJ, et al. Mature murine megakaryocytes present antigen-MHC class I molecules to T cells and transfer them to platelets. Blood Adv. (2017) 1:1773. doi: 10.1182/bloodadvances.2017007021
17. Kaser A, Brandacher G, Steurer W, Kaser S, Offner FA, Zoller H, et al. Interleukin-6 stimulates thrombopoiesis through thrombopoietin: role in inflammatory thrombocytosis. Blood. (2001) 98:2720. doi: 10.1182/blood.V98.9.2720
18. Tibbles HE, Navara CS, Hupke MA, Vassilev AO, Uckun FM. Thrombopoietin induces P-Selectin expression on platelets and subsequent platelet/leukocyte interactions. Biochem Biophy Res Commun. (2002) 292:987–91. doi: 10.1006/bbrc.2002.6759
19. Savage B, Saldivar E, Ruggeri ZM. Initiation of platelet adhesion by arrest onto fibrinogen or translocation on von Willebrand factor. Cell. (1996) 84:289–97. doi: 10.1016/S0092-8674(00)80983-6
20. Massberg S, Gawaz M, Gruner S, Schulte V, Konrad I, Zohlnhofer D, et al. A crucial role of glycoprotein VI for platelet recruitment to the injured arterial wall in vivo. J Exp Med. (2003) 197:41–9. doi: 10.1084/jem.20020945
21. Kato K, Kanaji T, Russell S, Kunicki TJ, Furihata K, Kanaji S, et al. The contribution of glycoprotein VI to stable platelet adhesion and thrombus formation illustrated by targeted gene deletion. Blood. (2003) 102:1701–7. doi: 10.1182/blood-2003-03-0717
22. Kuijpers MJ, Schulte V, Bergmeier W, Lindhout T, Brakebusch C, Offermanns S, et al. Complementary roles of glycoprotein VI and alpha2beta1 integrin in collagen-induced thrombus formation in flowing whole blood ex vivo. Faseb J. (2003) 17:685–7. doi: 10.1096/fj.02-0381fje
23. Shattil SJ, Kim C, Ginsberg MH. The final steps of integrin activation: the end game. Nat Rev Mol Cell Biol. (2010) 11:288. doi: 10.1038/nrm2871
24. Bombeli T, Schwartz BR, Harlan JM. Adhesion of activated platelets to endothelial cells: evidence for a GPIIbIIIa-dependent bridging mechanism and novel roles for endothelial intercellular adhesion molecule 1 (ICAM-1), alphavbeta3 integrin, and GPIbalpha. J Exp Med. (1998) 187:329–39. doi: 10.1084/jem.187.3.329
25. Platelet Receptor Inhibition in Ischemic Syndrome Management in Patients Limited by Unstable Signs and Symptoms (PRISM-PLUS) Study Investigators. Inhibition of the platelet glycoprotein IIb/IIIa receptor with tirofiban in unstable angina and non-Q-wave myocardial infarction. N Engl J Med. (1998) 338, 1488–1497. doi: 10.1056/NEJM199805213382102
26. Adams Harold P, Effron Mark B, Torner J, Dávalos A, Frayne J, Teal P, et al. Emergency administration of Abciximab for treatment of patients with acute ischemic stroke: results of an international phase III Trial. Stroke. (2008) 39:87–99. doi: 10.1161/STROKEAHA.106.476648
27. Gruner S, Prostredna M, Schulte V, Krieg T, Eckes B, Brakebusch C, et al. Multiple integrin-ligand interactions synergize in shear-resistant platelet adhesion at sites of arterial injury in vivo. Blood. (2003) 102:4021–7. doi: 10.1182/blood-2003-05-1391
28. Inoue O, Suzuki-Inoue K, Dean WL, Frampton J, Watson SP. Integrin alpha2beta1 mediates outside-in regulation of platelet spreading on collagen through activation of Src kinases and PLCgamma2. J Cell Biol. (2003) 160:769–80. doi: 10.1083/jcb.200208043
29. Flaumenhaft R. Chapter 18-Platelet secretion. In: Michelson AD, editor. Platelets, 3rd Edn. London: Academic Press (2013). p. 343–66.
30. Holmsen H, Weiss HJ. Secretable storage pools in platelets. Ann Rev Med. (1979) 30:119–34. doi: 10.1146/annurev.me.30.020179.001003
31. Maynard DM, Heijnen HFG, Horne MK, White JG, Gahl WA. Proteomic analysis of platelet α-granules using mass spectrometry. J Thromb Haemost. (2007) 5:1945–1955. doi: 10.1111/j.1538-7836.2007.02690.x
32. Cramer EM, Savidge GF, Vainchenker W, Berndt MC, Pidard D, Caen JP, et al. Alpha-granule pool of glycoprotein IIb-IIIa in normal and pathologic platelets and megakaryocytes. Blood. (1990) 75:1220–7.
