
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Immunol. , 26 June 2019
Sec. Vaccines and Molecular Therapeutics
Volume 10 - 2019 | https://doi.org/10.3389/fimmu.2019.01456
 Shivali Gupta1Berenice Salgado-Jiménez1†
Shivali Gupta1Berenice Salgado-Jiménez1† Nandadeva Lokugamage1†
Nandadeva Lokugamage1† Juan Carlos Vázquez-Chagoyán2
Juan Carlos Vázquez-Chagoyán2 Nisha Jain Garg1*
Nisha Jain Garg1*Background: Chagas cardiomyopathy is caused by Trypanosoma cruzi (Tc). Two antigenic candidates, TcG2 and TcG4, are recognized by antibodies in naturally infected dogs and humans; and these vaccine candidates provided protection from Tc infection in mice and dogs. Trypanosoma rangeli (Tr) is non-pathogenic to mammals and shown to elicit cross-reactive anti-Tc antibodies. In this study, we investigated if fixed Tr (fTr) can further enhance the efficacy of the TcG2/TcG4 DNA vaccine.
Methods and Results: C57BL/6 mice were immunized with TcG2/TcG4 DNA vaccine and fTr (delivered as an adjuvant or in prime-boost approach), and challenged with Tc. Serology studies showed that fTr (±quil-A) elicited Tc- and Tr-reactive IgGs that otherwise were not stimulated by TcG2/TcG4 vaccine only, and quil-A had suppressive effects on fTr-induced IgGs. After challenge infection, TcG2/TcG4-vaccinated mice exhibited potent expansion of antigen- and Tc-specific IgGs that were not boosted by fTr±quil-A. Flow cytometry analysis showed that TcG2/TcG4-induced dendritic cells (DC) and macrophages (Mφ) responded to challenge infection by expression of markers of antigen uptake, processing, and presentation, and production of pro-inflammatory cytokines. TcG2/TcG4-induced CD4+T cells acquired Th1 phenotype and expressed markers that orchestrate adaptive immunity. A fraction of vaccine-induced CD4+T cells exhibited iTreg phenotype responsible for aversion of self-injurious immune responses. Further, TcG2/TcG4-vaccinated mice exhibited potent expansion of poly-functional CD8+T cells with TNF-α/IFN-γ production and cytolytic phenotype post-infection. Subsequently, tissue parasites and pathology were hardly detectable in TcG2/TcG4-vaccinated/infected mice. Inclusion of fTr±quil-A had no clear additive effects in improving the Tc-specific adaptive immunity and parasite control than was noted in mice vaccinated with TcG2/TcG4 alone. Non-vaccinated mice lacked sufficient activation of Th1 CD4+/CD8+T cells, and exhibited >10-fold higher levels of tissue parasite burden than was noted in vaccinated/infected mice.
Conclusion: TcG2/TcG4 vaccine elicits highly effective immunity, and inclusion of fTr is not required to improve the efficacy of DNA vaccine against acute Tc infection in mice.
Chagas cardiomyopathy, caused by Trypanosoma cruzi, is a major health concern in Latin America and it is an emerging disease in the United States, Europe, Japan, and other countries (1). The majority of individuals exposed to T. cruzi remain seropositive for their life. In ~30% of the infected individuals, clinical symptoms progress from cardiac hypertrophic remodeling (i.e., wall thickening) to dilated cardiomyopathy, and ultimately result in cardiac arrest and death (2).
The sequencing of T. cruzi genome (3) and the development of approaches to produce recombinant proteins at low cost have made it feasible to produce, deliver, and test the efficacy of a variety of recombinant T. cruzi antigens as potential vaccine candidates in experimental models of infection and disease. We have screened several candidate antigens, and selected TcG1, TcG2, and TcG4 for further development as potential vaccine candidates. These antigens are phylogenetically conserved in clinically important T. cruzi strains, expressed in infective and intracellular stages of the parasite, and recognized by parasite-specific cellular and humoral immune responses in multiple T. cruzi-infected hosts (4–7). Further, we showed that prophylactic immunization with TcG1, TcG2, and TcG4 based subunit vaccine(s) elicited parasite-specific lytic antibodies and cytolytic T cell responses, and Th1 cytokines in mice and dogs (8–12). Recent studies have tested several other antigenic candidates as vaccine for their prophylactic and therapeutic efficacy against Chagas disease. Results of these vaccines are encouraging and summarized in recent reviews (13–15). However, till to date none of the anti-T. cruzi vaccines have produced sterile immunity in any of the experimental animal models.
The use of heterologous DNA-prime/inactivated microorganism-boost vaccine (13) has been previously reported with promising results. T. rangeli (Tr) exhibits significant homology (>60%) with T. cruzi proteome (15, 16), but Tr is not pathogenic for mammals (17, 18), and, therefore, it can be cultivated in large batches in clean, biosafety level one laboratory facilities. Studies in mice and dogs have suggested that immunization with fixed, lysed, or fractionated Tr can provide a degree of protection from T. cruzi infection and histopathological lesions produced during Chagas disease (19–21). When used in combination with a subunit vaccine, booster immunization with glutaraldehyde fixed T. rangeli (fTr) epimastigotes appeared to lower the blood parasitemia and tissue parasite foci in dogs (12). However, the immunological mechanisms that might be elicited by fTr to provide protection against T. cruzi infection are not known.
In this study, we aimed to determine if fTr enhances the efficacy of the DNA vaccine against T. cruzi. For this, we immunized mice with TcG2/TcG4 DNA and fTr (individually or in combination, or in a prime-boost approach), and then challenged with T. cruzi. We analyzed the reactivity of the vaccine-induced antibody responses against T. cruzi and T. rangeli, and profiled the activation of antigen presenting cells (APC), i.e., dendritic cells (DC) and macrophages (Mφ), and CD4+ and CD8+ T cells after vaccination and challenge infection. We also evaluated the efficacy of vaccine compositions in controlling parasite dissemination in tissues and examined histopathology of the heart and skeletal muscles, focusing on the efficacy of vaccination protocol in reducing the tissue injury by acute T. cruzi infection.
All animal experiments were conducted following the National Institutes of Health guidelines for housing and care of laboratory animals and in accordance with protocols approved by the Institutional Animal Care and Use Committee (protocol number 08-05-029) at The University of Texas Medical Branch at Galveston.
All experiments were conducted in ABSL2/BSL2-approved laboratory and all personnel have received appropriate ABSL2/BSL2 training.
The cDNAs for TcG2 and TcG4 (Genbank: AY727915 and AY727917, respectively) were cloned in pCDNA3.1 eukaryotic expression plasmid (10). Recombinant plasmids were transformed into E. coli DH5-alpha-competent cells, grown in Luria-Bertani broth containing 100 μg/ml ampicillin, and purified by anion exchange chromatography by using a Qiagen Endofree maxi prep kit (Qiagen, Chatsworth, CA) (4). Purified plasmids were used at a concentration of 25 μg each plasmid per vaccination.
T. rangeli (Tr, Guatemala strain) epimastigote cultures were propagated in LIT media (12). Tr epimastigotes (1 × 109/ml) were fixed with 0.1% glutaraldehyde solution (Sigma-Aldrich), washed three times with 1X cold Phosphate Buffered Saline (PBS). Fixed Tr (fTr) lysate (1 × 108 Tr equivalent in 100 μl PBS) was used with or without 5 μg quil A (QA) for vaccination (12).
C57BL/6 female mice (6-week-old) were obtained from Harlan Labs (Indianapolis, IN). To assess if fTr and Quil A (QA, saponin) adjuvant the DNA vaccine-induced responses, mice were vaccinated in following groups: (1) DNA vaccine only, two doses; (2) DNA vaccine + fTr, two doses; (3) DNA vaccine + QA, two doses; (4) DNA vaccine + fTr + QA, two doses. To determine if fTr boosts the DNA vaccine-induced immune responses, mice were vaccinated with DNA vaccine followed by fTr (gp5) or fTr+QA (gp6). Each dose of DNA vaccine was constituted of 25-μg of each plasmid (pCDNA3.TcG2 and pCDNA3.TcG4) and delivered in 100 μl PBS by intramuscular (im) injection in the hind thighs. When used, fTr (1 × 108 Tr in 100 μl PBS) was delivered by subcutaneous (sc) injection. When added, vaccine was emulsified with 5 μg QA per dose per mouse. Non-vaccinated (N) mice were used as controls. Prime and booster doses of vaccine were given at 21-day intervals. Immunized and control mice were euthanized at 21 days' post-vaccination (pv), and sera, splenocytes, and lymph node (LN) cells were collected to evaluate the vaccine-induced immunological responses.
For challenge infection, T. cruzi (Tc) trypomastigotes (Sylvio X10/4 strain) were maintained and propagated by continuous in vitro passage in C2C12 cells. We included gp1, gp5, and gp6 for challenge studies because mice in these groups exhibited maximal vaccine-induced T cell immunity. Mice were immunized as above, challenged with Tc (10,000 trypomastigotes/mouse, intraperitoneal) at 21 days after the 2nd vaccine dose, and euthanized at 21 days' post-infection (pi). Non-vaccinated mice infected with Tc (T) and euthanized at similar time-points were used as controls. The vaccination and challenge infection scheme is presented in Figure 1A.
