 Jennifer L. Hope
Jennifer L. Hope Christopher J. Stairiker
Christopher J. Stairiker Eun-Ah Bae
Eun-Ah Bae Dennis C. Otero
Dennis C. Otero Linda M. Bradley*
Linda M. Bradley*
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol. , 17 July 2019
Sec. Immunological Memory
Volume 10 - 2019 | https://doi.org/10.3389/fimmu.2019.01595
This article is part of the Research Topic Immune Responses to Persistent or Recurrent Antigens: Implications for Immunological Memory and Immunotherapy View all 8 articles
Effective adaptive immune responses are characterized by stages of development and maturation of T and B cell populations that respond to disturbances in the host homeostasis in cases of both infections and cancer. For the T cell compartment, this begins with recognition of specific peptides by naïve, antigen-inexperienced T cells that results in their activation, proliferation, and differentiation, which generates an effector population that clears the antigen. Loss of stimulation eventually returns the host to a homeostatic state, with a heterogeneous memory T cell population that persists in the absence of antigen and is primed for rapid responses to a repeat antigen exposure. However, in chronic infections and cancers, continued antigen persistence impedes a successful adaptive immune response and the formation of a stereotypical memory population of T cells is compromised. With repeated antigen stimulation, responding T cells proceed down an altered path of differentiation that allows for antigen persistence, but much less is known regarding the heterogeneity of these cells and the extent to which they can become “memory-like,” with a capacity for self-renewal and recall responses that are characteristic of bona fide memory cells. This review focuses on the differentiation of CD4+ and CD8+ T cells in the context of chronic antigen stimulation, highlighting the central observations in both human and mouse studies regarding the differentiation of memory or “memory-like” T cells. The importance of both the cellular and molecular drivers of memory T cell development are emphasized to better understand the consequences of persisting antigen on T cell fates. Integrating what is known and is common across model systems and patients can instruct future studies aimed at further understanding T cell differentiation and development, with the goal of developing novel methods to direct T cells toward the generation of effective memory populations.
T cells are essential for the adaptive immune system's responses to pathogens and tumors. They are vital for the clearance of host cells that become infected with viruses and intracellular bacteria as well as the elimination of tumor cells (1, 2). T cell memory is typically defined as a residual compartment of protective antigen-specific T cell that persists long after contraction of the effector pool and survives in the absence of antigen (3). It is an important distinction that antigenic withdrawal does not occur during chronic infections and cancer (Figure 1) despite prolonged survival of responding T cells. Therefore, further insights are required into the differentiation of T cells in these contexts and in settings where clearance of a once chronic antigen ultimately occurs. These include (1) identifying characteristic phenotypic markers and transcriptional profiles, (2) ascertaining the capacity for self-renewal, and (3) determining the ability for rapid re-activation and generation of polyfunctional responses (4, 5). The focus of this review is to highlight the known differences in memory CD4+ and CD8+ T cell development in the context of chronic pathogen infections or cancer progression as compared to acute infections in both mice and humans, with an emphasis on the cellular and molecular drivers of T cell memory development under these conditions.
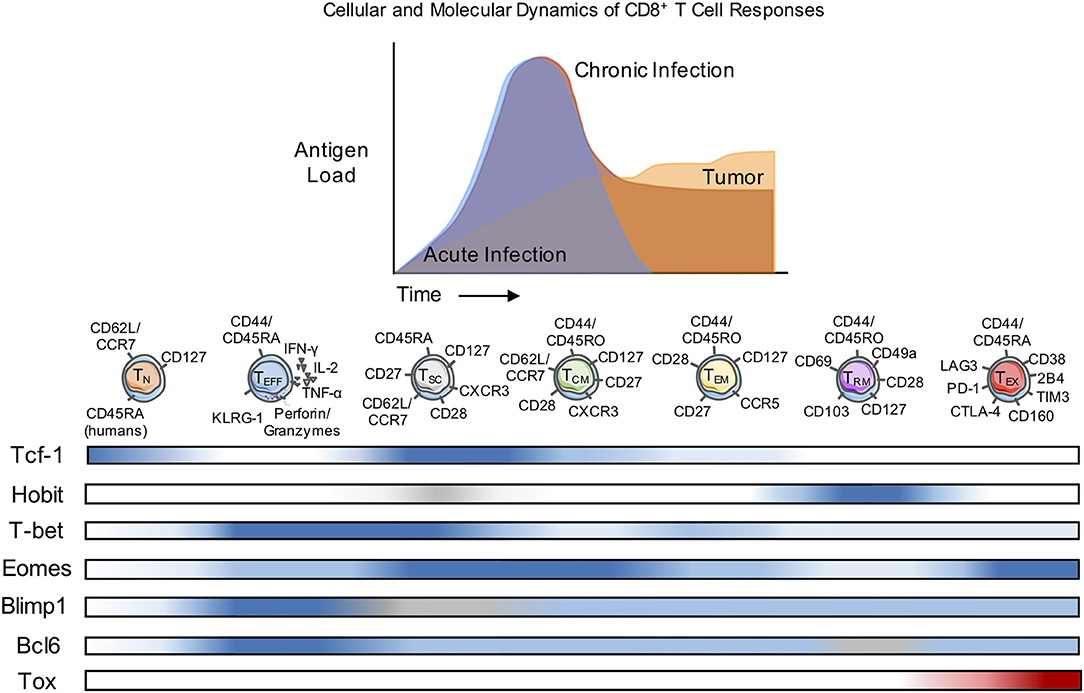
Figure 1. Cellular and Molecular Dynamics of CD8+ T Cell Responses. Naïve CD8+ T cells (TN) differentiate into diverse subsets with unique patterns of transcription factor (TF) expression and corresponding cell surface markers. The diversity of subset differentiation is highly influenced by the antigen or pathogen load in mice and patients. During acute infection, TN cells give rise to polyfunctional, highly-proliferative effector (TEFF) CD8+ T cells that clear intracellular pathogens. Following contraction of TEFF after antigen clearance, memory (TCM, TEM, and TRM) CD8+ T cells persist and rapidly respond upon re-infection. During chronic infection or in response to tumors, TEFF also arise from Naïve, but often fail to effectively clear the infection or tumor and in response to persistent antigen can develop into TEX with reduced function. In mice and patients, TSC have been identified as a population of CD8+ T cells that respond to checkpoint blockade therapy. The development of classically-defined TCM, TEM, and TRM during chronic infection or cancer remains contested and the differentiation of “memory-like” T cell populations is discussed within this review. Blue, level of expression of TF; white, no expression; gray, expression unknown; red, characteristic expression in TEX.
During the primary T cell response to infection or tumors, the antigen-specific T cell pool becomes highly heterogeneous, forming different subpopulations of CD4+ and CD8+ T cells defined by surface marker expression, transcription factors, cytokine production, and cytotoxic or memory-forming potential (Figure 1). Ultimately, with antigen clearance, the large majority of antigen-specific T cells die and a smaller pool of memory T cells that retain the capacity to respond to re-challenge can persist, often indefinitely (6, 7). However, memory T cells are also highly diverse, with substantial differences described for a variety of infections, implying the importance of contextual cues such as the duration of antigen exposure as well as the tissue localization and distribution of infection. Much less is known about the differentiation of memory CD4+ T cells compared to CD8+ T cells, in part because of the ability of naïve CD4+ T cells to adopt different effector cell fates that are uniquely regulated and are elicited by different infections. However, studies of circulating memory CD4+ (and CD8+) T cells in humans were the first to define effector memory T cells (TEM: CD45RA+CD127+CD62L−/CCR7−), and central memory cells (TCM: CD45RA+CD127+CD62L+/CCR7+) that primarily differ with respect to the circulation through secondary lymphoid organs and the capacity for self-renewal (TCM > TEM) (8). In contrast to the extensive literature on the development of T cell memory to infections, much less is known about the characteristics of memory T cells that develop in responses to tumors.
There is considerable diversity in the fates of a naïve T cell and the mechanisms regulating the formation and promotion of heterogeneity in effector and memory T cell pools are of great interest, particularly in the context of vaccine development. Several models of memory T cell differentiation have been proposed and previously discussed elsewhere, but there is currently robust evidence for the “one cell, multiple fates” model (9, 10). In mice, fate-mapping and memory differentiation of CD8+ T cells that were assessed by Dirk et al. by performing adoptive transfer of single naïve OT-I TCR transgenic CD8+ T cells into recipient mice, followed by infection with OVA-expressing bacterium Listeria monocytogenes (Lm-OVA) (11, 12). These studies conclusively demonstrated that a naïve T cell could subsequently differentiate into both effector and memory T cells. Another study from Schumacher et al also assessed memory CD8+ T cell differentiation by fate-mapping analysis of adoptively transferred T cells, but used DNA-barcoded, transduced thymocytes from OT-I mice that were injected intra-thymically into young recipients, followed by infection with Lm-OVA (10). This study showed that a single antigen-specific naive CD8+ T cell gave rise to daughter cells with multiple phenotypes, including TCM and TEM subsets. Furthermore, the progeny of a single naïve CD8+ T cell could efficiently seed the secondary lymphoid organs (bone marrow, blood, spleen, and lymph nodes) and were not restricted to a particular anatomical location. Importantly, barcoded memory CD8+ T cells that were transferred into tertiary hosts maintained barcode diversity upon re-challenge, indicating the potential for all clones to respond. The fundamental question of whether effector T cells can give rise to memory cells was also demonstrated by fate mapping studies in which effector CD8+ T cells, identified by acquisition of the cytotoxic protein granzyme B, were shown to form memory (13). A more recent study demonstrated the ability of effector CD8+ T cells to “de-differentiate” into memory T cells by epigenetic remodeling associated with alterations in the DNA methylation programs of the cells (14). Together, these groups and others have demonstrated that indeed one naïve CD8+ T cell has the potential to give rise to daughter cells with differing phenotypes and fates, and that effector differentiation does not preclude memory development. However, to our knowledge such comprehensive studies addressing CD4+ T cell memory development have yet to be published and is a significant gap in understanding overall T cell memory differentiation.
Signals determining memory T cell generation remain incompletely understood. It is evident that antigen availability and timing of entry into a response are important determinants for memory formation. In general, weaker TCR signals are thought to favor memory T cell development, which can be influenced by TCR affinity, tissue localization with respect to antigen distribution, or by progressive antigen clearance (15). For CD8+ T cells, there is evidence that unique TCR signaling and organization of the TCR signaling complex that engages NF-kB signaling dictates memory development (16). These findings along with observations that CD8+ memory T cell precursors can be distinguished early in responses to acute infections in some models (e.g., LCMV Armstrong and L. monocytogenes) support the concept that early events are critical for memory development. Other external signals such as from cytokines during the effector phase also contribute to memory T cell differentiation. For example, type I interferon (IFN) or IL-2 signaling is key to the sustained survival of CD8+, and CD4+ T cells, respectively, during memory formation during the primary response (17, 18). Signaling through CD28 is required for the re-activation of memory CD8+ T cells and optimal recall responses of memory CD4+ T cells (7, 19). Co-stimulation of T cells such as through CD28 enhances their survival and effector function by increasing expression of the anti-apoptosis regulator BCL-XL, as well as by inducing T cell expansion, by the production of IL-2 (20, 21). Cytokines that include type I IFNs and IL-12 induce changes in the transcription factors T-box expressed in T cells (T-bet) and Eomesodermin (Eomes) (22, 23), which play important roles in regulating effector and memory T cell differentiation (24, 25) as summarized below. CD4+ and CD8+ T cells also influence each other during memory T cell development. Although CD4+ T cells are dispensable for the generation of primary effector CD8+ T cell responses to some infections, CD27 on CD8+ T cells interacting with CD70 on APCs primed by CD4+ T cell “help” via CD40(APC)/CD40L(CD4+ T cell) activation is required for the generation of functional memory CD8+ T cells marked by reduced proliferative capacity during recall responses (26).
Following a primary adaptive immune response, distinct subsets of memory T cells are found within the lymphoid organs that include not only TCM or TEM, but also more recently defined memory cells that become resident in the initial site of the primary infection or tumor (TRM). All three subsets play roles in protective memory responses, although TRM are likely to provide a first line of defense against a tissue localized re-infection. The TRM compartment was first characterized in the skin where these cells control reinfection with herpes simplex virus (HSV), and have since been identified as key mediators of immunity in the lung, such as in response to RSV and influenza; and in the gut after infection with Lm or LCMV (27–29). Although TRM have been identified by phenotype in tumors, their functions are not yet established (30). The TRM pool contains two subsets distinguished by CD103 expression (also known as integrin alpha E), a receptor for E-cadherin (31). Current studies are focusing on possible functional differences between the CD103+ and CD103− subsets of TRM. Another recently-defined memory T cell subset is considered to have stem cell-like properties with respect to self-renewal, and has been designated stem cell memory T cells (TSCM). Unlike other memory CD8+ T cell subsets, these cells maintain a naïve-like phenotype yet have a high proliferative capacity (32). Surface marker expression remains one of the primary methods for the classification of these different memory T cell subpopulations in both humans and mice (Table 1), and several distinctions apply more narrowly to memory cells generated in responses to specific infections. The presence of many of phenotypic and functional distinctions of memory cells has been much less well-studied in anti-tumor responses. It is now evident that a spectrum of surface phenotypes can arise primarily because of contextual cues and that these may be fluid and insufficient to fully define memory T cell subsets. However, in combination with analyses of transcription factor expression, greater insights into distinct features of memory T cell fates emerge.
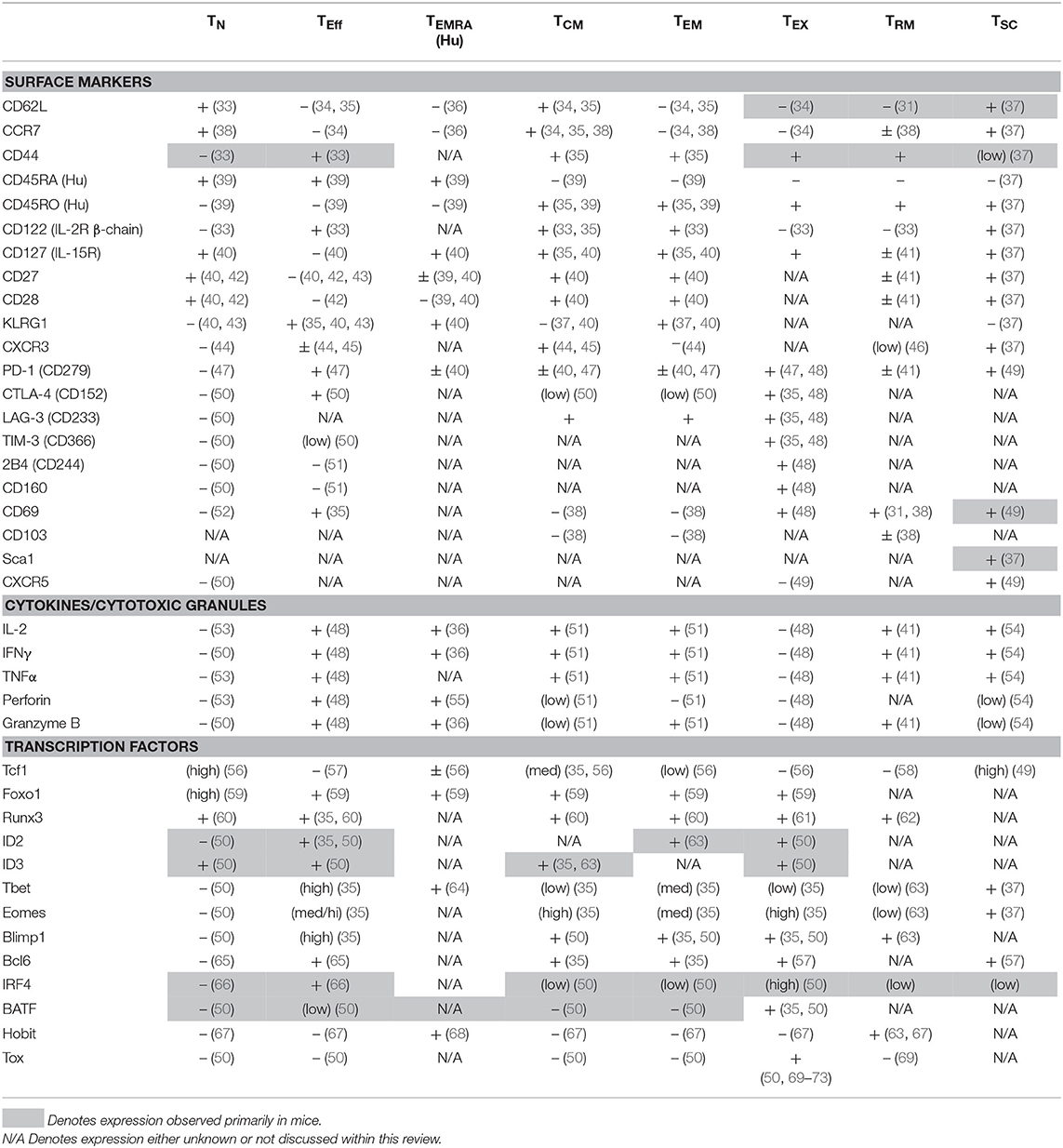
Table 1. Expression profiles of CD8+ T cell subsets.
Transcription factors are well-recognized as key regulators of T cell fate determination. In both CD4+ and CD8+ T cells, opposing levels of transcription factor pairs can strongly correlate with the T cell memory subsets. Examples of these gradients of transcription factors are T-bet vs. Eomes, B lymphocyte-induced maturation protein-1 (Blimp-1) vs. B-cell lymphoma 6 (Bcl-6), and Inhibitor of DNA binding 2 (Id2) vs. Inhibitor of DNA binding 3 (Id3) (65, 74, 75). At the memory stage, Eomes is more highly expressed than T-bet. Similarly, while Blimp-1 is highly expressed in effector cells and reciprocally Bcl-6 expression increases in memory cells; a similar relationship has been observed between Id2 and Id3. There is also a supportive role for transcription factor expression during early differentiation in memory formation and maintenance, specifically the Forkhead box proteins O-class proteins (Foxo) Foxo1 and Foxo3 (59, 76). Understanding how these transcription factors interact with each other remains an active area of research; for example, Foxo1 is known to regulate other transcription factors such as by increasing Tcf-1, Eomes, and Bcl-2 expression, while repressing the levels of T-bet. Within memory T cell subsets, there are also unique expression patterns of transcription factors, several of which are outlined in Table 1 and highlighted in Figure 1. These transcription factors are altered in exhausted T cells (TEX), and interestingly, expression patterns of some transcription factors associated with memory T cell formation are shared with TEX that persist during chronic infection or cancer suggesting that re-stimulation of more “memory-like” T cells could contribute to achieving a balance between terminal T cell differentiation and pathogen or tumor control and the extent of T cell exhaustion, which has been extensively reviewed elsewhere (77).
