 Paulus S. Rommer1*
Paulus S. Rommer1* Ron Milo2,3May H. Han4Sammita Satyanarayan4
Ron Milo2,3May H. Han4Sammita Satyanarayan4 Johann Sellner5,6Larissa Hauer7
Johann Sellner5,6Larissa Hauer7 Zsolt Illes8,9
Zsolt Illes8,9 Clemens Warnke10
Clemens Warnke10 Sarah Laurent10
Sarah Laurent10 Martin S. Weber11,12Yinan Zhang13
Martin S. Weber11,12Yinan Zhang13 Olaf Stuve6,13,14*
Olaf Stuve6,13,14*- 1Department of Neurology, Medical University of Vienna, Vienna, Austria
- 2Department of Neurology, Barzilai University Medical Center, Ashkelon, Israel
- 3Faculty of Health Sciences, Ben-Gurion University of the Negev, Beer-Sheva, Israel
- 4Neuroimmunology Division, Department of Neurology and Neurological Sciences, Stanford University, Stanford, CA, United States
- 5Department of Neurology, Christian Doppler Medical Center, Paracelsus Medical University, Salzburg, Austria
- 6Department of Neurology, Klinikum Rechts der Isar, Technische Universität, Munich, Germany
- 7Department of Psychiatry, Psychotherapy, and Psychosomatics, Christian Doppler Medical Center, Paracelsus Medical University, Salzburg, Austria
- 8Department of Neurology, Odense University Hospital, Odense, Denmark
- 9Institute of Clinical Research, University of Southern Denmark, Odense, Denmark
- 10Department of Neurology, Medical Faculty, University of Köln, Cologne, Germany
- 11Institute of Neuropathology, University Medical Center, Göttingen, Germany
- 12Department of Neurology, University Medical Center, Göttingen, Germany
- 13Department of Neurology and Neurotherapeutics, University of Texas Southwestern Medical Center, Dallas, TX, United States
- 14Neurology Section, VA North Texas Health Care System, Medical Service Dallas, VA Medical Center, Dallas, TX, United States
Multiple sclerosis (MS) is the most common neurological immune-mediated disease leading to disability in young adults. The outcome of the disease is unpredictable, and over time, neurological disabilities accumulate. Interferon beta-1b was the first drug to be approved in the 1990s for relapsing-remitting MS to modulate the course of the disease. Over the past two decades, the treatment landscape has changed tremendously. Currently, more than a dozen drugs representing 1 substances with different mechanisms of action have been approved (interferon beta preparations, glatiramer acetate, fingolimod, siponimod, mitoxantrone, teriflunomide, dimethyl fumarate, cladribine, alemtuzumab, ocrelizumab, and natalizumab). Ocrelizumab was the first medication to be approved for primary progressive MS. The objective of this review is to present the modes of action of these drugs and their effects on the immunopathogenesis of MS. Each agent's clinical development and potential side effects are discussed.
Introduction
Multiple sclerosis (MS) is the most common neuroinflammatory and neurodegenerative disease in young adults, with more than 2 million patients worldwide (1). Since the first insights into its pathogenesis were gained from anatomical studies on MS patients in the 19th century by Robert Carswell (2), Jean-Martin Charcot, and others (3), the understanding of pathophysiological concepts concerning MS has been broadened exceedingly. However, modification of the disease course was elusive until the approval of interferon beta-1b (IFN-ß) in 1993 (4). Over the past two decades, the treatment landscape has changed tremendously. Currently, more than a dozen drugs are approved for relapsing-remitting multiple sclerosis (RRMS), and one agent for primary progressive multiple sclerosis (PPMS) (5). These agents represent 10 different substance classes with different modes of action. Whereas, some drugs, including IFN-ß preparations and glatiramer acetate, have no clearly defined mechanisms of action, many of the other agents are target specific and the result of rational drug design. Consequently, much has been learnt about the pathogenesis of MS from the administration of these agents. Although T cells were thought to be the principal lymphocyte subset to initiate and perpetuate disease activity in MS (6), the efficacy of anti-CD20 agents, demonstrated in clinical trials, challenged that concept and pushed B cells together with T cells to the front stage of MS pathogenesis (7, 8).
The initial step in the immune cascade of MS seems to be the activation of T helper (Th) cells in lymph nodes through contact with antigens (either myelin antigens or non-self-antigens sharing similar epitopes to myelin antigens) presented by antigen-presenting cells (APC), including macrophages or dendritic cells (9). This results in the activation and differentiation of myelin-reactive T cells. Activated T cells exit the lymph nodes and circulate in peripheral blood (10), from where they can readily migrate into other tissues, including the central nervous system (CNS). There, these cells can proliferate and clonally expand if they encounter their cognate antigen. The level of chemokines and cytokines increases (e.g., interleukin [IL]-2, IL-1, interferon [IFN]-y, tumor necrosis factor [TNF]-α). As a consequence, additional T and B cells, as well as monocytes are recruited into the CNS and enhance the inflammatory cascade (11).
The monitoring of patients taking new and highly effective drugs that are associated with severe adverse events (AEs) or risks becomes increasingly important. Therefore, it is essential to understand the mode of action of MS drugs and their effects on the immune system. It supports remaining vigilant for unexpected novel AEs that in turn help to understand the mode of action more precisely, and explore the pathophysiology of MS.
This review aims to provide an overview of the approved MS drugs. The history of these drugs and their mode of action are presented considering the current understanding of the pathogenesis of MS.
The review starts with drugs for which the mechanisms of action are not entirely understood, followed by drugs with well-defined molecular and cellular targets.
Interferon Beta (IFN-ß)
Interferons are part of the cytokine family and are signaling proteins. They can be divided into three classes: type I (interferon alpha and beta), type II (interferon gamma), and type III (interferon delta), with different biological effects (12). IFN-ß belongs to the class of type I interferons and is produced by lymphocytes, fibroblasts, macrophages, and endothelial cells (12). Interferons play an important role in the regulation of the immune system. The effects modulated by IFN-ß are complex and have not been elucidated in detail. IFN-ß binds to the type I IFN receptors INFAR-1 and INFAR-2. Its affinity to INFAR-2 is higher than to INFAR-1. This binding activates the JAK/STAT (janus kinases/signal transducer and activator of transcription proteins) signaling pathway leading to the expression of cellular genes (e.g., Mx protein, ß2-microglobulin, 2′/5′-olygoadenylate synthetase, and neopterin) (13). Overall, the activation of signal-transduction pathways by IFN-ß leads to antiviral, immunomodulatory, and antiproliferative effects (14).
IFN-ß has a wide range of immunomodulatory effects. It reduces the number of dendritic cells and downregulates antigen presentation by APCs in the peripheral blood and also within the CNS (microglial cells and monocytes). The expression of Toll-like receptors (TLR) 3 and 7, as well as MyD88, on dendritic cells is upregulated, leading to altered immune responses. It induces CD4+, CD8+, CD25+, FOXP3+, and FoxA1+ T cells (Treg cells). IFN-ß decreases inflammatory T cell responses by inhibiting the stimulation and activation of T cells (e.g., by modulating costimulatory molecules on dendritic cells and inhibiting the expression of MHC class II molecules and co-stimulatory factors such as CD80 and CD28 on APCs) (15, 16). The secretion of cytokines and chemokines is altered (e.g., increased levels of IL-10 and IL-4, and decreased levels of IL-12 and TNF α), and the differentiation of CD4+ T cells shifts from a T helper-1 (Th1) to Th2 phenotype, resulting in a less pro-inflammatory and more anti-inflammatory cytokine milieu (17). The number of Th17 cells decreases, leading to a reduction in the release of Il-17 (12), and the apoptosis of auto-reactive T cells is induced (5). Effects on cytokines and chemokines, matrix metalloproteinases (MMP), and adhesion molecules (e.g., VLA-4 on T cells) have been suggested (15, 18–20), thus the migration of leukocytes into the CNS via the blood-brain-barrier (BBB) is reduced.
Currently, IFN-ß is available as IFN-ß-1a (Avonex®, Rebif®, and Plegridy®) and IFN-ß-1b (Betaferon® or Extavia®). IFN-ß-1-a differs from IFN-ß-1b in its amino-acid sequence, tertiary structure, and glycosylation status (21). IFN-ß-1b was the first drug approved by the US Food and Drug Administration (FDA) for the treatment of MS in 1993 (22) and was granted market authorization in 1995 in Europe (23). The preparations differ with respect to their frequency and route of administration. The frequency of administration ranges from every other day/thrice a week (tiw) (Betaferon® or Extavia®/Rebif® subcutaneously [SC]), to once a week (Avonex®, intramuscular [IM]), to biweekly (Plegridy®, SC). PEGylation led to more stable preparations and a longer half-life (24).
When tested in patients with MS, IFN-ß-1b significantly lowered relapse rates in RRMS by approximately one third (25), with more patients free of relapses after 2 years in the IFN-ß-1b cohort (26). No significant differences in disease progression or the relapse rate were confirmed at 6 months in patients with mild (Expanded Disability Status Scale [EDSS] ≤3.5) or moderate (EDSS >3.5) disability (27). In patients with clinically isolated syndrome (CIS) treated with IFN-ß, the conversion rate to MS was lower during the study period compared to the control group (28–30). However, in secondary progressive MS (SPMS), conflicting results were reported between a European (31) and a North American study (32), with positive effects on progression confirmed at 3 months in the European study, which were explained partly by the younger and clinically more active patient population in the European study than the North American study (33). A prospective study of 2,570 IFN-ß-treated MS patients revealed that an early start within the first years after diagnosis significantly lowered the risk of EDSS progression and long term disability (milestone: EDSS 4) (34). A 16-year follow up study of pivotal trials of IFN-ß-1b revealed different mortality rates for the study groups in the pivotal studies, with the highest mortality rate for the cohort initially treated with a placebo (18.3%), followed by the study group given 50 μg every other day (8.3%), and was lowest mortality rate was found in the group given the high and subsequently approved dose of 250 μg (5.4%). Standard long-term assessments did not show differences between the study groups except for mortality (35).
Since IFN-ß is immunogenic, allergic reactions might occur, and importantly, neutralizing antibodies (NAbs) can be formed in response to treatment. NAbs can lead to the decreased efficacy of IFN-ß preparations and a worsening disease outcome might be observed (36). NAbs were more frequently reported during treatment with IFN-ß-1b than with IFN-ß-1a. Based on the data from various trials, IFN-ß-1b seems to be more immunogenic than IFN-ß-1a (4, 37). This was confirmed by samples from 20,695 MS patients from 6 European Countries. IM administered IFN-ß-1a is the least immunogenic IFN-ß preparation followed by SC administered IFN-ß-1a preparations, with SC IFN-ß-1b preparations being the most immunogenic ones (37). The reason for this has not been elucidated.
The most common AEs include influenza-like symptoms, injection-site reactions, headache, thyroid disorders including autoimmunity, depression, allergic reactions, and elevated liver enzymes with the possibility of severe hepatic injury, which are all frequently reported. Hematological abnormalities (leukopenia, lymphopenia) can also be found (4, 5, 23, 26, 27).
Monitoring requirements include blood counts, liver-enzyme assessments and thyroid testing at regular intervals. NAbs should be tested when treatment failure is suspected (5). According to preclinical studies, harm to the fetus cannot be excluded. The data on IFN-ß during pregnancy in MS patients has so far revealed no association between treatment and an increased teratogenic or abortive potential. The data on treatment during the second and third trimester is limited (38). Table 1 shows data on all approved injectables.
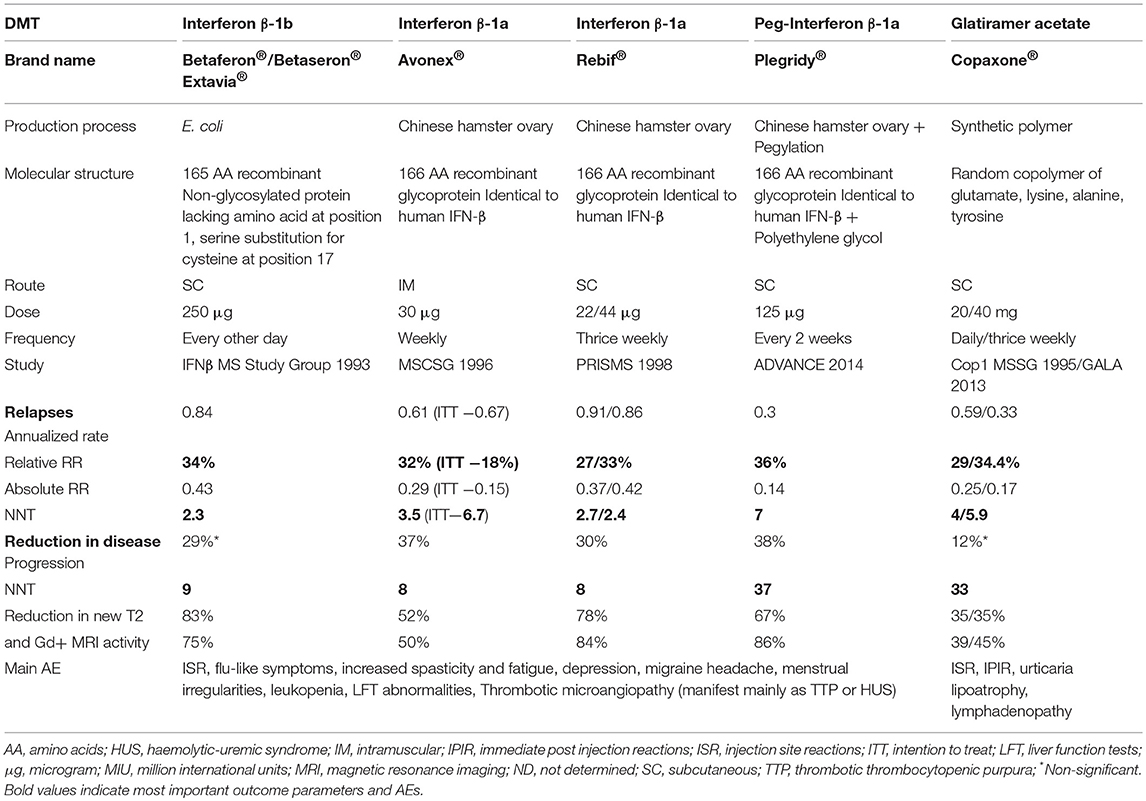
Table 1. Brand name as well as data on efficacy, dose, route of administration, adverse events of approved injectables.
Glatiramer Acetate
Glatiramer acetate (GA, Copaxone®, formerly known as copolymer-1 or Cop-1, and Glatopa®, a biosimilar) is a mixture of random synthetic polypeptides composed of 4 amino acids (glutamate, lysine, alanine, and tyrosine) in a pre-defined molar ratio. GA initially was developed at the Weizmann Institute in Israel as a chemical and immunological analog of the major myelin antigen, myelin basic protein (MBP), to induce experimental autoimmune encephalomyelitis (EAE). Surprisingly, GA did not prove to be encephalitogenic, nor did it induce EAE in susceptible animals, but rather showed high efficacy in suppressing, and even preventing EAE induced by MBP and other myelin antigens in a variety of species and models of EAE (39).
GA's exact mode of action in MS is not completely understood, but extensive research has shown that GA, initially considered to be specific for MBP-related T cell immune responses, affects a variety of immune and non-immune pathways. GA cross-reacts with MBP on the cellular and humoral levels (39) and probably functions as an altered peptide ligand that promotes regulatory T cells instead of stimulating adverse T cell autoreactivity (40). GA's immunomodulatory effects probably stem from strong and indiscriminate binding to MHC class II molecules on APCs, while competing with MBP (41) and probably other myelin antigens (42) for these binding sites. This binding effectively displaces MBP, proteolipid protein (PLP), and MOG-derived peptides from their MHC binding sites (43), resulting in altered T cell responses, as evidenced by the suppression of myelin-reactive T cells by GA (42, 44, 45) and the generation of regulatory Th2 cells recognizing both GA and MBP that can cross the BBB, secrete anti-inflammatory cytokines, and exert bystander suppression of auto-aggressive inflammatory T cells in the CNS (46–48). These GA-specific Th2 cells also secrete large amounts of brain-derived neurotrophic factor (BDNF) that might be neuroprotective (49). Other effects of GA on T cells include T cell receptor (TCR) antagonism via the specific engagement of TCR recognizing MBP through the GA-MHC complex in a manner that results in functional receptor inactivation (50) and induction of regulatory CD4+CD25+ T cells through activation of the transcription factor FoxP3 (51).
GA also modulates macrophages, microglia, and dendritic cells, and drives them into M2 phenotype and anti-inflammatory responses (52–54). Incubation of the human monocytic cell line THP-1 with GA results in down-regulation of the expression of MHC class II and molecules on the cell surface and reduced secretion of TNF-α and cathepsin-B (55). These effects may contribute to the modulation of CNS neuroinflammation (56).
Much attention has recently been drawn to the role of B cells in the pathogenesis of MS, and to the beneficial effects of anti-B cell therapy in both RRMS and PPMS (57). Several recent studies have shown that treatment with GA is associated with reductions in the number of B cells, plasmablasts, and memory B cells, as well as a shift from a pro-inflammatory to an anti-inflammatory B cell phenotype (58). It has been hypothesized that this may be mediated by the cross-reactivity of B cell receptors for GA with antigen (possibly myelin basic protein) expressed in the MS lesion (58).
GA has also been shown to promote repair mechanisms, remyelination, and neurogenesis in the EAE model by augmenting the proliferation, migration, and differentiation of oligodendroglial and neuronal progenitor cells (48, 59).
GA was initially tested on a small number of patients with advanced MS (n = 4) or acute disseminated encephalomyelitis (n = 3) in Israel (60) and in 16 MS patients in the US (61). No side effects or clinical deterioration were noted, and several patients even improved. These encouraging results prompted a pivotal phase-II clinical trial in 50 RRMS patients who were randomized to receive either daily SC injections of 20 mg GA or matching placebos. A marked reduction in the rate of relapses was noted in the GA group, especially in less-disabled patients (62). However, another 2-center randomized trial in 106 progressive MS patients failed to demonstrate a beneficial effect on disability progression resulting from 15 mg of GA injected SC twice daily. Nevertheless, two additional secondary disability endpoints and the primary endpoint in one center were met (63).
A pivotal phase-III clinical trial with GA was conducted in 11 US centers. In this trial, 251 RRMS patients were randomized to receive either 20 mg of GA or a placebo via daily SC injections for 2 years. A significant 29% reduction in the annual relapse rate (ARR) was observed in the GA group compared to the placebo group (p = 0.007). Significantly more patients on GA improved on the EDSS score, and significantly fewer patients worsened (64, 65). Unfortunately, no MRI scans were performed in this trial, except for at one center where patients on GA had significantly fewer gadolinium (Gd)-enhancing lesions and reduced brain volume loss compared to patients taking placebo (66). To better appreciate GA's effect on MRI parameters, 239 RRMS patients in Europe and Canada were randomized to daily GA or placebo treatment and had monthly MRI scans for 9 months. GA reduced the number of Gd-enhancing and new T2-weighted lesions (67) and the proportion of new Gd-enhancing lesions evolving into black holes (68). The daily dose of 20 mg of GA had similar efficacy as 40 mg GA administered daily (69) or thrice weekly (70), and both regimens (20 mg qd or 40 mg tiw) are approved for use in RRMS. Similarly to the interferons, GA has not been shown to reduce disability progression in PPMS (71), but is highly effective in delaying clinically definite MS after CIS (72). Long-term follow-up of patients with RRMS shows continuous efficacy with low relapse rates and minimal EDSS progression after 15 years (73).
In comparative trials with available interferons in RRMS, GA was as effective as IFN-β-1b (74) or SC IFN-β-1a (75), and superior to IM IFN-β-1a (76). The latter study also showed that the combination of GA and IM IFN-β-1a was not superior to either therapy alone (76).
GA's good safety profile has been established over many years of clinical use. Its principal side effects include local-injection-site reactions (tenderness, pruritus, erythema, or induration). Regional lymphadenopathy; local lipoatrophy, which may be permanent; allergic reactions and rare injection site skin necrosis may also occur. About 16% of patients experience a rare systemic post injection reaction comprising of various combinations of the following effects: chest tightness, dyspnea, flushing, palpitations, diaphoresis, and anxiety beginning immediately after GA injection and resolving spontaneously within a few minutes without any sequelae. Unlike IFN-β, treatment with GA is not associated with leukopenia, liver, or thyroid abnormalities; depression; or any additional systemic side effects. It is not associated with any serious AEs seen with other potent newer therapies for MS either, such as opportunistic infections, malignancy, or secondary autoimmunity. Virtually all patients develop binding antibodies, but not NAbs to GA, which do not impair its clinical efficacy (77). GA elicits no adverse effects on fertility, pregnancy, or fetal outcomes (78) and is the only MS drug that is no longer contraindicated during pregnancy in Europe.
Although only moderately effective in reducing disease activity, GA is registered worldwide as a first line platform therapy for patients with RRMS due to its long-term efficacy and safety.