33. Salter JW, Krieglstein CF, Issekutz AC, Granger DN. Platelets modulate ischemia/reperfusion-induced leukocyte recruitment in the mesenteric circulation. Am J Physiol Gastrointest Liver Physiol. (2001) 281:G1432–G1439. doi: 10.1152/ajpgi.2001.281.6.G1432
34. Wagner DD, Olmsted JB, Marder VJ. Immunolocalization of von Willebrand protein in Weibel-Palade bodies of human endothelial cells. J Cell Biol. (1982) 95:355. doi: 10.1083/jcb.95.1.355
35. Vandendries ER, Furie BC, Furie B. Role of P-selectin and PSGL-1 in coagulation and thrombosis. Thromb Haemost. (2004) 92:459–66. doi: 10.1160/TH04-05-0306
36. Frenette PS, Denis CV, Weiss L, Jurk K, Subbarao S, Kehrel B, et al. P-Selectin glycoprotein ligand 1 (PSGL-1) is expressed on platelets and can mediate platelet-endothelial interactions in vivo. J Exp Med. (2000) 191:1413–22. doi: 10.1084/jem.191.8.1413
37. Evangelista V, Manarini S, Sideri R, Rotondo S, Martelli N, Piccoli A, et al. Platelet/polymorphonuclear leukocyte interaction: p-selectin triggers protein-tyrosine phosphorylation–dependent CD11b/CD18 adhesion: role of PSGL-1 as a signaling molecule. Blood. (1999) 93:876.
38. Buttrum SM, Hatton R, Nash GB. Selectin-mediated rolling of neutrophils on immobilized platelets. Blood. (1993) 82:1165.
39. Romo GM, Dong JF, Schade AJ, Gardiner EE, Kansas GS, Li CQ, et al. The Glycoprotein Ib-IX-V complex is a platelet counterreceptor for P-selectin. J Exp Med. (1999) 190:803. doi: 10.1084/jem.190.6.803
40. Schulz C, Schafer A, Stolla M, Kerstan S, Lorenz M, von Bruhl ML, et al. Chemokine fractalkine mediates leukocyte recruitment to inflammatory endothelial cells in flowing whole blood: a critical role for P-selectin expressed on activated platelets. Circulation. (2007) 116:764–73. doi: 10.1161/CIRCULATIONAHA.107.695189
41. Langer HF, Daub K, Braun G, Schonberger T, May AE, Schaller M, et al. Platelets recruit human dendritic cells via Mac-1/JAM-C interaction and modulate dendritic cell function in vitro. Arterioscler Thromb Vasc Biol. (2007) 27:1463–70. doi: 10.1161/ATVBAHA.107.141515
42. Diacovo TG, Roth SJ, Buccola JM, Bainton DF, Springer TA. Neutrophil rolling, arrest, and transmigration across activated, surface-adherent platelets via sequential action of P-selectin and the beta 2-integrin CD11b/CD18. Blood. (1996) 88:146.
43. Wang Y., Gao H., Shi C., Erhardt P. W., Pavlovsky A. A., et al. (2017). Leukocyte integrin Mac-1 regulates thrombosis via interaction with platelet GPIbα. Nat Commun. 8:15559. doi: 10.1038/ncomms16124
44. Massberg S, Konrad I, Schürzinger K, Lorenz M, Schneider S, Zohlnhoefer D, et al. Platelets secrete stromal cell–derived factor 1α and recruit bone marrow–derived progenitor cells to arterial thrombi in vivo. J Exp Med. (2006) 203:1221. doi: 10.1084/jem.20051772
45. Grover Steven P, Mackman N. Tissue Factor. Arterioscler Thromb Vasc Biol. (2018) 38:709–25. doi: 10.1161/ATVBAHA.117.309846
46. Schwertz H, Tolley ND, Foulks JM, Denis MM, Risenmay BW, Buerke M, et al. Signal-dependent splicing of tissue factor pre-mRNA modulates the thrombogenicity of human platelets. J Exp Med. (2006) 203:2433–40. doi: 10.1084/jem.20061302
47. MÜLler I, Klocke A, Alex M, Kotzsch M, Luther T, Morgenstern E, et al. Intravascular tissue factor initiates coagulation via circulating microvesicles and platelets. FASEB J. (2003) 17:476–8. doi: 10.1096/fj.02-0574fje
48. Osterud B, Rapaport SI. Activation of factor IX by the reaction product of tissue factor and factor VII: additional pathway for initiating blood coagulation. Proc Natl Acad Sci USA. (1977) 74:5260–4. doi: 10.1073/pnas.74.12.5260
50. Bouchard BA, Mann KG, Butenas S. No evidence for tissue factor on platelets. Blood. (2010) 116:854. doi: 10.1182/blood-2010-05-285627
51. Verhoef JJ, Barendrecht AD, Nickel KF, Dijkxhoorn K, Kenne E, Labberton L, et al. Polyphosphate nanoparticles on the platelet surface trigger contact system activation. Blood. (2017) 129:1707–17. doi: 10.1182/blood-2016-08-734988
52. Renne T, Nieswandt B, Gailani D. The intrinsic pathway of coagulation is essential for thrombus stability in mice. Blood Cells Mol Dis. (2006) 36:148–51. doi: 10.1016/j.bcmd.2005.12.014
53. Bender L, Weidmann H, Rose John S, Renné T, Long AT. Factor XII-driven inflammatory reactions with implications for anaphylaxis. Front Immunol. (2017) 8:1115. doi: 10.3389/fimmu.2017.01115
54. Lammle B, Wuillemin WA, Huber I, Krauskopf M, Zurcher C, Pflugshaupt R, et al. Thromboembolism and bleeding tendency in congenital factor XII deficiency–a study on 74 subjects from 14 Swiss families. Thromb Haemost. (1991) 65:117–21. doi: 10.1055/s-0038-1647467
55. Pham M, Kleinschnitz C, Helluy X, Bartsch AJ, Austinat M, Behr VC, et al. Enhanced cortical reperfusion protects coagulation factor XII-deficient mice from ischemic stroke as revealed by high-field MRI. Neuroimage. (2010) 49:2907–14. doi: 10.1016/j.neuroimage.2009.11.061
56. Kleinschnitz C, Stoll G, Bendszus M, Schuh K, Pauer HU, Burfeind P, et al. Targeting coagulation factor XII provides protection from pathological thrombosis in cerebral ischemia without interfering with hemostasis. J Exp Med. (2006) 203:513–8. doi: 10.1084/jem.20052458
57. Renné T, Pozgajová M, Grüner S, Schuh KH, Pauer U, Burfeind P, et al. Defective thrombus formation in mice lacking coagulation factor XII. J Exp Med. (2005) 202:271. doi: 10.1084/jem.20050664
