
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Immunol. , 28 June 2019
Sec. Inflammation
Volume 10 - 2019 | https://doi.org/10.3389/fimmu.2019.01458
 Juliana P. Vago1,2
Juliana P. Vago1,2 Michelle A. Sugimoto2,3
Michelle A. Sugimoto2,3 Kátia M. Lima4
Kátia M. Lima4 Graziele L. Negreiros-Lima4
Graziele L. Negreiros-Lima4 Nagyung Baik1
Nagyung Baik1 Mauro M. Teixeira2
Mauro M. Teixeira2 Mauro Perretti3
Mauro Perretti3 Robert J. Parmer5
Robert J. Parmer5 Lindsey A. Miles1*†
Lindsey A. Miles1*† Lirlândia P. Sousa1,2,4*†
Lirlândia P. Sousa1,2,4*†Inflammation resolution is an active process that functions to restore tissue homeostasis. Clearance of apoptotic leukocytes by efferocytosis at inflammatory sites plays an important role in inflammation resolution and induces remarkable macrophage phenotypic and functional changes. Here, we investigated the effects of deletion of either plasminogen (Plg) or the Plg receptor, Plg-RKT, on the resolution of inflammation. In a murine model of pleurisy, the numbers of total mononuclear cells recruited to the pleural cavity were significantly decreased in both Plg−/− and Plg-RKT−/− mice, a response associated with decreased levels of the chemokine CCL2 in pleural exudates. Increased percentages of M1-like macrophages were determined in pleural lavages of Plg−/− and Plg-RKT−/− mice without significant changes in M2-like macrophage percentages. In vitro, Plg and plasmin (Pla) increased CD206/Arginase-1 expression and the levels of IL-10/TGF-β (M2 markers) while decreasing IFN/LPS-induced M1 markers in murine bone-marrow-derived macrophages (BMDMs) and human macrophages. Furthermore, IL4-induced M2-like polarization was defective in BMDMs from both Plg−/− and Plg-RKT−/− mice. Mechanistically, Plg and Pla induced transient STAT3 phosphorylation, which was decreased in Plg−/− and Plg-RKT−/− BMDMs after IL-4 or IL-10 stimulation. The extents of expression of CD206 and Annexin A1 (important for clearance of apoptotic cells) were reduced in Plg−/− and Plg-RKT−/− macrophage populations, which exhibited decreased phagocytosis of apoptotic neutrophils (efferocytosis) in vivo and in vitro. Taken together, these results suggest that Plg and its receptor, Plg-RKT, regulate macrophage polarization and efferocytosis, as key contributors to the resolution of inflammation.
Inflammation is the physiological response of the host to infectious or sterile injurious stimuli that ensures rapid and successful restoration of the tissue, and requires production of mediators and activation of signaling pathways (1). Anti-inflammatory/proresolving mechanisms are driven by a complex set of mediators that regulate cellular events required to clear inflammatory cells from sites of injury as a prerequisite for restoration of homeostasis (1–4). Phagocytic clearance of apoptotic cells (efferocytosis) at sites of inflammation also plays an important role in the resolution of inflammation by inducing remarkable macrophage phenotypic plasticity (5). Classically activated macrophages, M1 macrophages, are pro-inflammatory and enable host defense against infection, while M2 macrophages (alternatively activated macrophages) display anti-inflammatory and tissue remodeling properties, and play a key role in the resolution of inflammation (6, 7).
Plasminogen (Plg) is a zymogen (synthesized in liver) that subsequently is activated by plasminogen activators to generate plasmin (Pla), the major enzyme responsible for fibrin clot degradation in vivo (8, 9). In addition to fibrinolysis, the Plg/Pla system regulates the recruitment of mononuclear cells during the inflammatory response (9–12) and in non-phlogistic settings (13). The Plg receptor, Plg-RKT, is a unique transmembrane receptor with a C-terminal lysine exposed on the cell surface that interacts with Plg to promote Plg activation (14–16). Plg-RKT accounts for the majority of the Plg binding capacity of macrophages (17) and regulates macrophage recruitment in a model of sterile peritonitis (16).
While the functional role of the Plg/Pla system in the pro-inflammatory phase is well-established (18, 19) its involvement in the resolution of inflammation is an emerging area (10, 13, 20–24). We have shown previously that administration of Plg or Pla increases recruitment and migration of mononuclear cells (13), concomitantly with the induction of macrophage polarization toward anti-inflammatory and resolving phenotypes in vivo (24). It has been demonstrated that the key role that the Plg/Pla system plays in the phagocytosis of apoptotic cells requires interaction of Plg with the cell surface (21–24). However, specific plasminogen receptor(s) mediating this effect in vivo have not been identified. In the present study, we have investigated the effects of genetic deletion of either Plg, or its receptor Plg-RKT, on the resolution of inflammation. Deletion of either Plg or Plg-RKT impaired recruitment of mononuclear cells to the pleural cavity of mice in a self-resolving model of pleurisy, concomitantly with an increased percentage of inflammatory M1-like macrophages. Furthermore, Plg/Pla treatment increased M2 markers, and decreased IFN/LPS-induced M1 markers in vitro. Accordingly, IL4-induced polarization of macrophages to an M2-like phenotype was impaired in the absence of Plg and Plg-RKT, and was associated with decreased phosphorylated STAT3. Moreover, deletion of either Plg or Plg-RKT resulted in lower expression of pro-efferocytosis molecules in vivo resulting in lower engulfment of apoptotic neutrophils. Our results support a key role for Plg/Pla and Plg-RKT in the resolution of the inflammatory response.
Male Plg-RKT gene targeted mice (8–10 weeks of age) were backcrossed 10 generations into the C57Bl/6J background (17). Male mPlg-RKT−/− (Plg-RKT specifically deleted in myeloid cells) and Plg-RKTflox/flox mice (8–10 weeks of age) were generated and characterized as described in Figure S1. Breeding pairs of Plg-deficient mice were a kind gift from Dr. Victoria Ploplis, University of Notre Dame, Indiana, USA. Male mice (8–10 weeks of age) were used for all experiments.
Human Plg was from Enzyme Research Laboratories, San Diego, CA and human Pla was from Sigma-Aldrich (St. Louis, MO, USA). We have demonstrated previously that mouse Pla and human Pla have similar activity in the LPS-induced pleurisy model (13).
Mice received an intrapleural (i.pl.) injection of either LPS or PBS (vehicle) as described (24–26). Cells recruited to the cavity were recovered 24–48 h following injection by washing the cavity with 1 mL of PBS. Total cell counts were determined using Turk's stain in a modified Neubauer chamber. Differential cell counting was performed using standard morphological criteria to identify cell types on cyto-centrifuge preparations (Shandon Elliott) stained with May-Grünwald-Giemsa.
LPS from Escherichia coli (serotype O:111:B4) was from Sigma-Aldrich; IFN, IL-4, and IL-10 were from Biolegend (San Diego, CA, USA). Antibodies used for western blotting were anti-mouse arginase-1 and annexin A1 (Santa Cruz biotechnology, Dallas, TX, USA), p-STAT1, p-STAT3, and p-STAT6 (Cell Signaling Technology, Danvers, MA, USA) and anti-mouse β-actin and mouse/anti-rabbit secondary antibodies (LI-COR, Lincoln, NE, USA). Fluorescent monoclonal antibodies for flow cytometry were anti-mouse F4/80 (PE-Cy7 Invitrogen, Carlsbad, CA, USA), GR1 (APC-eBioscience/Thermo Fisher Scientific), CD11b (V500-BD Biosciences, Franklin Lakes, NJ, USA), CD206 (APC Biolegend), CD86 (PE, BD Biosciences), anti-rabbit secondary (alexa fluor 488-A11034 and alexa fluor 405-31556-Invitrogen); and anti-human HLADR/DP/DQ (FITC, BD Pharmingen) and anti-human CD86 (PE, Biolegend). ELISA kits for measurement of murine IL-10, TGF-β, CCL2, CXCL1, and TNF-α were from R&D Systems (Minneapolis, MN, USA); ELISA kits for measurement of human TNF-α and IL-10 were from eBioscience.
BMDMs were prepared as previously described (24) with modifications. Bone marrow was collected from tibias and femurs and washed with Dulbecco's Modified Eagles Medium (DMEM) containing penicillin 100 units/mL and streptomycin 100 μg/mL and the suspension obtained was then centrifuged for 5 min at 1,200 g. The pellet was resuspended with complete conditioned media for BMDM differentiation [DMEM with 10% heat-inactivated fetal bovine serum (FBS) and 20% L929 cell filtered supernatant media], seeded on tissue culture plates, and incubated at 37°C with 5% CO2. After 3 days, the medium was supplemented with additional complete conditioned media. At day 7 the supernatant was removed, and adherent macrophages were detached using a cell scraper and plated in 96-well-plates (Corning™ Costar™, Corning, NY, USA) (2 × 105 cells/well) for flow cytometry or in 6-well plates (Corning™ Costar™) (2 × 106 cells/well) for western blotting.
Human macrophages were prepared as described (27). Briefly, whole blood was centrifuged at 130 × g for 20 min and plasma was removed. For every 10 mL of whole blood, erythrocytes were depleted by sequentially layering 8 mL of 6% w/v dextran (Sigma-Aldrich, Poole, UK) and 10 mL of PBS. After 15 min, the leukocyte-rich fraction was layered over Histopaque 1077 (Sigma-Aldrich) and centrifuged for 30 min at 450 × g at room temperature to separate granulocytes from peripheral blood mononuclear cells (PBMC). PBMC (20 × 106 cells) were seeded in a petri dish. After 1 h incubation at 37 °C, cells were washed to remove lymphocytes and incubated with 50 ng/mL macrophage-colony stimulating factor (M-CSF, PeproTech, London, UK) in RPMI/FBS 10%. Medium was replaced on day 5. On day 7 macrophages were detached with cell detachment solution (Accutase™, Sigma-Aldrich) and seeded in 24-well plate (0.5 × 106 cells/well) for flow cytometric analysis. Experiments using healthy volunteers were approved by the local research ethics committee (P/00/029 East London and The City Local Research Ethics Committee 1). Informed written consent was provided according to the Declaration of Helsinki.
Murine leukocytes were stained with fluorescent monoclonal antibodies against F4/80 (labeled with PE-Cy7,), GR1 (labeled with APC,), CD11b (labeled with V500), CD206 (labeled with APC), CD86 (labeled with PE), anti-rabbit secondary (labeled with alexa 488 and with alexa 405). Stained cells were acquired in a NovoCyte (ACEA Biosciences, San Diego, CA, USA) and analyzed using FlowJo software (Tree Star Inc., Ashland, OR, USA). Macrophage populations were defined according to F4/80, GR1, and CD11b expression, as previously described (24, 28). For human cell analyses, macrophages were labeled with 1.25 μg/mL anti-HLADR/DP/DQ-FITC (BD Pharmingen) and 1 μg/mL anti-CD86-PE (BioLegend), at 4°C for 30 min. Cells were acquired on a LSRFortessa cytometer (BD Biosciences).