Fingolimod
Therapeutic concepts in MS include the down-regulation or depletion of pro-inflammatory T and B cells, the enhancement of anti-inflammatory immune responses (79, 80), the prevention of encephalitogenic lymphocytes from entering into the CNS, and the retention of auto-reactive lymphocytes within secondary lymphoid organs (as in the case of fingolimod) (81, 82). This recognition was based on the understanding of the interaction between sphingosine-1-phosphate (S1P), a signaling sphingolipid, and its receptors, S1PR1-5, essential for lymphocytes to egress from secondary lymphoid organs into the systemic circulation (83, 84). The search for molecules targeting the S1P pathway resulted in the discovery of the fungal metabolite myriocin, which eventually led to the development of FTY720 (fingolimod), an oral therapy for treating RRMS (85). FTY720, a functional antagonist of S1PR1-3,4,5 (S1PR1 being the dominant receptor in lymphocytes) (10) binds to the receptor, leading to internalization of the S1P/S1PR complex via the β-arrestin-mediated mechanism (86), thereby preventing lymphocytes' egress (10). This effect is primarily observed in the retention of CD4- and CD8-positive naïve lymphocytes and central memory (CD45RA−) T cells in the lymphoid organs. However, effector memory T cells (CD45 RA+/−), which primarily use a chemokine-based signaling mechanism, are spared (85, 87). Research dedicated to understanding the effect of fingolimod on lymphocyte subsets additionally identified CD4+CD25+ regulatory T cell populations as being up-regulated in-vitro, which could have implications in down-regulating pro-inflammatory lymphocyte reactivity (88). B lymphocytes are also sequestered in the spleen due to their unique dependence on the S1P pathway, although S1P alone is not sufficient for B cells to exit from the bone marrow (83). S1PRs are expressed at varying levels on endothelial cells, neurons, and CNS glia, however, their function and response to S1PR modulator therapies beyond the immune system are not well-understood (89, 90).
Two landmark, double-blind, randomized trials established the efficacy of fingolimod compared to a placebo or active comparator in treating RRMS. The FREEDOMS trial showed a decrease in the annualized relapse rate (ARR) (0.16 with a regimen of 1.25 mg daily, compared to 0.40 with a placebo), with a relative reduction of ~50% (91). Radiographically, a reduction in both new enhancing and non-enhancing lesions was reported. The results for FREEDOMS II were comparable (92). Subsequently, the TRANSFORMS trial compared two doses of oral fingolimod (0.5 mg daily and 1.25 mg daily) to weekly IM IFN-β-1a for 1 year, which showed a decrease in ARR to 0.16 with 0.5 mg fingolimod, 0.2 with 1.25 mg fingolimod, and 0.33 with IFNβ-1a therapy. Interestingly, this study did not show any difference in the progression of disability assessed using the EDSS (which might be partly explained by the trial design with a short trial duration) (93). The FREEDOMS trial also showed a reduction in the rate of whole-brain atrophy compared to the placebo, suggesting a potential neuro-protective effect of fingolimod (91). However, a recent trial with fingolimod showed no benefit on disability progression compared to placebo when tested for a primary composite endpoint, including EDSS, a 25-foot timed walk test, and a nine-hole peg test in PPMS; although a decrease in radiographic activity has been observed (86). Based on the trial results in RRMS, 0.5 mg daily fingolimod has been approved for the treatment of MS. Fingolimod has been tested in pediatric MS and was associated with a lower rate of relapses and lower accumulation of MRI lesions compared to patients treated with IFN-ß-1a (94). Based on these studies, it has been approved for the treatment of pediatric MS (95)1.
Fingolimod's most common AEs include bradycardia and less commonly first/second-degree atrioventricular block, likely due to effects on S1PRs in atrial myocytes (85). Notably, these are often asymptomatic, observed during the administration of the first dose, and they also might recur after an interruption of more than 2 weeks2 (96). Macular edema has been shown to occur in <1% of patients during the first 3 months of treatment and often resolves after treatment is discontinued. Disseminated varicella zoster infection occurred in one patient in previous clinical trials. Elevated liver enzymes (>3× upper limit of normal) were also observed in the FREEDOMS trial, though no cases indicated hepatotoxicity (92). Increased rates of lower respiratory tract infections, cutaneous malignancies (not only basal cell carcinoma, but also squamous cell carcinoma and cutaneous lymphoproliferative disorders), and opportunistic infections including cases of progressive multifocal leukoencephalopathy (PML), varicella-zoster-virus (VZV), and herpes-simplex-virus (HSV) associated encephalitis as well as cryptococcal infections have been reported (5, 97). Retrospective reviews of fingolimod's effects on pregnancy from clinical development trials and additional reports from smaller trials have shown few adverse fetal outcomes. However, the number of adverse outcomes and elective abortion were in the expected range of the general population. Data on fingolimod exposure beyond the first trimester is scarce (38). Since fingolimod exposure causes teratogenicity in rodents, a teratogenic potential cannot be ruled out (85). Similarly, since fingolimod can be detected in breast milk, it is also contraindicated in lactating women.
Based on potential untoward effects, screening before initiation of fingolimod treatment comprises baseline laboratory parameters (including a complete blood count, liver-function test, and varicella zoster antibody titers), electrocardiogram, spirometry (in cases of a previous respiratory disease, such as asthma), and an ophthalmologic examination to evaluate for macular edema. Patients are monitored closely for at least 6 h after the first dose (or at re-introduction) in a clinical setting for bradycardia and other cardiac-rhythm abnormalities. Patients with pre-existing cardiac abnormalities, such as conduction block or ischemic heart disease, or those taking medications that interfere with cardiac rhythm and conduction are advised to have a cardiology consultation if clinically indicated (92). Slight and mostly transient hypertension after initial doses of fingolimod also was observed in the FREEDOMS extension study, however, if it did not resolve, it remained stable over the treatment course (91, 98). Increased frequency of basal-cell carcinoma was reported in patients on long-term fingolimod therapy (91). Subsequently, periodic monitoring of blood counts is recommended given lymphopenia's association with fingolimod. The cessation of fingolimod treatment has been associated with cases of severe rebound syndrome leading to severe relapses or high MRI activity. Discontinuing MS treatments needs to be monitored and the sequence of the most suitable treatments needs to be assessed and planned (99).
Recent trials have investigated more receptor-specific agents targeting S1PRsin the hopes of mitigating side effects. Cardiac effects, lymphopenia, elevated liver enzymes, and macular edema still occur with these agents, though a dose-titration strategy was observed to diminish first dose-associated cardiac effects (100). Recently, a remarkable future path for SPMS treatment was revealed in the EXPAND trial, which showed that siponimod (a selective S1P1/S1P5 binding agent) was the first medication of utility in preventing disability progression at 3 months in SPMS (100). It has been approved as Mayzent© by the FDA for CIS, RRMS and active SPMS3.
Table 2 shows data on all approved oral drugs.
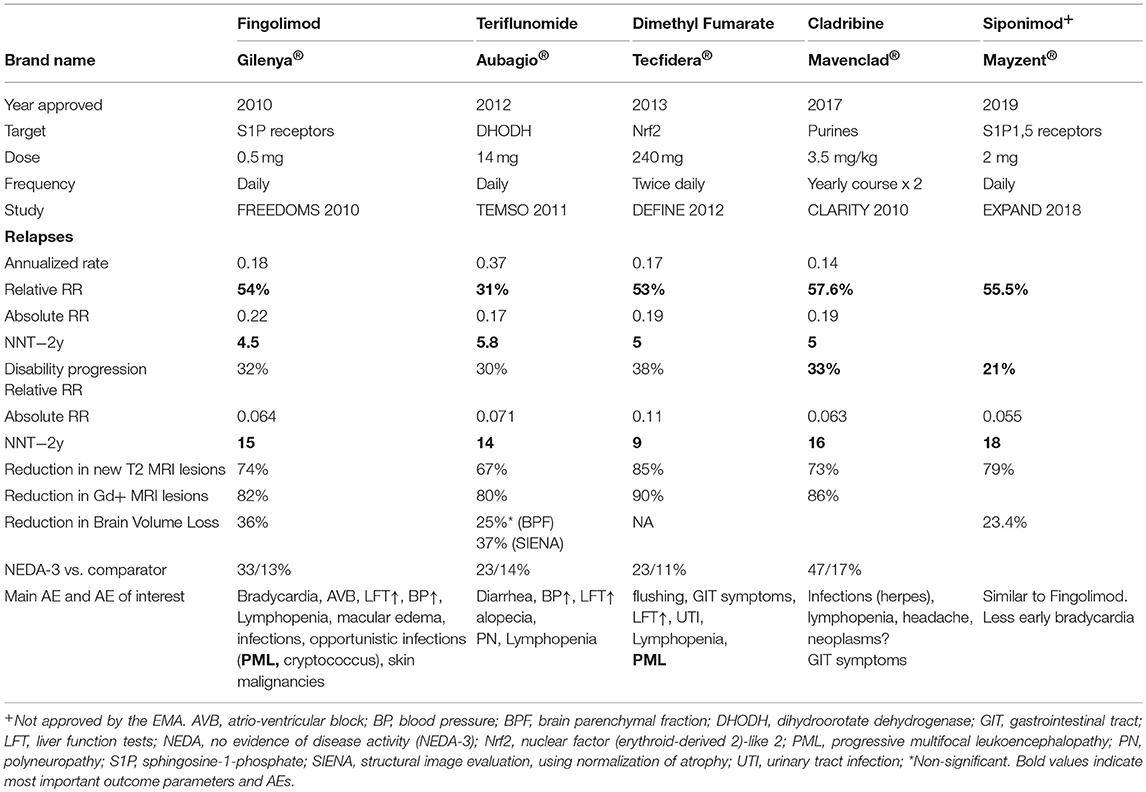
Table 2. Brand name as well as data on efficacy, dose, route of administration, adverse events of approved oral agents.
Mitoxantrone
Mitoxantrone is a synthetic anthracenedione derivate that initially was developed as a cytotoxic treatment for acute myeloid leukemia (101). As a type II topoisomerase inhibitor, the substance has potent anti-inflammatory and, to a lesser extent, immunomodulatory properties (102). The immunosuppressive effect is mediated by effects on proliferating B and T lymphocytes: induction of cell lysis and initiation of programmed cell death (103, 104). Mitoxantrone also demonstrates immunomodulatory effects and preferentially decreases the migratory capacity of monocytes into the CNS and enhances Th2 cytokine production in CD4+T cells (105). In-vitro, mitoxantrone interferes with antigen-presenting capabilities of dendritic cells (106).
Mitoxantrone was the first drug that the FDA and several European countries approved to treat worsening relapsing-remitting, secondary-progressive, and progressive-relapsing MS. Research evidence was generated from a phase-III clinical trial in Europe (107) and an earlier phase-II study (108). Both trials confirmed a significant reduction in the relapse rate and worsening of disability. Mitoxantrone is given intravenously (IV) at a dose of 12 mg/m2 at 3-month intervals. Some healthcare facilities prefer a fixed-dose regimen of mitoxantrone: 20 mg IV monthly together with methylprednisolone (1 g) (108).
Mitoxantrone has myelotoxic effects and reduces leukocyte counts; thus, its administration is not recommended when neutrophil numbers are below 1,500 mm3 (109). Reversible bone-marrow suppression and nausea are common side effects associated with mitoxantrone infusion (110). Dose adjustment based on leukocyte nadir is mandatory to minimize risks for infections, particularly of the urinary tract. Anemia occurred in 15% of patients (grade ≥ 1) (111). Amenorrhea was reported in up to 26% of mitoxantrone-treated women before the age of 45 (112, 113). Liver toxicity and alopecia have also been observed (111).
Severe AEs include therapy-related acute leukemia (TRAL), cardiotoxicity, and colon cancer (114). Acute promyelocytic leukemia (APL) is the most commonly seen TRAL after initiation of mitoxantrone treatment and is characterized by an aggressive, often fulminant disease course due to a life-threatening coagulopathy, e.g., CNS hemorrhages (115). A recent meta-analysis reveals a TRAL risk of ~0.81%, more than 10-fold higher than in previously reported meta-analyses (0.07%) (116). TRAL, in a mitoxantrone setting, has a mortality rate of ~40% (117).
Cardiotoxicity risk increases with cumulative doses of mitoxantrone (118). Therefore, the maximum cumulative dose is restricted to 100–140 mg/m2, however, cardiotoxicity can develop after doses well below the current maximum recommended levels (111). Re-evaluation by the European Medicine Agency (EMA) concluded that the ordinarily cumulative life-time doses for MS patients should not exceed 72 mg/m2 4. Systolic dysfunction occurs in approximately 12% and congestive heart failure in around 0.4% of treated patients (117). The authors of some studies, therefore, even suggest to limit mitoxantrone treatment to 1 year, or a cumulative dose to <60 mg/m2, to reduce the risk of TRAL and cardiotoxicity (119).
Treatment with mitoxantrone requires monitoring for possible cardiotoxicity and APL. Cardiac monitoring via regular echocardiography (measurement of left ventricular ejection fraction [LVEF]) is required before treatment begins, prior to each dose, and annually after the discontinuation of therapy (110). Close monitoring of full blood counts in patients with MS before and after mitoxantrone administration needs to be carried out to monitor leukocyte nadir (mostly after 10–14 days) and a return to normal levels (~21 days). Clinical vigilance and repeated full blood counts are necessary for 5 years after the termination of treatment (116). Mitoxantrone is contraindicated during pregnancy (38).
In recent years, the use of mitoxantrone has decreased due to the risk of severe AEs and the introduction of novel therapies. The agent should be restricted to selected patients with highly active relapsing multiple sclerosis associated with rapidly evolving disability for whom no alternative treatments are available5. In addition, clinical and laboratory vigilance is required both during and after mitoxantrone regimens.
Teriflunomide
Teriflunomide is the active metabolite of leflunomide, which has been used in the treatment of rheumatoid arthritis since 1988. Teriflunomide received approval for treating RRMS in 2012 in the US (7 and 14 mg daily) and in 2013 in Europe (14 mg daily) (120). Teriflunomide interferes with de-novo pyrimidine synthesis and DNA replication of highly proliferating T and B cells by reversibly inhibiting the mitochondrial enzyme dihydroorotate dehydrogenase (DHODH). Since resting T cells use nucleotides from degrading DNA and RNA and do not need DHODH, the protective immune responses are maintained, while the proliferation of activated T and B cells is reduced; thus, the viability of immune-cells is not affected. In the Teri-DYNAMIC study, a shift to regulatory T cell subtypes and a reduction in clonal diversity in the CD4+T cell repertoire were observed (121). An increase in Treg cells in gut associated lymphoid tissue also characterized protection in the autoimmune inflammatory model of MS (122). Teriflunomide crosses the blood-brain barrier (BBB) (121), decreases microglia proliferation, and induces IL-10 production by microglia in-vitro (123). Besides the anti-proliferative effect, leflunomide and potentially teriflunomide, inhibit the production of IL-17, TNF-alpha, protein tyrosine kinases, the NF-kB pathway, and the IgG secretion of activated B cells, and interfere with the kynurenine pathway (120, 124, 125).Teriflunomide induces apoptosis of EBV-transformed B cells (126). In a virus-induced animal model of MS, teriflunomide reduced glutamate levels and excitotoxicity (127). Teriflunomide also promotes oligodendrocyte differentiation in-vitro, ameliorated axonopathy by attenuating CD8+ T cell cytotoxicity and supported the proliferation of regulatory CD8+ T cells in the CNS of mice (128, 129). Despite these potent immunomodulatory and cytostatic effects, protective immune responses against foreign antigens are maintained. In the TERIVA study, more than 90% of the MS patients treated with teriflunomide achieved sufficient seroprotection rates when vaccinated against seasonal influenza (130).
Teriflunomide is administered as an oral drug once daily, and a steady-state concentration is reached after ~3 months. After withdrawal, serum levels are maintained above 0.02 μg/mL for 8 months, and in some individuals, up to 2 years, due to enterohepatic recirculation. Accelerated elimination can be achieved by administering 8 g of oral cholestyramine three times daily for 11 days, which can be reduced to 4 g three times daily in cases of intolerance. Alternatively, 50 g of activated charcoal powder every 12 h for 11 days can be used. To confirm proper elimination, the concentration should be below 0.02 μg/mL in two serum samples obtained 14 days apart (131).
Teriflunomide's efficacy and safety have been investigated extensively in one phase-II (132) and four phase-III (TEMSO, TOWER, TENERE, and TOPIC) clinical trials, all with long-term follow-up data involving several thousand patients (133–136). Over 90% of patients in the TEMSO and TOWER trials had RRMS (133, 134). Patients with a first single clinical episode were enrolled in the TOPIC trial (136). In the TENERE trial, teriflunomide was compared to SC IFN-ß-1a (135), whereas the other three phase-III trials were placebo-controlled.
In these clinical trials, teriflunomide showed a consistent effect on disease activity, measured by its impact on relapses, disability worsening, MRI outcomes, and combined measures such as no evidence of disease activity (NEDA). Compared to the placebo, 14 mg of teriflunomide daily reduced the ARR by 31–36% in the pivotal trials (133–136). Disability progression, confirmed after 3 months, was also reduced significantly by 29.8 and 31.5% in the 14 mg trial group in TOWER and TEMSO studies (133, 134). Similar efficacy data have been observed in real-life settings for up to 28 months (137). Comparison of the pooled phase-III trial data from 14 mg of teriflunomide and dimethyl-fumarate (DMF) (TEMSO/TOWER vs. DEFINE/CONFIRM) revealed similar numbers needed to treat (NNT) to prevent one relapse or worsening disability (121, 138). However, in a recent registry-based study, the ARR was ~49% lower in patients treated with DMF, and teriflunomide treatment was associated with an increased risk of first relapse and increased incidence of discontinuation due to disease breakthrough (139). In another recent registry-based study with large patient populations, the ARR was similar between teriflunomide and DMF, and discontinuation rate was also similar; nevertheless, ARR were lower in patients treated with fingolimod compared to both DMF and teriflunomide, while disability accumulation was the same (140). A recent Italian study did not observe differences in discontinuation either during the first 24 months (141). In the TEMSO study, the 14 mg daily dosage reduced the number of Gd-enhancing lesions by 80.4% and the total lesion volume by 67.4%; the effect of a 7 mg daily dose was less but still significant (133). The treatment (14 mg) of patients with a first clinical episode suggestive of MS, i.e., CIS, reduced the risk of conversion to clinically definite MS by 42.6%, however, only 44% of the patients completed the study due to early termination related to changes in MS diagnostic criteria (136).
The long-term outcomes in extension studies indicate that the effect of teriflunomide is maintained (class-III evidence), however, the dropout rates varied, ranging from 40 to 75% (121). The analysis of pooled TEMSO/TOWER long-term data (up to 9 and 5.5 years, respectively) indicated that more than 80% of the 122 patients with progressive relapsing MS did not experience worsening disability (121).
The AEs reported more frequently with teriflunomide than the placebo include hair thinning, ALT increase, nausea, diarrhea, paraesthesia, limb pain, arthralgia, nasopharyngitis, polyneuropathy, and menorrhagia. Hair thinning appeared in 10–14% of patients and led to discontinuation in 1.4–2% of cases in pivotal trials (133, 134, 142). The discontinuation of treatment was most commonly related to ALT elevation driven by the trial protocols. In real life, gastrointestinal AE was the most common cause of discontinuation (143). The pooled analysis of safety data from the phase-II, TEMSO, TOWER, TOPIC, extension of phase-II, and TEMSO (up to 12 years), in addition to the safety data from the TOWER, TOPIC, and TENERE extensions (up to 7 years), consistently supported the long-term beneficial AE profile (121, 143, 144). Deaths were not more common in the active arm compared to placebos in pivotal trials, and two deaths in the extension phases (pulmonary tuberculosis and suicide) were potentially related to treatment (121, 133–136, 143–145). A single case of PML after 3 months of teriflunomide treatment has been reported, but it most likely was carried over from preceding natalizumab therapy (146). During the 2.1 million patient years of leflunomide therapy since 1991, two cases of PML have been reported during monotherapy (120).
Teriflunomide is contraindicated during pregnancy and is classified as category X based on embryo-fetal toxicity and malformations in rats and rabbits (121). However, results from animal studies cannot be transferred unrestrictedly to humans, and among 26 reported live births, by human women taking the drug no abnormalities were present (147). The FDA suggests discontinuing teriflunomide in males who wish to father a child, however, this is not required in Europe. Accelerated elimination is necessary for women taking the drug before pregnancy who wish to become pregnant, and serum concentration must be <0.02 μg/mL in two serum samples obtained 14 days apart.
In summary, extensive and long-term data consistently indicate that teriflunomide's efficacy resembles that of injectables, and that it offers a beneficial AE profile. Its administration is convenient, however, frequent blood tests (blood count and liver transaminases) are required during the first 6 months of treatment.
Dimethyl Fumarate (DMF)
DMF has been used to treat psoriasis since 1959 and was approved to treat RRMS in 2013 (148). It is administered as a 240 mg tablet twice daily.