58. Warshaw AL, Laster L, Shulman NR. Protein synthesis by human platelets. J Biol Chem. (1967) 242:2094–7.
59. Zimmerman Guy A, Weyrich Andrew S. Signal-dependent protein synthesis by activated platelets. Arterioscl Thromb Vasc Biol. (2008) 28:s17–s24. doi: 10.1161/ATVBAHA.107.160218
60. Denis MM, Tolley ND, Bunting M, Schwertz H, Jiang H, Lindemann S, et al. Escaping the nuclear confines: signal-dependent pre-mRNA splicing in anucleate platelets. Cell. (2005) 122:379–91. doi: 10.1016/j.cell.2005.06.015
61. Palomo J, Dietrich D, Martin P, Palmer G, Gabay C. The interleukin (IL)-1 cytokine family – balance between agonists and antagonists in inflammatory diseases. Cytokine. (2015) 76:25–37. doi: 10.1016/j.cyto.2015.06.017
62. Coppinger JA, Cagney G, Toomey S, Kislinger T, Belton O, McRedmond JP, et al. Characterization of the proteins released from activated platelets leads to localization of novel platelet proteins in human atherosclerotic lesions. Blood. (2004). 103:2096 doi: 10.1182/blood-2003-08-2804
63. Stellos K, Langer H, Daub K, Schoenberger T, Gauss A, Geisler T, et al. Platelet-derived stromal cell–derived factor-1 regulates adhesion and promotes differentiation of human CD34+ cells to endothelial progenitor cells. Circulation. (2008) 117:206–15. doi: 10.1161/CIRCULATIONAHA.107.714691
64. Schober A, Karshovska E, Zernecke A, Weber C. SDF-1α-mediated tissue repair by stem cells: a promising tool in cardiovascular medicine? Trends Cardiovas Med. (2006) 16:103–8. doi: 10.1016/j.tcm.2006.01.006
65. Zernecke A, Schober A, Bot I, von Hundelshausen P, Liehn EA, Mopps B, et al. SDF-1alpha/CXCR4 axis is instrumental in neointimal hyperplasia and recruitment of smooth muscle progenitor cells. Circ Res. (2005) 96:784–91. doi: 10.1161/01.RES.0000162100.52009.38
66. Nakamura T, Teramoto H, Ichihara A. Purification and characterization of a growth factor from rat platelets for mature parenchymal hepatocytes in primary cultures. Proc Nat Acad Sci USA. (1986) 83:6489–93. doi: 10.1073/pnas.83.17.6489
67. Nakamura T, Matsumoto K, Mizuno S, Sawa Y, Matsuda H. Hepatocyte growth factor prevents tissue fibrosis, remodeling, and dysfunction in cardiomyopathic hamster hearts. Am J Physiol Heart Circ Physiol. (2005) 288:H2131-9 doi: 10.1152/ajpheart.01239.2003
68. Massberg S, Brand K, Gruner S, Page S, Muller E, Muller I, et al. A critical role of platelet adhesion in the initiation of atherosclerotic lesion formation. J Exp Med. (2002) 196:887–96. doi: 10.1084/jem.20012044
69. Langer HF, Choi EY, Zhou H, Schleicher R, Chung KJ, Tang Z, et al. Platelets contribute to the pathogenesis of experimental autoimmune encephalomyelitis. Circ Res. (2012) 110:1202–10. doi: 10.1161/CIRCRESAHA.111.256370
70. Yao SK, McNatt J, Cui K, Anderson HV, Maffrand JP, Buja LM, et al. Combined ADP and thromboxane A2 antagonism prevents cyclic flow variations in stenosed and endothelium-injured arteries in nonhuman primates. Circulation. (1993) 88:2888–93. doi: 10.1161/01.CIR.88.6.2888
71. Cloutier N, Allaeys I, Marcoux G, Machlus KR, Mailhot B, Zufferey A, et al. Platelets release pathogenic serotonin and return to circulation after immune complex-mediated sequestration. Proc Nat Acad Sci USA. (2018) 115:E1550. doi: 10.1073/pnas.1720553115
72. Baumgartner HR, Born GVR. Effects of 5-hydroxytryptamine on platelet aggregation. Nature. (1968) 218:137. doi: 10.1038/218137a0
73. Sauter RJ, Sauter M, Reis ES, Emschermann FN, Nording H, Ebenhoch S, et al. Functional relevance of the anaphylatoxin receptor C3aR for platelet function and arterial thrombus formation marks an intersection point between innate immunity and thrombosis. Circulation. (2018) 138:1720–35. doi: 10.1161/CIRCULATIONAHA.118.034600
