
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol., 25 July 2019
Sec. Cancer Immunity and Immunotherapy
Volume 10 - 2019 | https://doi.org/10.3389/fimmu.2019.01720
This article is part of the Research TopicRole of Metabolism in Regulating Immune Cell Fate DecisionsView all 17 articles
 Valentina Audrito1,2*
Valentina Audrito1,2* Antonella Managò1,2Federica Gaudino1,2
Antonella Managò1,2Federica Gaudino1,2 Leonardo Sorci3Vincenzo Gianluca Messana1,2
Leonardo Sorci3Vincenzo Gianluca Messana1,2 Nadia Raffaelli4
Nadia Raffaelli4 Silvia Deaglio1,2*
Silvia Deaglio1,2*Cancer cells, particularly in solid tumors, are surrounded by non-neoplastic elements, including endothelial and stromal cells, as well as cells of immune origin, which can support tumor growth by providing the right conditions. On the other hand, local hypoxia, and lack of nutrients induce tumor cells to reprogram their metabolism in order to survive, proliferate, and disseminate: the same conditions are also responsible for building a tumor-suppressive microenvironment. In addition to tumor cells, it is now well-recognized that metabolic rewiring occurs in all cellular components of the tumor microenvironment, affecting epigenetic regulation of gene expression and influencing differentiation/proliferation decisions of these cells. Nicotinamide adenine dinucleotide (NAD) is an essential co-factor for energy transduction in metabolic processes. It is also a key component of signaling pathways, through the regulation of NAD-consuming enzymes, including sirtuins and PARPs, which can affect DNA plasticity and accessibility. In addition, both NAD-biosynthetic and NAD-consuming enzymes can be present in the extracellular environment, adding a new layer of complexity to the system. In this review we will discuss the role of the “NADome” in the metabolic cross-talk between cancer and infiltrating immune cells, contributing to cancer growth and immune evasion, with an eye to therapeutic implications.
The solid tumor microenvironment (TME), as well as the lymphoid niche, is a dynamic and multicellular ecosystem with complex interactions (1, 2). Intercellular crosstalk within this niche is driven by multiple receptor-ligand systems, as well as by locally synthesized soluble proteins, including chemokines/cytokines, interleukins, interferons, growth, and angiogenic factors (3, 4). This unique environment is essential for tumor growth, metastatic dissemination, and drug-resistance. Furthermore, the cellular and soluble components of the TME have an important role in shaping metabolic reprogramming of cancer cells, an established hallmark of cancer, and in creating an immunosuppressive environment (5–8), as showed in Figure 1.
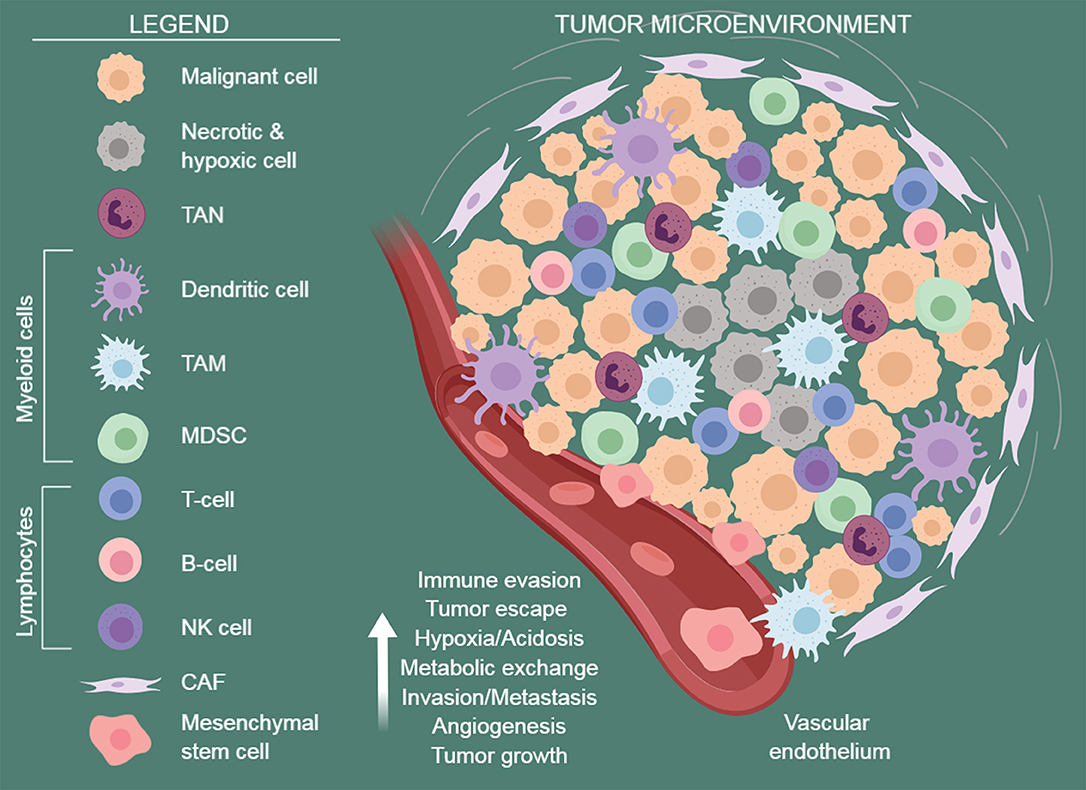
Figure 1. The tumor microenvironment. A schematic view of the tumor microenvironment components. Established cancers are usually surrounded by a wide array of stromal cells and infiltrating immune cells of both innate and acquired immunity, such as MDSCs, macrophages, dendritic cells, neutrophils, NK cells, and lymphocytes. They form a complex regulatory network that supports tumor growth by creating a tolerogenic environment that enables cancers to evade immune surveillance and destruction. TAN, tumor-associated neutrophils; TAM, tumor-associated macrophages; MDSC, myeloid-derived suppressive cells; CAF, cancer-associated fibroblasts. Figure arrange using BioRender software https://biorender.com/.
The formation of the TME and the regulation of immune responses are orchestrated by different types of host cells, including endothelial cells (ECs), mesenchymal stem/stromal cells [MSCs, including cancer-associated fibroblasts (CAFs) and tumor-associated MSCs (TA-MSCs)], and tumor-infiltrating immune cells [i.e., tumor-infiltrating lymphocytes (TILs), tumor-associated macrophages (TAMs), myeloid-derived suppressor cells (MDSCs), and tumor-associated neutrophils (TANs)]. Their concerted action promotes tumor growth and spreading (1, 2, 9, 10) (Figure 1).
ECs support blood supply, nutrient transport, metabolic homeostasis, and immune cell trafficking, and are involved in inflammatory response (11).
To provide nutrients to the growing tumor, ECs form tumor-associated (angiogenic) vessels originating from locally pre-existing vessels or recruiting bone marrow-derived endothelial progenitors. They also represent the first interface between circulating blood cells, tumor cells, and the extracellular matrix, thereby playing a central role in regulating leukocyte recruitment, tumor cell features, and metastasis dissemination (12). Tumor-associated EC are dysfunctional, partly as a consequence of local hypoxia, which induces the production of soluble factors promoting neo-angiogenesis and contributing to tumor dissemination and chemoresistance (13, 14). Among these factors, vascular endothelial growth factor A (VEGF-A) can also play a critical role in the control of immune tolerance, linking immune suppression with angiogenesis (15).
MSCs strongly affect the development and progression of various cancers (16). Stromal cells represent the main cell component with both supportive and immunoregulatory functions; they derived from multipotent cells of mesodermal origin which virtually reside in all tissues with an important role in tissue regeneration (16). MSCs have been found to migrate to tumors and to evolve into TA-MSCs and CAFs with an active role in tumor survival, proliferation, migration and drug resistance, and therefore, recently emerged as attractive targets or tools for anticancer approaches (17, 18).
CAFs are the most abundant resident cells of the TME. Numerous studies have demonstrated that CAFs have prominent roles in cancer pathogenesis (19, 20). Mechanistically, CAFs shape the extracellular matrix (ECM) structure, which supports the tumor cells (i) to invade and interact with stromal cells through the secretion of growth factors, cytokines and chemokines including interleukin-6 (IL-6), transforming growth factor-β (TGF-β) and CC-chemokine ligand 2 (CCL2); (ii) to amplify immune evasion recruiting immune cells, especially immunosuppressive cells into the tumor stroma; (iii) to promote the establishment of an intratumoral vascular network through proinflammatory and proangiogenic mediators (21). CAFs also activate epithelial-mesenchymal transition (EMT) in cancer cells, conferring their pro-invasive and stem-like features (22). In addition, CAFs are plastic cells that co-evolve with cancer cells and acquire a pro-tumor phenotype, contributing to tumor evolution (23). Due to the pro-tumor role of CAFs in support cancer development they become promising therapeutic targets for cancer therapy (21).
TILs are additional immune components, crucial in driving immune responses within the TME, adding more complexity in the composition of the TME (3). TILs are white blood cells, including T and B cells, that have left the bloodstream and migrated toward a tumor or tissue resident (1, 24). Their abundance varies according to tumor type and stage and in some cases relates to disease prognosis, tumor progression, and response to anticancer therapy (1, 25, 26). T cell differentiation status, survival, activation or “stemness properties” are determining factors of antitumor potency (27) and functions of TILs dynamically change within the TME (28). Sometimes TILs, specifically cytotoxic CD8+ memory T cells and CD4+ T helper 1 (Th1), which are normally antigen “experienced,” kill tumor cells (29), and the presence of lymphocytes in tumors is often associated with a better prognosis during immunotherapy treatment, including the adoptive transfer of naturally- TIL or genetically-engineered T cells and the use of immune-checkpoint inhibitors (26, 30). However, very often, during cancer progression and chronic inflammation, T cells become exhausted due to the persistent antigen exposure. T cell exhaustion is a state of T cell dysfunction defined by poor effector function, sustained expression of inhibitory receptors, such as programmed cell death protein 1 (PD1) and cytotoxic T lymphocyte antigen 4 (CTLA4), and transcriptional programs altered compared with functional effector or memory T cells (31).
Regulatory T (Treg) cells are another TME cell type that has immunosuppressive functions in cancer, inhibiting recognition, and clearance of tumor cells by the immune system (30, 32, 33). Tregs are characterized by the expression of CD4, CD25, and forkhead box P3 (FOXP3) as their master regulator. Foxp3 Treg can originate in the thymus (naturally occurring Treg) or can be induced (iTreg) in the periphery by soluble cytokines and cell-cell contact (34) and are essential for maintaining peripheral tolerance and limiting auto-immune diseases. However, the proportions of Tregs are much higher in the circulation of patients with solid and hematologic malignancies and accumulation of Tregs in the tumor microenvironment is associated with disease progression and reduced survival (35, 36). From a functional point of view, Tregs inhibit both cellular and humoral immune responses by suppressing expansion and activation of conventional CD4+ and cytotoxic CD8+ T cells, and natural killer cells, mainly through the secretion of suppressive cytokines, such as TGF-β and IL-10. The development of agents that specifically inhibit Treg functions or remove them from the TME will permit new approaches for anticancer immunotherapy (37).
TAMs are important mediators of tumorigenesis, resident in the tissue or deriving from peripheral reservoirs such as the bone marrow (BM) and spleen (2). Macrophages are functionally plastic and can be polarized into the immune stimulating and antitumor M1 subtype, or into “alternatively activated” M2 macrophages producing type II cytokines, promoting anti-inflammatory responses, and having pro-tumorigenic functions (38, 39). Macrophage polarization is finely tuned in response to different microenvironmental stimuli (40). For example, hypoxia may mediate this transition from tumor suppressing to tumor promoting macrophages (41). Furthermore, it has been shown a reciprocal regulation between CAFs and M2 macrophages: CAFs promote monocyte recruitment and polarization toward the M2 phenotype, leading to the enhancement of proangiogenic features, in parallel M2 macrophages are able to induce fibroblast activation (42). It is well-known that TAMs have a clear role in supporting multiple aspects of tumor progression (43). For example, TAMs promote tumor cell invasion through a paracrine loop that involves tumor-derived colony-stimulating factor 1 (CSF-1) and macrophage-derived epidermal growth factor (EGF) (43, 44). Moreover, TAMs induce immune suppression [reviewed in (45)] mediated by (i) expression of inhibitory receptors, including human leukocyte antigens (HLA)-E and HLA-G and T cell immune checkpoint ligands, such as PDL1, PDL2, CD80 and CD86, which directly inhibit T cell functions and NK cells; (ii) release of several cytokines, such as IL-10 and transforming growth factor-β (TGFβ), that contribute to feed a strong immunosuppressive microenvironment by inhibiting CD4+ (Th1 and Th2 cells) and CD8+ T cells and inducing Treg cell expansion and recruitment through CCL2, CCL3, and CCL20. Lastly, they induce depletion of essential amminoacids for cytotoxic activity of T cells including l-arginine and tryptophan, or production of kynurenine by indoleamine 2,3-dioxygenase (IDO) that inhibits T cell cytotoxicity.
Reversion of TAMs back to an M1 phenotype has also been reported (46), highlighting a potential therapeutic opportunity in which re-education of TME-resident macrophages might have beneficial anti-tumorigenic effects (45).
Along with TAMs, MDSCs are considered major promoters of tumor immune evasion (47). This population of myeloid cells, functionally defined as immunosuppressive, arises as a consequence of aberrant myelopoiesis typical of cancer (48). During tumorigenesis, MDSCs are mobilized from BM, via CXCR4/CXCL12 axis (49) and infiltrate tumors, where they promote tumor neoangiogenesis, producing endothelial growth factors [e.g., VEGF, basic fibroblast growth factor (bFGF)] (47). At the same time, they disrupt the major mechanisms of immunosurveillance, including antigen presentation by dendritic cells (DCs), T cell activation, M1 macrophage polarization and NK cell cytotoxicity, as reviewed in Safari et al. (50) and Wang et al. (51). Pharmacological inhibitors of CXCR4, are now under clinical investigation for the mobilization of immune and hematopoietic stem cells (52). Noteworthy, depletion of MDSCs by chemotherapeutic agents (e.g., gemcitabine, cyclophosphamide) can efficiently contribute to their anticancer action (48, 50, 53).
More recently, a population of neutrophils, known as TANs, has been identified as tumor supporter promoting growth, invasion, and angiogenesis of cancer cells, although they have been classically considered to exhibit a defensive response against tumor cells. Like all other leukocytes, they migrate into tissues under the effect of specific chemokines, cytokines and cell adhesion molecules for example TGF-β and IL-8 induce the formation of a pro-tumorigenic (N2) phenotype capable of supporting tumor growth and suppressing the antitumor immune responses (54, 55). Accordingly, TGF-β blocking results in the recruitment and activation of TAN with an anti-tumor phenotype (54). The main tumor-promoting mechanisms of TANs include secretion of chemokines and/or cytokines, reactive oxygen species (ROS), and matrix-degrading proteinases, among others, conditioning tumor immune surveillance, metastasis, invasion, angiogenesis, and cellular proliferation (55, 56).
It has been shown that the environment surrounding tumor cells is characterized by low oxygen tension (i.e., hypoxia) due to the abnormal blood vessel formation, defective blood perfusion, and unlimited cancer cell proliferation (14). The progression of hypoxia over time is a consequence of increased oxygen consumption and high glycolytic rate of aberrantly proliferating cancer cells (aerobic glycolysis or Warburg metabolism), leading to lactate dehydrogenase (LDH) activity, lactate excretion and TME acidosis, which alters the tumor-stroma “metabolic cross-talk” (Figure 1). Vice versa, hypoxia rapidly fosters energy production in tumor cells via glycolysis through hypoxia-inducible factor 1-alpha (HIF-1α)-mediated transcriptional control (57, 58). In addition, a hypoxic environment also modulates tumor-associated immune and stromal cells metabolism and fate. The rapid consumption of extracellular glucose and glutamine by tumor cells, especially in hypoxic conditions, leads to the accumulation of extracellular lactate, which was shown to affect several cell types within the TME (59). Increased lactate levels promote the insurance of an immune-permissive microenvironment by attenuating DCs and T cell activation, monocyte migration, and polarization of resident macrophages to TAMs (60–63). Furthermore, lactate accumulation promotes angiogenesis, stabilizes HIF-1α and activates NF-kB and PI-3 kinase signaling in endothelial cells, as well as inducing secretion of the proangiogenic factor VEGF from tumor-associated stromal cells (64–66). The secretion of lactate via the monocarboxylate transporter (MCT3) is coupled to the cotransport of H+, which supports acidification of the cellular microenvironment (59). The surplus of CO2 generated in mitochondrial decarboxylation reactions contributes to extracellular acidification as well (67). Then, a class of extracellular carbonic anhydrases (CA) can convert CO2 to H+ and HCO3−. Accordingly, expression of CAIX isoforms is elevated during hypoxia and can be considered a proxy for HIF-1α signaling (68). A consequence of increased extracellular acidification is the stimulation of the proteolytic activity of MMPs that promotes the degradation of the extracellular matrix components enhancing tumor invasion (69).
Lactate in TME can be also recycled, as occurs in the Cori cycle in the liver. In this reciprocal metabolite changes between cancer cells and immune/stromal cells, lactate produced under hypoxic conditions by glycolytic cells can be re-uptaken by aerobic cells, via MCT1, and utilized for mitochondrial tricarboxylic acid (TCA) cycle and oxidative phosphorylation (OXPHOS) (70, 71). This well characterized mechanism is known as the “reverse Warburg effect” (70, 72). In a model of epithelial cancer, tumor cells instruct the normal stroma to transform into a wound-healing stroma, providing the necessary energy-rich microenvironment for facilitating tumor growth and angiogenesis (72, 73). This metabolic cross-talk is evident in breast, prostate and ovarian cancer (74–76).
Both innate and adaptive immune cells increase their metabolic capacity upon stimulation, promoting energy generation, and biosynthesis supporting proliferation, effector molecule production, and differentiation (77). The impact of such altered metabolic state and levels of metabolites in TME on immune cell function is emerging. For example, a competition between tumor cells and T cells for the glucose pool in the aerobic microenvironment is linked to suppressed effector T-cell functions. In fact, activated T cells rely on glucose metabolism, up-regulating GLUT1 transporter via T cell receptor (TCR) and CD28-induced Akt activation (78, 79). Critical concentrations and/or lack of two amino acids, glutamine and arginine, necessary for T-cell activation, differentiation and proliferation, are therefore inhibitory to T cell functions (79).