One of the major mechanisms of DMF and its active metabolite mono-methyl fumarate (MMF) is an antioxidant response through activation of the Nrf-2 pathway, which might result in neuroprotective properties besides influencing NF-κB related cellular responses (149, 150). Activation of the Nrf-2 pathway leads to an expansion of FoxP3+ regulatory T cells and CD56bright natural killer cells, as well as to a reduced level of CD8+ T cells and B cells (151). DMF yields profound effects on immune responses in-vitro and in-vivo: It inhibits NF-κB activation and pro-inflammatory cytokine production by myeloid cells; reduces the generation of encephalitogenic T cells, partially by inhibiting antigen presentation; generates a shift from a Th1/Th17 to a Th2 profile; alters cytokine production by B cells; promotes apoptosis of B and T cells; and elicits an anti-proliferative effect (149, 152–156). In MS patients treated with DMF, the T and B cell subpopulations are reduced, and functional changes are observed in lymphocytes and APCs. Such reductions affect mostly cytotoxic T cells, effector/central memory T cells, Th1 cells, Th17 cells, mucosa-associated lymphoid tissue (MALT) cells follicular T cells with a Tfh1/17 phenotype, antigen experienced and memory B cells, and B cells producing TNF. Immunoregulatory CD56bright NK-cells, naïve T and B cells, Th2 cells, FoxP3+ Tregs, and follicular T cells with a Tfh2 phenotype are increased (151, 153–155, 157–162). Such a pro-tolerogenic shift is associated with NEDA in MS patients (158); higher levels of the NRF2 target gene NAD(P)H quinone dehydrogenase 1 (NQO1) was also associated with NEDA status after 1 year of DMF treatment (151). MMF crosses the BBB, and DMF/MMF alters the function of CNS resident cells in-vitro, suppresses inflammatory cytokine production by activated microglia and astrocytes, and increases the number of oligodendrocyte precursor cells (163–165).
DMF's clinical efficacy and safety as an MS drug have been investigated in two randomized placebo-controlled phase-III trials, DEFINE and CONFIRM (166, 167). An active agent, GA, was also included as a reference comparator in the CONFIRM trial. The ARR was reduced by 53% in the DEFINE study and 44% in the CONFIRM study, compared to the placebo (166, 167). The risk of confirmed disability progression sustained for 12 weeks was also reduced by 38% in the DEFINE study and 21% in the CONFIRM study (166, 167). The integrated analysis of the phase-III trials indicated a 32% (29%) risk reduction in 12 (24)-week confirmed disability progression (168). DMF reduced the number of new or enlarging hyperintense lesions on T2-weighted images by 71 and 85%, respectively, and reduced the odds of an increase in the number of Gd-enhancing lesions by 74% and 90% in the CONFIRM and DEFINE study, respectively (166, 167). Compared to the active comparator GA, DMF twice daily also significantly reduced T2-weighted hyperintense lesions in the CONFIRM trial, whereas the other efficacy outcomes were no different (167). The integrated analysis of CONFIRM and DEFINE demonstrated a 38.9% relative reduction in clinical disease activity (relapse and disability progression) over 2 years compared to placebo treated patients (169).
The phase-III trials, integrated analyses, and follow-up studies all indicated DMF's safety and beneficial AE profile. The frequency of AEs and serious AEs was similar to the placebo in the DEFINE and CONFIRM trials (serious AEs 17 and 18% vs. 21 and 22%, respectively) and GA in the CONFIRM study (17%) (166, 167). The most common AEs were flushing (31–38%), diarrhea (13–15%), nausea (11–13%), upper abdominal pain (10%), and vomiting (10%) (166, 167, 170). Increased liver enzymes were detected in 3–6% of patients treated with DMF (171). Overall, the incidence of AEs leading to discontinuation of the study drug was similar across groups. Discontinuations due to flushing and overall gastrointestinal events occurred more frequently in patients who received DMF (166, 167). Compared to the placebo group, at 1 year, the white-cell and lymphocyte counts decreased by ~10 and 28%, respectively (166). Grade 2 or 3 lymphopenia occurred in 4–10% of the patients compared to 1% or less in the placebo group and tends to persist in some patients (166, 167). Infections were common but the incidence was not significantly different between the DMF and placebo or GA groups (50–68%) (171). Although serious and opportunistic infections were not more common among patients treated with DMF, five cases of PML were reported by 2018 (172). Additional 14 cases were related to other DMF formulas used in psoriasis, and 13 out of the 19 cases had grade 3 lymphopenia (173). CD4+ and CD8+ T cell repopulation rates are delayed after switching to other disease modifying therapies (DMT) from DMF, and T cell counts may not recover or even continue to decline if DMF treatment is switched to fingolimod or alemtuzumab (174). Decrease of CD8+ and memory T cells is more likely compared to CD4+ and naïve T cells (156).
In addition to pivotal trials, a few studies have investigated DMF's efficacy and safety in real-life settings, and these recapitulated the findings from the pivotal trials. In two multicenter studies with 1,089 and 735 patients treated for up to 25 and 33 months, the ARR was reduced by 77 and 63% respectively (175, 176), whereas in another multicenter study, the ARR was reduced by 33% (177). NEDA status was achieved in 47.8% of patients after 1 year (176). In the first year, 11–19.5% of patients discontinued treatment, and 30% of patients stopped DMF after 2 years mainly due to poor tolerance (175, 176). Approximately one third of the patients had flushing or gastrointestinal AEs (176). Lymphocytopenia occurred in 16.5 and 18.7% of the patients, respectively (175, 176). Lower baseline lymphocyte counts, female gender and older age (>55 years) were associated with more severe lymphopenia (178). Several recent studies have highlighted the importance of early AE management to improve adherence. In a cohort of 400 patients, 34% stopped treatment within a year and 57% within 2 years (179). The data on treatment with DMF during pregnancy is limited, thus, no final assessment is possible. Generally, it is recommended to stop DMF when planning to conceive (5, 38). Several studies have investigated DMF's efficacy in relation to other DMTs. Fingolimod and DMF were evaluated based on the data from a pivotal study using a matching-adjusted comparison and revealed no significant differences in the effect on clinical parameters between the treatments (180). Similar results were shown in an Italian study based on real-world data. A propensity score-matched study revealed a similar NEDA-3 status for fingolimod (73%) and DMF (70%), however, in patients having switched from other therapeutics, fingolimod was superior to DMF (p = 0.007) (181). Another study measuring the indirect effectiveness of fingolimod vs. DMF vs. teriflunomide based on phase-III studies suggested that the probability of achieving NEDA-3 was highest for fingolimod (182). Similarly, a recent study including 3,728 patients from MSBase showed a superior effect of fingolimod on relapse rates and comparable results for disability progression in patients treated with fingolimod, DMF, and teriflunomide (140).
In the STRATEGY study, the risk of relapse after switching from natalizumab was 19.6%, and the ARR was lower in patients with <90 days of a washout period (183). Another study also indicated that DMF can be an option for patients discontinuing natalizumab: After 2 years of DMF treatment, 80% of the patients did not present clinical or MRI evidence of disease activity, and a post natalizumab rebound was observed in 1 out of 39 patients (184).
In summary, the data indicate DMF's efficacy and safety in treating RRMS. Whether its efficacy is higher than teriflunomide's and like fingolimod's is debatable. Sustained lymphopenia after stopping a DMF regimen might complicate the escalation to fingolimod and lymphocyte-depleting therapies. Whereas, pre-treatment with aspirin might mitigate flushing, gastrointestinal side effects are only slighthly mitigated by dose titration and are not worsened by pre-treatment with aspirin (185). Gastrointestinal side effects need early symptomatic treatments which may increase adherence significantly (186).
Cladribine
Cladribine (2-chloro-2′-deoxyadenosine) is a pro-drug that requires intracellular phosphorylation to become an active purine nucleoside analog that interferes with DNA synthesis and repair, and ultimately leads to cell death. The higher ratio of activity between certain enzymes that activate (desoxycytidine kinase) or deactivate (adenosine-monophosphate kinase and nukleoside-diphosphate kinase) the pro-drug explains the preferential and long-lasting depletion of peripheral B and T lymphocytes with a relative sparing of other hematogenic and immune cells. B and T cells are rapidly depleted. The slight recovery of Tregs before B and T cells repopulation might partly explain the long-lasting effects (187). A parenteral formulation of cladribine was first developed for therapy against hairy-cell leukemia, while the oral formulation of cladribine was developed later and tested in RRMS (188, 189).
Oral cladribine was studied in a phase-III trial (CLARITY) (190), a 96-week, placebo-controlled, double-blind, multicenter study. Patients with active RRMS (at least one relapse within 12 months prior to study entry) were included in the trial. Cladribine was administered based on body weight and tested in three groups: 3.5 mg/kg, 5.25 mg/kg, or a placebo. Compared to the placebo, the ARR at week 96 was reduced in both treatment groups by ~57%. The proportion of patients remaining free of relapses at week 96 increased from ~61 to 80%, resulting in an absolute benefit for approximately 19 out of 100 patients treated. In addition, the relative reduction in the risk of a 3-month sustained progression of disability in both cladribine groups, compared to the placebo group, was 31–33%, and patients treated with cladribine had a reduction of 77% in mean active T2 lesions on MRI (190). Furthermore, in patients with a first clinical attack, cladribine was shown to reduce the risk of a second attack, or three-month EDSS progression (191, 192).
The subsequent CLARITY EXTENSION study showed that treatment with cladribine for 2 years followed by 2 years' placebo treatment produced durable clinical benefits similar to 4 years of cladribine treatment, i.e., approximately 75% of patients treated with cladribine 3.5 mg/kg in CLARITY, remained relapse-free when given placebo during the extension (193, 194).
In the CLARITY study, at a dosage of 3.5 mg/kg, CD4+ T cells dropped by 40–45% and CD8+ T cells by 15–30% without significant recovery prior to the next treatment cycle. CD19+ B cells dropped by ~70–90%, slowly recovering to 15–25% of the baseline (195), suggesting a combined T and B cell-mediated mode of action.
Lymphopenia was dose-dependent (nadir at 4 months), with grade 3 lymphopenia (500–200 cells/uL) in ~25% of patients in the 3.5 mg/kg dose group, and grade 4 (<200 cells/uL) in <1% (194). The rate of common infections was similar when comparing placebo- and cladribine-treated patients. The rate of herpes zoster infections per 100 patient years was higher in the 3.5 mg/kg group than the placebo group (0.83 vs. 0.20) and associated with lymphopenia, explaining why patients with grade 4 lymphopenia should receive a prophylactic anti-herpes infection treatment. Furthermore, the incidence of severe infections was generally higher among patients with lymphopenia and who were taking cladribine at a dosage of 3.5 mg/kg compared to the placebo group (194). PML was not reported during an observational period of >8,500 patient years in the MS indication, whereas some PML cases have been observed with parenteral cladribine in lymphoma patients6. Three cases of tuberculosis were reported during the clinical trials, of which one case was fatal. Two cases of hepatitis B occurred, and one of those patients died (166).
Thus, not only clinical follow-up and standard laboratory tests, but also screening for HIV infection, active tuberculosis, and hepatitis are mandatory prior to a treatment course of cladribine. The malignancy rates were higher among cladribine-treated patients compared to the placebo cohort (33 vs. 4); these malignancies comprised of solid tumors with no specific patterns typical of tumors commonly seen during immunosuppression. No cases of leukemia, lymphoma, or lymphoproliferative disorders were reported (166). However, this imbalance explained the initial application rejection by the EMA in 2011, when additional safety data were requested. Such data then were obtained from clinical extension studies, meta-analyses of several other clinical studies with alternative MS drugs, and comparisons with epidemiological data, leading to EU approval in 2017, since the malignancy risk is comparable to other treatment options for MS. However, the EU approved cladribine only for “the treatment of adult patients with highly active relapsing MS as defined by clinical or imaging features”7, and it was approved by the FDA in 20198. Furthermore, due to potential teratogenic effects, both males and females must use effective contraception during therapy with cladribine, and for 6 months after a treatment cycle.
Overall, the registered dose of 3.5 mg/kg can be applied orally in short treatment cycles, which might lead to a high adherence to therapy, followed by a sustained therapeutic effect, with efficacy confirmed in highly active patients with the registered indication7. The downside of cladribine is that lymphopenia can be severe and frequently reaches grade 3, which is associated with a higher risk of infections. Data on lymphopenia in patients with prior immunosuppressive treatment is lacking (due to exclusion criteria in pivotal trials), thus more data needs to be collected. Furthermore, the long-term risks of malignancy and opportunistic infections remains to be established, as well as algorithms on how to treat patients with ongoing disease activity after a 2-year course of therapy. Finally, cladribine interferes with DNA synthesis and repair mechanisms, raising concerns in young adults of child-bearing age until additional safety data become available.
Alemtuzumab
Alemtuzumab is a humanized monoclonal IgG1-antibody that targets CD52, a surface molecule with largely unknown functions predominantly expressed at high levels on B and T cells (196, 197). Lower expression levels are found on monocytes, macrophages, and eosinophils. Mature NK cells, plasma cells, neutrophils, and, most importantly, hematological stem cells show little or no expression (198).
Alemtuzumab leads to a rapid and long-lasting depletion of CD52-positive cells by antibody-dependent, cell-mediated cytolysis (ADCC) and complement dependent cytolysis (CDC) (199), followed by a slow repopulation arising from unaffected hematopoietic precursor cells. Both, quantitative and qualitative changes in the immune-cell repertoire are observed, which might contribute to a rebalancing of autoimmune processes. While the exact mechanisms underlying the reprogramming of the immune system are only vaguely understood, a specific pattern exists to repopulate immune-cell subsets in peripheral blood (200, 201). Monocytes reach baseline levels after 3 months. B cell counts not only return to baseline numbers after 3 months, but also show an excess increase to 124–165% of baseline levels at 12 months. CD8+T lymphocytes are restored after 31 months, whereas CD4+T lymphocytes need ~60 months for complete repopulation. This rapid CD19+ B cell subset repopulation in the absence of effective T cell regulation might explain some of the AEs, e.g., secondary autoimmunities (202). Furthermore, an expansion in CD56bright NK cells also occurs (203). The effects of NK cells on disease progression are unclear, however, it is debated whether they will exhibit immunoregulatory properties (204).
Alemtuzumab's efficacy and safety have been evaluated in treatment naive RRMS patients in phase-II (205) and phase-III trials (206), and in RRMS patients who had an inadequate response (≥1 relapse after ≥6 months of treatment) to prior therapy (207). Due to different inclusion criteria, patients in CARE MS I were younger (mean age was 33.0 vs. 34.7 years), had a lower mean EDSS (2.0 vs. 2.7) and a shorter mean disease duration (2.1 vs. 4.5 years). In the phase-III CARE-MS trials, alemtuzumab demonstrated significantly lower disease activity over 2 years vs. SC IFN-ß-1a administered three times per week (206, 207). In both CARE-MS I and II studies, alemtuzumab significantly reduced the frequency of relapses over 2 years compared to SC IFN-ß-1a (54.9 and 49.4% reduction in relapses in the respective trials); significantly improved MRI outcomes including gd-enhancing lesions and new or enlarging T2 lesions in the alemtuzumab cohort compared to the IFN-ß-1a cohort, and significantly reduced the rate of brain-volume loss. Alemtuzumab also significantly reduced the rate of clinical disease worsening over 36 months in the phase-II CAMMS223 study (205). In CARE-MS II, patients treated with alemtuzumab were more likely to experience 6-month confirmed disability improvement than patients receiving SC IFN-ß-1a treatment (hazard ratio 2.57), whereas this outcome was not significant in CARE-MS I (206, 207).
AEs include infusion-associated reactions (IARs), serious infections, and autoimmune-adverse events, including thyroid disorders and, less frequently, immune thrombocytopenia (ITP) and nephropathies. Malignancies such as thyroid cancer, melanoma, and melanoma-in-situ as well as lymphoproliferative disorders have been reported9.
The IAR rate in the phase-III trials was >90%, mostly mild to moderate in severity and most frequently within the first 3 days of infusion (206, 207). The IARs, which are attributable mainly to cytokine-release syndrome, included headaches, rash, pyrexia, nausea, urticaria, pruritus, flushing, insomnia, fatigue, chills, chest discomfort, and dyspnea. The IARs decrease with successive infusions in a single course and in the second course (206). The clinical trials reported severe IARs ranging from 1 to 3%. Concomitant corticosteroids, antihistamines, and antipyretic drugs are applied with the infusion to avoid IAR. In addition, IARs might be reduced by slowing or temporarily stopping the infusion. Following a safety announcement by the FDA on the rare but serious risks of stroke and blood vessel wall tear (208), Azevedo et al. reported five patients that developed intracerebral hemorrhage within a few hours after administration of alemtuzumab (209).
Moreover, in all patients an increase in blood pressure or labile blood pressure was recognized. Labile hemodynamics under alemtuzumab treatment and infusion-associated reactions resulting in an activated immune system involving mast cells, basophils, complement, activation of platelet derived growth factor, and the release of interleukin-6 (Il-6), or tumor necrosis factor α (TNFα) are discussed as possible causes (210).
Infections are mostly mild to moderate and include oral herpes, herpes zoster, nasopharyngitis, urinary-tract infection, upper respiratory-tract infection, sinusitis, influenza, bronchitis, and localized superficial fungal infections. Serious infections were rare, although slightly elevated with alemtuzumab vs. SC IFN-ß-1a (205–207). Since herpes-virus infections increased in clinical trials with alemtuzumab, prophylactic treatment with an oral anti-herpes agent on the first day of alemtuzumab usage and for 1 month of each treatment cycle was introduced in the risk-management plan. Tuberculosis has been reported in patients treated with alemtuzumab; thus, before the initiation of therapy, all patients must be evaluated for both, active and inactive (“latent”) tuberculosis infection and treated according to local guidelines if required. Moreover, before receiving treatment with alemtuzumab, patients who have not contracted chickenpox and who have not been vaccinated against VZV should be tested for anti-VZV antibodies. Several cases of opportunistic infections including listeria meningitis, esophageal candidiasis, pyogenic granuloma, spirochetal gingivitis, nocardiosis, and cytomegalovirus were reported.
Listeria infections occur generally within 1 month of infusion. Thus, dietary recommendations require the exclusion of certain foods, such as unpasteurized milk and raw meat, during and for 1 month after treatment with alemtuzumab. Antibacterial treatment may be recommended depending on the different regulatory authorities.
Autoimmune AEs represent the most important risk associated with alemtuzumab treatment. These most commonly affect the thyroid; however, they can include rare cases of ITP and anti-glomerular basement membrane nephropathy. The exact pathomechanism leading to secondary autoimmunity remains to be determined. Currently, it is thought that the different temporal lymphocyte repopulation plays a role in this process (211, 212).
In the 5-year follow-up of CAMMS223, thyroid autoimmune AEs occurred in 39% of patients treated with alemtuzumab 12 mg (213). Onset ranged from 6 to 61 months after the first treatment course (207). Incidence peaked at year 3 and declined in subsequent years. Serious ITP events have been observed in ~1% of patients treated with alemtuzumab in the CARE-MS program, between 14 and 36 months after first exposure to alemtuzumab. The first ITP case, during the phase-II CAMMS223 trial, went unrecognized, and the patient died from intracerebral hemorrhage. Following this index case, a monitoring program was implemented to identify and manage ITP systematically, including education on the signs and symptoms for patients and physicians and monthly blood monitoring (142). Other autoimmune cytopenias such as neutropenia, hemolytic anemia, agranulocytosis, and pancytopenia have been reported in the CARE-MS trials with a lower incidence than ITP (206, 207). One patient experienced a recurrence of pancytopenia, which was associated with a lack of compliance with corticosteroid therapy, resulting in fatal sepsis 20 months after alemtuzumab treatment was completed w.
In pilot studies, two patients developed anti-glomerular basement membrane (anti-GBM) disease that ultimately required a kidney transplant (214). In phase-II and phase-III trials, four cases of glomerulonephritis occurred among 1,486 patients treated with alemtuzumab (0.3%). The onset ranged from 4 to 39 months after the last dose of alemtuzumab. Improvements in renal function were observed in two cases of anti-GBM disease after treatment with plasmapheresis, cyclophosphamide, and glucocorticosteroids, and in two cases of membranous glomerulonephritis after treatment with diuretics and/or lisinopril (215, 216).
Secondary autoimmunity is of special interest, since thyroid autoimmunity might affect almost half of the patients (217). A monitoring program was designed and implemented to facilitate the early detection of autoimmune events to ensure timely and adequate management (218): TSH measurements should be performed at baseline and every 3 months for 48 months following the last course (second or subsequent course) (219), and the patient should be monitored for any drug-induced ITP symptoms. Petechiae are uncommon and are usually observed on the lower limbs of patients with a platelet count <20 × 109/L (and often <10 × 109/L). Thus, a platelet count should be performed before the initial course of alemtuzumab, followed by monthly testing that should be continued until 48 months after the final course (220). The signs and symptoms of nephropathy often are non-specific. Routine creatinine testing should be performed before treatment, followed by monthly testing during treatment, continuing until 48 months after the last course (second or subsequent course) (221). Since it is given in cycles, there are no continuous levels of alemtuzumab in the blood. It is recommended that contraception should be used for four months after the last dose (221).
Alemtuzumab's high efficacy contrasts its considerable risks; thus, a thorough assessment of the benefits and risks, adherence to long-term monitoring requirements, and pharmacovigilance are mandatory. Long-term monthly monitoring for 48 months after the final alemtuzumab infusion poses a challenge to patient adherence and requires appropriate education of both physicians and patients. Due to the recently reported side effects including immune-mediated conditions and heart and vessels disorders, the EMA started a review on the medication and temporarily restricted it to patients with highly active disease despite treatment with at least two disease-modifying therapies or in cases when other therapies cannot be used (222). Nevertheless, it is a highly effective treatment option with a long-lasting clinical experience. It should be used in the appropriate patients with the appropriate monitoring schemes.
Table 3 shows data on all approved monoclonal antibodies.
