 Sarah Beth Nodder
Sarah Beth Nodder Suryaram Gummuluru
Suryaram Gummuluru- Department of Microbiology, Boston University School of Medicine, Boston, MA, United States
Vpr is a 14 kDa accessory protein conserved amongst extant primate lentiviruses that is required for virus replication in vivo. Although many functions have been attributed to Vpr, its primary role, and the function under selective pressure in vivo, remains elusive. The minimal importance of Vpr in infection of activated CD4+ T cells in vitro suggests that its major importance lies in overcoming restriction to virus replication in non-cycling myeloid cell populations, such as macrophages and dendritic cells. HIV-1 replication is attenuated in the absence of Vpr in myeloid cells such as monocyte-derived dendritic cells (MDDCs) and macrophages, and is correlated with the ability of Vpr to overcome a post-integration transcriptional defect in these cells. Intriguingly, recent identification of the human hub silencing (HUSH) complex as a target for DCAFCRL4-mediated degradation by numerous ancestral SIV Vpr alleles, and the Vpr paralog Vpx, signifies the potential function of HIV-1 Vpr to alter yet-to-be identified chromatin remodeling complexes and prevent host-mediated transcriptional repression of both invading viral genomes and pro-inflammatory responses. Myeloid cells constitute an important bridge between innate and adaptive immune responses to invading pathogens. Here, we seek to illustrate the numerous means by which Vpr manipulates the myeloid cellular environment and facilitates virus replication, myeloid cell-dependent HIV transmission, and systemic virus dissemination.
Introduction
Sexual transmission is the predominant means by which HIV is acquired (“Global AIDS Update” 2016). While the exact cell type targeted by HIV in the genital mucosa remains a matter of debate (1–3), various subsets of dendritic cells (DCs) and macrophages are present at high concentrations within the genital mucosa, and therefore may be early targets of HIV (4–6). Infection of DCs and macrophages is particularly important as they are uniquely poised to transmit HIV with high efficiency to CD4+ T cells during antigen presentation within the secondary lymphoid organs (5, 7, 8). As professional antigen presenting cells, DCs and macrophages have a unique cellular architecture to initiate and sustain robust interactions with CD4+ T cells. Virological synapse formation between DCs /macrophages and CD4+ T cells ensures directed delivery of HIV to its most permissive host: activated CD4+ T cells (9). In order for mucosal transmission and establishment of productive infection, HIV not only has to navigate tissue barriers (3), but also a number of cell-intrinsic immune defenses, or restriction factors, such as APOBEC3G (a cytidine deaminase that dramatically increases genome mutations), tetherin (which prevents HIV viral budding and enforces a positive type I IFN loop upon suppression of viral budding), and SAMHD1 (a dNTPase that limits dNTP levels within the cytoplasm to hinder reverse transcription), that prevent primate lentiviral infection of DCs, macrophages and resting T cells (10–13). However, primate lentiviruses have evolved to counteract these restriction factors by encoding accessory proteins that selectively inhibit their anti-viral function. Although postulated to act in this capacity, the function of Vpr has yet to be fully understood.
Vpr, or viral protein R, is a 14 kDa protein, encoded by all extant primate lentiviruses. Vpr is actively packaged into virions through its interaction with the p6 region of Gag (14, 15), and as such has roles in both pre- and post-integration steps of the viral life cycle. Although originally described as an accessory protein, and thus dispensable for virus replication in vitro, it has since been shown to play an important role in the infection of macrophages and dendritic cells (16–19). Importantly, Vpr function is necessary for viral pathogenesis in vivo. In 1993, six rhesus macaques were infected with pathogenic SIVmac239 isolate either containing or lacking Vpr (20). Within 16 weeks, wild type Vpr sequences were isolated from three of the five animals infected with Vpr null virus. Moreover, the remaining two animals infected with Vpr null virus displayed delayed pathogenesis, and reversion to Vpr-expressing virus by 66 weeks (20, 21). These studies validated retrospective work that found reversions of an internal Vpr stop codon to an open reading frame in an accidentally infected laboratory worker and experimentally infected chimpanzees (22, 23). Together, these studies were seminal in igniting research into the role of Vpr in the pathogenesis of HIV. Vpr is best known to induce an Ataxia-Telangiectasia and Rad3-related (ATR) dependent-DNA damage response, or DDR (24). Vpr-mediated DDR activation results in a G2 to M cell cycle arrest in cycling cells, which is perplexing as the cell populations whose infections are seemingly most reliant on the presence of Vpr are terminally differentiated and thus not susceptible to cell cycle arrest. The ability to induce a G2 to M cell cycle arrest is thought to be advantageous for viral transcription as the HIV LTR has been shown to be most active during this phase of the cell cycle (23, 25). What's more, the Vpr residues that confer G2 to M cell cycle arrest capabilities are under positive selection in vivo and are thus the most well-studied in the field (26). It remains to be determined if induction of DDR by Vpr, an evolutionarily conserved function amongst all primate lentiviral Vpr proteins (27–29), is necessary for establishment of virus replication in metabolically quiescent immune target cells, such as monocytes, macrophages and DCs.
Vpr/Vpx and the Importance of Co-opting the Ubiquitin-Ligase DCAFCRL4 in Myeloid Cells
Vpx, or Viral protein x, arose following the duplication of Vpr post-diversion of the primate lentiviral lineages that gave rise to HIV-1 and HIV-2 (30, 31). Vpx has a well-characterized role in degrading the retroviral restriction factor SAMHD1 (10, 11). In terminally differentiated or non-cycling cells, SAMHD1 reduces the concentration of dNTPs within the cytoplasm, thereby drastically limiting reverse transcription (32–35). Vpx bridges SAMHD1 to the E3-ubiquitin ligase CUL4A-DDB1 DCAF (DCAFCRL4) leading to its polyubiquitination and proteasomal degradation (10, 11, 36). There has been much interest in identifying the restriction factor(s) targeted by Vpr as it similarly co-opts the DCAFCRL4 complex to ubiquitinate target host proteins. Unlike Vpx however, infection of myeloid cells by either HIV-1 or HIV-2 still occurs in the absence of Vpr, albeit with significantly different outcomes (19, 24, 37–41). It is likely that the replication advantage conferred by Vpr lies in its ability to induce a DDR, though the mechanisms by which Vpr-induced DDR facilitates enhanced virus replication and spread in vivo are still to be determined. The multitude of DDR proteins associated with the Vpr-DCAFCRL4 complex (24, 37–39, 41), suggests that Vpr by co-opting a host protein complex involved in multiple cellular pathways, has managed to maximize its impact at the interface of virus and host to promote HIV spread.
Vpr Residues Involved in DCAFCRL4 Engagement
An NMR structure of HIV-1 Vpr provides insight into how it interacts with multiple proteins. Both N and C-termini are unstructured (nucleotides 1–16, and 77–96, respectively) and flank three α-helices from nucleotides 17–33, 38–50, and 56–77 (42). The HIV-2 Vpr, as well as the closely related Vpr alleles from SIVsmm and SIVmac, are predicted to be structurally homologous to that of HIV-1. Whilst the unstructured C- and N-terminal domains facilitate interactions with host targets, the DCAFCRL4- binding domain is isolated to the third α-helix (42–45). The HIV-1 Vpr mutants Q65R and H71R for example, and corresponding residues in HIV-2/SIVmac Vpr alleles fall within this region and fully abrogate Vpr-DCAFCRL4 interactions. These mutations prevent Vpr-mediated transcriptional enhancement in MDDCs (19), decrease degradation of multiple DNA damage response proteins (46–50), and prevent G2/M cell cycle arrest in CD4+ T cells (51). Furthermore, ablation of Vpr-DCAFCRL4 interaction, as occurs with a VprQ65R mutation (albeit not with the VprQ77R mutation), has been associated with long-term non-progression in vivo (52). Investigations into the DCAFCRL4-mediated enhancement of infection in myeloid cells use these select mutations to infer mechanisms of action and are thus worthy of mention.