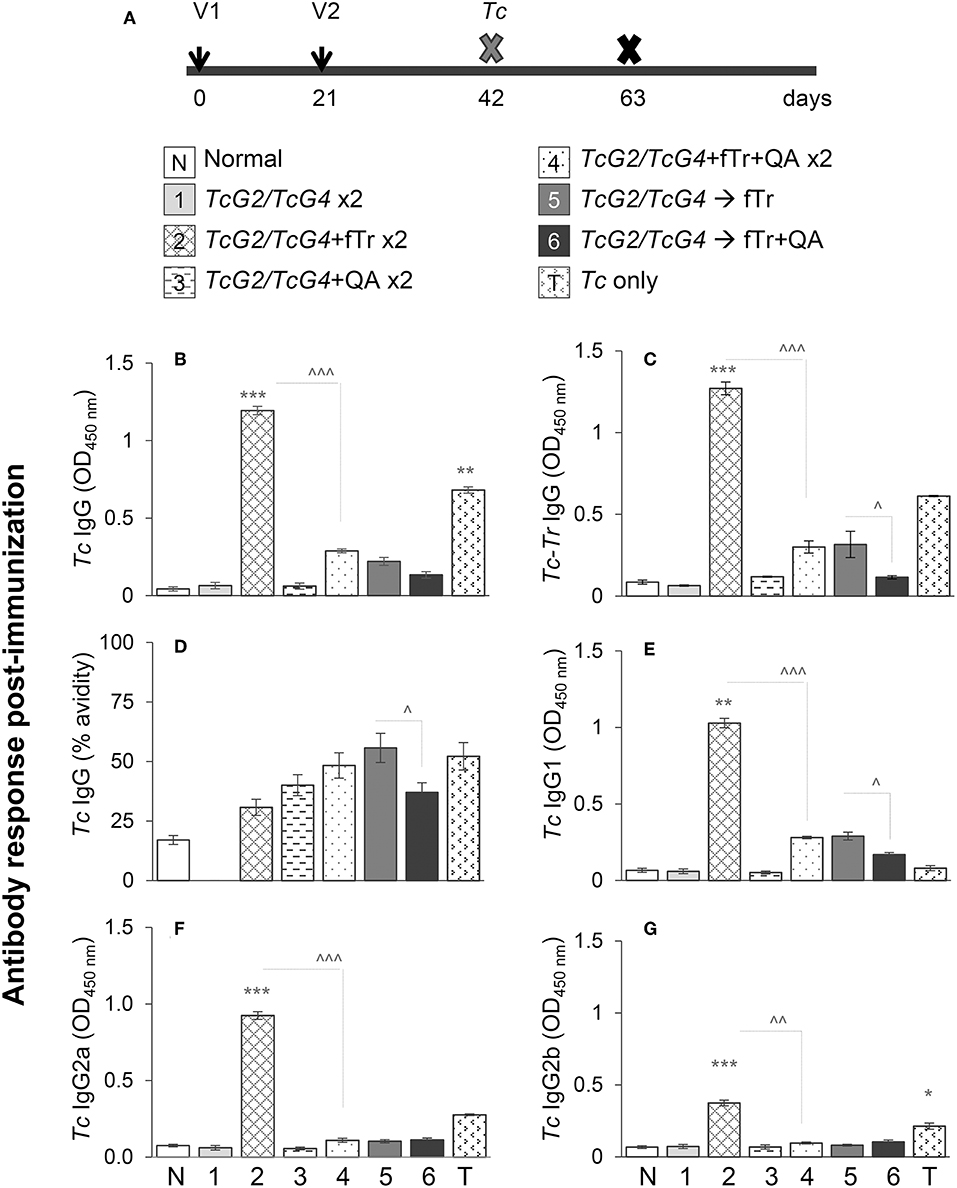
Figure 1. T. cruzi-specific antibody response in vaccinated mice. (A) Schematic of experimental plan is shown. C57BL/6 female mice were immunized with six different compositions of vaccines as described in Materials and Methods. Mice were immunized with dose 1 at day 0, dose 2 at day 21, and on day 42 euthanized to examine the vaccine-induced responses or challenged with Tc and then euthanized at day 63 to study the significance of vaccine in providing protection from infection. Vaccines (per dose) were constituted with 25-μg each of pCDNA3.TcG2 and pCDNA3.TcG4 plasmid DNA (intramuscular) and/or 1 × 108 T. rangeli fixed with 0.1% glutaraldehyde (fTr) with or without 5-μg Quil A (QA, subcutaneous). (B–G) T. cruzi-specific IgG (B), IgG1 (E), IgG2a (F), and IgG2b (G) antibodies in sera samples were measured by an ELISA. The vaccine-induced IgGs specificity for T. cruzi was determined after pre-adsorbing the sera samples with T. rangeli soluble lysate (C) and avidity of vaccine induced IgGs was calculated as [O.D. with urea/O.D. without urea treatment] × 100 (D). Sera samples from non-vaccinated or infected mice were used as negative and positive controls, respectively. Data (mean ± SD) are representative of two independent experiments (n = 4 mice per group per experiment, duplicate observations per sample), and significance is presented as * (none vs. vaccinated) or ∧ (comparison among vaccinated groups). The p-values of <0.05, <0.01, and p < 0.001 are annotated with one, two, and three symbols, respectively.
The cDNAs for TcG2 and TcG4 were cloned in-frame with a C-terminal His-tag into a pET-22b plasmid (Novagen, Gibbstown, NJ). Plasmids were transformed in BL21 (DE3) pLysS-competent cells, and recombinant proteins were purified by using the poly-histidine fusion, peptide-metal chelation chromatography system (8, 11). Tc trypomastigotes and Tr epimastigotes (2 × 107 each) were sonicated in 1 mL of 50 mM carbonate-bicarbonate buffer (pH 9.6), and centrifuged for 10 min at 10,000 × g at 4°C. The supernatants of Tc lysate (TcL) and Tr lysate (TrL) were used as soluble antigens for serology.
Serum immunoglobulin G (IgG) response was monitored by an enzyme linked immunosorbent assay (ELISA). Briefly, 96-well flat-bottom plates (Falcon, Becton Dickinson, Oxnard, CA) were coated for 2 h with TcL or TrL (5 × 105 protozoans' equivalent/well) or recombinant TcG2 and TcG4 proteins (10-μg/ml). Plates were blocked for 1 h with 5% non-fat dry milk (NFDM, Bio-Rad) in PBS-0.01% Tween 20 (PBST). Plates were washed and incubated for 2 h with sera from vaccinated, vaccinated/infected and control mice (1:100–1:1,000 dilutions in 0.5% NFDM, 100 μl/well). To detect the total IgG levels, plates were then directly incubated for 30 min with goat anti-mouse IgG–horseradish peroxidase (HRP) conjugate. To detect IgG subtypes, plates were sequentially incubated for 2 h with biotin-conjugated goat anti-mouse Ig subtypes (IgG1, IgG2a, IgG2b, or IgG2c), and for 30 min with streptavidin-HRP conjugate. All incubations were carried at 37°C, and plates were washed at each step with PBST and PBS. All antibodies were purchased from Southern Biotech, and used at 1: 5,000 dilutions in PBST-0.5% NFDM. Color was developed by incubation with 100 μl/well Sure Blue TMB substrate (Kirkegaard & Perry Labs) and recorded at 450 nm by using a SpectraMax M5 microplate reader (Molecular Devices). In some experiments, sera samples were pre-absorbed with TcL or TrL for 1 h before use in an ELISA assay (8, 16).
To examine the antigen-avidity index, plates were coated with TcL as above, blocked with 5% NFDM, and sequentially incubated with sera samples (1: 100 dilutions in 1% NFDM) for 1 h, 6 M urea for 30 min, and HRP-conjugated secondary antibody for 30 min. A colorimetric reaction was performed as above (% Avidity: [O.D. with urea/O.D. without urea treatment] × 100) (17).
Single-cell suspensions of spleen or lymph nodes (LN) from vaccinated, vaccinated/infected, non-vaccinated/infected, and non-vaccinated/non-infected mice were prepared by standard methods. Splenic and LN cell counts in vaccinated and control mice are presented in Table S1. Splenocytes or LN cells (105 cells/100 μl color-free RPMI) were used immediately for flow cytometry. In some experiments, splenocytes were in vitro stimulated with TcL (25-μg/ml) at 37°C, 5% CO2 for 48 h. The un-stimulated and in vitro- stimulated cells were washed in staining buffer (2% BSA/0.02% sodium azide in PBS) and incubated for 15 min with Fc Block (anti-CD16/CD32; BD Pharmingen). Cells were then incubated for 30 min at 4°C in dark with the fluorochrome-conjugated antibodies, washed twice in PBS, fixed in 2% paraformaldehyde, and washed again. Fluorescent cells were visualized by using a FACS-Calibur Cell Analyzer (BD Biosciences), acquiring >20,000 events in a live gate, and further analyzed by using FlowJo software (version 7.6.5, Tree-Star, San Carlo, CA) (22).
For the measurement of intracellular cytokines and immune cell activation markers, splenocytes were stimulated as above except that brefeldin A (10-μg/ml, Sigma) or monensin (5 μg/ml) (BD Pharmingen) was added for the final 6 h of culture to block protein secretion. Cells were then labeled with antibodies for surface markers, fixed with 2% paraformaldehyde, re-suspended in 100-μl permeabilization buffer (0.1% saponin/1% FBS in PBS), and utilized for intracellular staining of cytokines or specific markers. Samples were analyzed by flow cytometry as above. In all experiments, unstained cells and cells stained with isotype-matched IgGs and FMO were used as controls (13, 22).
All antibodies used for flow cytometry in this study are listed in Table S2. Briefly, to profile the antigen presenting cells, splenic and LN cells of all vaccinated and control groups before and challenge infection were first gated for Ly6GloCD11c+ dendritic cells (DC) and Ly6GloLy6C+CD11b+ monocytes/macrophages (Mφ). Each of the cell populations were further analyzed by flow cytometry for the expression of CD209 (DC-SIGN) C-type lectin receptor that recognizes PAMPs and activates phagocytosis and/or antigen presentation, CD205 that is involved in internalization, processing and presentation of antigens, MHCI/MHCII molecules that bind to peptide fragments from pathogens and present to the T cells, and CD80 that is the ligand for CD28 and CTLA4 and provides co-stimulatory signal for adaptive T cell response. The splenic CD11b+ Mφ cell subsets were also analyzed for intracellular expression of markers of classical/proinflammatory (IL-1βhi and TNF-αhi) and alternative/immunomodulatory (CD200+, CD206+) functional phenotype after in vitro stimulation with TcL (25-μg/mL) for 48 h (18).
To evaluate the T cell profile, splenocytes from vaccinated, vaccinated/infected, and control groups were in vitro stimulated with TcL (25-μg/mL) for 48 h. Splenic CD3+ lymphocytes were gated for CD4+ and CD8+ T cells. Each of the T cell subsets were further analyzed for the expression of CD69 the early activation marker of lymphocyte proliferation, CD11a (ITGAL) that along with CD18 forms lymphocyte function-associated antigen 1 (LFA-1) and binds with ICAMs to facilitate intercellular adhesion of leukocytes, CD95 (Fas) / CD95L (FasL) involved in immune elimination of infected cells, CD44 and CD62L molecules to distinguish naïve (CD62LhiCD44lo), effector (CD62LloCD44hi), and central memory (CD62LhiCD44hi) phenotypes, and intracellular production of Th1 cytokines (IFN-γ, TNF-α). The CD4+T cells were also examined for the surface expression of CD25 and FoxP3 to profile the splenic T regulatory cells. The CD8+T cells were also analyzed for the intracellular expression of perforin and CD107a that are markers of cytolytic activity (10, 11).