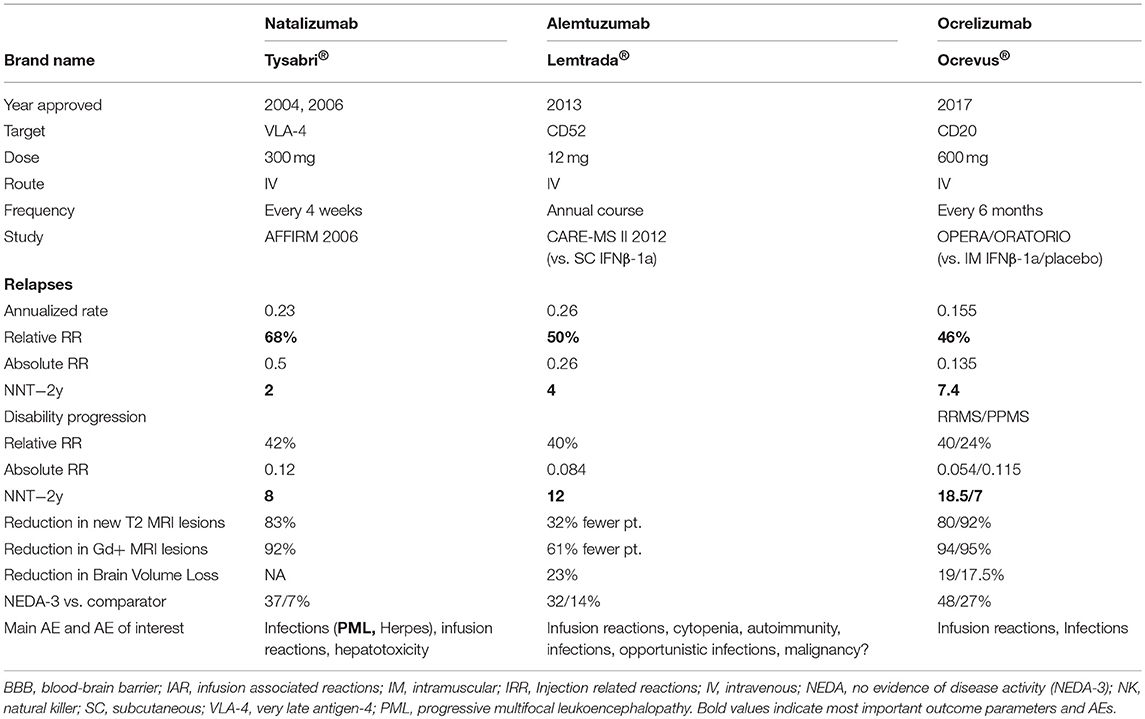
Table 3. Brand name as well as data on efficacy, dose, route of administration, adverse events of approved monoclonal antibodies.
Ocrelizumab
Within the past two decades, the pathogenic role of B cells has generated enormous interest in MS research. Traditionally, MS was primarily considered a T cell-mediated inflammatory disorder, although several findings, including, first and foremost, the development of oligoclonal bands (OCB) in the cerebrospinal fluid (CSF), have indicated a role for B cells. Besides being the source of antibody-producing plasma cells, B cells directly contribute to the development and progression of MS. Peripheral and CNS B cells show signs of chronic inflammation, along with a shift toward antigen-experienced memory B cells (223), indicative of an antigen-mediated activation of B cells in MS. Assumedly, as a consequence, MS patients' B cells show an increased expression of major histocompatibility complex (MHC) class II molecules (224), as well as a higher level of co-stimulatory molecules (225, 226), with the potential to promote the pro-inflammatory differentiation of responding T cells (227). Additional roles of B cells in MS pathogenesis are discussed: antigen presentation, driving T cell activation and auto-proliferation, unbalanced cytokine production, and the formation of ectopic lymphoid follicles (TLOs) under the meninges (228, 229).
Predominantly driven by the assumption that immunoglobulins reactive to a yet unknown self-antigen of the CNS are important drivers of MS pathogenesis, the concept of applying B cell-depleting therapies in MS has evolved. Monoclonal antibodies against CD20 deplete immature and mature B cells, but spare plasma cells and hematopoietic stem cells due to their lack of CD20 expression. Rituximab was the first anti-CD20 antibody to be tested in MS trials, and resulted in a rapid decline in the development of new CNS lesions in patients with RRMS (230, 231). In PPMS, a subgroup of young patients with ongoing CNS lesion formation experienced a slowing of disease progression (232). Testing the humanized successor of rituximab, ocrelizumab confirmed a substantial reduction in the frequency of clinical relapses and CNS lesion formation in RRMS (7, 233). Two identical, randomized, double-blind, double-dummy trials comparing IV ocrelizumab to an active comparator, SC IFN-ß-1a, demonstrated a substantially reduced ARR (0.16 vs. 0.29 p < 0.001 in both trials) in patients treated with ocrelizumab (7). Furthermore, ocrelizumab was superior to IFN-ß-1a with respect to disability progression confirmed at 12 and 24 weeks. In addition to these highly promising findings in RRMS, a placebo-controlled, phase-III trial in patients with PPMS revealed a significantly decelerated accumulation of disability, particularly in younger patients with MRI findings suggestive of ongoing inflammatory activity (8). Based on these phase-III clinical-trial findings, ocrelizumab has been approved recently for both MS indications: RRMS and PPMS. IARs have been observed, especially during the first administrations. Neoplasms (including breast carcinomas) were more often reported (2.3%) in the ocrelizumab cohort than in those patients receiving placebo (0.8%) in the PPMS trials (8). Long term effects of immunosuppression and B depletion are missing.
The anti-CD20 monoclonal antibodies rituximab and ocrelizumab differ from each other in certain aspects. Rituximab, which has not been brought to a phase-III trial mainly for strategic considerations, is a chimeric antibody and acts predominantly via CDC. Ocrelizumab is more humanized, and its B cell-depleting effector mechanism is mediated more by ADCC. A third anti-CD20 antibody currently tested in phase-III MS trials is ofatumumab (NCT02792231 and NCT02792218), a fully human anti-CD20 antibody (234).
Ocrelizumab is administered IV every 24 weeks at a maintenance dose of 600 mg. Within this interval, the vast majority of patients are continuously depleted of blood B cells. Very little is known about other bodily systems that might be more important immunologically, such as secondary lymphoid organs. In this regard, a recent experimental study revealed that a fraction of CD20+B cells in the spleen is resistant to systemic anti-CD20 treatment (235). After cessation of treatment, this population expanded in parallel to de novo B cell generation from bone marrow, resulting in an increased frequency of potentially pathogenic B cells in mice containing a B cell-stimulating immunization. This may be enormously important, since in classical autoimmune diseases, such as myasthenia gravis or AQP4-Ab+ NMO, the stimulating autoantigen may be present when B cells return after cessation of anti-CD20 treatment. Furthermore, the extinction and recovery of B cells may differ substantially, both quantitatively and qualitatively, when lower doses or other administration routes are used (236). In accordance with this, studies in rheumatoid-arthritis patients revealed that a single administration of 10 mg of ocrelizumab was sufficient to deplete B cells efficiently from blood, whereas B cell recovery started much earlier than with higher doses (237). Along with these lines, investigations have recently suggested that substantially lower doses of SC ofatumumab are sufficient to mediate a virtually complete removal of B cells from the blood (234, 238). Regarding other compartments, such as secondary lymphoid organs, experimental studies have suggested that a SC administration targets B cells most efficiently in draining lymph nodes, whereas the IV application of anti-CD20 exerts a more thorough effect on the removal of splenic B cells (238). These differences might have important clinical implications, since they might substantially impact both the clinical efficacy and safety of anti-CD20 treatment in MS patients. In pivotal trials, 12 patients developed anti-drug antibodies, and two of these were positive for neutralizing antibodies. Due to the low number and low incidence of these antibodies, no final assessment on their incidence and their impact possible10.
Data on administration of ocrelizumab during pregnancy is scarce, thus no final assessment is possible (239)10,11. However, the FDA (the EMA) requires 6 (12) months contraception after the last dose of ocrelizumab.
Natalizumab
A hallmark in the pathogenesis of MS is immune dysregulation, characterized by autoreactive lymphocytes penetrating the BBB, resulting in an inflammatory cascade that leads to demyelination, axonal transection, and neurologic deficits (240). The entry of lymphocytes into the CNS requires transmigration through the inflamed endothelium, and the prevention of this process should provide anti-inflammatory therapy in MS (241). Natalizumab was the first monoclonal antibody approved in 2004 for the treatment of RRMS. It is a humanized recombinant IgG4 monoclonal antibody that inhibits leukocyte extravasation into the CNS and intestinal tract by blocking the α4 subunit of integrin molecules on leukocytes (242). Integrins are cell-surface glycoproteins that facilitate cell-matrix adhesion and mediate leukocyte rolling and adhesion to the endothelium prior to extravasation (243). By inhibiting their interaction with vascular cell-adhesion molecule (VCAM)-1 expressed on endothelial cells, natalizumab prevents T lymphocytes from crossing the BBB, thereby reducing inflammation in the brain-tissue compartment (244). In 1992, a study by Yednock et al. concluded that antibodies against the α4-integrin substantially restricted the accumulation of leukocytes in the CNS and prevented the development of a model mimicking MS in rats known as experimental autoimmune encephalomyelitis (EAE) (245). These findings paved the way for early clinical trials with natalizumab. Perhaps equally important, natalizumab was later shown to sequester T cell and B cell subsets out of the CNS compartment (246–248), providing proof-of-concept evidence that a reduction in adaptive immune-cell access to the CNS benefits RRMS patients.
The efficacy of natalizumab for treating RRMS was shown in two phase-III trials: the AFFIRM and SENTINEL studies (249), In the AFFIRM study, 942 patients with RRMS were enrolled in a 2:1 ratio to receive natalizumab 300 mg every 4 weeks or a placebo. The primary outcomes included the clinical-relapse rate at 1 year and the sustained disability-progression rate at 2 years. The results showed that natalizumab reduced the ARR by 68% and lowered the risk of sustained disability progression at 2 years by 42% (249). The SENTINEL study enrolled 1,171 patients who had at least one relapse whilst on IFNβ-1a therapy in the previous 12 months. They received intramuscular IFNβ-1a in combination with 300 mg of natalizumab or a placebo. The outcome measures were identical to those of the AFFIRM study and showed that combination therapy with natalizumab yielded a 55% reduction in the ARR and a 24% reduction in the risk of sustained disability progression at 2 years (250). Both studies also showed significant reductions in the number of new or enlarging T2 lesions and enhancing lesions on MRI in patients receiving natalizumab. Natalizumab was studied further in patients with SPMS in the phase-III ASCEND trial, which did not meet the primary endpoint of disability progression (251), although its target α4-integrin is highly expressed in active lesions of patients who died in late secondary progressive MS (252). These results highlight the different disease pathophysiology that drives progressive forms of MS, which is characterized by axonal/neurodegeneration, innate immune responses mediated by CNS resident cells, development of meningeal lymphoid follicles contributing to expanding number of cortical lesions, and compartmentalized inflammation (137, 253).
Shortly after natalizumab was approved in 2004, the drug was withdrawn from the market after three patients developed PML, a life-threatening CNS-demyelinating disease caused by infection of oligodendrocytes with the John Cunningham virus (JCV). Natalizumab associated PML carries an average mortality rate of 23%, and survivors often develop debilitating neurological deficits from the disease and its treatment sequelae (254). In immunocompetent individuals, JCV almost never causes disease and remains latent in more than half of the world's population (255). PML more frequently affects immunosuppressed individuals, such as patients with acquired immune deficiency syndrome (AIDS), however, in patients taking natalizumab who are not systemically immunosuppressed, the disease is thought to be caused by forced migration of cells harboring JCV out of the bone marrow and the upregulation of gene products in B cell maturation that also promote JCV growth (256). For patients with a suspected PML diagnosis, natalizumab must be discontinued, and treatment with plasma exchange or immunoadsorption can help eliminate remaining circulating natalizumab (257). Patients might develop paradoxically worsening neurologic deficits due to an overwhelming inflammatory reaction due to the recovering immune system, a condition known as immune reconstitution inflammatory syndrome (IRIS), for which a corticosteroid can be given to provide modest benefits (258).
In 2006, natalizumab was reintroduced to the market with a black-box warning about PML risks. Prescribers and patients are required to enroll in a program that the FDA developed, known as Tysabri Outreach Unified Commitment to Health (TOUCH), which informs providers and patients about PML risks. The risks of developing PML have been identified in post-marketing analysis and include the presence of anti-JCV antibodies, prior exposure to immunosuppressants, and more than 2 years of natalizumab therapy (259, 260). In the absence of anti-JCV antibodies, the risk for PML is <0.1/1,000, but the risk increases up to 23/1,000 in patients with all three risk factors (261). Testing for JCV serology is recommended every 6 months for patients with negative and indeterminate results, given a seroconversion rate of 8.5–10.3% per year in natalizumab treated patients (262). Routine surveillance MRIs also can detect early stages of the disease (263, 264). Since there are no guidelines for quantifying the risk of PML, it is up to the clinician and the patient to be aware of risk factors and consider switching therapies based on the risks and benefits from continuing the drug.
Anti-idiotypic antibodies against natalizumab are known to reduce the clinical efficacy, as well as to increase the likelihood of infusion-related adverse events (265). The presence of anti-natalizumab antibodies may be transient or persistently positive, defined as present on at least two occasions 6 weeks apart (249). In the AFFIRM study, antibodies against natalizumab were detected in 9% of patients on natalizumab (6% persistently positive), and in the SENTINEL study, they were present in 12% of patients on natalizumab and IFNβ-1a (6% persistently positive). It is recommended that patients with ongoing disease activity or persistent adverse infusion reactions be tested for antibodies against natalizumab (265, 266). Natalizumab should not be given during pregnancy. However, available data from reports do not show a significant increase in abnormalities compared to other MS patients, and it might be administered based on the individual case. An individual plan should to be made for each female patient who wishes to conceive (38).
The risk of rebound MS disease activity exists for patients who discontinue natalizumab for reasons such as the increased risk of PML, pregnancy, or the presence of neutralizing antibodies. After cessation of natalizumab, it takes 3 months for serum natalizumab concentrations to fall below the threshold of 1 μg/mL for α4-integrin desaturation (267), and CSF lymphocyte counts remain suppressed for up to 6 months (246). Correlating with these laboratory findings, disease recurrence typically starts from 2 to 6 months after natalizumab discontinuation and peaks at 5–8 months (268). Clinical worsening of MS is seen in 20–30% of patients who stop taking natalizumab, and 50–60% show worsening MRI lesions with either new gadolinium-enhancing lesions or new or enlarging T2-hyperintense lesions (268). Restarting therapy with alternative agents—including GA, IFN-ß, and fingolimod—cannot completely prevent recurrence of disease, however, patients who have switched therapies generally have lower rebound activity (269–275). In addition, shortening the natalizumab washout period and tapering cessation have been shown to reduce the risk of rebound disease further (276–278).
Immunologically, disease reactivation after cessation of natalizumab has been associated with some degree of immune reconstitution in the CNS.
Table 4 summarizes proposed modes of action of all approved MS therapeutics.
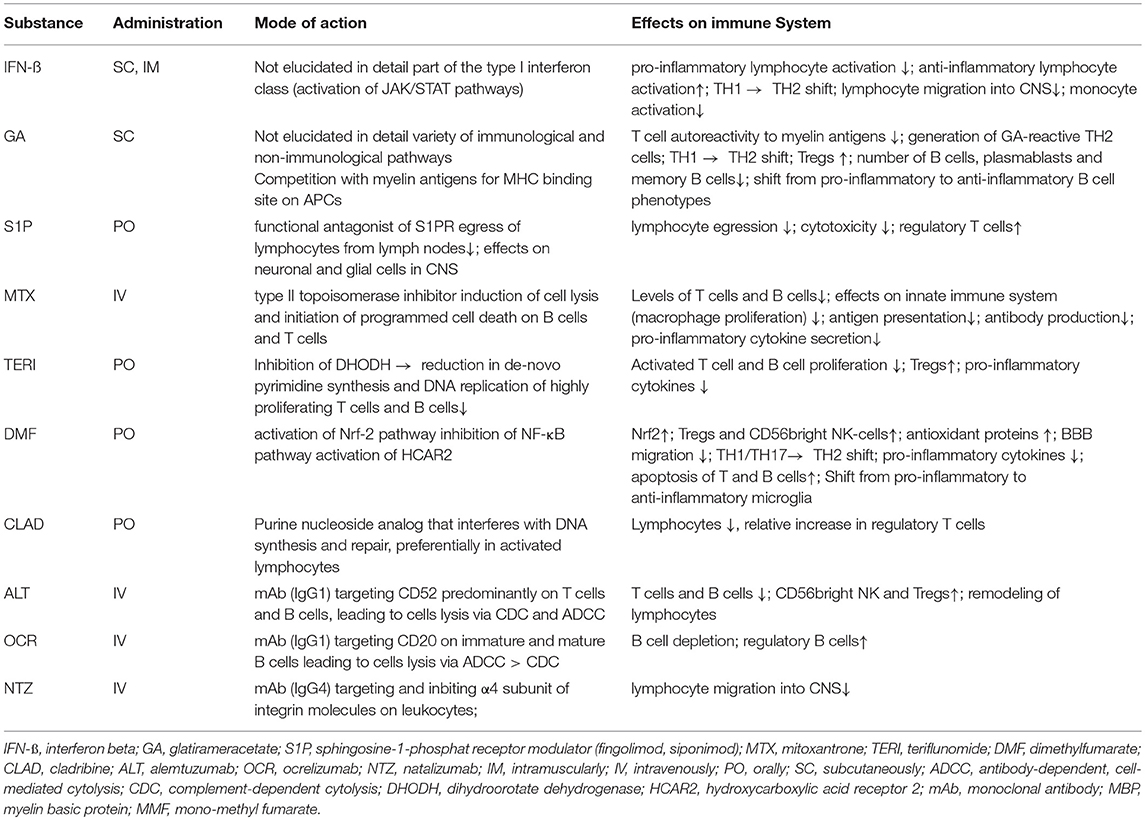
Table 4. Overview on supposed modes of action of approved therapeutics in MS and its proposed effects on the immune system.
Conclusion
The widening of the MS treatment landscape over the past few decades mirrors progress in our understanding of MS pathophysiology. Although the mode of action is not known in detail for some of the approved treatments, the known mechanisms for other agents support our current concepts of the pathophysiology of early MS. Moreover, some treatments, such as ocrelizumab, have broadened our perspective on pathogenic MS events (230), highlighting the importance of B cells in treating MS (279).
As stated, the progress that has been achieved over the past few decades regarding MS pharmacotherapies is tremendous, and perhaps unprecedented for any medical subspecialty. In addition to benefitting patients with RRMS and PPMS, many of the agents currently approved for MS patients have informed us about relevant molecular and cellular targets in this disorder. However, challenges remain, including the potentially serious AEs. Long-term safety data is needed to detect rare but serious AEs. Based on these data monitoring must be adapted to decrease the risk of severe or even life-threatening events (as JCV-Abs for PML in patients treated with natalizumab or secondary autoimmunities in alemtuzumab treated patients). Pharmacovigilance and a better understanding of the host factors that lead to these adverse events hopefully will reduce their incidence. MS is a heterogenous disease and the individual disease progression and prognosis is not predictable. Understanding the mode of action of the various treatment options is of utmost importance to (I) foresee side effects, and (II) to define the best possible treatment, especially for patients that have not responded to treatments. Further treatments such as autologous haematopoietic-stem-cell transplantation (aHCST) are assessed in trials (280). aHCST is not approved but shows promising results. Risks and benefits for patients must be balanced, and again the appropriate patient selection is of utmost importance.
Finally, there is no evidence that any of the currently approved agents benefit patients with non-active MS (281). Currently there is no agreement when to stop or de-escalate treatment. Re-activation of disease might be a risk (282, 283). Further studies are ongoing or needed. The definition of active and non-active MS needs to be improved by appropriate biomarkers to prevent patients from being treated with agents that provide no benefit to them yet expose them to potential AEs.
Author Contributions
All authors contributed at least one chapter to this review. All authors reviewed for intellectual content.
Conflict of Interest Statement
PR received honoraries from consultancy or lectures from AbbVie, Biogen, Celgene (institutional), Merck, Roche, Sanofi Genzyme, Teva. He served on advisory boards for Biogen, Merck, Roche, Sandoz. PR received research support from Biogen, Merck Serono and Roche. RM received honoraria/consulting fees: Actelion, Bayer Healthcare, Biogen-Idec, Medison, Merck-Serono, Neopharm, Novartis, Roche, Sanofi-Genzyme, Teva, and TG-Therapeutics. He served on advisory boards: Bayer Healthcare, Genzyme, Medison, Merck, Neopharm, Novartis, Roche Teva, and TG-Therapeutics. He received research grants: Bayer Healthcare, Medison, Merck-Serono, Novartis, and Teva. MH is on the steering committee for the Novartis FLUENT study. JS received personal compensation as a speaker or consultant from Bayer, Biogen, Merck, Gerot-Lannach, Roche, Sanofi, and Teva. He is member of advisory boards of Biogen, Merck, Sanofi, Roche, and MedDay. His institution received unrestricted grants from Biogen, Merck, Roche, and Sanofi. ZI has served on scientific advisory board for Biogen, Sanofi-Genzyme, Teva, Roche, Novartis, Merck; has received honoraria for lecturing from Biogen, Merck, Teva, Novartis, Sanofi-Genzyme; research support from Biogen, Sanofi-Genzyme, Merck; and support for congress participation from Biogen, Sanofi-Genzyme, Teva, Roche, Novartis, Merck. CW received speaker honoraria (institutional only) and/or research funding from Biogen, Novartis, Sanofi-Genzyme, and Roche.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Footnotes
1. ^Commissioner O of the. FDA Expands Approval of Gilenya to Treat Multiple Sclerosis in Pediatric Patients [Internet]. FDA (2018). Available online at: http://www.fda.gov/news-events/press-announcements/fda-expands-approval-gilenya-treat-multiple-sclerosis-pediatric-patients (accessed June 13, 2019).
