 Yu Wang
Yu Wang Hui Zhang
Hui Zhang You-Wen He
You-Wen He
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol. , 19 July 2019
Sec. Cancer Immunity and Immunotherapy
Volume 10 - 2019 | https://doi.org/10.3389/fimmu.2019.01574
This article is part of the Research Topic The Role of Complement in Tumors View all 10 articles
Cancer immunotherapy has made remarkable clinical advances in recent years. Antibodies targeting the immune checkpoint receptors PD-1 and CTLA-4 and adoptive cell therapy (ACT) based on ex vivo expanded peripheral CTLs, tumor infiltrating lymphocytes (TILs), gene-engineered TCR- and chimeric antigen receptor (CAR)-T cells have all shown durable clinical efficacies in multiple types of cancers. However, these immunotherapeutic approaches only benefit a small fraction of cancer patients as various immune resistance mechanisms and limitations make their effective use a challenge in the majority of cancer patients. For example, adaptive resistance to therapeutic PD-1 blockade is associated with an upregulation of some additional immune checkpoint receptors. The efficacy of transferred tumor-specific T cells under the current clinical ACT protocol is often limited by their inefficient engraftment, poor persistence, and weak capability to attack tumor cells. Recent studies demonstrate that the complement receptor C3aR and C5aR function as a new class of immune checkpoint receptors. Complement signaling through C3aR and C5aR expressed on effector T lymphocytes prevent the production of the cytokine interleukin-10 (IL-10). Removing C3aR/C5aR-mediated transcriptional suppression of IL-10 expression results in endogenous IL-10 production by antitumor effector T cells, which drives T cell expansion and enhances T cell-mediated antitumor immunity. Importantly, preclinical, and clinical data suggest that a signaling axis consisting of complement/C3aR/C5aR/IL-10 critically regulates T cell mediated antitumor immunity and manipulation of the pathway ex vivo and in vivo is an effective strategy for cancer immunotherapy. Furthermore, a combination of treatment strategies targeting the complement/C3aR/C5aR/IL-10 pathway with other treatment modalities may improve cancer therapeutic efficacy.
As a major component of the innate immunity, the complement system also directly regulates lymphocyte function (1, 2). Recent studies have shed important insights to the role of complement and its receptors in antitumor immunity. Clinical observations and animal studies suggest that complement signaling inhibits antitumor immunity. It was reported that tumor or circulating complement levels are positively correlated with tumor size and poor outcome in different types of cancers, such as neuroblastoma, colorectal, lung, ovarian cancer, chronic lymphocytic leukemia, and carcinomas of the digestive tract (3). Extensive animal studies have also demonstrated that the complement system functions to inhibit antitumor immunity (4–15). Mechanistically, complement may inhibit antitumor immunity by promoting recruitment of myeloid-derived suppressor cells (MDSCs) into the tumor microenvironment (TME) (4–6, 9, 12, 13) or by suppressing dendritic cells (DCs)/NK cell activation (7, 8). Recent studies suggest that a new mechanism plays an important role in complement signaling-mediated suppression of antitumor immunity: direct inhibition of IL-10 production in CD8+ tumor infiltrating lymphocytes (TILs) in TME (11, 14). Here, we summarize relevant findings and propose that C3aR and C5aR function as a new class of immune checkpoint receptors that should be targeted for cancer immunotherapy.
In addition to the many clinical reports positively correlating complement levels with tumor size and poor outcome in various human cancers [reviewed by Pio et al. (3)], animal studies testing different tumor types in different mouse models also show that the complement signaling pathway exerts potent inhibition on antitumor immunity (4–15) (Table 1). The tested mouse tumor models include TC-1 cervical cancer, Lewis lung cancer, RMA lymphoma, 4T1, and E0771 breast cancer, B16 melanoma, HPV16 skin cancer, and MC38 colon cancer with either transplanted tumor cells or spontaneously developed cancers. Complement signaling was disrupted in these animal studies by using genetic models including mice deficient for C3, C4, C3aR, or C5aR1 (4, 9–15) or inhibitors to complement C3, C3aR, and C5aR1 (4–6, 8, 10–12, 14). The reported results are highly consistent in that tumor growth is suppressed when complement-mediated signaling is inhibited or removed. In studying the underlying cellular mechanisms, Markiewski and colleagues first showed that C5a/C5aR1 interaction promotes the migration of MDSCs into tumors and enhances the suppressive capacity of tumor-associated MDSCs (4). The regulation of myeloid suppressor cells in tumors by complement signaling is also observed by several other studies (5, 6, 9, 12, 13). Thus, a major immune suppressive role by complement signaling may be mediated through recruitment of MDSCs into tumors. In addition to MDSCs, other innate cell populations such as neutrophils, DCs and NK cells are also involved in complement-mediated immune suppression of antitumor immunity (7, 8, 10). When complement C3 is exhausted using cobra venom factor, NK cells are greatly increased in tumors and depletion of NK cells nullifies the enhanced antitumor activity induced by cobra venom factor treatment (8). Although complement signaling modulates innate immune cell activities, the enhanced antitumor immunity exhibited in mice following disruption of complement signaling is T lymphocyte dependent. Not only effector CD4+ and CD8+ TILs are enhanced in these mice but also depletion of T cells through TCRα genetic deletion or antibodies against CD4+ or CD8+ T cells diminishes the enhanced antitumor immunity in the complement signaling deficient models (4, 8–15). These studies suggest that multiple immune suppressive mechanisms are induced by C3aR and C5aR1 signaling (Table 1).