For histological studies, heart and skeletal muscle tissue sections from vaccinated/infected and non-vaccinated/infected mice were fixed in 10% buffered formalin for 24 h, dehydrated in absolute alcohol, cleared in xylene, and embedded in paraffin. Paraffin-embedded 5-micron tissue-sections were stained with hematoxylin and eosin (H&E) and evaluated by light microscopy. Heart and skeletal muscle tissue slides (three mice per group, at least two slides per tissue, 10 microscopic fields per slide) were analyzed by light microscopy, and the presence of inflammatory cells was scored as (0)–absent/none, (1)–focal or mild with ≤ 1 foci, (2)–moderate with ≥2 inflammatory foci, (3)–extensive with generalized coalescing of inflammatory foci or disseminated inflammation (4)–severe with diffused inflammation, interstitial edema, and loss of tissue integrity (23).
To examine the parasite burden, heart and skeletal muscles tissues (10 mg) from vaccinated/infected and non-vaccinated/infected mice were subjected to proteinase K lysis, and total DNA was purified by phenol/chloroform extraction and ethanol precipitation. A real-time quantitative PCR was performed on an iCycler thermal cycler with SYBR Green Supermix (Bio-Rad), 50 ng of total DNA, and oligonucleotides specific for Tc18SrDNA (forward, 5′- TTTTGGGCAACAGCAGGTCT-3′; reverse, 5′- CTGCGCCTACGAGACATTCC-3′; amplicon size: 199 bp) and murine GAPDH (forward, 5′-AACTTTGGCATTGTGGAAGG-3′; reverse, 5′-ACACATTGGGGGTAGGAACA-3′; amplicon size: 223 bp). The threshold cycle (CT) values for Tc18SrDNA were normalized to GAPDH reference cDNA. The relative parasite burden (i.e., Tc18SrDNA level) was calculated by following the 2−ΔΔCt, where ΔCt represents the CT (sample)—Ct (GAPDH) and ΔΔCt represents ΔCt (sample)—ΔCt (no treatment control).
Data are expressed as mean ± SD (n = 4/group/experiment, minimum of duplicate observations per experiment). Data were analyzed by the Student's t-test (comparison of 2 groups) and 1-way analysis of variance (ANOVA) with Tukey's post-hoc test (comparison of multiple groups) by using an SPSS (version 14.0, SPSS Inc, Chicago, Illinois) or Graph Pad InStat ver.3 software. Significance is presented as * (vaccinated vs. non-vaccinated or vaccinated/infected vs. non-vaccinated/infected) and ∧ (comparison of vaccinated groups) and p-values of <0.05, <0.01, and 0.001 are annotated by one, two, or three symbols, respectively.
We first monitored if fTr, delivered as an adjuvant or as an antigen in prime-boost approach, modulates the TcG2/TcG4 DNA vaccine—induced antibody response in mice. For this, sera samples were obtained at 21 days after the 2nd vaccine dose, and analyzed by an ELISA. Mice immunized/boosted with TcG2/TcG4 (±QA, gp1, and gp3) exhibited no detectable levels of Tc-specific antibodies (Figures 1B,C). In comparison, all other vaccinated groups of mice that received fTr (±QA) as adjuvant or booster exhibited detectable, though variable, levels of Tc-specific IgGs with 25–51% avidity for Tc antigens (Figures 1B,C). Including fTr as an adjuvant with TcG2/TcG4 vaccine (gp2) elicited maximal levels of Tc-specific IgGs (*p < 0.001) and addition of QA suppressed the fTr-induced IgG (compare gp2 vs. gp4, ∧p < 0.001) (Figures 1B,C). Interestingly, avidity of fTr induced antibodies for Tc antigens was minimal among all vaccinated groups (compare gp2 with other groups, Figure 1D). Likewise, TcG2/TcG4 vaccine adjuvanted with fTr also resulted in maximal levels of Tc-specific IgG sub-types (IgG1, IgG2a, and IgG2b) (gp2 vs. all other groups, *p < 0.01) while co-delivery of QA had a suppressive effect on Tc-specific IgG subtypes stimulated by TcG2/TcG4 vaccine adjuvanted with fTr (∧p < 0.001, compare gp2 and gp4) or boosted with fTr (∧p < 0.05, compare gp5 and gp6) (Figures 1E–G).
Tr-specific IgGs constituted of IgG1, IgG2a, and IgG2b subtypes were also detected in mice that received fTr as an adjuvant with TcG2/TcG4 DNA or as a booster vaccine, and maximal Tr-specific antibodies were measured in sera of mice given fTr as an adjuvant with TcG2/TcG4 DNA vaccine (group 2 vs. control, *p < 0.001, Figures S1A–D). Low levels of Tr-reactive IgGs were also observed in mice infected with T. cruzi (Figure S1B). Normal mice and mice injected with vector only exhibited no Tc- and Tr-specific antibody response. Together, the results presented in Figure 1 and Figure S1 suggest that (a) TcG2/TcG4 DNA vaccine induced low levels of Tc- and antigen-specific antibodies, (b) co-delivery of fTr as an adjuvant elicited maximal levels of Tc- as well as Tr-reactive IgGs with minimal avidity for Tc antigens, and (c) addition of QA with fTr had an overall suppressive effect on antibody responses induced in vaccinated mice.
We evaluated the effect of vaccine compositions on the expansion of splenic and lymph node (LN) cells in mice. With respect to splenic cells, the overall frequency of splenocytes (range: 80–193-million cells) was increased by 62–141% in vaccinated (vs. control) mice (Table S1). Mice that received booster dose of TcG2/TcG4, fTr, or fTr+QA exhibited maximal expansion of splenocytes post-vaccination, while mice receiving TcG2/TcG4 adjuvanted with fTr, QA or fTr+QA exhibited minimal expansion of splenic immune cells (gp1, gp5, gp6, Table S1). With respect to lymph nodes, we noted up to 52% expansion of LN cells (range: 144-220-million cells) in vaccinated mice, and maximal expansion was noted in mice immunized with TcG2/TcG4 and boosted with fTr or fTr+QA (gp5 and gp6, Table S1). Based on the observations that fTr, QA, and fTr+QA adjuvants (gp2, gp3, gp4) did not enhance the TcG2/TcG4-induced expansion of splenic and LN cells (Table S1) as well as high avidity antibody responses (Figure 1), we excluded the gp2, gp3, and gp4 from further studies.
To examine if vaccine-induced IgGs expanded in response to infection, mice were challenged with T. cruzi at 21 days' pv, and sera samples were obtained at 21 days' pi (corresponds to acute parasitemia phase). All mice challenged with Tc, irrespective of the vaccination regimen, exhibited significant levels of Tc-, TcG2-, and TcG4-specific IgGs (vs. normal controls, *p < 0.05, Figures 2A–C). Booster immunization with fTr (±QA) slightly enhanced the hosts' capacity to respond to challenge infection with expansion of Tc-specific antibodies than was observed in TcG2/TcG4-immunized/infected mice (Figure 2A); however, booster immunization with fTr (±QA) did not enhance the TcG2- and TcG4-specific IgGs in DNA vaccinated mice (Figures 2B,C, compare gp5 and gp6 with gp1). The maximal levels of Tc- and antigen-specific IgGs were detected in infected mice that did not receive any vaccine (vs. vaccinated/infected, ∧p < 0.05, Figures 2A,C). We also monitored the Tc and antigen-specific antibody sub-types in immunized mice post challenge infection. Overall, all mice responded to challenge infection with a significant increase in Tc- TcG2- and TcG4-specific IgG1, IgG2a, and IgG2c antibody subtypes (vs. normal controls, *p < 0.01, Figures 2D–L). Maximal levels of Tc- and TcG2/TcG4-specific antibody subtypes (IgG2a+IgG2c > IgG1) were detected in non-vaccinated/infected mice (vs. normal controls, *p < 0.001, Figures 2D–L). Mice immunized with TcG2/TcG4 and boosted with TcG2/TcG4 also exhibited a predominance of Tc and antigen-specific IgG2a/2c (vs. IgG1) antibody sub-types post-challenge infection (compare Figures 2G–L with Figures 2D–F). Further, booster immunization with fTr (±QA) slightly, but non-significantly, enhanced the Tc- and antigen-specific antibody subtypes post-challenge infection than was observed in TcG2/TcG4-immunized/infected mice (Figures 2D–L). Together the results presented in Figure 2 suggest that (a) challenge infection elicits a predominance of antigen- and parasite-specific IgG2a/2c antibodies that are not enhanced by pre-immunization with DNA vaccine; and (b) fTr (±QA) did not improve the extent of antigen- and parasite-specific IgGs in TcG2/TcG4-vaccinated mice.