2. ^Fingolimod (Gilenya?): bradycardia and heart block [Internet]. GOV.UK. Available online at: https://www.gov.uk/drug-safety-update/fingolimod-gilenya-bradycardia-and-heart-block (accessed May 30, 2019).
3. ^Commissioner O of the. Press Announcements - FDA approves new oral drug to treat multiple sclerosis [Internet]. Available online at: https://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm634469.htm (accessed April 7, 2019).
4. ^novantrone-article-30-referral-annex-iii_en.pdf [Internet]. Available online at: https://www.ema.europa.eu/en/documents/referral/novantrone-article-30-referral-annex-iii_en.pdf (accessed April 7, 2019).
5. ^ml-concentrate-solution-infusion_de.pdf [Internet]. Available online at: https://www.ema.europa.eu/en/documents/referral/questions-answers-novantrone-associated-names-mitoxantrone-2-mg/ml-concentrate-solution-infusion_de.pdf (accessed April 7, 2019).
6. ^Foechterlen D. oder an das Bundesinstitut für Arzneimittel und Medizinprodukte (BfArM): elektronisch über das Internet www.bfarm.de – Arzneimittel – Pharmakovigilanz – Risiken melden; oder schriftlich an die Postadresse Kurt-Georg-Kiesinger-Allee 3, 53175 Bonn, oder per Fax 0228-207 5207.2.
7. ^Commissioner O of the. Press Announcements - FDA approves new oral treatment for multiple sclerosis [Internet]. Available online at: https://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm634837.htm (accessed April 7, 2019).
8. ^WC500234561.pdf [Internet]. Available online at: http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Product_Information/human/004230/WC500234561.pdf (cited September 26, 2018).
9. ^LEMTRADA® (alemtuzumab) | Official Healthcare Professional Site [Internet]. Available online at: https://www.lemtradahcp.com/safety-information (accesed June 2, 2019).
10. ^ocrevus-epar-product-information_en.pdf [Internet]. Available online at: https://www.ema.europa.eu/en/documents/product-information/ocrevus- epar-product-information_en.pdf (accessed June 1, 2019).
11. ^OCREVUS safely and effectively, 18.
References
1. Feigin VL, Abajobir AA, Abate KH, Abd-Allah F, Abdulle AM, Abera SF, et al. Global, regional, and national burden of neurological disorders during 1990–2015: a systematic analysis for the Global Burden of Disease Study 2015. Lancet Neurol. (2017) 16:877–97. doi: 10.1016/S1474-4422(17)30299-5
2. Fredrikson S, Kam-Hansen S. The 150-year anniversary of multiple sclerosis: does its early history give an etiological clue? Perspect Biol Med. (1989) 32:237–43. doi: 10.1353/pbm.1989.0000
3. Murray TJ. The history of multiple sclerosis: the changing frame of the disease over the centuries. J Neurol Sci. (2009) 277(Suppl. 1):S3–8. doi: 10.1016/S0022-510X(09)70003-6
4. Zettl UK, Hecker M, Aktas O, Wagner T, Rommer PS. Interferon β-1a and β-1b for patients with multiple sclerosis: updates to current knowledge. Expert Rev Clin Immunol. (2018) 14:137–53. doi: 10.1080/1744666X.2018.1426462
5. Rommer PS, Zettl UK. Managing the side effects of multiple sclerosis therapy: pharmacotherapy options for patients. Expert Opin Pharmacother. (2018) 19:483–98. doi: 10.1080/14656566.2018.1446944
6. Dendrou CA, Fugger L, Friese MA. Immunopathology of multiple sclerosis. Nat Rev Immunol. (2015) 15:545–58. doi: 10.1038/nri3871
7. Hauser SL, Bar-Or A, Comi G, Giovannoni G, Hartung H-P, Hemmer B, et al. Ocrelizumab versus interferon Beta-1a in relapsing multiple sclerosis. N Engl J Med. (2017) 376:221–34. doi: 10.1056/NEJMoa1601277
8. Montalban X, Hauser SL, Kappos L, Arnold DL, Bar-Or A, Comi G, et al. Ocrelizumab versus placebo in primary progressive multiple sclerosis. N Engl J Med. (2017) 376:209–20. doi: 10.1056/NEJMoa1606468
9. Frohman EM, Racke MK, Raine CS. Multiple sclerosis–the plaque and its pathogenesis. N Engl J Med. (2006) 354:942–55. doi: 10.1056/NEJMra052130
10. Schwab SR, Cyster JG. Finding a way out: lymphocyte egress from lymphoid organs. Nat Immunol. (2007) 8:1295–301. doi: 10.1038/ni1545
11. Ransohoff RM, Engelhardt B. The anatomical and cellular basis of immune surveillance in the central nervous system. Nat Rev Immunol. (2012) 12:623–35. doi: 10.1038/nri3265
12. Haji Abdolvahab M, Mofrad MRK, Schellekens H. Interferon beta: from molecular level to therapeutic effects. Int Rev Cell Mol Biol. (2016) 326:343–72. doi: 10.1016/bs.ircmb.2016.06.001
13. Madsen C. The innovative development in interferon beta treatments of relapsing-remitting multiple sclerosis. Brain Behav. (2017) 7:e00696. doi: 10.1002/brb3.696
14. De Andrea M, Ravera R, Gioia D, Gariglio M, Landolfo S. The interferon system: an overview. Eur J Paediatr Neurol EJPN Off J Eur Paediatr Neurol Soc. (2002) 6(Suppl. A):A41–46; discussion: A55–58. doi: 10.1053/ejpn.2002.0573
15. Markowitz CE. Interferon-beta: mechanism of action and dosing issues. Neurology. (2007) 68(24 Suppl. 4):S8–11. doi: 10.1212/01.wnl.0000277703.74115.d2
16. Zhang J, Hutton G, Zang Y. A comparison of the mechanisms of action of interferon beta and glatiramer acetate in the treatment of multiple sclerosis. Clin Ther. (2002) 24:1998–2021. doi: 10.1016/S0149-2918(02)80094-7
17. Wandinger KP, Stürzebecher CS, Bielekova B, Detore G, Rosenwald A, Staudt LM, et al. Complex immunomodulatory effects of interferon-beta in multiple sclerosis include the upregulation of T helper 1-associated marker genes. Ann Neurol. (2001) 50:349–57. doi: 10.1002/ana.1096
18. Schreiner B, Mitsdoerffer M, Kieseier BC, Chen L, Hartung H-P, Weller M, et al. Interferon-β enhances monocyte and dendritic cell expression of B7-H1 (PD-L1), a strong inhibitor of autologous T-cell activation: relevance for the immune modulatory effect in multiple sclerosis. J Neuroimmunol. (2004) 155:172–82. doi: 10.1016/j.jneuroim.2004.06.013
19. Yong VW, Chabot S, Stuve O, Williams G. Interferon beta in the treatment of multiple sclerosis: mechanisms of action. Neurology. (1998) 51:682–9. doi: 10.1212/WNL.51.3.682
20. Dhib-Jalbut S, Marks S. Interferon-β mechanisms of action in multiple sclerosis. Neurology. (2010) 74(Suppl. 1):S17–24. doi: 10.1212/WNL.0b013e3181c97d99
21. Neuhaus O, Kieseier BC, Hartung H-P. Pharmacokinetics and pharmacodynamics of the interferon-betas, glatiramer acetate, and mitoxantrone in multiple sclerosis. J Neurol Sci. (2007) 259:27–37. doi: 10.1016/j.jns.2006.05.071
22. Drug, Approval Package: Betaseron Interferon BETA-1B Subcutaneous(Generic Name) NDA # 103471 [Internet],. Available online at: https://www.accessdata.fda.gov/drugsatfda_docs/nda/pre96/103471s0000TOC.cfm (accessed August 8, 2018).
23. WC500053086.pdf [Internet]. Available online at: http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Summary_for_the_public/human/000081/WC500053086.pdf (accessed August 8, 2018).
24. White JT, Newsome SD, Kieseier BC, Bermel RA, Cui Y, Seddighzadeh A, et al. Incidence, characterization, and clinical impact analysis of peginterferon beta1a immunogenicity in patients with multiple sclerosis in the ADVANCE trial. Ther Adv Neurol Disord. (2016) 9:239–49. doi: 10.1177/1756285616633967
25. Interferon beta-1b in the treatment of multiple sclerosis: final outcome of the randomized controlled trial. The IFNB Multiple Sclerosis Study Group and The University of British Columbia MS/MRI Analysis Group. Neurology. (1995) 45:1277–85. doi: 10.1212/WNL.45.7.1277
26. Interferon beta-1b is effective in relapsing-remitting multiple sclerosis. I. Clinical results of a multicenter, randomized, double-blind, placebo-controlled trial. The IFNB Multiple Sclerosis Study Group. Neurology. (1993) 43:655–61. doi: 10.1212/WNL.43.4.655
27. Vermersch P, de Seze J, Stojkovic T, Hautecoeur P, G-SEP (Groupe Septentrional d'Etudes et de Recherches sur la SEP). Interferon beta1a (Avonex) treatment in multiple sclerosis: similarity of effect on progression of disability in patients with mild and moderate disability. J Neurol. (2002) 249:184–7. doi: 10.1007/PL00007862
28. Kappos L, Freedman MS, Polman CH, Edan G, Hartung H-P, Miller DH, et al. Effect of early versus delayed interferon beta-1b treatment on disability after a first clinical event suggestive of multiple sclerosis: a 3-year follow-up analysis of the BENEFIT study. Lancet. (2007) 370:389–97. doi: 10.1016/S0140-6736(07)61194-5
29. Comi G, De Stefano N, Freedman MS, Barkhof F, Polman CH, Uitdehaag BMJ, et al. Comparison of two dosing frequencies of subcutaneous interferon beta-1a in patients with a first clinical demyelinating event suggestive of multiple sclerosis (REFLEX): a phase 3 randomised controlled trial. Lancet Neurol. (2012) 11:33–41. doi: 10.1016/S1474-4422(11)70262-9
30. Jacobs LD, Beck RW, Simon JH, Kinkel RP, Brownscheidle CM, Murray TJ, et al. Intramuscular interferon beta-1a therapy initiated during a first demyelinating event in multiple sclerosis. CHAMPS Study Group. N Engl J Med. (2000) 343:898–904. doi: 10.1056/NEJM200009283431301
31. Placebo-controlled multicentre randomised trial of interferon beta-1b in treatment of secondary progressive multiple sclerosis. European Study Group on interferon beta-1b in secondary progressive MS. Lancet. (1998) 352:1491–7. doi: 10.1016/S0140-6736(98)10039-9
32. Panitch H, Miller A, Paty D, Weinshenker B, North American Study Group on Interferon beta-1b in Secondary Progressive MS. Interferon beta-1b in secondary progressive MS: results from a 3-year controlled study. Neurology. (2004) 63:1788–95. doi: 10.1212/01.WNL.0000146958.77317.3E
33. Rommer PS, Stüve O. Management of secondary progressive multiple sclerosis: prophylactic treatment-past, present, and future aspects. Curr Treat Options Neurol. (2013) 15:241–58. doi: 10.1007/s11940-013-0233-x
34. Trojano M, Pellegrini F, Paolicelli D, Fuiani A, Zimatore GB, Tortorella C, et al. Real-life impact of early interferon beta therapy in relapsing multiple sclerosis. Ann Neurol. (2009) 66:513–20. doi: 10.1002/ana.21757
35. Ebers GC, Traboulsee A, Li D, Langdon D, Reder AT, Goodin DS, et al. Analysis of clinical outcomes according to original treatment groups 16 years after the pivotal IFNB-1b trial. J Neurol Neurosurg Psychiatry. (2010) 81:907–12. doi: 10.1136/jnnp.2009.204123
36. Hemmer B, Stüve O, Kieseier B, Schellekens H, Hartung H-P. Immune response to immunotherapy: the role of neutralising antibodies to interferon beta in the treatment of multiple sclerosis. Lancet Neurol. (2005) 4:403–12. doi: 10.1016/S1474-4422(05)70117-4
37. Link J, Ramanujam R, Auer M, Ryner M, Hässler S, Bachelet D, et al. Clinical practice of analysis of anti-drug antibodies against interferon beta and natalizumab in multiple sclerosis patients in Europe: a descriptive study of test results. PLoS ONE. (2017) 12:e0170395. doi: 10.1371/journal.pone.0170395
38. Thöne J, Thiel S, Gold R, Hellwig K. Treatment of multiple sclerosis during pregnancy - safety considerations. Expert Opin Drug Saf. (2017) 16:523–34. doi: 10.1080/14740338.2017.1311321
39. Teitelbaum D, Arnon R, Sela M. Copolymer 1: from basic research to clinical application. Cell Mol Life Sci. (1997) 53:24–8. doi: 10.1007/PL00000576
40. Dhib-Jalbut S. Mechanisms of action of interferons and glatiramer acetate in multiple sclerosis. Neurology. (2002) 58(8 Suppl. 4):S3–9. doi: 10.1212/WNL.58.8_suppl_4.S3
41. Fridkis-Hareli M, Teitelbaum D, Gurevich E, Pecht I, Brautbar C, Kwon OJ, et al. Direct binding of myelin basic protein and synthetic copolymer 1 to class II major histocompatibility complex molecules on living antigen-presenting cells–specificity and promiscuity. Proc Natl Acad Sci USA. (1994) 91:4872–6. doi: 10.1073/pnas.91.11.4872
42. Ben-Nun A, Mendel I, Bakimer R, Fridkis-Hareli M, Teitelbaum D, Arnon R, et al. The autoimmune reactivity to myelin oligodendrocyte glycoprotein (MOG) in multiple sclerosis is potentially pathogenic: effect of copolymer 1 on MOG-induced disease. J Neurol. (1996) 243(4 Suppl. 1):S14–22. doi: 10.1007/BF00873697
43. Arnon R, Sela M, Teitelbaum D. New insights into the mechanism of action of copolymer 1 in experimental allergic encephalomyelitis and multiple sclerosis. J Neurol. (1996) 243(4 Suppl. 1):S8–13. doi: 10.1007/BF00873696
44. Teitelbaum D, Milo R, Arnon R, Sela M. Synthetic copolymer 1 inhibits human T-cell lines specific for myelin basic protein. Proc Natl Acad Sci USA. (1992) 89:137–41. doi: 10.1073/pnas.89.1.137
45. Teitelbaum D, Fridkis-Hareli M, Arnon R, Sela M. Copolymer 1 inhibits chronic relapsing experimental allergic encephalomyelitis induced by proteolipid protein (PLP) peptides in mice and interferes with PLP-specific T cell responses. J Neuroimmunol. (1996) 64:209–17. doi: 10.1016/0165-5728(95)00180-8
46. Neuhaus O, Farina C, Yassouridis A, Wiendl H, Then Bergh F, Dose T, et al. Multiple sclerosis: comparison of copolymer-1- reactive T cell lines from treated and untreated subjects reveals cytokine shift from T helper 1 to T helper 2 cells. Proc Natl Acad Sci USA. (2000) 97:7452–7. doi: 10.1073/pnas.97.13.7452
47. Chen M, Gran B, Costello K, Johnson K, Martin R, Dhib-Jalbut S. Glatiramer acetate induces a Th2-biased response and crossreactivity with myelin basic protein in patients with MS. Mult Scler.(2001) 7:209–19. doi: 10.1177/135245850100700401
48. Aharoni R. Immunomodulation neuroprotection and remyelination - the fundamental therapeutic effects of glatiramer acetate: a critical review. J Autoimmun. (2014) 54:81–92. doi: 10.1016/j.jaut.2014.05.005
49. Ziemssen T, Kümpfel T, Klinkert WEF, Neuhaus O, Hohlfeld R. Glatiramer acetate-specific T-helper 1- and 2-type cell lines produce BDNF: implications for multiple sclerosis therapy. Brain-derived neurotrophic factor. Brain J Neurol. (2002) 125(Pt 11):2381–91. doi: 10.1093/brain/awf252
50. Aharoni R, Teitelbaum D, Arnon R, Sela M. Copolymer 1 acts against the immunodominant epitope 82-100 of myelin basic protein by T cell receptor antagonism in addition to major histocompatibility complex blocking. Proc Natl Acad Sci USA. (1999) 96:634–9. doi: 10.1073/pnas.96.2.634
51. Hong J, Li N, Zhang X, Zheng B, Zhang JZ. Induction of CD4+CD25+ regulatory T cells by copolymer-I through activation of transcription factor Foxp3. Proc Natl Acad Sci USA. (2005) 102:6449–54. doi: 10.1073/pnas.0502187102
52. Weber MS, Starck M, Wagenpfeil S, Meinl E, Hohlfeld R, Farina C. Multiple sclerosis: glatiramer acetate inhibits monocyte reactivity in vitro and in vivo. Brain J Neurol. (2004) 127(Pt 6):1370–8. doi: 10.1093/brain/awh163
53. Kim HJ, Ifergan I, Antel JP, Seguin R, Duddy M, Lapierre Y, et al. Type 2 monocyte and microglia differentiation mediated by glatiramer acetate therapy in patients with multiple sclerosis. J Immunol. (2004) 172:7144–53. doi: 10.4049/jimmunol.172.11.7144
54. Vieira PL, Heystek HC, Wormmeester J, Wierenga EA, Kapsenberg ML. Glatiramer acetate (copolymer-1, copaxone) promotes Th2 cell development and increased IL-10 production through modulation of dendritic cells. J Immunol. (2003) 170:4483–8. doi: 10.4049/jimmunol.170.9.4483
55. Li Q, Milo R, Panitch H, Swoveland P, Bever CT. Glatiramer acetate blocks the activation of THP-1 cells by interferon-gamma. Eur J Pharmacol. (1998) 342:303–10. doi: 10.1016/S0014-2999(97)01509-4
56. Weber MS, Prod'homme T, Youssef S, Dunn SE, Rundle CD, Lee L, et al. Type II monocytes modulate T cell-mediated central nervous system autoimmune disease. Nat Med. (2007) 13:935–43. doi: 10.1038/nm1620
57. Milo R. Therapeutic strategies targeting B-cells in multiple sclerosis. Autoimmun Rev. (2016) 15:714–8. doi: 10.1016/j.autrev.2016.03.006
58. Kuerten S, Jackson LJ, Kaye J, Vollmer TL. Impact of glatiramer acetate on B cell-mediated pathogenesis of multiple sclerosis. CNS Drugs. (2018) 32:1039–51. doi: 10.1007/s40263-018-0567-8
59. Aharoni R, Arnon R, Eilam R. Neurogenesis and neuroprotection induced by peripheral immunomodulatory treatment of experimental autoimmune encephalomyelitis. J Neurosci Off J Soc Neurosci. (2005) 25:8217–28. doi: 10.1523/JNEUROSCI.1859-05.2005
60. Abramsky O, Teitelbaum D, Arnon R. Effect of a synthetic polypeptide (COP 1) on patients with multiple sclerosis and with acute disseminated encephalomeylitis. Preliminary report. J Neurol Sci. (1977) 31:433–8. doi: 10.1016/0022-510X(77)90220-9
61. Bornstein MB, Miller AI, Teitelbaum D, Arnon R, Sela M. Multiple sclerosis: trial of a synthetic polypeptide. Ann Neurol. (1982) 11:317–9. doi: 10.1002/ana.410110314
62. Bornstein MB, Miller A, Slagle S, Weitzman M, Crystal H, Drexler E, et al. A pilot trial of Cop 1 in exacerbating-remitting multiple sclerosis. N Engl J Med. (1987) 317:408–14. doi: 10.1056/NEJM198708133170703
63. Bornstein MB, Miller A, Slagle S, Weitzman M, Drexler E, Keilson M, et al. A placebo-controlled, double-blind, randomized, two-center, pilot trial of Cop 1 in chronic progressive multiple sclerosis. Neurology. (1991) 41:533–9. doi: 10.1212/WNL.41.4.533
64. Johnson KP, Brooks BR, Cohen JA, Ford CC, Goldstein J, Lisak RP, et al. Copolymer 1 reduces relapse rate and improves disability in relapsing-remitting multiple sclerosis: results of a phase III multicenter, double-blind placebo-controlled trial. The Copolymer 1 Multiple Sclerosis Study Group. Neurology. (1995) 45:1268–76. doi: 10.1212/WNL.45.7.1268
65. Johnson KP, Brooks BR, Cohen JA, Ford CC, Goldstein J, Lisak RP, et al. Extended use of glatiramer acetate (Copaxone) is well tolerated and maintains its clinical effect on multiple sclerosis relapse rate and degree of disability. Copolymer 1 Multiple Sclerosis Study Group. Neurology. (1998) 50:701–8. doi: 10.1212/WNL.50.3.701
66. Ge Y, Grossman RI, Udupa JK, Fulton J, Constantinescu CS, Gonzales-Scarano F, et al. Glatiramer acetate (Copaxone) treatment in relapsing-remitting MS: quantitative MR assessment. Neurology. (2000) 54:813–7. doi: 10.1212/WNL.54.4.813
67. Comi G, Filippi M, Wolinsky JS. European/Canadian multicenter, double-blind, randomized, placebo-controlled study of the effects of glatiramer acetate on magnetic resonance imaging–measured disease activity and burden in patients with relapsing multiple sclerosis. European/Canadian Glatiramer Acetate Study Group. Ann Neurol. (2001) 49:290–7. doi: 10.1002/ana.64
68. Filippi M, Rovaris M, Rocca MA, Sormani MP, Wolinsky JS, Comi G, et al. Glatiramer acetate reduces the proportion of new MS lesions evolving into ‘black holes'. Neurology. (2001) 57:731–3. doi: 10.1212/WNL.57.4.731
69. Comi G, Cohen JA, Arnold DL, Wynn D, Filippi M, FORTE Study Group. Phase III dose-comparison study of glatiramer acetate for multiple sclerosis. Ann Neurol. (2011) 69:75–82. doi: 10.1002/ana.22316
70. Khan O, Rieckmann P, Boyko A, Selmaj K, Zivadinov R, GALA Study Group. Three times weekly glatiramer acetate in relapsing-remitting multiple sclerosis. Ann Neurol. (2013) 73:705–13. doi: 10.1002/ana.23938
71. Wolinsky JS, Narayana PA, O'Connor P, Coyle PK, Ford C, Johnson K, et al. Glatiramer acetate in primary progressive multiple sclerosis: results of a multinational, multicenter, double-blind, placebo-controlled trial. Ann Neurol. (2007) 61:14–24. doi: 10.1002/ana.21079
72. Comi G, Martinelli V, Rodegher M, Moiola L, Bajenaru O, Carra A, et al. Effect of glatiramer acetate on conversion to clinically definite multiple sclerosis in patients with clinically isolated syndrome (PreCISe study): a randomised, double-blind, placebo-controlled trial. Lancet. (2009) 374:1503–11. doi: 10.1016/S0140-6736(09)61259-9
73. Ford C, Goodman AD, Johnson K, Kachuck N, Lindsey JW, Lisak R, et al. Continuous long-term immunomodulatory therapy in relapsing multiple sclerosis: results from the 15-year analysis of the US prospective open-label study of glatiramer acetate. Mult Scler. (2010) 16:342–50. doi: 10.1177/1352458509358088
74. O'Connor P, Filippi M, Arnason B, Comi G, Cook S, Goodin D, et al. 250 microg or 500 microg interferon beta-1b versus 20 mg glatiramer acetate in relapsing-remitting multiple sclerosis: a prospective, randomised, multicentre study. Lancet Neurol. (2009) 8:889–97. doi: 10.1016/S1474-4422(09)70226-1
75. Mikol DD, Barkhof F, Chang P, Coyle PK, Jeffery DR, Schwid SR, et al. Comparison of subcutaneous interferon beta-1a with glatiramer acetate in patients with relapsing multiple sclerosis (the REbif vs Glatiramer Acetate in Relapsing MS Disease [REGARD] study): a multicentre, randomised, parallel, open-label trial. Lancet Neurol. (2008) 7:903–14. doi: 10.1016/S1474-4422(08)70200-X
76. Lublin FD, Cofield SS, Cutter GR, Conwit R, Narayana PA, Nelson F, et al. Randomized study combining interferon and glatiramer acetate in multiple sclerosis. Ann Neurol. (2013) 73:327–40. doi: 10.1002/ana.23863
77. Karussis D, Teitelbaum D, Sicsic C, Brenner T, AC001 multi-center Israeli study group. Long-term treatment of multiple sclerosis with glatiramer acetate: natural history of the subtypes of anti-glatiramer acetate antibodies and their correlation with clinical efficacy. J Neuroimmunol. (2010) 220:125–30. doi: 10.1016/j.jneuroim.2010.01.009
78. Sandberg-Wollheim M, Neudorfer O, Grinspan A, Weinstock-Guttman B, Haas J, Izquierdo G, et al. Pregnancy outcomes from the branded glatiramer acetate pregnancy database. Int J MS Care. (2018) 20:9–14. doi: 10.7224/1537-2073.2016-079
79. Kalinke U, Prinz M. Endogenous, or therapeutically induced, type I interferon responses differentially modulate Th1/Th17-mediated autoimmunity in the CNS. Immunol Cell Biol. (2012) 90:505–9. doi: 10.1038/icb.2012.8
80. Lalive PH, Neuhaus O, Benkhoucha M, Burger D, Hohlfeld R, Zamvil SS, et al. Glatiramer acetate in the treatment of multiple sclerosis: emerging concepts regarding its mechanism of action. CNS Drugs. (2011) 25:401–14. doi: 10.2165/11588120-000000000-00000
81. Straus Farber R, Harel A, Lublin F. Novel agents for relapsing forms of multiple sclerosis. Annu Rev Med. (2016) 67:309–21. doi: 10.1146/annurev-med-052814-023415
82. Chun J, Brinkmann V. A mechanistically novel, first oral therapy for multiple sclerosis: the development of fingolimod (FTY720, Gilenya). Discov Med. (2011) 12:213–28.