Changes in the stimulatory conditions encountered by effector T cells could also impact the development of memory T cells, as reduced inflammation caused by increasing antigen control could limit the extent of the effector T cell response, particularly in the infection or tumor the draining lymphoid tissues. Thus, conditions that are highly influenced by: (1) the cellular microenvironment, and (2) changes in the molecular regulation of the responding cells are likely to be key in determining whether T cells become terminally differentiated or “memory-like” and thus lead to a spectrum of functionally heterogeneous T cells. An important consideration when contrasting T cell differentiation and development in responses to chronic infection or tumors is the influence of a highly systemic inflammatory response observed in some chronic infections and model systems compared to the more localized microenvironment typically associated with the early stages of cancer. However, there is evidence in both humans and mice of memory or “memory-like” CD4+ and CD8+ T cell formation under conditions of repeated stimulation. The following sections will outline the effects that persistent pathogenic infections or tumors have on CD4+ and CD8+ memory or “memory-like” T cell development and responses, and the interplay between the two.
The defining cellular environment of memory CD8+ T cells is a compilation of interactions with other cellular compartments and the localization of the cell (such as in circulation, lymphoid tissues, or non-lymphoid tissues). In both acute infections and upon re-challenge, secondary lymphoid organs such as the peripheral lymph nodes are important sites of naïve or memory CD8+ T cell activation during systemic viral infections such as LCMV. Here, dendritic cells (DC) that have captured and present LCMV antigens activate CD8+ T cells. Surrounding the interacting T cell-DC conjugates are fibroblastic reticular cells (FRC), which can either promote and accelerate T cell activation, or conversely can inhibit subsequent expansion within the lymph nodes via the production of nitric oxide (78). FRC also constitutively express PD-L1, the ligand for the T cell inhibitory receptor PD-1 (programmed cell death protein 1, CD279) (79). During chronic LCMV infection, PD-L1 expression on FRCs is elevated and the network that supports T cell migration is disrupted, leading to altered localization (80). These changes in the lymphoid tissue architecture are thought to promote T cell exhaustion and impede memory formation. Persistent viral infections, such as LCMV in mice and Human Immunodeficiency Virus-1 (HIV-1) and Hepatitis C Virus (HCV) in humans, can also result in the inhibition and loss of type 1 T helper (TH1) CD4+ T cell responses that play a major role in supporting memory CD8+ T cell development (81–83). During LCMV chronic infection, there is a progressive decline in virus-specific CD8+ T cells; however, reconstituting the TH1 CD4+ T cell compartment is sufficient to maintain the LCMV-specific CD8+ T cell population, providing greater evidence for a supportive role for CD4+ T cells in maintaining long-lasting CD8+ T cells that continue to undergo progressive differentiation (83).
Localization of virus-specific CD8+ T cells in other non-lymphoid sites such as the kidney, liver, and lungs during chronic LCMV infection has been previously described, with evidence that they too exhibit signs of functional exhaustion with decreased IFN-γ and TNF-α production upon ex vivo stimulation compared to conventional LCMV-specific memory CD8+ T cells (84). Similarly, in a chronic parasitic infection model of Trypanosoma cruzi in mice, muscle-resident CD8+ T cells have decreased effector function (85). The majority of long-lived CD8+ T cells in the lung, liver, and kidneys after chronic LCMV infection fail to express CD103; however, LCMV-specific intraepithelial CD8+ T cells found within the small intestine and lamina propria express both CD103 and CD69 (86), which establishes tissue localization via the G-protein-coupled receptor sphingosine-1-phosphate receptor (S1PR1) (87). However, whether these TEX in non-lymphoid tissues share features with TRM or provide a major role in maintaining chronic infection has not been studied.
While our understanding of CD8+ T cell differentiation and “memory-like” development during chronic infections has largely been derived from mouse model systems, several studies have focused on dissecting human virus-specific CD8+ T cell differentiation under persistent viral infections including HIV-1, HCV, Epstein Barr virus (EBV), and cytomegalovirus (CMV) through the use of Human Leukocyte Antigen class I (HLA-I) tetramers complexed with peptides of virus-derived CD8+ T cell-specific epitopes. Both CD27 and CD28 expression levels have been used to classify the differentiation state of CD8+ T cells and are regularly used in connection with CD45RA and CCR7 to distinguish effector and memory T cells. One study identified unique patterns of CD8+ T cell differentiation in the periphery based on the specific viral infection, finding a greater frequency of CD28+ virus-specific CD8+ T cells from HCV-infected patients compared to HIV, CMV, or EBV; conversely, the frequency of CD27+ virus-specific CD8+ T cells was lower in CMV (88). The expression of CD57, meanwhile, has been linked to both CD8+ T cell memory subsets (both TCM and TEM) but also senescent or functionally exhausted CD8+ T cells, adding to the complexity of differentiating between “memory-like” vs. exhausted human CD8+ T cell subsets (89, 90). In some patients, infection with Mycobacterium tuberculosis (TB) can result in latent infection (LTBI) where the bacteria remain quiescent until re-activation. Unlike during active TB infection, the differentiation of CD8+ T cells in LTBI patients is highly skewed toward terminally differentiated effector memory cells (TEMRA) as opposed to the TEM compartment (91). Together, these highlighted studies demonstrate how different infections, despite their chronicity or latency, can drive a highly heterogeneous memory CD8+ T cell population in patients.
An important consideration in defining TEX is the co-expression of inhibitory receptors including PD-1, CTLA-4, LAG3, TIM3, 2B4, and CD160. Expression of a single inhibitory receptor is insufficient to define TEX, as some inhibitory receptors such as PD-1 are upregulated upon T cell activation and therefore can also serve as activation markers. A major distinction between exhausted and memory CD8+ T cells is the ability to produce cytokines such as IFN-γ, TNF-α, and IL-2 upon TCR stimulation. Terminally exhausted T cells, initially described in the chronic LCMV infection model in mice, demonstrate a marked reduction or inability to produce these cytokines upon re-stimulation, and it is this concurrent loss of polyfunctionality (the ability to produce multiple cytokines and mediate toxicity) and increasing inhibitory receptor co-expression that is crucial when defining TEX. Greater insight into T cell exhaustion including molecular and cellular drivers of exhaustion is thoroughly reviewed in McLane et al. (77). In contrast, re-stimulation of memory CD8+ T cells results in a high level of cytokine production that is associated with low co-expression of inhibitory receptors. Insight into T cell responses during chronic infection was provided by the observation that antigen-load plays a crucial role in the development of an exhausted CD8+ T cell phenotype, as decreasing the abundance solely of GP33 (an LCMV-derived epitope recognized by CD8+ T cells) on the virus while maintaining viral loads and other LCMV-derived epitopes resulted in reduced expression of PD-1 and elevated dual-production of IFN-γ and TNF-α by P14 (GP33-specific TCR transgenic) CD8+ T cells (92). In support of “memory-like” CD8+ T cell development during this infection is the finding that virus-specific T cells transferred into naïve mice 4 weeks after initial infection with LCMV Cl13 were able to expand and control viral titers when recipient mice were infected with the acute virus LCMV Armstrong strain, despite retaining high PD-1 expression levels and reduced (but not absent) IFN-γ and TNF-α production (93). Further, these antigen-specific CD8+ T cells were maintained in the absence of antigen or infection, a foundational hallmark of memory T cells, through signaling from the homeostatic cytokines, IL-7 and IL-15, via their receptors CD127 (IL-7R) and CD122 (IL-15R). This is in contrast to an earlier finding which demonstrated that memory CD8+ T cells isolated from the very late time-point of 120 days post-infection fail to persist or respond to LCMV after transfer into naïve host mice (94). Moreover, with chronic LCMV infection, long-lived virus-specific CD8+ T cells during chronic LCMV infection show decreased expression of both CD127 and CD122 (95, 96). It is likely that fewer CD8+ T cells present in chronically infected hosts at day 28 post-infection have yet to be driven to terminal differentiation as compared to the very late time-point of 120 days.
A hallmark of memory CD8+ T cells is their capacity to proliferate upon TCR engagement, whereas TEX are ultimately driven toward apoptosis. Several studies have evaluated how chronic infection affects CD8+ T cell differentiation and impacts memory or “memory-like” T cell populations under these conditions. The discovery that checkpoint inhibitor blockade, notably through the use of anti-PD-1 and anti-CTLA-4 antibodies, reinvigorates exhausted T cells was a landmark finding that ultimately changed the landscape of cancer therapy. The important groundbreaking work by the pioneering studies on CTLA-4 and PD-1 by James Allison and Tasuku Honjo, respectively, was recently recognized by their receipt of the Nobel Prize in Medicine in 2018. Blockade of the PD-1/PD-L1 pathway was also found to abrogate T cell exhaustion when therapeutically administered to mice persistently infected with chronic LCMV (97, 98). While early interpretations of these data suggested the reversal of T cell exhaustion, more recent studies have identified a unique “memory-like” subset of exhausted T cells (TSC) that is responsible for the T cell response with PD-1 blockade therapy. One study has demonstrated that CD28 signaling is a cell-intrinsic requirement for LCMV-specific cells to proliferate in response to anti-PD-1 treatment (99). Further examination of PD-1+ cells in LCMV Cl13-infected mice identified CXCR5 expression as a distinguishing marker of PD-1 blockade responsiveness (49). Transcriptional profiling of CXCR5+ cells identified the expression of several Wnt signaling genes associated with self-renewal and hematopoietic stem cell maintenance (49). Importantly, this subset was also found to have high levels of Id3 over Id2, and high Eomes over T-bet—transcription factor profiles characteristic of memory precursor and memory CD8+ T cells (49). TRM also have unique transcriptional signatures from other memory T cell compartments, such as the expression patterns of transcription factors Blimp-1 and Hobit (Zfp683, “homolog of Blimp-1 in T cells”) which are co-expressed in TRM, with simultaneous low expression of Eomes and T-bet (86). In contrast, ZBTB32 (zinc finger and BTB domain containing 32) is another transcription factor co-expressed with Blimp-1 and limits CD8+ T cell memory development during both acute and chronic viral infections (100).
Further support for “memory-like” CD8+ T cell development during LCMV Cl13 infection was the identification of a role for the transcription factor T cell factor-1 (Tcf-1, encoded by the gene Tcf7) in a subpopulation of virus-specific CD8+ T cells. Previously described as a transcription factor co-activated by β-catenin downstream of canonical Wnt signaling, Tcf-1 was found to play a role in memory CD8+ T cell generation and function (101, 102). The use of Tcf-1 reporter mice identified that Tcf-1 expression in CD8+ T cells was associated with the maintenance and re-expansion of virus-specific CD8+ T cells in LCMV Cl13 infected mice. RNA-seq analysis of Tcf-1-expressing cells showed transcriptional characteristics that were shared with both memory and exhausted CD8+ T cells, but unique from effector T cells (103). In support of their characterization as a “memory-like” T cell compartment, Tcf-1-expressing CD8+ T cells show low levels of KLRG1, CX3CR1, T-bet, Blimp1, and granzymes while expressing high levels of IL-7R, CD62L, CCR7, Id3, and Bcl-6. However, Tcf-1-expressing cells share PD-1, LAG3, and c-Maf expression levels on par with exhausted T cells, supporting the concept that these cells are unique from archetypal memory cells. In humans, similar characteristics in Tcf-1-expressing cells including the ability to expand upon re-challenge stimulation were described in HCV-specific T cells (of which 20–60% were Tcf-1+), demonstrating that this is not a LCMV-specific phenomenon (103). Indeed, further studies involving HCV-infected patients attribute the heterogeneity of memory CD8+ T cells to differing levels of Tcf-1 expression (56). By assessing the graded expression levels of Tcf-1, a recent study found a reciprocal relationship between T-bet and Tcf-1, while Eomes expression was highest within the Tcf-1-intermediate compartment (56). While these studies have led to a greater understanding of CD8+ T cell biology, most importantly they led to an important connection between Tcf-1 expression and CD8+ T cell responsiveness to PD-1-targeted checkpoint blockade therapy in cancer, which is discussed below.
The vast heterogeneity of differentiated and “memory-like” CD8+ T cells that arise during persistent antigen exposure demonstrates the importance in understanding the cellular and molecular drivers of protective immunity. Importantly, we must better understand the conditions that give rise to “memory-like” exhausted TSC CD8+ T cells, as these appear to be the cells most responsive to checkpoint blockade therapy and therefore less sensitive to terminal exhaustion. Such insights are needed to instruct the future development of new immunomodulatory checkpoint blockade therapies, establish whether a patient would be responsive to therapy, and enhance vaccination strategies.
The importance of CD4+ T cells during persistent infections is highlighted by models in which this immune cell compartment is depleted. During chronic infection in humans and mice, decrease in helper CD4+ T cells or their functional capacity is associated with less pathogen control or the establishment of chronicity (104, 105). Further, the CD4+ T cell compartment plays a pivotal role by contributing to both the cellular and humoral arms of the immune response in chronic infection in both mice and humans (106). Despite the importance of the CD4+ T cell response during chronic infection, greater emphasis and research has focused on understanding their role in CD8+ T cell differentiation during chronic infection; much less is known about how chronic antigenic stimulation affects the differentiation and subsequent “memory-like” population of persisting CD4+ T cells in the context of persistent infection.
It is a well-defined phenomenon that CD8+ T cells become exhausted as a result of continuing antigenic exposure during chronic infections, as summarize above. Whether this is true for CD4+ T cells remains unclear, although the development of dysfunction clearly occurs. Using the LCMV models to compare acute vs. chronic viral infections, CD4+ T cells demonstrate a reduced characteristic TH1 cell cytokine profile, i.e., reduced production of IFN-γ, TNF-α, and IL-2 (107, 108). HCV infection is also known to cause an acute infection that can progress to chronicity if not controlled. In peripheral blood, broadly reactive CD4+ T cells were detected early during this infection but became undetectable in patient cohorts with chronic infection, even after viral loads diminished (109). Attempts to expand these cells in vitro were unsuccessful despite verification of their presence early in infection. In addition to the reduction of TH1 associated cytokine production, many studies have also identified altered cytokine expression by CD4+ T cells characterized by elevated IL-10 and IL-21 expression (107, 110–112). CD4+ T cells can also develop inhibitory receptor expression patterns associated with T cell exhaustion by expressing CTLA-4, which was observed in LCMV, HIV, and HBV chronic infections; PD-1; CD160; and BTLA (108, 112–115). CD4+ T cells are known to have high levels of PD-1 during chronic HIV infection and in one study this was observed to correlate with viral load (116). However, despite high levels of PD-1 expression, these cells retained the ability to produce IFN-γ (117).
Models of persistent parasite infection using T. gondii suggest that CD4+ T cells also become exhausted, with overlapping features of Blimp-1 expression, decreased expression of co-stimulatory molecules including OX40, ICOS, and 41BB, and increased inhibitory receptor expression such as 2B4; further, a reduction in cytokine expression was observed (118). These comparisons were made based upon the graded levels of PD-1, with cells that express greater PD-1 considered to be a more exhausted phenotype (118). Persistent antigenic exposure appears to be the main driver of this phenotype as suggested by infection models with Mycobacterium tuberculosis, which demonstrate that the temporal availability of antigen affects cytokine expression and magnitude of the CD4+ T cell response (119). Cells with limited exposure to antigens developed into cells that would be considered stereotypical memory cells, while continuously stimulated CD4+ T cells have a functionally altered phenotype. Still other studies using mouse models of malaria infection showed that malaria-specific CD4+ T cells exposed to chronic Plasmodium spp. infection had reduced cytokine production in comparison to those cells first deprived of antigen, then subsequently re-exposed in infected hosts (120, 121). Other studies have suggested exhaustion in the context of P. chabaudi infection, based on reduced cytokine production capacity and proliferation comparing early times points and later time points post infection (122). In humans, more evidence is needed to support this claim in Plasmodium infections as few studies have been performed and phenotypic analysis of CD4+ T cells only suggests exhaustion by inhibitory receptor expression (123). Disregarding whether CD4+ T cells are admittedly exhausted or dysfunctional, many studies in both mouse and human in multiple different chronic infections support the premise that blockade therapy of PD-1, PD-L1, or CTLA-4 augments CD4+ T cell cytokine production or proliferation (113, 115).
TH1 CD4+ T cells are commonly generated as a result of both acute and persistent viral infection and can be critical for CD8+ T cell function; but in the case of persistent infections, this population can be lost over time (83). For example in HIV patients, over long term treatment there is a discernable decrease in Gag293-tetramer specific CD4+ TH1 cells whereas CMV-specific CD4+ TH1 cells in the same patients remain unchanged (124). An important role for TH1 cells in chronic infection however is highlighted by the finding that in HIV controllers, individuals who control HIV replication without antiretroviral therapy, a polyfunctional TH1 CD4+ T cell population is maintained. As a note of caution, HIV-specific CD4+ T cells are often compared to CMV-specific CD4+ T cells in terms of function and phenotype, despite major differences in the course of infection and viral replication kinetics. Antigen availability and viral load may also play an important role in T cell differentiation. Therefore, the characteristics of different pathogens and antigen exposure clearly play a role in T cell differentiation and function (119). A similar phenomenon was described in HCV patients, in which patients who responded to interferon-α treatment had better maintenance of a polyfunctional HCV-specific TH1 CD4+ T cell population over non-responders (125). It was shown that CD4+ differentiation during chronic or prolonged antigenic stimulation in the context of infection skews CD4+ T cells toward a T follicular helper (TFH) cell lineage which may account for the loss of TH1 cell cytokine production. Recent studies have therefore focused on the contribution of the TFH cell population during chronic infection (126). Indeed, TH1 and TFH cells are generated early during infection with LCMV Cl-13 but the TH1 population is not maintained (127). This enrichment of TFH cells during chronic phases was also observed in SIV (simian immunodeficiency virus) models with rhesus macaques, as it was noted that chronically infected rhesus macaques had increased TFH cells and this correlated with elevated IL-6 levels, the cytokine known to induce TFH differentiation (128). Others had noticed a similar trend but suggested that CD4+ T cell differentiation was being redirected toward a TFH phenotype (129). TFH cell skewing is not only observable in viral infections as patients with chronic parasitic infection, Schistosoma, show increased numbers of T cells with a TFH phenotype that correlated with parasite-specific antibody levels (130). The development of a late TFH phenotype was also present in the chronic phase of Leishmania infantum infection in rhesus macaques where there was an elevation in transcripts of Bcl6, Cxcr5, and Il21, all molecules associated with a TFH response (131).