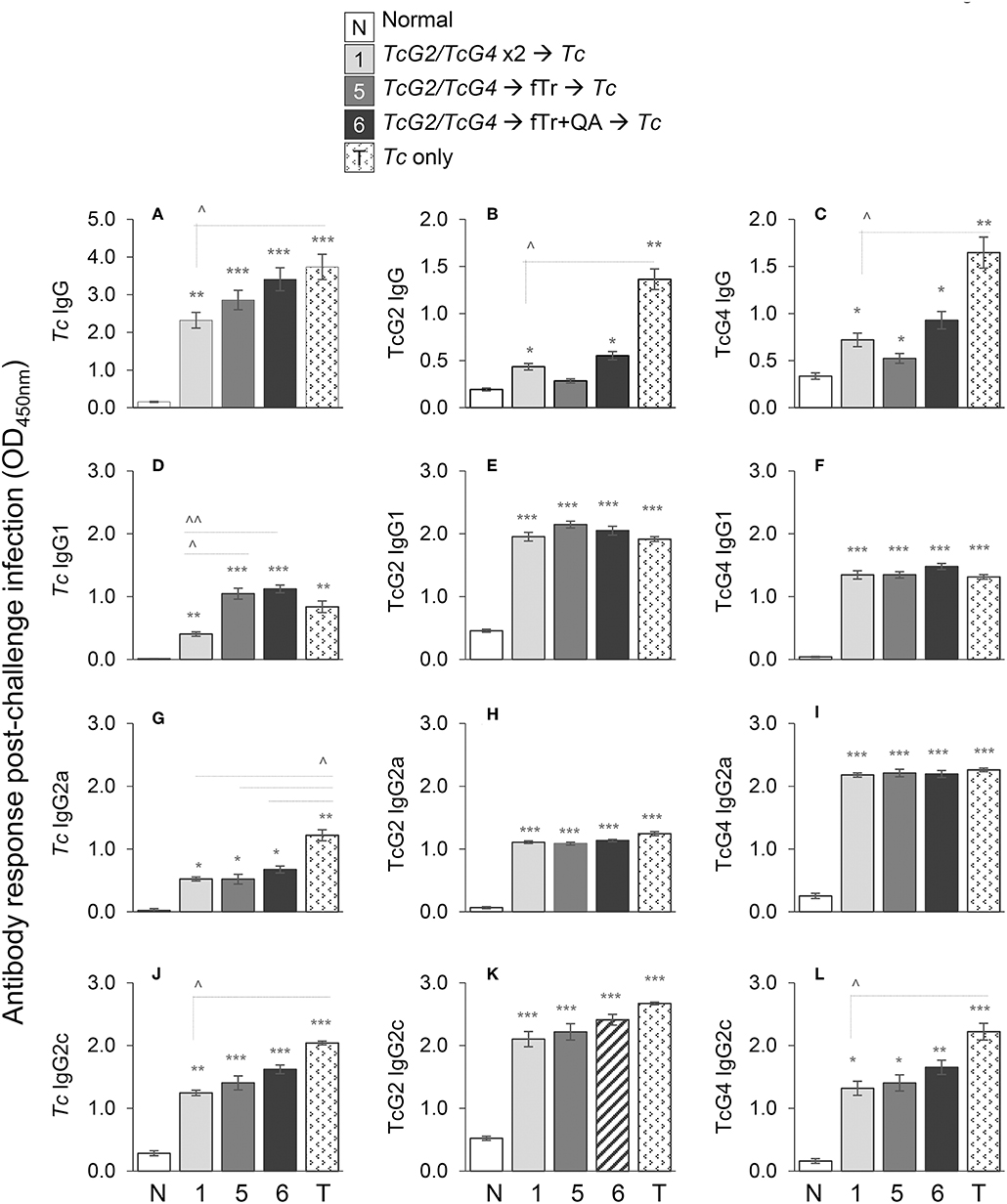
Figure 2. Antibody response in vaccinated mice after challenge infection. C57BL/6 female mice were immunized with subunit vaccine (pCDNA3.1 encoding TcG2 and TcG4) at day 0, boosted with subunit vaccine, fTr, or fTr+QA at day 21, and challenged with T. cruzi (10,000 parasites per mouse, intraperitoneal) at day 42. Mice were euthanized at day 63 (i.e., 21 days' post challenge infection). Sera samples were analyzed by an ELISA to evaluate the IgG (A–C), IgG1 (D–F), IgG2a (G–I), and IgG2c (J–L) levels captured with T. cruzi lysate (A,D,G,J) and recombinant TcG2 (B,E,H,K), and TcG4 (C,F,I,L) antigens. Sera samples from normal and infected mice were used as controls. Data (mean ± SD) are representative of duplicate observations per sample (n = 4 mice per group per experiment), and significance is annotated as * (none vs. vaccinated or infected vs. vaccinated/infected) and ∧ (comparison of vaccinated groups) (*,∧p < 0.05, **,∧∧p < 0.01, and ***p < 0.001).
The overall frequencies of DC (Ly6GloCD11c+, spleen: 4.4–6.2%, LN: 0.97–1.7%) as well as of CD209+ (maturation marker) and MHCI+ (antigen presentation marker) DC were either not changed or slightly decreased in spleen and LN of vaccinated (vs. non-vaccinated/control) mice (Figures 3A,B). However, the splenic frequencies of CD205+, MHCII+ and CD80+ DC, indicating an increase in antigen uptake, processing, and presentation to CD4+T cells, were increased by >2-fold in TcG2/TcG4 DNA vaccinated mice (all, *p < 0.01, Figure 3A). An increase in antigen presenting capacity of LN DC was also evidenced by ~2-fold increase in the frequency of CD205+ and MHCII+ DC in TcG2/TcG4 DNA vaccinated mice (*p < 0.05, Figure 3B). Importantly, boosting the TcG2/TcG4 DNA vaccine with fTr (±QA) had none or suppressive effects on the splenic and LN DC activation (Figures 3A,B).
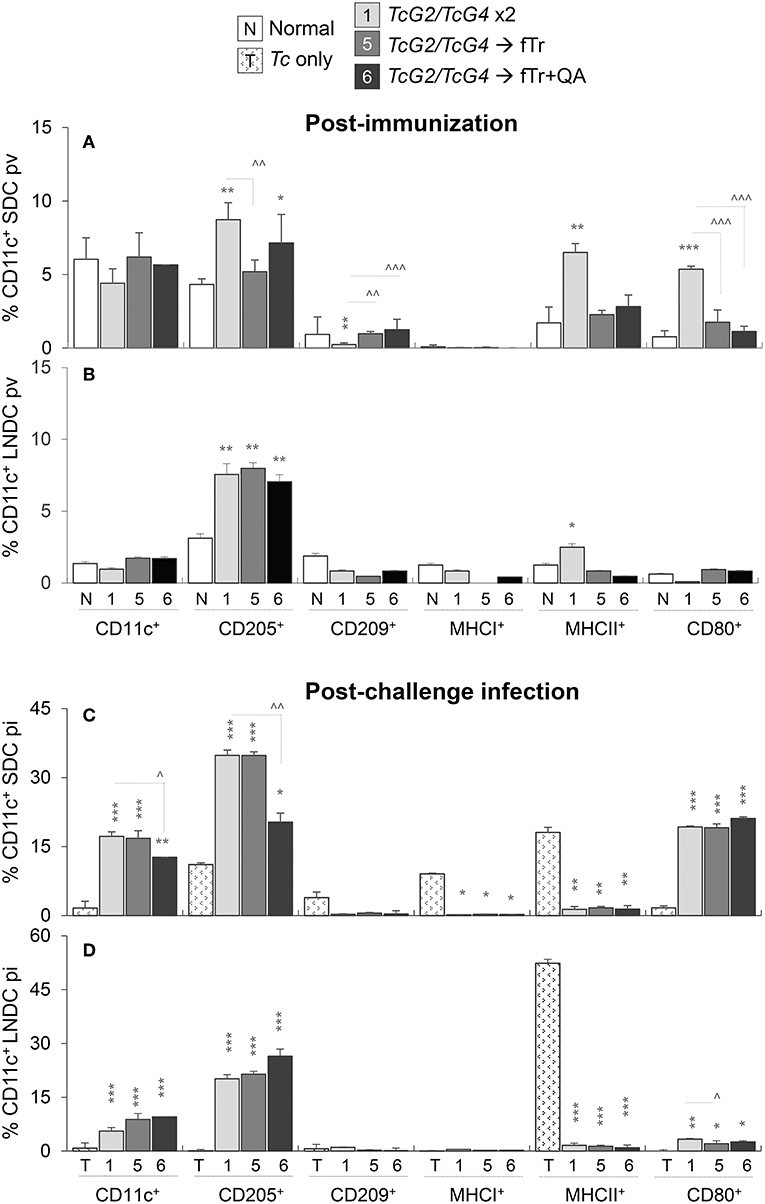
Figure 3. Antigen presenting capacity of dendritic cells (DC) in vaccinated mice (± T. cruzi). Mice were vaccinated and challenged with T. cruzi trypomastigotes (10,000 parasites/mouse), as in Figures 1, 2. Mice were euthanized at 21 days' post-vaccination (pv) or 21 days' post-infection (pi). Single cell suspensions of spleen (A,C) and lymph nodes (B,D) of vaccinated (A,B) and vaccinated/infected (C,D) mice were labeled with fluorochrome-conjugated antibodies and analyzed by flow cytometry. Bar graphs show ex vivo percentage of Ly6GloCD11c+ splenic DC (SDC) and lymph node DC (LNDC) subsets that expressed markers of maturation (CD209+), stimulation (CD80+), antigen uptake (CD205+), and antigen presentation (MHCI+, MHCII+) in vaccinated and vaccinated/infected mice. SDC and LNDC from non-vaccinated/non-infected (none) and non-vaccinated/infected mice were used as controls. Data are presented as mean ± SD and representative of duplicate observations per sample (n = 4 mice per group per experiment). Significance is annotated as * (none vs. vaccinated or infected vs. vaccinated/infected) and ∧ (comparison of vaccinated groups) (*,∧p < 0.05, **,∧∧p < 0.01, and ***,∧∧∧p < 0.001).
Challenge infection with T. cruzi led to > 3-fold increase in the frequency of CD11c+ DC and a comparable increase in the frequency of CD205+ and CD80+ DC in the splenic and LN compartment of mice that were immunized with TcG2/TcG4 DNA vaccine (compare Figures 3A,B with Figures 3C,D). Boosting with fTr or ftr+QA had non-significant or suppressive effects on TcG2/TcG4-induced DC activation post-challenge infection, and frequencies of MHCI+ and MHCII+ DC were not significantly changed in spleen and LN of vaccinated groups post-infection (Figures 3C,D). Non-vaccinated/infected (vs. normal control) mice exhibited an overall contraction of the splenic and LN levels of Ly6GloCD11c+ DC, and the low number of surviving DC primarily exhibited a MHCII+ phenotype in non-vaccinated/infected mice (Figures 3C,D). Together, the results presented in Figure 3 suggest that (a) TcG2/TcG4 DNA vaccine induced DC depot was capable of responding to Tc infection by increased proliferation and phenotypic (antigen uptake, processing, and presentation to CD4+T cells) activation, and (b) fTr booster was not useful in enhancing the DC response induced by DNA vaccine in mice before or after challenge infection.
The splenic and LN profile of Mφ are presented in Figure 4 and Figure S2. The overall percentages of Ly6GloLy6C+CD11b+ Mφ (range: 1.5–2.2% of total splenocytes) as well as of CD209+ and MHCI+ Mφs were either not changed or decreased in the splenic compartment of vaccinated mice (vs. non-vaccinated controls, Figure 4A). Mice immunized with TcG2/TcG4 (gp2) exhibited 60%, 170%, and 1350% increase in the frequencies of CD205+, MHCII+, and CD80+ Mφ, indicating an increase in antigen uptake, processing, and presentation, respectively, to CD4+T cells (all, **p < 0.01, Figure 4A), while booster immunization with fTr or fTr+QA (gp5 and gp6) resulted in increased expression of CD205 only (vs. normal controls, Figure 4A). Upon challenge infection, TcG2/TcG4 vaccinated mice exhibited a further 3-4-fold increase in splenic frequency of Mφ that were primarily CD205+ (compare Figures 4A,B). Including fTr or fTr+QA booster dose had either no effects or suppressive effects on CD209+, MHCI+, and MHCII+ Mφ responses after TcG2/TcG4 vaccination and challenge infection (∧p < 0.05, Figures 4A,B), and CD80+ Mφ were significantly enhanced by fTr+QA booster immunization in challenged mice, suggesting an increase in B7-1 costimulatory signal for T cell activation.