Total RNA from BMDMs was extracted using TRIzol Reagent (Invitrogen) according to the manufacturer's instructions. cDNA was synthetized using 500 ng of RNA with the SuperScript III Reverse Transcriptase (Invitrogen), according to the manufacturer's instructions. Real-time PCR was performed in duplicate, with obtained cDNA, specific primers and Power SYBR Green PCR Master Mix (Applied Biosystems, Foster City, CA, USA), using the StepOne™ System (Applied Biosystems). The data were analyzed using StepOne™ System software with a cycle threshold (Ct) in the linear range of amplification and then processed by the 2−ΔΔCt method. The dissociation step was always included to confirm the absence of unspecific products. Gapdh was used as an endogenous control to normalize the variability in expression levels and results were expressed as fold increase. Primers used are listed in Supplemental Materials.
Neutrophils were isolated from peripheral blood of healthy donors on a histopaque gradient (Histopaque 11191 and 10771, from Sigma-Aldrich) as previously described (24, 29). Neutrophil apoptosis was induced by treatment with 10 μM staurosporine (Sigma-Aldrich) for 1 h. Apoptotic neutrophils were labeled (for flow cytometric analysis) by incubation with 5 μM CFSE (carboxyfluorescein diacetate succinimidyl ester—Life Technologies, Carlsbad, CA, USA) at 37°C and 5% CO2 for 1 h. The percentage of apoptosis was determined in cytospin preparations, counted using oil immersion microscopy (100x objective) to determine the proportion of cells with high distinctive apoptotic morphology (25, 29, 30) and >90% were apoptotic. Apoptosis induction by staurosporine was also verified by flow cytometry using Annexin V-FITC and propidium iodide (BD Pharmingen).
The in vivo efferocytosis assay was performed as previously described (24, 31, 32). Mice received an intraperitoneal (i.p.) injection of zymosan (0.1 mg/mouse) to induce peritonitis. After 72 h, mice were injected i.p. with 3 × 106 apoptotic human neutrophils labeled with CFSE. One hour later, mice were euthanized and the cells in the peritoneal cavity were recovered by washing the cavity with 5 mL of PBS.
The in vitro efferocytosis assay was performed by co-culturing BMDMs with human or mouse apoptotic neutrophils labeled with CFSE in a proportion of 3 apoptotic neutrophils: 1 macrophage. Mouse neutrophils were obtained from bone marrow and isolated on a using histopaque gradient (Histopaque 11191 and 10771 from Sigma-Aldrich) as described previously (33). Neutrophils that had not been phagocytosed were removed 1 h later by vigorous washing of the wells with PBS 3 times. Efferocytosis by adherent macrophages was assessed by flow cytometric analyses using the frequency of F480+/CFSE+ cells, and the results were expressed as mean florescence intensity (MFI) of CFSE (flow cytometer laser set at 488). Efferocytosis was also assessed on cytospin preparations of cells after staining with May-Grunwald-Giemsa by determining the proportion of macrophages that ingested apoptotic neutrophils (500 cells/slides were counted). Results are presented as efferocytosis index.
Cells were lysed in RIPA buffer containing anti-protease and anti-phosphatase cocktail (Thermo Fisher Scientific). Cell lysates were electrophoresed on denaturing 4–15% polyacrylamide-SDS gels under reducing conditions and electrotransferred to nitrocellulose membranes. Membranes were incubated with primary antibodies, washed with PBS-Tween-20 0.1% and incubated with species specific IRDye®680RD/800CW-conjugated secondary antibodies. Immunoreactive bands were visualized using the Odyssey Imaging System (LI-COR), according to the manufacturer's instructions (Biosciences). For densitometric analysis, membranes were scanned and quantified using the software Image Studio™ Lite Software 5.2 (LI-COR).
Data are as mean ± SEM. The statistical significance between groups was determined by Student-Newman-Keuls post-hoc test, unless otherwise indicated. A P < 0.05 was considered significant. Calculations were performed using the prism 5.0 software program for Windows (GraphPad software, San Diego, CA, USA).
All animal experiments were approved by the Institutional Animal Care and Use Committee of The Scripps Research Institute.
To examine the impact of Plg and Plg-RKT deficiency on the resolution of inflammation, we utilized a well-established model of LPS-induced inflammation. In this model, intrapleural injection of LPS induces a time-dependent influx of leukocytes into the pleural cavity, characterized by early neutrophilic infiltration, with resolution at 48 h, when neutrophils are scarce and the number of mononuclear cells is maximal (25, 26). Plg−/− and Plg-RKT−/− mice and their respective wild type (WT) littermate controls (Plg+/+ and Plg-RK+/+ mice) were challenged with LPS or PBS and pleural cavities were washed 4, 8, 24, and 48 h later. In Plg+/+ and Plg-RK+/+ mice, the number of mononuclear cells recruited to the pleural cavity in response to LPS injection increased with time (Figures 1A,B). At 48 h, the number of mononuclear cells recruited to the pleural cavity was significantly lower in Plg−/− and Plg-RKT−/− mice than their respective WT controls (Figures 1A,B). Pleural levels of the chemokine CCL2 [a chemoattractant for monocytes and lymphocytes (34)] were significantly decreased at 8 h in Plg−/− and Plg-RKT−/− mice (Figures 1C,D), consistent with the decreased mononuclear cell recruitment in these mice. There was no significant effect of either genotype on neutrophil recruitment (Figures 1E,F), although there was a trend for increased neutrophil recruitment in Plg−/− mice (Figure 1E), consistent with increased levels of the neutrophil chemoattractant, CXCL1 (Figure 1G). However, and in accordance with the similar profile of neutrophil recruitment in Plg-RKT+/+ and Plg-RKT−/− mice, no difference in CXCL1 levels between these two genotypes was detected (Figure 1H). Interestingly, mPlg-RKT−/− mice in which Plg-RKT was specifically deleted in myeloid cells (Figure S1), did not differ in the number of mononuclear cells recruited into the pleural cavity or in levels of CCL2 compared with Plg-RKTflox/flox controls (Figures S2A,B). Thus, Plg-RKT deficiency in other cell types in addition to myeloid cells is likely to impact Plg-RKT-dependent monocyte recruitment. There was also no effect of specific deletion of Plg-RKT in myeloid cells on the number of recruited neutrophils or pleural levels of CXCL1 (Figures S2C,D), consistent with the results obtained with mice with global Plg-RKT deletion.
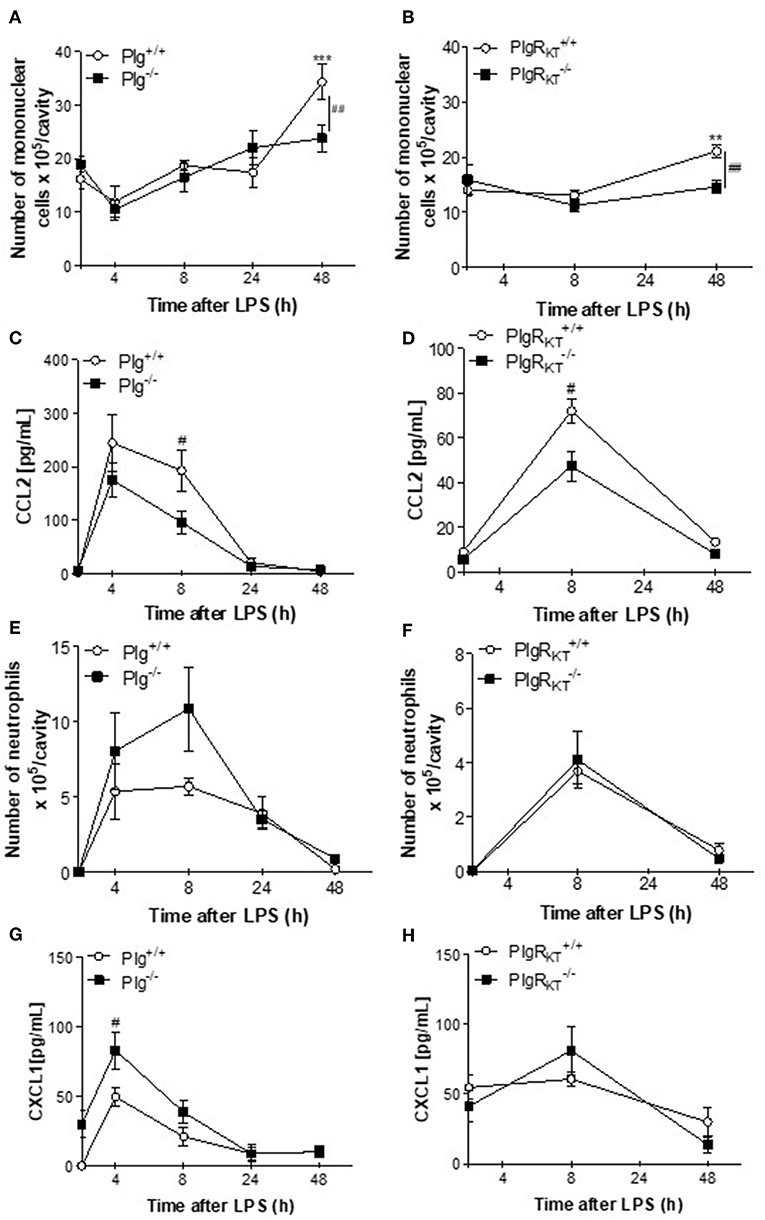
Figure 1. Time-course of leukocyte recruitment during LPS-induced pleurisy in Plg−/− and Plg-RKT−/− mice and their respective wild type littermates. Plg+/+, Plg−/−, Plg-RKT+/+, and Plg-RKT−/− mice were injected intrapleurally with LPS (250 ng/cavity, i.pl.) or PBS. Cells present in the pleural cavity were harvested 4, 8, 24, and 48 h after LPS challenge. The number of mononuclear cells (A,B) and neutrophils (E,F) were evaluated by counting cytospin slides stained with May-Grunwald-Giemsa. The levels of the monocyte chemoattractant CCL2 (C,D) and the neutrophil chemoattractant, CXCL1 (G,H) were quantified in cell-free pleural lavages by ELISA. Results are shown as the mean ± SEM of at least five mice per group. **P < 0.01 and ***P < 0.001, when comparing LPS-injected mice with PBS-injected mice. #P < 0.05 and ##P < 0.01, when comparing wild type and knockout mice injected with LPS.