The TME shows high levels of immunosuppressive metabolic byproducts, including a turnover in the TME release of adenosine triphosphate (ATP) and nicotinamide dinucleotide (NAD) which are metabolized by the ectoenzymes CD39, CD73, and the NADase CD38 to adenosine (80, 81). Adenosine binds to the T-cell adenosine A2R receptor inhibiting effector T-cell functions and stimulating Treg cells (82, 83). Furthermore, the adenosinergic axis is over-functional in hypoxic conditions, connecting adenosine-mediated immunesuppression to low oxygen tension (84, 85).
Overall, a better understanding of the critical players within the TME and their specific roles in immune regulation will help design of metabolism-targeted therapeutic strategies for improving immunotherapy regimens in cancer.
Recently, NAD pathway enzymes and metabolites were shown to affect immune-cell functions and fate and alter the cancer cell-TME crosstalk. The following paragraphs are focused on describing these molecular circuits and their therapeutic implications.
NAD is a vital molecule governing many metabolic processes. It is used as a redox coenzyme by several dehydrogenases, and as a co-substrate by various NAD-consuming enzymes (86, 87). Among them are (i) mono- or poly-ADP ribosyltransferases (including ARTs and PARPs), which transfer the ADP ribose moiety to acceptor proteins resulting in their modification and function regulation, (ii) sirtuins, which catalyze the NAD-dependent deacetylation of metabolic enzymes and transcription factors, thus controlling their activity; (iii) NAD glycohydrolase that generates different NAD metabolites, including ADP ribose (ADPR), cyclic ADP ribose (cADPR) and nicotinic acid adenine dinucleotide phosphate (NAADP), with calcium (Ca+2) mobilizing activity. These enzymes are involved in the control of a wide range of biological processes, including transcription, DNA repair, cell adaptation to stress signals, and immune response (88). By catalyzing their reactions, they render NAD continuous re-synthesis an indispensable process. Various NAD biosynthetic routes guarantee the coenzyme regeneration, in different combination and with different efficiency depending on the cell-type and metabolic status (89, 90). A schematic overview of NAD homeostasis is shown in Figure 2 and reviewed in Sharif et al. (87), Magni et al. (91), and Houtkooper et al. (92).
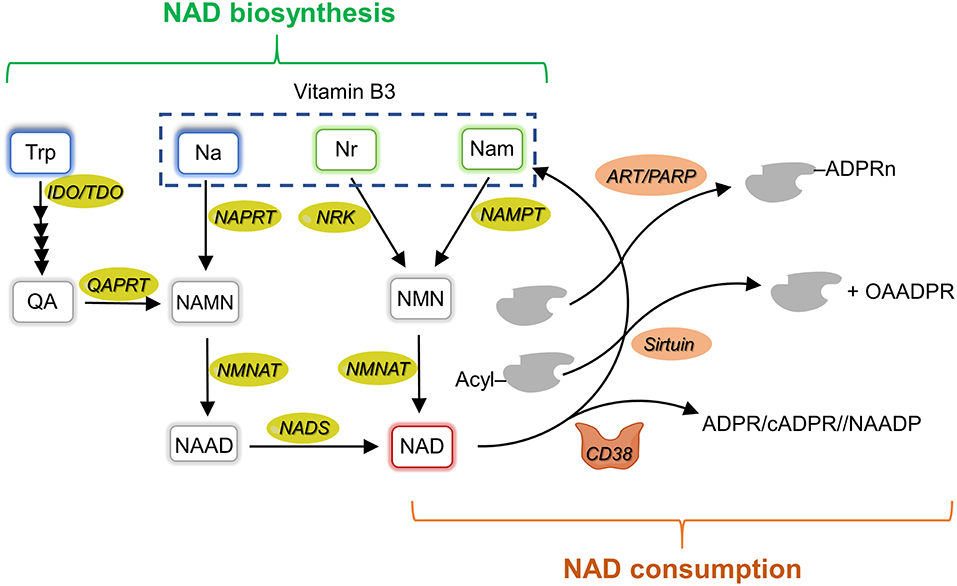
Figure 2. NAD metabolism overview. Schematic representation of mammalian NAD metabolism including biosynthetic (left side, in green) and consuming (right side, in orange) pathways. Na, nicotinic acid; NAD, nicotinamide adenine dinucleotide; NAPRT, nicotinate phosphoribosyltransferase; NAMN, nicotinate mononucleotide; NAAD, nicotinate adenine dinucleotide; Nam, nicotinamide; NAMPT, nicotinamide phosphoribosyltransferase; NADS, NAD synthetase; NMN, nicotinamide mononucleotide; NMNAT, NMN adenylyltransferase; Nr, nicotinamide riboside; NRK, nicotinamide riboside kinase; QA, quinolinic acid; QAPRT, quinolinate phosphoribosyltransferase; IDO, indoleamine 2,3-dioxygenase; TDO, tryptophan 2,3-dioxygenase; Trp, tryptophan; OAADPR, 2'-O-acetyl-ADP ribose; ART, ADP-ribosyltransferases; PARP, poly-ADP-ribose polymerase; ADPR, ADP-ribose; cADPR, cyclic ADPR; NAADP, nicotinic acid adenine dinucleotide phosphate.
The route which recycles nicotinamide (Nam), produced by the breakage of the N-glyosidic bond in the various NAD-consuming reactions, back to NAD that is considered the major pathway ensuring NAD homeostasis. It involves the phosphoribosylation of Nam to nicotinamide mononucleotide (NMN) by the enzyme Nam phosphoribosyltransferase (NAMPT) and the subsequent adenylation of NMN to NAD by NMN adenylyltransferase (NMNATs). This same route also salvages extracellular Nam that can be of dietary origin or can be formed in the extracellular space by the NAD glycohydrolase activity of the CD38 ectoenzyme acting on extracellular NAD and/or NMN. NAD can also be synthetized from exogenous nicotinamide riboside (NR) and nicotinic acid (NA) through distinct routes that are initiated by NR kinase (NRK) and NA phosphoribosyltransferase (NAPRT), respectively. The former enzyme phosphorylates NR to NMN, whereas the latter enzyme phosphoribosylates NA to nicotinate mononucleotide (NAMN). NMNATs convert NMN to NAD, and NAMN to nicotinate adenine dinucleotide (NAAD). NAAD is finally amidated to NAD by the enzyme NAD synthetase. A de novo biosynthetic route, which starts from tryptophan and enters the amidated route from NA, is also operative in several tissues and cell-types. The first and rate- limiting step in this pathway is the conversion of tryptophan to N-formylkynurenine by either IDO or tryptophan 2,3 -dioxygenase (TDO). Four reactions are then required to transform N-formylkynurenine to an unstable intermediate, α-amino-β-carboxymuconate-ε-semialdehyde (ACMS), which undergoes either decarboxylation, directed toward oxidation, or spontaneous cyclization to quinolinic acid (QA) directed toward NAD formation. Indeed, QA is phosphoribosylated to NAMN by the enzyme QA phosphoribosyltransferase (QAPRT), and the formed NAMN enters the NA salvage pathway. Among the enzymes involved in NAD homeostasis, NAMPT, CD38, sirtuins, and IDO are overexpressed in different types of cancer (93) and have been shown to play a role in cancer immune tolerance (94, 95). In the following sections, we will review what is known about their expression and function in the TME.
As the first and rate-limiting enzyme, NAMPT plays a pivotal role in the biosynthesis pathway of NAD from its nicotinamide precursor. It converts Nam and 5-phosphoribosyl-1-pyrophosphate (PRPP) into NMN in a complex reaction that can be significantly improved by a non-stoichiometric ATP hydrolysis (96). NAMPT is found both intracellularly and extracellularly (97, 98). Intracellular NAMPT (iNAMPT) is primarily located in the nucleus and cytosol. Previous studies reported NAMPT in mitochondria as well (99), but this remains a controversial finding (100, 101). As one of the main regulators of NAD intracellular level, NAMPT plays a crucial role in cellular metabolism (102). Conversely, the extracellular form of NAMPT (eNAMPT) has emerged as an important mediator of inflammatory programs (103). eNAMPT has been found in plasma and other extracellular fluids, including the supernatants of numerous cell types (103); however, while the mechanisms behind eNAMPT secretion remain unknown, they do not seem to rely on the classic pathway (104). Notably, the cytokine-like functions appear independent of the protein catalytic activity (105). In keeping with this view, NAMPT's substrates PRPP and ATP are apparently unavailable in the extracellular space to sustain the enzymatic activity (106).
eNAMPT was originally found to be secreted by activated lymphocytes and bone marrow stromal cells by Samal et al. (107) and called pre-B-cell colony enhancing factor [PBEF (107). In 2005, Fukuhara (108) identified eNAMPT as an adipokine and called it visfatin. These different names reflect its role in immune system and adipose tissue regulation.
Independent studies have conclusively shown that NAMPT expression and secretion can be induced by inflammatory signals in immune cells, in particular neutrophils, monocytes and macrophages (109). Both pathogen-derived lipopolysaccharide (LPS) and host-derived inflammatory stimuli, including tumor necrosis factor-α (TNF-α), IL-1β, IL-6, and leptin, can up-regulate NAMPT transcription in macrophages and other several types of cells (110–113). Several studies showed stimulation of cytokine release after exposure of cells to exogenous NAMPT, highlighting a role of eNAMPT as an inflammatory mediator as reviewed in Garten et al. (103). Following NAMPT treatment, IL-1β, IL-6, TNF-α, and IL-10 are up-regulated in peripheral blood mononuclear cells (PBMCs) and CD14+ monocytes (114). Co-stimulatory molecules such as CD54, CD40, and CD80 are also up-regulated in response to NAMPT treatment, an effect mediated through PI3-kinase and MAPKs p38, MEK1, and JNK (114). Furthermore, in macrophages NAMPT increases MMPs expression and activity (115). In vitro, eNAMPT promotes cell survival in macrophages subjected to endoplasmic reticulum (ER) stress, a frequent event in obesity and obesity-associated diseases. eNAMPT induces IL-6 secretion, followed by IL-6-mediated autocrine/paracrine activation of the prosurvival signal transducer STAT3, with a mechanism that is independent of the enzymatic activity (112).
Emerging evidence supports a role of NAMPT in regulating the differentiation program and the metabolic adaptation of myeloid cells. As described previously, activated macrophages can be divided in two subgroups in vitro: those with pro-inflammatory activity (M1) involved in first line of defense against bacterial infection, and those with anti-inflammatory activity (M2) that regulate tissue repair and wound healing (116), even if this is an oversimplification of the functional diversity occurring in vivo. Metabolic reprogramming of immune cells is required for both pro- and anti-inflammatory responses and a vast spectrum of metabolic statuses accompanies the complexity of phenotypes [reviewed in (117, 118)]. In general, an increase in glycolysis and in glucose uptake is typically associated to an M1 phenotype (119), while M2 macrophages rely on intact TCA cycle and OXPHOS as major source of ATP via electron transport chain and ATP synthase (120, 121). However, in addition to an augmented mitochondrial metabolism, alternatively activated macrophages can also use glycolysis when OXPHOS is disrupted (122). Another important pathway is the pentose phosphate pathway (PPP), which generates pentoses, 5-ribose phosphate and nicotinamide adenine dinucleotide phosphate (NADPH). NADPH is essential in activated M1 macrophages because it fuels ROS production by NADPH oxidase (123), even if other groups demonstrated that NADPH and NADPH oxidase play a role even in M2 differentiation (124). Concerning lipid metabolism, fatty acid synthesis is coupled to pro-inflammatory activity of macrophages, while beta-oxidation is typical of anti-inflammatory macrophages (117).
The increase of glycolysis associated with M1 activation of macrophages is orchestrated by the transcription factor HIF-1α. When cells experience low oxygen levels HIF-1α is stabilized and, upon binding of the HIF-1β subunit, initiates the transcription of genes such as glucose transporter and glycolytic enzymes (125, 126). NF-kB is required for transcriptional activation of HIF-1α (127); whereas, in M2 macrophages, genes involved in metabolic reprogramming are largely controlled by STAT6 and peroxisome proliferator-activated receptor gamma coactivator-1 beta (PGC-1β) (128).
Both iNAMPT and eNAMPT influence fundamental monocyte/macrophages processes such as differentiation, polarization and migration, even if the exact role of iNAMPT/eNAMPT in the process of myelopoiesis is incompletely elucidated so far (129–131) as summarized in Figure 3. For example, NAMPT has a role in the induction of an immunosuppressive and tumor-promoting microenvironment in chronic lymphocytic leukemia, where eNAMPT is important for the differentiation of monocytes toward tumor-supporting immunosuppresive M2 macrophage, promoting their differentiation, and polarization in tumor-supportive cells including TAMs (130). Recently, it was demonstrated that iNAMPT acts also on MDSCs, where NAMPT inhibits CXCR4 transcription, via NAD/SIRT1/HIF-1α axis, and this, in turn, leads to a mobilization of MDSCs and enhances their production of suppressive nitric oxide (132).
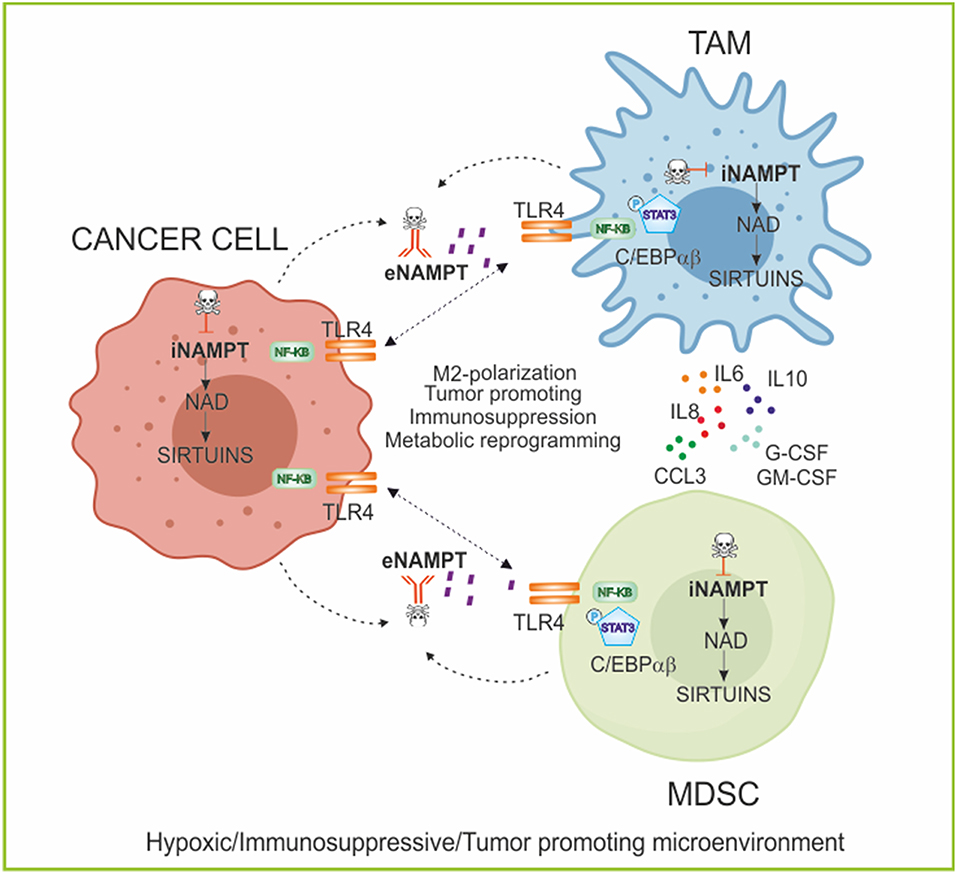
Figure 3. NAMPT in regulating myeloid cell fate and immunometabolism. Role of iNAMPT/eNAMPT in skewing myeloid populations into tumor-supporting M2-like macrophages and myeloid suppressive cells. Specifically, the iNAMPT/sirtuins axis regulates the metabolic reprogramming of cancer and myeloid cells in condition of low oxygen tension; while eNAMPT/TLR4 axis activates intracellular signaling promoting differentiation of myeloid cells and secretion of anti-inflammatory and pro-tumor cytokines creating an immunosuppressive microenvironment. The block of NAMPT functions, using iNAMPT pharmacological inhibitors and/or neutralizing antibodies, can repolarize the myeloid populations and inhibit tumor growth. TLR4, Toll-like receptor 4; C/EBPα/β, CCAAT/enhancer-binding protein α/β; G-CSF, Granulocyte Colony-Stimulating Factor; GM-CSF, Granulocytes-Macrophage Colony-Stimulating Factor; TAM, tumor-associated macrophages; MDSC, myeloid-derived suppressive cells.
Changes in NAD levels characterize different stage of macrophage polarization: in general, higher levels of NAD are typical of classically activated pro-inflammatory macrophages (M1), while NAD levels are lower in alternatively activated anti-inflammatory macrophages (M2). The NAMPT/NAD/SIRT1 axis seems to play a relevant role in myeloid cell functions as shown by the fact that efficient activation of M1 macrophages needs an increase of both NAMPT expression and cytosolic NAD (133). NAMPT-dependent generation of NAD is also crucial in the metabolic switch characterizing the transition from the early initiation phase of acute inflammation, which is anabolic and primarily requires glycolysis, to the later adaptation phase which is catabolic and relies on fatty acid oxidation (FAO) for energy (134). During these processes, also NAD-consuming deacetylases enzymes SIRT1 and SIRT6 have a role in regulating metabolism, increasing fatty oxidation and reducing glycolysis, respectively, coupling metabolic polarity with the inflammatory response, as described with more details later (135, 136). These data support the notion that NAD homeostasis has a crucial role in connecting bioenergetics and inflammation (134). A further feedback loop that links NAD to polarization of myeloid component has been suggested in monocytes, where NAMPT expression is induced by TNF-α via HIF-1α. In turn, NAMPT signaling involving NF-kB pathway activates activating protein 1 (AP1), inducing IL6 and TNFA transcription modulating myeloid cell activation (137).