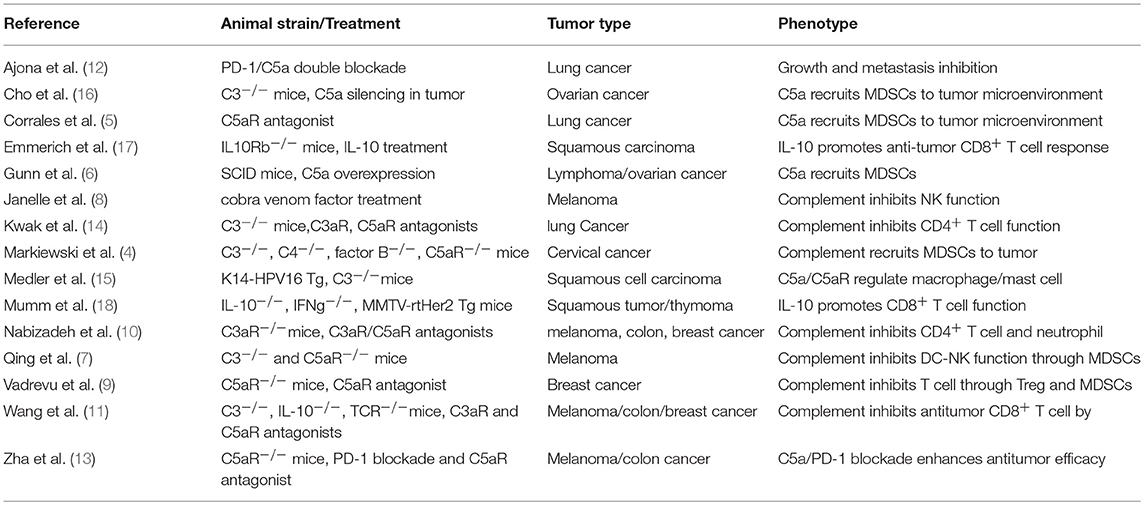
Table 1. Mouse models on the complement/C3aR/C5aR1/IL-10 pathway in antitumor immunity.
Three small cationic peptides, C3a, C4a, and C5a, generated by complement activation are termed as anaphylatoxins. These peptides induce chemotaxis, cell activation, and inflammatory signaling by binding to their respective G-protein-coupled receptors (GPCR), referred to as C3aR and C5aR1. The models for anaphylatoxins binding to their cognitive receptors have been proposed after the molecular cloning of C3aR and C5aR1 (19). In the immune system, C3aR is predominantly distributed on leukocytes of myeloid lineages, such as neutrophils, basophils, eosinophils, mast cells, monocytes/macrophages (20–23). Ligand-receptor engagement induced receptor phosphorylation leads to receptor desensitization, internalization, and activation of diverse downstream signaling pathways in different cell types. C3aR is highly expressed on neutrophils, and C3a induces calcium influx in response to C3a (24); however, C3aR inhibits neutrophil mobilization in vivo in an intestinal ischemia-reperfusion model (25). In mast cells, C3a activates PI3K signaling pathways and subsequent Akt-phosphorylation, as well as MAP kinases Erk1/Erk2 to promote cytokine expression (26). In human monocyte/macrophage, engagement of C3a to Ca3R, together with TLR signaling induces secretion of proinflammatory cytokines such as IL-1β, IL-6, and TNFα (27, 28). C3aR signaling modulates IL-1β secretion through NLRP3 inflammasome activation by regulating ATP efflux (29). Similar to C3aR, C5aR is abundantly expressed in neutrophils, eosinophils and basophils, monocytes/macrophages, and mast cells (30–33). C5a binding to C5aR causes calcium flux as well as activation of several components of different signaling pathways, including PI3K-γ kinase, phospholipase C, phospholipase D and Raf-1/B-Raf mediated activation of MEK-1 (34–37). In addition to a similar proinflammatory function of C3aR, C5aR1 is also a chemotactic receptor. Upon engagement with C5aR1, C5a serves as a chemoattractant for monocytes, neutrophils, eosinophils, and basophils (38).
It is well established that complement components and their receptors C3aR and C5aR1 are expressed in not only myeloid and tumor cells but also CD4+ T lymphocytes (39–44). Furthermore, endogenously or locally produced C3a and C5a bind to C3aR and C5aR on CD4+ T cells and regulate T cell function, such as differentiation, survival and cytokine production (40, 41, 45, 46). Interestingly, in contrast to the lack of C3aR and C5aR1 expression on peripheral CD8+ T cells in naive mice, both receptors are strongly upregulated on CD8+ TILs from mouse and human tumors (11). Overall, ~20% of CD8+ TILs are C3aR and C5aR double positive. To determine the source of complement that mediates immune suppression on CD8+ TILs, chimeric mice with either lymphocytes or host cells lacking C3 were used as tumor-bearing hosts. C3-deletion in CD8+ T cells was sufficient to remove complement-mediated suppression on antitumor immunity (11), suggesting that autocrine C3 production and the interaction of activation products with Ca3R/C5aR plays a critical role in suppressing effector CD8+ TIL function.