In the LPS-induced pleurisy model, proinflammatory macrophages are the predominant macrophage phenotype present in the pleural space 8 h after LPS injection (the productive phase of inflammation). Anti-inflammatory macrophages are the predominant macrophage phenotype present in the resident pleural macrophage population (PBS-injected) and are also predominant at 48 h following LPS treatment (the resolving phase) (28). Interestingly, Plg expression as well as Pla activity are increased in the resolving phase of this model (24). Therefore, we analyzed whether deletion of either Plg or Plg-RKT would affect the proportion of macrophage phenotype at different time points. In flow cytometric analyses, we found increased proportion of proinflammatory M1 macrophages (F4/80low GR1+ CD11bmed) in the pleural cavities of Plg−/− and Plg-RKT−/− mice during the active phase of inflammation (8 h) compared with their respective WT controls (Figures 2A,B). There were no significant genotype-dependent effect on the proportion of M2-like macrophage (F4/80high GR1− CD11bhigh) (Figures 2C,D), suggesting that deletion of either Plg or Plg-RKT causes impairment in polarization to the anti-inflammatory (M2) phenotype and thus, macrophages retain the M1-like phenotype.
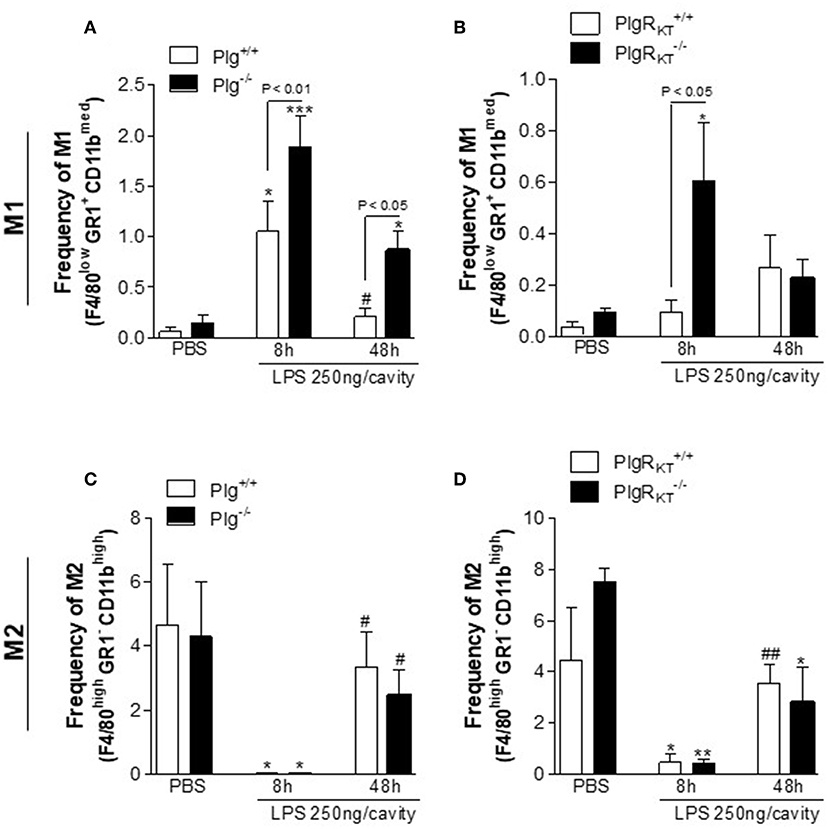
Figure 2. Characterization of macrophage polarization in Plg−/− and Plg-RKT−/− mice and their respective wild type littermates during LPS-induced pleurisy. Plg+/+, Plg−/−, Plg-RKT+/+, and Plg-RKT−/− mice were injected with either LPS (250 ng/cavity, i.pl.) or PBS. Cells present in the pleural cavity were harvested at 8 and 48 h after LPS challenge or PBS injection. The percentage of M1-like [F4/80low GR1+ CD11bmed] (A,B) and M2-like [F4/80high GR1− CD11bhigh] (C,D) macrophage populations were determined by flow cytometry. Results are shown as the mean ± SEM of at least four mice in each group. *P < 0.05, **P < 0.01, and ***P < 0.001, when comparing 8 h LPS-injected with PBS-injected mice; #P < 0.05 and ##P < 0.01, when comparing cells harvested 48 h after LPS injection with cells harvested with 8 h after LPS injection mice.
Based on the results above, we investigated the effect of either Plg or Pla treatment on macrophage polarization in vitro, using WT C57Bl/6J BMDMs. Significantly increased expression of the M2 markers CD206 (Figures 3A,B) and arginase-1 (Figure 3C) was observed following cell incubation with Plg/Pla. Also, in response to Plg/Pla treatment, conditioned media harvested from treated BMDMs had significantly increased levels of cytokines typically secreted by M2-like macrophages, TGF-β (Figure 3D) and IL-10 (Figure 3E). In contrast, Plg/Pla downregulated BMDM expression of the M1 marker, iNOS (Figure 3F). No effect of Plg/Pla treatment was observed on secretion of the proinflammatory cytokine TNF-α (Figure 3G). Interestingly, pretreatment of BMDMs with Plg/Pla reduced IFN/LPS-induced upregulation of the M1 marker, CD86 (Figure 3H). In parallel experiments with human macrophages, pretreatment with Pla inhibited IFN/LPS-induced expression of the M1 markers, CD86 and HLA (Figures 3I,J) and secretion of the M1-like product, TNF-α (Figure 3K), while increasing secretion of the M2-like secretory product, IL-10 (Figure 3L). Taken together these results suggest that Plg and Pla promote macrophage polarization to the M2-like phenotype while decreasing expression of IFN/LPS-induced M1 phenotypic markers in both mouse and human macrophages.
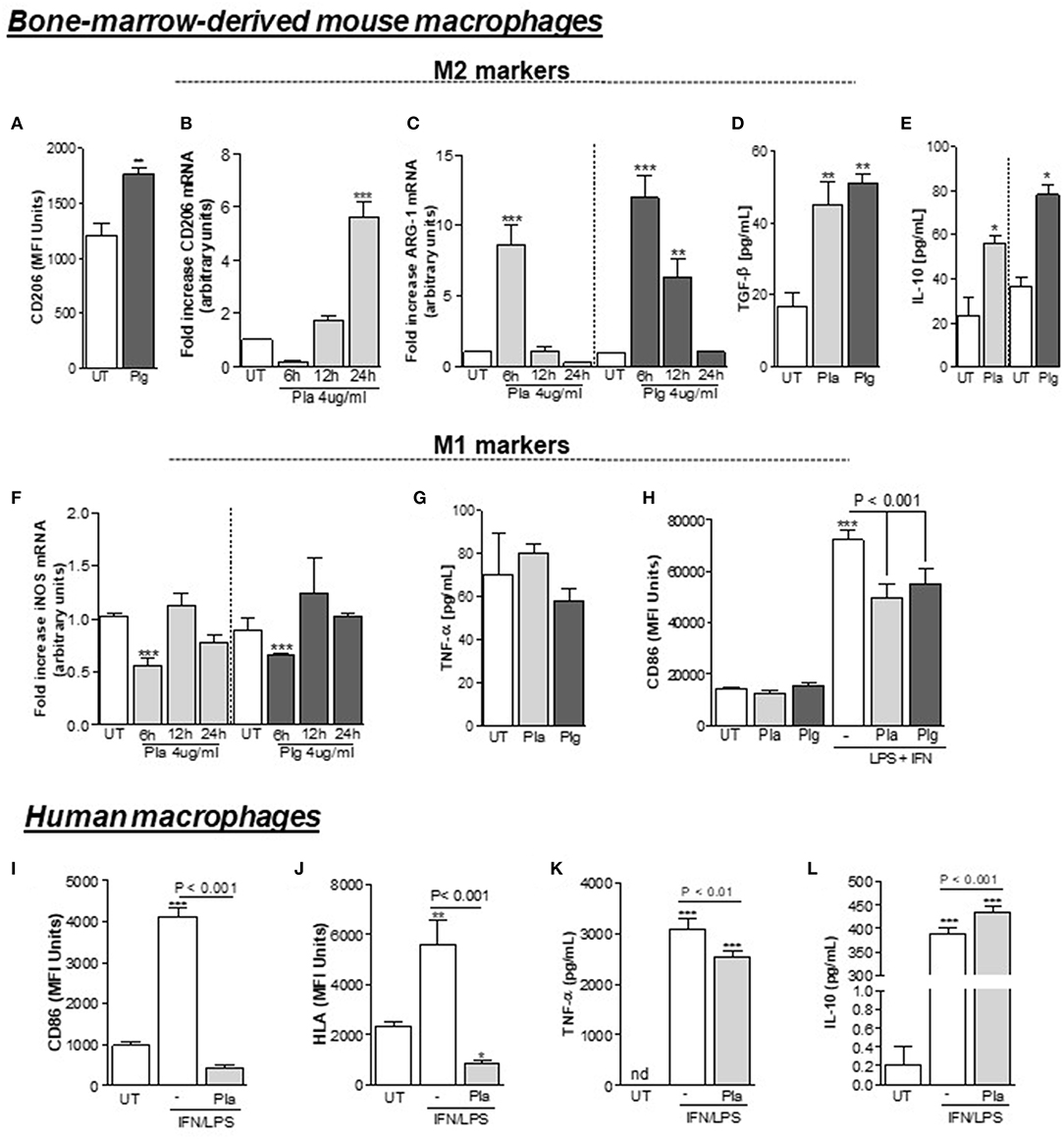
Figure 3. Effect of Plg and Pla on macrophage polarization. BMDMs from C57BL/6J WT mice were washed 3 times with serum free DMEM, treated with Plg (4 μg/mL) or Pla (4 μg/mL) or untreated (UT) for the indicated times and analyzed by flow cytometry (A) or qPCR (B) for expression of the M2 marker CD206, and by qPCR for expression of the M2 marker, arginase-1 (C). Levels of the secretory products of M2-like macrophages, TGF-β (D) and IL-10 (E) were quantified 24 h after treatments in conditioned media by ELISA. Treated and untreated BMDMs were also analyzed for expression of the M1 marker iNOS by qPCR (F) and conditioned media collected at 24 h were analyzed by ELISA for concentrations of the secretory product of M1-like macrophages, TNF-α (G). BMDMs (H) and human macrophages (I–L) were pre-treated with Plg or Pla (both at 4 μg/mL) for 1 h, and then, stimulated with LPS (10 ng/mL) + IFN (10 ng/mL) and with LPS (10 ng/mL) + IFN (20 ng/mL) for 24 h, respectively. The expression of the M1 markers CD86 (H,I) and HLA (J) was measured using flow cytometry and conditioned media were subjected to ELISA for detection of human TNF-α (K) or IL-10 (L). nd, not-detected. *P < 0.05, **P < 0.01, and ***P < 0.001 when comparing Plg- or Pla-treated with untreated (UT) BMDMs or untreated human macrophages.