T helper cell differentiation is also observed during SIV infection, however new designations of “type 1 induced TFH cells” have been adopted to account for those TFH cells which have features of TH1 cells, including expression of CXCR3 and IFN-γ, but are more phenotypically TFH by transcription factor and surface cell marker expression (132). An interesting study in SIV-infected rhesus macaques probed the question from the opposite perspective and sought to determine the kinetics of IL-21 expression during infection (133). IL-21 was produced by multiple TH cell subsets, but predominantly TH1 cells and this early expression of IL-21 in TH1 cells negatively correlated with viral load, demonstrating the importance of a polyfunctional CD4+ T cell response in the early stages of a chronic infection (133). Variability in cytokine expression of CD4+ T cells suggests that the initial classification of CD4+ T cells into subsets based on cytokine production and transcription factor expression will likely need to be revisited in the context of chronic antigen stimulation in infections. What is unclear is whether persisting TFH exhibit features of effector, memory, or exhausted T cells. As RNA-seq becomes more widely used as well as the ability to obtain transcriptomes of fewer cells using single-cell RNA-sequencing (scRNA-seq), the degree of heterogeneity in the CD4+ T cell population is becoming more apparent and will likely lead to new insights into CD4+ T cell responses to chronic antigen stimulation (112, 134).
At present, with the observation that there may be skewing of CD4+ T cell subsets during chronic or prolonged antigenic exposure, there are a few explanations as to the mechanism by which this occurs. Due to constant replication, studies with LCMV suggest that the exposure to type I IFN inhibits the de novo TH1 differentiation; this was first only surmised to be an indirect effect on CD4+ T cells as IFN receptor deficient CD4+ T cells did not augment the number of TH1 cells (127). Later experiments would support this claim, demonstrating that type I IFN induced IL-10 and PD-L1 on dendritic cells that would then suppress TH1 differentiation, and the subsequent loss of TH1 help would contribute to CD8+ T cell dysfunction (83). TFH cell differentiation is likely driven by IL-6 that is produced later during the course of chronic LCMV infection (135). Recent studies in mice lacking the TCR scaffolding protein CD2AP, thus resulting in altered TCR signal strength, demonstrated increased TFH generation and a concomitant increase in neutralizing antibody activity in LCMV which implies a role for TCR signaling in TFH generation during infection (136).
Glucocorticoid-induced tumor necrosis factor related protein (GITR) is another molecule that was demonstrated to be important for CD4+ T cell differentiation during chronic LCMV infection (137). GITR-deficiency was shown to inhibit CD4+ T cells in the early TH1 production of IL-2 that is needed to support CD8+ T cell proliferation as well as the late TFH cell response to promote humoral immunity through provision of B cell help. Thus, it remains possible that TH1 and TFH are both generated during the initial infection and TH1 cells are not maintained during chronic infection. This differentiation toward a sustained TFH cell presence during chronic infection appears to provide many benefits to the immune response. TFH are named for their role in providing help to B cells and orchestrating the germinal center reaction (138). Importantly, resolution of chronic viral infection with LCMV is dependent on antibody production promoted by TFH cells (139, 140). The importance of TFH in HIV is also well-noted as the number of these circulating cells positively correlated with the presence of broadly neutralizing antibodies (141). During chronic or prolonged infections, many have observed the production of IL-21 by additional CD4+ T cell subsets including TFH and TH17 cells (112, 142). Although typically associated with its importance in the germinal center reaction, in the context of chronic or prolonged infection, this cytokine has been shown to support CD8+ T cell function. Early studies in the LCMV chronic infection model noted the importance of CD4+ help to CD8+ T cells in the form of IL-21, however this appeared to come at a cost of reduced TH1 cytokine production in CD4+ T cells (143).
In the LCMV model, IL-21 signaling was linked to the induction of the transcription factor BATF in CD8+ T cells, which is important for maintenance of CD8+ T cell effector function (106). Similar evidence for IL-21 production preventing CD8+ T cell exhaustion during chronic infection was observed in a mouse model of parasitic infection using T. gondii (144). Beyond its role in the CD8+ T cell response, IL-21 deficiency was also observed to compromise the humoral arm in T.gondii infections, leaving mice more susceptible to toxoplasmic encephalitis (145). Lack of IL-21 signaling by global deletion of the IL-21 receptor (IL-21R) brought about increased inhibitory receptor expression on CD8+ T cells concomitant with greater parasite burden and reactivation (144). This susceptibility due to IL-21 insensitivity was also observed in a mouse model of tuberculosis (146, 147). When considering HIV in humans, small populations of IL-21-producing CD4+ T cells were present in the blood of patients with acute and chronic HIV and a greater frequency of HIV-specific CD8+ T cells expressed the IL-21R when compared to CMV-specific T cells (148). Combined with data suggesting that IL-21 ligation of IL-21R on HIV-specific CD8+ T cells enhanced effector molecule production, these findings support the role of CD4+ T cell derived IL-21 in providing necessary help to sustain CD8+ T cells during chronic infection (149). In studies of HIV/HCV co-infected individuals, these IL-21 producing CD4+ T cells were also associated with viral control, further supporting the role of this cytokine in antiviral immunity (150). Although attributed to TH17 cells, in SIV infection of rhesus macaques, IL-21 supported CD8+ T cell responses and prevented exhaustion (151).
Compared to CD8+ T cells, more information on CD4+ T cell differentiation during chronic infection is needed to accurately determine what effect chronic antigenic stimulation has on T helper cell differentiation and function. Whether TFH or IL-21-producing CD4+ T cells that persist with time after chronic infection form “memory-like” cells has yet to be studied. Of note, this review does not discuss the implications chronic antigenic stimulation has on the development or differentiation of regulatory CD4+ T cells, or the levels of inhibitory receptor expression and suppressive cytokine production expressed by these cells. Many of the studies discussed, however, highlight the plasticity and heterogeneity present within the helper CD4+ T cell population as an adaptive immune cell that appears to be dynamically regulated by temporal and environmental dimensions. As noted above, future transcriptome studies utilizing scRNA-seq will enable further insight into the regulators that determine CD4+ T cell fate during chronic infection but also the profile of these cells. These studies can also help answer the question of whether the different antigen specific CD4+ T cell subpopulations are selectively lost as a result of chronic infection or their differentiation is skewed toward alternative differentiation lineages as the “memory-like” compartment develops. More recent studies have already hinted at the limitations of staining for a few markers and the possibility that CD4+ populations are much more polyfunctional than previously anticipated (133). This polyfunctionality of CD4+ T cell subsets, as demonstrated the ability of different cells to contribute to both humoral and cellular immunity (e.g., TH1 and TFH), highlights the importance of the different CD4+ T cell compartments and warrants further research to understand the dynamics and differentiation during chronic infections, and whether “memory-like” CD4+ T cells contribute to the sustained responses to chronic infections.
Although our knowledge of effector, memory, and exhausted T cell differentiation largely comes from studies using virus and other infection models, it is crucial to better understand the extent of memory T cell formation in response to tumors as this can instruct the development of novel cancer treatments and aid in the development of vaccine strategies against cancer, particularly as recent studies have similarities between T cell subsets derived from chronic infection and tumors (152). From studies in both mice and humans, it is becoming more appreciated that the efficacy of anti-tumor responses is enhanced by the generation of both CD4+ and CD8+ “memory-like” T cell compartments.
The priming of tumor-specific CD8+ T cells occurs in the lymph nodes by DCs that take up and cross-present neoantigens from the tumor, and activated tumor-specific T cells migrate into tumors guided by cytokine gradients (153, 154). Highly cytotoxic tumor-specific effector CD8+ T cells are a fundamental component of protective tumor infiltrating lymphocytes (TIL), and strongly correlate with patient survival (155, 156). After tumorigenesis, as with chronic infections, tumor-specific CD8+ T cells can become progressively dysfunctional and further persistence of the tumor can ultimately lead to the establishment of a permanent state of exhaustion (157, 158). At this stage, exhaustion cannot be reversed by anti-PD-1 therapy due to epigenetic modifications that prevent transcription of genes associated with effector function (158). As found with chronic virus infections, a major defining characteristic of TEX in tumors is the increased expression and co-expression of multiple inhibitory receptors that include PD-1, Tim3, LAG3, CD160, and TIGIT, the absence of the transcription factor Tcf-1 with high expression of TOX, and progressive reduction in effector functions that are linked in part to dysregulated metabolism (77, 159). The critical role of TOX in the development of CD8+ TEX in both chronic virus infections and cancer has only recently been described, with several studies identifying the necessity for TOX in TEX development. These studies show a role for TOX in regulating chromatin accessibility/epigenetic modifications associated with TEX, and its expression is driven by NFAT and chronic TCR stimulation (70–73). However, dysfunctional tumor-specific CD8+ T cells can display two different chromatin states: a plastic and fixed dysfunctional state (160). Those cells within the fixed dysfunctional chromatin state are resistant to reprogramming and express high levels of CD38 and CD101, whereas PD-1+ TIL lacking CD38 and CD101 can undergo reprogramming to develop into effector cells (160).
Alterations in surface marker expression are determined by the transcriptional profiles of tumor-specific CD8+ T cells that define the differentiation states of the cells including “memory-like” CD8+ T cell compartments. Transcriptome analysis of tumor-specific CD8+ T cells from non-small cell lung carcinoma (NSCLC) and melanoma patients has identified the altered expression patterns of several transcription factors known to be major regulators of effector and memory CD8+ T cell differentiation, including Blimp1, Id2, T-bet, and Eomes (65, 161, 162). Phenotypically, in addition to expression of various inhibitory receptors, PD-1hi CD44int Eomeshi CD8+ T cells exhibited a terminal TEX cell phenotype, whereas PD-1low CD44hi Eomeslo T-bethi CD8+ T cells could form effector cells. Terminal TEX cells are characterized by high expression of Eomes and decreased levels of T-bet. First defined in chronic LCMV infection, PD-1+CXCR5+Tim3− CD8+ T cells in the lymphoid organs were found to be responsive to PD-1 blockade therapy and express the transcription factor Tcf-1 while sharing a common gene signature with CD8+ memory precursors and were subsequently denoted as TSC (49). Similar to virus-specific TSC, intratumoral melanoma tumor-antigen-specific Tcf-1+PD-1+CD8+ T cells exhibit stem-like properties that include self-renewal and proliferation and expanded in response to checkpoint blockade were found to have characteristics of both TEX and TSC (163). In melanoma patients, the Tcf-1+PD-1+CD8+ T cell population increased in response to anti-CTLA-4 and/or anti-PD-1 treatment and there is the potential that detection of this population can predict patient survival. CX3CR1 expression is also associated with increased responsiveness to PD-1 checkpoint blockade therapy, as increased expression of CX3CR1 on CD11a+CD8+ T cells in NSCLC patients strongly correlated with a positive clinical response to treatment (164).
Because of the recognized heterogeneity of CD8+ T cells within tumors, the use of single-cell analysis techniques is yielding important new insights into the unique properties of tumor-specific T cells. A recent study from Sade-Feldman et al. used scRNA-seq to address whether patterns in the tumor transcriptome could predict patient responses to checkpoint blockade therapy (162). In comparing the transcriptomes of tumors from 48 melanoma patients, their study highlighted the heterogeneity of the CD8+ T cell compartment and identified a strong correlation between the expression of Tcf-1 in CD8+ T cells and clinical responses to checkpoint blockade (162). Although they did not detect an association with CXCR5 expression and T cell responsiveness in their patient population as was found in previous studies, they showed that expression of CD39 was indicative of CD8+ TEX cells. Several recent studies have also shown that TILs are a highly diverse T cell pool. Li et al. found that CD8+ TILs from melanoma patients form a gradient of dysfunction as indicated by transcription factor and inhibitory receptor expression (165). Furthermore, dysfunctional CD8+ T cells maintained the ability to clonally expand in the early phase of tumor progression (165). In a study of hepatocellular carcinoma (HCC), scRNA-seq analysis not only highlighted an enrichment of CD8+ TEX in HCC, but also identified a CX3CR1 cluster of effector “memory-like” CD8+ T cells, drawing parallels to the findings in NSCLC (166). At this junction, it does appear that these cells, which are only found in the context of chronic antigen stimulation, can be considered to be memory cells despite some overlap in gene signatures with TEX.
In both humans and mice, there is evidence supporting the development of tumor-specific “memory-like” CD8+ T cells, which may be favored at the early stages of tumor growth when the extent of inflammation and levels of antigen exposure are reduced compared to later stages of cancer progression. In melanoma patients that received adoptive T cell therapy, it was shown that the infused CD8+ T cells developed a TCM phenotype in vivo (167). Further, in some patients with colorectal cancer, TEM- and TCM-like populations have been identified, demonstrating the possibility that memory CD8+ T cells may naturally develop in response to cancer antigens. In one study, CD8+CD45RO+CCR7−CD28+CD27+ effector memory phenotype T cells were detected within colorectal tumor resections and were associated with increased survival in patients and noted a positive correlation between the infiltration of “memory-like” CD8+ T cells and patient survival (168). In particular, high levels of “memory-like” CD45RO+ cells within the tumor strongly correlated with the absence of early metastatic disease. In breast cancer patients, it has been shown that the ratio of the “memory” T cell compartment (CD45RO+) compared to naïve T cells in the bone marrow was significantly increased for both CD4+ and CD8+ T cells in patients compared to healthy controls, with the greatest increase in memory phenotype CD4+ T cells in bone marrow of patients where disseminated tumor cells were detected. Although the significance of these findings is unclear, this study found that despite an initial increase in HLA-A2/Her-2/neu369−377 tetramer-binding tumor specific “memory-like” T cells in the bone marrow, as the tumor advanced to later stages, this population ultimately decreased (169). This could potentially indicate a role for antigen load in the deletion or distribution of “memory-like” tumor-specific T cells. From mouse studies, it is thought that TCM may be more protective and effective against cancer compared to TEM, in part due to their high levels of IL-2 production and capacity for proliferation (170). One study found that, on a per-cell basis, in vitro-generated tumor-specific TCM-like CD8+ T cells were able to mount a strong recall response to tumors greater than that of their TEM-like cultured counterparts and were capable of eradicating established tumors when combined with both exogenous IL-2 and a cancer-antigen vaccination strategy (170).
Vaccination strategies have also been employed to promote the development of tumor-specific memory CD8+ T cells, recognizing the importance of these T cells in achieving long-term tumor control. In one very promising study in mice, vaccination was applied after tumor excision. Following excision of primary B16 melanoma tumors, mice were vaccinated with optTRP1455 peptide and also given TGF-β blockade to reverse the tumor and regulatory CD4+ (Treg) cell TGF-β-mediated suppression of CD8+ T cells (171). Strikingly, upon re-challenge with B16 tumors, mice that had received both treatments showed increased protection with 50% of mice failing to develop tumors. This was attributed to the development of a protective CD8+ T cell population characterized by the stronger prevalence of tumor-infiltrating CD8+ T cells with a memory precursor phenotype. Another study aimed at exploring the impact of Tregs on limiting the development of effective CD8+ T cell responses to B16 melanoma. This study found that prophylactic depletion of Tregs by anti-CD25 treatment prior to primary tumor engraftment and followed by primary tumor resection resulted in protection of 80% of mice against secondary tumor growth re-challenge (172). Further, deletion of the bulk CD4+ T cell population allowed for long-lived antigen-specific CD8+ T cells in secondary lymphoid organs and were protective after primary tumor resection against both localized and systemic secondary tumor challenges. While these studies demonstrate the potential for “memory-like” CD8+ T cell formation in response to tumors, it is unclear how long these “memory-like” populations persist in patients and their efficacy in protecting against relapse. It is equally important to recognize that these populations may only arise in cancers that are more localized (e.g., breast cancer, melanoma) rather than systemic (e.g., leukemia or lymphoma) and that the rate of disease progression may play a major role in determining if “memory-like” CD8+ T cells will form.
Recently, there has been great interest in the TRM compartment in cancer due to their function in local protection against repeat infections (173). Previous studies have shown that human lung tumor-infiltrating CD8+ T cells express high levels of CD103 and CD69, and low levels of CD62L and CCR7, suggestive of TRM which retain characteristics of activated cells and induce rapid and effective responses against disease (174). In the tumor microenvironment, abundant TGF-β and T cell receptor signaling through the tumor antigen/MHC class I (MHC-I) complex has been shown to induce the formation of tumor-specific CD8+CD103+ T cells (175). TGF-β signaling triggers CD103 expression on T cells, and enhances the lytic function of anti-tumor CD8+ T cells (176). TRM cells in human lung cancers express high levels of granzyme B, perforin, CD107a, and IFN-γ (177). Further, CD103 interactions with E-cadherin induces CCR5-mediated recruitment of CD8+ T cells into tumor as well as polarization and exocytosis of cytolytic granules, ultimately leading to tumor cell lysis (178). Tumor-infiltrating cells with a TRM phenotype from advanced melanoma and lung cancer patients express higher inhibitory receptors such as PD-1, Tim3, and LAG3, which opens up the possibility that checkpoint blockade might promote the greater anti-tumor immunity by TRM cells (177, 179, 180). Studies in mice have provided encouraging evidence for the ability of PD-1 blockade therapy to promote the infiltration of TRM-like (CD69+CD103+/−) CD8+ OT-I T cells generated from transferred vaccination-derived TCM (CD44+CD62L+) into both B16-OVA and MC38-OVA (181). In both model systems, the addition of PD-1 blockade resulted in better tumor control and increased numbers of TRM-like donor OT-I cells per gram of tumor. As the prevalence of checkpoint blockade therapy in patients grows, it will be important to evaluate how these therapies contribute to the development of “memory-like” CD8+ T cells in patients that are in remission. Losing the potentially beneficial contribution of TGF-β to TRM formation in the tumor microenvironment must therefore be considered when thinking about therapeutic TGF-β to limit CD8+ T cell inhibition.