In congenital neutropenia, a disorder in which patients display accumulation of granulocytic progenitors and no mature neutrophils in bone marrow, it has been shown that granulocyte colony-stimulating factor (G-CSF) is effective as it up-regulates NAMPT, which in turn triggers NAD/SIRT1 dependent granulopoiesis via CCAAT/enhancer-binding protein α/β (C/EBPα/β) up-regulation (129). On the contrary, GM-CSF is not effective in congenital neutropenia because it is unable to activate iNAMPT upregulation and NAD/SIRT1 axis (138). Following the induction of myeloid differentiation with G-CSF, the NAD-consuming enzyme SIRT1 deacetylase C/EBPα at position Lys 161 (129, 138). NAMPT inhibition with FK866 led to the dramatic elevation of acetylated C/EBPα levels and reduced amounts of total C/EBPα protein, accompanied by diminished mRNA expression of C/EBPα target genes (G-CSF, G-CSFR, and ELANE). Moreover, treatment of acute myeloid leukemia cell line HL-60 with recombinant NAMPT or transduction of HL-60 cells with NAMPT-expressing lentiviral construct induced myeloid differentiation of these cells per sé (138).
An open question is whether the cytokine-like actions that eNAMPT exerts on myeloid cells are related to its enzymatic activity or are mediated by the binding to a cell surface receptor. The fact that treatment with low concentrations of recombinant eNAMPT is sufficient to activate specific intracellular signaling pathways suggests that eNAMPT has cytokine-like properties and binds to and activates a cell surface receptor. In 2015, Camp et al. identified eNAMPT as a new ligand of the Toll-like receptor 4 (TLR4) (105). The authors demonstrated that in human lung endothelial cells, eNAMPT activates an inflammatory response via activation of NF-kB signaling pathway by binding TLR4-MD2 (105). However, the fact that recombinant eNAMPT is often produced in E. Coli strains renders the interpretation of these results controversial for the possible contamination of LPS, the natural ligand of TLR4, and activator of inflammatory programs. New studies have to confirm the TLR4 engagement by eNAMPT and correlate this with myeloid differentiation and plasticity.
The evidence linking myeloid cell fate and NAD/NAMPT could open the way to pharmacological inhibition of either iNAMPT and/or eNAMPT for re-education of myeloid cells. This could be useful in the context of acute inflammation, but also in cancer to force a reversion of immunosuppressive microenvironment, in combination with immunotherapy, as summarized in Figure 3.
For iNAMPT specific small molecules inhibitors exist, most known FK866 (also known as APO866) and GMX1778 (also known as CHS-828), among others (Table 1) (139–143, 159–161). However, most of the data on these drugs describe their effect on the tumor itself, and not on cells of the microenvironment (141, 161). Whether these inhibitors could also affect also eNAMPT activity is unknown, even if, as mentioned before, the enzymatic activity of eNAMPT is controversial. On the other hand, for eNAMPT, the group of Garcia, in order to block only the cytokine-like activity of eNAMPT, has devised a polyclonal eNAMPT neutralizing antibody (130, 144), that could be useful in those condition in which only the extracellular form of eNAMPT is detrimental and intracellular enzymatic activity needs to be preserved.
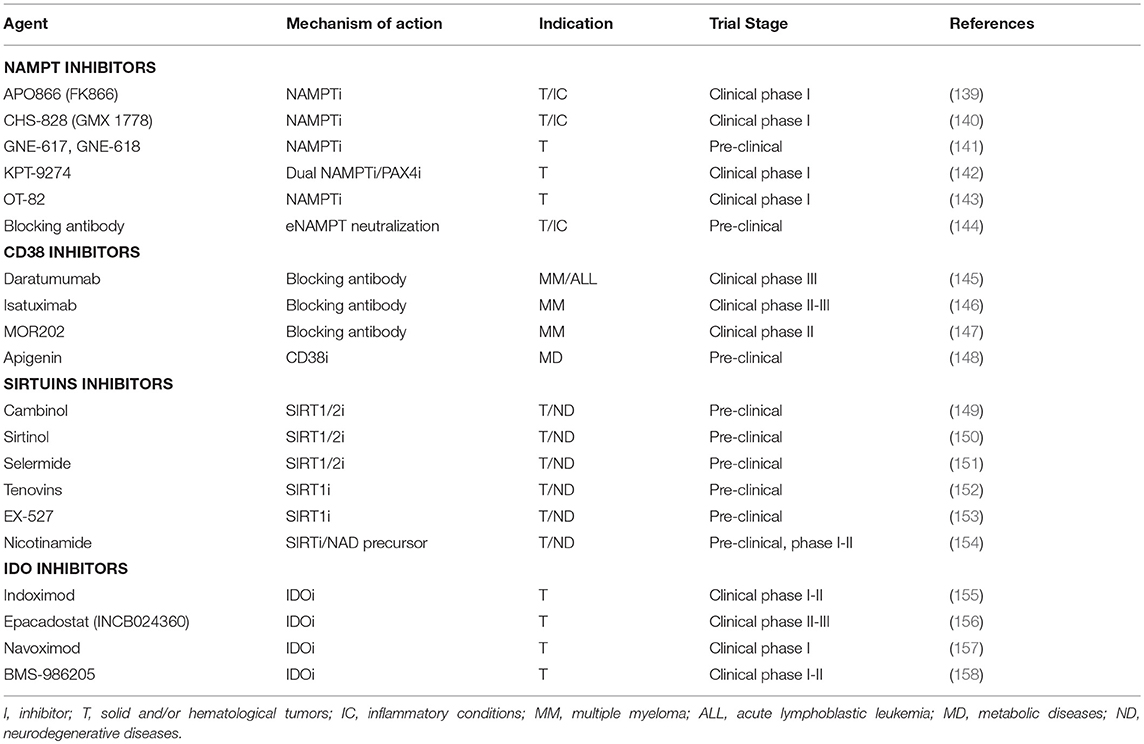
Table 1. Pharmacologic tools currently undergoing pre- or clinical evaluation to block NADome enzymes.
Cluster of differentiation (CD) protein CD38, first identified as a lymphocyte antigen, is a cell surface glycohydrolase that cleaves a glycosidic bond within NAD to yield Nam, ADP-ribose (ADPR), and cyclic ADPR (cADPR), and converts NAD phosphate (NADP) to NAADP, all calcium (Ca2+) mobilizing molecules (162, 163). These molecules bind specific receptors, like the ryanodine receptor on endoplasmic reticulum, the lysosomal two-pore channel and the plasma membrane calcium channel transient receptor (TRPM2), activating calcium signaling, which in turn affects gene expression, cell cycle control, cell survival, energy metabolism, leukocyte trafficking, and inflammation (87).
CD38 is a transmembrane protein with four different forms, according to the cellular localization (164). The most common form of CD38 has a type II membrane orientation, i.e., with the catalytic domain facing the extracellular space. By contrast, the less abundant type III transmembrane form has its catalytic site facing the inside. Intriguingly, soluble intracellular and extracellular forms of CD38 have also been ascribed (165, 166). CD38 is widely expressed both in immune cell types (bone marrow progenitors, natural killer cells, monocytes, and activated T- and B-lymphocytes) and in non-hematopoietic cells (167).
CD38 is also an unquestionable contributor to intracellular NAD homeostasis (168, 169) and this apparent “paradox” has been in part reconciled by recent reports demonstrating that CD38 can also degrade circulating NAD precursors such as NMN and NR, thus preventing their fueling of NAD biosynthesis (170, 171). Notably, CD38 enzymatic activity mediates many roles which include metabolism regulation and pathogenesis of heart disease, obesity, aging and inflammation, among the others. Nevertheless, it is well-established that CD38 overexpression is correlated to different hematological malignances including myelomas and leukemias (172). In this contest, a broad immune regulatory role for NAD and CD38 on T cell behavior has been reported (87, 145, 173) and summarized in Figure 4. In order to elucidate the impact of CD38 modulation of NAD homeostasis in T cell, a brief synthesis of T cell metabolism is necessary, as metabolism drives T cell life (36, 174, 175). One of the main challenges of the field in a translational perspective is to manipulate T cell metabolism in order to improve their immune response capacity. Defined metabolic pathways orchestrate T cell development, differentiation, function and persistence (176). TCA/OXPHOS-mediated ATP production is instrumental for the maturation of Naïve T (TN) lymphocytes, a population of quiescent non-proliferative cells, in primary lymphoid organs (177). T cell activation is initiated after antigen recognition and TCR ligation. This step, requiring major histocompatibility complex, and co-stimulatory molecules, activates T lymphocytes inducing both a rapid proliferation rate and a differentiation program toward effector functions (176). To sustain both clonal expansion and active immune response, T cells shift to an anabolic metabolism which provides faster ATP production and nutrients supply. While cytolytic CD8+ T (Tc) cells dominantly shift metabolism to glycolysis, activated CD4+ T helper (Th) cells increase both glycolysis and FAO (178). FAO also supports metabolism of iTreg and long living memory T-cell (Tm) (178). All these T cell subsets, to achieve their metabolic profile, require a coordinated transcriptional program together with a specific system of nutrient uptake. T cells depend on the import of substrates such as glucose, amino-acids (especially glutamine), and glycerol. In TN and Tm cells, increased expression of glucose and glutamine transporters is controlled by the transcription factor c-Myc (36) and regulated by a specific cytokine, IL-7 (175). AKT-mTOR and TLR signaling, as well as the transcription factors HIF-1α, c-Myc and FoxP3, have been shown to directly regulate Treg metabolic programming and development, while HIF-1α and mTOR control the glycolytic phenotype and activation (IFN-γ production) of effector T-cells, Th1, Th2, and Th17 lineages (36). Metabolism underpins T cell cycle through quiescence and activation states and T-cells failure to engage specific metabolic programs is a biological phenomenon accompanying tumor aggressiveness and T cell exhaustions (176). The crosstalk between cancer cell and tumor TILs is played at different levels. As already mentioned, it has been shown that the establishments of nutrients competition between tumor cells and TILs has a primary role in influencing T cell fate and dysfunctions (34, 179–181). Malignant cells push their metabolism toward a Warburg phenotype. The consequent induction of a hypoxic and nutrient-deprived environment (low glucose, glutamine, glycine, and serine) shapes a tumor sustaining microenvironment and immune tolerance (179). Indeed, T cells migrating to tumors sites must adapt to both (i) nutrient-depleted environments (182) and contemporarily to (ii) the presence of hypoxic tumor-derived metabolites including lactate, adenosine, cyclic adenosine monophosphate (cAMP), IDO/kynurenine.
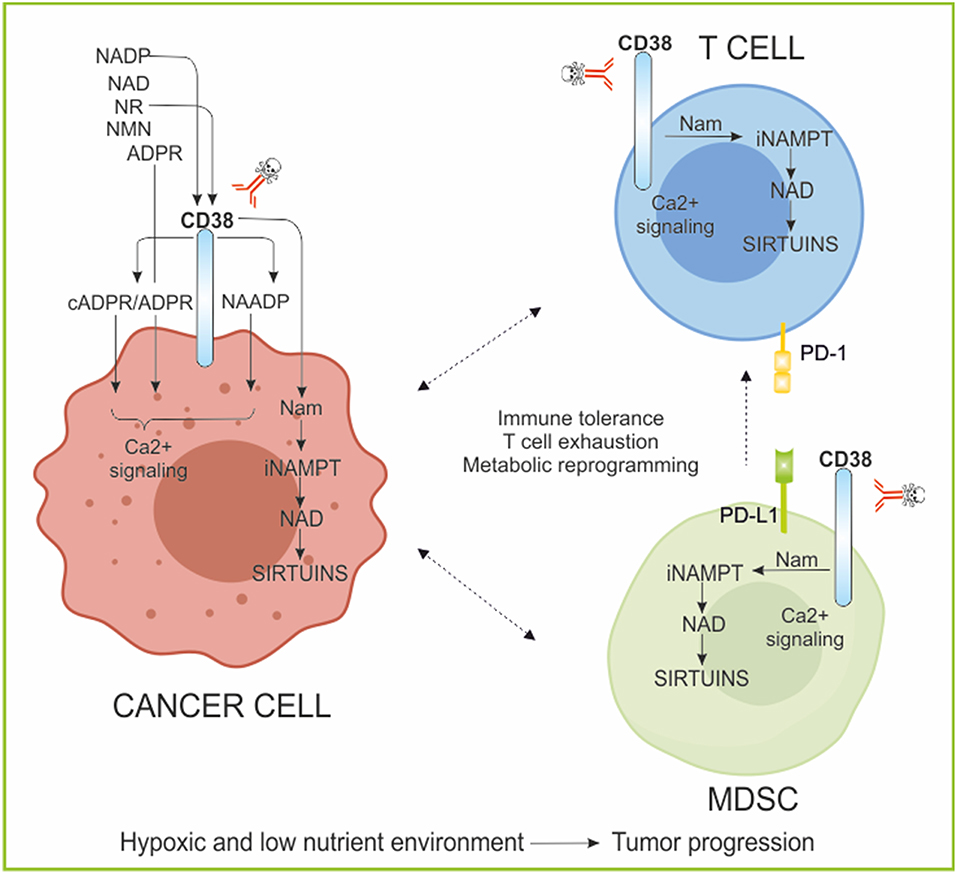
Figure 4. CD38/NAD axis regulates T cell phenotype and responses. The ectoenzyme CD38, expressed by tumor cells and immune cells is involved in the activation of calcium (Ca2+) signaling through the generated metabolite cADPR/ADPR/NAADP. Moreover, it metabolizes NAD, releasing Nam, rendering the substrate of NAMPT available for continuous NAD regeneration. These reactions occur also in immune cells modifying NAD concentrations and affecting sirtuins activities. The NAD/CD38/SIRTUINS axis regulates T cell immune cell fate, metabolism, and gene transcription. Evidence of high CD38 expressing immune suppressive cells have been reported in several tumors. CD38 inhibition was sufficient to re-establish T cell proliferation, antitumor cytokine secretion, and killing capability. ADPR, ADP-ribose; cADPR, cyclic ADPR; NADP, NAD phosphate; NAADP, nicotinic acid adenine dinucleotide phosphate; Ca2+, calcium; NR, nicotinamide riboside; NMN, nicotinamide mononucleotide; Nam, nicotinamide; MDSC, myeloid-derived suppressive cells; PD-1, programmed cell death protein 1; PD-L1, PD-1 ligand.
In this context, CD38-mediated Ca2+ mobilization can directly affect T cell metabolism. In the physiology of a T lymphocyte, Ca2+ controls T cell gene expression and consequently differentiation, development and cytotoxicity (183). Alteration of Ca2+ signaling affects immune deregulation and consequently tumor initiation and progression (183–186). A second level of T cell metabolic reprogramming control by the CD38/NAD axis also involves sirtuins (173). Indeed, a lot of literature has been produced on the role of SIRT1, as a key modulator of immune cell functions, as described in a dedicated section of this review (166, 187, 188). In this case, the inverse correlation between expression of CD38 and intracellular NAD contents, act on SIRT1-mediated post-transcriptional control of key genes involved in T cell functions (173). Furthermore, it was recently shown that CD38 is highly expressed by specific subsets of immunosuppressive TILs (i.e., Treg and Th17) (34, 36, 173, 189) and by MDSC, another key immunosuppressive cellular component of tumor milieu represented (190). Both, CD38highMDSC cells-mediated suppression of activated T-cells and the concomitant expression of CD38 with exhaustion markers on T cells, for example PD1, pointed to an active role of CD38 in modulating T cell metabolism and fate toward the generation of an immune tolerant landscape in tumor (173). Evidence of high CD38 expressing Treg have been reported for multiple myeloma and acute lymphoblastic leukemia, where the use of mAbs against CD38 (daratumumab, isatuximab, and MOR202, Table 1) is more than a promising therapeutic option to reestablish a functional immune surveillance (145–147, 189, 191, 192). In these tumor models, suppression of CD38+ cancer cells associate with an increase in T-helper and cytotoxic T lymphocytes, T-cell functional response and TCR clonality (191, 192). A functional relationship between CD38 and Th17 has also been highlighted (173). Th17 is a CD4+ T cell subpopulation secreting IL17, which gained interest in the field of immunotherapy due to their self-renewal, plasticity and hematopoietic stem-like phenotype (173, 193). Adoptive T cell transfer (ACT) therapy is a powerful strategy developed for controlling cancer (194, 195). The emerged staminal potential of Th17, together with their ability to persist for long times at tumor sites, made of this T cell subset an ideal candidate to improve ACT efficacy (173, 196, 197). Chatterjee et al. recently demonstrated that, SIRT1-dependent deacetylation of the transcription factor forkhead box O1 (FOXO1) drives the functional homing in different organs of a hybrid Th1/Th17 population 24 h after ACT. Most importantly, they reported that, the decrease of CD38 expression on Th17 cells leads to the increase of intracellular NAD concentration, reinforcing the SIRT1-dependent immune efficacy of this T cell population (173). For these reasons, the inhibition of CD38 has been proposed not only to specifically target CD38high immune suppressive cell populations (MDSCs, Treg), but also to improve tumor control via ACT therapy or using immunomodulatory drugs (173, 191, 192).
Lastly, very recently CD38 was considered as major acquired mechanism of resistance to PD-1/PD-L1 blockade, causing CD8+ T cell suppression. Co-targeting of CD38 and PD-L1 improves anti-tumor immune response. CD38 manipulation was sufficient to regulate CD8+ T cell proliferation, antitumor cytokine secretion, and killing capability (198).
Sirtuins, initially described as transcriptional silencers in yeast (199), represent a class of NAD-dependent enzymes with deacetylase activity. So far, seven isoforms (SIRT1-7) constitute the family of mammalian sirtuins, which differ in subcellular compartmentation, enzymatic activity, and in vivo substrate selectivity (200). As a primary cellular location, SIRT1, SIRT6, and SIRT7 are found in the nucleus, SIRT2 in the cytoplasm, and SIRT3-SIRT5 in mitochondria (201). However, recent reports have shown that sirtuins are not anchored to precise subcellular compartments, and may shuttle between them, depending on cell type or physio-pathological conditions (202–205). The canonical reaction catalyzed by sirtuins is the transfer of an acetyl group from protein lysine residues to the ADPR moiety of NAD. As a result, the reaction produces Nam, first released, the deacetylated lysine, and 2'-O-acetyl-ADP ribose (206). Although lysine deacetylation is the primary activity of sirtuins, recent studies have shown that these enzymes can remove a variety of other acyl-lysine groups (207). Some sirtuins act as ADP-ribosyltransferases, although the biological relevance of such activity is incompletely understood. Mammalian sirtuins target different proteins in an isoform-specific fashion (207, 208), allowing their regulation of multiple processes like energy metabolism, epigenetic regulation of gene expression, DNA repair, inflammation, cellular stress resistance, healthy aging, tumorigenesis, autophagy, and apoptosis as reviewed in Haigis and Sinclair (208), Finkel et al. (209), and Houtkooper et al. (210).