74. Sauter RJ, Sauter M, Obrich M, Emschermann FN, Nording H, Patzelt J, et al. Anaphylatoxin receptor C3aR contributes to platelet function, thrombus formation and in vivo haemostasis. Thromb Haemost. (2019) 119:179–82. doi: 10.1055/s-0038-1676349
75. Nording H, Giesser A, Patzelt J, Sauter R, Emschermann F, Stellos K, et al. Platelet bound oxLDL shows an inverse correlation with plasma anaphylatoxin C5a in patients with coronary artery disease. Platelets. (2016) 27:593–7. doi: 10.3109/09537104.2016.1148807
76. Battinelli EM, Markens BA, Italiano JE Jr. Release of angiogenesis regulatory proteins from platelet alpha granules: modulation of physiologic and pathologic angiogenesis. Blood. (2011) 118:1359–69. doi: 10.1182/blood-2011-02-334524
77. Schleicher RI, Reichenbach F, Kraft P, Kumar A, Lescan M, Todt F, et al. Platelets induce apoptosis via membrane-bound Fas L., Blood. (2015) 126:1483–93. doi: 10.1182/blood-2013-12-544445
78. Angénieux C, Maître B, Eckly A, Lanza F, Gachet C, de la Salle H. Time-dependent decay of mRNA and ribosomal RNA during platelet aging and its correlation with translation activity. PLoS ONE. (2016) 11:e0148064. doi: 10.1371/journal.pone.0148064
79. Mills EW, Green R, Ingolia NT. Slowed decay of mRNAs enhances platelet specific translation. Blood. (2017) 129:e38. doi: 10.1182/blood-2016-08-736108
80. Montenont E, Echagarruga C, Allen N, Araldi E, Suarez Y, Berger JS. Platelet WDR1 suppresses platelet activity and is associated with cardiovascular disease. Blood. (2016) 128:2033. doi: 10.1182/blood-2016-03-703157
81. Kueh HY, Charras GT, Mitchison TJ, Brieher WM. Actin disassembly by cofilin, coronin, and Aip1 occurs in bursts and is inhibited by barbed-end cappers. J Cell Biol. (2008) 182:341. doi: 10.1083/jcb.200801027
82. Bender M, Eckly A, Hartwig JH, Elvers M, Pleines I, Gupta S, et al. ADF/n-cofilin–dependent actin turnover determines platelet formation and sizing. Blood. (2010) 116:1767. doi: 10.1182/blood-2010-03-274340
83. Kile BT, Panopoulos AD, Stirzaker RA, Hacking DF, Tahtamouni LH, Willson TA, et al. Mutations in the cofilin partner Aip1/Wdr1 cause autoinflammatory disease and macrothrombocytopenia. Blood. (2007) 110:2371. doi: 10.1182/blood-2006-10-055087
84. Stoll G, Kleinschnitz C, Nieswandt B. Combating innate inflammation: a new paradigm for acute treatment of stroke? Ann N Y Acad Sci. (2010) 1207:149–54. doi: 10.1111/j.1749-6632.2010.05730.x
85. Pham M, Helluy X, Kleinschnitz C, Kraft P, Bartsch AJ, Jakob P, et al. Sustained reperfusion after blockade of glycoprotein-receptor-ib in focal cerebral ischemia: an MRI study at 17.6 Tesla. PLoS ONE. (2011) 6:e18386. doi: 10.1371/journal.pone.0018386
86. De Meyer Simon F, Schwarz T, Deckmyn H, Denis Cécile V, Nieswandt B, Stoll G, et al. Binding of von willebrand factor to collagen and glycoprotein Ibα, but not to glycoprotein IIb/IIIa, contributes to ischemic stroke in mice—brief report. Arterioscl Thromb Vascu Biol. (2010) 30:1949–51. doi: 10.1161/ATVBAHA.110.208918
87. Kleinschnitz C, De Meyer SF, Schwarz T, Austinat M, Vanhoorelbeke K, Nieswandt B, et al. Deficiency of von Willebrand factor protects mice from ischemic stroke. Blood. (2009) 113:3600–3. doi: 10.1182/blood-2008-09-180695
88. Akca S, Haji-Michael P, de Mendonca A, Suter P, Levi M, Vincent JL. Time course of platelet counts in critically ill patients. Crit Care Med. (2002) 30:753–6. doi: 10.1097/00003246-200204000-00005
89. Winning J, Neumann J, Kohl M, Claus RA, Reinhart K, Bauer M, et al. Antiplatelet drugs and outcome in mixed admissions to an intensive care unit. Crit Care Med. (2010) 38:32–7. doi: 10.1097/CCM.0b013e3181b4275c