DCAFCRL4-Dependent Roles of Vpr in Myeloid Cells
DNA Damage Response Proteins
Human Uracil DNA Glycosylase
Monocyte-derived macrophages (MDMs) have a high ratio of dUTP/TTP in their cytoplasm that can lead to the mis-incorporation of uracil in the reverse transcribed genome. The ratio of dUTP/TTP in macrophages was found to be as high as 60 (53, 54). Human uracil DNA glycosylase (hUNG) excises mis-incorporated UTP and recruits additional repair enzymes to the site of genome mutation. Thus, HIV-1 Vpr-mediated DCAFCRL4-dependent ubiquitination and proteasomal degradation of hUNG was hypothesized to restrict virus replication through either degradation of uracilated viral DNA prior to integration or via transcriptional interference of the uracilated provirus (53, 55, 56). However, due to the intrinsically low levels of hUNG in MDMs (56), the utility of uracil-dependent restriction of HIV-1 in MDMs is limited. Furthermore, infection of MDDCs with HIV-1 expressing hUNG-binding deficient VprW54R mutant does not result in transcriptional attenuation nor deficiency in viral spread (19). Thus, the significance of hUNG degradation by the Vpr-DCAFCRL4 complex (39, 57) remains unclear.
SLX4-SLX/MUS81-EME1
The importance of Vpr in the Holliday junction repair pathway has been of great interest as it promised to provide insight into the role of Vpr at the viral integration step. Original reports suggested that HIV-1 Vpr-DCAFCRL4-mediated ubiquitination of MUS81 which, in the presence of phosphorylated EME1 and kinase-active PLK1, prematurely activates the quaternary endonuclease complex SLX4com (47). This activation was shown to precede G2/M cell cycle arrest and result in the formation of FANCD2 foci as a result of activation of the Fanconi anemia pathway (47). Notably, virion-associated Vpr-mediated activation of SLX4com was shown to prevent type I IFN production which is of great importance due to the myriad of interferon stimulated genes (ISGs) that modulate myeloid cell function and determine the dissemination efficiency of virus through the host (47). However, subsequent studies have only shown a Vpr-DCAFCRL4 dependent degradation of the SLX4com subunits MUS81-EME1 (50, 58–60) and have not addressed whether the active SLX4com suppressed innate immune detection of HIV-1 in myeloid cells. Additionally, interaction of SLX4com with Vpr is not conserved amongst all primate lentiviral Vpr alleles (59). Together, published findings so far, suggest that the Vpr-mediated activation of SLX4com does not have a conserved role in suppressing innate immune detection of primate lentiviruses in myeloid cells.
Helicase-Like Transcription Factor (HLTF)
Helicase like transcription factor, or HLTF, is a target of Vpr-mediated DCAFCRL4 degradation (46, 48). Like UNG2 and SLX4-SLX1/MUS81-EME1, HLTF is involved in DNA damage repair. Specifically, HLTF is critical to the remodeling and repair of stalled replication forks (61). Although HLTF is degraded in macrophages in a Vpr- DCAFCRL4 dependent manner, it is unclear whether HLTF antagonizes viral replication in myeloid cells.
Exonuclease 1
Exonuclease 1, or Exo1, is a Rad2/XPG 5′ to 3′ exonuclease involved in numerous DNA repair processes that ensures genome stability throughout the cell cycle (62). Exo1 has recently been identified as a substrate for Vpr-DCAFCRL4 polyubiquitination and proteasomal degradation in CD4+ T cells (49). The authors speculate that Exo1 antagonism prevents deleterious processing of reverse transcription- and viral integration-intermediates, and thereby attribute Exo1 restriction to virion-associated Vpr rather than its de novo synthesized partner (49). As of yet, Exo1 has not been shown to play a role in promoting HIV-1 infection of macrophages or dendritic cells.
While these published studies highlight the numerous interactions of Vpr with diverse DDR proteins, contribution of these interactions to viral pathogenesis have remained unclear. Although understudied in the case of HIV-1 infection, there is a robust literature tying innate immune signaling and DDR (63, 64). It should be noted that manipulation of the DDR is not unique to HIV. Rather, it is a shared pathogenic strategy used extensively at the interface of hosts with both bacteria and viruses that can promote pathogen replication and pathogenesis (65). Kaposi sarcoma herpesvirus, for example, encodes a protein (Latency-Associated Nuclear Antigen or LANA) that sequesters Rad50, Mre11, and NBS1, all members of the DDR signaling activator MRN complex to prevent cytoplasmic sensing of viral DNA and innate immune activation (66). Another example of virus subversion of DDR pathway includes murine γ-herpesvirus which specifically encodes orf36 whose role is to induce an ATM-dependent DDR and H2AX phosphorylation (67). In the absence of orf36 or ATM activation, virus replication is attenuated, pointing toward a role for the DDR in facilitating virus replication (67). Overall, it is evident that Vpr uses DCAFCRL4 to induce a DDR, with potentially divergent outcomes in different cell populations. What remains unclear is how activation of the DDR and interaction of Vpr with DNA repair proteins allows viral evasion of immune detection in myeloid cells. Since the kinetics of reverse transcription of HIV-1 in myeloid cells is relatively slow, it is tempting to speculate that manipulation of diverse DDR pathways is a conserved strategy by primate lentiviral Vpr alleles to overcome premature host repair of viral reverse transcription intermediates (63), though definitive evidence for this hypothesis has been lacking. Rather, it is likely that activation of DDR promotes multiple discrete stages of the virus life cycle. For example, Vpr can induce DDR through both the ATM and ATR pathways (24, 68). Unresolved ATM activity can lead to activation of NF-κB (69) and increased production of inflammatory cytokines, such as IL-6, both of which can result in enhanced viral gene expression and macrophage-dependent HIV-1 transmission to CD4+ T cells (70).
Vpr Functions in Transcriptional De-repression
Transcriptional Enhancement
Previous work by our group has shown a post-integration defect in monocyte-derived dendritic cells (MDDCs), infected with Vpr deficient HIV-1 (19). Infections in the absence of virion associated Vpr were characterized by low proviral transcription despite similar levels of integration, and reduced infection of CD4+ T cells in co-cultures (19). This defect is dependent on Vpr binding to DCAFCRL4 as it is fully abrogated upon infection with Vpr mutants (Q65R or H71R) lacking DCAFCRL4 interactions. It should be noted that numerous viruses besides HIV-1, most notably Hepatitis B virus, can also manipulate the E3 ubiquitin ligase DCAFCRL4 to enhance transcription (71). While, the mechanism of HIV-1 Vpr-mediated transcriptional enhancement remains unclear, previous research has shown Vpr-mediated degradation of HDACs (38) and members of the NuRD chromatin remodeling complex (72) which may globally enhance transcription. Furthermore, DCAFCRL4 also has a well-known role in the degradation of a transcriptional repressor, ATF3, which is necessary to correct UV-damage (73). This explanation is not satisfactory given the cell type dependency of the transcriptional enhancement. Whether a MDDC-specific repressor/activator is degraded or sequestered remains unknown and warrants further investigation.