Emerging evidence demonstrated that sirtuins are key regulators of inflammatory stress response in immune and non-immune cells (95, 211–213). Sirtuins are involved in epigenetic regulation, through deacetylation of histones and/or non-histone proteins, of metabolic, phenotypic, and bioenergetics reprogramming of immune cells (immuno-metabolism) (210, 212–214).
SIRT1 is the most extensively studied sirtuins, especially for its role in aging (210, 214). In addition, SIRT1 is involved in controlling stem cell development, cell differentiation and autophagy, metabolic reprogramming and inflammation (209, 215). SIRT1 is also the most studied among sirtuins involved in immune regulation and here we summarized some SIRT1 activities in epigenetic regulation of metabolism and immune response (Figure 5).
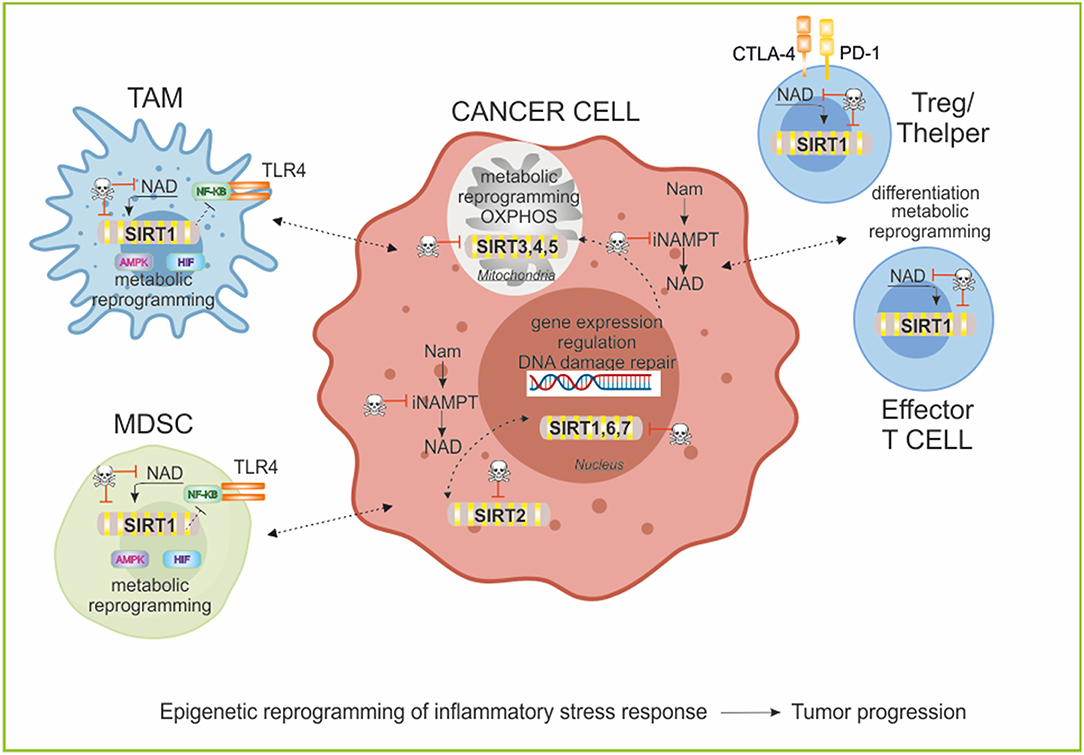
Figure 5. Sirtuins and epigenetic regulation of immune cell functions. Sirtuins are a family of 7 members with different subcellular localization. These NAD-dependent deacetylases are involved in epigenetic regulation of metabolic reprogramming of cancer and immune cells and in promoting T cell differentiation and function. Particularly, in myeloid cells SIRT1 decreases inflammation negatively regulating NF-KB pathway, and inducing a metabolic rewiring mediated by its activity on AMPK/PGC-1α and HIF-1α stabilization. In T cell populations, SIRT1 modifies the phenotypic plasticity of Thelper and Treg and induces T cell tolerance. The manipulation of the Sirtuins/NAD axis is an important area of study for therapeutic implications in cancer research to repolarization of immune cell responses and to block tumor progression. TLR4, Toll-like receptor 4; AMPK, AMP-activated protein kinase; HIF, Hypoxia-inducible factor; PD-1, programmed cell death protein 1; CTLA-4, Cytotoxic T-Lymphocyte Antigen 4; TAM, tumor-associated macrophages; MDSC, myeloid-derived suppressive cells.
Epigenetic mechanisms are essential to the development and differentiation of the immune system, as well as in related pathologies (216–218). Epigenetic mechanisms include multilevel intracellular events that influence chromatin structure and gene expression such as histone methylation and acetylation, as well as DNA methylation, non-coding RNAs and chromatin remodeling (219). Further, numerous signals (i.e., TCR, TLRs, inhibitory receptors, and cytokines) drive changes in the epigenome that result in downstream modulation of immune responses (77). TLR signaling in macrophages regulates differentiation/polarization and activation in response to pathogens affecting gene expression and metabolic reprogramming (220, 221). In particular, TLR4 engagement by LPS in macrophages drives a shift toward a glycolytic metabolism impairing mitochondrial respiration (222), resulting in a marked shifts in NAD/NADH ratios, which influence the activities of SIRT1, potentially altering deacetylation of histone and non-histone substrates (134, 223). Liu et al. found in TLR4-stimulated THP-1 promonocytes that SIRT1 support a switch from increased glycolysis to increased FAO as early inflammation converts to late inflammation (134). The shift to late acute inflammation and elevated FAO required peroxisome proliferator-activated receptor gamma coactivator (PGC-1α),a known target of SIRT1 (187, 224–227). A circuit of AMP-activated protein kinase [(AMPK)/SIRT1/PGC-1α] results in the deacetylation and modulation of the activity of downstream SIRT1 targets that include the PGC-1α and the FOXO1 and FOXO3a transcription factors. The AMPK-induced SIRT1-mediated deacetylation of these targets explains many of the convergent biological effects of these two energy sensors, AMPK and SIRT1, on cellular metabolism (225, 226).
Recent studies have showed that the regulation of innate immunity and energy metabolism are connected through antagonistic crosstalk between NF-κB and SIRT1 signaling pathways (228). NF-κB signaling has a major role in innate immunity defense, while SIRT1 regulates the oxidative respiration and cellular survival (229). However, NF-κB activation can stimulate glycolysis during acute inflammation, whereas SIRT1 activation inhibits NF-κB signaling and enhances oxidative metabolism and the resolution of inflammation. SIRT1 inhibits NF-κB signaling directly by deacetylating the p65 subunit of NF-κB complex (230). SIRT1 stimulates oxidative energy production via the activation of AMPK, peroxisome proliferator activated receptor (PPARα) and PGC-1α and simultaneously, these factors inhibit NF-κB pathway and suppress inflammation (225, 226, 231). Using a myeloid cell-specific SIRT1 knockout (Mac-SIRT1 KO) mouse model, Schug et al. show that ablation of SIRT1 in macrophages renders NF-κB hyperacetylated, resulting in increased transcription of proinflammatory target genes. Consistent with increased proinflammatory gene expression, Mac-SIRT1 KO mice challenged with a high-fat diet display high levels of activated macrophages in liver and adipose tissue, predisposing the animals to development of systemic insulin resistance and metabolic derangement (232). In some cases, the effects of SIRT1 in regulating metabolism of immune cells are mediated by HIF-1α (233). SIRT1 can bind and deacetylate HIF-1α resulting in a stabilization or in an inhibition of the protein, depending on the cells and context (234, 235). The SIRT1-HIF-1α axis bridges the innate immune signal to an adaptive immune response by directing affecting metabolism, cytokines production, and differentiation of immune cells (236). For example, (i) SIRT1 can limit the function and differentiation of MDSCs through HIF-1α-induced glycolytic metabolic reprogramming (237), (ii) SIRT1 can regulate T helper 9 (Th9) cell differentiation through the mTOR/HIF-1α-dependent glycolytic pathway (238). The interplay between HIFs and sirtuins may also extend to stress settings such as hypoxic tumors, in which cellular redox balance is perturbed (64, 239).
For adaptive immune cells, SIRT1 has a key role in mediating the differentiation of T cell subsets in a NAD-dependent manner. T cells exhibit remarkable phenotypic and functional plasticity during immune responses (240). SIRT1 is involved in (i) Th and Treg cell differentiation (238, 241); (ii) SIRT1 signals in DCs can repress PPARγ activity and promote T helper 2 (Th2) cell responses in airway allergy through metabolism-independent manners (242); (iii) SIRT1 interacts with c- Jun and inhibits CD4 T cells to mediate T cell tolerance (243); (iv) SIRT1 regulates CD8 T-cell differentiation interacting with basic leucine zipper transcription factor ATF-like (BATF) and regulating both epigenetic remodeling and energy metabolism of T cells (244). Furthermore, (v) SIRT1/FOXO1 axis regulates metabolic reprogramming of terminally differentiated memory T cells, as previously described (188).
Finally, SIRT1 has been shown to play also important roles in physiological processes affecting organismal longevity as well as stem cell function and self-renewal (245, 246). In macrophages, SIRT1 is emerging as critical positive modulator of self-renewal, regulating G1/S transition, cell cycle progression and a network of self-renewal genes (247).
Similar functions in regulating inflammation and metabolism are exerted also by SIRT2 and SIRT6 (134, 213, 248).
Interactions of cellular metabolic and epigenetic pathways and how these two key biological processes interplay to feedback modulate immune cell function is attracting in cancer therapy. Sirtuins/NAD axis has proven to be a crucial link between epigenetics and metabolism, and hence, it is an important area of study for therapeutic implications (215). While there are only specific activators or inhibitors for SIRT1 exist, drugs that affect NAD levels or NAD precursors offer the possibility to regulate all seven sirtuins coordinately (239). These compounds can be used alone or in combination with existing cancer therapies. The effects of SIRT1 inhibitors (e.g., cambinol, sirtinol, tenovins, Ex-527, Table 1) are currently studied mainly in the context of cancer (239). Very recent data show the impact of SIRT1 inhibition or genetic deletion on T cell responses, particularly on Treg differentiation. Genetic deletion or pharmacologic inhibition of SIRT1 through EX-527 improves Foxp3+ Treg number and function through increased Foxp3 transcription its acetylation, leading to decreased Foxp3 turnover from ubiquitination and poly(ADP)ribosylation. As a result, targeting SIRT1 increases both central and inducible Foxp3+ Tregs and promotes their suppressive functions, as summarized in Chadha et al. (249). SIRT1 inhibition is therefore useful in the context of graft-vs.-host disease (GVHD), to extend allograft survival (249–251). However, there are a number of studies in which SIRT1 deletion or inhibition led to proinflammatory conditions, indicating that regulation of the system is still incompletely understood (249, 252). Interestingly, the Nam generated in deacetylase reactions by SIRTs acts as a negative feedback regulator of SIRT activity (253, 254). This Nam is converted back to NAD by the action of NAMPT and NMNATs. Hence, NAD-biosynthetic enzymes, in particular NAMPT, also regulate sirtuins signaling (255) providing the rational to use NAMPT inhibitors to interfere with Sirtuins functions.
Overall these results indicate that sirtuins broadly coordinate innate and adaptive immune reprogramming and represent druggable immunometabolic enhancement targets, useful also to repolarize immune cells in TME.
Amino acid catabolism is a key effector in driving immune tolerance. IDO is a cytosolic, heme-dependent enzyme responsible for the rate-limiting step of de novo NAD synthesis from tryptophan in extrahepatic tissues. The catalyzed-reaction yields N-formylkynurenine and commits the aminoacid toward its conversion to QA through the kynurenine pathway (90), which accounts for >90% of tryptophan catabolism (256). Tryptophan is an essential amino acid in protein metabolism, a precursor for the synthesis of the neurotransmitter serotonin and tryptamine, as well as for the synthesis of NAD and the hormone melatonin (257, 258).
In recent years, IDO has drawn enormous attention due to its immune regulatory functions (259–261) and summarized in Figure 6. IDO is not constitutively expressed in immune cells. Rather, various stimuli, and signaling pathways induce transcription and translation of metabolically-active IDO enzyme protein. Among them, TLRs, tumor necrosis factor superfamily members (TNFRs), interferon beta receptor (IFNBR), the interferon gamma receptor (IFNGR), transforming growth factor beta receptors (TGFBRs) and the aryl hydrocarbon receptor (AhR) all can activate signaling mechanisms that either induce or maintain IDO expression. NF-KB activation is a central downstream signal of these pathways regulating IDO expression (262).
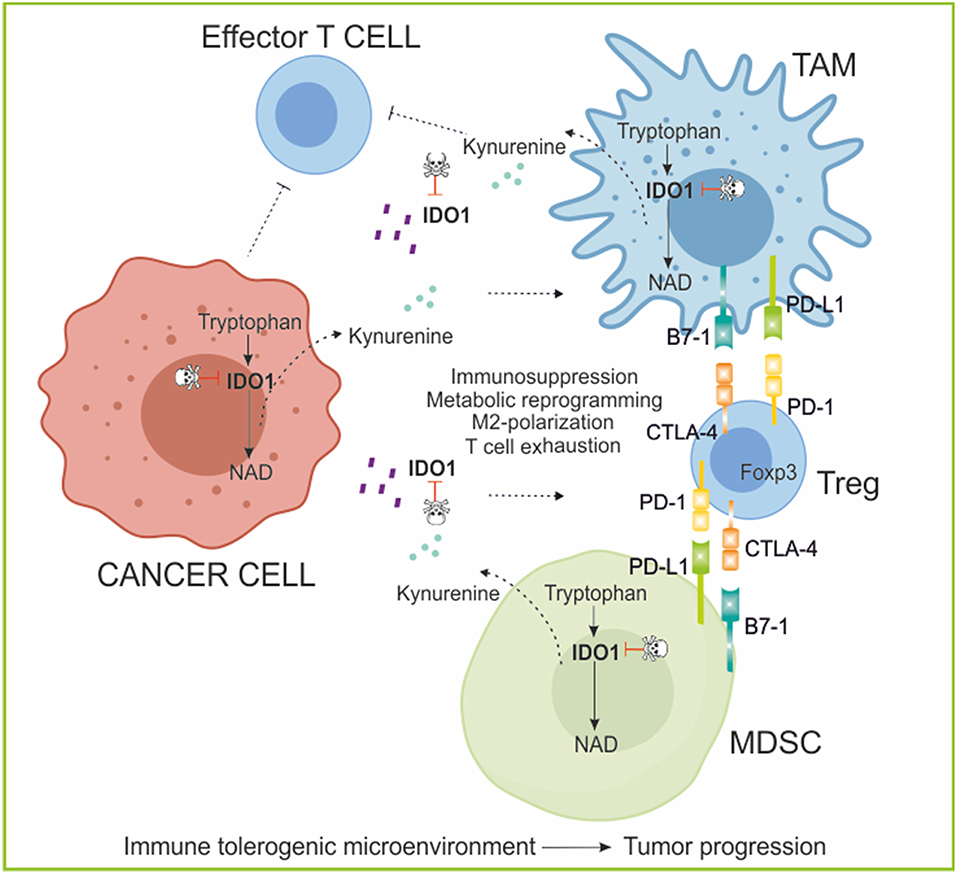
Figure 6. The role of IDO/kynurenine in cancer immunoediting. IDO1 is an enzyme involved in the catabolism of tryptophan (kynurenine pathway). IDO and kynurenine can be secreted by tumor and tolerogenic immune cells in the microenvironment where exert an immunosuppressive function polarizing myeloid cell toward M2 phenotype (TAM; MDSC) and suppressing effector T cell functions, while stimulating expansion and activation of permissive Treg population, increasing immune escape mechanisms (PD-1/PD-L1; CTLA-4/B7-1 crosstalk). Moreover, in the extracellular space, IDO1 depletes the essential amino acid tryptophan from the tumor microenvironment, favoring tumor growth. IDO1 inhibitors in combination with Immunotherapy aim to reverse immunoediting (backward arrow) by inhibiting and activating local immunosuppressive and tumor eradication mechanisms, respectively. PD-1, programmed cell death protein 1; PD-L1, PD-1 ligand; CTLA-4, Cytotoxic T-Lymphocyte Antigen 4; Foxp3, forkhead box P3; TAM, tumor-associated macrophages; MDSC, myeloid-derived suppressive cells.
By catalyzing the initial and rate-limiting step of tryptophan degradation, IDO reduces the local tryptophan concentration and produces immune modulatory tryptophan metabolites (263). In particular, cells expressing IDO and TDO produce the tryptophan catabolite kynurenine that, by interacting with the aryl hydrocarbon receptor expressed by T cells, Tregs and DCs, regulates immunity (264). Additionally, inhibition of CD8+ T-cell-mediated cytotoxic function was found to be an important mechanism behind IDO's immune-modulating property (264). Due to the role in regulating T cell response and fate, IDO function is critical in organ and tissue graft survival, in viral infection, in tissue-specific autoimmunity and the promotion of cancer cell survival (265). The biologic function of the IDO pathway was originally described as both counter-regulatory (controlling inflammation) and tolerogenic (creating acquired antigen-specific tolerance in T cells) (257).
Escape from the immune response is essential for cancer progression, however, mechanisms underlying this process remain unclear. Kynurenine in the tumor microenvironment was recently shown to favor immunosuppression (265, 266). Tryptophan catabolism was shown to create an immuno-suppressive milieu in tumors and in tumor-draining lymph nodes through accumulation and secretion of immunosuppressive tryptophan catabolites that bind and activate AhR (267), leading to induction of T-cell anergy, apoptosis, increased conversion of naïve CD4+ T cells into Tregs and polarization of DCs and macrophages toward an immunosuppressive phenotype (190, 261, 265, 268).
Clinically, studies of ovarian, lung, colorectal, breast cancer, brain tumors, melanoma, and others have shown that increased expression of IDO was associated with poor survival outcomes (258, 265, 269, 270). In most studies, the ratio of kynurenine to tryptophan was measured in patient plasma as a measure of IDO and TDO activity (156, 267). Moreover, not only tumor can express IDO, but also immune cells including both TAM and MDSC express high levels of IDO, in response to inflammatory cytokines, of which IFN-γ is the most potent inducer, amplifying the circuit of immunosuppression (190, 271, 272).