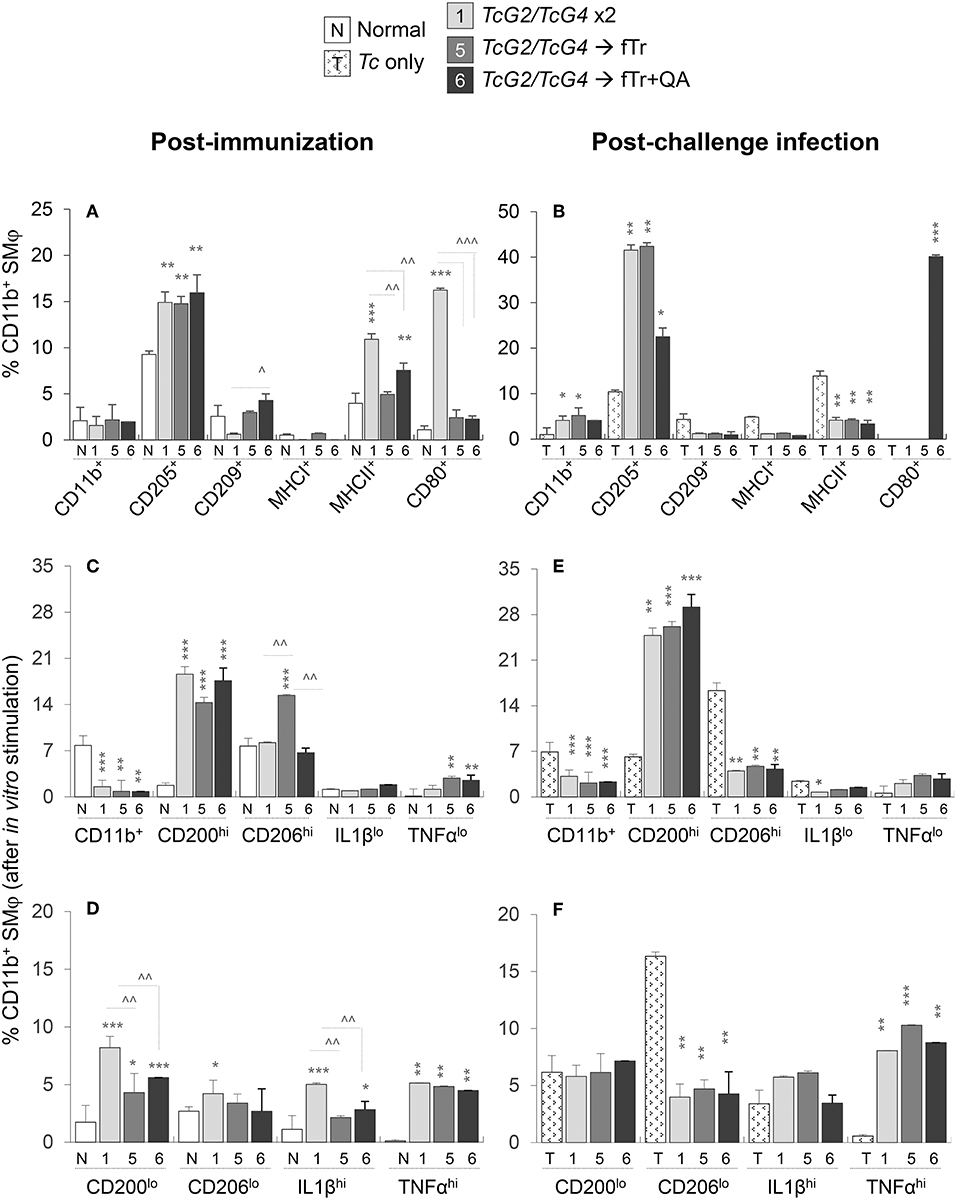
Figure 4. Antigen presenting capacity and functional profile of splenic macrophages (Mφ) in vaccinated mice (± T. cruzi). Mice were vaccinated and challenged with T. cruzi trypomastigotes as in Figures 1, 2. (A,B) Splenocytes were labeled with fluorochrome-conjugated antibodies and analyzed by flow cytometry. Bar graphs show ex vivo percentages of the splenic Ly6GloCD11b+ Mφ that exhibited surface expression of markers of maturation, antigen uptake, and antigen presentation (CD205+, CD209+, MHCI+, MHCII+, CD80+) in vaccinated (A) and vaccinated/infected (B) mice. (C–F) Splenic cells from vaccinated (C,D) and vaccinated/infected (E,F) mice were in vitro stimulated for 48 h in the presence of soluble T. cruzi lysate (TcL), and then labeled with fluorochrome-conjugated antibodies. Shown are the mean percentages of CD11b+ Mφ that responded to TcL stimulation with expression of markers of classical/proinflammatory (IL-1βhi, TNF-αhi) and alternative/immunomodulatory (CD200hi, CD206hi) phenotype. Splenic cells from non-vaccinated/non-infected and non-vaccinated/infected mice were used as controls. Data (mean ± SD) are representative of two independent experiments (n = 3 mice per group per experiment, duplicate observations per mouse), and significance is annotated as *none vs. vaccinated or infected vs. vaccinated/infected, and ∧comparison of vaccinated groups (*,∧p < 0.05, **,∧∧p < 0.01, and ***,∧∧∧p < 0.001).
Functional analysis of splenic Mφ after in vitro stimulation with Tc lysate for 48 h showed 5-9-fold increase in the frequencies of proinflammatory (CD200lo, IL-1βhi, and TNF-αhi) and immunomodulatory (CD200hi and TNF-αlo) CD11b+ Mφ in TcG2/TcG4-vaccinated mice (vs. normal controls, all, ***p < 0.05 Figures 4C,D). Splenic Mφ of vaccinated/infected mice responded to in vitro stimulation with Tc lysate with a further >2-fold increase in the splenic expansion of CD200hi and TNF-αhi Mφ (compare Figures 4C,D with Figures 4E,F). Except for an increase in CD206hiCD11b+ Mφ population (Figure 4C), inclusion of fTr booster had non-significant effects on TcG2/TcG4-induced Mφ activation before or after challenge infection, and co-delivery of QA had an overall suppressive effect on the splenic CD11b+ Mφ in vaccinated mice (Figures 4C–F). Non-vaccinated mice exhibited an increase in CD206lo Mφ sub-population post-challenge infection (Figure 4F).
In the LN compartment, non-significant changes or a decline in the percentages of CD205+, CD209+, MHCI+ MHCII+, and CD80+ Mφ were detected in TcG2/TcG4-vaccinated (vs. control) mice (Figure S2A). Inclusion of fTr or fTr+QA booster had an overall suppressive effect on all of the TcG2/TcG4-induced sub-populations of LN Mφ, except that fTr+QA booster enhanced the frequency of MHCII+CD11b+ Mφ in DNA-vaccinated mice (Figure S2A ). Further, challenge infection resulted in no increase in the overall percentages of CD11b+ LN Mφ in any of the vaccinated groups (Figure S2B). Yet, a substantial phenotypic activation, evidenced by an increase in the frequency of CD205+, MHCI+, and CD80+ Mφ, was noted in LN of TcG2/TcG4 vaccinated/infected mice (compare Figures S2A,B). Inclusion of fTr (±QA) booster increased the CD205 and CD80 expression and had no significant effect on the CD209 and MHCII expression than that noted in TcG2/TcG4 immunized mice post-challenge infection (Figure S2B). Non-vaccinated/infected (vs. normal control) mice continued to exhibit a predominance of antigen presenting Mφ (MHCII+) in the LN compartment (Figure S2B). Together, the results presented in Figure 4 and Figure S2 suggest that (a) TcG2/TcG4 DNA vaccine induced Mφ depot was capable of responding to T. cruzi infection by increased proliferation, and proinflammatory activation (antigen uptake, processing, and presentation to CD4+ T cells; production of IL-1β+ and TNF-α cytokines), and (b) fTr based booster immunization had minimal-to-none effects in further enhancing the Mφ response induced by DNA vaccine in mice before or after challenge infection.
Next, we examined if the TcG2/TcG4-induced activation of antigen presenting cells modulate the splenic CD4+T cell responses in mice after immunization and challenge infection. For this, splenocytes from vaccinated, vaccinated/infected, and control mice were in vitro stimulated with Tc lysate, CD3+ lymphocytes were gated for CD4+T and CD8+T cell subsets, and each of the T cell subsets were further analyzed by flow cytometry. Vaccinated (vs. normal control) mice exhibited a significant increase in splenic population of CD4+T cells (range: 9.76–18.8% of total) and CD4+TNF-α+T cells (range: 2.1–7.4% of CD4+T cells), respectively (all, *p < 0.05, Figure 5A). Maximal expansion in CD4+T cells was noted in mice immunized with TcG2/TcG4 and boosted with fTr±QA. No significant differences in the expression of other markers of CD4+T cell activation (CD69, CD95, CD95L) were noted in any of the vaccinated (vs. normal control) groups (Figure 5B). Interestingly, fTr booster skewed the CD4+T cells from effector (CD62LloCD44hi) to central memory (CD62LloCD44hi) phenotype and addition of QA reversed this effect in vaccinated mice (Figure 5C).