TET2
Members of the TET DNA dioxygenase family have recently been shown to be degraded in a Vpr-DCAFCRL4 dependent manner (70). In myeloid cells TET2 is naturally monoubiquitinated. TET2 N-terminal monoubiquitylation allows for efficient binding to chromatin and subsequent recruitment of chromatin remodeling machinery and transcription factors. However, in the presence of Vpr, TET2 is rapidly polyubiquitinated at a site independent of its natural monoubiquitylation site and undergoes DCAFCRL4- dependent proteasomal degradation (70). This is relevant in myeloid cells as TET2 is an upstream suppressor of IL-6 expression. TET2 recruits HDAC1 and HDAC2 to the IL-6 promotor thereby repressing IL-6 transcription. Importantly, in monocyte-derived macrophages and the monocytic cell line THP-1, the lack of Vpr-mediated degradation of TET2 was associated with reduced viral particle release and slower spread of HIV-1 infection. Upon TET2 knockout, the differences in infection between Vpr-competent and Vpr-deficient viruses was lost. Since IL-6 has long been recognized as a transcriptional enhancer of HIV in monocytes (70, 74–76), these findings are further suggestive of a direct link between Vpr, TET2 degradation, and persistent IL-6 production, which might result in enhanced efficiency of viral spread from myeloid cells to CD4+ T cells.
Epigenetic Regulation of Provirus
Until recently, it was not known whether there were host-intrinsic mechanisms to restrict retroviral replication following integration. However, recent investigations have identified a novel method of cell-intrinsic restriction: that of deposition of transcriptionally suppressive methylation marks at proviral LTRs. Following reverse transcription and integration, the LTR of proviruses within heterochromatin are methylated through the sequential recruitment of HP1 and the methyltransferase Suv39H1 (77) Tri-methylated H3K9 recruits the HUSH (HUman Silencing Hub) complex of which there are three components; TASOR, MPP8, and periphilin (78). Although the HUSH complex does not harbor methyltransferase activity itself, HUSH recruits the methylase SETDB1 which induces further H3K9me3 methylation of the provirus. Notably, shRNA-mediated knockdown of each HUSH complex protein rescues endogenous and exogenous retroviral gene expression, thereby signifying the importance of its quaternary assembly for transcriptional repression (78). Interestingly, TASOR is targeted by the SIVmac/HIV-2 lineage Vpx for DCAFCRL4-mediated polyubiquitination and proteasomal degradation, thereby increasing the transcriptional activity of proviruses that would otherwise be suppressed (78–80). While HUSH complex can repress transcription from integrated HIV-1 LTR (78, 80, 81), surprisingly, HIV-1 Vpr does not target the HUSH complex proteins for degradation (79). However, multiple Vpr alleles from ancestral primate lentiviruses to HIV-1, including alleles derived from SIVAGM, SIVMUS2, and SIVSAB, have been shown to prevent HUSH-mediated silencing (79, 80). These studies mark the beginning of investigations into Vpx/Vpr antagonism of antiviral host proteins at the proviral DNA level. Although HUSH-mediated transcriptional silencing is not a myeloid specific anti-viral mechanism, the HUSH complex and its associated facilitators are active in myeloid lineages. Recent studies in the literature provide evidence for epigenetic control of proinflammatory cytokine responses in macrophages (82, 83). For instance, the histone methyltransferases, SETDB1 and Smyd2, potently suppress TLR4- mediated induction of IL-6 and TNFα production, and mice with macrophage-specific SetDB1 deficiency are hyper-responsive to endotoxin challenge (82). Whilst antagonism of HUSH complex has not been attributed to HIV-1 Vpr, transcriptional silencing of the HIV-1 LTR in MDDCs in the absence of Vpr (19) suggests the existence of additional mechanisms of myeloid cell-intrinsic transcriptional repression that are targeted by HIV-1 Vpr.
Dicer and miRNA Processing
Modulation of the RNA interference (RNAi) and microRNA (miRNA) pathways is an integral means by which pathogens usurp host functions to their advantage (84, 85). MicroRNAs in particular have long been known to play a role in HIV replication in multiple cell populations. For instance, miR-29a, has been implicated in the suppression of HIV mRNA levels through its binding to the 3′-UTR of HIV RNA and subsequent attachment to P body proteins and RISC complexes (86). Dicer is required for processing pre-miRNA substrates to reveal a double-stranded miRNA complex, which then binds to the RISC complex and represses target mRNA expression, either via translation inhibition or via mRNA degradation (87). Recent studies have also demonstrated the ability of miRNAs to negatively regulate proinflammatory responses in macrophages by restricting chromatin remodeling and enforcing transcriptional silencing of promoters of select inflammatory genes (88). Interestingly, Dicer has been identified in complex with Vpr-DCAFCRL4 prior to its degradation and depletion of Dicer within infected MDMs has been shown to increase viral replication via unknown mechanisms (89). We posit that Vpr-Dicer dependent modulation of select miRNA expression might contribute to the de-repression of inflammatory responses. It should be noted that Vpr-mediated Dicer depletion has also been shown in CD4+ T cells and as such, is not a myeloid-specific antagonist of innate restriction (89). The role of Dicer degradation has yet to be fully understood, particularly as research into the function of miRNA and RNAi in HIV pathogenesis is increasing (90).
DCAFCRL4-Independent Roles of Vpr in Myeloid Cells
Envelope Trafficking
Myeloid cells often populate mucosal tissues and as such are poised to disseminate HIV from the periphery to sites harboring abundant activated CD4+ T cells. Macrophages and dendritic cells are capable of transferring virus via cis or trans infection. Both methods facilitate the concentration of virions to the infectious synapse, and in doing so greatly increase the probability of CD4+ T cell infection (9, 91, 92). Two studies have investigated the role of Vpr in the concentration and delivery of virus at the virological synapse. Both of these studies show that in the absence of Vpr, Env-positive virions are trafficked to the lysosome for degradation, thereby reducing the efficiency of macrophage to CD4+ T cell virus spread at low multiplicity of infection (93, 94). In contrast, our investigations into whether Vpr facilitates evasion of this Env-dependent reduction in virus release in other cells types, such as MDDCs, have not yielded similar results (19), suggesting that the effect of Vpr on Env expression might be restricted to specific cell types and not universally observed.
Type I IFN and Pro-Inflammatory Responses
There is mounting evidence to suggest that Vpr modulates the immune response of myeloid cells to favor viral replication and dissemination throughout the host. Early studies suggested a possible defect in the activation of MDMs and MDDCs upon treatment with recombinant Vpr (95). This defect was characterized by low CD33 surface expression, poor CD80/86 upregulation, and impaired antigen presentation to activated CD4+ T cells (95). In contrast to studies utilizing exogenous addition of recombinant Vpr, there has been a preponderance of research investigating the role of Vpr in the context of a viral infection. For instance, de novo expression of Vpr in productively infected MDDCs induced pro-inflammatory cytokine (TNF-α, IL-6 and IL-8) production (70, 96, 97). Previous work by our group has shown enhanced proviral transcription in MDDCs in the presence of Vpr (19). Other studies have similarly shown a role for Vpr in proviral transcription. Liu et al. showed that Vpr alters the availability of the NF- κB p50-p65 heterodimer and AP1 (98), both of which are necessary for the initiation of HIV transcription from the 5′ LTR (99, 100) and expression of pro-inflammatory cytokines. In this study, Vpr was shown to facilitate the polyubiquitination and subsequent phosphorylation (activation) of TAK1, an upstream regulator of NF-κB and AP1 (98). Interruption of TAK1 phosphorylation, and thus inhibiting its activation, significantly reduced proviral transcription (98).
Both our study (19) and the work showing Vpr-mediated modification of TAK1 (98) are important in light of the recent identification of a novel viral detection pathway: one in which host sensing of de novo expressed intron-containing HIV-1 RNA (HIV icRNA) in MDMs and MDDCs results in ISG expression and proinflammatory chemokine and cytokine production, including IP-10 and IL-15 (101). IP-10 is an inflammatory chemokine and is a ligand for the receptor CXCR3 (102), while IL-15 is a γ-chain cytokine critical for the proliferation and homeostasis of T cells (103). CXCR3 is expressed on activated CD4+ T cells, and thus secretion of IP-10 from productively infected myeloid cells may result in additional recruitment of virus-susceptible cells to sites of viral replication in the peripheral tissues. Furthermore, IL-15 exposure can result in SAMHD1 phosphorylation and inactivation of its dNTPase activity, thus alleviating restrictions to viral replication in quiescent CD4+ T cells (104). Interestingly, a TAK1 inhibitor reduced production of IP-10 from HIV-1 infected macrophages (101). Thus, in this model, Vpr-dependent enhanced proviral transcription potentially increases the pool of viral icRNAs subject to cell-intrinsic innate immune sensing, resulting in the establishment of a pro-inflammatory state and enhanced virus dissemination.