We next examined the response of BMDMs from Plg−/− and Plg-RKT−/− mice to standard macrophage polarizing agents to induce either the M1-like (IFN+LPS) or the M2-like phenotype (IL-4). Expression of CD206 and arginase-1 following stimulation with IL-4 was lower in Plg−/− and Plg-RKT−/− BMDMs compared with their WT controls (Figures 4A,B,H,I, respectively), consistent with a requirement for both Plg and its receptor, Plg-RKT, in macrophage polarization to the M2-like phenotype. In contrast, in these settings TGF-β and IL-10 levels were not affected by deletion of either Plg or Plg-RKT (Figures 4C,D,J,K, respectively). No impairment of expression of markers of polarization to the M1-like phenotype CD86 (Figure 4E) and TNF-α (Figure 4F) was observed in Plg−/− BMDMs following treatment with IFN/LPS. Interestingly, we found an increased expression of CD86 without modification in TNF-α levels after IFN+LPS stimulation of Plg-RKT−/− BMDMs (Figures 4L,M). Levels of IL-10 were reduced in IFN/LPS-stimulated Plg−/− BMDMs (Figure 4G) with a trend for reduction in IL-10 levels in Plg-RKT−/− BMDMs (Figure 4N).
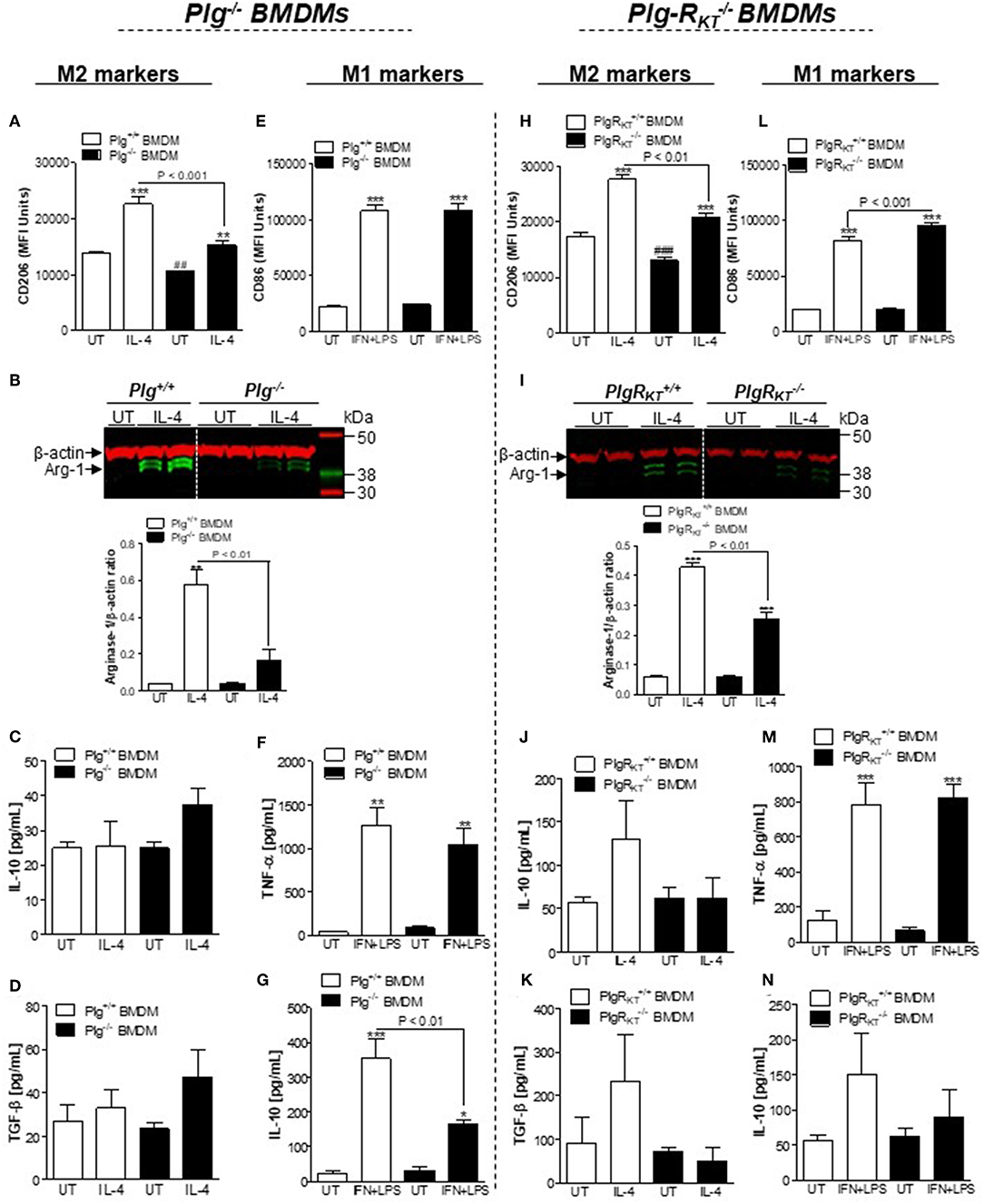
Figure 4. Effects of deletion of either Plg or Plg-RKT on macrophage polarization. BMDMs from Plg+/+, Plg−/−, Plg-RKT+/+, and Plg-RKT−/− mice were separately washed 3 times with serum free media DMEM and then were either untreated (UT) or stimulated with IL-4 (20 ng/mL) or LPS (10 ng/mL) + IFN (10 ng/mL) for 24 h. Cells were analyzed by flow cytometry for expression of the M2 markers CD206 (A,H) and the M1 marker CD86 (E,L). Expression of the M2 marker, arginase-1 (B,I), was determined by Western blotting with anti-β-actin used as loading control. The levels of secretory products of M2-like macrophages TGF-β (D,K) and IL-10 (C,J,G,N), and M1-like macrophages TNF-α (F,M) were determined in conditioned media by ELISA. *P < 0.05, **P < 0.01, and ***P < 0.001 when comparing treated with untreated (UT) BMDMs and ##P < 0.001 and ###P < 0.001 when comparing UT wild type with UT knockout BMDMs.
We also examined the responses to either IFN+LPS or to IL-4 by BMDMs from mPlg-RKT−/− mice. Expression of M1-like and M2-like markers by mPlg-RKT−/− BMDMs mirrored that observed with Plg-RKT−/− BMDMs. In response to IL-4 treatment, expression of CD206 was impaired (Figure S3A), while expression of the M2 secretory products IL-10 and TGF-β, were not significantly different than with Plg-RKTflox/flox controls (Figures S3B,C). Results with LPS+IFN stimulation also were in parallel with those of Plg-RKT−/− BMDMs with an increase in expression of CD86 (Figure S3D) without modification in IL-10 levels (Figure S3F). In contrast, TNF-α levels were increased in conditioned media of mPlg-RKT−/− BMDMs (Figure S3E). Taken together, these results suggest that deficiency of either Plg or Plg-RKT impairs macrophage polarization toward an M2-like phenotype.
Standard macrophage polarizing agents such as IL-4 induce an M2-like phenotype through both STAT6 (canonical pathway) and STAT3 (non-canonical pathway), and IFN+LPS induces macrophage polarization toward the M1-like phenotype via STAT1 (35). Therefore, we investigated the effect of deletion of either Plg or Plg-RKT on levels of pSTAT3, pSTAT6, or pSTAT1 during polarization of BMDMs. Levels of pSTAT3 were increased following IL-4 treatment in Plg+/+ and Plg-RKT+/+ BMDMs, while the response was markedly and significantly decreased in Plg−/− and Plg-RKT−/− BMDMs (Figures 5A,B,E,F). There was no genotype effect on IL4-induced levels of pSTAT6 or IFN+LPS-induced levels of pSTAT1 (Figures 5A,B,E,F). Results with mPlg-RKT−/− BMDMs mirrored those with Plg-RKT−/− BMDMs (Figure S4A).
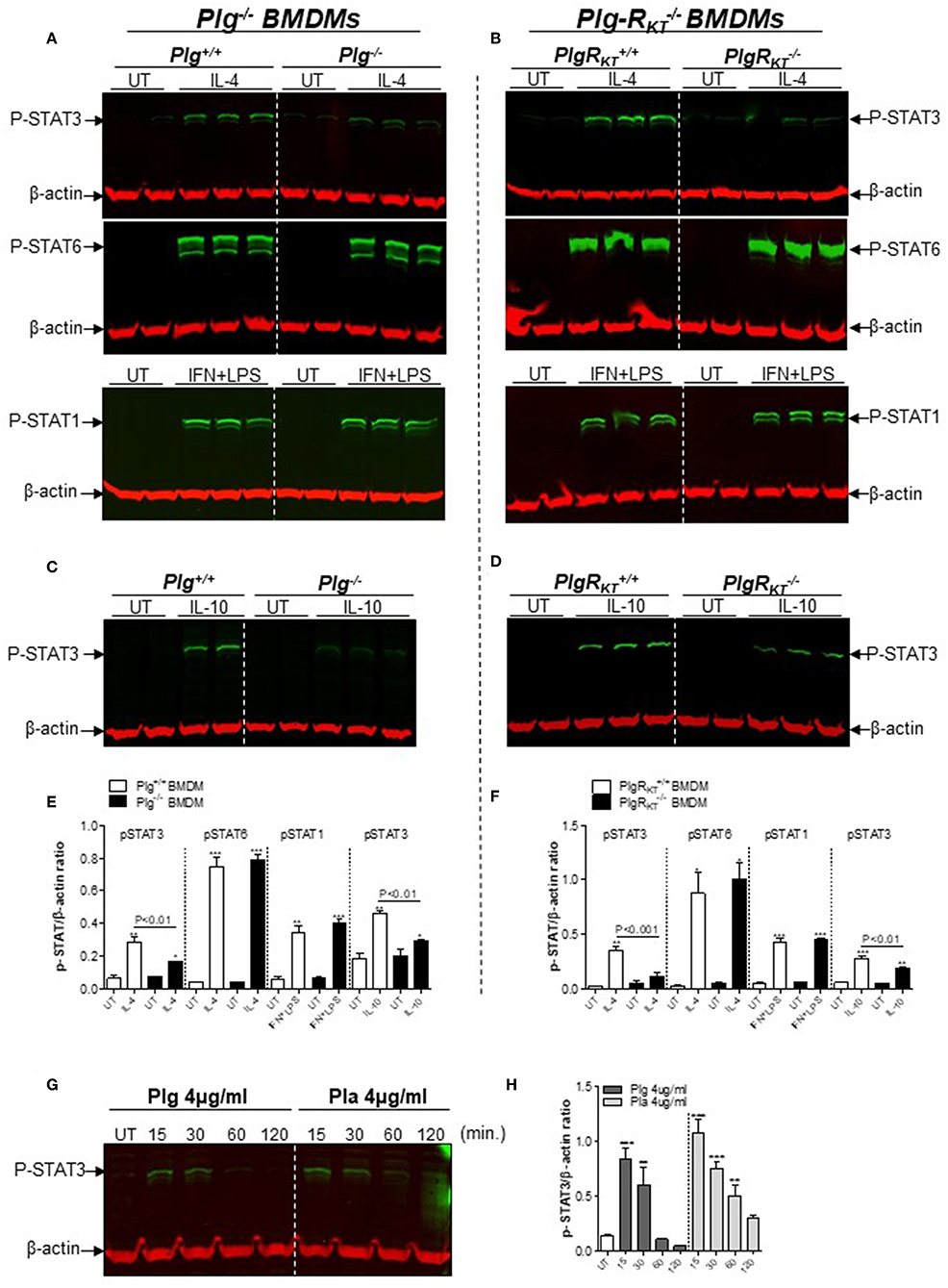
Figure 5. Effect of plasmin(ogen) treatment and deletion of either Plg or Plg-RKT on STAT signaling pathways. BMDMs from Plg+/+, Plg−/−, Plg-RKT+/+, and Plg-RKT−/− mice were washed 3 times with serum free media and then treated with either IL-4 (20 ng/mL), LPS (10 ng/mL) + IFN (10 ng/mL), IL-10 (20 ng/mL), or untreated (UT) for 30 min (A–D). BMDMs from wild type C57Bl/6J mice were washed 3 times with serum free media and then treated with Plg or Pla (each at 4 μg/mL) for 15–120 min or untreated (UT) for 30 min (G,H). Cell lysates were analyzed by Western blotting for the indicated antigens. β-actin was used as loading control. β-actin was used as a loading control. Densitometry analyses are shown (E,F,H). *P < 0.05, **P < 0.01, and ***P < 0.001 when comparing treated with UT BMDMs.