How does autocrine complement signaling inhibit effector CD8+ T cell function? Several clues suggest a possible mechanism underlying C3aR/C5aR signaling-mediated immune checkpoint function: complement signaling may inhibit IL-10 production in effector T lymphocytes given the role of IL-10 in CD8+ TIL expansion and immune activating function in antitumor immunity (see Discussion in next section). First, it was shown that a fraction of CD8+ effectors expresses IL-10 at the peak of coronavirus infection and the IL-10+CD8+ T cells show superior CTL activity and in vivo protection against chronic infection (47). Second, we found that complement pathway related genes are enriched in the IL-10+CD8+ T cells (11), suggesting a possibility of mutual or reciprocal regulation. Indeed, in C3−/− Il10 reporter (Tiger) mice, CD8+ TILs within B16 tumors but not peripheral blood readily express IL-10 (11). Kwak and colleagues also observed enhanced IL-10 expression in CD4+ and CD8+ T lymphocytes in lungs of tumor-bearing C3-deficient mice (14). Furthermore, antagonists to C3aR and C5aR1 also promote IL-10 production in CD8+ TILs as well as in vitro activated CD8+ T cells (11). Importantly, the enhanced antitumor immunity in complement-deficient mice or wildtype mice treated with antagonists to C3aR and C5aR1 depend on IL-10. Depletion of the IL-10 gene in these mice completely abolishes the enhanced antitumor immunity in both melanoma and breast cancer tumor-bearing C3-deficient mice (11). The suppression of IL-10 production in CD8+ TILs is mediated through endogenously produced complement and its autocrine interaction with C3aR and C5aR on CD8+ T cells. The inhibition on IL-10 production by signaling through C3aR and C5aR is redundant as antagonism to one of these receptors alone does not promote IL-10 production. Accordingly, antagonism to C3aR and C5aR1, but not to a single receptor, suppresses tumor growth and the antitumor effect depends on IL-10 in vivo (11). Therefore, inhibition of antitumor immunity through suppression of IL-10 production in CD8+ TILs in response to complement/C3aR/C5aR signaling represents a new mechanism of complement-mediated immune supression (48).
IL-10 is a pleiotropic cytokine produced by many cell populations, including but not limited to activated T cells, B cells, macrophages as well as mast cells (49, 50). Although it was initially identified as a cofactor for thymocytes growth and T cell activation, IL-10 was further recognized as a regulatory cytokine due to its anti-inflammatory functions. IL-10 impairs the maturation of dendritic cells and macrophages by interfering with upregulation of costimulatory molecules such as CD80, CD86, MHCII, and CD83 on activated dendritic cells and macrophages (51, 52). In addition, IL-10 skews the Th1/Th2 balance to Th2 by selectively blocking IL-12 synthesis in activated dendritic cells (53). Macrophages can be polarized to M1 (inflammatory) or M2 (anti-inflammatory) phenotypes depending on the microenvironmental stimuli. IL-10 inhibits the activation and proliferation through Stat3-dependent and -independent pathways and polarizes macrophage to a M2 like phenotype (54, 55). IL-10 directly acts on CD4+ T cells to differentiate T helper cells into inducible regulatory T cells and maintain the expression of key transcription factor Foxp3 (56, 57). Regulatory T cells also express IL-10 and mice deficient for IL-10 in regulatory T cells did not display systemic autoimmunity; however, these mice developed spontaneous colitis, skin and lung hyperreactivity, suggesting an organ specific role of IL-10 on regulatory T cells (58).
Although IL-10 is often associated with an immune suppressive function, recent clinical studies have unequivocally shown that IL-10 is an immune activating cytokine promoting antitumor immunity (59–61). In a phase I clinical trial, pegylated recombinant human IL-10 (rhIL-10) has shown encouraging clinical efficacy in several types of solid tumors (59). Among the 24 patients treated with rhIL-10 monotherapy at 20–40 μg/kg active dose, the overall objective response rate is 21%. Furthermore, IL-10 treatment increases serum levels of pro-inflammatory cytokines IL-18 and IFNγ as well as FasL in cancer patients and the induced cytokine levels are strongly correlated with clinical responses (59, 61). Pegylated rhIL-10 treatment dramatically expands PD-1+LAG-3+ activated CD8+ T cells in the blood of cancer patients. Importantly, both the number and effector function of CD8+ TILs from these patients are increased (61). These results support that the major function of rhIL-10 is to expand the number as well as enhance the effector function of antitumor CD8+ T cells in cancer patients. A Phase 3 clinical trial (NCT02923921) in patients with metastatic pancreatic cancer is being conducted based on promising efficacy data from early clinical studies. Mechanistically, checkpoint inhibition, and IL-10 treatment together enhances the number and quality of pre-existing TILs. The efficacy of PD-1/PD-L1 inhibitors is highly associated with tumor microenvironment such as TIL density, PD-1/PD-L1 expression; tumor intrinsic feature, such as tumor mutational burden, microsatellite instability; as well as gut microbiota (62). The clinical trials of pegylated recombinant human IL-10 are focused on several solid tumor types. Its efficacy on solid and blood tumor types needs to be tested clinically in the future.
Another potential application for IL-10 and C3aR/C5aR antagonists is to incorporate them into in vitro expansion protocols of T cells for ACT. IL-2 is the primary cytokine used in the in vitro expansion of TILs and gene-engineered T cells for clinical use, however, the in vivo efficacy of expanded T cells under the current clinical ACT protocol is often limited by their inefficient engraftment, poor persistence, and weak capability to attack tumor cells (63–68). It was shown long ago that IL-10 augments IL-2-induced proliferation and promotes CTL activity of activated CD8+ T cells (69–72). Consistent with animal studies and human clinical trial data showing that IL-10 promotes CD8+ TIL proliferation (11, 17, 18, 61), addition of IL-10 to in vitro culture of TILs from human lung cancer with IL-2 drastically enhances the quantity and quality of the expanded human TILs and upregulates genes related to several signaling pathways, such TCR signaling, Notch signaling, cell cycle and CTL killing (11). Furthermore, pegylated rhIL-10 also prevents continuous TCR-stimulation induced apoptosis of activated human T cells (61). Interestingly, remissions in lymphoma patients treated with anti-CD19 chimeric antigen receptor (CAR-T) cells are associated with high serum levels of IL-10 and IL-15 (73). These results strongly suggest that the addition of IL-10 to the IL-2-supported in vitro T cell expansion protocol may improve the clinical efficacy of adoptive T cell therapy. In addition to IL-10, antagonists to C3aR/C5aR1 may also be used in such protocol as the in vitro culture of activated CD8+ T cells in the presence of C3aR/C5aR1 antagonists induces IL-10 production (11).