It should be noted that an increase in viral transcription in myeloid cells is a double-edged sword for HIV in that there is also induction of ISG expression and establishment of a putative anti-viral state. While the nucleic acid sensing mechanism responsible for detection of HIV icRNA is yet to be determined, Vpr can block phosphorylation and nuclear translocation of interferon regulatory factor 3 (IRF3) via inducing ubiquitination and proteosomal degradation of IRF3, though this has only been shown in CD4+ T cell lines (105). Additionally, Vpr has been shown to bind to TBK1 in myeloid cells and prevent its phosphorylation, thereby preventing induction of type I IFN production (106). Thus, we posit that Vpr might function to promote NF-κB-dependent pro-inflammatory responses while contributing to the suppression of induction of anti-viral host defenses. Furthermore, induction of ISGs such as CD169 in MDMs and MDDCs upon host sensing of HIV icRNA in myeloid cells (101) might further tip the balance toward enhanced virus dissemination as opposed to virus restriction. For instance, induced CD169 expression on HIV-infected macrophages and dendritic cells can facilitate cell-to-cell transmission of CD4+ T cells across infectious synapses (101, 107, 108). Together, these studies point to the role of Vpr as a protein that carefully navigates multiple viral sensing systems to induce recruitment of additional cellular targets of virus, whilst evading antiviral immunity.
Conclusion
It is clear that Vpr plays an important role in the infection of myeloid cells (see Figure 1). A number of tissue-resident macrophages, such as microglia, kupfer cells, alveolar, intestinal, testicular and vaginal macrophages harbor proviral DNA (109–113), and tissue-resident macrophages are estimated to compromise up to 4% of infected cells in vivo (114), and importantly, can remain persistently infected with HIV-1 even in the presence of cART (109–112, 115). It is possible that the Vpr-mediated DDR activates a pro-inflammatory state that promotes the establishment of a tissue-resident myeloid cell reservoir, whereby virus spreads efficiently due to persistent virion production and enhanced cell-to-cell contacts between HIV-infected myeloid cells and CD4+ T cells. In this way, the infection of myeloid cells is the bridge between the relatively hostile sites of virus acquisition (most notably the peripheral mucosal tissues) and the key target of HIV; CD4+ T cells. It seems likely that the true value of Vpr in vivo is its versatility, allowing for evasion of viral restriction both prior to and post integration in myeloid cells. Future studies will need to address the relative importance of each of the known Vpr functions in vivo.
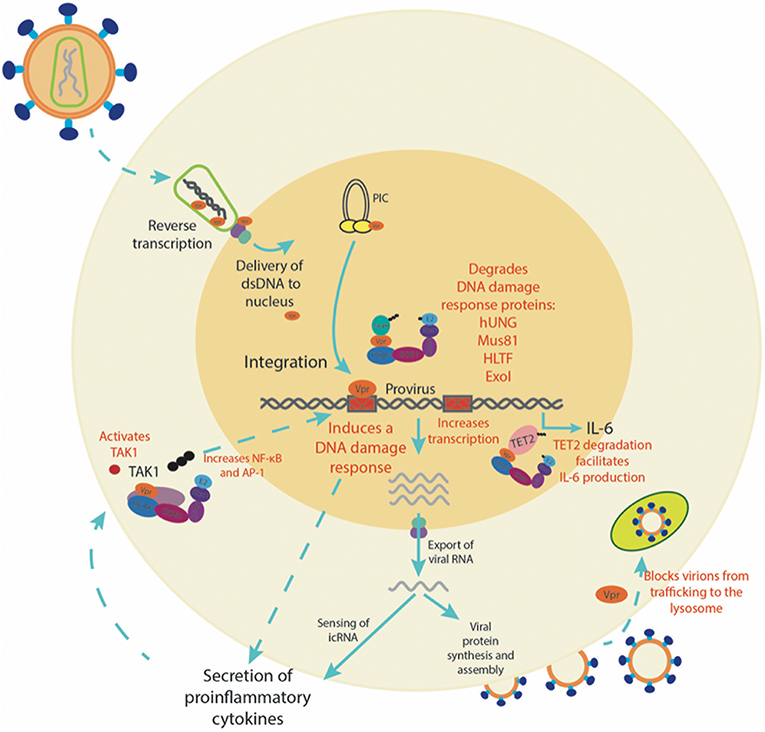
Figure 1. The role of Vpr in the infection of myeloid cells. A summary of the multiple functions Vpr plays in myeloid cells, from enhancing transcription and inducing a DDR to the secretion of pro-inflammatory cytokines. In red are processes in which Vpr has been directly investigated.
Author Contributions
All authors listed have made a substantial, direct and intellectual contribution to the work, and approved it for publication.
Funding
This work was supported by NIH grants R01AI064099, P30AI042853, and R01AG060890 (SG).
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Anderson DJ, Le Grand R. Cell-associated HIV mucosal transmission: the neglected pathway. J Infect Dis. (2014) 210:S606–S08. doi: 10.1093/infdis/jiu538
2. Barreto-de-Souza V, Arakelyan A, Margolis L, Vanpouille C. HIV-1 vaginal transmission: cell-free or cell-associated virus? Am J Reproduc Immunol. (2014) 71:589–99. doi: 10.1111/aji.12240
3. Burgener A, McGowan I, Klatt NR. HIV and mucosal barrier interactions: consequences for transmission and pathogenesis. Curr Opin Immunol. (2015) 36:22–30. doi: 10.1016/j.coi.2015.06.004
4. Pena-Cruz V, Agosto LM, Akiyama H, Olson A, Moreau Y, Larrieux JR, et al. HIV-1 replicates and persists in vaginal epithelial dendritic cells. J Clin Invest. (2018) 128:3439–44. doi: 10.1172/JCI98943
5. Rodriguez-Garcia M, Shen Z, Barr FD, Boesch AW, Ackerman ME, Kappes JC, et al. Dendritic cells from the human female reproductive tract rapidly capture and respond to HIV. Mucosal Immunol. (2017) 10:531–44. doi: 10.1038/mi.2016.72
6. Shen R, Kappes JC, Smythies LE, Richter HE, Novak L, Smith PD. Vaginal myeloid dendritic cells transmit founder HIV-1. J Virol. (2014) 88:7683–8. doi: 10.1128/JVI.00766-14
7. Peressin M, Proust A, Schmidt S, Su B, Lambotin M, Biedma ME, et al. Efficient transfer of HIV-1 in trans and in cis from Langerhans dendritic cells and macrophages to autologous T lymphocytes. AIDS. (2014) 28:667–77. doi: 10.1097/QAD.0000000000000193
8. Turville SG, Santos JJ, Frank I, Cameron PU, Wilkinson J, Miranda-Saksena M, et al. Immunodeficiency virus uptake, turnover, and 2-phase transfer in human dendritic cells. Blood. (2004) 103:2170–9. doi: 10.1182/blood-2003-09-3129
9. Wu L, KewalRamani VN. Dendritic-cell interactions with HIV: infection and viral dissemination. Nat Rev Immunol. (2006) 6:859–68. doi: 10.1038/nri1960
10. Hrecka K, Hao C, Gierszewska M, Swanson SK, Kesik-Brodacka M, Srivastava S, et al. Vpx relieves inhibition of HIV-1 infection of macrophages mediated by the SAMHD1 protein. Nature. (2011) 474:658–61. doi: 10.1038/nature10195
11. Laguette N, Sobhian B, Casartelli N, Ringeard M, Chable-Bessia C, Ségéral E, et al. SAMHD1 is the dendritic- and myeloid-cell-specific HIV-1 restriction factor counteracted by Vpx. Nature. (2011) 474:654–7. doi: 10.1038/nature10117
12. Neil SJ, Zang T, Bieniasz PD. Tetherin inhibits retrovirus release and is antagonized by HIV-1 Vpu. Nature. (2008) 451:425–30. doi: 10.1038/nature06553
13. Sheehy AM, Gaddis NC, Choi JD, Malim MH. Isolation of a human gene that inhibits HIV-1 infection and is suppressed by the viral Vif protein. Nature. (2002) 418:646–50. doi: 10.1038/nature00939
14. Selig L, Pages JC, Tanchou V, Prévéral S, Berlioz-Torrent C, Liu LX, et al. Interaction with the p6 domain of the gag precursor mediates incorporation into virions of Vpr and Vpx proteins from primate lentiviruses. J Virol. (1999) 73:592–600.