Because STAT3 is a component of the canonical pathway induced during M2 polarization in response to IL-10 treatment (36), we measured levels of pSTAT3 following IL-10 treatment of BMDMs. pSTAT3 levels were markedly lower in BMDMs from Plg−/−, Plg-RKT−/− and mPlg-RKT−/− mice compared with BMDMs from control littermates (Figures 5C,D and Figure S4B). In a complementary manner, we measured secreted TGF-β as an additional readout of IL10-induced M2-like polarization and found lower levels in conditioned media from Plg−/− and mPlg-RKT−/− BMDMs compared to their respective WT controls (Figures S5A,B). Since phosphorylated-STAT3 levels were lower in Plg−/− and Plg-RKT−/− BMDMs (Figures 5A–F), we tested whether treatment with either Plg or Pla would affect pSTAT3. Treatment of WT C57Bl/6J BMDMs with either Plg or Pla resulted in transient increases in levels of pSTAT3 within 15–30 min, decreasing at 60–120 min (Figures 5G,H). Taken together these results suggest that Plg and Plg-RKT mediate polarization of macrophages to the M2-like phenotype via STAT3 signaling.
M2 macrophages play a key role in efferocytosis (6, 7). Therefore, we examined the impact of Plg or Plg-RKT deficiency on expression by macrophages of representative molecules known to regulate efferocytosis of apoptotic cells, CD206 and Annexin A1 (AnxA1) (37, 38). During the resolving phase of LPS-induced pleurisy (48 h), the frequency of cells doubly positive for F4/80/CD206 (Figures 6A,B) and doubly positive for F4/80/AnxA1 (Figures 6C,D) was markedly decreased in the pleural exudates of both Plg−/− and Plg-RKT−/− mice compared to their respective WT littermate controls. These results suggest that Plg and Plg-RKT contribute to the expression of molecules associated with M2-like macrophages and with efferocytosis during the resolution phase of inflammation.
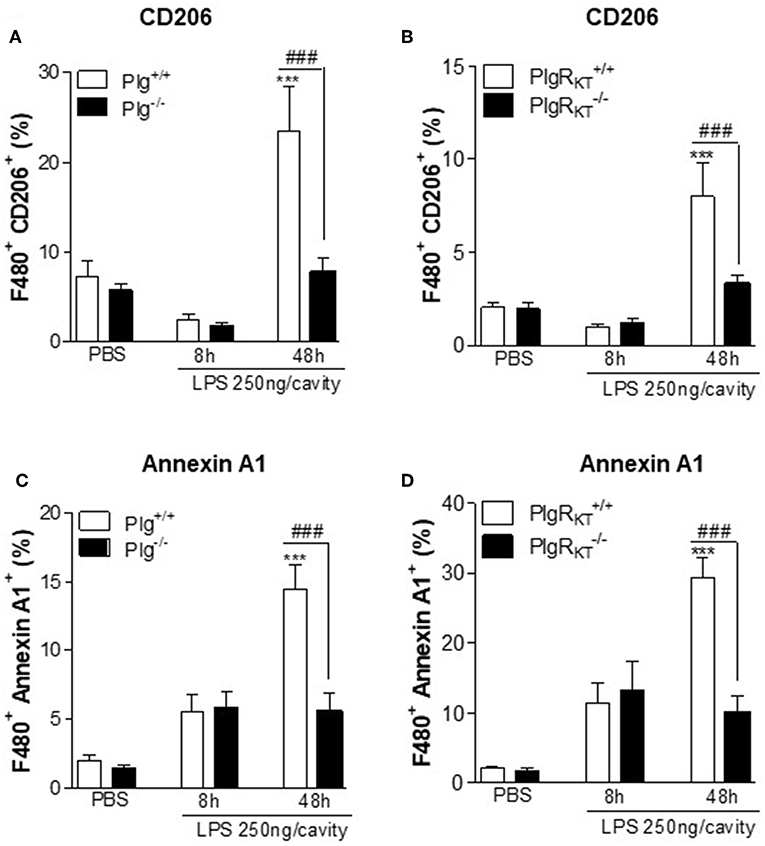
Figure 6. Expression of engulfment molecules during LPS-induced pleurisy. Plg+/+, Plg−/−, Plg-RKT+/+, and Plg-RKT−/− mice were injected with LPS (250 ng/cavity, i.pl.) or PBS. Cells present in the pleural cavity were harvested 8 and 48 h after LPS challenge or after PBS injection (48 h). The frequency of F4/80+ CD206+ (A,B) and F4/80+ Annexin A1+ (C,D) double positives cells was determined by flow cytometry. Results are shown as the mean ± SEM of at least four mice in each group. ***P < 0.001, when comparing LPS-injected mice with PBS-injected mice and ###P < 0.01 when comparing LPS-injected wild type with knockout mice.
Next, by using an in vivo model of efferocytosis (24, 31, 32) we tested whether deletion of either Plg or Plg-RKT would affect the ability of macrophages to phagocytose apoptotic neutrophils (efferocytosis). Mice received an intraperitoneal injection of zymosan (0.1 mg/cavity) to induce macrophage recruitment and were then injected intraperitoneally with human apoptotic neutrophils labeled with fluorescent CFSE as described (24, 31, 32). Cells were collected from the peritoneal exudate 1 h later for flow cytometry. Representative histograms are shown in Figure 7A. Engulfment of apoptotic neutrophils was lower in Plg−/−, Plg-RKT−/−, and mPlg-RKT−/− mice compared to their littermate controls, as seen by flow cytometry (Figures 7B–D) or by counting the percentage of macrophages that had ingested apoptotic neutrophils, on cytospin slides (Figures 7F,G). Representative images are shown in Figure 7E. Interestingly, the frequency of macrophages expressing CD206 during efferocytosis was also significantly decreased in Plg−/− and mPlg-RKT−/− mice relative to their WT controls (Figures 7H,I).
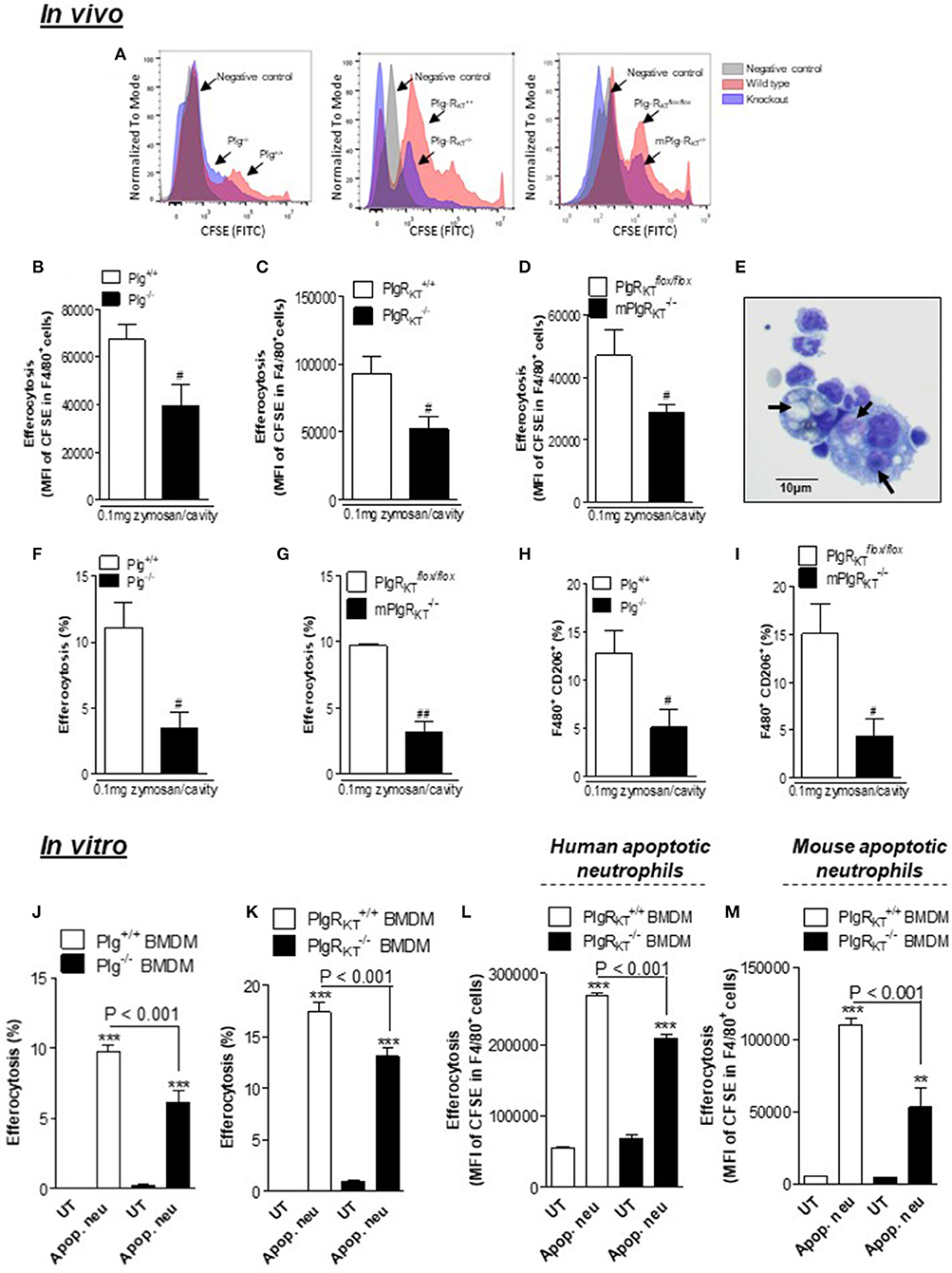
Figure 7. Effect of deletion of Plg or its receptor, Plg-RKT, on efferocytosis. To determine efferocytosis in vivo, Plg+/+, Plg−/−, Plg-RKT+/+, Plg-RKT−/−, mPlg-RKT−/−, and Plg-RKT-flox/flox mice received an i.p. injection of 0.1 mg of zymosan and 71 h later received an i.p. injection of 3 × 106 apoptotic human neutrophils labeled with fluorescent CFSE. The cells from the peritoneal cavity were collected 1 h later. The in vitro efferocytosis assay was performed by co-culturing BMDMs with either human or mouse apoptotic neutrophils labeled with CFSE in a proportion of 3 neutrophils per macrophage. Efferocytosis was assessed by flow cytometry analyzing MFI (mean fluorescence intensity) of F4/80+ FITC+ (B–D,L,M) and by counting cytospin slides (F,G,J,K). Representative histograms (A) and images of neutrophils inside macrophages (E) are shown (arrows). Magnification 40X. The frequency of F4/80+ CD206+ (H,I) double positives cells were determined by flow cytometry. Flow cytometry data are expressed as MFI or frequency, and are shown as the mean ± SEM of at least four mice in each group. **P < 0.01 and ***P < 0.001, when comparing BMDMs treated with apoptotic neutrophils with untreated (UT) BMDMs and #P < 0.05 and ##P < 0.01 when comparing zymosan-injected knockout with control mice.