According to the role of IDO in driving immunosuppression, in the last years IDO became a valid target in cancer therapy (273, 274). Competitive inhibitors of IDO are currently being tested in clinical trials in patients with solid cancer, with the aim of enhancing the efficacy of conventional chemotherapy, vaccine or checkpoint inhibitors (275). Agents currently account for the majority of the trials: indoximod (1-methyl-D-tryptophan), an inhibitor of the IDO pathway (155, 276), epacadostat (INCB024360) (156) and BMS-986205 (158) (Table 1), with encouraging results. Importantly, the use of IDO inhibitors can be also overcome the resistance to immunotherapies targeting immune checkpoints, strongly supporting the combination therapies with IDO inhibitors irrespective of IDO expression by the tumor cells (277). Additional IDO inhibitors are in the development pipeline, as well as agents that may target TDO, or a second isoform of IDO (IDO2) (275).
Anticancer strategies targeting simultaneously oncogenic and metabolic pathways, de-regulated in cancer cells, seem to be ideal and have shown some promising results. Interestingly, local conditions in the tumor microenvironment affect also metabolic responses of immune cells, favoring immune-tolerance, and immune-escape mechanisms. One of the goals of immunotherapy could be to re-educate the immune system to kill tumors, by reprogramming their metabolism. The network of immunosuppressive mechanisms in the TME is complex, multifactorial, and mutually reinforcing. A better knowledge of the main players of this cross-talk can help in designing more effective combination therapies. In this picture, NAD-metabolizing enzymes are receiving increasing attention to due to their role in conditioning several aspects of immune cell fate and functions. It is foreseeable that modulators/inhibitors of the NADome (summarized in Table 1) will become useful alone or in combination with current anti-cancer therapeutic strategies to regulate both tumor growth and immune populations of TME.
All authors listed have made a substantial, direct and intellectual contribution to the work, and approved it for publication.
This work was supported by the Italian Institute for Genomic Medicine Institutional funds, by Gilead Fellowship program 2018, by the Ministry of Education University and Research-MIUR, PRIN Project 2017CBNCYT and Progetto strategico di Eccellenza Dipartimentale #D15D18000410001 (the latter awarded to the Dept. of Medical Sciences, University of Turin) and ITN INTEGRATA program (grant agreeement 813284).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
1. Hanahan D, Coussens LM. Accessories to the crime: functions of cells recruited to the tumor microenvironment. Cancer Cell. (2012) 21:309–22. doi: 10.1016/j.ccr.2012.02.022
2. Quail DF, Joyce JA. Microenvironmental regulation of tumor progression and metastasis. Nat Med. (2013) 19:1423–37. doi: 10.1038/nm.3394
3. Fridman WH, Pages F, Sautes-Fridman C, Galon J. The immune contexture in human tumours: impact on clinical outcome. Nat Rev Cancer. (2012) 12:298–306. doi: 10.1038/nrc3245
4. Nagarsheth N, Wicha MS, Zou W. Chemokines in the cancer microenvironment and their relevance in cancer immunotherapy. Nat Rev Immunol. (2017) 17:559–72. doi: 10.1038/nri.2017.49
5. Cairns RA, Harris IS, Mak TW. Regulation of cancer cell metabolism. Nat Rev Cancer. (2011) 11:85–95. doi: 10.1038/nrc2981
6. Kareva I, Hahnfeldt P. The emerging “hallmarks” of metabolic reprogramming and immune evasion: distinct or linked? Cancer Res. (2013) 73:2737–42. doi: 10.1158/0008-5472.CAN-12-3696
7. Boroughs LK, Deberardinis RJ. Metabolic pathways promoting cancer cell survival and growth. Nat Cell Biol. (2015) 17:351–9. doi: 10.1038/ncb3124
8. Morandi A, Giannoni E, Chiarugi P. Nutrient exploitation within the tumor-stroma metabolic crosstalk. Trends Cancer. (2016) 2:736–46. doi: 10.1016/j.trecan.2016.11.001
9. Kerkar SP, Restifo NP. Cellular constituents of immune escape within the tumor microenvironment. Cancer Res. (2012) 72:3125–30. doi: 10.1158/0008-5472.CAN-11-4094
10. Schiavoni G, Gabriele L, Mattei F. The tumor microenvironment: a pitch for multiple players. Front Oncol. (2013) 3:90. doi: 10.3389/fonc.2013.00090
11. Armulik A, Abramsson A, Betsholtz C. Endothelial/pericyte interactions. Circ Res. (2005) 97:512–23. doi: 10.1161/01.RES.0000182903.16652.d7
12. Chouaib S, Kieda C, Benlalam H, Noman MZ, Mami-Chouaib F, Ruegg C. Endothelial cells as key determinants of the tumor microenvironment: interaction with tumor cells, extracellular matrix and immune killer cells. Crit Rev Immunol. (2010) 30:529–45. doi: 10.1615/CritRevImmunol.v30.i6.30
13. Cao Z, Ding BS, Guo P, Lee SB, Butler JM, Casey SC, et al. Angiocrine factors deployed by tumor vascular niche induce B cell lymphoma invasiveness and chemoresistance. Cancer Cell. (2014) 25:350–65. doi: 10.1016/j.ccr.2014.02.005
14. Petrova V, Annicchiarico-Petruzzelli M, Melino G, Amelio I. The hypoxic tumour microenvironment. Oncogenesis. (2018) 7:10. doi: 10.1038/s41389-017-0011-9
15. Hegde PS, Wallin JJ, Mancao C. Predictive markers of anti-VEGF and emerging role of angiogenesis inhibitors as immunotherapeutics. Semin Cancer Biol. (2018) 52:117–24. doi: 10.1016/j.semcancer.2017.12.002
16. Uccelli A, Moretta L, Pistoia V. Mesenchymal stem cells in health and disease. Nat Rev Immunol. (2008) 8:726–36. doi: 10.1038/nri2395
17. Shah K. Mesenchymal stem cells engineered for cancer therapy. Adv Drug Deliv Rev. (2012) 64:739–48. doi: 10.1016/j.addr.2011.06.010
18. Shi Y, Du L, Lin L, Wang Y. Tumour-associated mesenchymal stem/stromal cells: emerging therapeutic targets. Nat Rev Drug Discov. (2017) 16:35–52. doi: 10.1038/nrd.2016.193
19. Quante M, Tu SP, Tomita H, Gonda T, Wang SS, Takashi S, et al. Bone marrow-derived myofibroblasts contribute to the mesenchymal stem cell niche and promote tumor growth. Cancer Cell. (2011) 19:257–72. doi: 10.1016/j.ccr.2011.01.020
20. Su S, Chen J, Yao H, Liu J, Yu S, Lao L, et al. CD10(+)GPR77(+) Cancer-associated fibroblasts promote cancer formation and chemoresistance by sustaining cancer stemness. Cell. (2018) 172:841–56.e816. doi: 10.1016/j.cell.2018.01.009
21. Chen X, Song E. Turning foes to friends: targeting cancer-associated fibroblasts. Nat Rev Drug Discov. (2019) 18:99–115. doi: 10.1038/s41573-018-0004-1
22. Giannoni E, Bianchini F, Masieri L, Serni S, Torre E, Calorini L, et al. Reciprocal activation of prostate cancer cells and cancer-associated fibroblasts stimulates epithelial-mesenchymal transition and cancer stemness. Cancer Res. (2010) 70:6945–56. doi: 10.1158/0008-5472.CAN-10-0785
23. Ohlund D, Elyada E, Tuveson D. Fibroblast heterogeneity in the cancer wound. J Exp Med. (2014) 211:1503–23. doi: 10.1084/jem.20140692
24. Molodtsov A, Turk MJ. Tissue resident CD8 memory T cell responses in cancer and autoimmunity. Front Immunol. (2018) 9:2810. doi: 10.3389/fimmu.2018.02810
25. Gentles AJ, Newman AM, Liu CL, Bratman SV, Feng W, Kim D, et al. The prognostic landscape of genes and infiltrating immune cells across human cancers. Nat Med. (2015) 21:938–45. doi: 10.1038/nm.3909
26. Speiser DE, Ho PC, Verdeil G. Regulatory circuits of T cell function in cancer. Nat Rev Immunol. (2016) 16:599–611. doi: 10.1038/nri.2016.80
27. Zhao S, Xu W, Jiang W, Yu W, Lin Y, Zhang T, et al. Regulation of cellular metabolism by protein lysine acetylation. Science. (2010) 327:1000–4. doi: 10.1126/science.1179689
28. Gooden MJ, De Bock GH, Leffers N, Daemen T, Nijman HW. The prognostic influence of tumour-infiltrating lymphocytes in cancer: a systematic review with meta-analysis. Br J Cancer. (2011) 105:93–103. doi: 10.1038/bjc.2011.189
29. Klebanoff CA, Gattinoni L, Torabi-Parizi P, Kerstann K, Cardones AR, Finkelstein SE, et al. Central memory self/tumor-reactive CD8+ T cells confer superior antitumor immunity compared with effector memory T cells. Proc Natl Acad Sci USA. (2005) 102:9571–6. doi: 10.1073/pnas.0503726102
30. Whiteside TL. Disarming suppressor cells to improve immunotherapy. Cancer Immunol Immunother. (2012) 61:283–8. doi: 10.1007/s00262-011-1171-7
31. Wherry EJ, Kurachi M. Molecular and cellular insights into T cell exhaustion. Nat Rev Immunol. (2015) 15:486–99. doi: 10.1038/nri3862
32. Sakaguchi S, Yamaguchi T, Nomura T, Ono M. Regulatory T cells and immune tolerance. Cell. (2008) 133:775–87. doi: 10.1016/j.cell.2008.05.009
33. Hsieh CS, Lee HM, Lio CW. Selection of regulatory T cells in the thymus. Nat Rev Immunol. (2012) 12:157–67. doi: 10.1038/nri3155
34. Newton R, Priyadharshini B, Turka LA. Immunometabolism of regulatory T cells. Nat Immunol. (2016) 17:618–25. doi: 10.1038/ni.3466
35. Shang B, Liu Y, Jiang SJ, Liu Y. Prognostic value of tumor-infiltrating FoxP3+ regulatory T cells in cancers: a systematic review and meta-analysis. Sci Rep. (2015) 5:15179. doi: 10.1038/srep15179
36. Huang L, Xu H, Peng G. TLR-mediated metabolic reprogramming in the tumor microenvironment: potential novel strategies for cancer immunotherapy. Cell Mol Immunol. (2018) 15:428–37. doi: 10.1038/cmi.2018.4
37. Von Boehmer H, Daniel C. Therapeutic opportunities for manipulating T(Reg) cells in autoimmunity and cancer. Nat Rev Drug Discov. (2013) 12:51–63. doi: 10.1038/nrd3683
38. Sica A, Larghi P, Mancino A, Rubino L, Porta C, Totaro MG, et al. Macrophage polarization in tumour progression. Semin Cancer Biol. (2008) 18:349–55. doi: 10.1016/j.semcancer.2008.03.004
39. Qian BZ, Pollard JW. Macrophage diversity enhances tumor progression and metastasis. Cell. (2010) 141:39–51. doi: 10.1016/j.cell.2010.03.014
40. Biswas SK, Mantovani A. Macrophage plasticity and interaction with lymphocyte subsets: cancer as a paradigm. Nat Immunol. (2010) 11:889–96. doi: 10.1038/ni.1937
41. Escribese MM, Casas M, Corbi AL. Influence of low oxygen tensions on macrophage polarization. Immunobiology. (2012) 217:1233–40. doi: 10.1016/j.imbio.2012.07.002
42. Comito G, Giannoni E, Segura CP, Barcellos-De-Souza P, Raspollini MR, Baroni G, et al. Cancer-associated fibroblasts and M2-polarized macrophages synergize during prostate carcinoma progression. Oncogene. (2014) 33:2423–31. doi: 10.1038/onc.2013.191
43. Condeelis J, Pollard JW. Macrophages: obligate partners for tumor cell migration, invasion, and metastasis. Cell. (2006) 124:263–6. doi: 10.1016/j.cell.2006.01.007
44. Goswami S, Sahai E, Wyckoff JB, Cammer M, Cox D, Pixley FJ, et al. Macrophages promote the invasion of breast carcinoma cells via a colony-stimulating factor-1/epidermal growth factor paracrine loop. Cancer Res. (2005) 65:5278–83. doi: 10.1158/0008-5472.CAN-04-1853
45. Cassetta L, Pollard JW. Targeting macrophages: therapeutic approaches in cancer. Nat Rev Drug Discov. (2018) 17:887–904. doi: 10.1038/nrd.2018.169
46. Motz GT, Coukos G. Deciphering and reversing tumor immune suppression. Immunity. (2013) 39:61–73. doi: 10.1016/j.immuni.2013.07.005
47. Talmadge JE, Gabrilovich DI. History of myeloid-derived suppressor cells. Nat Rev Cancer. (2013) 13:739–52. doi: 10.1038/nrc3581
48. Gabrilovich DI, Ostrand-Rosenberg S, Bronte V. Coordinated regulation of myeloid cells by tumours. Nat Rev Immunol. (2012) 12:253–68. doi: 10.1038/nri3175
49. Foudi A, Jarrier P, Zhang Y, Wittner M, Geay JF, Lecluse Y, et al. Reduced retention of radioprotective hematopoietic cells within the bone marrow microenvironment in CXCR4-/- chimeric mice. Blood. (2006) 107:2243–51. doi: 10.1182/blood-2005-02-0581
50. Safari E, Ghorghanlu S, Ahmadi-Khiavi H, Mehranfar S, Rezaei R, Motallebnezhad M. Myeloid-derived suppressor cells and tumor: current knowledge and future perspectives. J Cell Physiol. (2018) 234:9966–81. doi: 10.1002/jcp.27923
51. Wang Y, Ding Y, Guo N, Wang S. MDSCs: key criminals of tumor pre-metastatic niche formation. Front Immunol. (2019) 10:172. doi: 10.3389/fimmu.2019.00172
52. Peled A, Abraham M, Avivi I, Rowe JM, Beider K, Wald H, et al. The high-affinity CXCR4 antagonist BKT140 is safe and induces a robust mobilization of human CD34+ cells in patients with multiple myeloma. Clin Cancer Res. (2014) 20:469–79. doi: 10.1158/1078-0432.CCR-13-1302
53. Serafini P, Borrello I, Bronte V. Myeloid suppressor cells in cancer: recruitment, phenotype, properties, and mechanisms of immune suppression. Semin Cancer Biol. (2006) 16:53–65. doi: 10.1016/j.semcancer.2005.07.005
54. Fridlender ZG, Sun J, Kim S, Kapoor V, Cheng G, Ling L, et al. Polarization of tumor-associated neutrophil phenotype by TGF-beta: “N1” versus “N2” TAN. Cancer Cell. (2009) 16:183–94. doi: 10.1016/j.ccr.2009.06.017
55. Gregory AD, Houghton AM. Tumor-associated neutrophils: new targets for cancer therapy. Cancer Res. (2011) 71:2411–6. doi: 10.1158/0008-5472.CAN-10-2583
56. Hurt B, Schulick R, Edil B, El Kasmi KC, Barnett C Jr. Cancer-promoting mechanisms of tumor-associated neutrophils. Am J Surg. (2017) 214:938–44. doi: 10.1016/j.amjsurg.2017.08.003
57. Semenza GL. HIF-1 mediates metabolic responses to intratumoral hypoxia and oncogenic mutations. J Clin Invest. (2013) 123:3664–71. doi: 10.1172/JCI67230
58. Sormendi S, Wielockx B. Hypoxia pathway proteins as central mediators of metabolism in the tumor cells and their microenvironment. Front Immunol. (2018) 9:40. doi: 10.3389/fimmu.2018.00040
59. Pavlova NN, Thompson CB. The emerging hallmarks of cancer metabolism. Cell Metab. (2016) 23:27–47. doi: 10.1016/j.cmet.2015.12.006
60. Gottfried E, Kunz-Schughart LA, Ebner S, Mueller-Klieser W, Hoves S, Andreesen R, et al. Tumor-derived lactic acid modulates dendritic cell activation and antigen expression. Blood. (2006) 107:2013–21. doi: 10.1182/blood-2005-05-1795
61. Fischer K, Hoffmann P, Voelkl S, Meidenbauer N, Ammer J, Edinger M, et al. Inhibitory effect of tumor cell-derived lactic acid on human T cells. Blood. (2007) 109:3812–9. doi: 10.1182/blood-2006-07-035972
62. Goetze K, Walenta S, Ksiazkiewicz M, Kunz-Schughart LA, Mueller-Klieser W. Lactate enhances motility of tumor cells and inhibits monocyte migration and cytokine release. Int J Oncol. (2011) 39:453–63. doi: 10.3892/ijo.2011.1055
63. Colegio OR, Chu NQ, Szabo AL, Chu T, Rhebergen AM, Jairam V, et al. Functional polarization of tumour-associated macrophages by tumour-derived lactic acid. Nature. (2014) 513:559–63. doi: 10.1038/nature13490
64. Majmundar AJ, Wong WJ, Simon MC. Hypoxia-inducible factors and the response to hypoxic stress. Mol Cell. (2010) 40:294–309. doi: 10.1016/j.molcel.2010.09.022
65. Belaiba RS, Bonello S, Zahringer C, Schmidt S, Hess J, Kietzmann T, et al. Hypoxia up-regulates hypoxia-inducible factor-1alpha transcription by involving phosphatidylinositol 3-kinase and nuclear factor kappaB in pulmonary artery smooth muscle cells. Mol Biol Cell. (2007) 18:4691–7. doi: 10.1091/mbc.e07-04-0391
66. Porporato PE, Payen VL, De Saedeleer CJ, Preat V, Thissen JP, Feron O, et al. Lactate stimulates angiogenesis and accelerates the healing of superficial and ischemic wounds in mice. Angiogenesis. (2012) 15:581–92. doi: 10.1007/s10456-012-9282-0
67. Kato Y, Ozawa S, Miyamoto C, Maehata Y, Suzuki A, Maeda T, et al. Acidic extracellular microenvironment and cancer. Cancer Cell Int. (2013) 13:89. doi: 10.1186/1475-2867-13-89
68. Swietach P, Hulikova A, Vaughan-Jones RD, Harris AL. New insights into the physiological role of carbonic anhydrase IX in tumour pH regulation. Oncogene. (2010) 29:6509–21. doi: 10.1038/onc.2010.455
69. Chiche J, Brahimi-Horn MC, Pouyssegur J. Tumour hypoxia induces a metabolic shift causing acidosis: a common feature in cancer. J Cell Mol Med. (2010) 14:771–94. doi: 10.1111/j.1582-4934.2009.00994.x
70. Pavlides S, Whitaker-Menezes D, Castello-Cros R, Flomenberg N, Witkiewicz AK, Frank PG, et al. The reverse Warburg effect: aerobic glycolysis in cancer associated fibroblasts and the tumor stroma. Cell Cycle. (2009) 8:3984–4001. doi: 10.4161/cc.8.23.10238
71. Gladden LB. Lactate metabolism: a new paradigm for the third millennium. J Physiol. (2004) 558:5–30. doi: 10.1113/jphysiol.2003.058701
72. Jia D, Park JH, Jung KH, Levine H, Kaipparettu BA. Elucidating the metabolic plasticity of cancer: mitochondrial reprogramming and hybrid metabolic states. Cells. (2018) 7:21. doi: 10.3390/cells7030021
73. Fu Y, Liu S, Yin S, Niu W, Xiong W, Tan M, et al. The reverse Warburg effect is likely to be an Achilles' heel of cancer that can be exploited for cancer therapy. Oncotarget. (2017) 8:57813–25. doi: 10.18632/oncotarget.18175