90. Erlich JM, Talmor DS, Cartin-Ceba R, Gajic O, Kor DJ. Prehospitalization antiplatelet therapy is associated with a reduced incidence of acute lung injury: a population-based cohort study. Chest. (2011) 139:289–95. doi: 10.1378/chest.10-0891
91. Goerge T, Ho-Tin-Noe B, Carbo C, Benarafa C, Remold-O'Donnell E, Zhao BQ, et al. Inflammation induces hemorrhage in thrombocytopenia. Blood. (2008) 111:4958–64. doi: 10.1182/blood-2007-11-123620
92. Hillgruber C, Pöppelmann B, Weishaupt C, Steingräber AK, Wessel F, Berdel WE, et al. Blocking neutrophil diapedesis prevents hemorrhage during thrombocytopenia. J Exp Med. (2015) 212:1255. doi: 10.1084/jem.20142076
93. Gros A, Syvannarath V, Lamrani L, Ollivier V, Loyau S, Goerge T, et al. Single platelets seal neutrophil-induced vascular breaches via GPVI during immune-complex–mediated inflammation in mice. Blood. (2015) 126:1017. doi: 10.1182/blood-2014-12-617159
94. Rayes J, Jadoui S, Lax S, Gros A, Wichaiyo S, Ollivier V, et al. The contribution of platelet glycoprotein receptors to inflammatory bleeding prevention is stimulus and organ dependent. Haematologica. (2018) 103:e256. doi: 10.3324/haematol.2017.182162
95. Xiang B, Zhang G, Guo LX, Li A, Morris AJ, Daugherty A, et al. Platelets protect from septic shock by inhibiting macrophage-dependent inflammation via the cyclooxygenase 1 signalling pathway. Nat Commun. (2013) 4:2657. doi: 10.1038/ncomms3657
96. Luo S, Wang Y, An Q, Chen H, Zhao J, Zhang J, et al. Platelets protect lung from injury induced by systemic inflammatory response. Sci Rep. (2017) 7:42080. doi: 10.1038/srep42080
97. Radoshevich L, Cossart P. Listeria monocytogenes: towards a complete picture of its physiology and pathogenesis. Nat Rev Microbiol. (2017) 16:32. doi: 10.1038/nrmicro.2017.126
98. McLauchlin J. Human listeriosis in Britain, 1967-85, a summary of 722 cases. 1. Listeriosis during pregnancy and in the newborn. Epidemiol Infect. (1990) 104:181–9. doi: 10.1017/S0950268800059343
99. Verschoor A, Neuenhahn M, Navarini AA, Graef P, Plaumann A, Seidlmeier A, et al. A platelet-mediated system for shuttling blood-borne bacteria to CD8α+ dendritic cells depends on glycoprotein GPIb and complement C3. Nat Immunol. (2011) 12:1194. doi: 10.1038/ni.2140
100. Sharron M, Hoptay CE, Wiles AA, Garvin LM, Geha M, Benton AS, et al. Platelets induce apoptosis during sepsis in a contact-dependent manner that is inhibited by GPIIb/IIIa blockade. PLoS ONE. (2012) 7:e41549. doi: 10.1371/journal.pone.0041549
101. Merle NS, Church SE, Fremeaux-Bacchi V, Roumenina LT. Complement system part I – molecular mechanisms of activation and regulation. Front Immunol. (2015) 6:262. doi: 10.3389/fimmu.2015.00262
102. Merle NS, Noe R, Halbwachs-Mecarelli L, Fremeaux-Bacchi V, Roumenina LT. Complement system part II: role in immunity. Front Immunol. (2015) 6:257. doi: 10.3389/fimmu.2015.00257
103. Conway EM. HUS and the case for complement. Blood. (2015) 126:2085. doi: 10.1182/blood-2015-03-569277
104. Ståhl AL, Vaziri-Sani F, Heinen SA, Kristoffersson CK, Gydell H, Raafat R, et al. Factor H dysfunction in patients with atypical hemolytic uremic syndrome contributes to complement deposition on platelets and their activation. Blood. (2008) 111:5307. doi: 10.1182/blood-2007-08-106153
105. Frémeaux-Bacchi V, Miller EC, Liszewski MK, Strain L, Blouin J, Brown AL, et al. Mutations in complement C3 predispose to development of atypical hemolytic uremic syndrome. Blood. (2008) 112:4948. doi: 10.1182/blood-2008-01-133702
106. Del Conde I, Crúz MA, Zhang H, López JA, Afshar-Kharghan V. Platelet activation leads to activation and propagation of the complement system. J Exp Med. (2005) 201:871–9. doi: 10.1084/jem.20041497
107. Suades R, Padro T, Alonso R, Mata P, Badimon L. High levels of TSP1+/CD142+ platelet-derived microparticles characterise young patients with high cardiovascular risk and subclinical atherosclerosis. Thromb Haemost. (2015) 114:1310–21. doi: 10.1160/TH15-04-0325