At the molecular level, TRM cells do not express Eomes and Tcf-1, which are expressed by other memory T cell subsets [Table 1, (182, 183)]. Absence of Eomes expression is required for CD103 induction and low expression of T-bet is necessary for expression of CD122 and maintaining IL-15 responsiveness by TRM cells (175). On the other hand, expression of the transcription factors Hobit (homolog of Blimp1 in T cells) and Blimp1 promote the retention of TRM cells in multiple organs and suppress genes related to egress from tissues (86). Runx3 is required to form TRM cells in various tissues and tumors (62), and the transcription factors BATF (which is essential in the differentiation of effector T cells) and NAB1 (which is proposed to prevent apoptosis of TILs) are also upregulated in TRM cells in tumors (177). Although the function of TRM cells in anti-tumor immunity has not yet been fully addressed, accumulating data indicates that the cells can have a crucial role in anti-tumor responses (30). Malik et al. showed that skin-resident TRM induced by vitiligo have a CD103+CD69+ phenotype and are beneficial in protecting against melanoma (126). In untreated lung cancer patients, the density of CD103+ TRM cells among tumor-infiltrating CD8+ T cells shows a high potential as a prognostic markers for increased patient survival (177). Similarly, CD103+ TILs from high-grade serous ovarian cancer (HGSC) correlate with better patient survival (184).
Taken together, the studies of CD8+ T cells in anti-tumor responses support the possibility of generating bona fide tumor-specific memory particularly in the context of localized tumors and as a consequence of vaccination strategies with tumor-specific epitopes that can be generated by cancers with frequent mutations. Moreover, with adoptive cell therapies such as those based on TILs, it may ultimately be possible to preselect memory cells to develop infusion products that can become established as memory cells and thereafter maintained to protect against re-emergence of tumors such as observed with the persistence of the chimeric antigen receptor (CAR) T cell therapies (185).
While the main focus of basic and clinical research has been on improving CD8+ T cell-mediated eradication of tumor cells, the role of CD4+ T cells in tumor immunotherapy is much less developed. Moreover, evidence for the involvement of CD4+ T cells in tumor eradication extends beyond the canonical function of helper T cells and their ability to promote CD8+ T cell and B cell responses. These include direct effects on tumor cells by cytokines produced by CD4+ T cells such as IFN-γ, TNF-α, and IL-2, modulation of DCs and other antigen presenting cells in the tumor microenvironment as well as direct killing of tumor cells by cytolytic CD4+ cells. As such, promoting CD4+ responses to tumors and the generation of CD4+ T cell memory are crucial to developing an effective anti-tumor immune response.
It has been known for some time that MHC class II-restricted (MHC-II) tumor antigens were capable of initiating CD4+ T cell responses critical for maintenance of anti-tumor immunity (186). More recently, MHC II-restricted neoantigens were found to possibly be more effective targets for cancer immunotherapy (187). Using these neoantigens in tumor targeted vaccine-based strategies is thus an important consideration for promoting memory development. In certain tumors such as breast cancer, the presence of memory phenotype T cells are a prognostic indicator for anti-tumor responses, with an increase in TCM-like and decrease in TEM-like CD4+ cells in the lymph nodes of patients progressing from stage I to stage III disease (188). Similarly, an increase in intratumoral CD4+ TEM in colorectal tumors correlated with disease-free and survival rates in patients (155, 189). In the case of immune checkpoint blockade therapy, it was recently shown that an increase in a subset of central “memory-like” (CD27+Fas−CD45RA−CCR7+) CD4+ T cells in patients with malignant melanoma could be used as a predictor of clinical response to PD-1 blockade therapy (190, 191). In fact, CD4+ T cell memory could be induced by tri-specific antibody treatment targeting immune checkpoint inhibitors to the tumor and activating tumor-specific both CD4+ and CD8+ T cells simultaneously, with the greatest effect observed in the CD4+ TEM and TCM compartments in mice (192). Thus, there is great therapeutic potential in harnessing the power of memory CD4+ T cells to promote the most effective anti-tumor immune responses.
Although cytotoxic CD8+ T cells have been the focus of eliciting an anti-tumor response, it is clear that this response benefits from CD4+ T cell help and it has been shown that cross-priming of CD8+ T cells by DCs requires CD4+ T cell help for effective cytotoxic CD8+ T cell responses (193–195). DCs involved in the initiation of the anti-tumor T cell response also benefit from CD4+ T cell help, as CD40/CD40L interaction with CD4+ T cells is required to fully activate DCs that can subsequently generate CD8+ TEFF and long-lasting CD8+ T cell memory (196). Further, it has been shown that TH1 cells can induce cytotoxic DCs that can kill tumor cells (197). Conversely, inhibition MHC-II antigen presentation by DCs to CD4+ T cells also promotes the development of anergic anti-tumor CD8+ T cells (198). PD-1+ tumor-specific CD8+ T cells are found in the blood of melanoma patients, indicating that priming of these T cells has occurred, although these cells are largely dysfunctional and resemble TEX cells that develop during chronic infections (158, 199). Interestingly, these TEX cells also are very similar to T cells which have not received CD4+ T cell help, suggesting that the tumor specific CD8+ T cells identified following initial priming by DCs did not see CD4+ T cell help at that time. These cytotoxic CD8+ T cells have been shown to be excluded from the tumor microenvironment in part due to TGF-β signaling (200). This exclusion is associated with poor clinical outcome as well as poor response to immune checkpoint blockade therapy (155, 201). Moreover, CD4+ help during priming can provide the signals needed to promote invasiveness of cytotoxic CD8+ T cells (202, 203). In addition, polyclonal CD4+ T cells from MHC-II-negative ovarian cancer tumor-bearing mice were able to secrete CCL5 and recruit CCR5+ DCs to the tumor (204). This was also shown to be important to optimize CD4+ T cell help to cytotoxic CD8+ cells as CCR5 ligands can improve the anti-tumor response (205, 206). Although some tumor cells do not express MHC-II, it has previously been shown that CD4+ T cells can still mediate rejection of these MHC-II-deficient tumors through indirect mechanisms and there is also evidence for the development of a CD4+ T cell anti-tumor memory compartment in breast cancer patients and in the B16 melanoma mouse model (198, 207–209). In breast cancer patients, analysis of bone marrow detected both TCM and TEM phenotype CD4+ T cells, and the adoptive transfer of these cells into NOD scid mice with patient tumor transplants showed infiltration of these cells into the tumors (210). This suggests that CD4+ T cell help promotes CTL responses through the recruitment of functional CD8+ T cells primed by DCs and capable of migrating into the tumor. In a Her2-positive breast cancer model in mice, one study found that bulk “memory” CD4+ T cells from viral immune-oncotherapy cured tumor-bearing mice proliferated upon either in vivo or in vitro challenge (211). In B16 melanoma, administration of DCs loaded with apoptotic B16 cells to mice promoted the development of a long-lived functional anti-tumor CD4+ T cell compartment that produced IFN-γ upon stimulation. Importantly, this compartment was highly protective as mice subsequently challenged with B16 tumors were protected unless CD4+ (or CD8+) T cells were depleted prior to tumor challenge (212). Taken together, these studies demonstrate that generating the formation of a long-lived, functional “memory-like” CD4+ T cell compartment can provide anti-tumor immunity. In addition, the long-lived and highly proliferative population resembling TSC cells can be generated in vitro by activating CD4+ T cells by co-culture with stromal cells expressing Notch ligands (213). Importantly, these cells can expand and develop into tumor-specific effector cells after restimulation, a promising prospect for adoptive cell immunotherapy.
Thus far, the development of cancer vaccines solely focusing on CD8+ T cell epitopes has not been particularly successful without considering CD4+ T cell help (214, 215). Immune adjuvant therapy, the administration of an immune stimulant in connection with treatment, has been found to be beneficial in generating anti-tumor immunity by promoting T cell memory (216, 217). As an example, in breast cancer patients, peptide vaccination using the E75 peptide in combination with GM-CSF in breast cancer patients was able to activate both naïve CD4+ T cells as well as memory-phenotype CD4+ T cells specific for the tumor. Sustained anti-tumor CD4+ T cell “memory-like” formation was also shown in a vaccine trial of prostate cancer patients utilizing the AE37 vaccine and the DR11/AE37 tetramer to identify AE37 specific T cells. AE37 specific CD4+ T cells were detected up to 4 years following vaccination, and retained responsiveness as shown by peptide stimulation (218). Work by Bergman et al. has shown the effectiveness of generating potent anti-tumor CD4+ memory response (211, 219). These studies utilized viral oncolytic immunotherapy to prime T cell responses that were otherwise suppressed by chemotherapy-based regimens. Memory recall capability was shown by adoptive cell therapy and while transferred CD8+ T cells were poor in controlling tumor growth, transfer of memory CD4+ T cells was capable of resolving established tumors, albeit when injected in high numbers. Therefore, any consideration of adoptive immune cell therapy or cancer vaccines should include promoting the development of antigen-specific memory CD4+ T cells.
Even though providing help is a major role for CD4+ T cells in anti-tumor immune responses, CD4+ T cells can contribute directly to regulation of the tumor microenvironment and to killing of cancer cells (220, 221). It was suggested that CD4+ cells kill tumor cells through a mechanism that did not involve Fas/FasL or TNF-α, but was dependent on the TNF-α related apoptosis inducing ligand (TRAIL) (222). TH1 CD4+ T cell responses can support anti-tumor immunity, in part due to the direct impact IFN-γ has on tumor cells (194). One study described TEM CD4+ T cells that were capable of tumor elimination and this was dependent on IFN-γ (223). Strikingly, tumor reactive cytotoxic CD4+ T cells could be induced following checkpoint blockade therapy (224). These CD4+ T cells expressed Eomes but not T-bet, secreted IFN-γ, expressed granzyme B and perforin, and were capable of lysing autologous tumor cells (224). Similarly, it was shown that OX40 engagement induced both cytotoxic and memory CD4+ T cells characterized by Eomes expression (221). These cells were capable of controlling tumors in mice and lysing human tumor cells in vitro. Thus, independent of their function in providing help, CD4+ T cells can be generated that can directly target cancer cells for elimination.
Taken together, these studies in both humans and mice identify not only the potential for memory anti-tumor CD4+ and CD8+ T cell development, but also highlight their strong anti-tumor potential. Moreover, developing new strategies aimed at generating optimal CD4+ T cell responses and memory in the context of chronic antigen exposure may offer treatments for cancers that are resistant to current immunotherapies.
The many studies discussed within this review demonstrate the possibility of memory CD4+ and CD8+ T cell generation under conditions of chronic or persistent antigenic stimulation. Perhaps most importantly, they highlight the high degree of diversity and heterogeneity of long-lasting and persisting memory or “memory-like” T cells generated in patients. The importance of this diversity is shown by the reproducible formation of highly heterogeneous memory T cells within genetically identical mice and with TCR transgenic T cell models where the T cell repertoire is defined (225). Memory T cell diversity, in part, reflects an array of persisting antigen-experienced T cells that have progressed through various stages of differentiation in different contexts of antigen exposure in different tissues. Indeed, a process of tissue “imprinting” can govern the migration and maintenance of memory T cells in sites such as the gut associated tissues, skin, and lung. A major contribution to memory T cell fate determination is the antigen dose and extent of the inflammatory milieu, which can drive the development of terminal effectors that are lost during the contraction phase as antigen becomes cleared (226). Indeed, exposure of T cells to lower levels of antigen at this stage of a response can favor the generation of memory cells with the capacity for self-renewal (227). A robust immune response and rapid pathogen clearance by the T cell response favors greater generation of such memory T cells, and it is cells with similar properties (e.g., Tcf-1 expression) that can respond to immune checkpoint blockade in the settings of chronic antigen exposure (49, 228). These observations underscore the concept that antigen-experienced memory T cells that retain functional and protective capabilities are generated during chronic exposure to antigens but are unable to respond because of suppressive mechanisms in the local environment. Factors like impaired antigen-presentation, limited T cell activation in response to TCR signaling, and metabolic suppression that impair to differentiate into secondary effectors and elicit control of chronic infections and cancers also inhibit the generation of memory T cells. Although we have identified some of the parameters that distinguish subsets of memory T cells and are beginning to clinically exploit properties that promote their function, it is clear that identifying strategies that promote the development of memory in the context of chronic antigen-exposure will be crucial.
Many publications also highlight the perhaps long-standing misconception that CD4+ and CD8+ T cells follow similar differentiation pathways or develop similar characteristics as a result of chronic antigenic stimulation. This may very well be a result of differences in peptide-stimulation itself, as CD8+ T cells encounter peptide on nearly all nucleated cells in the context of MHC-I while CD4+ T cells are somewhat more protected from this constant bombardment of antigen by the more restricted expression of MHC-II. Indeed, many autologous T cell transfer strategies aim to expand T cells ex vivo and in turn provide them with a period of antigen deprivation whereby T cells can be rested from these debilitating environments. These models also highlight not only the effect that the degree of antigen exposure has on T cell development, but also introduce a temporal aspect. Dysfunction is favored by longer duration of exposure to persistent antigen-stimulating environments that decreases the likelihood of “rescuing” these cells from dysfunctional differentiation states. Limiting antigen or reducing the time of exposure may reveal key aspects to direct future avenues for restoring the proper differentiation pathway of T cells exposed to chronic antigenic stimulation.
Another concept not fully discussed within this review is that of memory inflation, or the temporal increase in a T cell population with a virus-specific (tetramer+) “effector-memory” phenotype (CCR7lowCD62LlowCD28lowCD27low) and accumulation of these cells in many non-lymphoid tissues. First defined in mouse models of murine CMV (MCMV), memory inflation has also been observed humans following CMV infection, parvoviruses B19 and PARV4, chronic norovirus, extreme responses to EBV, and to adenovirus-based vaccinations (229). It is currently understood that antigenic persistence is a requirement for memory T cell inflation and is believed to be driven by sites of latent virus infection, as removal of the primary site of viral replication (e.g., the salivary glands) does not stop the phenomenon of memory inflation (230). An important distinction however between “classical” TEX formed under persistent antigenic conditions such as with LCMV Cl13 or HIV infections compared to T cells generated via memory inflation is the retention of effector cytokine production and an overall lack of TEX hallmark features such as co-expression of inhibitory receptors. The localization of inflationary memory T cells in non-lymphoid peripheral tissues is a hallmark they share with TRM; however, while TRM are confined to the tissue in which they were generated, a high number of inflationary memory T cells can be found in circulation after MCMV and CMV infection (229). Transcriptional profiling of both inflationary T cells and TRM identified some commonalities between the two T cell types (e.g., upregulation of chemokine receptors and T-bet), but also showed significant transcriptional diversity (e.g., upregulation of AP-1 family members in TRM, and IRF8 and EZH2 in inflationary T cells) (231). This distinction further highlights the high potential for diversity in T cell differentiation and stresses the importance of understanding how antigen availability and persistence can influence the development of functional memory or “memory-like” T cells compared to exhaustion.
Evaluation of the contribution of CD4+ T cells is often neglected as CD8+ T cells have a more direct role in cell elimination; however, CD4+ T cells are important for both cellular and humoral immunity. As cells supporting both arms of immunity, they warrant further study into the roles they play during persistent antigen exposure and what affect they could have on promoting memory T cell formation. A loss of CD4+ T cell help contributes to CD8+ T cell dysfunction in chronic viral infection, but the potential for CD4+ T cells to form memory during chronic infection remains unconfirmed. In addition, more studies into the direct effects of CD4+ T cells on the tumor microenvironment and their contribution to the killing of cancer cells are needed. An important question that remains unanswered is whether the presence of a memory CD4+ T cell pool limits the establishment of secondary/metastatic tumors by affecting the tumor microenvironment. And further, to what extent is the maintenance CD4+ T cell help required to sustain the cytotoxic effector functions of CD8+ T cells within tumors? Further studies on CD8+ T cells are also needed to define parameters that limit CD8+ T cells development into true memory compartments, to address how dysfunctional differentiation pathways can be skewed toward successful memory, and to identify possible interventions to establish functional memory. As sequencing techniques have become more robust and with the advent of methods that allow for RNA transcriptome analysis to be performed on smaller cell numbers, a large emphasis has been placed on understanding the molecular determinants of memory T cell formation. Perhaps the greatest aide in understanding the complex and heterogeneous memory T cell pool has been the development of scRNA-seq, as this allows for the first time the evaluation of transcription factor co-expression and relative expression levels on the single-cell level. It also raises the question as to how different subsets arise from the same inflammatory environment and antigenic stimuli.
Not fully discussed in this review are the important findings regarding changes in epigenetics and their contributing role in T cell differentiation and particularly dysfunction, as these have been extensively and recently reviewed elsewhere (77). It is clear that changes in DNA methylation and chromatin structure play an important role in both CD4+ and CD8+ T cell fate decisions, and studies aimed at deciphering patterns in epigenetic remodeling of T cells during chronic infection and cancer have provided key insight into the regulation of T cells that are effective in killing infected or malignant cells (232, 233). Future studies combining evaluation of memory and exhausted T cells that arise during chronic antigen stimulation at both the epigenetic and transcriptome level may provide key insight into targets for therapies that promote the formation of beneficial T cell responses.