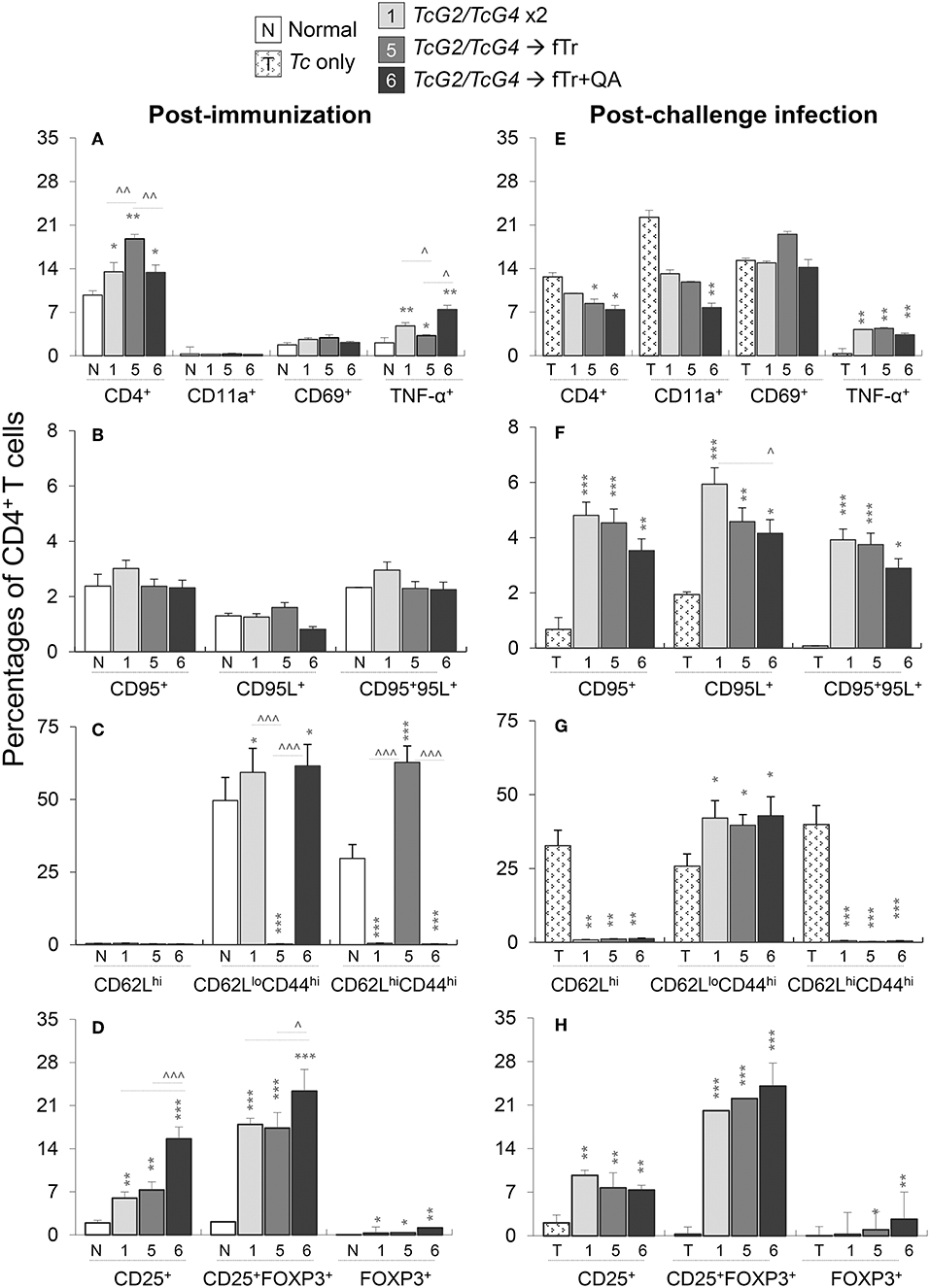
Figure 5. CD4+T cell functional profile in vaccinated mice (± T. cruzi). Mice were vaccinated and challenged with T. cruzi trypomastigotes as in Figures 1, 2. Splenocytes from vaccinated (A–D) and vaccinated/infected (E–H) mice were in vitro stimulated for 48 h in the presence of soluble T. cruzi lysate, labeled with fluorochrome-conjugated antibodies, and analyzed by flow cytometry. Shown are the percentages of PE+CD4+ splenic T cells exhibiting the expression of markers of (A,E) recruitment (CD11a+, CD69+) and activation (TNF-α+); (B,F) cell death (CD95+, CD95L+); (C,G) effector/memory phenotype (CD62L+, CD44+); and (D,H) T regulatory phenotype (CD25+ ± FoxP3+). Data (mean ± SD) are representative of two independent experiments (n = 3 mice per group per experiment, duplicate observations per sample), and significance is annotated as *none vs. vaccinated or infected vs vaccinated/infected, and ∧comparison of vaccinated groups (*,∧p < 0.05, **,∧∧p < 0.01, and ***,∧∧∧p < 0.001).
Upon challenge infection, CD4+T cells constituted 7.4–12.6% of the total splenocytes, and a large proportion of the CD4+T cells exhibited a CD69+/CD11a+ phenotype (range: 7.7–22%) indicative of T cell recruitment and early activation (Figure 4E). Importantly, CD4+T cells in vaccinated/infected (vs. non-vaccinated/infected) mice exhibited >10-fold and 2-6-fold increase in Th1 cytokine (TNF-α) production and Fas/FasL (CD95+/CD95L+) expression, respectively (*p < 0.01, Figures 5E,F), that are required to support CD8+T cells in eliminating intracellular infection. Moreover, up to 24% of the naïve CD4+T cells also differentiated to induced T regulatory (iTreg) phenotype (CD25+FoxP3+) in vaccinated mice before and challenge infection (*p < 0.01, Figures 5D,H). While adjuvanting with fTr±QA had no effect (data not shown), delivery of fTr±QA as booster dose substantially increased the expansion of CD4+T cells and iTreg population when compared to that noted in mice immunized with TcG2/TcG4 only (all, ∧p < 0.05, Figures 5A,D). However, fTr±QA booster did not enhance the DNA vaccine induced CD4+T cell responses after challenge infection (Figures 5E–H). In non-vaccinated/infected (vs. normal control) mice, the CD4+T cells exhibited an increase in the expression of CD11a and CD69, but maintained a predominance of naïve (CD62Lhi) or central memory (CD62LhiCD44hi) phenotype (Figures 5E–G). Together, the results presented in Figure 5 suggest that (a) TcG2/TcG4 DNA vaccine induced CD4+T cells acquired the Th1 phenotype evidenced by TNF-α production and expression of markers that orchestrate adaptive immunity to kill intracellular pathogen; (b) a fraction of the vaccine-induced CD4+T cells acquired iTreg phenotype to potentially avert the over-reactivity of the immune responses and prevent self-tissue injury; and (c) including fTr booster immunization slightly shifted the CD4+T cell response toward iTreg population in vaccinated mice. In comparison, (d) non-vaccinated mice failed to respond to challenge infection with a potent activation of Th1 CD4+T cell response.
The splenic CD8+T cell response to vaccination and challenge infection is presented in Figure 6. The overall CD8+T cell population (range: 17.7–20.1% of total splenocytes) was only slightly increased in vaccinated (vs. control) mice (Figure 6A). Yet, the frequencies of CD8+T cells expressing the phenotypic markers of early activation (CD69, 3.29–8.05% of CD8+T cells) and cell migration/adhesion (CD11a: 0.69–2.78% of CD8+T cells) were increased by 43–144% and 104–302%, respectively, in vaccinated (vs. control) mice (*p < 0.05, Figure 6A), the maximal increase being noted in TcG2/TcG4-immunized mice. Flow cytometry also revealed that vaccination primed type 1 immunity evidenced by a significant increase in the frequency of IFN-γ+ (~1.2% of total, up to 8-fold increase) and TNF-α+ (3.2–7.5%, 0.5–2.5-fold increase) CD8+T cells with a polyfunctional, cytolytic phenotype (Perforin+CD107+: 8.4–27.5% of CD8+T cells, 1.7–7.8-fold increase; Perforin+TNF-α+: 2.5–5.4% of CD8+T cells, 1.3–3.9-fold increase) when compared to non-vaccinated controls (Figures 6A,B, p < 0.05). The maximal increase in CD8+T cell frequency, activation, and poly-functional phenotype was noted in mice immunized with TcG2/TcG4 DNA vaccine only or boosted with fTr+QA. Splenic CD8+T cells exhibited no changes in the expression of other markers of activation (CD95/CD95L, Figure 6C). As was noted for CD4+T cells, immunization with fTr booster skewed the CD8+ T cells from effector (CD62LloCD44hi) toward central memory (CD62LloCD44hi) phenotype and addition of QA reversed this effect in vaccinated mice (Figure 6D).
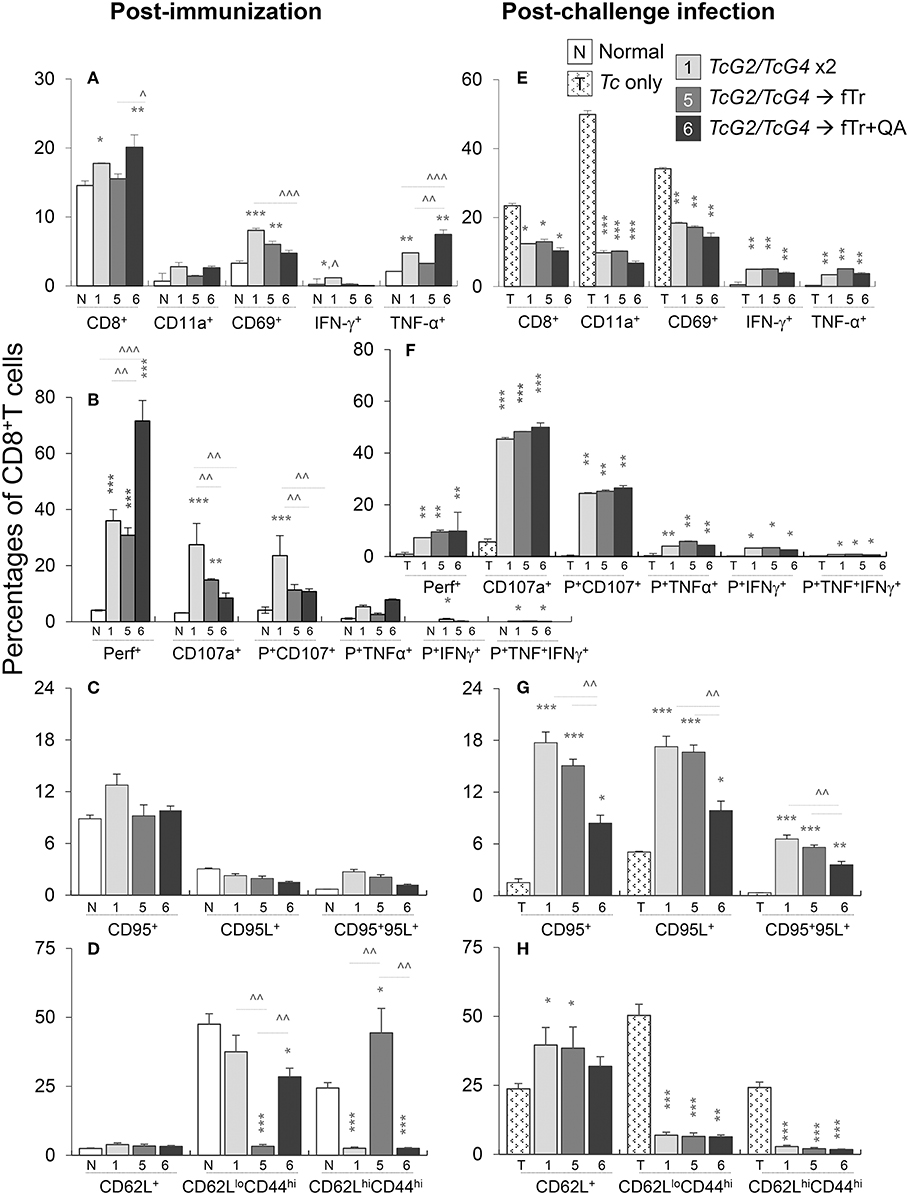
Figure 6. CD8+T cell functional profile in vaccinated mice (± T. cruzi). Mice were vaccinated and challenged with T. cruzi as in Figures 1, 2. Splenocytes from vaccinated (A–D) and vaccinated/infected (E–H) mice were in vitro stimulated for 48 h in the presence of Tc antigenic lysate, and then labeled with fluorochrome-conjugated antibodies. The FITC+CD8+T cells were analyzed by flow cytometry. Shown are the mean percentages of (A,E) CD8+T cells expressing adhesion and migration markers (CD11a+, CD69+) and type 1 cytokines (IFN-γ and TNF-α) production; (B,F) poly-functional CD8+T cells that exhibited several markers of cytolytic activity (perforin+, CD107a+, TNF-α+, and IFN-γ+); (C,D) CD8+T cells with expression of cell death markers (CD95+, CD95L+); and (C,G) CD8+T cells expressing effector/memory markers (CD44+, CD62L+). Data (mean ± SD) are representative of two independent experiments (n = 3 mice per group per experiment, duplicate observations per sample). Significance is annotated as *none vs. vaccinated or infected vs vaccinated/infected, and ∧comparison of vaccinated groups (*,∧p < 0.05, **,∧∧p < 0.01, and ***,∧∧∧p < 0.001).