108. Diehl P, Aleker M, Helbing T, Sossong V, Beyersdorf F, Olschewski M, et al. Enhanced microparticles in ventricular assist device patients predict platelet, leukocyte and endothelial cell activation. Inter CardioVasc Thora Surg. (2010) 11:133–7. doi: 10.1510/icvts.2010.232603
109. Shah P, Tantry US, Bliden KP, Gurbel PA. Bleeding and thrombosis associated with ventricular assist device therapy. J Heart Lung Transplant. (2017) 36:1164–73. doi: 10.1016/j.healun.2017.05.008
110. Vogel S, Chatterjee M, Metzger K, Borst O, Geisler T, Seizer P, et al. Activated platelets interfere with recruitment of mesenchymal stem cells to apoptotic cardiac cells via high mobility group box 1/Toll-like receptor 4-mediated down-regulation of hepatocyte growth factor receptor MET. J Biol Chem. (2014) 289:11068–82. doi: 10.1074/jbc.M113.530287
111. Vogel S, Trapp T, Börger V, Peters C, Lakbir D, Dilloo D, et al. Hepatocyte growth factor-mediated attraction of mesenchymal stem cells for apoptotic neuronal and cardiomyocytic cells. Cell Mol Life Sci. (2010) 67:295–303. doi: 10.1007/s00018-009-0183-3
112. Langer HF, Stellos K, Steingen C, Froihofer A, Schonberger T, Kramer B, et al. Platelet derived bFGF mediates vascular integrative mechanisms of mesenchymal stem cells in vitro. J Mol Cell Cardiol. (2009) 47:315–25. doi: 10.1016/j.yjmcc.2009.03.011
113. Rath D, Chatterjee M, Bongartz A, Muller K, Droppa M, Stimpfle F, et al. Platelet surface expression of SDF-1 is associated with clinical outcomes in the patients with cardiovascular disease. Platelets. (2017) 28:34–9. doi: 10.1080/09537104.2016.1203399
114. Sonia D'Souza C, Li Z, Luke Maxwell D, Trusler O, Murphy M, Crewther S, et al. Platelets drive inflammation and target gray matter and the retina in autoimmune-mediated encephalomyelitis. J Neuropathol Exp Neurol. (2018) 77:567–576. doi: 10.1093/jnen/nly032
115. Constantinescu CS, Farooqi N, O'Brien K, Gran B. Experimental autoimmune encephalomyelitis (EAE) as a model for multiple sclerosis (MS). Br J Pharmacol. (2011) 164:1079–106. doi: 10.1111/j.1476-5381.2011.01302.x
116. Lock C, Hermans G, Pedotti R, Brendolan A, Schadt E, Garren H, et al. Gene-microarray analysis of multiple sclerosis lesions yields new targets validated in autoimmune encephalomyelitis. Nat Med. (2002) 8:500–8. doi: 10.1038/nm0502-500
117. Carmeliet P. Angiogenesis in life, disease and medicine. Nature. (2005) 438:932–6. doi: 10.1038/nature04478
118. Carmeliet P. Angiogenesis in health and disease. Nat Med. (2003) 9:653–60. doi: 10.1038/nm0603-653
119. Mor F, Quintana FJ, Cohen IR. Angiogenesis-inflammation cross-talk: vascular endothelial growth factor is secreted by activated T cells and induces Th1 polarization. J Immunol. (2004) 172:4618–23. doi: 10.4049/jimmunol.172.7.4618
120. Arnaoutova I, Kleinman HK. In vitro angiogenesis: endothelial cell tube formation on gelled basement membrane extract. Nat Protocols. (2010) 5:628. doi: 10.1038/nprot.2010.6
121. Pipili-Synetos E, Papadimitriou E, Maragoudakis ME. Evidence that platelets promote tube formation by endothelial cells on matrigel. Br J Pharmacol. (1998) 125:1252–7. doi: 10.1038/sj.bjp.0702191
122. Chatterjee M, Huang Z, Zhang W, Jiang L, Hultenby K, Zhu L, et al. Distinct platelet packaging, release, and surface expression of proangiogenic and antiangiogenic factors on different platelet stimuli. Blood. (2011) 117:3907–11. doi: 10.1182/blood-2010-12-327007
123. Verheul HM, Hoekman K, Luykx-de Bakker S, Eekman CA, Folman CC, Broxterman HJ, et al. Platelet: transporter of vascular endothelial growth factor. Clin Cancer Res. (1997) 3(12 Pt 1), 2187–190.