15. Paxton W, Connor RI, Landau NR. Incorporation of Vpr into human immunodeficiency virus type 1 virions: requirement for the p6 region of gag and mutational analysis. J Virol. (1993) 67:7229–37.
16. Campbell BJ, Hirsch VM. Vpr of simian immunodeficiency virus of African green monkeys is required for replication in macaque macrophages and lymphocytes. J Virol. (1997) 71:5593–602.
17. Connor RI, Chen BK, Choe S, Landau NR. Vpr is required for efficient replication of human immunodeficiency virus type-1 in mononuclear phagocytes. Virology. (1995) 206:935–44. doi: 10.1006/viro.1995.1016
18. Eckstein DA, Sherman MP, Penn ML, Chin PS, Greene WC, Goldsmith MA. HIV-1 Vpr enhances viral burden by facilitating infection of tissue macrophages but not nondividing CD4+ T Cells. J Exp Med. (2001) 194:1407–19. doi: 10.1084/jem.194.10.1407
19. Miller CM, Akiyama H, Agosto LM, Emery A, Ettinger CR, Swanstrom RI, et al. Virion-associated Vpr alleviates a postintegration block to HIV-1 infection of dendritic cells. J Virol. (2017) 91:e00051-17. doi: 10.1128/JVI.00051-17
20. Lang SMM, Weeger C, Stahl-Hennig C, Coulibaly G, Hunsmann J, Müller H, et al. Importance of Vpr for infection of rhesus monkeys with simian immunodeficiency virus. J Virol. (1993) 67:902–12.
21. Hoch J SM, Lang M, Weeger C, Stahl-Hennig C, Coulibaly U, Dittmer G, et al. vpr deletion mutant of simian immunodeficiency virus induces AIDS in rhesus monkeys. J Virol. (1995) 69:4807–13.
22. Beaumont T, van Nuenen A, Broersen S, Blattner WA, Lukashov VV, Schuitemaker H. Reversal of Human Immunodeficiency Virus type 1 IIIB to a neutralization-resistant phenotype in an accidentally infected laboratory worker with a progressive clinical course. J Virol. (2001) 75:2246–52. doi: 10.1128/JVI.75.5.2246-2252.2001
23. Goh WC, Rogel ME, Kinsey CM, Michael SF, Fultz PN, Nowak MA, et al. HIV-1 Vpr increases viral expression by manipulation of the cell cycle: A mechanism for selection of Vpr in vivo. Nat Med. (1998) 4:65. doi: 10.1038/nm0198-065
24. Roshal M, Kim B, Zhu Y, Nghiem P, Planelles V. Activation of the ATR-mediated DNA damage response by the HIV-1 viral protein R. J Biol Chem. (2003) 278:25879–86. doi: 10.1074/jbc.M303948200
25. Gummuluru S, Emerman M. Cell cycle- and Vpr-mediated regulation of human immunodeficiency virus type 1 expression in primary and transformed T-cell lines. J Virol. (1999) 73:5422–30.
26. Yedavalli VR, Chappey C, Ahmad N. Maintenance of an Intact Human Immunodeficiency Virus Type 1 vpr Gene following Mother-to-Infant Transmission. J Virol. (1998) 72:6937–43.
27. Malim MH, Emerman M. HIV-1 accessory proteins–ensuring viral survival in a hostile environment. Cell Host Microbe. (2008) 3:388–98. doi: 10.1016/j.chom.2008.04.008
28. Planelles V, Jowett JB, Li QX, Xie Y, Hahn B, Chen IS. Vpr-induced cell cycle arrest is conserved among primate lentiviruses. J Virol. (1996) 70:2516–24.
29. Stivahtis GL, Soares MA, Vodicka MA, Hahn BH, Emerman M. Conservation and host specificity of Vpr-mediated cell cycle arrest suggest a fundamental role in primate lentivirus evolution and biology. J Virol. (1997) 71:4331–8.
30. Fregoso OI, Ahn J, Wang C, Mehrens J, Skowronski J, Emerman M. Evolutionary toggling of Vpx/Vpr specificity results in divergent recognition of the restriction factor SAMHD1. PLoS Pathog. (2013) 9:e1003496. doi: 10.1371/journal.ppat.1003496
31. Lim ES, Fregoso OI, McCoy CO, Matsen FA, Malik HS, Emerman M. The ability of primate lentiviruses to degrade the monocyte restriction factor SAMHD1 preceded the birth of the viral accessory protein Vpx. Cell Host Microbe. (2012) 11:194–204. doi: 10.1016/j.chom.2012.01.004
32. Baldauf HM, Pan X, Erikson E, Schmidt S, Daddacha W, Burggraf M, et al. SAMHD1 restricts HIV-1 infection in resting CD4(+) T cells. Nat Med. (2012) 18:1682–7. doi: 10.1038/nm.2964
33. Goujon C, Arfi V, Pertel T, Luban J, Lienard J, Rigal D, et al. Characterization of simian immunodeficiency virus SIVSM/human immunodeficiency virus type 2 Vpx function in human myeloid cells. J Virol. (2008) 82:12335–45. doi: 10.1128/JVI.01181-08
34. Lahouassa H, Daddacha W, Hofmann H, Ayinde D, Logue EC, Dragin L, et al. SAMHD1 restricts the replication of human immunodeficiency virus type 1 by depleting the intracellular pool of deoxynucleoside triphosphates. Nat Immunol. (2012) 13:223–8. doi: 10.1038/ni.2236
35. Descours B, Cribier A, Chable-Bessia C, Ayinde D, Rice G, Crow Y, et al. SAMHD1 restricts HIV-1 reverse transcription in quiescent CD4(+) T-cells, Retrovirology. (2012) 9:87. doi: 10.1186/1742-4690-9-87
36. Hofmann H, Logue EC, Bloch N, Daddacha W, Polsky SB, Schultz ML, et al. The Vpx lentiviral accessory protein targets SAMHD1 for degradation in the nucleus. J Virol. (2012) 86:12552–60. doi: 10.1128/JVI.01657-12
37. Belzile JP, Richard J, Rougeau N, Xiao Y, Cohen EA. HIV-1 Vpr induces the K48-linked polyubiquitination and proteasomal degradation of target cellular proteins to activate ATR and promote G2 arrest. J Virol. (2010) 84:3320–30. doi: 10.1128/JVI.02590-09
38. Romani B, Baygloo NS, Hamidi-Fard M, Aghasadeghi MR, Allahbakhshi E. HIV-1 Vpr protein induces proteasomal degradation of chromatin-associated class I HDACs to overcome latent infection of macrophages. J Biol Chem. (2016) 291:2696–711. doi: 10.1074/jbc.M115.689018
39. Schröfelbauer B, Yu Q, Zeitlin SG, Landau NR. Human immunodeficiency virus type 1 Vpr induces the degradation of the UNG and SMUG uracil-DNA glycosylases. J Virol. (2005) 79:10978–87. doi: 10.1128/JVI.79.17.10978-10987.2005
40. Ueno F, Shiota H, Miyaura M, Yoshida A, Sakurai A, Tatsuki J, et al. Vpx and Vpr proteins of HIV-2 up-regulate the viral infectivity by a distinct mechanism in lymphocytic cells. Microbes Infect. (2003) 5:387–95. doi: 10.1016/S1286-4579(03)00042-X
41. Wang H, Guo H, Su J, Rui Y, Zheng W, Gao W, et al. Inhibition of Vpx-mediated SAMHD1 and Vpr-mediated host helicase transcription factor degradation by selective disruption of viral CRL4 (DCAF1) E3 ubiquitin ligase assembly. J Virol. (2017) 91:e00225-17 doi: 10.1128/JVI.00225-17
42. Morellet N, Bouaziz S, Petitjean P, Roques BP. NMR structure of the HIV-1 regulatory protein VPR. J Mol Biol. (2003) 327:215–27. doi: 10.1016/S0022-2836(03)00060-3
43. Morellet N, Roques BP, Bouaziz S. Structure-function relationship of Vpr: biological implications. Curr HIV Res. (2009) 7:184–210. doi: 10.2174/157016209787581490
44. Wu Y, Zhou X, Barnes CO, DeLucia M, Cohen AE, Gronenborn AM, et al. The DDB1-DCAF1-Vpr-UNG2 crystal structure reveals how HIV-1 Vpr steers human UNG2 toward destruction. Nat Struc Mol Biol. (2016) 23:933–40. doi: 10.1038/nsmb.3284
45. Zhao LJ, Mukherjee S, Narayan O. Biochemical mechanism of HIV-I Vpr function. Specific interaction with a cellular protein. J Biol Chem. (1994) 269:15577–82.