Greater consideration is now being given to the influence of metabolism on T cell differentiation and memory T cell development, particularly under the conditions of chronic or persistent antigen. Several groups have now demonstrated the unique metabolic requirements of the different T cell subsets, such as the glycolytic switch that occurs upon TCR stimulation and the subsequent switch back to fatty acid oxidation (FAO) and oxidative phosphorylation (OXPHOS) by bona fide memory T cells (234, 235). In chronic virus infections, it has already been demonstrated that exhausted T cells develop altered metabolism compared to functional virus-specific T cells, specifically an increased reliance on glycolysis and inability to use oxidative phosphorylation when exhausted (236). This may be a crucial distinction, since in the case of cancer, tumor cells can outcompete T cells for glucose (237). More recently, it has been shown that checkpoint blockade therapy can affect T cell metabolism, as both PD-1 and CTLA-4 signaling have been shown to inhibit glycolysis and PD-1 signaling promotes FAO in T cells (238, 239). As glycolysis has previously been linked to the production of inflammatory cytokines by T cells (240), this is an important consideration when trying to reverse T cell exhaustion in patients and promote memory T cell development as memory T cells have unique metabolic requirements as previously stated. Further, we are only now beginning to understand how the tumor's metabolism can impact T cell metabolism beyond nutrient deprivation and competition for glucose. The highly hypoxic tumor microenvironment promotes HIF1α expression in TILs, which further promotes glycolysis and decreased reliance on OXPHOS by the T cells. In general, TILs demonstrate major alterations in metabolism including defects in mitochondrial biogenesis and oxidative function (237, 241). Work from Delgoffe demonstrates how the state of the tumor (e.g., oxidative metabolism) can influence T cell responses to checkpoint blockade therapy and provide a predictive indicator to anti-PD-1 therapy responsiveness (241). Although tumor heterogeneity is often discussed in the context of antigen availability and “hot vs. cold” in terms of the presence of TILs, we may be overlooking the metabolic complexity of different tumor microenvironments and this significant contribution to T cell responsiveness. Better understanding the metabolic requirements of a highly effective TIL response in cancer and concurrently how the tumor metabolic requirements can be altered to generate a favorable TIL response could lead to an important convergence of anti-cancer therapies with a two-pronged approach.
Taken together, the studies summarized in this review highlight the complexities that must be considered when discussing and evaluating alterations in T cell responses and particularly when comparing memory formation with acute infection to conditions of chronic antigen stimulation. We are rapidly gaining greater insight into the molecular regulators of T cell dysfunction, effector generation, and memory development at both the transcriptional and epigenetic levels. Addressing how T cells interact with their microenvironment and the role of subsequent metabolic changes in the context of these important findings will be key in unlocking new strategies aimed at improving patient responses to chronic infections as well as cancer.
All authors listed have made a substantial, direct and intellectual contribution to the work, and approved it for publication.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Figure 1 was generated in part by modifying and adapting individual Servier Medical Art (https://smart.servier.com/) in accordance with the Servier Medical Art by Service Creative Commons Attribution 3.0 Unported License.
TN, Naive T cell; TEFF, Effector T cell; TEX, Exhausted T cell; TCM, Central memory T cell; TEM, Effector memory T cell; TRM, Resident memory T cell; TSC, Stem-cell like T cell; TEMRA, Terminally differentiated effector memory T cell; OVA, Ovalbumin; Lm-OVA, Listeria monocytogenes-OVA; OT-I, Transgenic mouse CD8+ T cells that recognize OVA (SIINFEKL); TCR, T cell receptor; IFN, Intereferon; IL, Interleukin; T-bet, T-box transcription factor TBX21; Eomes, Eomesodermin; HSV, Herpes simplex virus; RSV, Respiratory syncytial virus; Blimp-1, B lymphocyte-induced maturation protein-1; Bcl-6, B-cell lymphoma 6; Bcl-2, B-cell lymphoma 2; Foxo, Class O of forkhead box transcription factors; FRC, Fibroblastic reticular cells; PD-L1, Programmed death-ligand 1; PD-1, Programmed cell death protein 1; CTLA-4, Cytotoxic T-lymphocyte-associated protein 4; HIV-1, Human immunodeficiency virus 1; HCV, Hepatitis C virus; TH1, Type 1 helper cell; IFN-γ, Interferon gamma; TNF-α, Tumor necrosis factor alpha; LCMV, Lymphocytic choriomeningitis virus; EBV, Epstein-Barr virus; CMV, Cytomegalovirus; HLA-I, Human leukocyte antigen, class I; CD, Cluster of differentiation; TB, Tuberculosis; LTBI, Latent tuberculosis infection; Hobit, Homolog of Blimp-1 in T cells; Tcf-1, T-cell factor 1; KLRG1, Killer cell lectin like receptor G1; CX3CR1, C-X3-C motif chemokine receptor 1; LAG3, Lymphocyte activating 3; ICOS, Inducible T-cell costimulator; TFH, T follicular helper cells; T17, T-helper 17 cells; SIV, Simian immunodeficiency virus; TIM3, T-cell immunoglobulin and mucin-domain containing-3; TIGIT, T cell immunoreceptor with Ig and ITIM domains; NSCLC, Non-small-cell lung carcinoma; scRNA-seq, Single cell RNA-sequencing; HCC, Hepatocellular carcinoma; TGF-β, Transforming growth factor beta 1; DC, Dendritic cell; MHC-I, Major histocompatibility complex class I; MHC-II, Major histocompatibility complex class II; GM-CSF, Granulocyte-macrophages colony-stimulating factor; FAO, Fatty acid oxidation; OXPHOS, Oxidative phosphorylation.
1. Bender BS, Croghan T, Zhang L, Small PA Jr. Transgenic mice lacking class I major histocompatibility complex-restricted T cells have delayed viral clearance and increased mortality after influenza virus challenge. J Exp Med. (1992) 175:1143–5. doi: 10.1084/jem.175.4.1143
2. Klebanoff CA, Gattinoni L, Restifo NP. CD8+ T-cell memory in tumor immunology and immunotherapy. Immunol Rev. (2006) 211:214–24. doi: 10.1111/j.0105-2896.2006.00391.x
3. Mckinstry KK, Strutt TM, Swain SL. The potential of CD4 T-cell memory. Immunology. (2010) 130:1–9. doi: 10.1111/j.1365-2567.2010.03259.x
4. Veiga-Fernandes H, Walter U, Bourgeois C, Mclean A, Rocha B. Response of naive and memory CD8+ T cells to antigen stimulation in vivo. Nat Immunol. (2000) 1:47–53. doi: 10.1038/76907
5. Pennock ND, White JT, Cross EW, Cheney EE, Tamburini BA, Kedl RM. T cell responses: naive to memory and everything in between. Adv Physiol Educ. (2013) 37:273–83. doi: 10.1152/advan.00066.2013
6. Razvi ES, Jiang Z, Woda BA, Welsh RM. Lymphocyte apoptosis during the silencing of the immune response to acute viral infections in normal, lpr, and Bcl-2-transgenic mice. Am J Pathol. (1995) 147:79–91.
7. Fuse S, Zhang W, Usherwood EJ. Control of memory CD8+ T cell differentiation by CD80/CD86-CD28 costimulation and restoration by IL-2 during the recall response. J Immunol. (2008) 180:1148–57. doi: 10.4049/jimmunol.180.2.1148
8. Sallusto F, Lenig D, Forster R, Lipp M, Lanzavecchia A. Two subsets of memory T lymphocytes with distinct homing potentials and effector functions. Nature. (1999) 401:708–12. doi: 10.1038/44385
9. Ahmed R, Bevan MJ, Reiner SL, Fearon DT. The precursors of memory: models and controversies. Nat Rev Immunol. (2009) 9:662–8. doi: 10.1038/nri2619
10. Gerlach C, Van Heijst JW, Swart E, Sie D, Armstrong N, Kerkhoven RM, et al. One naive T cell, multiple fates in CD8+ T cell differentiation. J Exp Med. (2010) 207:1235–46. doi: 10.1084/jem.20091175
11. Stemberger C, Huster KM, Koffler M, Anderl F, Schiemann M, Wagner H, et al. A single naive CD8+ T cell precursor can develop into diverse effector and memory subsets. Immunity. (2007) 27:985–97. doi: 10.1016/j.immuni.2007.10.012
12. Buchholz VR, Flossdorf M, Hensel I, Kretschmer L, Weissbrich B, Graf P, et al. Disparate individual fates compose robust CD8+ T cell immunity. Science. (2013) 340:630–5. doi: 10.1126/science.1235454
13. Bannard O, Kraman M, Fearon DT. Secondary replicative function of CD8+ T cells that had developed an effector phenotype. Science. (2009) 323:505–9. doi: 10.1126/science.1166831
14. Youngblood B, Hale JS, Kissick HT, Ahn E, Xu X, Wieland A, et al. Effector CD8 T cells dedifferentiate into long-lived memory cells. Nature. (2017) 552:404–9. doi: 10.1038/nature25144
15. Daniels MA, Teixeiro E. TCR signaling in T cell memory. Front Immunol. (2015) 6:617. doi: 10.3389/fimmu.2015.00617
16. Teixeiro E, Daniels MA, Hamilton SE, Schrum AG, Bragado R, Jameson SC, et al. Different T cell receptor signals determine CD8+ memory versus effector development. Science. (2009) 323:502–5. doi: 10.1126/science.1163612
17. Goldrath AW, Sivakumar PV, Glaccum M, Kennedy MK, Bevan MJ, Benoist C, et al. Cytokine requirements for acute and Basal homeostatic proliferation of naive and memory CD8+ T cells. J Exp Med. (2002) 195:1515–22. doi: 10.1084/jem.20020033
18. Bradley LM, Haynes L, Swain SL. IL-7: maintaining T-cell memory and achieving homeostasis. Trends Immunol. (2005) 26:172–6. doi: 10.1016/j.it.2005.01.004
19. Borowski AB, Boesteanu AC, Mueller YM, Carafides C, Topham DJ, Altman JD, et al. Memory CD8+ T cells require CD28 costimulation. J Immunol. (2007) 179:6494–503. doi: 10.4049/jimmunol.179.10.6494
20. Boise LH, Minn AJ, Noel PJ, June CH, Accavitti MA, Lindsten T, et al. CD28 costimulation can promote T cell survival by enhancing the expression of Bcl-XL. Immunity. (1995) 3:87–98. doi: 10.1016/1074-7613(95)90161-2
21. Riley JL, June CH. The CD28 family: a T-cell rheostat for therapeutic control of T-cell activation. Blood. (2005) 105:13–21. doi: 10.1182/blood-2004-04-1596
22. Shedlock DJ, Shen H. Requirement for CD4 T cell help in generating functional CD8 T cell memory. Science. (2003) 300:337–9. doi: 10.1126/science.1082305
23. Cox MA, Kahan SM, Zajac AJ. Anti-viral CD8 T cells and the cytokines that they love. Virology. (2013) 435:157–69. doi: 10.1016/j.virol.2012.09.012
24. Cruz-Guilloty F, Pipkin ME, Djuretic IM, Levanon D, Lotem J, Lichtenheld MG, et al. Runx3 and T-box proteins cooperate to establish the transcriptional program of effector CTLs. J Exp Med. (2009) 206:51–9. doi: 10.1084/jem.20081242
25. Lazarevic V, Glimcher LH, Lord GM. T-bet: a bridge between innate and adaptive immunity. Nat Rev Immunol. (2013) 13:777–89. doi: 10.1038/nri3536
26. Feau S, Garcia Z, Arens R, Yagita H, Borst J, Schoenberger SP. The CD4(+) T-cell help signal is transmitted from APC to CD8(+) T-cells via CD27-CD70 interactions. Nat Commun. (2012) 3:948. doi: 10.1038/ncomms1948
27. Gebhardt T, Mackay L. Local immunity by tissue-resident CD8+ memory T cells. Front Immunol. (2012) 3:340. doi: 10.3389/fimmu.2012.00340
28. Sheridan BS, Pham QM, Lee YT, Cauley LS, Puddington L, Lefrancois L. Oral infection drives a distinct population of intestinal resident memory CD8(+) T cells with enhanced protective function. Immunity. (2014) 40:747–57. doi: 10.1016/j.immuni.2014.03.007
29. Jozwik A, Habibi MS, Paras A, Zhu J, Guvenel A, Dhariwal J, et al. RSV-specific airway resident memory CD8+ T cells and differential disease severity after experimental human infection. Nat Commun. (2015) 6:10224. doi: 10.1038/ncomms10224
30. Amsen D, van Gisbergen KPJM, Hombrink P, van Lier RAW. Tissue-resident memory T cells at the center of immunity to solid tumors. Nat Immunol. (2018) 19:538–46. doi: 10.1038/s41590-018-0114-2
31. Kumar BV, Ma W, Miron M, Granot T, Guyer RS, Carpenter DJ, et al. Human tissue-resident memory T cells are defined by core transcriptional and functional signatures in lymphoid and mucosal sites. Cell Rep. (2017) 20:2921–34. doi: 10.1016/j.celrep.2017.08.078
32. Lugli E, Dominguez MH, Gattinoni L, Chattopadhyay PK, Bolton DL, Song K, et al. Superior T memory stem cell persistence supports long-lived T cell memory. J Clin Invest. (2013) 123:594–9. doi: 10.1172/JCI66327
33. Berard M, Tough DF. Qualitative differences between naive and memory T cells. Immunology. (2002) 106:127–38. doi: 10.1046/j.1365-2567.2002.01447.x
34. Nolz JC, Starbeck-Miller GR, Harty JT. Naive, effector and memory CD8 T-cell trafficking: parallels and distinctions. Immunotherapy. (2011) 3:1223–33. doi: 10.2217/imt.11.100
35. Reiser J, Banerjee A. Effector, memory, and dysfunctional CD8(+) T cell fates in the antitumor immune response. J Immunol Res. (2016) 2016:8941260. doi: 10.1155/2016/8941260
36. Willinger T, Freeman T, Hasegawa H, Mcmichael AJ, Callan MF. Molecular signatures distinguish human central memory from effector memory CD8 T cell subsets. J Immunol. (2005) 175:5895–903. doi: 10.4049/jimmunol.175.9.5895
37. Gattinoni L, Lugli E, Ji Y, Pos Z, Paulos CM, Quigley MF, et al. A human memory T cell subset with stem cell-like properties. Nat Med. (2011) 17:1290–7. doi: 10.1038/nm.2446
38. Sathaliyawala T, Kubota M, Yudanin N, Turner D, Camp P, Thome JJ, et al. Distribution and compartmentalization of human circulating and tissue-resident memory T cell subsets. Immunity. (2013) 38:187–97. doi: 10.1016/j.immuni.2012.09.020
39. Sallusto F, Geginat J, Lanzavecchia A. Central memory and effector memory T cell subsets: function, generation, and maintenance. Annu Rev Immunol. (2004) 22:745–63. doi: 10.1146/annurev.immunol.22.012703.104702
40. Larbi A, Fulop T. From “truly naive” to “exhausted senescent” T cells: when markers predict functionality. Cytometry A. (2014) 85:25–35. doi: 10.1002/cyto.a.22351
41. Kumar BV, Kratchmarov R, Miron M, Carpenter DJ, Senda T, Lerner H, et al. Functional heterogeneity of human tissue-resident memory T cells based on dye efflux capacities. JCI Insight. (2018) 3:123568. doi: 10.1172/jci.insight.123568