Significant progress has been made on testing the synergistic effect of combined treatment targeting the complement/C3aR/C5aR/IL-10 pathway and other cancer treatment modalities. The first combined strategy is dual blockade of complement signaling and immune checkpoint receptor PD-1 (Figure 1). The complement signaling/IL-10 pathway is independent of the PD-1/PD-L1 pathway as modulation of this pathway does not affect the expression of PD-1 on T cells and PD-L1 on tumor cells (11). Two different experimental systems in which (1) PD-L1-silenced B16F10 tumors were inoculated in C3-deficient mice or (2) B16F10 tumor-bearing wildtype mice were treated with anti-PD1 and antagonists to C3aR/C5aR clearly show that blockade of complement signaling and PD-1/PD-L1 interaction has dramatic synergistic antitumor effect (11). This synergistic antitumor effect is subsequently confirmed by two other studies (12, 13). These data provide important clues to rational design of future clinical trials.
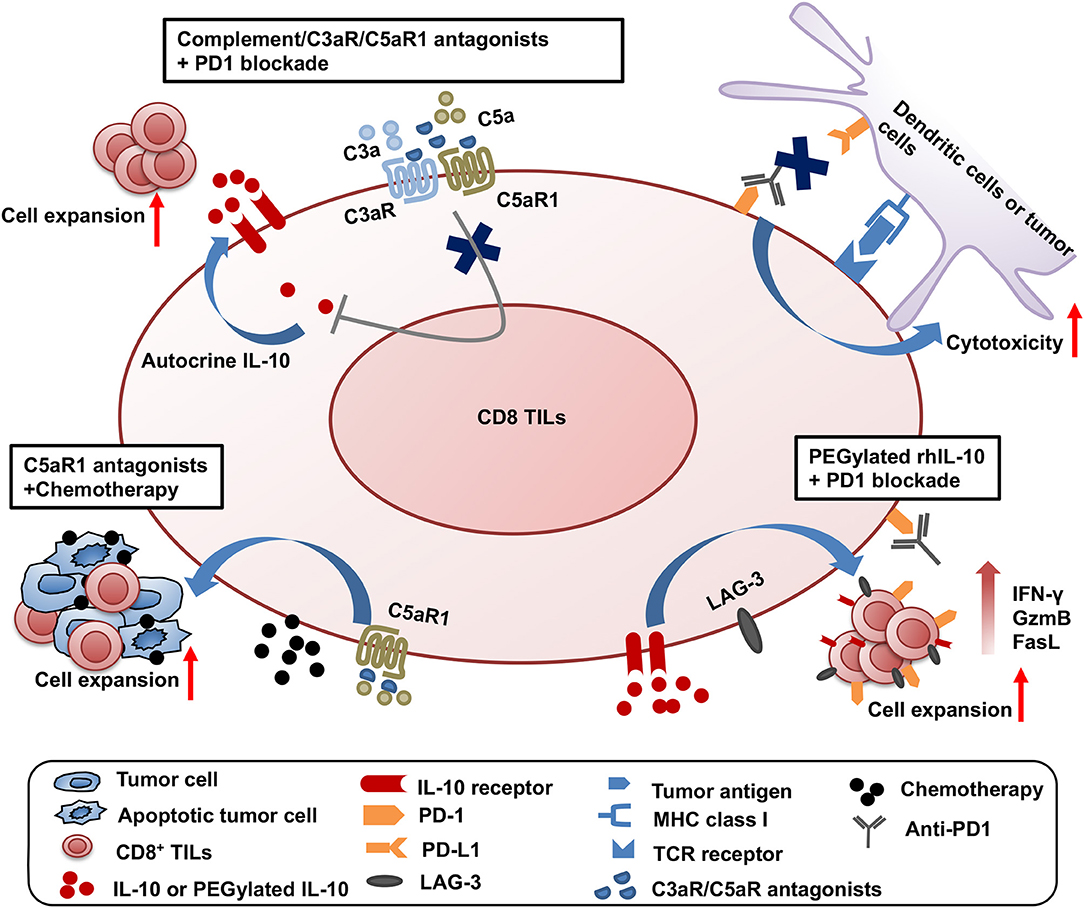
Figure 1. Synergistic effect of three combined strategies by targeting complement/C3aR /C5aR1/IL-10 pathway and other treatment modalities. The three combined strategies were shown as follows: (1) dual blockade of complement signaling and immune checkpoint receptor PD-1; (2) complement signaling blockade and chemotherapy; (3) the clinical use of pegylated rhIL-10 with anti-PD-1 antibody.
The second combined treatment strategy uses complement signaling blockade and chemotherapy (Figure 1). In a squamous cell carcinoma (SCC) model, antagonist to C5aR1 enhances the treatment efficacy of paclitaxel chemotherapy and the synergistic effect depends on CD8+ T lymphocytes (15). Increased CD8+ TILs and the expansion of specific T cell clones were associated with enhanced efficacy (15).