124. Potente M, Carmeliet P. The link between angiogenesis and endothelial metabolism. Annu Rev Physiol. (2017) 79:43–66. doi: 10.1146/annurev-physiol-021115-105134
125. Herzog S, Sager H, Khmelevski E, Deylig A, Ito WD. Collateral arteries grow from preexisting anastomoses in the rat hindlimb. Am J Physiol Heart Circul Physiol. (2002) 283:H2012–H2020. doi: 10.1152/ajpheart.00257.2002
126. Feng W, Madajka M, Kerr BA, Mahabeleshwar GH, Whiteheart SW, Byzova TV. A novel role for platelet secretion in angiogenesis: mediating bone marrow–derived cell mobilization and homing. Blood. (2011) 117:3893. doi: 10.1182/blood-2010-08-304808
127. Rhee JS, Black M, Schubert U, Fischer S, Morgenstern E, Hammes HP, et al. The functional role of blood platelet components in angiogenesis. Thromb Haemost. (2004) 92:394–402. doi: 10.1160/TH03-04-0213
128. Mazzucco L, Medici D, Serra M, Panizza R, Rivara G, Orecchia S, et al. The use of autologous platelet gel to treat difficult-to-heal wounds: a pilot study. Transfusion. (2004) 44:1013–8. doi: 10.1111/j.1537-2995.2004.03366.x
129. Martinez-Zapata MJ, Martí-Carvajal AJ, Solà I, Expósito JA, Bolíbar I, Rodríguez L, et al. Autologous platelet-rich plasma for treating chronic wounds. Cochrane Database Syst Rev. (2016) CD006899. doi: 10.1002/14651858.CD006899.pub3
130. Nagata S. Apoptosis and clearance of apoptotic cells. Ann Rev Immunol. (2018) 36:489–517. doi: 10.1146/annurev-immunol-042617-053010
131. d'Arminio A, Sabin CA, Phillips AN, Reiss P, Weber R, Kirk O, et al. (2004). Cardio- and cerebrovascular events in HIV-infected persons. Aids 18:1811–7. doi: 10.1097/00002030-200409030-00010
132. Mesquita EC, Hottz ED, Amancio RT, Carneiro AB, Palhinha L, Coelho LE, et al. Persistent platelet activation and apoptosis in virologically suppressed HIV-infected individuals. Sci Rep. (2018) 8:14999. doi: 10.1038/s41598-018-33403-0
133. Hauguel-Moreau M, Boccara F, Boyd AJ, Salem E, Brugier D, Curjol A, et al. Platelet reactivity in human immunodeficiency virus infected patients on dual antiplatelet therapy for an acute coronary syndrome: the EVERE2ST-HIV study. Eur Heart J. (2017) 38:1676–86. doi: 10.1093/eurheartj/ehw583
134. Sheth RA, Niekamp A, Quencer KB, Shamoun F, Knuttinen MG, Naidu S, et al. Thrombosis in cancer patients: etiology, incidence, and management. Cardiovasc Diagn Ther. (2017) 7:S178–S185. doi: 10.21037/cdt.2017.11.02
135. Labelle M, Begum S, Hynes RO. Direct signaling between platelets and cancer cells induces an epithelial-mesenchymal-like transition and promotes metastasis. Cancer Cell. (2011) 20:576–90. doi: 10.1016/j.ccr.2011.09.009
136. Velez J, Enciso LJ, Suarez M, Fiegl M, Grismaldo A, Lopez C, et al. Platelets promote mitochondrial uncoupling and resistance to apoptosis in leukemia cells: a novel paradigm for the bone marrow microenvironment. Cancer Microenviron. (2014) 7:79–90. doi: 10.1007/s12307-014-0149-3
137. Janowska-Wieczorek A, Wysoczynski M, Kijowski J, Marquez-Curtis L, Machalinski B, Ratajczak J, et al. Microvesicles derived from activated platelets induce metastasis and angiogenesis in lung cancer. Int J Cancer. (2005) 113:752–60. doi: 10.1002/ijc.20657
138. Palumbo JS, Talmage KE, Massari JV, La Jeunesse CM, Flick MJ, Kombrinck KW, et al. Platelets and fibrin(ogen) increase metastatic potential by impeding natural killer cell-mediated elimination of tumor cells. Blood. (2005) 105:178–85. doi: 10.1182/blood-2004-06-2272
139. Nieswandt B, Hafner M, Echtenacher B, Mannel DN. Lysis of tumor cells by natural killer cells in mice is impeded by platelets. Cancer Res. (1999) 59:1295–300.
140. Demers M, Ho-Tin-Noe B, Schatzberg D, Yang JJ, Wagner DD. Increased efficacy of breast cancer chemotherapy in thrombocytopenic mice. Cancer Res. (2011) 71:1540–9. doi: 10.1158/0008-5472.CAN-10-2038
141. Ho-Tin-Noé B, Goerge T, Cifuni SM, Duerschmied D, Wagner DD. platelet granule secretion continuously prevents intratumor hemorrhage. Cancer Res. (2008) 68:6851. doi: 10.1158/0008-5472.CAN-08-0718
142. Stone RL, Nick AM, McNeish IA, Balkwill F, Han HD, Bottsford-Miller J, et al. Paraneoplastic thrombocytosis in ovarian cancer. N Engl J Med. (2012) 366:610–8. doi: 10.1056/NEJMoa1110352
143. Bottsford-Miller J, Choi HJ, Dalton HJ, Stone RL, Cho MS, Haemmerle M, et al. Differential platelet levels affect response to taxane-based therapy in ovarian cancer. Clin Cancer Res. (2015) 21:602–10. doi: 10.1158/1078-0432.CCR-14-0870
144. Lee AY, Levine MN, Baker RI, Bowden C, Kakkar AK, Prins M, et al. Low-molecular-weight heparin versus a coumarin for the prevention of recurrent venous thromboembolism in patients with cancer. N Engl J Med. (2003) 349:146–53. doi: 10.1056/NEJMoa025313
145. Elwood PC, Pickering JE, Morgan G, Galante J, Weightman AL, Morris D, et al. Systematic review update of observational studies further supports aspirin role in cancer treatment: Time to share evidence and decision-making with patients? PLoS ONE. (2018) 13:e0203957. doi: 10.1371/journal.pone.0203957
146. Reed GL, Fitzgerald ML, Polgar J. Molecular mechanisms of platelet exocytosis: insights into the “secrete” life of thrombocytes. Blood. (2000) 96:3334–42.