83. Cyster JG, Schwab SR. Sphingosine-1-phosphate and lymphocyte egress from lymphoid organs. Annu Rev Immunol. (2012) 30:69–94. doi: 10.1146/annurev-immunol-020711-075011
84. Matloubian M, Lo CG, Cinamon G, Lesneski MJ, Xu Y, Brinkmann V, et al. Lymphocyte egress from thymus and peripheral lymphoid organs is dependent on S1P receptor 1. Nature. (2004) 427:355–60. doi: 10.1038/nature02284
85. Pelletier D, Hafler DA. Fingolimod for multiple sclerosis. N Engl J Med. (2012) 366:339–47. doi: 10.1056/NEJMct1101691
86. Reeves PM, Kang Y-L, Kirchhausen T. Endocytosis of ligand-activated sphingosine 1-phosphate receptor 1 mediated by the clathrin-pathway. Traffic Cph Den. (2016) 17:40–52. doi: 10.1111/tra.12343
87. Mehling M, Brinkmann V, Antel J, Bar-Or A, Goebels N, Vedrine C, et al. FTY720 therapy exerts differential effects on T cell subsets in multiple sclerosis. Neurology. (2008) 71:1261–7. doi: 10.1212/01.wnl.0000327609.57688.ea
88. Zhou PJ, Wang H, Shi GH, Wang XH, Shen ZJ, Xu D. Immunomodulatory drug FTY720 induces regulatory CD4(+)CD25(+) T cells in vitro. Clin Exp Immunol. (2009) 157:40–7. doi: 10.1111/j.1365-2249.2009.03942.x
89. Yamagata K, Tagami M, Torii Y, Takenaga F, Tsumagari S, Itoh S, et al. Sphingosine 1-phosphate induces the production of glial cell line-derived neurotrophic factor and cellular proliferation in astrocytes. Glia. (2003) 41:199–206. doi: 10.1002/glia.10180
90. Edsall LC, Pirianov GG, Spiegel S. Involvement of sphingosine 1-phosphate in nerve growth factor-mediated neuronal survival and differentiation. J Neurosci Off J Soc Neurosci. (1997) 17:6952–60. doi: 10.1523/JNEUROSCI.17-18-06952.1997
91. Kappos L, O'Connor P, Radue E-W, Polman C, Hohlfeld R, Selmaj K, et al. Long-term effects of fingolimod in multiple sclerosis: the randomized FREEDOMS extension trial. Neurology. (2015) 84:1582–91. doi: 10.1212/WNL.0000000000001462
92. Calabresi PA, Radue E-W, Goodin D, Jeffery D, Rammohan KW, Reder AT, et al. Safety and efficacy of fingolimod in patients with relapsing-remitting multiple sclerosis (FREEDOMS II): a double-blind, randomised, placebo-controlled, phase 3 trial. Lancet Neurol. (2014) 13:545–56. doi: 10.1016/S1474-4422(14)70049-3
93. Cohen JA, Barkhof F, Comi G, Hartung H-P, Khatri BO, Montalban X, et al. Oral fingolimod or intramuscular interferon for relapsing multiple sclerosis. N Engl J Med. (2010) 362:402–15. doi: 10.1056/NEJMoa0907839
94. Chitnis T, Arnold DL, Banwell B, Brück W, Ghezzi A, Giovannoni G, et al. Trial of fingolimod versus interferon Beta-1a in pediatric multiple sclerosis. N Engl J Med. (2018) 379:1017–27. doi: 10.1056/NEJMoa1800149
95. Anonymous. Gilenya [Internet]. European Medicines Agency (2018). Available online at: https://www.ema.europa.eu/en/medicines/human/EPAR/gilenya (cited June 13, 2019).
96. Meissner A, Limmroth V. Update on the cardiovascular profile of fingolimod in the therapy of relapsing-remitting multiple sclerosis (MS). Mult Scler Relat Disord. (2016) 8:19–26. doi: 10.1016/j.msard.2016.04.002
97. Winkelmann A, Loebermann M, Reisinger EC, Hartung H-P, Zettl UK. Disease-modifying therapies and infectious risks in multiple sclerosis. Nat Rev Neurol. (2016) 12:217–33. doi: 10.1038/nrneurol.2016.21
98. Druart C, El Sankari S, van Pesch V. Long-term safety and real-world effectiveness of fingolimod in relapsing multiple sclerosis. Patient Relat Outcome Meas. (2017) 9:1–10. doi: 10.2147/PROM.S122401
99. Hatcher SE, Waubant E, Nourbakhsh B, Crabtree-Hartman E, Graves JS. Rebound syndrome in patients with multiple sclerosis after cessation of fingolimod treatment. JAMA Neurol. (2016) 73:790–4. doi: 10.1001/jamaneurol.2016.0826
100. Kappos L, Bar-Or A, Cree BAC, Fox RJ, Giovannoni G, Gold R, et al. Siponimod versus placebo in secondary progressive multiple sclerosis (EXPAND): a double-blind, randomised, phase 3 study. Lancet. (2018) 391:1263–73. doi: 10.1016/S0140-6736(18)30475-6
101. Thomas X, Archimbaud E. Mitoxantrone in the treatment of acute myelogenous leukemia: a review. Hematol Cell Ther. (1997) 39:63–74. doi: 10.1007/s00282-997-0163-8
102. Putzki N, Kumar M, Kreuzfelder E, Grosse-Wilde H, Diener HC, Limmroth V. Mitoxantrone does not restore the impaired suppressive function of natural regulatory T cells in patients suffering from multiple sclerosis. A longitudinal ex vivo and in vitro study. Eur Neurol. (2009) 61:27–32. doi: 10.1159/000165346
103. Chan A, Weilbach FX, Toyka KV, Gold R. Mitoxantrone induces cell death in peripheral blood leucocytes of multiple sclerosis patients. Clin Exp Immunol. (2005) 139:152–8. doi: 10.1111/j.1365-2249.2005.02653.x
104. Neuhaus O, Wiendl H, Kieseier BC, Archelos JJ, Hemmer B, Stüve O, et al. Multiple sclerosis: mitoxantrone promotes differential effects on immunocompetent cells in vitro. J Neuroimmunol. (2005) 168:128–37. doi: 10.1016/j.jneuroim.2005.01.024
105. Kopadze T, Dehmel T, Hartung H-P, Stüve O, Kieseier BC. Inhibition by mitoxantrone of in vitro migration of immunocompetent cells: a possible mechanism for therapeutic efficacy in the treatment of multiple sclerosis. Arch Neurol. (2006) 63:1572–8. doi: 10.1001/archneur.63.11.1572
106. Neuhaus O, Kieseier BC, Hartung H-P. Mechanisms of mitoxantrone in multiple sclerosis–what is known? J Neurol Sci. (2004) 223:25–7. doi: 10.1016/j.jns.2004.04.015
107. Hartung H-P, Gonsette R, König N, Kwiecinski H, Guseo A, Morrissey SP, et al. Mitoxantrone in progressive multiple sclerosis: a placebo-controlled, double-blind, randomised, multicentre trial. Lancet. (2002) 360:2018–25. doi: 10.1016/S0140-6736(02)12023-X
108. Edan G, Miller D, Clanet M, Confavreux C, Lyon-Caen O, Lubetzki C, et al. Therapeutic effect of mitoxantrone combined with methylprednisolone in multiple sclerosis: a randomised multicentre study of active disease using MRI and clinical criteria. J Neurol Neurosurg Psychiatry. (1997) 62:112–8. doi: 10.1136/jnnp.62.2.112
109. Martinelli V, Cocco E, Capra R, Salemi G, Gallo P, Capobianco M, et al. Acute myeloid leukemia in Italian patients with multiple sclerosis treated with mitoxantrone. Neurology. (2011) 77:1887–95. doi: 10.1212/WNL.0b013e318238ee00
110. Cocco E, Marrosu MG. The current role of mitoxantrone in the treatment of multiple sclerosis. Expert Rev Neurother. (2014) 14:607–16. doi: 10.1586/14737175.2014.915742
111. Kingwell E, Koch M, Leung B, Isserow S, Geddes J, Rieckmann P, et al. Cardiotoxicity and other adverse events associated with mitoxantrone treatment for MS. Neurology. (2010) 74:1822–6. doi: 10.1212/WNL.0b013e3181e0f7e6
112. Le Page E, Leray E, Edan G, French Mitoxantrone Safety Group. Long-term safety profile of mitoxantrone in a French cohort of 802 multiple sclerosis patients: a 5-year prospective study. Mult Scler.(2011) 17:867–75. doi: 10.1177/1352458511398371
113. Cocco E, Sardu C, Gallo P, Capra R, Amato MP, Trojano M, et al. Frequency and risk factors of mitoxantrone-induced amenorrhea in multiple sclerosis: the FEMIMS study. Mult Scler.(2008) 14:1225–33. doi: 10.1177/1352458508094882
114. Wingerchuk DM, Weinshenker BG. Disease modifying therapies for relapsing multiple sclerosis. BMJ. (2016) 354:i3518. doi: 10.1136/bmj.i3518
115. Ammatuna E, Montesinos P, Hasan SK, Ramadan SM, Esteve J, Hubmann M, et al. Presenting features and treatment outcome of acute promyelocytic leukemia arising after multiple sclerosis. Haematologica. (2011) 96:621–5. doi: 10.3324/haematol.2010.036657
116. Chan A, Lo-Coco F. Mitoxantrone-related acute leukemia in MS: an open or closed book? Neurology. (2013) 80:1529–33. doi: 10.1212/WNL.0b013e31828cf891
117. Soelberg Sorensen P. Safety concerns and risk management of multiple sclerosis therapies. Acta Neurol Scand. (2017) 136:168–86. doi: 10.1111/ane.12712
118. Fleischer V, Salmen A, Kollar S, Weyer V, Siffrin V, Chan A, et al. Cardiotoxicity of mitoxantrone treatment in a german cohort of 639 multiple sclerosis patients. J Clin Neurol Seoul Korea. (2014) 10:289–95. doi: 10.3988/jcn.2014.10.4.289
119. Ellis R, Brown S, Boggild M. Therapy-related acute leukaemia with mitoxantrone: four years on, what is the risk and can it be limited? Mult Scler. (2015) 21:642–5. doi: 10.1177/1352458514541508
120. Aly L, Hemmer B, Korn T. From leflunomide to teriflunomide: drug development and immunosuppressive oral drugs in the treatment of multiple sclerosis. Curr Neuropharmacol. (2017) 15:874–91. doi: 10.2174/1570159X14666161208151525
121. Miller AE. Oral teriflunomide in the treatment of relapsing forms of multiple sclerosis: clinical evidence and long-term experience. Ther Adv Neurol Disord. (2017) 10:381–96. doi: 10.1177/1756285617722500
122. Ochoa-Repáraz J, Colpitts SL, Kircher C, Kasper EJ, Telesford KM, Begum-Haque S, et al. Induction of gut regulatory CD39+ T cells by teriflunomide protects against EAE. Neurol Neuroimmunol Neuroinflamm. (2016) 3:e291. doi: 10.1212/NXI.0000000000000291
123. Wostradowski T, Prajeeth CK, Gudi V, Kronenberg J, Witte S, Brieskorn M, et al. In vitro evaluation of physiologically relevant concentrations of teriflunomide on activation and proliferation of primary rodent microglia. J Neuroinflamm. (2016) 13:250. doi: 10.1186/s12974-016-0715-3
124. Manna SK, Aggarwal BB. Immunosuppressive leflunomide metabolite (A77 1726) blocks TNF-dependent nuclear factor-kappa B activation and gene expression. J Immunol Baltim. (1999) 162:2095–102.
125. González-Alvaro I, Ortiz AM, Domínguez-Jiménez C, Aragón-Bodi A, Díaz Sánchez B, Sánchez-Madrid F. Inhibition of tumour necrosis factor and IL-17 production by leflunomide involves the JAK/STAT pathway. Ann Rheum Dis. (2009) 68:1644–50. doi: 10.1136/ard.2008.096743
126. Bilger A, Plowshay J, Ma S, Nawandar D, Barlow EA, Romero-Masters JC, et al. Leflunomide/teriflunomide inhibit Epstein-Barr virus (EBV)- induced lymphoproliferative disease and lytic viral replication. Oncotarget. (2017) 8:44266–80. doi: 10.18632/oncotarget.17863
127. Modica CM, Schweser F, Sudyn ML, Bertolino N, Preda M, Polak P, et al. Effect of teriflunomide on cortex-basal ganglia-thalamus (CxBGTh) circuit glutamatergic dysregulation in the Theiler's Murine Encephalomyelitis Virus mouse model of multiple sclerosis. PLoS ONE. (2017) 12:e0182729. doi: 10.1371/journal.pone.0182729
128. Göttle P, Manousi A, Kremer D, Reiche L, Hartung H-P, Küry P. Teriflunomide promotes oligodendroglial differentiation and myelination. J Neuroinflamm. (2018) 15:76. doi: 10.1186/s12974-018-1110-z
129. Groh J, Hörner M, Martini R. Teriflunomide attenuates neuroinflammation-related neural damage in mice carrying human PLP1 mutations. J Neuroinflamm. (2018) 15:194. doi: 10.1186/s12974-018-1228-z
130. Bar-Or A, Freedman MS, Kremenchutzky M, Menguy-Vacheron F, Bauer D, Jodl S, et al. Teriflunomide effect on immune response to influenza vaccine in patients with multiple sclerosis. Neurology. (2013) 81:552–8. doi: 10.1212/WNL.0b013e31829e6fbf
131. Freedman MS. Teriflunomide in relapsing multiple sclerosis: therapeutic utility. Ther Adv Chronic Dis. (2013) 4:192–205. doi: 10.1177/2040622313492810
132. O'Connor PW, Li D, Freedman MS, Bar-Or A, Rice GPA, Confavreux C, et al. A Phase II study of the safety and efficacy of teriflunomide in multiple sclerosis with relapses. Neurology. (2006) 66:894–900. doi: 10.1212/01.wnl.0000203121.04509.31
133. O'Connor P, Wolinsky JS, Confavreux C, Comi G, Kappos L, Olsson TP, et al. Randomized trial of oral teriflunomide for relapsing multiple sclerosis. N Engl J Med. (2011) 365:1293–303. doi: 10.1056/NEJMoa1014656
134. Confavreux C, O'Connor P, Comi G, Freedman MS, Miller AE, Olsson TP, et al. Oral teriflunomide for patients with relapsing multiple sclerosis (TOWER): a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet Neurol. (2014) 13:247–56. doi: 10.1016/S1474-4422(13)70308-9
135. Vermersch P, Czlonkowska A, Grimaldi LME, Confavreux C, Comi G, Kappos L, et al. Teriflunomide versus subcutaneous interferon beta-1a in patients with relapsing multiple sclerosis: a randomised, controlled phase 3 trial. Mult Scler. (2014) 20:705–16. doi: 10.1177/1352458513507821
136. Miller AE, Wolinsky JS, Kappos L, Comi G, Freedman MS, Olsson TP, et al. Oral teriflunomide for patients with a first clinical episode suggestive of multiple sclerosis (TOPIC): a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet Neurol. (2014) 13:977–86. doi: 10.1016/S1474-4422(14)70191-7
137. Elkjaer ML, Molnar T, Illes Z. Teriflunomide for multiple sclerosis in real-world setting. Acta Neurol Scand. (2017) 136:447–53. doi: 10.1111/ane.12755
138. Freedman MS, Montalban X, Miller AE, Dive-Pouletty C, Hass S, Thangavelu K, et al. Comparing outcomes from clinical studies of oral disease-modifying therapies (dimethyl fumarate, fingolimod, and teriflunomide) in relapsing MS: assessing absolute differences using a number needed to treat analysis. Mult Scler Relat Disord. (2016) 10:204–12. doi: 10.1016/j.msard.2016.10.010
139. Buron MD, Chalmer TA, Sellebjerg F, Frederiksen J, Góra MK, Illes Z, et al. Comparative effectiveness of teriflunomide and dimethyl fumarate: a nationwide cohort study. Neurology. (2019) 92:e1811–20. doi: 10.1212/WNL.0000000000007314
140. Kalincik T, Kubala Havrdova E, Horakova D, Izquierdo G, Prat A, Girard M, et al. Comparison of fingolimod, dimethyl fumarate and teriflunomide for multiple sclerosis. J Neurol Neurosurg Psychiatry. (2019) 90:458–68. doi: 10.1136/jnnp-2018-319831
141. D'Amico E, Zanghì A, Sciandra M, Borriello G, Callari G, Gallo A, et al. Discontinuation of teriflunomide and dimethyl fumarate in a large Italian multicentre population: a 24-month real-world experience. J Neurol. (2019) 266:411–6. doi: 10.1007/s00415-018-9144-9
142. Rommer PS, Zettl UK, Kieseier B, Hartung H-P, Menge T, Frohman E, et al. Requirement for safety monitoring for approved multiple sclerosis therapies: an overview. Clin Exp Immunol. (2014) 175:397–407. doi: 10.1111/cei.12206
143. Comi G, Freedman MS, Kappos L, Olsson TP, Miller AE, Wolinsky JS, et al. Pooled safety and tolerability data from four placebo-controlled teriflunomide studies and extensions. Mult Scler Relat Disord. (2016) 5:97–104. doi: 10.1016/j.msard.2015.11.006
144. O'Connor P, Comi G, Freedman MS, Miller AE, Kappos L, Bouchard J-P, et al. Long-term safety and efficacy of teriflunomide: nine-year follow-up of the randomized TEMSO study. Neurology. (2016) 86:920–30. doi: 10.1212/WNL.0000000000002441
145. Confavreux C, Li DK, Freedman MS, Truffinet P, Benzerdjeb H, Wang D, et al. Long-term follow-up of a phase 2 study of oral teriflunomide in relapsing multiple sclerosis: safety and efficacy results up to 8.5 years. Mult Scler.(2012) 18:1278–89. doi: 10.1177/1352458512436594
146. Lorefice L, Fenu G, Gerevini S, Frau J, Coghe G, Barracciu MA, et al. PML in a person with multiple sclerosis: is teriflunomide the felon? Neurology. (2018) 90:83–5. doi: 10.1212/WNL.0000000000004804
147. Kieseier BC, Benamor M. Pregnancy outcomes following maternal and paternal exposure to teriflunomide during treatment for relapsing-remitting multiple sclerosis. Neurol Ther. (2014) 3:133–8. doi: 10.1007/s40120-014-0020-y
149. Dubey D, Kieseier BC, Hartung HP, Hemmer B, Warnke C, Menge T, et al. Dimethyl fumarate in relapsing-remitting multiple sclerosis: rationale, mechanisms of action, pharmacokinetics, efficacy and safety. Expert Rev Neurother. (2015) 15:339–46. doi: 10.1586/14737175.2015.1025755
150. Clausen BH, Lundberg L, Yli-Karjanmaa M, Martin NA, Svensson M, Alfsen MZ, et al. Fumarate decreases edema volume and improves functional outcome after experimental stroke. Exp Neurol. (2017) 295:144–54. doi: 10.1016/j.expneurol.2017.06.011
151. Hammer A, Waschbisch A, Kuhbandner K, Bayas A, Lee D-H, Duscha A, et al. The NRF2 pathway as potential biomarker for dimethyl fumarate treatment in multiple sclerosis. Ann Clin Transl Neurol. (2018) 5:668–76. doi: 10.1002/acn3.553
152. Mills EA, Ogrodnik MA, Plave A, Mao-Draayer Y. Emerging understanding of the mechanism of action for dimethyl fumarate in the treatment of multiple sclerosis. Front Neurol. (2018) 9:5. doi: 10.3389/fneur.2018.00005
153. Holm Hansen R, Højsgaard Chow H, Sellebjerg F, Rode von Essen M. Dimethyl fumarate therapy suppresses B cell responses and follicular helper T cells in relapsing-remitting multiple sclerosis. Mult Scler. (2018). doi: 10.1177/1352458518790417. [Epub ahead of print].