The third combination is the clinical use of pegylated rhIL-10 with the anti-PD-1 antibody pembrolizumab in a cohort of heavily pretreated patients with melanoma, no-squamous cell lung cancer or renal cell carcinoma (61) (Figure 1). This combination achieved a 42% objective response rate, in contrast to the 21% objective response rate by pegylated rhIL-10 monotherapy (59). A combination of pegylated rhIL-10 with anti-PD-1 promotes persistent proliferation and expansion of LAG-3+PD-1+ CD8+ T cells in the cancer patients. These exciting clinical trial results have opened new avenues for effective cancer immunotherapy.
In summary, we have identified that tumor infiltrating CD8+ T cells express complement receptors C3aR and C5aR and complement signaling inhibits anti-tumor functions through repression of endogenous IL-10 production in CD8+ TILs. We and other groups have also confirmed that endogenous and exogenous IL-10 enhances anti-tumor functions of CD8+ T cells in human and mouse in vitro and in vivo. The independence of complement/C3aR/C5aR/IL-10 from the PD-1/PD-L1 signaling pathway makes it possible to block complement receptors and PD-1/PD-L1 as a combined therapy to treat cancer patients clinically. Results from other groups also suggest that the combined blockade of complement and PD-1/PD-L1 signaling with antibodies improves the efficacy of treatment through other mechanisms. Together, we and other groups provide clear evidences that complement receptors C3aR and C5aR are a new class of immune checkpoint receptors.
YW, HZ, and Y-WH co-wrote the review.
YW was supported by Innovation Talents Project LR2017019.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
1. Carroll MC. The complement system in regulation of adaptive immunity. Nat Immunol. (2004) 5:981–6. doi: 10.1038/ni1113
2. Heeger PS, Kemper C. Novel roles of complement in T effector cell regulation. Immunobiology. (2012) 217:216–24. doi: 10.1016/j.imbio.2011.06.004
3. Pio R, Corrales L, Lambris JD. The role of complement in tumor growth. Adv Exp Med Biol. (2014) 772:229–62. doi: 10.1007/978-1-4614-5915-6_11
4. Markiewski MM, DeAngelis RA, Benencia F, Ricklin-Lichtsteiner SK, Koutoulaki A, Gerard C, et al. Modulation of the antitumor immune response by complement. Nat Immunol. (2008) 9:1225–35. doi: 10.1038/ni.1655
5. Corrales L, Ajona D, Rafail S, Lasarte JJ, Riezu-Boj JI, Lambris JD, et al. Anaphylatoxin C5a creates a favorable microenvironment for lung cancer progression. J Immunol. (2012) 189:4674–83. doi: 10.4049/jimmunol.1201654
6. Gunn L, Ding C, Liu M, Ma Y, Qi C, Cai Y, et al. Opposing roles for complement component C5a in tumor progression and the tumor microenvironment. J Immunol. (2012) 189:2985–94. doi: 10.4049/jimmunol.1200846
7. Qing X, Koo GC, Salmon JE. Complement regulates conventional DC-mediated NK-cell activation by inducing TGF-β1 in Gr-1+ myeloid cells. Eur J Immunol. (2012) 42:1723–34. doi: 10.1002/eji.201142290
8. Janelle V, Langlois MP, Tarrab E, Lapierre P, Poliquin L, Lamarre A. Transient complement inhibition promotes a tumor-specific immune response through the implication of natural killer cells. Cancer Immunol Res. (2014) 2:200–6. doi: 10.1158/2326-6066.CIR-13-0173
9. Vadrevu SK, Chintala NK, Sharma SK, Sharma P, Cleveland C, Riediger L, et al. Complement c5a receptor facilitates cancer metastasis by altering T-cell responses in the metastatic niche. Cancer Res. (2014) 74:3454–65. doi: 10.1158/0008-5472.CAN-14-0157
10. Nabizadeh JA, Manthey HD, Steyn FJ, Chen W, Widiapradja A, Md Akhir FN, et al. The complement C3a receptor contributes to melanoma tumorigenesis by inhibiting neutrophil and CD4+ T cell responses. J Immunol. (2016) 196:4783–92. doi: 10.4049/jimmunol.1600210
11. Wang Y, Sun SN, Liu Q, Yu YY, Guo J, Wang K, et al. Autocrine complement inhibits IL10-dependent T-cell-mediated antitumor immunity to promote tumor progression. Cancer Discov. (2016) 6:1–11. doi: 10.1158/2159-8290.CD-15-1412
12. Ajona D, Ortiz-Espinosa S, Moreno H, Lozano T, Pajares MJ, Agorreta J, et al. A combined PD-1/C5a blockade synergistically protects against lung cancer growth and metastasis. Cancer Discov. (2017) 7:694–703. doi: 10.1158/2159-8290.CD-16-1184
13. Zha H, Han X, Zhu Y, Yang F, Li Y, Li Q, et al. Blocking C5aR signaling promotes the anti-tumor efficacy of PD-1/PD-L1 blockade. Oncoimmunology. (2017) 6:e1349587. doi: 10.1080/2162402X.2017.1349587
14. Kwak JW, Laskowski J, Li HY, McSharry MV, Sippel TR, Bullock BL, et al. Complement activation via a C3a receptor pathway alters CD4(+) T lymphocytes and mediates lung cancer progression. Cancer Res. (2018) 78:143–56. doi: 10.1158/0008-5472.CAN-17-0240