74. Fiaschi T, Marini A, Giannoni E, Taddei ML, Gandellini P, De Donatis A, et al. Reciprocal metabolic reprogramming through lactate shuttle coordinately influences tumor-stroma interplay. Cancer Res. (2012) 72:5130–40. doi: 10.1158/0008-5472.CAN-12-1949
75. Suh DH, Kim HS, Kim B, Song YS. Metabolic orchestration between cancer cells and tumor microenvironment as a co-evolutionary source of chemoresistance in ovarian cancer: a therapeutic implication. Biochem Pharmacol. (2014) 92:43–54. doi: 10.1016/j.bcp.2014.08.011
76. Penkert J, Ripperger T, Schieck M, Schlegelberger B, Steinemann D, Illig T. On metabolic reprogramming and tumor biology: a comprehensive survey of metabolism in breast cancer. Oncotarget. (2016) 7:67626–49. doi: 10.18632/oncotarget.11759
77. Phan AT, Goldrath AW, Glass CK. Metabolic and epigenetic coordination of T cell and macrophage Immunity. (2017) 46:714–29. doi: 10.1016/j.immuni.2017.04.016
78. Ho PC, Bihuniak JD, Macintyre AN, Staron M, Liu X, Amezquita R, et al. Phosphoenolpyruvate is a metabolic checkpoint of anti-tumor T cell responses. Cell. (2015) 162:1217–28. doi: 10.1016/j.cell.2015.08.012
79. Scharping NE, Menk AV, Moreci RS, Whetstone RD, Dadey RE, Watkins SC, et al. The tumor microenvironment represses T cell mitochondrial biogenesis to drive intratumoral T cell metabolic insufficiency and dysfunction. Immunity. (2016) 45:701–3. doi: 10.1016/j.immuni.2016.08.009
80. Beckermann KE, Dudzinski SO, Rathmell JC. Dysfunctional T cell metabolism in the tumor microenvironment. Cytokine Growth Factor Rev. (2017) 35:7–14. doi: 10.1016/j.cytogfr.2017.04.003
81. Di Virgilio F, Sarti AC, Falzoni S, De Marchi E, Adinolfi E. Extracellular ATP and P2 purinergic signalling in the tumour microenvironment. Nat Rev Cancer. (2018) 18:601–18. doi: 10.1038/s41568-018-0037-0
82. Deaglio S, Dwyer KM, Gao W, Friedman D, Usheva A, Erat A, et al. Adenosine generation catalyzed by CD39 and CD73 expressed on regulatory T cells mediates immune suppression. J Exp Med. (2007) 204:1257–65. doi: 10.1084/jem.20062512
83. Vijayan D, Young A, Teng MWL, Smyth MJ. Targeting immunosuppressive adenosine in cancer. Nat Rev Cancer. (2017) 17:765. doi: 10.1038/nrc.2017.86
84. Serra S, Vaisitti T, Audrito V, Bologna C, Buonincontri R, Chen SS, et al. Adenosine signaling mediates hypoxic responses in the chronic lymphocytic leukemia microenvironment. Blood Adv. (2016) 1:47–61. doi: 10.1182/bloodadvances.2016000984
85. Vaupel P, Multhoff G. Accomplices of the hypoxic tumor microenvironment compromising antitumor immunity: adenosine, lactate, acidosis, vascular endothelial growth factor, potassium ions, and phosphatidylserine. Front Immunol. (2017) 8:1887. doi: 10.3389/fimmu.2017.01887
86. Gossmann TI, Ziegler M, Puntervoll P, De Figueiredo LF, Schuster S, Heiland I. NAD(+) biosynthesis and salvage–a phylogenetic perspective. Febs J. (2012) 279:3355–63. doi: 10.1111/j.1742-4658.2012.08559.x
87. Sharif T, Martell E, Dai C, Ghassemi-Rad MS, Kennedy BE, Lee PWK, et al. Regulation of cancer and cancer-related genes via NAD(). Antioxid Redox Signal. (2018) 30:906–23. doi: 10.1089/ars.2017.7478
88. Canto C, Menzies KJ, Auwerx J. NAD(+) Metabolism and the control of energy homeostasis: a balancing act between mitochondria and the nucleus. Cell Metab. (2015) 22:31–53. doi: 10.1016/j.cmet.2015.05.023
89. Nikiforov A, Kulikova V, Ziegler M. The human NAD metabolome: functions, metabolism and compartmentalization. Crit Rev Biochem Mol Biol. (2015) 50:284–97. doi: 10.3109/10409238.2015.1028612
90. Ruggieri S, Orsomando G, Sorci L, Raffaelli N. Regulation of NAD biosynthetic enzymes modulates NAD-sensing processes to shape mammalian cell physiology under varying biological cues. Biochim Biophys Acta. (2015) 1854:1138–49. doi: 10.1016/j.bbapap.2015.02.021
91. Magni G, Orsomando G, Raffelli N, Ruggieri S. Enzymology of mammalian NAD metabolism in health and disease. Front Biosci. (2008) 13:6135–54. doi: 10.2741/3143
92. Houtkooper RH, Canto C, Wanders RJ, Auwerx J. The secret life of NAD+: an old metabolite controlling new metabolic signaling pathways. Endocr Rev. (2010) 31:194–223. doi: 10.1210/er.2009-0026
93. Chiarugi A, Dolle C, Felici R, Ziegler M. The NAD metabolome–a key determinant of cancer cell biology. Nat Rev Cancer. (2012) 12:741–52. doi: 10.1038/nrc3340
94. Haag F, Adriouch S, Brass A, Jung C, Moller S, Scheuplein F, et al. Extracellular NAD and ATP: partners in immune cell modulation. Purinergic Signal. (2007) 3:71–81. doi: 10.1007/s11302-006-9038-7
95. Grahnert A, Klein C, Schilling E, Wehrhahn J, Hauschildt S. Review: NAD +: a modulator of immune functions. Innate Immun. (2011) 17:212–33. doi: 10.1177/1753425910361989
96. Burgos ES, Schramm VL. Weak coupling of ATP hydrolysis to the chemical equilibrium of human nicotinamide phosphoribosyltransferase. Biochemistry. (2008) 47:11086–96. doi: 10.1021/bi801198m
97. Kitani T, Okuno S, Fujisawa H. Growth phase-dependent changes in the subcellular localization of pre-B-cell colony-enhancing factor. FEBS Lett. (2003) 544:74–8. doi: 10.1016/S0014-5793(03)00476-9
98. Revollo JR, Korner A, Mills KF, Satoh A, Wang T, Garten A, et al. Nampt/PBEF/Visfatin regulates insulin secretion in beta cells as a systemic NAD biosynthetic enzyme. Cell Metab. (2007) 6:363–75. doi: 10.1016/j.cmet.2007.09.003
99. Yang H, Yang T, Baur JA, Perez E, Matsui T, Carmona JJ, et al. Nutrient-sensitive mitochondrial NAD+ levels dictate cell survival. Cell. (2007) 130:1095–107. doi: 10.1016/j.cell.2007.07.035
100. Pittelli M, Formentini L, Faraco G, Lapucci A, Rapizzi E, Cialdai F, et al. Inhibition of nicotinamide phosphoribosyltransferase: cellular bioenergetics reveals a mitochondrial insensitive NAD pool. J Biol Chem. (2010) 285:34106–14. doi: 10.1074/jbc.M110.136739
101. Davila A, Liu L, Chellappa K, Redpath P, Nakamaru-Ogiso E, Paolella LM, et al. Nicotinamide adenine dinucleotide is transported into mammalian mitochondria. Elife. (2018) 7. doi: 10.7554/eLife.33246
102. Kennedy BE, Sharif T, Martell E, Dai C, Kim Y, Lee PW, et al. NAD(+) salvage pathway in cancer metabolism and therapy. Pharmacol Res. (2016) 114:274–83. doi: 10.1016/j.phrs.2016.10.027
103. Garten A, Schuster S, Penke M, Gorski T, De Giorgis T, Kiess W. Physiological and pathophysiological roles of NAMPT and NAD metabolism. Nat Rev Endocrinol. (2015) 11:535–46. doi: 10.1038/nrendo.2015.117
104. Tanaka M, Nozaki M, Fukuhara A, Segawa K, Aoki N, Matsuda M, et al. Visfatin is released from 3T3-L1 adipocytes via a non-classical pathway. Biochem Biophys Res Commun. (2007) 359:194–201. doi: 10.1016/j.bbrc.2007.05.096
105. Camp SM, Ceco E, Evenoski CL, Danilov SM, Zhou T, Chiang ET, et al. Unique toll-like receptor 4 activation by NAMPT/PBEF induces NFkappaB signaling and inflammatory lung injury. Sci Rep. (2015) 5:13135. doi: 10.1038/srep13135
106. Hara N, Yamada K, Shibata T, Osago H, Tsuchiya M. Nicotinamide phosphoribosyltransferase/visfatin does not catalyze nicotinamide mononucleotide formation in blood plasma. PLoS ONE. (2011) 6:e22781. doi: 10.1371/journal.pone.0022781
107. Samal B, Sun Y, Stearns G, Xie C, Suggs S, Mcniece I. Cloning and characterization of the cDNA encoding a novel human pre-B-cell colony-enhancing factor. Mol Cell Biol. (1994) 14:1431–7. doi: 10.1128/MCB.14.2.1431
108. Fukuhara A, Matsuda M, Nishizawa M, Segawa K, Tanaka M, Kishimoto K, et al. Visfatin: a protein secreted by visceral fat that mimics the effects of insulin. Science. (2005) 307:426–30. doi: 10.1126/science.1097243
109. Sun Z, Lei H, Zhang Z. Pre-B cell colony enhancing factor. (PBEF), a cytokine with multiple physiological functions. Cytokine Growth Factor Rev. (2013) 24:433–42. doi: 10.1016/j.cytogfr.2013.05.006
110. Ognjanovic S, Bao S, Yamamoto SY, Garibay-Tupas J, Samal B, Bryant-Greenwood GD. Genomic organization of the gene coding for human pre-B-cell colony enhancing factor and expression in human fetal membranes. J Mol Endocrinol. (2001) 26:107–17. doi: 10.1677/jme.0.0260107
111. Iqbal J, Zaidi M. TNF regulates cellular NAD+ metabolism in primary macrophages. Biochem Biophys Res Commun. (2006) 342:1312–8. doi: 10.1016/j.bbrc.2006.02.109
112. Nowell MA, Richards PJ, Fielding CA, Ognjanovic S, Topley N, Williams AS, et al. Regulation of pre-B cell colony-enhancing factor by STAT-3-dependent interleukin-6 trans-signaling: implications in the pathogenesis of rheumatoid arthritis. Arthritis Rheum. (2006) 54:2084–95. doi: 10.1002/art.21942
113. Gosset M, Berenbaum F, Salvat C, Sautet A, Pigenet A, Tahiri K, et al. Crucial role of visfatin/pre-B cell colony-enhancing factor in matrix degradation and prostaglandin E2 synthesis in chondrocytes: possible influence on osteoarthritis. Arthritis Rheum. (2008) 58:1399–409. doi: 10.1002/art.23431
114. Moschen AR, Kaser A, Enrich B, Mosheimer B, Theurl M, Niederegger H, et al. Visfatin, an adipocytokine with proinflammatory and immunomodulating properties. J Immunol. (2007) 178:1748–58. doi: 10.4049/jimmunol.178.3.1748
115. Fan Y, Meng S, Wang Y, Cao J, Wang C. Visfatin/PBEF/Nampt induces EMMPRIN and MMP-9 production in macrophages via the NAMPT-MAPK. (p38, ERK1/2)-NF-kappaB signaling pathway. Int J Mol Med. (2011) 27:607–15. doi: 10.3892/ijmm.2011.621
116. Murray PJ, Allen JE, Biswas SK, Fisher EA, Gilroy DW, Goerdt S, et al. Macrophage activation and polarization: nomenclature and experimental guidelines. Immunity. (2014) 41:14–20. doi: 10.1016/j.immuni.2014.06.008
117. Diskin C, Palsson-Mcdermott EM. Metabolic modulation in macrophage effector function. Front Immunol. (2018) 9:270. doi: 10.3389/fimmu.2018.00270
118. Stunault MI, Bories G, Guinamard RR, Ivanov S. Metabolism plays a key role during macrophage activation. Mediators Inflamm. (2018) 2018:2426138. doi: 10.1155/2018/2426138
119. Suzuki H, Hisamatsu T, Chiba S, Mori K, Kitazume MT, Shimamura K, et al. Glycolytic pathway affects differentiation of human monocytes to regulatory macrophages. Immunol Lett. (2016) 176:18–27. doi: 10.1016/j.imlet.2016.05.009
120. Kelly B, O'neill LA. Metabolic reprogramming in macrophages and dendritic cells in innate immunity. Cell Res. (2015) 25:771–84. doi: 10.1038/cr.2015.68
121. Van Den Bossche J, Baardman J, De Winther MP. Metabolic characterization of polarized M1 and M2 bone marrow-derived macrophages using real-time extracellular flux analysis. J Vis Exp. (2015) 53424. doi: 10.3791/53424
122. Hamers AJ, Pillai AB. A sweet alternative: maintaining M2 macrophage polarization. Sci Immunol. (2018) 3:eaav7759. doi: 10.1126/sciimmunol.aav7759
123. Jha AK, Huang SC, Sergushichev A, Lampropoulou V, Ivanova Y, Loginicheva E, et al. Network integration of parallel metabolic and transcriptional data reveals metabolic modules that regulate macrophage polarization. Immunity. (2015) 42:419–30. doi: 10.1016/j.immuni.2015.02.005
124. Xu Q, Choksi S, Qu J, Jang J, Choe M, Banfi B, et al. NADPH oxidases are essential for macrophage differentiation. J Biol Chem. (2016) 291:20030–41. doi: 10.1074/jbc.M116.731216
125. Huang LE, Arany Z, Livingston DM, Bunn HF. Activation of hypoxia-inducible transcription factor depends primarily upon redox-sensitive stabilization of its alpha subunit. J Biol Chem. (1996) 271:32253–9. doi: 10.1074/jbc.271.50.32253
126. Kim JW, Tchernyshyov I, Semenza GL, Dang CV. HIF-1-mediated expression of pyruvate dehydrogenase kinase: a metabolic switch required for cellular adaptation to hypoxia. Cell Metab. (2006) 3:177–85. doi: 10.1016/j.cmet.2006.02.002
127. Rius J, Guma M, Schachtrup C, Akassoglou K, Zinkernagel AS, Nizet V, et al. NF-kappaB links innate immunity to the hypoxic response through transcriptional regulation of HIF-1alpha. Nature. (2008) 453:807–11. doi: 10.1038/nature06905
128. Vats D, Mukundan L, Odegaard JI, Zhang L, Smith KL, Morel CR, et al. Oxidative metabolism and PGC-1beta attenuate macrophage-mediated inflammation. Cell Metab. (2006) 4:13–24. doi: 10.1016/j.cmet.2006.05.011
129. Skokowa J, Lan D, Thakur BK, Wang F, Gupta K, Cario G, et al. NAMPT is essential for the G-CSF-induced myeloid differentiation via a NAD(+)-sirtuin-1-dependent pathway. Nat Med. (2009) 15:151–8. doi: 10.1038/nm.1913
130. Audrito V, Serra S, Brusa D, Mazzola F, Arruga F, Vaisitti T, et al. Extracellular nicotinamide phosphoribosyltransferase. (NAMPT) promotes M2 macrophage polarization in chronic lymphocytic leukemia. Blood. (2015) 125:111–23. doi: 10.1182/blood-2014-07-589069
131. Travelli C, Colombo G, Mola S, Genazzani AA, Porta C. NAMPT: a pleiotropic modulator of monocytes and macrophages. Pharmacol Res. (2018) 135:25–36. doi: 10.1016/j.phrs.2018.06.022
132. Travelli C, Consonni FM, Sangaletti S, Storto M, Morlacchi S, Grolla AA, et al. Nicotinamide phosphoribosyltransferase. (NAMPT) acts as a metabolic gate for mobilization of myeloid-derived suppressor cells. Cancer Res. (2019) 79:1938–51 doi: 10.1158/0008-5472.CAN-18-1544
133. Fernandes CA, Fievez L, Neyrinck AM, Delzenne NM, Bureau F, Vanbever R. Sirtuin inhibition attenuates the production of inflammatory cytokines in lipopolysaccharide-stimulated macrophages. Biochem Biophys Res Commun. (2012) 420:857–61. doi: 10.1016/j.bbrc.2012.03.088
134. Liu TF, Vachharajani VT, Yoza BK, Mccall CE. NAD+-dependent sirtuin 1 and 6 proteins coordinate a switch from glucose to fatty acid oxidation during the acute inflammatory response. J Biol Chem. (2012) 287:25758–69. doi: 10.1074/jbc.M112.362343
135. Weidemann A, Johnson RS. Biology of HIF-1alpha. Cell Death Differ. (2008) 15:621–7. doi: 10.1038/cdd.2008.12
136. Gleyzer N, Scarpulla RC. PGC-1-related coactivator. (PRC), a sensor of metabolic stress, orchestrates a redox-sensitive program of inflammatory gene expression. J Biol Chem. (2011) 286:39715–25. doi: 10.1074/jbc.M111.291575
137. Li Y, Zhang Y, Dorweiler B, Cui D, Wang T, Woo CW, et al. Extracellular Nampt promotes macrophage survival via a nonenzymatic interleukin-6/STAT3 signaling mechanism. J Biol Chem. (2008) 283:34833–43. doi: 10.1074/jbc.M805866200
138. Koch C, Samareh B, Morishima T, Mir P, Kanz L, Zeidler C, et al. GM-CSF treatment is not effective in congenital neutropenia patients due to its inability to activate NAMPT signaling. Ann Hematol. (2017) 96:345–53. doi: 10.1007/s00277-016-2894-5
139. Galli U, Travelli C, Massarotti A, Fakhfouri G, Rahimian R, Tron GC, et al. Medicinal chemistry of nicotinamide phosphoribosyltransferase. (NAMPT) inhibitors. J Med Chem. (2013) 56:6279–96. doi: 10.1021/jm4001049
140. Watson M, Roulston A, Belec L, Billot X, Marcellus R, Bedard D, et al. The small molecule GMX1778 is a potent inhibitor of NAD+ biosynthesis: strategy for enhanced therapy in nicotinic acid phosphoribosyltransferase 1-deficient tumors. Mol Cell Biol. (2009) 29:5872–88. doi: 10.1128/MCB.00112-09
141. Dalamaga M, Christodoulatos GS, Mantzoros CS. The role of extracellular and intracellular Nicotinamide phosphoribosyl-transferase in cancer: diagnostic and therapeutic perspectives and challenges. Metabolism. (2018) 82:72–87. doi: 10.1016/j.metabol.2018.01.001
142. Abu Aboud O, Chen CH, Senapedis W, Baloglu E, Argueta C, Weiss RH. Dual and specific inhibition of NAMPT and PAK4 By KPT-9274 decreases kidney cancer growth. Mol Cancer Ther. (2016) 15:2119–29. doi: 10.1158/1535-7163.MCT-16-0197
143. Gibson AE, Mendoza A, Korotchkina L, Chernova O, Heste CM. Targeting nicotinamide phosphoribosyltransferase (NAMPT) with OT-82 in Ewing sarcoma [abstract]. In: Proceedings of the American Association for Cancer Research Annual Meeting 2018; 2018 Apr 14-18. Chicago, IL; Philadelphia (PA): AACR. Cancer Res. (2018) 78(13 Suppl.):5477. doi: 10.1158/1538-7445.AM2018-5477
144. Pillai VB, Sundaresan NR, Kim G, Samant S, Moreno-Vinasco L, Garcia JG, et al. Nampt secreted from cardiomyocytes promotes development of cardiac hypertrophy and adverse ventricular remodeling. Am J Physiol Heart Circ Physiol. (2013) 304:H415–26. doi: 10.1152/ajpheart.00468.2012
145. Krejcik J, Casneuf T, Nijhof IS, Verbist B, Bald J, Plesner T, et al. Daratumumab depletes CD38+ immune regulatory cells, promotes T-cell expansion, and skews T-cell repertoire in multiple myeloma. Blood. (2016) 128:384–94. doi: 10.1182/blood-2015-12-687749
146. Martin T, Baz R, Benson DM, Lendvai N, Wolf J, Munster P, et al. A phase 1b study of isatuximab plus lenalidomide and dexamethasone for relapsed/refractory multiple myeloma. Blood. (2017) 129:3294–303. doi: 10.1182/blood-2016-09-740787
147. Boxhammer R, Weirather J, Steidl S, Endell J. MOR202, a human anti-CD38 monoclonal antibody, mediates potent tumoricidal activity in vivo and shows synergistic efficacy in combination with different antineoplastic compounds. Blood. (2015) 126:3015.