46. Hrecka K, Hao C, Shun M–C, Kaur S, Swanson SK, Florens L, et al. HIV-1 and HIV-2 exhibit divergent interactions with HLTF and UNG2 DNA repair proteins. Proceedings of the National Academy of Sciences. (2016) 113, E3921–E30. doi: 10.1073/pnas.1605023113
47. Laguette N, Brégnard C, Hue P, Basbous J, Yatim A, Larroque M, et al. Premature activation of the SLX4 complex by Vpr promotes G2/M arrest and escape from innate immune sensing. Cell. (2014) 156:134–45. doi: 10.1016/j.cell.2013.12.011
48. Lahouassa H, Blondot ML, Chauveau L, Chougui G, Morel M, Leduc M, et al. HIV-1 Vpr degrades the HLTF DNA translocase in T cells and macrophages. Proceedings of the National Academy of Sciences. (2016) 113:5311–6. doi: 10.1073/pnas.1600485113
49. Yan J, Shun M-C, Hao C, Zhang Y, Qian J, Hrecka K, et al. (2018). HIV-1 Vpr reprograms CLR4DCAF1 E3 ubiquitin ligase to antagonize Exonuclease 1-mediated restriction of HIV-1 infection. mBio. 9. doi: 10.1128/mBio.01732-18
50. Zhou X, DeLucia M, Ahn J. SLX4-SLX1 protein-independent down-regulation of MUS81-EME1 protein by HIV-1 Viral protein R (Vpr). Journal of Biological Chemistry. (2016) 291:16936–47. doi: 10.1074/jbc.M116.721183
51. Rouzic EL, Belaïdouni N, Estrabaud E, Morel M, Rain J-C, Transy C, et al. (2007). HIV1 Vpr arrests the cell cycle by recruiting DCAF1/VprBP. a receptor of the Cul4-DDB1 ubiquitin ligase.Cell Cycle, 6:182–188. doi: 10.4161/cc.6.2.3732
52. Jacquot G, Le Rouzic E, Maidou-Peindara P, Maizy M, Lefrère J-J, Daneluzzi V, et al. (2009). Characterization of the Molecular Determinants of Primary HIV-1 Vpr Proteins: Impact of the Q65R and R77Q Substitutions on Vpr Functions. PLOS ONE. 4:e7514. doi: 10.1371/journal.pone.0007514
53. Hansen EC, Ransom M, Hesselberth JR, Hosmane NN, Capoferri AA, Bruner KM, et al. Diverse fates of uracilated HIV-1 DNA during infection of myeloid lineage cells. eLife. (2016) 5:e18447. doi: 10.7554/eLife.18447
54. Kennedy EM, Daddacha W, Slater R, Gavegnano C, Fromentin E, Schinazi RF, et al. Abundant non-canonical dUTP found in primary human macrophages drives its frequent incorporation by HIV-1 reverse transcriptase. J Biol Chem. (2011) 286:25047–55. doi: 10.1074/jbc.M111.234047
55. Langevin C, Maidou-Peindara P, Aas PA, Jacquot G, Otterlei M, Slupphaug G, et al. Human immunodeficiency virus type 1 Vpr modulates cellular expression of UNG2 via a negative transcriptional effect. J Virol. (2009) 83:10256–63. doi: 10.1128/JVI.02654-08
56. Weil AF, Ghosh D, Zhou Y, Seiple L, McMahon MA, Spivak AM, et al. Uracil DNA glycosylase initiates degradation of HIV-1 cDNA containing misincorporated dUTP and prevents viral integration. Proceedings of the National Academy of Sciences. (2013) 110:E448–E57. doi: 10.1073/pnas.1219702110
57. Ahn J, Vu T, Novince Z, Guerrero-Santoro J, Rapic-Otrin V, Gronenborn AM. HIV-1 Vpr loads Uracil DNA Glycosylase-2 onto DCAF1, a substrate recognition subunit of a Cullin 4A-RING E3 ubiquitin ligase for proteasome-dependent degradation. Journal of Biological Chemistry. (2010) 285:37333–41. doi: 10.1074/jbc.M110.133181
58. Berger G, Lawrence M, Hué S, Neil SJ. (2014). G2/M cell cycle arrest correlates with primate lentiviral Vpr interaction with the SLX4 complex. J Virol. 89:230–240. doi: 10.1128/JVI.02307-14
59. Fregoso OI, Emerman M. (2016). Activation of the DNA Damage Response Is a Conserved Function of HIV-1 and HIV-2 Vpr That Is Independent of SLX4 Recruitment. mBio.7. doi: 10.1128/mBio.01433-16
60. Greenwood EJD, Williamson JC, Sienkiewicz A, Naamati A, Matheson NJ, Lehner PJ. Promiscuous targeting of cellular proteins by Vpr drives systems-level proteomic remodeling in HIV-1 infection. Cell Rep. (2019) 27:1579–96e7. doi: 10.1016/j.celrep.2019.04.025
61. Achar YJ, Balogh D, Haracska L. Coordinated protein and DNA remodeling by human HLTF on stalled replication fork. Proc Natl Acad Sci USA. (2011) 108:14073–8. doi: 10.1073/pnas.1101951108
62. Keijzers G, Bakula D, Petr MA, Madsen NGK, Teklu A, Mkrtchyan G, et al. Human Exonuclease 1 (EXO1) regulatory functions in DNA replication with putative roles in cancer. Int J Mol Sci. (2018) 20:74. doi: 10.3390/ijms20010074
63. Brégnard C, Benkirane M, Laguette N. DNA damage repair machinery and HIV escape from innate immune sensing. Front Microbiol. (2014) 5:176. doi: 10.3389/fmicb.2014.00176
64. Weitzman MD, Fradet-Turcotte A. Virus DNA replication and the host DNA damage response. Annu Rev Virol. (2018) 5:141–64. doi: 10.1146/annurev-virology-092917-043534
65. Hossain H, Tchatalbachev S, Chakraborty T. Host gene expression profiling in pathogen–host interactions. Curr Opin Immunol. (2006) 18:422–9. doi: 10.1016/j.coi.2006.05.018
66. Mariggio G, Koch S, Zhang G, Weidner-Glunde M, Ruckert J, Kati S, et al. Kaposi Sarcoma Herpesvirus (KSHV) Latency-Associated Nuclear Antigen (LANA) recruits components of the MRN (Mre11-Rad50-NBS1) repair complex to modulate an innate immune signaling pathway and viral latency. PLoS Pathog. (2017) 13:e1006335. doi: 10.1371/journal.ppat.1006335
67. Tarakanova VL, Leung-Pineda V, Hwang S, Yang CW, Matatall K, Basson M, et al. Gamma-herpesvirus kinase actively initiates a DNA damage response by inducing phosphorylation of H2AX to foster viral replication. Cell Host Microbe. (2007) 1:275–86. doi: 10.1016/j.chom.2007.05.008
68. Nakai-Murakami C, Shimura M, Kinomoto M, Takizawa Y, Tokunaga K, Taguchi T, et al. HIV-1 Vpr induces ATM-dependent cellular signal with enhanced homologous recombination. Oncogene. (2007) 26:477–86. doi: 10.1038/sj.onc.1209831