15. Medler TR, Murugan D, Horton W, Kumar S, Cotechini T, Forsyth AM, et al. Complement C5a fosters squamous carcinogenesis and limits T cell response to chemotherapy. Cancer Cell. (2018) 34:e566. doi: 10.1016/j.ccell.2018.09.003
16. Cho MS, Vasquez HG, Rupaimoole R, Pradeep S, Wu S, Zand B, et al. Autocrine effects of tumor-derived complement. Cell Rep. (2014) 6:1085–95. doi: 10.1016/j.celrep.2014.02.014
17. Emmerich J, Mumm JB, Chan IH, LaFace D, Truong H, McClanahan T, et al. IL-10 directly activates and expands tumor-resident CD8(+) T cells without de novo infiltration from secondary lymphoid organs. Cancer Res. (2012) 72:3570–81. doi: 10.1158/0008-5472.CAN-12-0721
18. Mumm JB, Emmerich J, Zhang X, Chan I, Wu L, Mauze S, et al. IL-10 elicits IFNγ-dependent tumor immune surveillance. Cancer Cell. (2011) 20:781–96. doi: 10.1016/j.ccr.2011.11.003
19. Ember JA, Hugli TE. Complement factors and their receptors. Immunopharmacology. (1997) 38:3–15. doi: 10.1016/S0162-3109(97)00088-X
20. Dahinden CA, Bischoff SC, Brunner T, Krieger M, Takafuji S, de Weck AL. Regulation of mediator release by human basophils: importance of the sequence and time of addition in the combined action of different agonists. Int Arch Allergy Appl Immunol. (1991) 94:161–4. doi: 10.1159/000235350
21. Elsner J, Oppermann M, Czech W, Dobos G, Schopf E, Norgauer J, et al. C3a activates reactive oxygen radical species production and intracellular calcium transients in human eosinophils. Eur J Immunol. (1994) 24:518–22. doi: 10.1002/eji.1830240304
22. Martin U, Bock D, Arseniev L, Tornetta MA, Ames RS, Bautsch W, et al. The human C3a receptor is expressed on neutrophils and monocytes, but not on B or T lymphocytes. J Exp Med. (1997) 186:199–207. doi: 10.1084/jem.186.2.199
23. Zwirner J, Gotze O, Sieber A, Kapp A, Begemann G, Zuberbier T, et al. The human mast cell line HMC-1 binds and responds to C3a but not C3a(desArg). Scand J Immunol. (1998) 47:19–24. doi: 10.1046/j.1365-3083.1998.00250.x
24. Norgauer J, Dobos G, Kownatzki E, Dahinden C, Burger R, Kupper R, et al. Complement fragment C3a stimulates Ca2+ influx in neutrophils via a pertussis-toxin-sensitive G protein. Eur J Biochem. (1993) 217:289–94. doi: 10.1111/j.1432-1033.1993.tb18245.x
25. Wu MC, Brennan FH, Lynch JP, Mantovani S, Phipps S, Wetsel RA, et al. The receptor for complement component C3a mediates protection from intestinal ischemia-reperfusion injuries by inhibiting neutrophil mobilization. Proc Natl Acad Sci USA. (2013) 110:9439–44. doi: 10.1073/pnas.1218815110
26. Venkatesha RT, Berla Thangam E, Zaidi AK, Ali H. Distinct regulation of C3a-induced MCP-1/CCL2 and RANTES/CCL5 production in human mast cells by extracellular signal regulated kinase and PI3 kinase. Mol Immunol. (2005) 42:581–7. doi: 10.1016/j.molimm.2004.09.009
27. Takabayashi T, Vannier E, Clark BD, Margolis NH, Dinarello CA, Burke JF, et al. A new biologic role for C3a and C3a desArg: regulation of TNF-alpha and IL-1 beta synthesis. J Immunol. (1996) 156:3455–60.
28. Fischer WH, Jagels MA, Hugli TE. Regulation of IL-6 synthesis in human peripheral blood mononuclear cells by C3a and C3a(desArg). J Immunol. (1999) 162:453–9.
29. Asgari E, Le Friec G, Yamamoto H, Perucha E, Sacks SS, Kohl J, et al. C3a modulates IL-1β secretion in human monocytes by regulating ATP efflux and subsequent NLRP3 inflammasome activation. Blood. (2013) 122:3473–81. doi: 10.1182/blood-2013-05-502229
30. Chenoweth DE, Goodman MG. The C5a receptor of neutrophils and macrophages. Agents Actions Suppl. (1983) 12:252–73. doi: 10.1007/978-3-0348-9352-7_15
31. Gerard NP, Hodges MK, Drazen JM, Weller PF, Gerard C. Characterization of a receptor for C5a anaphylatoxin on human eosinophils. J Biol Chem. (1989) 264:1760–6.
32. Kretzschmar T, Jeromin A, Gietz C, Bautsch W, Klos A, Kohl J, et al. Chronic myelogenous leukemia-derived basophilic granulocytes express a functional active receptor for the anaphylatoxin C3a. Eur J Immunol. (1993) 23:558–61. doi: 10.1002/eji.1830230239
33. Werfel T, Oppermann M, Begemann G, Gotze O, Zwirner J. C5a receptors are detectable on mast cells in normal human skin and in psoriatic plaques but not in weal and flare reactions or in uticaria pigmentosa by immunohistochemistry. Arch Dermatol Res. (1997) 289:83–6. doi: 10.1007/s004030050159
34. Mullmann TJ, Siegel MI, Egan RW, Billah MM. Complement C5a activation of phospholipase D in human neutrophils. A major route to the production of phosphatidates and diglycerides. J Immunol. (1990) 144:1901–8.