154. Smith MD, Martin KA, Calabresi PA, Bhargava P. Dimethyl fumarate alters B-cell memory and cytokine production in MS patients. Ann Clin Transl Neurol. (2017) 4:351–5. doi: 10.1002/acn3.411
155. Li R, Rezk A, Ghadiri M, Luessi F, Zipp F, Li H, et al. Dimethyl fumarate treatment mediates an anti-inflammatory shift in B cell subsets of patients with multiple sclerosis. J Immunol Baltim. (2017) 198:691–8. doi: 10.4049/jimmunol.1601649
156. Ghadiri M, Rezk A, Li R, Evans A, Luessi F, Zipp F, et al. Dimethyl fumarate-induced lymphopenia in MS due to differential T-cell subset apoptosis. Neurol Neuroimmunol Neuroinflamm. (2017) 4:e340. doi: 10.1212/NXI.0000000000000340
157. Wu Q, Wang Q, Mao G, Dowling CA, Lundy SK, Mao-Draayer Y. Dimethyl fumarate selectively reduces memory T cells and shifts the balance between Th1/Th17 and Th2 in multiple sclerosis patients. J Immunol Baltim. (2017) 198:3069–80. doi: 10.4049/jimmunol.1601532
158. Medina S, Villarrubia N, Sainz de la Maza S, Lifante J, Costa-Frossard L, Roldán E, et al. Optimal response to dimethyl fumarate associates in MS with a shift from an inflammatory to a tolerogenic blood cell profile. Mult Scler. (2018) 24:1317–27. doi: 10.1177/1352458517717088
159. Diebold M, Sievers C, Bantug G, Sanderson N, Kappos L, Kuhle J, et al. Dimethyl fumarate influences innate and adaptive immunity in multiple sclerosis. J Autoimmun. (2018) 86:39–50. doi: 10.1016/j.jaut.2017.09.009
160. Smith MD, Calabresi PA, Bhargava P. Dimethyl fumarate treatment alters NK cell function in multiple sclerosis. Eur J Immunol. (2018) 48:380–3. doi: 10.1002/eji.201747277
161. Longbrake EE, Cantoni C, Chahin S, Cignarella F, Cross AH, Piccio L. Dimethyl fumarate induces changes in B- and T-lymphocyte function independent of the effects on absolute lymphocyte count. Mult Scler. (2018) 24:728–38. doi: 10.1177/1352458517707069
162. Cunill V, Massot M, Clemente A, Calles C, Andreu V, Núñez V, et al. Relapsing-remitting multiple sclerosis is characterized by a T follicular cell pro-inflammatory shift, reverted by dimethyl fumarate treatment. Front Immunol. (2018) 9:1097. doi: 10.3389/fimmu.2018.01097
163. Galloway DA, Williams JB, Moore CS. Effects of fumarates on inflammatory human astrocyte responses and oligodendrocyte differentiation. Ann Clin Transl Neurol. (2017) 4:381–91. doi: 10.1002/acn3.414
164. Brennan MS, Matos MF, Richter KE, Li B, Scannevin RH. The NRF2 transcriptional target, OSGIN1, contributes to monomethyl fumarate-mediated cytoprotection in human astrocytes. Sci Rep. (2017) 7:42054. doi: 10.1038/srep42054
165. Paraiso HC, Kuo P-C, Curfman ET, Moon HJ, Sweazey RD, Yen J-H, et al. Dimethyl fumarate attenuates reactive microglia and long-term memory deficits following systemic immune challenge. J Neuroinflamm. (2018) 15:100. doi: 10.1186/s12974-018-1125-5
166. Gold R, Kappos L, Arnold DL, Bar-Or A, Giovannoni G, Selmaj K, et al. Placebo-controlled phase 3 study of oral BG-12 for relapsing multiple sclerosis. N Engl J Med. (2012) 367:1098–107. doi: 10.1056/NEJMoa1114287
167. Fox RJ, Miller DH, Phillips JT, Hutchinson M, Havrdova E, Kita M, et al. Placebo-controlled phase 3 study of oral BG-12 or glatiramer in multiple sclerosis. N Engl J Med. (2012) 367:1087–97. doi: 10.1056/NEJMoa1206328
168. Viglietta V, Miller D, Bar-Or A, Phillips JT, Arnold DL, Selmaj K, et al. Efficacy of delayed-release dimethyl fumarate in relapsing-remitting multiple sclerosis: integrated analysis of the phase 3 trials. Ann Clin Transl Neurol. (2015) 2:103–18. doi: 10.1002/acn3.148
169. Havrdova E, Giovannoni G, Gold R, Fox RJ, Kappos L, Phillips JT, et al. Effect of delayed-release dimethyl fumarate on no evidence of disease activity in relapsing-remitting multiple sclerosis: integrated analysis of the phase III DEFINE and CONFIRM studies. Eur J Neurol. (2017) 24:726–33. doi: 10.1111/ene.13272
170. Fernández Ó, Giovannoni G, Fox RJ, Gold R, Phillips JT, Potts J, et al. Efficacy and safety of delayed-release dimethyl fumarate for relapsing-remitting multiple sclerosis in prior interferon users: an integrated analysis of DEFINE and CONFIRM. Clin Ther. (2017) 39:1671–9. doi: 10.1016/j.clinthera.2017.06.012
171. Guarnera C, Bramanti P, Mazzon E. Comparison of efficacy and safety of oral agents for the treatment of relapsing–remitting multiple sclerosis. Drug Des Dev Ther. (2017) 11:2193–207. doi: 10.2147/DDDT.S137572
172. Mills EA, Mao-Draayer Y. Understanding progressive multifocal leukoencephalopathy risk in multiple sclerosis patients treated with immunomodulatory therapies: a bird's eye view. Front Immunol. (2018) 9:138. doi: 10.3389/fimmu.2018.00138
173. Gieselbach R-J, Muller-Hansma AH, Wijburg MT, de Bruin-Weller MS, van Oosten BW, Nieuwkamp DJ, et al. Progressive multifocal leukoencephalopathy in patients treated with fumaric acid esters: a review of 19 cases. J Neurol. (2017) 264:1155–64. doi: 10.1007/s00415-017-8509-9
174. Khatri BO, Tarima SS, Essig B, Sesing J, Olapo T. Delayed lymphocyte re-population following discontinuation of dimethyl fumarate and after switching to other disease modifying drug therapies. Mult Scler Relat Disord. (2017) 18:60–4. doi: 10.1016/j.msard.2017.09.014
175. Mirabella M, Prosperini L, Lucchini M, Boffa L, Borriello G, Buscarinu MC, et al. Safety and efficacy of dimethyl fumarate in multiple sclerosis: an Italian, multicenter, real-world study. CNS Drugs. (2018) 32:963–70. doi: 10.1007/s40263-018-0543-3
176. Mallucci G, Annovazzi P, Miante S, Torri-Clerici V, Matta M, La Gioia S, et al. Two-year real-life efficacy, tolerability and safety of dimethyl fumarate in an Italian multicentre study. J Neurol. (2018) 265:1850–9. doi: 10.1007/s00415-018-8916-6
177. Miclea A, Leussink VI, Hartung HP, Gold R, Hoepner R. Safety and efficacy of dimethyl fumarate in multiple sclerosis: a multi-center observational study. J Neurol. (2016) 263:1626–32. doi: 10.1007/s00415-016-8175-3
178. Baharnoori M, Gonzalez CT, Chua A, Diaz-Cruz C, Healy BC, Stankiewicz J, et al. Predictors of hematological abnormalities in multiple sclerosis patients treated with fingolimod and dimethyl fumarate and impact of treatment switch on lymphocyte and leukocyte count. Mult Scler Relat Disord. (2018) 20:51–7. doi: 10.1016/j.msard.2017.12.003
179. Eriksson I, Cars T, Piehl F, Malmström RE, Wettermark B, von Euler M. Persistence with dimethyl fumarate in relapsing-remitting multiple sclerosis: a population-based cohort study. Eur J Clin Pharmacol. (2018) 74:219–26. doi: 10.1007/s00228-017-2366-4
180. Fox RJ, Chan A, Zhang A, Xiao J, Levison D, Lewin JB, et al. Comparative effectiveness using a matching-adjusted indirect comparison between delayed-release dimethyl fumarate and fingolimod for the treatment of multiple sclerosis. Curr Med Res Opin. (2017) 33:175–83. doi: 10.1080/03007995.2016.1248380
181. Prosperini L, Lucchini M, Haggiag S, Bellantonio P, Bianco A, Buscarinu MC, et al. Fingolimod vs dimethyl fumarate in multiple sclerosis: a real-world propensity score-matched study. Neurology. (2018) 91:e153–61. doi: 10.1212/WNL.0000000000005772
182. Nixon R, Bergvall N, Tomic D, Sfikas N, Cutter G, Giovannoni G. No evidence of disease activity: indirect comparisons of oral therapies for the treatment of relapsing-remitting multiple sclerosis. Adv Ther. (2014) 31:1134–54. doi: 10.1007/s12325-014-0167-z
183. Cohan SL, Moses H, Calkwood J, Tornatore C, LaGanke C, Smoot KE, et al. Clinical outcomes in patients with relapsing-remitting multiple sclerosis who switch from natalizumab to delayed-release dimethyl fumarate: a multicenter retrospective observational study (STRATEGY). Mult Scler Relat Disord. (2018) 22:27–34. doi: 10.1016/j.msard.2018.02.028
184. Calabrese M, Pitteri M, Farina G, Bajrami A, Castellaro M, Magliozzi R, et al. Dimethyl fumarate: a possible exit strategy from natalizumab treatment in patients with multiple sclerosis at risk for severe adverse events. J Neurol Neurosurg Psychiatry. (2017) 88:1073–8. doi: 10.1136/jnnp-2017-316236
185. O'Gorman J, Russell HK, Li J, Phillips G, Kurukulasuriya NC, Viglietta V. Effect of aspirin pretreatment or slow dose titration on flushing and gastrointestinal events in healthy volunteers receiving delayed-release dimethyl fumarate. Clin Ther. (2015) 37:1402–1419.e5. doi: 10.1016/j.clinthera.2015.03.028
186. Sejbaek T, Nybo M, Petersen T, Illes Z. Real-life persistence and tolerability with dimethyl fumarate. Mult Scler Relat Disord. (2018) 24:42–6. doi: 10.1016/j.msard.2018.05.007
187. Ceronie B, Jacobs BM, Baker D, Dubuisson N, Mao Z, Ammoscato F, et al. Cladribine treatment of multiple sclerosis is associated with depletion of memory B cells. J Neurol. (2018) 265:1199–209. doi: 10.1007/s00415-018-8830-y
188. Warnke C, Leussink VI, Goebels N, Aktas O, Boyko A, Kieseier BC, et al. Cladribine as a therapeutic option in multiple sclerosis. Clin Immunol. (2012) 142:68–75. doi: 10.1016/j.clim.2011.05.009
189. Warnke C, Wiendl H, Hartung H-P, Stüve O, Kieseier BC. Identification of targets and new developments in the treatment of multiple sclerosis–focus on cladribine. Drug Des Devel Ther. (2010) 4:117–26. doi: 10.2147/DDDT.S6627
190. Giovannoni G, Comi G, Cook S, Rammohan K, Rieckmann P, Soelberg Sørensen P, et al. A placebo-controlled trial of oral cladribine for relapsing multiple sclerosis. N Engl J Med. (2010) 362:416–26. doi: 10.1056/NEJMoa0902533
191. Leist TP, Comi G, Cree BAC, Coyle PK, Freedman MS, Hartung H-P, et al. Effect of oral cladribine on time to conversion to clinically definite multiple sclerosis in patients with a first demyelinating event (ORACLE MS): a phase 3 randomised trial. Lancet Neurol. (2014) 13:257–67. doi: 10.1016/S1474-4422(14)70005-5
192. Freedman MS, Leist TP, Comi G, Cree BA, Coyle PK, Hartung H-P, et al. The efficacy of cladribine tablets in CIS patients retrospectively assigned the diagnosis of MS using modern criteria: results from the ORACLE-MS study. Mult Scler J. (2017) 3:2055217317732802. doi: 10.1177/2055217317732802
193. Comi G, Cook S, Rammohan K, Soelberg Sorensen P, Vermersch P, Adeniji AK, et al. Long-term effects of cladribine tablets on MRI activity outcomes in patients with relapsing-remitting multiple sclerosis: the CLARITY Extension study. Ther Adv Neurol Disord. (2018) 11:175628561775(3365). doi: 10.1177/1756285617753365
194. Giovannoni G, Soelberg Sorensen P, Cook S, Rammohan K, Rieckmann P, Comi G, et al. Safety and efficacy of cladribine tablets in patients with relapsing-remitting multiple sclerosis: results from the randomized extension trial of the CLARITY study. Mult Scler. (2017) 24:1594–604. doi: 10.1177/1352458517727603
195. Baker D, Herrod SS, Alvarez-Gonzalez C, Zalewski L, Albor C, Schmierer K. Both cladribine and alemtuzumab may effect MS via B-cell depletion. Neurol Neuroimmunol Neuroinflamm. (2017) 4:e360. doi: 10.1212/NXI.0000000000000360
196. Ruck T, Bittner S, Wiendl H, Meuth SG. Alemtuzumab in multiple sclerosis: mechanism of action and beyond. Int J Mol Sci. (2015) 16:16414–39. doi: 10.3390/ijms160716414
197. Hale G. The CD52 antigen and development of the CAMPATH antibodies. Cytotherapy. (2001) 3:137–43. doi: 10.1080/146532401753174098
198. Ginaldi L, De Martinis M, Matutes E, Farahat N, Morilla R, Dyer MJ, et al. Levels of expression of CD52 in normal and leukemic B and T cells: correlation with in vivo therapeutic responses to Campath-1H. Leuk Res. (1998) 22:185–91. doi: 10.1016/S0145-2126(97)00158-6
199. Rao SP, Sancho J, Campos-Rivera J, Boutin PM, Severy PB, Weeden T, et al. Human peripheral blood mononuclear cells exhibit heterogeneous CD52 expression levels and show differential sensitivity to alemtuzumab mediated cytolysis. PLoS ONE. (2012) 7:e39416. doi: 10.1371/journal.pone.0039416
200. Coles AJ, Cox A, Le Page E, Jones J, Trip SA, Deans J, et al. The window of therapeutic opportunity in multiple sclerosis: evidence from monoclonal antibody therapy. J Neurol. (2006) 253:98–108. doi: 10.1007/s00415-005-0934-5
201. Thompson SAJ, Jones JL, Cox AL, Compston DAS, Coles AJ. B-cell reconstitution and BAFF after alemtuzumab (Campath-1H) treatment of multiple sclerosis. J Clin Immunol. (2010) 30:99–105. doi: 10.1007/s10875-009-9327-3
202. Baker D, Herrod SS, Alvarez-Gonzalez C, Giovannoni G, Schmierer K. Interpreting lymphocyte reconstitution data from the pivotal phase 3 trials of alemtuzumab. JAMA Neurol. (2017) 74:961–9. doi: 10.1001/jamaneurol.2017.0676
203. Gross CC, Ahmetspahic D, Ruck T, Schulte-Mecklenbeck A, Schwarte K, Jörgens S, et al. Alemtuzumab treatment alters circulating innate immune cells in multiple sclerosis. Neurol Neuroimmunol Neuroinflamm. (2016) 3:e289. doi: 10.1212/NXI.0000000000000289
204. Gross CC, Schulte-Mecklenbeck A, Wiendl H, Marcenaro E, Kerlero de Rosbo N, Uccelli A, et al. Regulatory functions of natural killer cells in multiple sclerosis. Front Immunol. (2016) 7:606. doi: 10.3389/fimmu.2016.00606
205. CAMMS223 Trial Investigators, Coles AJ, Compston DAS, Selmaj KW, Lake SL, Moran S, et al. Alemtuzumab vs. interferon beta-1a in early multiple sclerosis. N Engl J Med. (2008) 359:1786–801. doi: 10.1056/NEJMoa0802670
206. Cohen JA, Coles AJ, Arnold DL, Confavreux C, Fox EJ, Hartung H-P, et al. Alemtuzumab versus interferon beta 1a as first-line treatment for patients with relapsing-remitting multiple sclerosis: a randomised controlled phase 3 trial. Lancet. (2012) 380:1819–28. doi: 10.1016/S0140-6736(12)61769-3
207. Coles AJ, Twyman CL, Arnold DL, Cohen JA, Confavreux C, Fox EJ, et al. Alemtuzumab for patients with relapsing multiple sclerosis after disease-modifying therapy: a randomised controlled phase 3 trial. Lancet. (2012) 380:1829–39. doi: 10.1016/S0140-6736(12)61768-1
208. FDA Drug Safety Communication. FDA Warns About Rare But Serious Risks of Stroke and Blood Vessel Wall Tears With Multiple Sclerosis Drug Lemtrada (alemtuzumab). FDA [Internet]. (2019). Available online at: http://www.fda.gov/drugs/drug-safety-and-availability/fda-warns-about-rare-serious-risks-stroke-and-blood-vessel-wall-tears-multiple-sclerosis-drug (accessed June 2, 2019).