42. Tomiyama H, Matsuda T, Takiguchi M. Differentiation of human CD8(+) T cells from a memory to memory/effector phenotype. J Immunol. (2002) 168:5538–50. doi: 10.4049/jimmunol.168.11.5538
43. Hinrichs CS, Borman ZA, Gattinoni L, Yu Z, Burns WR, Huang J, et al. Human effector CD8+ T cells derived from naive rather than memory subsets possess superior traits for adoptive immunotherapy. Blood. (2011) 117:808–14. doi: 10.1182/blood-2010-05-286286
44. Hu JK, Kagari T, Clingan JM, Matloubian M. Expression of chemokine receptor CXCR3 on T cells affects the balance between effector and memory CD8 T-cell generation. Proc Natl Acad Sci USA. (2011) 108:E118–27. doi: 10.1073/pnas.1101881108
45. Kobayashi N, Kondo T, Takata H, Yokota S, Takiguchi M. Functional and phenotypic analysis of human memory CD8+ T cells expressing CXCR3. J Leukoc Biol. (2006) 80:320–9. doi: 10.1189/jlb.1205725
46. Gilchuk P, Hill TM, Guy C, Mcmaster SR, Boyd KL, Rabacal WA, et al. A distinct lung-interstitium-resident memory CD8(+) T cell subset confers enhanced protection to lower respiratory tract infection. Cell Rep. (2016) 16:1800–9. doi: 10.1016/j.celrep.2016.07.037
47. Yi JS, Cox MA, Zajac AJ. T-cell exhaustion: characteristics, causes and conversion. Immunology. (2010) 129:474–81. doi: 10.1111/j.1365-2567.2010.03255.x
48. Wherry EJ, Kurachi M. Molecular and cellular insights into T cell exhaustion. (2015) Nat Rev Immunol. 15:486–99. doi: 10.1038/nri3862
49. Im SJ, Hashimoto M, Gerner MY, Lee J, Kissick HT, Burger MC, et al. Defining CD8+ T cells that provide the proliferative burst after PD-1 therapy. Nature. (2016) 537:417–21. doi: 10.1038/nature19330
50. Man K, Gabriel SS, Liao Y, Gloury R, Preston S, Henstridge DC, et al. Transcription factor IRF4 promotes CD8(+) T cell exhaustion and limits the development of memory-like T cells during chronic infection. Immunity. (2017) 47:1129–41 e1125. doi: 10.1016/j.immuni.2017.11.021
51. Wherry EJ, Ahmed R. Memory CD8 T-cell differentiation during viral infection. J Virol. (2004) 78:5535–45. doi: 10.1128/JVI.78.11.5535-5545.2004
52. Cho BK, Wang C, Sugawa S, Eisen HN, Chen J. Functional differences between memory and naive CD8 T cells. Proc Natl Acad Sci USA. (1999) 96:2976–81. doi: 10.1073/pnas.96.6.2976
53. Joshi NS, Kaech SM. Effector CD8 T cell development: a balancing act between memory cell potential and terminal differentiation. J Immunol. (2008) 180:1309–15. doi: 10.4049/jimmunol.180.3.1309
54. Schmueck-Henneresse M, Sharaf R, Vogt K, Weist BJ, Landwehr-Kenzel S, Fuehrer H, et al. Peripheral blood-derived virus-specific memory stem T cells mature to functional effector memory subsets with self-renewal potency. J Immunol. (2015) 194:5559–67. doi: 10.4049/jimmunol.1402090
55. Rock MT, Yoder SM, Wright PF, Talbot TR, Edwards KM, Crowe JE Jr. Differential regulation of granzyme and perforin in effector and memory T cells following smallpox immunization. J Immunol. (2005) 174:3757–64. doi: 10.4049/jimmunol.174.6.3757
56. Kratchmarov R, Magun AM, Reiner SL. TCF1 expression marks self-renewing human CD8(+) T cells. Blood Adv. (2018) 2:1685–90. doi: 10.1182/bloodadvances.2018016279
57. Wu T, Ji Y, Moseman EA, Xu HC, Manglani M, Kirby M, et al. The TCF1-Bcl6 axis counteracts type I interferon to repress exhaustion and maintain T cell stemness. Sci Immunol. (2016) 1:eaai8593. doi: 10.1126/sciimmunol.aai8593
58. Behr FM, Chuwonpad A, Stark R, Van Gisbergen K. Armed and ready: transcriptional regulation of tissue-resident memory CD8 T cells. Front Immunol. (2018) 9:1770. doi: 10.3389/fimmu.2018.01770
59. Utzschneider DT, Delpoux A, Wieland D, Huang X, Lai C-Y, Hofmann M, et al. Active maintenance of T cell memory in acute and chronic viral infection depends on continuous expression of FOXO1. Cell Rep. (2018) 22:3454–67. doi: 10.1016/j.celrep.2018.03.020
60. Egawa T, Tillman RE, Naoe Y, Taniuchi I, Littman DR. The role of the Runx transcription factors in thymocyte differentiation and in homeostasis of naive T cells. J Exp Med. (2007) 204:1945–57. doi: 10.1084/jem.20070133
61. Shan Q, Zeng Z, Xing S, Li F, Hartwig SM, Gullicksrud JA, et al. The transcription factor Runx3 guards cytotoxic CD8(+) effector T cells against deviation towards follicular helper T cell lineage. Nat Immunol. (2017) 18:931–9. doi: 10.1038/ni.3773
62. Milner JJ, Toma C, Yu B, Zhang K, Omilusik K, Phan AT, et al. Runx3 programs CD8(+) T cell residency in non-lymphoid tissues and tumours. Nature. (2017) 552:253–7. doi: 10.1038/nature24993
63. Martin MD, Badovinac VP. Defining Memory CD8 T Cell. Front Immunol. (2018) 9:2692. doi: 10.3389/fimmu.2018.02692
64. Gordon CL, Lee LN, Swadling L, Hutchings C, Zinser M, Highton AJ, et al. Induction and maintenance of CX3CR1-intermediate peripheral memory CD8(+) T cells by persistent viruses and vaccines. Cell Rep. (2018) 23:768–82. doi: 10.1016/j.celrep.2018.03.074
65. Kaech SM, Cui W. Transcriptional control of effector and memory CD8+ T cell differentiation. Nat Rev Immunol. (2012) 12:749–61. doi: 10.1038/nri3307
66. Nayar R, Schutten E, Bautista B, Daniels K, Prince AL, Enos M, et al. Graded levels of IRF4 regulate CD8+ T cell differentiation and expansion, but not attrition, in response to acute virus infection. J Immunol. (2014) 192:5881–93. doi: 10.4049/jimmunol.1303187
67. Zundler S, Becker E, Spocinska M, Slawik M, Parga-Vidal L, Stark R, et al. Hobit- and Blimp-1-driven CD4(+) tissue-resident memory T cells control chronic intestinal inflammation. Nat Immunol. (2019) 20:288–300. doi: 10.1038/s41590-018-0298-5
68. Vieira Braga FA, Hertoghs KM, Kragten NA, Doody GM, Barnes NA, Remmerswaal EB, et al. Blimp-1 homolog Hobit identifies effector-type lymphocytes in humans. Eur J Immunol. (2015) 45:2945–58. doi: 10.1002/eji.201545650
69. Doedens AL, Rubinstein MP, Gross ET, Best JA, Craig DH, Baker MK, et al. Molecular programming of tumor-infiltrating CD8+ T cells and IL15 resistance. Cancer Immunol Res. (2016) 4:799–811. doi: 10.1158/2326-6066.CIR-15-0178
70. Alfei F, Kanev K, Hofmann M, Wu M, Ghoneim HE, Roelli P, et al. TOX reinforces the phenotype and longevity of exhausted T cells in chronic viral infection. Nature. (2019) 571:265–69. doi: 10.1038/s41586-019-1326-9
71. Khan O, Giles JR, Mcdonald S, Manne S, Ngiow SF, Patel KP, et al. TOX transcriptionally and epigenetically programs CD8(+) T cell exhaustion. Nature. (2019) 571:211–18. doi: 10.1038/s41586-019-1325-x
72. Scott AC, Dundar F, Zumbo P, Chandran SS, Klebanoff CA, Shakiba M, et al. TOX is a critical regulator of tumour-specific T cell differentiation. Nature. (2019) 571:270–74. doi: 10.1038/s41586-019-1324-y
73. Seo H, Chen J, Gonzalez-Avalos E, Samaniego-Castruita D, Das A, Wang YH, et al. TOX and TOX2 transcription factors cooperate with NR4A transcription factors to impose CD8(+) T cell exhaustion. Proc Natl Acad Sci USA. (2019) 116:12410–5. doi: 10.1073/pnas.1905675116
74. Yang CY, Best JA, Knell J, Yang E, Sheridan AD, Jesionek AK, et al. The transcriptional regulators Id2 and Id3 control the formation of distinct memory CD8+ T cell subsets. Nat Immunol. (2011) 12:1221–9. doi: 10.1038/ni.2158
75. Kaech SM, Wherry EJ. Heterogeneity and cell-fate decisions in effector and memory CD8+ T cell differentiation during viral infection. Immunity. (2007) 27:393–405. doi: 10.1016/j.immuni.2007.08.007
76. Hess Michelini R, Doedens AL, Goldrath AW, Hedrick SM. Differentiation of CD8 memory T cells depends on Foxo1. J Exp Med. (2013) 210:1189–200. doi: 10.1084/jem.20130392
77. Mclane LM, Abdel-Hakeem MS, Wherry EJ. CD8 T Cell exhaustion during chronic viral infection and cancer. Annu Rev Immunol. (2019) 37:457–95. doi: 10.1146/annurev-immunol-041015-055318
78. Siegert S, Huang HY, Yang CY, Scarpellino L, Carrie L, Essex S, et al. Fibroblastic reticular cells from lymph nodes attenuate T cell expansion by producing nitric oxide. PLoS ONE. (2011) 6:e27618. doi: 10.1371/journal.pone.0027618
79. Siegert S, Luther SA. Positive and negative regulation of T cell responses by fibroblastic reticular cells within paracortical regions of lymph nodes. Front Immunol. (2012) 3:285. doi: 10.3389/fimmu.2012.00285
80. Mueller SN, Matloubian M, Clemens DM, Sharpe AH, Freeman GJ, Gangappa S, et al. Viral targeting of fibroblastic reticular cells contributes to immunosuppression and persistence during chronic infection. Proc Natl Acad Sci USA. (2007) 104:15430–5. doi: 10.1073/pnas.0702579104
81. Ekkens MJ, Shedlock DJ, Jung E, Troy A, Pearce EL, Shen H, et al. Th1 and Th2 cells help CD8 T-cell responses. Infect Immun. (2007) 75:2291–6. doi: 10.1128/IAI.01328-06
82. Okoye AA, Picker LJ. CD4(+) T-cell depletion in HIV infection: mechanisms of immunological failure. Immunol Rev. (2013) 254:54–64. doi: 10.1111/imr.12066
83. Snell LM, Osokine I, Yamada DH, De La Fuente JR, Elsaesser HJ, Brooks DG. Overcoming CD4 Th1 cell fate restrictions to sustain antiviral CD8 T cells and control persistent virus infection. Cell Rep. (2016) 16:3286–96. doi: 10.1016/j.celrep.2016.08.065
84. Wherry EJ, Blattman JN, Murali-Krishna K, Van Der Most R, Ahmed R. Viral persistence alters CD8 T-cell immunodominance and tissue distribution and results in distinct stages of functional impairment. J Virol. (2003) 77:4911–27. doi: 10.1128/JVI.77.8.4911-4927.2003
85. Leavey JK, Tarleton RL. Cutting edge: dysfunctional CD8+ T cells reside in nonlymphoid tissues during chronic Trypanosoma cruzi infection. J Immunol. (2003) 170:2264–8. doi: 10.4049/jimmunol.170.5.2264
86. Mackay LK, Minnich M, Kragten NA, Liao Y, Nota B, Seillet C, et al. Hobit and Blimp1 instruct a universal transcriptional program of tissue residency in lymphocytes. Science. (2016) 352:459–63. doi: 10.1126/science.aad2035
87. Takamura S, Yagi H, Hakata Y, Motozono C, Mcmaster SR, Masumoto T, et al. Specific niches for lung-resident memory CD8+ T cells at the site of tissue regeneration enable CD69-independent maintenance. J Exp Med. (2016) 213:3057–73. doi: 10.1084/jem.20160938
88. Appay V, Dunbar PR, Callan M, Klenerman P, Gillespie GM, Papagno L, et al. Memory CD8+ T cells vary in differentiation phenotype in different persistent virus infections. Nat Med. (2002) 8:379–85. doi: 10.1038/nm0402-379
89. Verma K, Ogonek J, Varanasi PR, Luther S, Bunting I, Thomay K, et al. Human CD8+ CD57- TEMRA cells: too young to be called “old”. PLoS ONE. (2017) 12:e0177405. doi: 10.1371/journal.pone.0177405
90. Cura Daball P, Ventura Ferreira MS, Ammann S, Klemann C, Lorenz MR, Warthorst U, et al. CD57 identifies T cells with functional senescence before terminal differentiation and relative telomere shortening in patients with activated PI3 kinase delta syndrome. Immunol Cell Biol. (2018) 96:1060–71. doi: 10.1111/imcb.12169
91. Rozot V, Vigano S, Mazza-Stalder J, Idrizi E, Day CL, Perreau M, et al. Mycobacterium tuberculosis-specific CD8+ T cells are functionally and phenotypically different between latent infection and active disease. Eur J Immunol. (2013) 43:1568–77. doi: 10.1002/eji.201243262
92. Utzschneider DT, Alfei F, Roelli P, Barras D, Chennupati V, Darbre S, et al. High antigen levels induce an exhausted phenotype in a chronic infection without impairing T cell expansion and survival. J Exp Med. (2016) 213:1819–34. doi: 10.1084/jem.20150598
93. Utzschneider DT, Legat A, Fuertes Marraco SA, Carrie L, Luescher I, Speiser DE, et al. T cells maintain an exhausted phenotype after antigen withdrawal and population reexpansion. Nat Immunol. (2013) 14:603–10. doi: 10.1038/ni.2606
94. Wherry EJ, Barber DL, Kaech SM, Blattman JN, Ahmed R. Antigen-independent memory CD8 T cells do not develop during chronic viral infection. Proc Natl Acad Sci USA. (2004) 101:16004–9. doi: 10.1073/pnas.0407192101
95. Fuller MJ, Hildeman DA, Sabbaj S, Gaddis DE, Tebo AE, Shang L, et al. Cutting edge: emergence of CD127high functionally competent memory T cells is compromised by high viral loads and inadequate T cell help. J Immunol. (2005) 174:5926–30. doi: 10.4049/jimmunol.174.10.5926
96. Lang KS, Recher M, Navarini AA, Harris NL, Lohning M, Junt T, et al. Inverse correlation between IL-7 receptor expression and CD8 T cell exhaustion during persistent antigen stimulation. Eur J Immunol. (2005) 35:738–45. doi: 10.1002/eji.200425828
97. Lee J, Ahn E, Kissick HT, Ahmed R. Reinvigorating exhausted T cells by blockade of the PD-1 pathway. Immunopathol Dis Ther. (2015) 6:7–17. doi: 10.1615/ForumImmunDisTher.2015014188
98. Penaloza-Macmaster P, Provine NM, Blass E, Barouch DH. CD4 T Cell depletion substantially augments the rescue potential of PD-L1 blockade for deeply exhausted CD8 T cells. J Immunol. (2015) 195:1054–63. doi: 10.4049/jimmunol.1403237
99. Kamphorst AO, Wieland A, Nasti T, Yang S, Zhang R, Barber DL, et al. Rescue of exhausted CD8 T cells by PD-1-targeted therapies is CD28-dependent. Science. (2017) 355:1423–7. doi: 10.1126/science.aaf0683
100. Shin HM, Kapoor VN, Kim G, Li P, Kim HR, Suresh M, et al. Transient expression of ZBTB32 in anti-viral CD8+ T cells limits the magnitude of the effector response and the generation of memory. PLoS Pathog. (2017) 13:e1006544. doi: 10.1371/journal.ppat.1006544
101. Jeannet G, Boudousquie C, Gardiol N, Kang J, Huelsken J, Held W. Essential role of the Wnt pathway effector Tcf-1 for the establishment of functional CD8 T cell memory. Proc Natl Acad Sci USA. (2010) 107:9777–82. doi: 10.1073/pnas.0914127107
102. Zhou X, Yu S, Zhao DM, Harty JT, Badovinac VP, Xue HH. Differentiation and persistence of memory CD8(+) T cells depend on T cell factor 1. Immunity. (2010) 33:229–40. doi: 10.1016/j.immuni.2010.08.002
103. Utzschneider DT, Charmoy M, Chennupati V, Pousse L, Ferreira DP, Calderon-Copete S, et al. T cell factor 1-expressing memory-like CD8(+) T cells sustain the immune response to chronic viral infections. Immunity. (2016) 45:415–27. doi: 10.1016/j.immuni.2016.07.021
104. Matloubian M, Concepcion RJ, Ahmed R. CD4+ T cells are required to sustain CD8+ cytotoxic T-cell responses during chronic viral infection. J Virol. (1994) 68:8056–63.