35. Buhl AM, Avdi N, Worthen GS, Johnson GL. Mapping of the C5a receptor signal transduction network in human neutrophils. Proc Natl Acad Sci USA. (1994) 91:9190–4. doi: 10.1073/pnas.91.19.9190
36. Jiang H, Kuang Y, Wu Y, Smrcka A, Simon MI, Wu D. Pertussis toxin-sensitive activation of phospholipase C by the C5a and fMet-Leu-Phe receptors. J Biol Chem. (1996) 271:13430–4. doi: 10.1074/jbc.271.23.13430
37. Perianayagam MC, Balakrishnan VS, King AJ, Pereira BJ, Jaber BL. C5a delays apoptosis of human neutrophils by a phosphatidylinositol 3-kinase-signaling pathway. Kidney Int. (2002) 61:456–63. doi: 10.1046/j.1523-1755.2002.00139.x
38. Springer TA. Traffic signals for lymphocyte recirculation and leukocyte emigration: the multistep paradigm. Cell. (1994) 76:301–14. doi: 10.1016/0092-8674(94)90337-9
39. Werfel T, Kirchhoff K, Wittmann M, Begemann G, Kapp A, Heidenreich F, et al. Activated human T lymphocytes express a functional C3a receptor. J Immunol. (2000) 165:6599–605. doi: 10.4049/jimmunol.165.11.6599
40. Lalli PN, Strainic MG, Yang M, Lin F, Medof ME, Heeger PS. Locally produced C5a binds to T cell-expressed C5aR to enhance effector T-cell expansion by limiting antigen-induced apoptosis. Blood. (2008) 112:1759–66. doi: 10.1182/blood-2008-04-151068
41. Strainic MG, Liu J, Huang D, An F, Lalli PN, Muqim N, et al. Locally produced complement fragments C5a and C3a provide both costimulatory and survival signals to naive CD4+ T cells. Immunity. (2008) 28:425–35. doi: 10.1016/j.immuni.2008.02.001
42. Kwan WH, van der Touw W, Paz-Artal E, Li MO, Heeger PS. Signaling through C5a receptor and C3a receptor diminishes function of murine natural regulatory T cells. J Exp Med. (2013) 210:257–68. doi: 10.1084/jem.20121525
43. Strainic MG, Shevach EM, An F, Lin F, Medof ME. Absence of signaling into CD4(+) cells via C3aR and C5aR enables autoinductive TGF-β1 signaling and induction of Foxp3(+) regulatory T cells. Nat Immunol. (2013) 14:162–71. doi: 10.1038/ni.2499
44. van der Touw W, Cravedi P, Kwan WH, Paz-Artal E, Merad M, Heeger PS. Cutting edge: receptors for C3a and C5a modulate stability of alloantigen-reactive induced regulatory T cells. J Immunol. (2013) 190:5921–5. doi: 10.4049/jimmunol.1300847
45. Liszewski MK, Kolev M, Le Friec G, Leung M, Bertram PG, Fara AF, et al. Intracellular complement activation sustains T cell homeostasis and mediates effector differentiation. Immunity. (2013) 39:1143–57. doi: 10.1016/j.immuni.2013.10.018
46. Arbore G, West EE, Spolski R, Robertson AA, Klos A, Rheinheimer C, et al. T helper 1 immunity requires complement-driven NLRP3 inflammasome activity in CD4(+) T cells. Science. (2016) 352:aad1210. doi: 10.1126/science.aad1210
47. Trandem K, Zhao J, Fleming E, Perlman S. Highly activated cytotoxic CD8 T cells express protective IL-10 at the peak of coronavirus-induced encephalitis. J Immunol. (2011) 186:3642–52. doi: 10.4049/jimmunol.1003292
48. Peng W, McKenzie JA, Hwu P. Complementing T-cell function: an inhibitory role of the complement system in T-cell-mediated antitumor immunity. Cancer Discov. (2016) 6:953–5. doi: 10.1158/2159-8290.CD-16-0698
49. Ouyang W, Rutz S, Crellin NK, Valdez PA, Hymowitz SG. Regulation and functions of the IL-10 family of cytokines in inflammation and disease. Annu Rev Immunol. (2011) 29:71–109. doi: 10.1146/annurev-immunol-031210-101312
50. Mannino MH, Zhu Z, Xiao H, Bai Q, Wakefield MR, Fang Y. The paradoxical role of IL-10 in immunity and cancer. Cancer Lett. (2015) 367:103–7. doi: 10.1016/j.canlet.2015.07.009
51. McBride JM, Jung T, de Vries JE, Aversa G. IL-10 alters DC function via modulation of cell surface molecules resulting in impaired T-cell responses. Cell Immunol. (2002) 215:162–72. doi: 10.1016/S0008-8749(02)00007-2
52. Mittal SK, Cho KJ, Ishido S, Roche PA. Interleukin 10 (IL-10)-mediated Immunosuppression: MARCH-I INDUCTION REGULATES ANTIGEN PRESENTATION BY MACROPHAGES BUT NOT DENDRITIC CELLS. J Biol Chem. (2015) 290:27158–67. doi: 10.1074/jbc.M115.682708
53. De Smedt T, Van Mechelen M, De Becker G, Urbain J, Leo O, Moser M. Effect of interleukin-10 on dendritic cell maturation and function. Eur J Immunol. (1997) 27:1229–35. doi: 10.1002/eji.1830270526
54. O'Farrell AM, Liu Y, Moore KW, Mui AL. IL-10 inhibits macrophage activation and proliferation by distinct signaling mechanisms: evidence for Stat3-dependent and -independent pathways. EMBO J. (1998) 17:1006–18. doi: 10.1093/emboj/17.4.1006