Upon challenge infection, the overall splenic percentage of CD8+T cells (range: 10.3–13%) contracted in vaccinated mice, yet, CD8+T cells in vaccinated mice responded to challenge infection with an increase in the expression of phenotypic markers of T cell recruitment and early activation (CD69: 14.3–18.3% of CD8+T cells, CD11a: 6.7–10.2% of CD8+T cells, compare Figures 6A,E). Transport of integral membrane proteins' (CD107a and CD107b) to plasma membrane, expression of perforin and granzyme or Fas/FasL (CD95/CD95L), and release of IFN-γ and other cytokines offer effector CD8+T cell function through direct killing of the infected cells and/or pleiotropic effects that suppress intracellular pathogen (19). The flow cytometry data showed that vaccinated (vs. non-vaccinated) mice responded to challenge infection with >8-fold increase in the frequency of Th1 cytokine producing (IFN-γ: 3.9–5.1%, TNF-α: 3.5–5.2%) CD8+T cells (all, *p < 0.05, Figure 6F). Further, the frequencies of CD8+T cells responding to challenge infection with a polyfunctional, cytolytic activation (Perforin+CD107+: 24–26.5%, Perforin+TNF-α+: 4.0–5.8%; CD95+CD95L+: 3.6–6.6%) were significantly increased in vaccinated/infected (vs. non-vaccinated/infected) mice (all, *p < 0.05, Figures 6E–G). The potent increase in CD8+T cell frequency, activation, and functional phenotype noted in mice immunized with TcG2/TcG4 DNA vaccine only was not boosted when fTr±QA were added as adjuvant (data not shown) or as a heterologous booster vaccine (Figure 6). The CD8+T cells of non-vaccinated mice responded to challenge infection with an increase in the markers of early activation (CD11a and CD69, Figure 6E), and effector (CD62LloCD44hi) or central memory (CD62LhiCD44hi) phenotype (Figure 6H) but failed to develop polyfunctional cytolytic response (Figures 6E–G). Together, the results presented in Figure 6 suggest that TcG2/TcG4 vaccine elicited type 1 CD8+T cell activation. The vaccine-induced CD8+T cells were polyfunctional, and, thus, had a potential to act as cytolytic, effector T cells against T. cruzi. The heterologous vaccination with fTr and QA had no clear additive effect in enhancing the TcG2/TcG4 DNA vaccine induced Tc-specific functional CD8+ T cell profile.
Finally, we determined if fTr (±QA) booster immunization enhanced the TcG2/TcG4 vaccine efficacy in controlling inflammatory pathology associated with tissue parasite burden. Histological studies revealed low level of infiltration of inflammatory cells in the heart (Figures 7B–D, histological score: 0–1) and skeletal muscle (Figures 7G–I, histological score: 0–1) of all groups of vaccinated/infected mice. The extent of inflammatory infiltrate in heart and skeletal muscle tissues of non-vaccinated/infected mice (Figures 7E,J, histological score: 2–4) was significantly higher than that detected in tissues of vaccinated/infected mice. Extensive inflammatory foci as well as diffused inflammation, interstitial edema, and loss of tissue integrity were visible in all tissue sections of non-vaccinated/infected mice (Figures 7E,J). In comparison, tissue sections from normal mice showed no inflammatory infiltrate (Figures 7A,F).
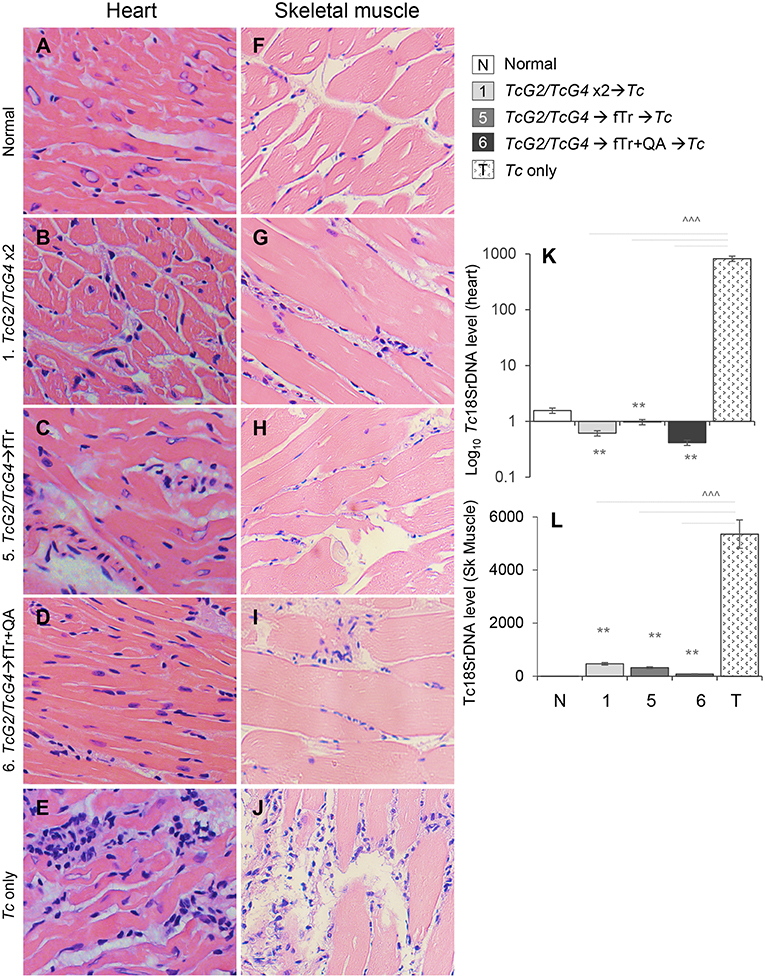
Figure 7. Tissue inflammation and parasite burden in vaccinated mice post-challenge infection. Mice were vaccinated as in Figure 1, challenged with T. cruzi at 21 days' post-vaccination and euthanized at 21 days post-infection. (A–J) Paraffin-embedded left ventricular heart (A–E) and skeletal muscle (F–J) tissue sections (5 μM) were examined by hematoxylin/eosin staining (blue: nuclear; pink: muscle/cytoplasm). Shown are representative H&E images of tissue sections from vaccinated/infected and non-vaccinated/infected mice. Non-treated/non-infected mice were used as controls. (K,L) Total DNA was isolated from heart (K) and skeletal muscle (L) tissue sections and submitted to real-time qPCR amplification of Tc18SrDNA sequence (normalized to murine GAPDH). Data (mean ± SD) are representative of two independent experiments (n = 4 mice per group per experiment, triplicate observations per sample). Significance is annotated as *none vs. vaccinated or infected vs vaccinated/infected, and ∧comparison of vaccinated groups (*,∧p < 0.05, **,∧∧p < 0.01, and ***,∧∧∧p < 0.001).
The control of tissue parasite burden by immunization was validated by real time qPCR. All vaccinated/infected mice exhibited non-detectable levels of Tc18SrDNA in the heart tissue (Figure 7K). The skeletal muscle levels of Tc18SrDNA were also barely detectable in vaccinated mice (Figure 7L). A slightly better control of parasite burden in the skeletal muscle was noted in mice vaccinated with TcG2/TcG4 DNA followed by fTr+QA than in mice given other vaccine compositions (Figure 7L). In comparison, non-vaccinated/infected mice exhibited >10-fold higher levels Tc18SrDNA in the myocardial and skeletal muscle tissues post-challenge infection (∧p < 0.001, Figures 7K,L). Together, these results suggest that TcG2/TcG4-based DNA vaccine is highly effective in reducing the acute tissue parasite burden and associated inflammatory pathology, and boosting the TcG2/TcG4-based immunity with fTr (±QA) provides minimal benefits in enhancing the vaccine efficacy against acute T. cruzi persistence and tissue injury in mice.
In this study, we have conducted detailed immunological analysis to determine how a homologous DNA-prime/DNA-boost vaccine provides protection from T. cruzi infection and examined if the DNA vaccine's efficacy can be complemented with T. rangeli as an adjuvant or antigen.