147. Herijgers P, Leunens V, Tjandra-Maga TB, Mubagwa K, Flameng W. Changes in organ perfusion after brain death in the rat and its relation to circulating catecholamines. Transplantation. (1996) 62:330–5. doi: 10.1097/00007890-199608150-00005
148. Sternberg DI, Shimbo D, Kawut SM, Sarkar J, Hurlitz G, D'Ovidio F, et al. Platelet activation in the postoperative period after lung transplantation. J Thorac Cardiovasc Surg. (2008) 135:679–84. doi: 10.1016/j.jtcvs.2007.09.058
149. Xu H, Zhang X, Mannon RB, Kirk AD. Platelet-derived or soluble CD154 induces vascularized allograft rejection independent of cell-bound CD154. J Clin Invest. (2006) 116:769–74. doi: 10.1172/JCI27155
150. Ashman N, Macey MG, Fan SL, Azam U, Yaqoob MM. Increased platelet-monocyte aggregates and cardiovascular disease in end-stage renal failure patients. Nephrol Dial Transplant. (2003) 18:2088–96. doi: 10.1093/ndt/gfg348
151. Ballotta A, Saleh HZ, El Baghdady HW, Gomaa M, Belloli F, Kandil H, et al. Comparison of early platelet activation in patients undergoing on-pump versus off-pump coronary artery bypass surgery. J Thorac Cardiovasc Surg. (2007) 134:132–8. doi: 10.1016/j.jtcvs.2007.01.055
152. Parolari A, Mussoni L, Frigerio M, Naliato M, Alamanni F, Polvani GL, et al. The role of tissue factor and P-selectin in the procoagulant response that occurs in the first month after on-pump and off-pump coronary artery bypass grafting. J Thorac Cardiovasc Surg. (2005) 130:1561–6. doi: 10.1016/j.jtcvs.2005.07.049
153. Henn V, Slupsky JR, Grafe M, Anagnostopoulos I, Forster R, Muller-Berghaus G, et al. CD40 ligand on activated platelets triggers an inflammatory reaction of endothelial cells. Nature. (1998) 391:591–4. doi: 10.1038/35393
154. Choi MH, Lee IK, Kim GW, Kim BU, Han YH, Yu DY, et al. Regulation of PDGF signalling and vascular remodelling by peroxiredoxin I. Nature. (2005) 435:347–53. doi: 10.1038/nature03587
155. Wang Catherine Y, Mazer Sean P, Minamoto K, Takuma S, Homma S, Yellin M, et al. Suppression of Murine Cardiac Allograft Arteriopathy by Long-Term Blockade of CD40-CD154 Interactions. Circulation. (2002) 105:1609–1614 doi: 10.1161/01.CIR.0000013022.11250.30
156. Abele S, Weyand M, Wollin M, Hiemann NE, Harig F, Fischlein T, et al. Clopidogrel reduces the development of transplant arteriosclerosis. J Thorac Cardiovasc Surg. (2006) 131:1161–6. doi: 10.1016/j.jtcvs.2006.01.010
157. Eckl S, Heim C, Abele-Ohl S, Hoffmann J, Ramsperger-Gleixner M, Weyand M, et al. Combination of clopidogrel and everolimus dramatically reduced the development of transplant arteriosclerosis in murine aortic allografts. Transplant Int. (2010) 23:959–66. doi: 10.1111/j.1432-2277.2010.01072.x
158. Heim C, Eckl S, Preidl R, Ramsperger-Gleixner M, Koch N, Goldmann K, et al. Delayed therapy with clopidogrel and everolimus prevents progression of transplant arteriosclerosis and impairs humoral alloimmunity in murine aortic allografts. Eur J Cardiothorac Surg. (2015) 47:180–7. doi: 10.1093/ejcts/ezu098
Keywords: platelets, innate immunity, complement, inflammation, stroke, infection, tissue remodeling, EAE
Citation: Mezger M, Nording H, Sauter R, Graf T, Heim C, von Bubnoff N, Ensminger SM and Langer HF (2019) Platelets and Immune Responses During Thromboinflammation. Front. Immunol. 10:1731. doi: 10.3389/fimmu.2019.01731
Received: 15 March 2019; Accepted: 09 July 2019;
Published: 26 July 2019.
Edited by:
Benoît Ho-Tin-Noé, Institut National de la Santé et de la Recherche Médicale (INSERM), FranceReviewed by:
Robert H. Lee, University of North Carolina at Chapel Hill, United StatesFrederik Denorme, The University of Utah, United States
Copyright © 2019 Mezger, Nording, Sauter, Graf, Heim, von Bubnoff, Ensminger and Langer. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Harald F. Langer, aGFyYWxkLmxhbmdlckB1a3NoLmRl