69. Wu ZH, Shi Y, Tibbetts RS, Miyamoto S. Molecular linkage between the kinase ATM and NF-kappaB signaling in response to genotoxic stimuli. Science. (2006) 311:1141–6. doi: 10.1126/science.1121513
70. Lv L, Wang Q, Xu Y, Tsao LC, Nakagawa T, Guo H, et al. Vpr targets TET2 for degradation by CRL4VprBP E3 ligase to sustain IL-6 expression and enhance HIV-1 replication. Mol Cell. (2018) 70:961–70.e5. doi: 10.1016/j.molcel.2018.05.007
71. van Breugel PC, Robert EI, Mueller H, Decorsière A, Zoulim F, Hantz O, et al. Hepatitis B virus X protein stimulates gene expression selectively from extrachromosomal DNA templates. Hepatology. (2012) 56:2116–24. doi: 10.1002/hep.25928
72. Maudet C, Sourisce A, Dragin L, Lahouassa H, Rain J-C, Bouaziz S, et al. HIV-1 Vpr induces the degradation of ZIP and sZIP, adaptors of the NuRD chromatin remodeling complex, by hijacking DCAF1/VprBP. PLoS ONE. (2013) 8:e77320. doi: 10.1371/journal.pone.0077320
73. Epanchintsev A, Costanzo F, Rauschendorf MA, Caputo M, Ye T, Donnio LM, et al. Cockaynes syndrome A and B proteins regulate transcription arrest after genotoxic stress by promoting ATF3 degradation. Mol Cell. (2017) 68:1054–66.e6. doi: 10.1016/j.molcel.2017.11.009
74. Poli G, Bressler P, Kinter A, Duh E, Timmer WC, Rabson A, et al. Interleukin 6 induces human immunodeficiency virus expression in infected monocytic cells alone and in synergy with tumor necrosis factor alpha by transcriptional and post-transcriptional mechanisms. J Exp Med. (1990) 172:151–8. doi: 10.1084/jem.172.1.151
75. Weissman D, Poli G, Fauci AS. Interleukin 10 blocks HIV replication in macrophages by inhibiting the autocrine loop of Tumor Necrosis Factor α and Interleukin 6 induction of virus. AIDS Res Hum Retroviruses. (1994) 10:1199–206. doi: 10.1089/aid.1994.10.1199
76. Zhang Q, Zhao K, Shen Q, Han Y, Gu Y, Li X, et al. Tet2 is required to resolve inflammation by recruiting Hdac2 to specifically repress IL-6. Nature. (2015) 525:389–93. doi: 10.1038/nature15252
77. Chéné Id, Basyuk E, Lin YL, Triboulet R, Knezevich A, Chable-Bessia C, et al. Suv39H1 and HP1γ are responsible for chromatin-mediated HIV-1 transcriptional silencing and post-integration latency. EMBO J. (2007) 26:424–35. doi: 10.1038/sj.emboj.7601517
78. Tchasovnikarova IA, Timms RT, Matheson NJ, Wals K, Antrobus R, Göttgens B, et al. GENE SILENCING. Epigenetic silencing by the HUSH complex mediates position-effect variegation in human cells. Science. (2015) 348:1481–5. doi: 10.1126/science.aaa7227
79. Chougui G, Munir-Matloob S, Matkovic R, Martin MM, Morel M, Lahouassa H, et al. HIV-2/SIV viral protein X counteracts HUSH repressor complex. Nat Microbiol. (2018) 3:891–7. doi: 10.1038/s41564-018-0179-6
80. Yurkovetskiy L, Guney MH, Kim K, Goh SL, McCauley S, Dauphin A, et al. Primate immunodeficiency virus proteins Vpx and Vpr counteract transcriptional repression of proviruses by the HUSH complex. Nat Microbiol. (2018) 3:1354. doi: 10.1038/s41564-018-0256-x
81. Zhu Y, Wang GZ, Cingöz O, Goff SP. NP220 mediates silencing of unintegrated retroviral DNA. Nature. (2018) 564:278–82. doi: 10.1038/s41586-018-0750-6
82. Hachiya R, Shiihashi T, Shirakawa I, Iwasaki Y, Matsumura Y, Oishi Y, et al. The H3K9 methyltransferase Setdb1 regulates TLR4-mediated inflammatory responses in macrophages. Sci Rep. (2016) 6:28845. doi: 10.1038/srep28845
83. Xu G, Liu G, Xiong S, Liu H, Chen X, Zheng B. The histone methyltransferase Smyd2 is a negative regulator of macrophage activation by suppressing interleukin 6 (IL-6) and tumor necrosis factor alpha (TNF-alpha) production. J Biol Chem. (2015) 290:5414–23. doi: 10.1074/jbc.M114.610345
84. Triboulet R, Mari B, Lin YL, Chable-Bessia C, Bennasser Y, Lebrigand K, et al. Suppression of microRNA-silencing pathway by HIV-1 during virus replication. Science. (2007) 315:1579–82. doi: 10.1126/science.1136319
85. Umbach JL, Cullen BR. The role of RNAi and microRNAs in animal virus replication and antiviral immunity. Genes Dev. (2009) 23:1151–64. doi: 10.1101/gad.1793309
86. Nathans R, Chu CY, Serquina AK, Lu CC, Cao H, Rana TM. Cellular microRNA and P bodies modulate host-HIV-1 interactions. Mol Cell. (2009) 34:696–709. doi: 10.1016/j.molcel.2009.06.003
87. Valencia-Sanchez MA, Liu J, Hannon GJ, Parker R. Control of translation and mRNA degradation by miRNAs and siRNAs. Genes Dev. (2006) 20:515–24. doi: 10.1101/gad.1399806
88. Seeley JJ, Baker RG, Mohamed G, Bruns T, Hayden MS, Deshmukh SD, et al. Induction of innate immune memory via microRNA targeting of chromatin remodelling factors. Nature. (2018) 559:114–9. doi: 10.1038/s41586-018-0253-5
89. Casey Klockow L, Sharifi HJ, Wen X, Flagg M, Furuya AK, Nekorchuk M, et al. The HIV-1 protein Vpr targets the endoribonuclease Dicer for proteasomal degradation to boost macrophage infection. Virology. (2013) 444:191–202. doi: 10.1016/j.virol.2013.06.010
90. Balasubramaniam M, Pandhare J, Dash C. Are microRNAs important players in HIV-1 infection? An update. Viruses. (2018) 10:110. doi: 10.3390/v10030110
91. Agosto LM, Uchil PD, Mothes W. HIV cell-to-cell transmission: effects on pathogenesis and antiretroviral therapy. Trends Microbiol. (2015) 23:289–95. doi: 10.1016/j.tim.2015.02.003
92. Gummuluru S, Pina Ramirez NG, Akiyama H. CD169-dependent cell-associated HIV-1 transmission: a driver of virus dissemination. J Infect Dis. (2014) 210(Suppl. 3):S641–7. doi: 10.1093/infdis/jiu442
93. Collins DR, Lubow J, Lukic Z, Mashiba M, Collins KL. Vpr promotes macrophage-dependent HIV-1 infection of CD4+ T lymphocytes. PLOS Pathog. (2015) 11:e1005054. doi: 10.1371/journal.ppat.1005054