We also examined efferocytosis in vitro. By counting cytospin slides we found that % efferocytosis by both Plg−/− and Plg-RKT−/− BMDMs was decreased relative to BMDMs from their respective WT controls (Figures 7J,K). By FACS, we observed similar effects with Plg-RKT−/− BMDMs when using either human or mouse neutrophils as prey (Figures 7L,M). These results indicate that Plg and Plg-RKT function to regulate macrophage polarization and efferocytosis of apoptotic cells both in vivo and in vitro.
The role of the Plg/Pla system in the resolving phase of inflammation and the receptor-dependence of key steps in the resolution of inflammation is an emerging area (24). In the current study, utilizing both Plg and Plg-RKT-deficient mice, we found that: (1) Optimal mononuclear cell recruitment in response to LPS-induced pleurisy required both Plg and the Plg receptor, Plg-RKT; (2) Optimal polarization of macrophages to the anti-inflammatory M2-like phenotype required both Plg/Pla and Plg-RKT; (3) Stimulation of key intracellular signaling events for macrophage polarization required both Plg/Pla and Plg-RKT; (4) Expression of engulfment molecules and effective efferocytosis required both Plg/Pla and Plg-RKT. Therefore, we provide evidence that endogenous Plg and Plg-RKT regulate key steps in the resolution of inflammation by regulating monocyte/macrophage migration, reprogramming macrophages toward an M2-like phenotype, regulating cytokine release and promoting efferocytosis.
Monocyte migration leading to macrophage infiltration at inflammatory sites is a hallmark of the resolution of inflammation (39, 40). These cells promote clearance of the neutrophilic infiltrate, to prevent apoptotic neutrophils from becoming necrotic and pro-inflammatory (5). Plg/Pla is required for optimal macrophage recruitment in response to a diverse array of inflammatory stimuli (10, 13, 41, 42). This requires association of Plg/Pla with C-terminal lysines exposed on the surfaces of monocytes/macrophages (11, 13, 16, 43, 44) and the proteolytic activity of Pla (10, 13). We have shown that Pla-induced macrophage chemotaxis in vitro and mononuclear cell recruitment in vivo are dependent on lysine binding sites and Pla activity (13). Here, utilizing a self-resolving model of LPS-induced pleurisy, we observed impaired recruitment of mononuclear cells in both Plg−/− and Plg-RKT−/− mice, consistent with previous studies in a model of sterile peritonitis (10, 16, 41). Moreover, our results provide the first evidence for decreased levels of the macrophage chemoattractant, CCL2, in Plg-RKT−/− mice after inflammatory stimulation, consistent with the impaired mononuclear cell recruitment. Surprisingly, mononuclear cell recruitment and peritoneal CCL2 levels were not impaired in mice with Plg-RKT deleted specifically in myeloid cells, suggesting that Plg-RKT deficiency in other cell types such as mesothelial cells (45) may impact the production and/or processing of CCL2 and contribute to decreased monocyte migration in mice with globally deleted Plg-RKT. CCL2 can be produced by several cell types including endothelial, mesothelial, fibroblasts, epithelial, smooth muscle, mesangial, astrocytic, monocyte/macrophage, and microglial cells (45–49). Cailhier et al. have suggested that mesothelial cells are responsible for the production of this chemokine in the model of carrageenan-induced pleurisy (50). The results in the carrageenan-induced pleurisy model may be applicable to LPS-induced pleurisy as well. Pla has been shown to stimulate cytokine release by several cell types (19) and this may be dependent on plasminogen binding to Plg-RKT.
In Plg−/− mice impaired macrophage migration also was associated with reduced pleural levels of CCL2 in LPS-induced pleurisy. A previous study found decreased peritoneal levels of CCL2 (~50% lower), associated with decreased macrophage recruitment after biomaterial implantation in Plg−/− mice (42). We have previously shown that Plg/Pla treatment increases CCL2 levels and promotes monocyte migration to the pleural cavity of mice in a manner dependent on the CCL2/CCR2 axis (13), an effect also reported in vitro with human monocytes (51). Previous studies showed that Pla increases CCL2 chemotactic potency by cleavage at lysine 104 (52), and Pla may be indispensable to full activation of this chemokine in vivo (53). However, Plg-RKT-bound Pla proteolytic activity is likely necessary, but not sufficient for optimal recruitment of mononuclear cells, because mPlg-RKT−/− mice are expected to have impaired surface expression of Pla, yet there is no effect on mononuclear cell recruitment in this model in mPlg-RKT−/− mice. Although Plg−/− and Plg-RKT−/− mice exhibited increased polarization to M1-like macrophages following LPS challenge they did not exhibit prolonged inflammation, as determined by the presence of similar numbers of pleural neutrophil over time, compared to their wild-type littermate controls. This is consistent with previous studies in which Plg−/− mice exhibited reduced mononuclear cell recruitment into the peritoneal cavity of thioglycolate-challenged mice, while the extent of neutrophil recruitment was similar to that of wild-type mice (10, 41) and with our previous study in which injection of Pla into the pleural cavity selectively induced the recruitment of mononuclear cells, but not neutrophils (13, 24). With regard to effects on other neutrophil functions, previous studies have demonstrated that, in contrast to monocytes, macrophages and dendritic cells, Pla does not directly activate neutrophils (19).
Coexistence of macrophages in different activation states and mixed phenotypes is observed at inflammatory sites. The distinct functional phenotypes can be induced in vivo or in vitro by inducers either alone, or in combination, including IFN, immune complexes, helminth infections, LPS, complement components, apoptotic cells, macrophage colony stimulating factor (M-CSF), glucocorticoids, IL-4, IL-13, IL-10, and TGF-beta (35, 36). Exposure to different activators results in a spectrum of macrophages phenotypes distinguished by variations in functions and expression/secretion of distinct molecules. For example, macrophages can differentiate to M1-like or a spectrum of M2-like subtypes (M2a, M2b, M2c, and M2d) depending on the applied stimuli (54). During the resolving phase of LPS-induced inflammation, macrophages are reprogramed toward resolving phenotypes (28). Here, in the model of LPS-induced pleurisy, we observed a greater percentage of M1-like macrophages in pleural exudates of both Plg−/− and Plg-RKT−/− mice, compared with their respective WT littermate controls. Interestingly, treatment with Plg/Pla reduced the expression of CD86 by M1-like BMDMs stimulated with IFN+LPS and decreased the expression of CD86 and HLA in M1-like human macrophages. Similarly, Borg et al. found decreased expression of CD86 and HLA by monocyte-derived dendritic cells in response to Pla treatment (23). Correspondingly, we found increased expression of CD86 in M1-like BMDMs from Plg-RKT-deficient mice. Also, Plg/Pla treatment decreased iNOS expression in BMDMs as previously observed in vivo in cells harvested from the pleural cavity (24). In addition, Plg/Pla treatment did not affect TNF-α levels, whereas Pla decreased TNF-α secretion and increased IL-10 secretion in human M1-like cells, suggesting a broad spectrum of Pla action to downregulate markers of pro-inflammatory macrophages. Moreover, Plg/Pla treatment increased expression of the M2 markers, Arginase-1/CD206, and secretion of IL-10 and TGF-β by BMDMs, similar to the effect of Plg/Pla treatment on TGF-β secretion by dendritic cells in vitro (23) and similar to the effect of intrapleural injection of Plg/Pla in vivo (24). Akin to these findings, polarization of BMDMs from Plg−/− and Plg-RKT−/− toward an M2-like phenotype with either IL-4 or IL-10 was impaired. Previous studies have shown that urokinase-type plasminogen activator (uPA) induces polarization of macrophages to an M2-like phenotype and prevents the M0 to M1 transition (55). In addition, uPA receptor (uPAR) deficient mice show increased expression of M1 markers and decreased M2 markers in gut tissue in a model of colitis (56). Taken together with our current data, effects of Pla on macrophage polarization are likely to require both proteolysis and receptor-dependent signaling.
The signal transduction and activators of transcription (STATs) convey the signaling of many inducers of the M2-like phenotype, including the cytokines IL-4, IL-10, and IL-6 (57–59), and the proresolving molecules AnxA1 (60) and maresins (61). Pla induces rapid phosphorylation of STAT3 in monocytes (51). And, Pla triggers activation and nuclear translocation of STAT3, but not of STAT1 or STAT5 in macrophages (62). In a murine model of sepsis, pSTAT3 expression is reduced in Plg−/− mice compared to wild type control mice (63). IL-4 induces macrophage polarization toward the M2-like phenotype through both STAT6 (canonical pathway) and STAT3 (non-canonical pathway), and IFN/LPS treatment induces macrophage polarization toward the M1-like phenotype via STAT1 (35). Here we found decreased levels of pSTAT3, but not of pSTAT6, following treatment of either Plg−/− or Plg-RKT−/− BMDMs with inducers of M2 polarization (IL-4 or IL-10) compared to WT BMDMs. Mechanistically, our results suggest that Plg/Pla and Plg-RKT mediate macrophage polarization to the M2 phenotype via STAT3 signaling. We suggest that Plg/Pla interacts with Plg-RKT to mediate such polarization. This is the first report of a role for Plg-RKT in intracellular signaling. As only a 4 amino acid sequence of Plg-RKT has a cytoplasmic location, we expect that signaling will require interaction with other adjacent molecules that then become phosphorylated, as in the case of uPAR that mediates intracellular signaling despite being a GPI-linked receptor (64).