209. Azevedo CJ, Kutz C, Dix A, Boster A, Sanossian N, Kaplan J. Intracerebral haemorrhage during alemtuzumab administration. Lancet Neurol. (2019) 18:329–31. doi: 10.1016/S1474-4422(19)30076-6
210. Doessegger L, Banholzer ML. Clinical development methodology for infusion-related reactions with monoclonal antibodies. Clin Transl Immunol. (2015) 4:e39. doi: 10.1038/cti.2015.14
211. Jones JL, Thompson SAJ, Loh P, Davies JL, Tuohy OC, Curry AJ, et al. Human autoimmunity after lymphocyte depletion is caused by homeostatic T-cell proliferation. Proc Natl Acad Sci USA. (2013) 110:20200–5. doi: 10.1073/pnas.1313654110
212. Jones JL, Phuah C-L, Cox AL, Thompson SA, Ban M, Shawcross J, et al. IL-21 drives secondary autoimmunity in patients with multiple sclerosis, following therapeutic lymphocyte depletion with alemtuzumab (Campath-1H). J Clin Invest. (2009) 119:2052–61. doi: 10.1172/JCI37878
213. Daniels GH, Vladic A, Brinar V, Zavalishin I, Valente W, Oyuela P, et al. Alemtuzumab-related thyroid dysfunction in a phase 2 trial of patients with relapsing-remitting multiple sclerosis. J Clin Endocrinol Metab. (2014) 99:80–9. doi: 10.1210/jc.2013-2201
214. Meyer D, Coles A, Oyuela P, Purvis A, Margolin DH. Case report of anti-glomerular basement membrane disease following alemtuzumab treatment of relapsing-remitting multiple sclerosis. Mult Scler Relat Disord. (2013) 2:60–3. doi: 10.1016/j.msard.2012.07.002
215. Cuker A, Coles AJ, Sullivan H, Fox E, Goldberg M, Oyuela P, et al. A distinctive form of immune thrombocytopenia in a phase 2 study of alemtuzumab for the treatment of relapsing-remitting multiple sclerosis. Blood. (2011) 118:6299–305. doi: 10.1182/blood-2011-08-371138
216. Clatworthy MR, Wallin EF, Jayne DR. Anti-glomerular basement membrane disease after alemtuzumab. N Engl J Med. (2008) 359:768–9. doi: 10.1056/NEJMc0800484
217. Tuohy O, Costelloe L, Hill-Cawthorne G, Bjornson I, Harding K, Robertson N, et al. Alemtuzumab treatment of multiple sclerosis: long-term safety and efficacy. J Neurol Neurosurg Psychiatry. (2015) 86:208–15. doi: 10.1136/jnnp-2014-307721
218. Devonshire V, Phillips R, Wass H, Da Roza G, Senior P. Monitoring and management of autoimmunity in multiple sclerosis patients treated with alemtuzumab: practical recommendations. J Neurol. (2018) 265:2494-2505. doi: 10.1007/s00415-018-8822-y
219. Decallonne B, Bartholomé E, Delvaux V, D'haeseleer M, El Sankari S, Seeldrayers P, et al. Thyroid disorders in alemtuzumab-treated multiple sclerosis patients: a Belgian consensus on diagnosis and management. Acta Neurol Belg. (2018) 118:153–9. doi: 10.1007/s13760-018-0883-2
220. Lambert C, Dubois B, Dive D, Lysandropoulos A, Selleslag D, Vanopdenbosch L, et al. Management of immune thrombocytopenia in multiple sclerosis patients treated with alemtuzumab: a Belgian consensus. Acta Neurol Belg. (2018) 118:7–11. doi: 10.1007/s13760-018-0882-3
221. Sprangers B, Decoo D, Dive D, Lysandropoulos A, Vanopdenbosch L, Bovy C. Management of adverse renal events related to alemtuzumab treatment in multiple sclerosis: a Belgian consensus. Acta Neurol Belg. (2018) 118:143–51. doi: 10.1007/s13760-017-0864-x
222. FRANCISCO EM. Lemtrada [Internet]. European Medicines Agency. (2019). Available online at: https://www.ema.europa.eu/en/medicines/human/referrals/lemtrada (cited June 2, 2019).
223. Duddy M, Niino M, Adatia F, Hebert S, Freedman M, Atkins H, et al. Distinct effector cytokine profiles of memory and naive human B cell subsets and implication in multiple sclerosis. J Immunol. (2007) 178:6092–9. doi: 10.4049/jimmunol.178.10.6092
224. Mathias A, Perriard G, Canales M, Soneson C, Delorenzi M, Schluep M, et al. Increased ex vivo antigen presentation profile of B cells in multiple sclerosis. Mult Scler. (2017) 23:802–9. doi: 10.1177/1352458516664210
225. Aung LL, Balashov KE. Decreased Dicer expression is linked to increased expression of co-stimulatory molecule CD80 on B cells in multiple sclerosis. Mult Scler. (2015) 21:1131–8. doi: 10.1177/1352458514560923
226. Genç K, Dona DL, Reder AT. Increased CD80(+) B cells in active multiple sclerosis and reversal by interferon beta-1b therapy. J Clin Invest. (1997) 99:2664–71. doi: 10.1172/JCI119455
227. Harp CT, Ireland S, Davis LS, Remington G, Cassidy B, Cravens PD, et al. Memory B cells from a subset of treatment-naïve relapsing-remitting multiple sclerosis patients elicit CD4(+) T-cell proliferation and IFN-γ production in response to myelin basic protein and myelin oligodendrocyte glycoprotein. Eur J Immunol. (2010) 40:2942–56. doi: 10.1002/eji.201040516
228. Wekerle H. B cells in multiple sclerosis. Autoimmunity. (2017) 50:57–60. doi: 10.1080/08916934.2017.1281914
229. Häusser-Kinzel S, Weber MS. The role of B cells and antibodies in multiple sclerosis, neuromyelitis optica, and related disorders. Front Immunol. (2019) 10:201. doi: 10.3389/fimmu.2019.00201
230. Hauser SL, Waubant E, Arnold DL, Vollmer T, Antel J, Fox RJ, et al. B-cell depletion with rituximab in relapsing-remitting multiple sclerosis. N Engl J Med. (2008) 358:676–88. doi: 10.1056/NEJMoa0706383
231. Bar-Or A, Calabresi PAJ, Arnold D, Arnlod D, Markowitz C, Shafer S, et al. Rituximab in relapsing-remitting multiple sclerosis: a 72-week, open-label, phase I trial. Ann Neurol. (2008) 63:395–400. doi: 10.1002/ana.21363
232. Hawker K, O'Connor P, Freedman MS, Calabresi PA, Antel J, Simon J, et al. Rituximab in patients with primary progressive multiple sclerosis: results of a randomized double-blind placebo-controlled multicenter trial. Ann Neurol. (2009) 66:460–71. doi: 10.1002/ana.21867
233. Kappos L, Li D, Calabresi PA, O'Connor P, Bar-Or A, Barkhof F, et al. Ocrelizumab in relapsing-remitting multiple sclerosis: a phase 2, randomised, placebo-controlled, multicentre trial. Lancet. (2011) 378:1779–87. doi: 10.1016/S0140-6736(11)61649-8
234. Sorensen PS, Lisby S, Grove R, Derosier F, Shackelford S, Havrdova E, et al. Safety and efficacy of ofatumumab in relapsing-remitting multiple sclerosis: a phase 2 study. Neurology. (2014) 82:573–81. doi: 10.1212/WNL.0000000000000125
235. Häusler D, Häusser-Kinzel S, Feldmann L, Torke S, Lepennetier G, Bernard CCA, et al. Functional characterization of reappearing B cells after anti-CD20 treatment of CNS autoimmune disease. Proc Natl Acad Sci USA. (2018) 115:9773–8. doi: 10.1073/pnas.1810470115
236. Greenberg BM, Graves D, Remington G, Hardeman P, Mann M, Karandikar N, et al. Rituximab dosing and monitoring strategies in neuromyelitis optica patients: creating strategies for therapeutic success. Mult Scler. (2012) 18:1022–6. doi: 10.1177/1352458511432896
237. Genovese MC, Kaine JL, Lowenstein MB, Del Giudice J, Baldassare A, Schechtman J, et al. Ocrelizumab, a humanized anti-CD20 monoclonal antibody, in the treatment of patients with rheumatoid arthritis: a phase I/II randomized, blinded, placebo-controlled, dose-ranging study. Arthritis Rheum. (2008) 58:2652–61. doi: 10.1002/art.23732
238. Bar-Or A, Grove RA, Austin DJ, Tolson JM, VanMeter SA, Lewis EW, et al. Subcutaneous ofatumumab in patients with relapsing-remitting multiple sclerosis: the MIRROR study. Neurology. (2018) 90:e1805–14. doi: 10.1212/WNL.0000000000005516
239. Juanatey A, Blanco-Garcia L, Tellez N. Ocrelizumab: its efficacy and safety in multiple sclerosis. Rev Neurol. (2018) 66:423–33. doi: 10.33588/rn.6612.2018132
240. Reich DS, Lucchinetti CF, Calabresi PA. Multiple sclerosis. N Engl J Med. (2018) 378:169–80. doi: 10.1056/NEJMra1401483
241. Luster AD, Alon R, von Andrian UH. Immune cell migration in inflammation: present and future therapeutic targets. Nat Immunol. (2005) 6:1182–90. doi: 10.1038/ni1275
242. Stüve O, Bennett JL. Pharmacological properties, toxicology and scientific rationale for the use of natalizumab (Tysabri) in inflammatory diseases. CNS Drug Rev. (2007) 13:79–95. doi: 10.1111/j.1527-3458.2007.00003.x
243. Sheremata WA, Minagar A, Alexander JS, Vollmer T. The role of alpha-4 integrin in the aetiology of multiple sclerosis: current knowledge and therapeutic implications. CNS Drugs. (2005) 19:909–22. doi: 10.2165/00023210-200519110-00002
244. Ali R, Nicholas RSJ, Muraro PA. Drugs in development for relapsing multiple sclerosis. Drugs. (2013) 73:625–50. doi: 10.1007/s40265-013-0030-6
245. Yednock TA, Cannon C, Fritz LC, Sanchez-Madrid F, Steinman L, Karin N. Prevention of experimental autoimmune encephalomyelitis by antibodies against alpha 4 beta 1 integrin. Nature. (1992) 356:63–6. doi: 10.1038/356063a0
246. Stüve O, Marra CM, Jerome KR, Cook L, Cravens PD, Cepok S, et al. Immune surveillance in multiple sclerosis patients treated with natalizumab. Ann Neurol. (2006) 59:743–7. doi: 10.1002/ana.20858
247. Stüve O, Marra CM, Bar-Or A, Niino M, Cravens PD, Cepok S, et al. Altered CD4+/CD8+ T-cell ratios in cerebrospinal fluid of natalizumab-treated patients with multiple sclerosis. Arch Neurol. (2006) 63:1383–7. doi: 10.1001/archneur.63.10.1383
248. Stüve O, Cravens PD, Frohman EM, Phillips JT, Remington GM, von Geldern G, et al. Immunologic, clinical, and radiologic status 14 months after cessation of natalizumab therapy. Neurology. (2009) 72:396–401. doi: 10.1212/01.wnl.0000327341.89587.76
249. Polman CH, O'Connor PW, Havrdova E, Hutchinson M, Kappos L, Miller DH, et al. A randomized, placebo-controlled trial of natalizumab for relapsing multiple sclerosis. N Engl J Med. (2006) 354:899–910. doi: 10.1056/NEJMoa044397
250. Rudick RA, Stuart WH, Calabresi PA, Confavreux C, Galetta SL, Radue E-W, et al. Natalizumab plus interferon beta-1a for relapsing multiple sclerosis. N Engl J Med. (2006) 354:911–23. doi: 10.1056/NEJMoa044396
251. Kapoor R, Ho P-R, Campbell N, Chang I, Deykin A, Forrestal F, et al. Effect of natalizumab on disease progression in secondary progressive multiple sclerosis (ASCEND): a phase 3, randomised, double-blind, placebo-controlled trial with an open-label extension. Lancet Neurol. (2018) 17:405–15. doi: 10.1016/S1474-4422(18)30069-3
252. Frisch T, Elkjaer ML, Reynolds R, Michel TM, Kacprowski T, Burton M, et al. MS Atlas – A molecular map of brain lesion stages in progressive multiple sclerosis. bioRxiv [Preprint]. (2019). doi: 10.1101/584920
253. Lassmann H. Pathogenic mechanisms associated with different clinical courses of multiple sclerosis. Front Immunol. (2018) 9:3116. doi: 10.3389/fimmu.2018.03116
254. Clerico M, Artusi CA, Di Liberto A, Rolla S, Bardina V, Barbero P, et al. Long-term safety evaluation of natalizumab for the treatment of multiple sclerosis. Expert Opin Drug Saf. (2017) 16:963–72. doi: 10.1080/14740338.2017.1346082
255. Berger JR, Khalili K. The pathogenesis of progressive multifocal leukoencephalopathy. Discov Med. (2011) 12:495–503.
256. Major EO, Yousry TA, Clifford DB. Pathogenesis of progressive multifocal leukoencephalopathy and risks associated with treatments for multiple sclerosis: a decade of lessons learned. Lancet Neurol. (2018) 17:467–80. doi: 10.1016/S1474-4422(18)30040-1
257. Wenning W, Haghikia A, Laubenberger J, Clifford DB, Behrens PF, Chan A, et al. Treatment of progressive multifocal leukoencephalopathy associated with natalizumab. N Engl J Med. (2009) 361:1075–80. doi: 10.1056/NEJMoa0810257
258. Tan IL, McArthur JC, Clifford DB, Major EO, Nath A. Immune reconstitution inflammatory syndrome in natalizumab-associated PML. Neurology. (2011) 77:1061–7. doi: 10.1212/WNL.0b013e31822e55e7
259. Bloomgren G, Richman S, Hotermans C, Subramanyam M, Goelz S, Natarajan A, et al. Risk of natalizumab-associated progressive multifocal leukoencephalopathy. N Engl J Med. (2012) 366:1870–80. doi: 10.1056/NEJMoa1107829
260. Gorelik L, Lerner M, Bixler S, Crossman M, Schlain B, Simon K, et al. Anti-JC virus antibodies: implications for PML risk stratification. Ann Neurol. (2010) 68:295–303. doi: 10.1002/ana.22128
261. Berger JR, Fox RJ. Reassessing the risk of natalizumab-associated PML. J Neurovirol. (2016) 22:533–5. doi: 10.1007/s13365-016-0427-6
262. Schwab N, Schneider-Hohendorf T, Pignolet B, Breuer J, Gross CC, Göbel K, et al. Therapy with natalizumab is associated with high JCV seroconversion and rising JCV index values. Neurol Neuroimmunol Neuroinflamm. (2016) 3:e195. doi: 10.1212/NXI.0000000000000195
263. Lindå H, von Heijne A, Major EO, Ryschkewitsch C, Berg J, Olsson T, et al. Progressive multifocal leukoencephalopathy after natalizumab monotherapy. N Engl J Med. (2009) 361:1081–7. doi: 10.1056/NEJMoa0810316
264. McGuigan C, Craner M, Guadagno J, Kapoor R, Mazibrada G, Molyneux P, et al. Stratification and monitoring of natalizumab-associated progressive multifocal leukoencephalopathy risk: recommendations from an expert group. J Neurol Neurosurg Psychiatry. (2016) 87:117–25. doi: 10.1136/jnnp-2015-311100
265. Calabresi PA, Giovannoni G, Confavreux C, Galetta SL, Havrdova E, Hutchinson M, et al. The incidence and significance of anti-natalizumab antibodies: results from AFFIRM and SENTINEL. Neurology. (2007) 69:1391–403. doi: 10.1212/01.wnl.0000277457.17420.b5
266. Fox EJ, Vartanian TK, Zamvil SS. The immunogenicity of disease-modifying therapies for multiple sclerosis: clinical implications for neurologists. Neurologist. (2007) 13:355–62. doi: 10.1097/NRL.0b013e318148c08e
267. Khatri BO, Man S, Giovannoni G, Koo AP, Lee J-C, Tucky B, et al. Effect of plasma exchange in accelerating natalizumab clearance and restoring leukocyte function. Neurology. (2009) 72:402–9. doi: 10.1212/01.wnl.0000341766.59028.9d
268. Rasenack M, Derfuss T. Disease activity return after natalizumab cessation in multiple sclerosis. Expert Rev Neurother. (2016) 16:587–94. doi: 10.1586/14737175.2016.1168295
269. Havla J, Gerdes LA, Meinl I, Krumbholz M, Faber H, Weber F, et al. De-escalation from natalizumab in multiple sclerosis: recurrence of disease activity despite switching to glatiramer acetate. J Neurol. (2011) 258:1665–9. doi: 10.1007/s00415-011-5996-y
270. Clerico M, Schiavetti I, De Mercanti SF, Piazza F, Gned D, Brescia Morra V, et al. Treatment of relapsing-remitting multiple sclerosis after 24 doses of natalizumab: evidence from an Italian spontaneous, prospective, and observational study (the TY-STOP Study). JAMA Neurol. (2014) 71:954–60. doi: 10.1001/jamaneurol.2014.1200
271. Jokubaitis VG, Li V, Kalincik T, Izquierdo G, Hodgkinson S, Alroughani R, et al. Fingolimod after natalizumab and the risk of short-term relapse. Neurology. (2014) 82:1204–11. doi: 10.1212/WNL.0000000000000283
272. Rinaldi F, Seppi D, Calabrese M, Perini P, Gallo P. Switching therapy from natalizumab to fingolimod in relapsing-remitting multiple sclerosis: clinical and magnetic resonance imaging findings. Mult Scler. (2012) 18:1640–3. doi: 10.1177/1352458512464282
273. Prosperini L, Annovazzi P, Capobianco M, Capra R, Buttari F, Gasperini C, et al. Natalizumab discontinuation in patients with multiple sclerosis: profiling risk and benefits at therapeutic crossroads. Mult Scler. (2015) 21:1713–22. doi: 10.1177/1352458515570768
274. Sangalli F, Moiola L, Ferrè L, Radaelli M, Barcella V, Rodegher M, et al. Long-term management of natalizumab discontinuation in a large monocentric cohort of multiple sclerosis patients. Mult Scler Relat Disord. (2014) 3:520–6. doi: 10.1016/j.msard.2014.04.003
275. Capobianco M, di Sapio A, Malentacchi M, Malucchi S, Matta M, Sperli F, et al. No impact of current therapeutic strategies on disease reactivation after natalizumab discontinuation: a comparative analysis of different approaches during the first year of natalizumab discontinuation. Eur J Neurol. (2015) 22:585–7. doi: 10.1111/ene.12487
276. Kappos L, Radue E-W, Comi G, Montalban X, Butzkueven H, Wiendl H, et al. Switching from natalizumab to fingolimod: a randomized, placebo-controlled study in RRMS. Neurology. (2015) 85:29–39. doi: 10.1212/WNL.0000000000001706
277. Cohen M, Maillart E, Tourbah A, De Sèze J, Vukusic S, Brassat D, et al. Switching from natalizumab to fingolimod in multiple sclerosis: a French prospective study. JAMA Neurol. (2014) 71:436–41. doi: 10.1001/jamaneurol.2013.6240
278. Weinstock-Guttman B, Hagemeier J, Kavak KS, Saini V, Patrick K, Ramasamy DP, et al. Randomised natalizumab discontinuation study: taper protocol may prevent disease reactivation. J Neurol Neurosurg Psychiatry. (2016) 87:937–43. doi: 10.1136/jnnp-2015-312221
279. Li R, Patterson KR, Bar-Or A. Reassessing B cell contributions in multiple sclerosis. Nat Immunol. (2018) 19:696–707. doi: 10.1038/s41590-018-0135-x
280. Collins F, Kazmi M, Muraro PA. Progress and prospects for the use and the understanding of the mode of action of autologous hematopoietic stem cell transplantation in the treatment of multiple sclerosis. Expert Rev Clin Immunol. (2017) 13:611–22. doi: 10.1080/1744666X.2017.1297232
281. Lublin FD, Reingold SC, Cohen JA, Cutter GR, Sørensen PS, Thompson AJ, et al. Defining the clinical course of multiple sclerosis: the 2013 revisions. Neurology. (2014) 83:278–86. doi: 10.1212/WNL.0000000000000560
282. Sellner J, Rommer PS. A review of the evidence for a natalizumab exit strategy for patients with multiple sclerosis. Autoimmun Rev. (2019) 18:255–61. doi: 10.1016/j.autrev.2018.09.012
Keywords: multiple sclerosis, immunotherapeutics, immunomodulation, immunosuppression, monoclonal antibodies
Citation: Rommer PS, Milo R, Han MH, Satyanarayan S, Sellner J, Hauer L, Illes Z, Warnke C, Laurent S, Weber MS, Zhang Y and Stuve O (2019) Immunological Aspects of Approved MS Therapeutics. Front. Immunol. 10:1564. doi: 10.3389/fimmu.2019.01564
Received: 29 September 2018; Accepted: 24 June 2019;
Published: 11 July 2019.
Edited by:
Björn Tackenberg, University of Marburg, GermanyReviewed by:
Michael Levy, Massachusetts General Hospital and Harvard Medical School, United StatesJeannette Lechner-Scott, Hunter New England Health, Australia
Copyright © 2019 Rommer, Milo, Han, Satyanarayan, Sellner, Hauer, Illes, Warnke, Laurent, Weber, Zhang and Stuve. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Paulus S. Rommer, cGF1bHVzLnJvbW1lckBtZWR1bml3aWVuLmFjLmF0; Olaf Stuve, b2xhZi5zdHV2ZUB1dHNvdXRod2VzdGVybi5lZHU=