148. Escande C, Nin V, Price NL, Capellini V, Gomes AP, Barbosa MT, et al. Flavonoid apigenin is an inhibitor of the NAD+ase CD38: implications for cellular NAD+ metabolism, protein acetylation, and treatment of metabolic syndrome. Diabetes. (2013) 62:1084–93. doi: 10.2337/db12-1139
149. Heltweg B, Gatbonton T, Schuler AD, Posakony J, Li H, Goehle S, et al. Antitumor activity of a small-molecule inhibitor of human silent information regulator 2 enzymes. Cancer Res. (2006) 66:4368–77. doi: 10.1158/0008-5472.CAN-05-3617
150. Wang J, Kim TH, Ahn MY, Lee J, Jung JH, Choi WS, et al. Sirtinol, a class III HDAC inhibitor, induces apoptotic and autophagic cell death in MCF-7 human breast cancer cells. Int J Oncol. (2012) 41:1101–9. doi: 10.3892/ijo.2012.1534
151. Lara E, Mai A, Calvanese V, Altucci L, Lopez-Nieva P, Martinez-Chantar ML, et al. Salermide, a Sirtuin inhibitor with a strong cancer-specific proapoptotic effect. Oncogene. (2009) 28:781–91. doi: 10.1038/onc.2008.436
152. Lain S, Hollick JJ, Campbell J, Staples OD, Higgins M, Aoubala M, et al. Discovery, in vivo activity, and mechanism of action of a small-molecule p53 activator. Cancer Cell. (2008) 13:454–63. doi: 10.1016/j.ccr.2008.03.004
153. Peck B, Chen CY, Ho KK, Di Fruscia P, Myatt SS, Coombes RC, et al. SIRT inhibitors induce cell death and p53 acetylation through targeting both SIRT1 and SIRT2. Mol Cancer Ther. (2010) 9:844–55. doi: 10.1158/1535-7163.MCT-09-0971
154. Avalos JL, Bever KM, Wolberger C. Mechanism of sirtuin inhibition by nicotinamide: altering the NAD(+) cosubstrate specificity of a Sir2 enzyme. Mol Cell. (2005) 17:855–68. doi: 10.1016/j.molcel.2005.02.022
155. Wainwright DA, Chang AL, Dey M, Balyasnikova IV, Kim CK, Tobias A, et al. Durable therapeutic efficacy utilizing combinatorial blockade against IDO, CTLA-4, and PD-L1 in mice with brain tumors. Clin Cancer Res. (2014) 20:5290–301. doi: 10.1158/1078-0432.CCR-14-0514
156. Liu X, Shin N, Koblish HK, Yang G, Wang Q, Wang K, et al. Selective inhibition of IDO1 effectively regulates mediators of antitumor immunity. Blood. (2010) 115:3520–30. doi: 10.1182/blood-2009-09-246124
157. Prendergast GC, Malachowski WJ, Mondal A, Scherle P, Muller AJ. Indoleamine 2,3-Dioxygenase and its therapeutic inhibition in cancer. Int Rev Cell Mol Biol. (2018) 336:175–203. doi: 10.1016/bs.ircmb.2017.07.004
158. Tabernero J, Luke JJ, Joshua AM, Varga AI, Moreno V, Desai J. BMS-986205, an indoleamine 2,3-dioxygenase 1 inhibitor (IDO1i), in combination with nivolumab (NIVO): Updated safety across all tumor cohorts and efficacy in pts with advanced bladder cancer (advBC). J Clin Oncol. (2018) 36(15_suppl):4512. doi: 10.1200/JCO.2018.36.15_suppl.4512
159. Hasmann M, Schemainda I. FK866, a highly specific noncompetitive inhibitor of nicotinamide phosphoribosyltransferase, represents a novel mechanism for induction of tumor cell apoptosis. Cancer Res. (2003) 63:7436–42.
160. Olesen UH, Christensen MK, Bjorkling F, Jaattela M, Jensen PB, Sehested M, et al. Anticancer agent CHS-828 inhibits cellular synthesis of NAD. Biochem Biophys Res Commun. (2008) 367:799–804. doi: 10.1016/j.bbrc.2008.01.019
161. Audrito V, Manago A, Gaudino F, Deaglio S. Targeting metabolic reprogramming in metastatic melanoma: the key role of nicotinamide phosphoribosyltransferase. (NAMPT). Semin Cell Dev Biol. (2019). doi: 10.1016/j.semcdb.2019.05.001. [Epub ahead of print].
162. Howard M, Grimaldi JC, Bazan JF, Lund FE, Santos-Argumedo L, Parkhouse RM, et al. Formation and hydrolysis of cyclic ADP-ribose catalyzed by lymphocyte antigen CD38. Science. (1993) 262:1056–9. doi: 10.1126/science.8235624
163. Sauve AA, Munshi C, Lee HC, Schramm VL. The reaction mechanism for CD38. A single intermediate is responsible for cyclization, hydrolysis, and base-exchange chemistries. Biochemistry. (1998) 37:13239–49. doi: 10.1021/bi981248s
164. Chini EN, Chini CCS, Espindola Netto JM, De Oliveira GC, Van Schooten W. The pharmacology of CD38/NADase: an emerging target in cancer and diseases of aging. Trends Pharmacol Sci. (2018) 39:424–36. doi: 10.1016/j.tips.2018.02.001
165. Malavasi F, Deaglio S, Funaro A, Ferrero E, Horenstein AL, Ortolan E, et al. Evolution and function of the ADP ribosyl cyclase/CD38 gene family in physiology and pathology. Physiol Rev. (2008) 88:841–86. doi: 10.1152/physrev.00035.2007
166. Chini EN. CD38 as a regulator of cellular NAD: a novel potential pharmacological target for metabolic conditions. Curr Pharm Des. (2009) 15:57–63. doi: 10.2174/138161209787185788
167. Hartman WR, Pelleymounter LL, Moon I, Kalari K, Liu M, Wu TY, et al. CD38 expression, function, and gene resequencing in a human lymphoblastoid cell line-based model system. Leuk Lymphoma. (2010) 51:1315–25. doi: 10.3109/10428194.2010.483299
168. Aksoy P, White TA, Thompson M, Chini EN. Regulation of intracellular levels of NAD: a novel role for CD38. Biochem Biophys Res Commun. (2006) 345:1386–92. doi: 10.1016/j.bbrc.2006.05.042
169. Young GS, Choleris E, Lund FE, Kirkland JB. Decreased cADPR and increased NAD+ in the Cd38-/- mouse. Biochem Biophys Res Commun. (2006) 346:188–92. doi: 10.1016/j.bbrc.2006.05.100
170. Grozio A, Sociali G, Sturla L, Caffa I, Soncini D, Salis A, et al. CD73 protein as a source of extracellular precursors for sustained NAD+ biosynthesis in FK866-treated tumor cells. J Biol Chem. (2013) 288:25938–49. doi: 10.1074/jbc.M113.470435
171. Camacho-Pereira J, Tarrago MG, Chini CCS, Nin V, Escande C, Warner GM, et al. CD38 dictates age-related NAD decline and mitochondrial dysfunction through an SIRT3-dependent mechanism. Cell Metab. (2016) 23:1127–39. doi: 10.1016/j.cmet.2016.05.006
172. Malavasi F, Deaglio S, Damle R, Cutrona G, Ferrarini M, Chiorazzi N. CD38 and chronic lymphocytic leukemia: a decade later. Blood. (2011) 118:3470–8. doi: 10.1182/blood-2011-06-275610
173. Chatterjee S, Daenthanasanmak A, Chakraborty P, Wyatt MW, Dhar P, Selvam SP, et al. CD38-NAD(+)axis regulates immunotherapeutic anti-tumor T cell response. Cell Metab. (2018) 27:85–100.e108. doi: 10.1016/j.cmet.2017.10.006
174. Kaelin WG Jr., Mcknight SL. Influence of metabolism on epigenetics and disease. Cell. (2013) 153:56–69. doi: 10.1016/j.cell.2013.03.004
175. Dugnani E, Pasquale V, Bordignon C, Canu A, Piemonti L, Monti P. Integrating T cell metabolism in cancer immunotherapy. Cancer Lett. (2017) 411:12–8. doi: 10.1016/j.canlet.2017.09.039
176. Buck MD, O'sullivan D, Pearce EL. T cell metabolism drives immunity. J Exp Med. (2015) 212:1345–60. doi: 10.1084/jem.20151159
177. Williams MA, Bevan MJ. Effector and memory CTL differentiation. Annu Rev Immunol. (2007) 25:171–92. doi: 10.1146/annurev.immunol.25.022106.141548
178. Sinclair LV, Rolf J, Emslie E, Shi YB, Taylor PM, Cantrell DA. Control of amino-acid transport by antigen receptors coordinates the metabolic reprogramming essential for T cell differentiation. Nat Immunol. (2013) 14:500–8. doi: 10.1038/ni.2556
179. Chang CH, Qiu J, O'sullivan D, Buck MD, Noguchi T, Curtis JD, et al. Metabolic competition in the tumor microenvironment is a driver of cancer progression. Cell. (2015) 162:1229–41. doi: 10.1016/j.cell.2015.08.016
180. Sukumar M, Roychoudhuri R, Restifo NP. Nutrient competition: a new axis of tumor immunosuppression. Cell. (2015) 162:1206–8. doi: 10.1016/j.cell.2015.08.064
181. Chang CH, Pearce EL. Emerging concepts of T cell metabolism as a target of immunotherapy. Nat Immunol. (2016) 17:364–8. doi: 10.1038/ni.3415
182. Pearce EL, Poffenberger MC, Chang CH, Jones RG. Fueling immunity: insights into metabolism and lymphocyte function. Science. (2013) 342:1242454. doi: 10.1126/science.1242454
183. Feske S. Calcium signalling in lymphocyte activation and disease. Nat Rev Immunol. (2007) 7:690–702. doi: 10.1038/nri2152
185. Ouyang K, Leandro Gomez-Amaro R, Stachura DL, Tang H, Peng X, Fang X, et al. Loss of IP3R-dependent Ca2+ signalling in thymocytes leads to aberrant development and acute lymphoblastic leukemia. Nat Commun. (2014) 5:4814. doi: 10.1038/ncomms5814
186. Cui C, Merritt R, Fu L, Pan Z. Targeting calcium signaling in cancer therapy. Acta Pharm Sin B. (2017) 7:3–17. doi: 10.1016/j.apsb.2016.11.001
187. Zhang T, Kraus WL. SIRT1-dependent regulation of chromatin and transcription: linking NAD(+) metabolism and signaling to the control of cellular functions. Biochim Biophys Acta. (2010) 1804:1666–75. doi: 10.1016/j.bbapap.2009.10.022
188. Jeng MY, Hull PA, Fei M, Kwon HS, Tsou CL, Kasler H, et al. Metabolic reprogramming of human CD8(+) memory T cells through loss of SIRT1. J Exp Med. (2018) 215:51–62. doi: 10.1084/jem.20161066
189. Feng X, Zhang L, Acharya C, An G, Wen K, Qiu L, et al. Targeting CD38 suppresses induction and function of T regulatory cells to mitigate immunosuppression in multiple myeloma. Clin Cancer Res. (2017) 23:4290–300. doi: 10.1158/1078-0432.CCR-16-3192
190. Sica A, Strauss L, Consonni FM, Travelli C, Genazzani A, Porta C. Metabolic regulation of suppressive myeloid cells in cancer. Cytokine Growth Factor Rev. (2017) 35:27–35. doi: 10.1016/j.cytogfr.2017.05.002
191. Franssen LE, Mutis T, Lokhorst HM, Van De Donk N. Immunotherapy in myeloma: how far have we come? Ther Adv Hematol. (2019) 10:2040620718822660. doi: 10.1177/2040620718822660
192. Naik J, Themeli M, De Jong-Korlaar R, Ruiter RWJ, Poddighe PJ, Yuan H, et al. CD38 as a therapeutic target for adult acute myeloid leukemia and T-cell acute lymphoblastic leukemia. Haematologica. (2019) 104:e100–103. doi: 10.3324/haematol.2018.192757
193. Bailey SM, Udoh US, Young ME. Circadian regulation of metabolism. J Endocrinol. (2014) 222:R75–96. doi: 10.1530/JOE-14-0200
194. Al-Khami AA, Mehrotra S, Nishimura MI. Adoptive immunotherapy of cancer: gene transfer of T cell specificity. Self Nonself. (2011) 2:80–4. doi: 10.4161/self.2.2.15832
195. Rosenberg SA, Restifo NP. Adoptive cell transfer as personalized immunotherapy for human cancer. Science. (2015) 348:62–8. doi: 10.1126/science.aaa4967
196. Paulos CM, Carpenito C, Plesa G, Suhoski MM, Varela-Rohena A, Golovina TN, et al. The inducible costimulator. (ICOS) is critical for the development of human T(H)17 cells. Sci Transl Med. (2010) 2:55ra78. doi: 10.1126/scitranslmed.3000448
197. Muranski P, Borman ZA, Kerkar SP, Klebanoff CA, Ji Y, Sanchez-Perez L, et al. Th17 cells are long lived and retain a stem cell-like molecular signature. Immunity. (2011) 35:972–85. doi: 10.1016/j.immuni.2011.09.019
198. Chen L, Diao L, Yang Y, Yi X, Rodriguez BL, Li Y, et al. CD38-mediated immunosuppression as a mechanism of tumor cell escape from PD-1/PD-L1 blockade. Cancer Discov. (2018) 8:1156–75. doi: 10.1158/2159-8290.CD-17-1033
199. Rine J, Herskowitz I. Four genes responsible for a position effect on expression from HML and HMR in Saccharomyces cerevisiae. Genetics. (1987) 116:9–22.