105. Klenerman P, Thimme R. T cell responses in hepatitis C: the good, the bad and the unconventional. Gut. (2012) 61:1226–34. doi: 10.1136/gutjnl-2011-300620
106. Xin G, Schauder DM, Lainez B, Weinstein JS, Dai Z, Chen Y, et al. A critical role of IL-21-induced BATF in sustaining CD8-T-cell-mediated chronic viral control. Cell Rep. (2015) 13:1118–24. doi: 10.1016/j.celrep.2015.09.069
107. Brooks DG, Teyton L, Oldstone MBA, Mcgavern DB. Intrinsic functional dysregulation of CD4 T cells occurs rapidly following persistent viral Infection. J Virol. (2005) 79:10514–27. doi: 10.1128/JVI.79.16.10514-10527.2005
108. Tinoco R, Carrette F, Barraza ML, Otero DC, Magana J, Bosenberg MW, et al. PSGL-1 is an immune checkpoint regulator that promotes T cell exhaustion. Immunity. (2016) 44:1190–203. doi: 10.1016/j.immuni.2016.04.015
109. Schulze Zur Wiesch J, Ciuffreda D, Lewis-Ximenez L, Kasprowicz V, Nolan BE, Streeck H, et al. Broadly directed virus-specific CD4+ T cell responses are primed during acute hepatitis C infection, but rapidly disappear from human blood with viral persistence. J Exp Med. (2012) 209:61–75. doi: 10.1084/jem.20100388
110. Brooks DG, Mcgavern DB, Oldstone MBA. Reprogramming of antiviral T cells prevents inactivation and restores T cell activity during persistent viral infection. J Clin Invest. (2006) 116:1675–85. doi: 10.1172/JCI26856
111. Ejrnaes M, Filippi CM, Martinic MM, Ling EM, Togher LM, Crotty S, et al. Resolution of a chronic viral infection after interleukin-10 receptor blockade. J Exp Med. (2006) 203:2461–72. doi: 10.1084/jem.20061462
112. Crawford A, Angelosanto JM, Kao C, Doering TA, Odorizzi PM, Barnett BE, et al. Molecular and transcriptional basis of CD4(+) T cell dysfunction during chronic infection. Immunity. (2014) 40:289–302. doi: 10.1016/j.immuni.2014.01.005
113. Kaufmann DE, Kavanagh DG, Pereyra F, Zaunders JJ, Mackey EW, Miura T, et al. Upregulation of CTLA-4 by HIV-specific CD4+ T cells correlates with disease progression and defines a reversible immune dysfunction. Nat Immunol. (2007) 8:1246–54. doi: 10.1038/ni1515
114. Kassu A, Marcus RA, D'souza MB, Kelly-Mcknight EA, Golden-Mason L, Akkina R, et al. Regulation of virus-specific CD4+ T cell function by multiple costimulatory receptors during chronic HIV infection. J Immunol. (2010) 185:3007–18. doi: 10.4049/jimmunol.1000156
115. Raziorrouh B, Heeg M, Kurktschiev P, Schraut W, Zachoval R, Wendtner C, et al. Inhibitory phenotype of HBV-specific CD4+ T-cells is characterized by high PD-1 expression but absent coregulation of multiple inhibitory molecules. PLoS ONE. (2014) 9:e105703. doi: 10.1371/journal.pone.0105703
116. D'souza M, Fontenot AP, Mack DG, Lozupone C, Dillon S, Meditz A, et al. Programmed death 1 expression on HIV-specific CD4andlt;supandgt;+andlt;/supandgt; T cells is driven by viral replication and associated with T cell dysfunction. J Immunol. (2007) 179:1979. doi: 10.4049/jimmunol.179.3.1979
117. Teigler JE, Zelinskyy G, Eller MA, Bolton DL, Marovich M, Gordon AD, et al. Differential inhibitory receptor expression on t cells delineates functional capacities in chronic viral infection. J Virol. (2017) 91:e01263–17. doi: 10.1128/JVI.01263-17
118. Hwang S, Cobb DA, Bhadra R, Youngblood B, Khan IA. Blimp-1-mediated CD4 T cell exhaustion causes CD8 T cell dysfunction during chronic toxoplasmosis. J Exp Med. (2016) 213:1799–818. doi: 10.1084/jem.20151995
119. Moguche AO, Musvosvi M, Penn-Nicholson A, Plumlee CR, Mearns H, Geldenhuys H, et al. Antigen availability shapes T cell differentiation and function during tuberculosis. Cell Host Microbe. (2017) 21:695–706.e695. doi: 10.1016/j.chom.2017.05.012
120. Butler NS, Moebius J, Pewe LL, Traore B, Doumbo OK, Tygrett LT, et al. Therapeutic blockade of PD-L1 and LAG-3 rapidly clears established blood-stage Plasmodium infection. Nat Immunol. (2011) 13:188–95. doi: 10.1038/ni.2180
121. Opata MM, Stephens R. Chronic Plasmodium chabaudi infection generates CD4 memory T cells with increased T cell receptor sensitivity but poor secondary expansion and increased apoptosis. Infect Immun. (2017) 85:e00744–e00716. doi: 10.1128/IAI.00744-16
122. Horne-Debets JM, Faleiro R, Karunarathne DS, Liu X, Lineburg KE, Poh CM, et al. PD-1 dependent exhaustion of CD8+ T cells drives chronic malaria. Cell Rep. (2013) 5:1204–13. doi: 10.1016/j.celrep.2013.11.002
123. Illingworth J, Butler NS, Roetynck S, Mwacharo J, Pierce SK, Bejon P, et al. Chronic exposure to Plasmodium falciparum is associated with phenotypic evidence of B and T cell exhaustion. J Immunol. (2013) 190:1038–47. doi: 10.4049/jimmunol.1202438
124. Vingert B, Benati D, Lambotte O, De Truchis P, Slama L, Jeannin P, et al. HIV controllers maintain a population of highly efficient Th1 effector cells in contrast to patients treated in the long term. J Virol. (2012) 86:10661–74. doi: 10.1128/JVI.00056-12
125. Flynn JK, Dore GJ, Hellard M, Yeung B, Rawlinson WD, White PA, et al. Maintenance of Th1 hepatitis C virus (HCV)-specific responses in individuals with acute HCV who achieve sustained virological clearance after treatment. J. Gastroenterol Hepatol. (2013) 28:1770–81. doi: 10.1111/jgh.12265
126. Malik BT, Byrne KT, Vella JL, Zhang P, Shabaneh TB, Steinberg SM, et al. Resident memory T cells in skin mediate durable immunity to melanoma. Sci Immunol. (2017) 2:eaam6346. doi: 10.1126/sciimmunol.aam6346
127. Osokine I, Snell LM, Cunningham CR, Yamada DH, Wilson EB, Elsaesser HJ, et al. Type I interferon suppresses de novo virus-specific CD4 Th1 immunity during an established persistent viral infection. Proc Natl Acad Sci USA. (2014) 111:7409–14. doi: 10.1073/pnas.1401662111
128. Petrovas C, Yamamoto T, Gerner MY, Boswell KL, Wloka K, Smith EC, et al. CD4 T follicular helper cell dynamics during SIV infection. J Clin Invest. (2012) 122:3281–94. doi: 10.1172/JCI63039
129. Fahey LM, Wilson EB, Elsaesser H, Fistonich CD, Mcgavern DB, Brooks DG. Viral persistence redirects CD4 T cell differentiation toward T follicular helper cells. J Exp Med. (2011) 208:987–99. doi: 10.1084/jem.20101773
130. Zhang Y, Jiang Y, Wang Y, Liu H, Shen Y, Yuan Z, et al. Higher frequency of circulating PD-1high CXCR5+CD4+ Tfh cells in patients with chronic schistosomiasis. Int J Biol Sci. (2015) 11:1049–55. doi: 10.7150/ijbs.12023
131. Rodrigues V, Laforge M, Campillo-Gimenez L, Soundaramourty C, Correia-De-Oliveira A, Dinis-Oliveira RJ, et al. Abortive T follicular helper development is associated with a defective humoral response in Leishmania infantum-infected macaques. PLoS Pathog. (2014) 10:e1004096. doi: 10.1371/journal.ppat.1004096
132. Velu V, Mylvaganam GH, Gangadhara S, Hong JJ, Iyer SS, Gumber S, et al. Induction of Th1-biased T follicular helper (Tfh) cells in lymphoid tissues during chronic simian immunodeficiency virus infection defines functionally distinct germinal center tfh cells. J Immunol. (2016) 197:1832–1842. doi: 10.4049/jimmunol.1600143
133. Shi S, Seki S, Matano T, Yamamoto H. IL-21-producer CD4+ T cell kinetics during primary simian immunodeficiency virus infection. Microb Infec. (2013) 15:697–707. doi: 10.1016/j.micinf.2013.06.004
134. Xin G, Zander R, Schauder DM, Chen Y, Weinstein JS, Drobyski WR, et al. Single-cell RNA sequencing unveils an IL-10-producing helper subset that sustains humoral immunity during persistent infection. Nat Commun. (2018) 9:5037. doi: 10.1038/s41467-018-07492-4
135. Harker JA, Lewis GM, Mack L, Zuniga EI. Late interleukin-6 escalates T follicular helper cell responses and controls a chronic viral infection. Science. (2011) 334:825–9. doi: 10.1126/science.1208421
136. Raju S, Kometani K, Kurosaki T, Shaw AS, Egawa T. The adaptor molecule CD2AP in CD4 T cells modulates differentiation of follicular helper T cells during chronic LCMV infection. PLoS Pathog. (2018) 14:e1007053. doi: 10.1371/journal.ppat.1007053
137. Clouthier DL, Zhou AC, Wortzman ME, Luft O, Levy GA, Watts TH. GITR intrinsically sustains early type 1 and late follicular helper CD4 T cell accumulation to control a chronic viral infection. PLoS Pathog. (2015) 11:e1004517. doi: 10.1371/journal.ppat.1004517
138. Greczmiel U, Oxenius A. The janus face of follicular T helper cells in chronic viral infections. Front Immunol. (2018) 9:1162. doi: 10.3389/fimmu.2018.01162
139. Cook KD, Shpargel KB, Starmer J, Whitfield-Larry F, Conley B, Allard DE, et al. T follicular helper cell-dependent clearance of a persistent virus infection requires T cell expression of the histone demethylase UTX. Immunity. (2015) 43:703–14. doi: 10.1016/j.immuni.2015.09.002
140. Greczmiel U, Krautler NJ, Pedrioli A, Bartsch I, Agnellini P, Bedenikovic G, et al. Sustained T follicular helper cell response is essential for control of chronic viral infection. Sci Immunol. (2017) 2:eaam8686. doi: 10.1126/sciimmunol.aam8686
141. Locci M, Havenar-Daughton C, Landais E, Wu J, Kroenke MA, Arlehamn CL, et al. Human circulating PD-1+CXCR3-CXCR5+ memory Tfh cells are highly functional and correlate with broadly neutralizing HIV antibody responses. Immunity. (2013) 39:758–69. doi: 10.1016/j.immuni.2013.08.031
142. Tian Y, Zajac AJ. IL-21 and T cell differentiation: consider the context. Trends Immunol. (2016) 37:557–68. doi: 10.1016/j.it.2016.06.001
143. Elsaesser H, Sauer K, Brooks DG. IL-21 is required to control chronic viral infection. Science. (2009) 324:1569–1572. doi: 10.1126/science.1174182
144. Moretto MM, Hwang S, Khan IA. Downregulated IL-21 response and T follicular helper cell exhaustion correlate with compromised CD8 T cell immunity during chronic toxoplasmosis. Front Immunol. (2017) 8:1436. doi: 10.3389/fimmu.2017.01436
145. Stumhofer JS, Silver JS, Hunter CA. IL-21 is required for optimal antibody production and T cell responses during chronic Toxoplasma gondii infection. PLoS ONE. (2013) 8:e62889. doi: 10.1371/journal.pone.0062889
146. Booty MG, Barreira-Silva P, Carpenter SM, Nunes-Alves C, Jacques MK, Stowell BL, et al. IL-21 signaling is essential for optimal host resistance against Mycobacterium tuberculosis infection. Sci Rep. (2016) 6:36720. doi: 10.1038/srep36720
147. Cheekatla SS, Tripathi D, Venkatasubramanian S, Paidipally P, Welch E, Tvinnereim AR, et al. IL-21 receptor signaling is essential for optimal CD4(+) T cell function and control of Mycobacterium tuberculosis infection in mice. J Immunol. (2017) 199:2815–22. doi: 10.4049/jimmunol.1601231
148. Yue FY, Lo C, Sakhdari A, Lee EY, Kovacs CM, Benko E, et al. HIV-specific IL-21 producing CD4+ T cells are induced in acute and chronic progressive HIV infection and are associated with relative viral control. J Immunol. (2010) 185:498–506. doi: 10.4049/jimmunol.0903915
149. Chevalier MF, Julg B, Pyo A, Flanders M, Ranasinghe S, Soghoian DZ, et al. HIV-1-specific interleukin-21+ CD4+ T cell responses contribute to durable viral control through the modulation of HIV-specific CD8+ T cell function. J Virol. (2011) 85:733–41. doi: 10.1128/JVI.02030-10
150. Macparland SA, Fadel SM, Mihajlovic V, Fawaz A, Kim C, Rahman AK, et al. HCV specific IL-21 producing T cells but not IL-17A producing T cells are associated with HCV viral control in HIV/HCV coinfection. PLoS ONE. (2016) 11:e0154433. doi: 10.1371/journal.pone.0154433
151. Mendez-Lagares G, Lu D, Merriam D, Baker CA, Villinger F, Van Rompay KKA, et al. IL-21 therapy controls immune activation and maintains antiviral CD8(+) T cell responses in acute simian immunodeficiency virus infection. AIDS Res Hum Retroviruses. (2017) 33:S81–s92. doi: 10.1089/aid.2017.0160
152. Miller BC, Sen DR, Al Abosy R, Bi K, Virkud YV, Lafleur MW, et al. Subsets of exhausted CD8+ T cells differentially mediate tumor control and respond to checkpoint blockade. Nat Immunol. (2019) 20:326–36. doi: 10.1038/s41590-019-0312-6
153. Baxevanis CN, Voutsas IF, Tsitsilonis OE, Gritzapis AD, Sotiriadou R, Papamichail M. Tumor-specific CD4+ T lymphocytes from cancer patients are required for optimal induction of cytotoxic T cells against the autologous tumor. J Immunol. (2000) 164:3902–12. doi: 10.4049/jimmunol.164.7.3902
154. Peng W, Liu C, Xu C, Lou Y, Chen J, Yang Y, et al. PD-1 blockade enhances T cell migration to tumors by elevating IFN-γ inducible chemokines. Canres Res. (2012) 72:5209–18. doi: 10.1158/0008-5472.CAN-12-1187
155. Galon J, Costes A, Sanchez-Cabo F, Kirilovsky A, Mlecnik B, Lagorce-Pages C, et al. Type, density, and location of immune cells within human colorectal tumors predict clinical outcome. Science. (2006) 313:1960–4. doi: 10.1126/science.1129139
156. Mahmoud SM, Paish EC, Powe DG, Macmillan RD, Grainge MJ, Lee AH, et al. Tumor-infiltrating CD8+ lymphocytes predict clinical outcome in breast cancer. J Clin Oncol. (2011) 29:1949–55. doi: 10.1200/JCO.2010.30.5037
157. Mumprecht S, Schurch C, Schwaller J, Solenthaler M, Ochsenbein AF. Programmed death 1 signaling on chronic myeloid leukemia-specific T cells results in T-cell exhaustion and disease progression. Blood. (2009) 114:1528–36. doi: 10.1182/blood-2008-09-179697
158. Schietinger A, Philip M, Krisnawan VE, Chiu EY, Delrow JJ, Basom RS, et al. Tumor-specific T cell dysfunction is a dynamic antigen-driven differentiation program initiated early during tumorigenesis. Immunity. (2016) 45:389–401. doi: 10.1016/j.immuni.2016.07.011
160. Philip M, Fairchild L, Sun L, Horste EL, Camara S, Shakiba M, et al. Chromatin states define tumour-specific T cell dysfunction and reprogramming. Nature. (2017) 545:452–6. doi: 10.1038/nature22367
161. Brummelman J, Mazza EMC, Alvisi G, Colombo FS, Grilli A, Mikulak J, et al. High-dimensional single cell analysis identifies stem-like cytotoxic CD8 T cells infiltrating human tumors. J Exp Med. (2018) 215:2520–35. doi: 10.1084/jem.20180684
162. Sade-Feldman M, Yizhak K, Bjorgaard SL, Ray JP, De Boer CG, Jenkins RW, et al. Defining T cell states associated with response to checkpoint immunotherapy in melanoma. Cell. (2018) 175:998–1013.e1020. doi: 10.1016/j.cell.2018.10.038
163. Siddiqui I, Schaeuble K, Chennupati V, Marraco SAF, Calderon-Copete S, Pais Ferreira D, et al. Intratumoral Tcf1+ PD-1+ CD8+ T cells with stem-like properties promote tumor control in response to vaccination and checkpoint blockade immunotherapy. Immunity. (2019) 50:195–211.e10. doi: 10.1016/j.immuni.2018.12.021
164. Yan Y, Cao S, Liu X, Harrington SM, Bindeman WE, Adjei AA, et al. CX3CR1 identifies PD-1 therapy-responsive CD8+ T cells that withstand chemotherapy during cancer chemoimmunotherapy. JCI Insight. (2018) 3:97828. doi: 10.1172/jci.insight.97828
165. Li H, Van Der Leun AM, Yofe I, Lubling Y, Gelbard-Solodkin D, Van Akkooi AC, et al. Dysfunctional CD8 T cells form a proliferative, dynamically regulated compartment within human melanoma. Cell. (2018) 176:775–89.e18. doi: 10.1016/j.cell.2018.11.043
166. Zheng C, Zheng L, Yoo JK, Guo H, Zhang Y, Guo X, et al. Landscape of infiltrating T cells in liver cancer revealed by single-cell sequencing. Cell. (2017) 169:1342–56 e1316. doi: 10.1016/j.cell.2017.05.035
167. Chapuis AG, Thompson JA, Margolin KA, Rodmyre R, Lai IP, Dowdy K, et al. Transferred melanoma-specific CD8+ T cells persist, mediate tumor regression, and acquire central memory phenotype. Proc Natl Acad Sci USA. (2012) 109:4592–7. doi: 10.1073/pnas.1113748109
168. Pages F, Berger A, Camus M, Sanchez-Cabo F, Costes A, Molidor R, et al. Effector memory T cells, early metastasis, and survival in colorectal cancer. N Engl J Med. (2005) 353:2654–66. doi: 10.1056/NEJMoa051424
169. Feuerer M, Rocha M, Bai L, Umansky V, Solomayer EF, Bastert G, et al. Enrichment of memory T cells and other profound immunological changes in the bone marrow from untreated breast cancer patients. Int J Cancer. (2001) 92:96–105. doi: 10.1002/1097-0215(200102)9999:9999<::AID-IJC1152>3.0.CO;2-Q
170. Klebanoff CA, Gattinoni L, Torabi-Parizi P, Kerstann K, Cardones AR, Finkelstein SE, et al. Central memory self/tumor-reactive CD8+ T cells confer superior antitumor immunity compared with effector memory T cells. Proc Natl Acad Sci USA. (2005) 102:9571–6. doi: 10.1073/pnas.0503726102
171. Bellavance EC, Kohlhapp FJ, Zloza A, O'sullivan JA, Mccracken J, Jagoda MC, et al. Development of tumor-infiltrating CD8+ T cell memory precursor effector cells and antimelanoma memory responses are the result of vaccination and TGF-beta blockade during the perioperative period of tumor resection. J Immunol. (2011) 186:3309–16. doi: 10.4049/jimmunol.1002549
172. Zhang P, Cote AL, De Vries VC, Usherwood EJ, Turk MJ. Induction of postsurgical tumor immunity and T-cell memory by a poorly immunogenic tumor. Cancer Res. (2007) 67:6468–76. doi: 10.1158/0008-5472.CAN-07-1264
173. Mcmaster SR, Wein AN, Dunbar PR, Hayward SL, Cartwright EK, Denning TL, et al. Pulmonary antigen encounter regulates the establishment of tissue-resident CD8 memory T cells in the lung airways and parenchyma. Mucosal Immunol. (2018) 11:1071–8. doi: 10.1038/s41385-018-0003-x
174. Djenidi F, Adam J, Goubar A, Durgeau A, Meurice G, De Montpreville V, et al. CD8+CD103+ tumor-infiltrating lymphocytes are tumor-specific tissue-resident memory T cells and a prognostic factor for survival in lung cancer patients. J Immunol. (2015) 194:3475–86. doi: 10.4049/jimmunol.1402711
175. Mackay LK, Wynne-Jones E, Freestone D, Pellicci DG, Mielke LA, Newman DM, et al. T-box transcription factors combine with the cytokines TGF-beta and IL-15 to control tissue-resident memory T cell fate. Immunity. (2015) 43:1101–11. doi: 10.1016/j.immuni.2015.11.008
176. Franciszkiewicz K, Le Floc'h A, Jalil A, Vigant F, Robert T, Vergnon I, et al. Intratumoral induction of CD103 triggers tumor-specific CTL function and CCR5-dependent T-cell retention. Cancer Res. (2009) 69:6249–255. doi: 10.1158/0008-5472.CAN-08-3571
177. Ganesan, A. -P., Clarke J, Wood O, Garrido-Martin EM, Chee SJ, Mellows T, et al. Tissue-resident memory features are linked to the magnitude of cytotoxic T cell responses in human lung cancer. Nat Immunol. (2017) 18:940. doi: 10.1038/ni.3775
178. Le Floc'h A, Jalil A, Vergnon I, Le Maux Chansac B, Lazar V, Bismuth G, et al. Alpha E beta 7 integrin interaction with E-cadherin promotes antitumor CTL activity by triggering lytic granule polarization and exocytosis. J Exp Med. (2007) 204:559–70. doi: 10.1084/jem.20061524
179. Webb JR, Milne K, Nelson BH. PD-1 and CD103 are widely coexpressed on prognostically favorable intraepithelial CD8 T cells in human ovarian cancer. Cancer Immunol Res. (2015) 3:926–35. doi: 10.1158/2326-6066.CIR-14-0239
180. Boddupalli CS, Bar N, Kadaveru K, Krauthammer M, Pornputtapong N, Mai Z, et al. Interlesional diversity of T cell receptors in melanoma with immune checkpoints enriched in tissue-resident memory T cells. JCI Insight. (2016) 1:e88955. doi: 10.1172/jci.insight.88955
181. Enamorado M, Iborra S, Priego E, Cueto FJ, Quintana JA, Martínez-Cano S, et al. Enhanced anti-tumour immunity requires the interplay between resident and circulating memory CD8+ T cells. Nat Commun. (2017) 8:16073. doi: 10.1038/ncomms16073
182. Intlekofer AM, Takemoto N, Wherry EJ, Longworth SA, Northrup JT, Palanivel VR, et al. Effector and memory CD8+ T cell fate coupled by T-bet and eomesodermin. Nat Immunol. (2005) 6:1236–44. doi: 10.1038/ni1268
183. Milner JJ, Goldrath AW. Transcriptional programming of tissue-resident memory CD8(+) T cells. Curr Opin Immunol. (2018) 51:162–9. doi: 10.1016/j.coi.2018.03.017
184. Webb JR, Milne K, Watson P, Deleeuw RJ, Nelson BH. Tumor-infiltrating lymphocytes expressing the tissue resident memory marker CD103 are associated with increased survival in high-grade serous ovarian cancer. Clin Cancer Res. (2014) 20:434–44. doi: 10.1158/1078-0432.CCR-13-1877
185. Fraietta JA, Lacey SF, Orlando EJ, Pruteanu-Malinici I, Gohil M, Lundh S, et al. Determinants of response and resistance to CD19 chimeric antigen receptor (CAR) T cell therapy of chronic lymphocytic leukemia. Nat Med. (2018) 24:563–71. doi: 10.1038/s41591-018-0010-1
186. Matsuzaki J, Tsuji T, Luescher IF, Shiku H, Mineno J, Okamoto S, et al. Direct tumor recognition by a human CD4(+) T-cell subset potently mediates tumor growth inhibition and orchestrates anti-tumor immune responses. Sci Rep. (2015) 5:14896. doi: 10.1038/srep14896
187. Gao Y, Barmada MA, Bergman I. Antitumor memory T-cells become functionally mature from 30 to 100 days in a mouse model of neoplasia. Anticancer Res. (2018) 38:147–57. doi: 10.21873/anticanres.12202
188. Vahidi Y, Faghih Z, Talei AR, Doroudchi M, Ghaderi A. Memory CD4(+) T cell subsets in tumor draining lymph nodes of breast cancer patients: a focus on T stem cell memory cells. Cell Oncol. (2018) 41:1–11. doi: 10.1007/s13402-017-0352-6
189. Jia Q, Yang Y, Wan Y. Tumor-infiltrating memory T-lymphocytes for prognostic prediction in cancer patients: a meta-analysis. Int J Clin Exp Med. (2015) 8:1803–13.