Engulfment of apoptotic cells is crucial for the resolution of inflammation and resumption of tissue homeostasis and prevents chronic inflammation and autoimmunity (5). The process of efferocytosis is mediated predominantly by M2-like macrophages (6, 65). Plg promotes clearance of apoptotic cells and this requires Plg activation, Pla activity, interaction of the lysine binding sites of plasmin(ogen) with C-terminal lysines expressed on the cell surface as well as de novo protein synthesis (21–24). Our data implicate, a specific receptor, Plg-RKT (a plasminogen receptor exposing a C-terminal lysine on the cell surface) as a regulator of plasmin-mediated efferocytosis. Impaired efferocytosis in Plg−/− and Plg-RKT−/− mice was observed in parallel with decreased expression of AnxA1 and CD206, known to be involved in recognition and engulfment, respectively, of phagocytic material (38, 66). AnxA1 is one of many phagocytosis-related genes dysregulated in spleen and liver of Plg−/− mice (22). Recently, we showed that treatment with or injection of Plg/Pla increases the efferocytic capacity of murine macrophages in vitro and in vivo, in an AnxA1-dependent manner (24). Plg and Pla increase AnxA1 expression in resolving macrophages, and AnxA1−/− mice are refractory to Plg/Pla stimulation of efferocytosis, suggesting AnxA1 as a key molecule regulating Plg/Pla-mediated efferocytosis (24). Our current data implicating Plg-RKT−/− in efferocytosis are in agreement with the requirement for cell surface lysine-binding sites for the phagocytosis-inducing effect of Plg/Pla (22). Thus, our results are consistent with a mechanism in which Pla bound to Plg-RKT stimulates expression of AnxA1 and further efferocytosis. Here, we have shown that optimal induction of pSTAT3 and of IL-10 expression require both Plg/Pla and Plg-RKT, as an additional potential mechanism by which these molecules regulate efferocytosis. Indeed, it was recently shown that macrophage efferocytosis during inflammation resolution involves an IL-10 triggered STAT3 pathway (67). Taken together our results support the concept that Plg/Pla is required for macrophage reprogramming and efferocytosis, and these effects are dependent on Plg/Pla association with its receptor, Plg-RKT. Considering that these events are crucial steps of inflammation resolution, our results provide an important role for Plg/Pla and the receptor Plg-RKT in the resolution of the inflammatory response.
All datasets generated for this study are included in the manuscript and/or the Supplementary Files.
All animal experiments were approved by the Institutional Animal Care and Use Committee of The Scripps Research Institute. Experiments using healthy volunteers were approved by the local research ethics committee (P/00/029 East London and The City Local Research Ethics Committee 1). Informed written consent was provided according to the Declaration of Helsinki.
JV, RP, LS, and LM designed research and analyzed data. JV, LS, and LM wrote the paper. JV, LS, MS, KL, GN-L, and NB performed experiments. MT and MP provided essential tools and expertise.
This work was supported by grants from Coordenação de Aperfeiçoamento de Pessoal de Nível Superior- Brazil (CAPES)- Finance code 001, Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq, Brazil), Fundação de Amparo a Pesquisa do Estado de Minas Gerais (FAPEMIG, Brazil), National Institutes of Health (grant HL 081046 to LM) and by Merit Review Award #5I01BX002026 and I01BX003933 from the U.S. Department of Veterans Affairs (to RP). National Institute of Science and Technology in Dengue and host-pathogen interactions (465425/2014-3). MP acknowledges the support of the William Harvey Research Foundation.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2019.01458/full#supplementary-material
1. Sousa LP, Alessandri AL, Pinho V, Teixeira MM. Pharmacological strategies to resolve acute inflammation. Curr Opin Pharmacol. (2013) 13:625–31. doi: 10.1016/j.coph.2013.03.007
2. Serhan CN, Brain SD, Buckley CD, Gilroy DW, Haslett C, O'Neill LA, et al. Resolution of inflammation: state of the art, definitions and terms. FASEB J. (2007) 21:325–32. doi: 10.1096/fj.06-7227rev
3. Alessandri AL, Sousa LP, Lucas CD, Rossi AG, Pinho V, Teixeira MM. Resolution of inflammation: mechanisms and opportunity for drug development. Pharmacol Ther. (2013) 139:189–212. doi: 10.1016/j.pharmthera.2013.04.006
4. Sugimoto MA, Sousa LP, Pinho V, Perretti M, Teixeira MM. Resolution of inflammation: what controls its onset? Front Immunol. (2016) 7:160. doi: 10.3389/fimmu.2016.00160
5. Poon IK, Lucas CD, Rossi AG, Ravichandran KS. Apoptotic cell clearance: basic biology and therapeutic potential. Nat Rev Immunol. (2014) 14:166–80. doi: 10.1038/nri3607
6. Ariel A, Serhan CN. New lives given by cell death: macrophage differentiation following their encounter with apoptotic leukocytes during the resolution of inflammation. Front Immunol. (2012) 3:4. doi: 10.3389/fimmu.2012.00004
7. Lichtnekert J, Kawakami T, Parks WC, Duffield JS. Changes in macrophage phenotype as the immune response evolves. Curr Opin Pharmacol. (2013) 13:555–64. doi: 10.1016/j.coph.2013.05.013
8. Bugge TH, Flick MJ, Daugherty CC, Degen JL. Plasminogen deficiency causes severe thrombosis but is compatible with development and reproduction. Genes Dev. (1995) 9:794–807. doi: 10.1101/gad.9.7.794
9. Ploplis VA, Carmeliet P, Vazirzadeh S, Van Vlaenderen I, Moons L, Plow EF, et al. Effects of disruption of the plasminogen gene on thrombosis, growth, and health in mice. Circulation. (1995) 92:2585–93. doi: 10.1161/01.CIR.92.9.2585
10. Gong Y, Hart E, Shchurin A, Hoover-Plow J. Inflammatory macrophage migration requires MMP-9 activation by plasminogen in mice. J Clin Investig. (2008) 118:3012–24. doi: 10.1172/JCI32750
11. Wygrecka M, Marsh LM, Morty RE, Henneke I, Guenther A, Lohmeyer J, et al. Enolase-1 promotes plasminogen-mediated recruitment of monocytes to the acutely inflamed lung. Blood. (2009) 113:5588–98. doi: 10.1182/blood-2008-08-170837
12. Gong Y, Hoover-Plow J. The plasminogen system in regulating stem cell mobilization. J Biomed Biotechnol. (2012) 2012:437920. doi: 10.1155/2012/437920
13. Carmo AA, Costa BR, Vago JP, de Oliveira LC, Tavares LP, Nogueira CR, et al. Plasmin induces in vivo monocyte recruitment through protease-activated receptor-1-, MEK/ERK-, and CCR2-mediated signaling. J Immunol. (2014) 193:3654–63. doi: 10.4049/jimmunol.1400334
14. Andronicos NM, Chen EI, Baik N, Bai H, Parmer CM, Kiosses WB, et al. Proteomics-based discovery of a novel, structurally unique, and developmentally regulated plasminogen receptor, Plg-RKT, a major regulator of cell surface plasminogen activation. Blood. (2010) 115:1319–30. doi: 10.1182/blood-2008-11-188938
15. Bai H, Baik N, Kiosses WB, Krajewski S, Miles LA, Parmer RJ. The novel plasminogen receptor, plasminogen receptor(KT) [Plg-R(KT)], regulates catecholamine release. J Biol Chem. (2011) 286:33125–33. doi: 10.1074/jbc.M111.218693
16. Lighvani S, Baik N, Diggs JE, Khaldoyanidi S, Parmer RJ, Miles LA. Regulation of macrophage migration by a novel plasminogen receptor Plg-R KT. Blood. (2011) 118:5622–30. doi: 10.1182/blood-2011-03-344242
17. Miles LA, Baik N, Lighvani S, Khaldoyanidi S, Varki NM, Bai H, et al. Deficiency of plasminogen receptor, Plg-RKT, causes defects in plasminogen binding and inflammatory macrophage recruitment in vivo. J Thromb Haemost. (2017) 15:155–62. doi: 10.1111/jth.13532
18. Del Rosso M, Fibbi G, Pucci M, Margheri F, Serrati S. The plasminogen activation system in inflammation. Front Biosci. (2008) 13:4667–86. doi: 10.2741/3032
19. Syrovets T, Lunov O, Simmet T. Plasmin as a proinflammatory cell activator. J Leukoc Biol. (2012) 92:509–19. doi: 10.1189/jlb.0212056
20. Shen Y, Guo Y, Mikus P, Sulniute R, Wilczynska M, Ny T, et al. Plasminogen is a key proinflammatory regulator that accelerates the healing of acute and diabetic wounds. Blood. (2012) 119:5879–87. doi: 10.1182/blood-2012-01-407825
21. Rosenwald M, Koppe U, Keppeler H, Sauer G, Hennel R, Ernst A, et al. Serum-derived plasminogen is activated by apoptotic cells and promotes their phagocytic clearance. J Immunol. (2012) 189:5722–8. doi: 10.4049/jimmunol.1200922
22. Das R, Ganapathy S, Settle M, Plow EF. Plasminogen promotes macrophage phagocytosis in mice. Blood. (2014) 124:679–88. doi: 10.1182/blood-2014-01-549659
23. Borg RJ, Samson AL, Au AE, Scholzen A, Fuchsberger M, Kong YY, et al. Dendritic cell-mediated phagocytosis but not immune activation is enhanced by plasmin. PLoS ONE. (2015) 10:e0131216. doi: 10.1371/journal.pone.0131216
24. Sugimoto MA, Ribeiro ALC, Costa BRC, Vago JP, Lima KM, Carneiro FS, et al. Plasmin and plasminogen induce macrophage reprogramming and regulate key steps of inflammation resolution via annexin A1. Blood. (2017) 129:2896–907. doi: 10.1182/blood-2016-09-742825
25. Sousa LP, Lopes F, Silva DM, Tavares LP, Vieira AT, Rezende BM, et al. PDE4 inhibition drives resolution of neutrophilic inflammation by inducing apoptosis in a PKA-PI3K/Akt-dependent and NF-kappaB-independent manner. J Leukoc Biol. (2010) 87:895–904. doi: 10.1189/jlb.0809540
26. Vago JP, Nogueira CR, Tavares LP, Soriani FM, Lopes F, Russo RC, et al. Annexin A1 modulates natural and glucocorticoid-induced resolution of inflammation by enhancing neutrophil apoptosis. J Leukoc Biol. (2012) 92:249–58. doi: 10.1189/jlb.0112008
27. Rhys HI, Dell'Accio F, Pitzalis C, Moore A, Norling LV, Perretti M. Neutrophil microvesicles from healthy control and rheumatoid arthritis patients prevent the inflammatory activation of macrophages. Ebio Med. (2018) 29:60–9. doi: 10.1016/j.ebiom.2018.02.003
28. Vago JP, Tavares LP, Garcia CC, Lima KM, Perucci LO, Vieira EL, et al. The role and effects of glucocorticoid-induced leucine zipper in the context of inflammation resolution. J Immunol. (2015) 194:4940–50. doi: 10.4049/jimmunol.1401722
29. Vago JP, Tavares LP, Sugimoto MA, Lima GL, Galvao I, de Caux TR, et al. Proresolving actions of synthetic and natural protease inhibitors are mediated by annexin A1. J Immunol. (2016) 196:1922–32. doi: 10.4049/jimmunol.1500886
30. Ward C. NF-kappa B Activation is a critical regulator of human granulocyte apoptosis in vitro. J Biol Chem. (1999) 274:4309–18. doi: 10.1074/jbc.274.7.4309
31. Montero-Melendez T, Patel HB, Seed M, Nielsen S, Jonassen TEN, Perretti M. The melanocortin agonist AP214 exerts anti-inflammatory and proresolving properties. Am J Pathol. (2011) 179:259–69. doi: 10.1016/j.ajpath.2011.03.042
32. Newson J, Stables M, Karra E, Arce-Vargas F, Quezada S, Motwani M, et al. Resolution of acute inflammation bridges the gap between innate and adaptive immunity. Blood. (2014) 124:1748–64. doi: 10.1182/blood-2014-03-562710
33. Swamydas M, Lionakis MS. Isolation, purification and labeling of mouse bone marrow neutrophils for functional studies and adoptive transfer experiments. J Vis Exp. (2013) 77:e50586. doi: 10.3791/50586
34. Griffith JW, Sokol CL, Luster AD. Chemokines and chemokine receptors: positioning cells for host defense and immunity. Annu Rev Immunol. (2014) 32:659–702. doi: 10.1146/annurev-immunol-032713-120145
35. Murray PJ, Allen JE, Biswas SK, Fisher EA, Gilroy DW, Goerdt S, et al. Macrophage activation and polarization: nomenclature and experimental guidelines. Immunity. (2014) 41:14–20. doi: 10.1016/j.immuni.2014.07.009
36. Sica A, Mantovani A. Macrophage plasticity and polarization: in vivo veritas. J Clin Invest. (2012) 122:787–95. doi: 10.1172/JCI59643
37. Perretti M, D'Acquisto F. Annexin A1 and glucocorticoids as effectors of the resolution of inflammation. Nat Rev Immunol. (2009) 9:62–70. doi: 10.1038/nri2470
38. Sugimoto MA, Vago JP, Teixeira MM, Sousa LP. Annexin A1 and the resolution of inflammation: modulation of neutrophil recruitment, apoptosis, and clearance. J Immunol Res. (2016) 2016:8239258. doi: 10.1155/2016/8239258
39. Norling LV, Perretti M. Control of myeloid cell trafficking in resolution. J Innate Immun. (2013) 5:367–76. doi: 10.1159/000350612
40. Dalli J, Serhan CN. Pro-resolving mediators in regulating and conferring macrophage function. Front Immunol. (2017) 8:1400. doi: 10.3389/fimmu.2017.01400
41. Ploplis VA, French EL, Carmeliet P, Collen D, Plow EF. Plasminogen deficiency differentially affects recruitment of inflammatory cell populations in mice. Blood. (1998) 91:2005–9.