200. Frye RA. Phylogenetic classification of prokaryotic and eukaryotic Sir2-like proteins. Biochem Biophys Res Commun. (2000) 273:793–8. doi: 10.1006/bbrc.2000.3000
201. Michishita E, Park JY, Burneskis JM, Barrett JC, Horikawa I. Evolutionarily conserved and nonconserved cellular localizations and functions of human SIRT proteins. Mol Biol Cell. (2005) 16:4623–35. doi: 10.1091/mbc.e05-01-0033
202. North BJ, Verdin E. Interphase nucleo-cytoplasmic shuttling and localization of SIRT2 during mitosis. PLoS ONE. (2007) 2:e784. doi: 10.1371/journal.pone.0000784
203. Tanno M, Kuno A, Yano T, Miura T, Hisahara S, Ishikawa S, et al. Induction of manganese superoxide dismutase by nuclear translocation and activation of SIRT1 promotes cell survival in chronic heart failure. J Biol Chem. (2010) 285:8375–82. doi: 10.1074/jbc.M109.090266
204. Iwahara T, Bonasio R, Narendra V, Reinberg D. SIRT3 functions in the nucleus in the control of stress-related gene expression. Mol Cell Biol. (2012) 32:5022–34. doi: 10.1128/MCB.00822-12
205. Yanagisawa S, Baker JR, Vuppusetty C, Koga T, Colley T, Fenwick P, et al. The dynamic shuttling of SIRT1 between cytoplasm and nuclei in bronchial epithelial cells by single and repeated cigarette smoke exposure. PLoS ONE. (2018) 13:e0193921. doi: 10.1371/journal.pone.0193921
206. Sauve AA, Wolberger C, Schramm VL, Boeke JD. The biochemistry of sirtuins. Annu Rev Biochem. (2006) 75:435–65. doi: 10.1146/annurev.biochem.74.082803.133500
207. Bheda P, Jing H, Wolberger C, Lin H. The substrate specificity of sirtuins. Annu Rev Biochem. (2016) 85:405–29. doi: 10.1146/annurev-biochem-060815-014537
208. Haigis MC, Sinclair DA. Mammalian sirtuins: biological insights and disease relevance. Annu Rev Pathol. (2010) 5:253–95. doi: 10.1146/annurev.pathol.4.110807.092250
209. Finkel T, Deng CX, Mostoslavsky R. Recent progress in the biology and physiology of sirtuins. Nature. (2009) 460:587–91. doi: 10.1038/nature08197
210. Houtkooper RH, Pirinen E, Auwerx J. Sirtuins as regulators of metabolism and healthspan. Nat Rev Mol Cell Biol. (2012) 13:225–38. doi: 10.1038/nrm3293
211. Natoli G. When sirtuins and NF-kappaB collide. Cell. (2009) 136:19–21. doi: 10.1016/j.cell.2008.12.034
212. Preyat N, Leo O. Sirtuin deacylases: a molecular link between metabolism and immunity. J Leukoc Biol. (2013) 93:669–80. doi: 10.1189/jlb.1112557
213. Vachharajani VT, Liu T, Wang X, Hoth JJ, Yoza BK, Mccall CE. Sirtuins link inflammation and metabolism. J Immunol Res. (2016) 2016:8167273. doi: 10.1155/2016/8167273
214. Verdin E. The many faces of sirtuins: Coupling of NAD metabolism, sirtuins and lifespan. Nat Med. (2014) 20:25–7. doi: 10.1038/nm.3447
215. Chen X, Lu Y, Zhang Z, Wang J, Yang H, Liu G. Intercellular interplay between Sirt1 signalling and cell metabolism in immune cell biology. Immunology. (2015) 145:455–67. doi: 10.1111/imm.12473
216. Reiner SL. Epigenetic control in the immune response. Hum Mol Genet. (2005) 14 Spec 1:R41–6. doi: 10.1093/hmg/ddi115
217. Busslinger M, Tarakhovsky A. Epigenetic control of immunity. Cold Spring Harb Perspect Biol. (2014) 6:a024174. doi: 10.1101/cshperspect.a024174
218. Jasiulionis MG. Abnormal epigenetic regulation of immune system during aging. Front Immunol. (2018) 9:197. doi: 10.3389/fimmu.2018.00197
219. Jaenisch R, Bird A. Epigenetic regulation of gene expression: how the genome integrates intrinsic and environmental signals. Nat Genet. (2003) 33:245–54. doi: 10.1038/ng1089
220. Glass CK, Natoli G. Molecular control of activation and priming in macrophages. Nat Immunol. (2016) 17:26–33. doi: 10.1038/ni.3306
221. O'neill LA, Pearce EJ. Immunometabolism governs dendritic cell and macrophage function. J Exp Med. (2016) 213:15–23. doi: 10.1084/jem.20151570
222. Rodriguez-Prados JC, Traves PG, Cuenca J, Rico D, Aragones J, Martin-Sanz P, et al. Substrate fate in activated macrophages: a comparison between innate, classic, and alternative activation. J Immunol. (2010) 185:605–14. doi: 10.4049/jimmunol.0901698
223. Wang X, Buechler NL, Woodruff AG, Long DL, Zabalawi M, Yoza BK, et al. Sirtuins and immuno-metabolism of sepsis. Int J Mol Sci. (2018) 19:2738. doi: 10.3390/ijms19092738
224. Rodgers JT, Lerin C, Haas W, Gygi SP, Spiegelman BM, Puigserver P. Nutrient control of glucose homeostasis through a complex of PGC-1alpha and SIRT1. Nature. (2005) 434:113–8. doi: 10.1038/nature03354
225. Canto C, Auwerx J. PGC-1alpha, SIRT1 and AMPK, an energy sensing network that controls energy expenditure. Curr Opin Lipidol. (2009) 20:98–105. doi: 10.1097/MOL.0b013e328328d0a4
226. Canto C, Gerhart-Hines Z, Feige JN, Lagouge M, Noriega L, Milne JC, et al. AMPK regulates energy expenditure by modulating NAD+ metabolism and SIRT1 activity. Nature. (2009) 458:1056–60. doi: 10.1038/nature07813
227. Simmons GE, Pruitt WM, Pruitt K. Diverse roles of SIRT1 in cancer biology and lipid metabolism. Int J Mol Sci. (2015) 16:950–65. doi: 10.3390/ijms16010950
228. Kauppinen A, Suuronen T, Ojala J, Kaarniranta K, Salminen A. Antagonistic crosstalk between NF-kappaB and SIRT1 in the regulation of inflammation and metabolic disorders. Cell Signal. (2013) 25:1939–48. doi: 10.1016/j.cellsig.2013.06.007
229. Salminen A, Kaarniranta K, Kauppinen A. Crosstalk between oxidative stress and SIRT1: impact on the aging process. Int J Mol Sci. (2013) 14:3834–59. doi: 10.3390/ijms14023834
230. Yeung F, Hoberg JE, Ramsey CS, Keller MD, Jones DR, Frye RA, et al. Modulation of NF-kappaB-dependent transcription and cell survival by the SIRT1 deacetylase. EMBO J. (2004) 23:2369–80. doi: 10.1038/sj.emboj.7600244
231. Yang H, Zhang W, Pan H, Feldser HG, Lainez E, Miller C, et al. SIRT1 activators suppress inflammatory responses through promotion of p65 deacetylation and inhibition of NF-kappaB activity. PLoS ONE. (2012) 7:e46364. doi: 10.1371/journal.pone.0046364
232. Schug TT, Xu Q, Gao H, Peres-Da-Silva A, Draper DW, Fessler MB, et al. Myeloid deletion of SIRT1 induces inflammatory signaling in response to environmental stress. Mol Cell Biol. (2010) 30:4712–21. doi: 10.1128/MCB.00657-10
233. Leiser SF, Kaeberlein M. A role for SIRT1 in the hypoxic response. Mol Cell. (2010) 38:779–80. doi: 10.1016/j.molcel.2010.06.015
234. Lim JH, Lee YM, Chun YS, Chen J, Kim JE, Park JW. Sirtuin 1 modulates cellular responses to hypoxia by deacetylating hypoxia-inducible factor 1alpha. Mol Cell. (2010) 38:864–78. doi: 10.1016/j.molcel.2010.05.023
235. Joo HY, Yun M, Jeong J, Park ER, Shin HJ, Woo SR, et al. SIRT1 deacetylates and stabilizes hypoxia-inducible factor-α. (HIF-1α) via direct interactions during hypoxia. Biochem Biophys Res Commun. (2015) 462:294–300. doi: 10.1016/j.bbrc.2015.04.119
236. Yu Q, Dong L, Li Y, Liu G. SIRT1 and HIF1α signaling in metabolism and immune responses. Cancer Lett. (2018) 418:20–6. doi: 10.1016/j.canlet.2017.12.035
237. Liu G, Bi Y, Shen B, Yang H, Zhang Y, Wang X, et al. SIRT1 limits the function and fate of myeloid-derived suppressor cells in tumors by orchestrating HIF-1α-dependent glycolysis. Cancer Res. (2014) 74:727–37. doi: 10.1158/0008-5472.CAN-13-2584
238. Wang Y, Bi Y, Chen X, Li C, Li Y, Zhang Z, et al. Histone deacetylase SIRT1 negatively regulates the differentiation of interleukin-9-producing CD4(+) T cells. Immunity. (2016) 44:1337–49. doi: 10.1016/j.immuni.2016.05.009
239. Chalkiadaki A, Guarente L. The multifaceted functions of sirtuins in cancer. Nat Rev Cancer. (2015) 15:608–24. doi: 10.1038/nrc3985
240. Henning AN, Roychoudhuri R, Restifo NP. Epigenetic control of CD8(+) T cell differentiation. Nat Rev Immunol. (2018) 18:340–56. doi: 10.1038/nri.2017.146
241. Zhang X, Liu J, Cao X. Metabolic control of T-cell immunity via epigenetic mechanisms. Cell Mol Immunol. (2018) 15:203–5. doi: 10.1038/cmi.2017.115
242. Legutko A, Marichal T, Fievez L, Bedoret D, Mayer A, De Vries H, et al. Sirtuin 1 promotes Th2 responses and airway allergy by repressing peroxisome proliferator-activated receptor-γ activity in dendritic cells. J Immunol. (2011) 187:4517–29. doi: 10.4049/jimmunol.1101493
243. Zhang J, Lee SM, Shannon S, Gao B, Chen W, Chen A, et al. The type III histone deacetylase Sirt1 is essential for maintenance of T cell tolerance in mice. J Clin Invest. (2009) 119:3048–58. doi: 10.1172/JCI38902
244. Kuroda S, Yamazaki M, Abe M, Sakimura K, Takayanagi H, Iwai Y. Basic leucine zipper transcription factor, ATF-like. (BATF) regulates epigenetically and energetically effector CD8 T-cell differentiation via Sirt1 expression. Proc Natl Acad Sci USA. (2011) 108:14885–9. doi: 10.1073/pnas.1105133108
245. Han MK, Song EK, Guo Y, Ou X, Mantel C, Broxmeyer HE. SIRT1 regulates apoptosis and Nanog expression in mouse embryonic stem cells by controlling p53 subcellular localization. Cell Stem Cell. (2008) 2:241–51. doi: 10.1016/j.stem.2008.01.002
246. Boutant M, Canto C. SIRT1 metabolic actions: integrating recent advances from mouse models. Mol Metab. (2014) 3:5–18. doi: 10.1016/j.molmet.2013.10.006
247. Imperatore F, Maurizio J, Vargas Aguilar S, Busch CJ, Favret J, Kowenz-Leutz E, et al. SIRT1 regulates macrophage self-renewal. EMBO J. (2017) 36:2353–72. doi: 10.15252/embj.201695737
248. Gomes P, Fleming Outeiro T, Cavadas C. Emerging role of sirtuin 2 in the regulation of mammalian metabolism. Trends Pharmacol Sci. (2015) 36:756–68. doi: 10.1016/j.tips.2015.08.001
249. Chadha S, Wang L, Hancock WW, Beier UH. Sirtuin-1 in immunotherapy: a Janus-headed target. J Leukoc Biol. (2019). doi: 10.1002/JLB.2RU1118-422R
250. Beier UH, Wang L, Bhatti TR, Liu Y, Han R, Ge G, et al. Sirtuin-1 targeting promotes Foxp3+ T-regulatory cell function and prolongs allograft survival. Mol Cell Biol. (2011) 31:1022–9. doi: 10.1128/MCB.01206-10
251. Daenthanasanmak A, Iamsawat S, Chakraborty P, Nguyen HD, Bastian D, Liu C, et al. Targeting Sirt-1 controls GVHD by inhibiting T-cell allo-response and promoting Treg stability in mice. Blood. (2019) 133:266–79. doi: 10.1182/blood-2018-07-863233
252. Liu TF, Mccall CE. Deacetylation by SIRT1 reprograms inflammation and cancer. Genes Cancer. (2013) 4:135–47. doi: 10.1177/1947601913476948
253. Bitterman KJ, Anderson RM, Cohen HY, Latorre-Esteves M, Sinclair DA. Inhibition of silencing and accelerated aging by nicotinamide, a putative negative regulator of yeast sir2 and human SIRT1. J Biol Chem. (2002) 277:45099–107. doi: 10.1074/jbc.M205670200
254. Audrito V, Vaisitti T, Rossi D, Gottardi D, D'arena G, Laurenti L, et al. Nicotinamide blocks proliferation and induces apoptosis of chronic lymphocytic leukemia cells through activation of the p53/miR-34a/SIRT1 tumor suppressor network. Cancer Res. (2011) 71:4473–83. doi: 10.1158/0008-5472.CAN-10-4452
255. Revollo JR, Grimm AA, Imai S. The NAD biosynthesis pathway mediated by nicotinamide phosphoribosyltransferase regulates Sir2 activity in mammalian cells. J Biol Chem. (2004) 279:50754–63. doi: 10.1074/jbc.M408388200
256. Geng J, Liu A. Heme-dependent dioxygenases in tryptophan oxidation. Arch Biochem Biophys. (2014) 544:18–26. doi: 10.1016/j.abb.2013.11.009
257. Munn DH, Zhou M, Attwood JT, Bondarev I, Conway SJ, Marshall B, et al. Prevention of allogeneic fetal rejection by tryptophan catabolism. Science. (1998) 281:1191–3. doi: 10.1126/science.281.5380.1191
258. Mbongue JC, Nicholas DA, Torrez TW, Kim NS, Firek AF, Langridge WH. The role of indoleamine 2, 3-dioxygenase in immune suppression and autoimmunity. Vaccines. (2015) 3:703–29. doi: 10.3390/vaccines3030703
259. Takikawa O. Biochemical and medical aspects of the indoleamine 2,3-dioxygenase-initiated L-tryptophan metabolism. Biochem Biophys Res Commun. (2005) 338:12–9. doi: 10.1016/j.bbrc.2005.09.032
260. King NJ, Thomas SR. Molecules in focus: indoleamine 2,3-dioxygenase. Int J Biochem Cell Biol. (2007) 39:2167–72. doi: 10.1016/j.biocel.2007.01.004
261. Katz JB, Muller AJ, Prendergast GC. Indoleamine 2,3-dioxygenase in T-cell tolerance and tumoral immune escape. Immunol Rev. (2008) 222:206–21. doi: 10.1111/j.1600-065X.2008.00610.x
262. Puccetti P, Grohmann U. IDO and regulatory T cells: a role for reverse signalling and non-canonical NF-kappaB activation. Nat Rev Immunol. (2007) 7:817–23. doi: 10.1038/nri2163
263. Mellor AL, Munn DH. IDO expression by dendritic cells: tolerance and tryptophan catabolism. Nat Rev Immunol. (2004) 4:762–74. doi: 10.1038/nri1457
264. Munn DH, Mellor AL. Indoleamine 2,3 dioxygenase and metabolic control of immune responses. Trends Immunol. (2013) 34:137–43. doi: 10.1016/j.it.2012.10.001
265. Munn DH, Mellor AL. IDO in the tumor microenvironment: inflammation, counter-regulation, and tolerance. Trends Immunol. (2016) 37:193–207. doi: 10.1016/j.it.2016.01.002
266. Platten M, Wick W, Van Den Eynde BJ. Tryptophan catabolism in cancer: beyond IDO and tryptophan depletion. Cancer Res. (2012) 72:5435–40. doi: 10.1158/0008-5472.CAN-12-0569
267. Opitz CA, Litzenburger UM, Sahm F, Ott M, Tritschler I, Trump S, et al. An endogenous tumour-promoting ligand of the human aryl hydrocarbon receptor. Nature. (2011) 478:197–203. doi: 10.1038/nature10491
268. Uyttenhove C, Pilotte L, Theate I, Stroobant V, Colau D, Parmentier N, et al. Evidence for a tumoral immune resistance mechanism based on tryptophan degradation by indoleamine 2,3-dioxygenase. Nat Med. (2003) 9:1269–74. doi: 10.1038/nm934
269. Hornyak L, Dobos N, Koncz G, Karanyi Z, Pall D, Szabo Z, et al. The role of indoleamine-2,3-dioxygenase in cancer development, diagnostics, and therapy. Front Immunol. (2018) 9:151. doi: 10.3389/fimmu.2018.00151
270. Yu CP, Fu SF, Chen X, Ye J, Ye Y, Kong LD, et al. The clinicopathological and prognostic significance of IDO1 expression in human solid tumors: evidence from a systematic review and meta-analysis. Cell Physiol Biochem. (2018) 49:134–43. doi: 10.1159/000492849
271. Zhao Q, Kuang DM, Wu Y, Xiao X, Li XF, Li TJ, et al. Activated CD69+ T cells foster immune privilege by regulating IDO expression in tumor-associated macrophages. J Immunol. (2012) 188:1117–24. doi: 10.4049/jimmunol.1100164
272. Pinton L, Solito S, Damuzzo V, Francescato S, Pozzuoli A, Berizzi A, et al. Activated T cells sustain myeloid-derived suppressor cell-mediated immune suppression. Oncotarget. (2016) 7:1168–84. doi: 10.18632/oncotarget.6662
273. Lob S, Konigsrainer A, Rammensee HG, Opelz G, Terness P. Inhibitors of indoleamine-2,3-dioxygenase for cancer therapy: can we see the wood for the trees? Nat Rev Cancer. (2009) 9:445–52. doi: 10.1038/nrc2639
274. Brochez L, Chevolet I, Kruse V. The rationale of indoleamine 2,3-dioxygenase inhibition for cancer therapy. Eur J Cancer. (2017) 76:167–82. doi: 10.1016/j.ejca.2017.01.011
275. Prendergast GC, Malachowski WP, Duhadaway JB, Muller AJ. Discovery of IDO1 inhibitors: from bench to bedside. Cancer Res. (2017) 77:6795–811. doi: 10.1158/0008-5472.CAN-17-2285
276. Hou DY, Muller AJ, Sharma MD, Duhadaway J, Banerjee T, Johnson M, et al. Inhibition of indoleamine 2,3-dioxygenase in dendritic cells by stereoisomers of 1-methyl-tryptophan correlates with antitumor responses. Cancer Res. (2007) 67:792–801. doi: 10.1158/0008-5472.CAN-06-2925
Keywords: immunometabolism, metabolic reprogramming, immune cell regulation, NAD, tumor microenvironment
Citation: Audrito V, Managò A, Gaudino F, Sorci L, Messana VG, Raffaelli N and Deaglio S (2019) NAD-Biosynthetic and Consuming Enzymes as Central Players of Metabolic Regulation of Innate and Adaptive Immune Responses in Cancer. Front. Immunol. 10:1720. doi: 10.3389/fimmu.2019.01720
Received: 25 April 2019; Accepted: 09 July 2019;
Published: 25 July 2019.
Edited by:
Anna Ohradanova-Repic, Medical University of Vienna, AustriaReviewed by:
Armando A. Genazzani, University of Eastern Piedmont, ItalyCopyright © 2019 Audrito, Managò, Gaudino, Sorci, Messana, Raffaelli and Deaglio. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Valentina Audrito, dmFsZW50aW5hLmF1ZHJpdG9AdW5pdG8uaXQ=; Silvia Deaglio, c2lsdmlhLmRlYWdsaW9AdW5pdG8uaXQ=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.