55. Lopes RL, Borges TJ, Zanin RF, Bonorino C. IL-10 is required for polarization of macrophages to M2-like phenotype by mycobacterial DnaK (heat shock protein 70). Cytokine. (2016) 85:123–9. doi: 10.1016/j.cyto.2016.06.018
56. Murai M, Turovskaya O, Kim G, Madan R, Karp CL, Cheroutre H, et al. Interleukin 10 acts on regulatory T cells to maintain expression of the transcription factor Foxp3 and suppressive function in mice with colitis. Nat Immunol. (2009) 10:1178–84. doi: 10.1038/ni.1791
57. Hsu P, Santner-Nanan B, Hu M, Skarratt K, Lee CH, Stormon M, et al. IL-10 potentiates differentiation of human induced regulatory T cells via STAT3 and Foxo1. J Immunol. (2015) 195:3665–74. doi: 10.4049/jimmunol.1402898
58. Rubtsov YP, Rasmussen JP, Chi EY, Fontenot J, Castelli L, Ye X, et al. Regulatory T cell-derived interleukin-10 limits inflammation at environmental interfaces. Immunity. (2008) 28:546–58. doi: 10.1016/j.immuni.2008.02.017
59. Naing A, Papadopoulos KP, Autio KA, Ott PA, Patel MR, Wong DJ, et al. Safety, antitumor activity, and immune activation of pegylated recombinant human interleukin-10 (AM0010) in patients with advanced solid tumors. J Clin Oncol. (2016) 290:27158–97. doi: 10.1200/JCO.2016.68.1106
60. Zhang H, Wang Y, Hwang ES, He YW. Interleukin-10: an immune-activating cytokine in cancer immunotherapy. J Clin Oncol. (2016) 34:3576–8. doi: 10.1200/JCO.2016.69.6435
61. Naing A, Infante JR, Papadopoulos KP, Chan IH, Shen C, Ratti NP, et al. PEGylated IL-10 (Pegilodecakin) induces systemic immune activation, CD8(+) T cell invigoration and polyclonal T cell expansion in cancer patients. Cancer Cell. (2018) 34:e773. doi: 10.1016/j.ccell.2018.10.007
62. Yi M, Jiao D, Xu H, Liu Q, Zhao W, Han X, et al. Biomarkers for predicting efficacy of PD-1/PD-L1 inhibitors. Mol Cancer. (2018) 17:129. doi: 10.1186/s12943-018-0864-3
63. Feldman SA, Assadipour Y, Kriley I, Goff SL, Rosenberg SA. Adoptive cell therapy–tumor-infiltrating lymphocytes, T-cell receptors, and chimeric antigen receptors. Semin Oncol. (2015) 42:626–39. doi: 10.1053/j.seminoncol.2015.05.005
64. Rosenberg SA, Restifo NP. Adoptive cell transfer as personalized immunotherapy for human cancer. Science. (2015) 348:62–8. doi: 10.1126/science.aaa4967
65. Fesnak AD, June CH, Levine BL. Engineered T cells: the promise and challenges of cancer immunotherapy. Nat Rev Cancer. (2016) 16:566–81. doi: 10.1038/nrc.2016.97
66. Khalil DN, Smith EL, Brentjens RJ, Wolchok JD. The future of cancer treatment: immunomodulation, CARs and combination immunotherapy. Nat Rev Clin Oncol. (2016) 13:273–90. doi: 10.1038/nrclinonc.2016.25
67. Klebanoff CA, Rosenberg SA, Restifo NP. Prospects for gene-engineered T cell immunotherapy for solid cancers. Nat Med. (2016) 22:26–36. doi: 10.1038/nm.4015
68. Hammerl D, Rieder D, Martens JWM, Trajanoski Z, Debets R. Adoptive T cell therapy: new avenues leading to safe targets and powerful allies. Trends Immunol. (2018) 39:921–36. doi: 10.1016/j.it.2018.09.004
69. Chen WF, Zlotnik A. IL-10: a novel cytotoxic T cell differentiation factor. J Immunol. (1991) 147:528–34.
70. Yang G, Hellstrom KE, Mizuno MT, Chen L. In vitro priming of tumor-reactive cytolytic T lymphocytes by combining IL-10 with B7-CD28 costimulation. J Immunol. (1995) 155:3897–903.
71. Groux H, Bigler M, de Vries JE, Roncarolo MG. Inhibitory and stimulatory effects of IL-10 on human CD8+ T cells. J Immunol. (1998) 160:3188–93.
72. Santin AD, Hermonat PL, Ravaggi A, Bellone S, Pecorelli S, Roman JJ, et al. Interleukin-10 increases Th1 cytokine production and cytotoxic potential in human papillomavirus-specific CD8(+) cytotoxic T lymphocytes. J Virol. (2000) 74:4729–37. doi: 10.1128/JVI.74.10.4729-4737.2000
Keywords: complement, cancer immuno therapy, complement receptor C3aR, complement receptor C5aR, IL-10 (interleukin-10), PD-1 - PDL-1 axis, immune check point
Citation: Wang Y, Zhang H and He Y-W (2019) The Complement Receptors C3aR and C5aR Are a New Class of Immune Checkpoint Receptor in Cancer Immunotherapy. Front. Immunol. 10:1574. doi: 10.3389/fimmu.2019.01574
Received: 29 November 2018; Accepted: 24 June 2019;
Published: 19 July 2019.
Edited by:
Barbara Rolfe, University of Queensland, AustraliaReviewed by:
Cristian Smerdou, University of Navarra, SpainCopyright © 2019 Wang, Zhang and He. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: You-Wen He, eW91d2VuLmhlQGR1a2UuZWR1
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.