42. Busuttil SJ, Ploplis VA, Castellino FJ, Tang L, Eaton JW, Plow EF. A central role for plasminogen in the inflammatory response to biomaterials. J Thromb Haemost. (2004) 2:1798–805. doi: 10.1111/j.1538-7836.2004.00916.x
43. Swaisgood CM, Schmitt D, Eaton D, Plow EF. In vivo regulation of plasminogen function by plasma carboxypeptidase B. J Clin Invest. (2002) 110:1275–82. doi: 10.1172/JCI0215082
44. O'Connell PA, Surette AP, Liwski RS, Svenningsson P, Waisman DM. S100A10 regulates plasminogen-dependent macrophage invasion. Blood. (2010) 116:1136–46. doi: 10.1182/blood-2010-01-264754
45. Park JH, Kim YG, Shaw M, Kanneganti TD, Fujimoto Y, Fukase K, et al. Nod1/RICK and TLR signaling regulate chemokine and antimicrobial innate immune responses in mesothelial cells. J Immunol. (2007) 179:514–21. doi: 10.4049/jimmunol.179.1.514
46. Cushing SD, Berliner JA, Valente AJ, Territo MC, Navab M, Parhami F, et al. Minimally modified low density lipoprotein induces monocyte chemotactic protein 1 in human endothelial cells and smooth muscle cells. Proc Natl Acad Sci USA. (1990) 87:5134–8. doi: 10.1073/pnas.87.13.5134
47. Standiford TJ, Kunkel SL, Phan SH, Rollins BJ, Strieter RM. Alveolar macrophage-derived cytokines induce monocyte chemoattractant protein-1 expression from human pulmonary type II-like epithelial cells. J Biol Chem. (1991) 266:9912–8.
48. Brown Z, Strieter RM, Neild GH, Thompson RC, Kunkel SL, Westwick J. IL-1 receptor antagonist inhibits monocyte chemotactic peptide 1 generation by human mesangial cells. Kidney Int. (1992) 42:95–101. doi: 10.1038/ki.1992.266
49. Barna BP, Pettay J, Barnett GH, Zhou P, Iwasaki K, Estes ML. Regulation of monocyte chemoattractant protein-1 expression in adult human non-neoplastic astrocytes is sensitive to tumor necrosis factor (TNF) or antibody to the 55-kDa TNF receptor. J Neuroimmunol. (1994) 50:101–7. doi: 10.1016/0165-5728(94)90220-8
50. Cailhier JF, Partolina M, Vuthoori S, Wu S, Ko K, Watson S, et al. Conditional macrophage ablation demonstrates that resident macrophages initiate acute peritoneal inflammation. J Immunol. (2005) 174:2336–42. doi: 10.4049/jimmunol.174.4.2336
51. Burysek L, Syrovets T, Simmet T. The serine protease plasmin triggers expression of MCP-1 and CD40 in human primary monocytes via activation of p38 MAPK and janus kinase (JAK)/STAT signaling pathways. J Biol Chem. (2002) 277:33509–17. doi: 10.1074/jbc.M201941200
52. Yao Y, Tsirka SE. Truncation of monocyte chemoattractant protein 1 by plasmin promotes blood-brain barrier disruption. J Cell Sci. (2011) 124(Pt 9):1486–95. doi: 10.1242/jcs.082834
53. Yao Y, Tsirka SE. The CCL2-CCR2 system affects the progression and clearance of intracerebral hemorrhage. Glia. (2012) 60:908–18. doi: 10.1002/glia.22323
54. Mantovani A, Sica A, Sozzani S, Allavena P, Vecchi A, Locati M. The chemokine system in diverse forms of macrophage activation and polarization. Trends Immunol. (2004) 25:677–86. doi: 10.1016/j.it.2004.09.015
55. Meznarich J, Malchodi L, Helterline D, Ramsey SA, Bertko K, Plummer T, et al. Urokinase plasminogen activator induces pro-fibrotic/m2 phenotype in murine cardiac macrophages. PLoS ONE. (2013) 8:e57837. doi: 10.1371/journal.pone.0057837
56. Genua M, D'Alessio S, Cibella J, Gandelli A, Sala E, Correale C, et al. The urokinase plasminogen activator receptor (uPAR) controls macrophage phagocytosis in intestinal inflammation. Gut. (2015) 64:589–600. doi: 10.1136/gutjnl-2013-305933
57. Luzina IG, Keegan AD, Heller NM, Rook GA, Shea-Donohue T, Atamas SP. Regulation of inflammation by interleukin-4: a review of “alternatives.” J Leukoc Biol. (2012) 92:753–64. doi: 10.1189/jlb.0412214
58. Casella G, Garzetti L, Gatta AT, Finardi A, Maiorino C, Ruffini F, et al. IL4 induces IL6-producing M2 macrophages associated to inhibition of neuroinflammation in vitro and in vivo. J Neuroinflammation. (2016) 13:139. doi: 10.1186/s12974-016-0596-5
59. Garlet GP, Giannobile WV. Macrophages: The bridge between inflammation resolution and tissue repair? J Dent Res. (2018) 97:1079–81. doi: 10.1177/0022034518785857
60. Pupjalis D, Goetsch J, Kottas DJ, Gerke V, Rescher U. Annexin A1 released from apoptotic cells acts through formyl peptide receptors to dampen inflammatory monocyte activation via JAK/STAT/SOCS signalling. EMBO Mol Med. (2011) 3:102–14. doi: 10.1002/emmm.201000113
61. Tang S, Wan M, Huang W, Stanton RC, Xu Y. Maresins: specialized proresolving lipid mediators and their potential role in inflammatory-related diseases. Mediat Inflamm. (2018) 2018:2380319. doi: 10.1155/2018/2380319
62. Li Q, Laumonnier Y, Syrovets T, Simmet T. Plasmin triggers cytokine induction in human monocyte-derived macrophages. Arterioscler Thromb Vasc Biol. (2007) 27:1383–9. doi: 10.1161/ATVBAHA.107.142901
63. Guo Y, Li J, Hagstrom E, Ny T. Beneficial and detrimental effects of plasmin(ogen) during infection and sepsis in mice. PLoS ONE. (2011) 6:e24774. doi: 10.1371/journal.pone.0024774
64. Blasi F, Carmeliet P. uPAR: a versatile signalling orchestrator. Nat Rev Mol Cell Biol. (2002) 3:932–43. doi: 10.1038/nrm977
65. Bystrom J, Evans I, Newson J, Stables M, Toor I, van Rooijen N, et al. Resolution-phase macrophages possess a unique inflammatory phenotype that is controlled by cAMP. Blood. (2008) 112:4117–27. doi: 10.1182/blood-2007-12-129767
66. N AG, Quintana JA, Garcia-Silva S, Mazariegos M, Gonzalez de la Aleja A, Nicolas-Avila JA, et al. Phagocytosis imprints heterogeneity in tissue-resident macrophages. J Exp Med. (2017) 214:1281–96. doi: 10.1084/jem.20161375
Keywords: resolution of inflammation, plasminogen system, plasminogen receptor KT, macrophages reprogramming, efferocytosis
Citation: Vago JP, Sugimoto MA, Lima KM, Negreiros-Lima GL, Baik N, Teixeira MM, Perretti M, Parmer RJ, Miles LA and Sousa LP (2019) Plasminogen and the Plasminogen Receptor, Plg-RKT, Regulate Macrophage Phenotypic, and Functional Changes. Front. Immunol. 10:1458. doi: 10.3389/fimmu.2019.01458
Received: 07 March 2019; Accepted: 10 June 2019;
Published: 28 June 2019.
Edited by:
Haichao Wang, Feinstein Institute for Medical Research, United StatesReviewed by:
Laurie Kay McCauley, University of Michigan, United StatesCopyright © 2019 Vago, Sugimoto, Lima, Negreiros-Lima, Baik, Teixeira, Perretti, Parmer, Miles and Sousa. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Lindsey A. Miles, bG1pbGVzQHNjcmlwcHMuZWR1; Lirlândia P. Sousa, bGlwc291c2E3MkBnbWFpbC5jb20=
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.