94. Mashiba M, Collins DR, Terry VH, Collins KL. Vpr overcomes macrophage-specific restriction of HIV-1 Env expression and virion production. Cell Host Microbe. (2014) 16:722–35. doi: 10.1016/j.chom.2014.10.014
95. Muthumani K, Hwang DS, Choo AY, Mayilvahanan S, Dayes NS, Thieu KP, et al. HIV-1 Vpr inhibits the maturation and activation of macrophages and dendritic cells in vitro. Int Immunol. (2005) 17:103–16. doi: 10.1093/intimm/dxh190
96. Majumder B, Janket ML, Schafer EA, Schaubert K, Huang XL, Kan-Mitchell J, et al. Human immunodeficiency virus type 1 Vpr impairs dendritic cell maturation and T-cell activation: implications for viral immune escape. J Virol. (2005) 79:7990–8003. doi: 10.1128/JVI.79.13.7990-8003.2005
97. Roux P, Alfieri C, Hrimech M, Cohen EA, Tanner JE. Activation of transcription factors NF-κB and NF-IL-6 by human immunodeficiency virus type 1 protein R (Vpr) induces Interleukin-8 expression. J Virol. (2000) 74:4658–65. doi: 10.1128/JVI.74.10.4658-4665.2000
98. Liu R, Lin Y, Jia R, Geng Y, Liang C, Tan J, et al. HIV-1 Vpr stimulates NF-kappaB and AP-1 signaling by activating TAK1. Retrovirology. (2014) 11:45. doi: 10.1186/1742-4690-11-45
99. Kaczmarek K, Morales A, Henderson AJ. T Cell Transcription Factors and Their Impact on HIV Expression. Virology. (2013) 2013:41–7. doi: 10.4137/VRT.S12147
100. Schiralli Lester GM, Henderson AJ. Mechanisms of HIV transcriptional regulation and their contribution to latency. Mol Biol Int. (2012) 2012:614120. doi: 10.1155/2012/614120
101. Akiyama H, Miller CM, Ettinger CR, Belkina AC, Snyder-Cappione JE, Gummuluru S. HIV-1 intron-containing RNA expression induces innate immune activation and T cell dysfunction. Nat Commun. (2018) 9:3450. doi: 10.1038/s41467-018-05899-7
102. Groom JR, Luster AD. CXCR3 ligands: redundant, collaborative and antagonistic functions. Immunol Cell Biol. (2011) 89:207–15. doi: 10.1038/icb.2010.158
103. Lin JX, Leonard WJ. The common cytokine receptor gamma chain family of cytokines. Cold Spring Harb Perspect Biol. (2018) 10:a028449. doi: 10.1101/cshperspect.a028449
104. Manganaro L, Hong P, Hernandez MM, Argyle D, Mulder LCF, Potla U, et al. IL-15 regulates susceptibility of CD4+ T cells to HIV infection. Proc Natl Acad Sci USA. (2018) 115:E9659–E67. doi: 10.1073/pnas.1806695115
105. Okumura A, Alce T, Lubyova B, Ezelle H, Strebel K, Pitha PM. HIV-1 accessory proteins VPR and Vif modulate antiviral response by targeting IRF-3 for degradation. Virology. (2008) 373:85–97. doi: 10.1016/j.virol.2007.10.042
106. Harman AN, Nasr N, Feetham A, Galoyan A, Alshehri AA, Rambukwelle D, et al. HIV blocks interferon induction in human dendritic cells and macrophages by dysregulation of TBK1. J Virol. (2015) 89:6575–84. doi: 10.1128/JVI.00889-15
107. Akiyama H, Ramirez NG, Gudheti MV, Gummuluru S. CD169-mediated trafficking of HIV to plasma membrane invaginations in dendritic cells attenuates efficacy of anti-gp120 broadly neutralizing antibodies. PLoS Pathog. (2015) 11:e1004751. doi: 10.1371/journal.ppat.1004751
108. Puryear WB, Akiyama H, Geer SD, Ramirez NP, Yu X, Reinhard BM, et al. Interferon-inducible mechanism of dendritic cell-mediated HIV-1 dissemination is dependent on Siglec-1/CD169. PLoS Pathog. (2013) 9:e1003291. doi: 10.1371/journal.ppat.1003291
109. Cribbs SK, Lennox J, Caliendo AM, Brown LA, Guidot DM. Healthy HIV-1-infected individuals on highly active antiretroviral therapy harbor HIV-1 in their alveolar macrophages. AIDS Res Hum Retroviruses. (2015) 31:64–70. doi: 10.1089/aid.2014.0133
110. Estes JD, Kityo C, Ssali F, Swainson L, Makamdop KN, Del Prete GQ, et al. Defining total-body AIDS-virus burden with implications for curative strategies. Nat Med. (2017) 23:1271–6. doi: 10.1038/nm.4411
111. Honeycutt JB, Thayer WO, Baker CE, Ribeiro RM, Lada SM, Cao Y, et al. HIV persistence in tissue macrophages of humanized myeloid-only mice during antiretroviral therapy. Nat Med. (2017) 23:638–43. doi: 10.1038/nm.4319
112. Lamers SL, Rose R, Maidji E, Agsalda-Garcia M, Nolan DJ, Fogel GB, et al. HIV DNA is frequently present within pathologic tissues evaluated at autopsy from combined antiretroviral therapy-treated patients with undetectable viral loads. J Virol. (2016) 90:8968–83. doi: 10.1128/JVI.00674-16
113. McElrath MJ, Smythe K, Randolph-Habecker J, Melton KR, Goodpaster TA, Hughes SM, et al. Comprehensive assessment of HIV target cells in the distal human gut suggests increasing HIV susceptibility toward the anus. J Acquir Immune Defic Syndr. (2013) 63:263–71. doi: 10.1097/QAI.0b013e3182898392
114. Yukl SA, Sinclair E, Somsouk M, Hunt PW, Epling L, Killian M, et al. (2014). A comparison of methods for measuring rectal HIV levels suggests that HIV DNA resides in cells other than CD4+ T cells, including myeloid cells. AIDS. 28:439–442. doi: 10.1097/QAD.0000000000000166
Keywords: HIV, Vpr, myeloid, DDR, DCAF, VprBP, ubiquitin ligase
Citation: Nodder SB and Gummuluru S (2019) Illuminating the Role of Vpr in HIV Infection of Myeloid Cells. Front. Immunol. 10:1606. doi: 10.3389/fimmu.2019.01606
Received: 22 March 2019; Accepted: 27 June 2019;
Published: 23 July 2019.
Edited by:
Paul Urquhart Cameron, The University of Melbourne, AustraliaReviewed by:
Monsef Benkirane, Centre National de la Recherche Scientifique (CNRS), FranceNathaniel Roy Landau, NYU School of Medicine, United States
Copyright © 2019 Nodder and Gummuluru. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Suryaram Gummuluru, rgummulu@bu.edu