190. Goding SR, Wilson KA, Xie Y, Harris KM, Baxi A, Akpinarli A, et al. Restoring immune function of tumor-specific CD4+ T cells during recurrence of melanoma. J Immunol. (2013) 190:4899–909. doi: 10.4049/jimmunol.1300271
191. Takeuchi Y, Tanemura A, Tada Y, Katayama I, Kumanogoh A, Nishikawa H. Clinical response to PD-1 blockade correlates with a sub-fraction of peripheral central memory CD4+ T cells in patients with malignant melanoma. Int Immunol. (2018) 30:13–22. doi: 10.1093/intimm/dxx073
192. Deppisch N, Ruf P, Eissler N, Lindhofer H, Mocikat R. Potent CD4+ T cell-associated antitumor memory responses induced by trifunctional bispecific antibodies in combination with immune checkpoint inhibition. Oncotarget. (2017) 8:4520–9. doi: 10.18632/oncotarget.13888
193. Bennett SR, Carbone FR, Karamalis F, Miller JF, Heath WR. Induction of a CD8+ cytotoxic T lymphocyte response by cross-priming requires cognate CD4+ T cell help. J Exp Med. (1997) 186:65–70. doi: 10.1084/jem.186.1.65
194. Knocke S, Fleischmann-Mundt B, Saborowski M, Manns MP, Kuhnel F, Wirth TC, et al. Tailored tumor immunogenicity reveals regulation of CD4 and CD8 T cell responses against cancer. Cell Rep. (2016) 17:2234–46. doi: 10.1016/j.celrep.2016.10.086
195. Malandro N, Budhu S, Kuhn NF, Liu C, Murphy JT, Cortez C, et al. Clonal abundance of tumor-specific CD4(+) T cells potentiates efficacy and alters susceptibility to exhaustion. Immunity. (2016) 44:179–93. doi: 10.1016/j.immuni.2015.12.018
196. Pardoll DM, Topalian SL. The role of CD4+ T cell responses in antitumor immunity. Curr Opin Immunol. (1998) 10:588–94. doi: 10.1016/S0952-7915(98)80228-8
197. Lacasse CJ, Janikashvili N, Larmonier CB, Alizadeh D, Hanke N, Kartchner J, et al. Th-1 lymphocytes induce dendritic cell tumor killing activity by an IFN-γ-dependent mechanism. J Immunol. (2011) 187:6310–7. doi: 10.4049/jimmunol.1101812
198. Gerner MY, Casey KA, Mescher MF. Defective MHC class II presentation by dendritic cells limits CD4 T cell help for antitumor CD8 T cell responses. J Immunol. (2008) 181:155–64. doi: 10.4049/jimmunol.181.1.155
199. Gros A, Parkhurst MR, Tran E, Pasetto A, Robbins PF, Ilyas S, et al. Prospective identification of neoantigen-specific lymphocytes in the peripheral blood of melanoma patients. Nat Med. (2016) 22:433–8. doi: 10.1038/nm.4051
200. Mariathasan S, Turley SJ, Nickles D, Castiglioni A, Yuen K, Wang Y, et al. TGFbeta attenuates tumour response to PD-L1 blockade by contributing to exclusion of T cells. Nature. (2018) 554:544–8. doi: 10.1038/nature25501
201. Tumeh PC, Harview CL, Yearley JH, Shintaku IP, Taylor EJ, Robert L, et al. PD-1 blockade induces responses by inhibiting adaptive immune resistance. Nature. (2014) 515:568–71. doi: 10.1038/nature13954
202. Bos R, Sherman LA. CD4+ T-cell help in the tumor milieu is required for recruitment and cytolytic function of CD8+ T lymphocytes. Cancer Res. (2010) 70:8368–77. doi: 10.1158/0008-5472.CAN-10-1322
203. Ahrends T, Spanjaard A, Pilzecker B, Babala N, Bovens A, Xiao Y, et al. CD4(+) T cell help confers a cytotoxic t cell effector program including coinhibitory receptor downregulation and increased tissue invasiveness. Immunity. (2017) 47:848–61 e845. doi: 10.1016/j.immuni.2017.10.009
204. Nesbeth YC, Martinez DG, Toraya S, Scarlett UK, Cubillos-Ruiz JR, Rutkowski MR, et al. CD4+ T cells elicit host immune responses to MHC class II-negative ovarian cancer through CCL5 secretion and CD40-mediated licensing of dendritic cells. J Immunol. (2010) 184:5654–62. doi: 10.4049/jimmunol.0903247
205. Castellino F, Huang AY, Altan-Bonnet G, Stoll S, Scheinecker C, Germain RN. Chemokines enhance immunity by guiding naive CD8+ T cells to sites of CD4+ T cell-dendritic cell interaction. Nature. (2006) 440:890–5. doi: 10.1038/nature04651
206. Gonzalez-Martin A, Gomez L, Lustgarten J, Mira E, Manes S. Maximal T cell-mediated antitumor responses rely upon CCR5 expression in both CD4(+) and CD8(+) T cells. Cancer Res. (2011) 71:5455–66. doi: 10.1158/0008-5472.CAN-11-1687
207. Hung K, Hayashi R, Lafond-Walker A, Lowenstein C, Pardoll D, Levitsky H. The central role of CD4(+) T cells in the antitumor immune response. J Exp Med. (1998) 188:2357–68. doi: 10.1084/jem.188.12.2357
208. Thibodeau J, Bourgeois-Daigneault MC, Lapointe R. Targeting the MHC Class II antigen presentation pathway in cancer immunotherapy. Oncoimmunology. (2012) 1:908–16. doi: 10.4161/onci.21205
209. Seliger B, Kloor M, Ferrone S. HLA class II antigen-processing pathway in tumors: molecular defects and clinical relevance. Oncoimmunology. (2017) 6:e1171447. doi: 10.1080/2162402X.2016.1171447
210. Beckhove P, Feuerer M, Dolenc M, Schuetz F, Choi C, Sommerfeldt N, et al. Specifically activated memory T cell subsets from cancer patients recognize and reject xenotransplanted autologous tumors. J Clin Invest. (2004) 114:67–76. doi: 10.1172/JCI200420278
211. Gao Y, Whitaker-Dowling P, Barmada MA, Basse PH, Bergman I. Viral infection of implanted meningeal tumors induces antitumor memory T-cells to travel to the brain and eliminate established tumors. Neuro Oncol. (2015) 17:536–44. doi: 10.1093/neuonc/nou231
212. Goldszmid RS, Idoyaga J, Bravo AI, Steinman R, Mordoh J, Wainstok R. Dendritic cells charged with apoptotic tumor cells induce long-lived protective CD4 and CD8 T cell immunity against B16 melanoma. J Immunol. (2003) 171:5940–7. doi: 10.4049/jimmunol.171.11.5940
213. Kondo T, Morita R, Okuzono Y, Nakatsukasa H, Sekiya T, Chikuma S, et al. Notch-mediated conversion of activated T cells into stem cell memory-like T cells for adoptive immunotherapy. Nat Commun. (2017) 8:15338. doi: 10.1038/ncomms15338
214. Rosenberg SA, Yang JC, Restifo NP. Cancer immunotherapy: moving beyond current vaccines. Nat Med. (2004) 10:909–15. doi: 10.1038/nm1100
215. Klebanoff CA, Acquavella N, Yu Z, Restifo NP. Therapeutic cancer vaccines: are we there yet? Immunol Rev. (2011) 239:27–44. doi: 10.1111/j.1600-065X.2010.00979.x
216. Hueman MT, Stojadinovic A, Storrer CE, Dehqanzada ZA, Gurney JM, Shriver CD, et al. Analysis of naive and memory CD4 and CD8 T cell populations in breast cancer patients receiving a HER2/neu peptide (E75) and GM-CSF vaccine. Cancer Immunol Immunother. (2007) 56:135–46. doi: 10.1007/s00262-006-0188-9
217. Chevaleyre C, Benhamouda N, Favry E, Fabre E, Mhoumadi A, Nozach H, et al. The tumor antigen cyclin B1 hosts multiple CD4 T cell epitopes differently recognized by pre-existing naive and memory cells in both healthy and cancer donors. J Immunol. (2015) 195:1891–901. doi: 10.4049/jimmunol.1402548
218. Anastasopoulou EA, Voutsas IF, Papamichail M, Baxevanis CN, Perez SA. MHC class II tetramer analyses in AE37-vaccinated prostate cancer patients reveal vaccine-specific polyfunctional and long-lasting CD4(+) T-cells. Oncoimmunology. (2016) 5:e1178439. doi: 10.1080/2162402X.2016.1178439
219. Gao Y, Whitaker-Dowling P, Griffin JA, Bergman I. Treatment with targeted vesicular stomatitis virus generates therapeutic multifunctional anti-tumor memory CD4 T cells. Cancer Gene Ther. (2012) 19:282–91. doi: 10.1038/cgt.2011.90
220. Quezada SA, Simpson TR, Peggs KS, Merghoub T, Vider J, Fan X, et al. Tumor-reactive CD4(+) T cells develop cytotoxic activity and eradicate large established melanoma after transfer into lymphopenic hosts. J Exp Med. (2010) 207:637–50. doi: 10.1084/jem.20091918
221. Hirschhorn-Cymerman D, Budhu S, Kitano S, Liu C, Zhao F, Zhong H, et al. Induction of tumoricidal function in CD4+ T cells is associated with concomitant memory and terminally differentiated phenotype. J Exp Med. (2012) 209:2113–26. doi: 10.1084/jem.20120532
222. Thomas WD, Hersey P. TNF-related apoptosis-inducing ligand (TRAIL) induces apoptosis in Fas ligand-resistant melanoma cells and mediates CD4 T cell killing of target cells. J Immunol. (1998) 161:2195–200.
223. Broderick L, Yokota SJ, Reineke J, Mathiowitz E, Stewart CC, Barcos M, et al. Human CD4+ effector memory T cells persisting in the microenvironment of lung cancer xenografts are activated by local delivery of IL-12 to proliferate, produce IFN-gamma, and eradicate tumor cells. J Immunol. (2005) 174:898–906. doi: 10.4049/jimmunol.174.2.898
224. Kitano S, Tsuji T, Liu C, Hirschhorn-Cymerman D, Kyi C, Mu Z, et al. Enhancement of tumor-reactive cytotoxic CD4+ T cell responses after ipilimumab treatment in four advanced melanoma patients. Cancer Immunol Res. (2013) 1:235–44. doi: 10.1158/2326-6066.CIR-13-0068
225. Wagner DH. Re-shaping the T cell repertoire: TCR editing and TCR revision for good and for bad. Clin Immunol. (2007) 123:1–6. doi: 10.1016/j.clim.2006.08.006
226. Kim MT, Harty JT. Impact of inflammatory cytokines on effector and memory CD8+ T cells. Front Immunol. (2014) 5:295. doi: 10.3389/fimmu.2014.00295
227. Park SO, Han YW, Aleyas AG, George JA, Yoon HA, Lee JH, et al. Low-dose antigen-experienced CD4+ T cells display reduced clonal expansion but facilitate an effective memory pool in response to secondary exposure. Immunology. (2008) 123:426–37. doi: 10.1111/j.1365-2567.2007.02707.x
228. Kurtulus S, Madi A, Escobar G, Klapholz M, Nyman J, Christian E, et al. Checkpoint blockade immunotherapy induces dynamic changes in PD-1(-)CD8(+) tumor-infiltrating T Cells. Immunity. (2019) 50:181–94 e186. doi: 10.1016/j.immuni.2018.11.014
229. Klenerman P. The (gradual) rise of memory inflation. Immunol Rev. (2018) 283:99–112. doi: 10.1111/imr.12653
230. Loewendorf AI, Arens R, Purton JF, Surh CD, Benedict CA. Dissecting the requirements for maintenance of the CMV-specific memory T-cell pool. Viral Immunol. (2011) 24:351–5. doi: 10.1089/vim.2010.0140
231. Welten SPM, Sandu I, Baumann NS, Oxenius A. Memory CD8 T cell inflation vs. tissue-resident memory T cells: same patrollers, same controllers? Immunol Rev. (2018) 283:161–75. doi: 10.1111/imr.12649
232. Youngblood B, Oestreich KJ, Ha SJ, Duraiswamy J, Akondy RS, West EE, et al. Chronic virus infection enforces demethylation of the locus that encodes PD-1 in antigen-specific CD8(+) T cells. Immunity. (2011) 35:400–12. doi: 10.1016/j.immuni.2011.06.015
233. Schmidl C, Delacher M, Huehn J, Feuerer M. Epigenetic mechanisms regulating T-cell responses. J Allergy Clin Immunol. (2018) 142:728–43. doi: 10.1016/j.jaci.2018.07.014
234. Van Der Windt GJ, Pearce EL. Metabolic switching and fuel choice during T-cell differentiation and memory development. Immunol Rev. (2012) 249:27–42. doi: 10.1111/j.1600-065X.2012.01150.x
235. Zhang L, Romero P. Metabolic control of CD8(+) T cell fate decisions and antitumor immunity. Trends Mol Med. (2018) 24:30–48. doi: 10.1016/j.molmed.2017.11.005
236. Schurich A, Pallett LJ, Jajbhay D, Wijngaarden J, Otano I, Gill US, et al. Distinct metabolic requirements of exhausted and functional virus-specific CD8 T cells in the same host. Cell Rep. (2016) 16:1243–52. doi: 10.1016/j.celrep.2016.06.078
237. Chang CH, Qiu J, O'sullivan D, Buck MD, Noguchi T, Curtis JD, et al. Metabolic competition in the tumor microenvironment is a driver of cancer progression. Cell. (2015) 162:1229–41. doi: 10.1016/j.cell.2015.08.016
238. Parry RV, Chemnitz JM, Frauwirth KA, Lanfranco AR, Braunstein I, Kobayashi SV, et al. CTLA-4 and PD-1 receptors inhibit T-cell activation by distinct mechanisms. Mol Cell Biol. (2005) 25:9543–53. doi: 10.1128/MCB.25.21.9543-9553.2005
239. Patsoukis N, Bardhan K, Chatterjee P, Sari D, Liu B, Bell LN, et al. PD-1 alters T-cell metabolic reprogramming by inhibiting glycolysis and promoting lipolysis and fatty acid oxidation. Nat Commun. (2015) 6:6692. doi: 10.1038/ncomms7692
240. Menk AV, Scharping NE, Moreci RS, Zeng X, Guy C, Salvatore S, et al. Early TCR signaling induces rapid aerobic glycolysis enabling distinct acute t cell effector functions. Cell Rep. (2018) 22:1509–21. doi: 10.1016/j.celrep.2018.01.040
Keywords: T cell memory, cancer, chronic infection, CD4 T cells, CD8 T cells
Citation: Hope JL, Stairiker CJ, Bae E-A, Otero DC and Bradley LM (2019) Striking a Balance—Cellular and Molecular Drivers of Memory T Cell Development and Responses to Chronic Stimulation. Front. Immunol. 10:1595. doi: 10.3389/fimmu.2019.01595
Received: 01 March 2019; Accepted: 26 June 2019;
Published: 17 July 2019.
Edited by:
Stefano Caserta, University of Hull, United KingdomReviewed by:
Thomas Poiret, Karolinska Institute (KI), SwedenCopyright © 2019 Hope, Stairiker, Bae, Otero and Bradley. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Linda M. Bradley, bGJyYWRsZXlAc2JwZGlzY292ZXJ5Lm9yZw==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.