Subunit vaccines are the safest form of vaccine (17, 18). Pre-clinical and clinical trials have demonstrated significant efficacy of subunit vaccines, e.g., THYB-03 (tuberculosis), influvac (influenza), rts,s/AS01 (malaria), gD2gB2-MF59 (HSV2), and RV144 (HIV) (20). In the context of Chagas disease, several candidate antigens (e.g., ASP2, CRP, cruzipain, GP90, GP82, GP56, Tc24, Tc52) have been tested as subunit vaccine, delivered in the form of DNA, recombinant protein, or a host of viral and bacterial expression vectors, in small animal models [reviewed in Padilla et al. (19) and Zak and Aderem (20)]. However, in a majority of the studies, the selected vaccine candidates were chosen based on the preferences or biases of the research teams and investigators. To prevent the investigator bias in vaccine design, we developed a computational/bioinformatic algorithm for screening the Tc sequence database for the vaccine candidates (4). We identified 11 potential candidates that were submitted to rigorous analysis for eliciting immunity against Tc (5, 8, 21, 24). Eventually, we narrowed down to TcG1, TcG2, and TcG4 candidates that exhibited relevant characteristics as vaccine candidates. These antigens were (1) highly conserved in clinically relevant Tc isolates (4), (2) expressed (mRNA/protein) in trypomastigotes and amastigotes (mammalian stages) of Tc (4), and (3) recognized by IgGs and type 1 CD8+T cells in experimentally or naturally infected mice (5), dogs (6), and humans (7). In a series of studies, we have tested the vaccine potential of TcG1, TcG2, and TcG4 through the use of recombinant eukaryotic plasmid DNA or viral vectors or as a recombinant protein, in eliciting resistance to Tc infection (7–11, 16, 18). The homologous DNA-prime/DNA-boost was simplest in composition, and most cost-effective and field-ready, and therefore tested in several compositions (individual candidate antigens, co-delivery of antigenic candidates, different doses and time-intervals, with or without cytokine adjuvants etc.) (4, 5, 23, 25). We found that each of the three candidates elicit antigen-specific IgG and CD8+T cell responses and co-delivery of the vaccine candidates elicited additive immune responses (8). The TcG1 candidate was least immunogenic; it stimulated antibody and T cell responses only when it was delivered with IL-12 and GM-CSF adjuvants, and, therefore, it was removed from the later formulations (11). Our published studies discussed above and the present results also suggest that two-dose vaccine delivered at 21-day interval is the most efficacious approach for eliciting anti-parasite protective immune responses.
Our choice of fixed Tr as an adjuvant and booster was based on the literature findings that show that Tr shares significant antigenic homology with T. cruzi (26, 27); Tr infects triatomines and mammals but does not induce disease in humans (22, 28), and Tr does not circulate in some of the endemic countries (29). Studies in rodents and dogs have shown that exposure to T. rangeli or use of heat or chemically attenuated Tr elicits antibodies that are reactive against Tc antigens, and antibody response was associated with a degree of resistance to infection with T. cruzi (30–32). It is also proposed that Tr can adjuvant the innate and adaptive immunity against Tc infection, though this is not experimentally shown.
Monocytes (Mo), Mφ, and DCs are the mononuclear phagocytic cells that together constitute monocyte phagocyte system (MPS) (33). Mo/Mφ cells elicit the primary immune defense to invading Tc by proinflammatory cytokines' expression; and superoxide and nitric oxide (NO) production by the NADPH oxidase (NOX2) and inducible nitric oxide synthase (iNOS) enzymes, respectively (34–36). Along with the cytokines and other mediators they produce, Mφ and DC also serve an important function of antigen presentation to adaptive immune system, and thus strongly influence the magnitude and quality of the vaccine induced protection. Our characterization of DC and Mφ responses in this study showed that TcG2/TcG4 DNA immunization increased the frequencies of splenic and LN DCs and Mφ expressing markers of antigen uptake, processing, and presentation (e.g., CD205, CD209, MHCII, CD80). The DNA vaccine induced Mφ, despite the immunomodulatory (CD200hi) phenotype, exhibited proinflammatory functional activation (TNFαhi/IL-1βhi) that was further amplified upon in vitro stimulation with Tc lysate as well as upon challenge infection. The observations that the TcG2/TcG4-induced innate immune cells expanded both the antigen presentation and functional profile in response to 2nd stimulation (Figures 3, 4) suggest that DNA vaccine-induced innate immune cells retained memory of Tc-specific antigen exposure. Indeed, there is growing appreciation for Mφs' contribution to innate immune memory; a recent study showed Mφ memory to S. aureus was tissue specific and transferable between individual animals (37). Also referred as trained immunity, innate immune memory favors the production and release of proinflammatory cytokines (e.g., TNF-α, IL-6, and IL-1β) (38), as we have noted in vaccinated/challenged mice, and it is also defined by metabolic and epigenetic hallmarks. Recent studies have shown that metabolic reprogramming through a shift from oxidative phosphorylation to aerobic glycolysis mediated by Akt/mTOR/HIF-1α pathway is a key mechanism for trained immunity (39); while glycolysis, glutaminolysis, and cholesterol synthesis pathways in Mo/Mφ were suggested to support the epigenetic wiring and induction of improved innate immunity (39, 40). Further studies will be needed to mechanistically demonstrate if TcG2/TcG4 DNA vaccine promoted the metabolic and epigenetic shifts, and evaluate the longevity of the trained innate immunity and its effects on T cell responses. Yet, our results allow us to surmise that the subunit vaccine design harnessed both the effector response of innate immune cells as well as activation state to enhance adaptive T cell response to the antigens.
Quality and quantity of T cell response, and their cytokine polyfunctionality are critical factors in defining the protective efficacy of vaccine(s) against intracellular pathogens. Immunodominant peptides binding stably to MHC class II on the surface of APCs enhance the recruitment and proliferation of effector CD4+ T cells, and secretion of IFN-γ/TNF-α and chemokines by activated APCs and CD4+T cells enhance the CD8+T cells' accumulation (41). Of a number of immune mediators produced by T cells, IFN-γ and TNF-α play a major role in parasite clearance (10, 12), and production of these cytokines together leads to enhanced killing than either cytokines alone. Additionally, CD8+ (and to some degree CD4+) T cells mediate cytolytic activity through the release of perforin/granzymes (10). We have noted that DNA immunization of mice enhanced the frequency of Th1 effector CD4+T cells and poly-functional, cytotoxic CD8+T cells that had the capability to respond to challenge infection with further expansion. The control of tissue parasite burden in immunized mice by >90% suggest that the vaccine induced T cells were highly effective in killing the intracellular pathogen and consequently pathological tissue inflammation was also controlled. CD4+ T cells also support immunoglobulin switch and affinity maturation of B cells that enhances the production of neutralizing antibodies; however, we did not notice an increase in quality or quantity of antibody production in vaccinated mice, thus, suggesting that B cell response was not a major feature of the current vaccine design. Another essential feature of the T cells is the type and homing properties of the memory T cell subsets. Whether the current DNA vaccine will provoke long-term/sustained memory T cell response remains to be seen in future studies.
Our results in this study support the existing literature and clearly demonstrate that use of fTr as an adjuvant to subunit DNA vaccine or as an antigen in a booster vaccine dose elicits Tc-reactive antibodies that are not elicited by DNA vaccine only. However, fTr did not improve the host outcomes evidenced by the findings that the subunit DNA vaccine delivered by a homologous prime/boost approach was capable of providing >90% control of tissue parasite burden, and inclusion of fTr in a heterologous prime/boost approach did not significantly enhance the efficacy of the DNA vaccine. Further, the inclusion of fTr complicates the efficacy of treatment with anti-parasite drugs or vaccines, as seroconversion is routinely taken as a measure of parasite clearance, and presence of fTr-induced antibodies can hinder the development and efficacy testing of the new drugs and vaccines. Thus, considering the hurdle of facilities, resources and costs associated with the production of fTr, we surmise that inclusion of fTr in subunit vaccine is not needed. These results also provide an explanation for why the first generation of vaccines prepared from the T. cruzi parasites (attenuated, killed, fractionated etc.) were not rigorously followed for designing of a commercially viable Chagas vaccine.
With the advancement of CRISPR and other technologies to knockdown genes in trypanosomes, the interest in the development of genetically attenuated live T. cruzi vaccine has rekindled (42, 43). The proponents of the live attenuated vaccine believe that such a vaccine will mimic the natural course of infection and provide an appropriate microenvironment of antigen processing and presentation, create a subclinical infection, provide a full-spectrum of antigenic epitopes, and ensure antigen persistence for stimulation of long-lasting immunological responses (44). Yet, other investigators are concerned that an attenuated live vaccine may trigger a full-blown infection and disease in the immunocompromised individuals. Further, a live vaccine, even if protective against one lineage of T. cruzi, may not be efficacious in providing protection from infection from other five lineages of T. cruzi. Indeed, experimental studies have shown that mice infected with T. cruzi and cleared of parasite by treatment with anti-parasite drugs are not protected from re-challenge infection (45). Thus, the concerns in the use of Tr- or Tc-based attenuated vaccines appear to outweigh the benefits offered by whole organism vaccine in the context of Chagas disease, though this remains to be experimentally proven.
In summary, we have demonstrated that a DNA-prime/DNA-boost vaccine using relatively simple immunogens provides protection from challenge infection. The TcG2/TcG4 DNA vaccine (a) stimulated pro-inflammatory cytokines' production by DC, Mφ, and T cells, and (b) provided antigen uptake, processing, and presentation capacity to cells of the innate immune system that led to the (c) activation of CD4+ helper T cells and CD8+ polyfunctional, cytolytic T cells. Our data suggest that (d) induction of antibody responses by vaccine is not important for providing protection from infection, and instead an interaction between APCs and adaptive T cell immunity capable of expanding upon challenge infection is sufficient to control T. cruzi dissemination. We also conclude that TcG2/TcG4 vaccine elicits highly effective anti-parasite immunity, and inclusion of attenuated Tr (± QA) is not required to improve the DNA vaccine's efficacy against acute Tc infection. Future studies should utilize parasites of different lineages, different parasite inoculation size, and monitor all different tissues to ensure that vaccine is fully protective. Our results also encourage the designing of recombinant nano-plasmids that lack antibiotic genes, can incorporate both TcG2 and TcG4 genes in one plasmid, and are suitable for large-scale production; and testing of the recombinant nano-plasmids as prophylactic or therapeutic vaccine against T. cruzi in multiple hosts.
All data generated or analyzed during this study are included in this article (and its Supplementary Information Files).
All animal experiments were conducted following the National Institutes of Health guidelines for housing and care of laboratory animals and in accordance with protocols approved by the Institutional Animal Care and Use Committee (protocol number 08-05-029) at The University of Texas Medical Branch at Galveston.
Chagas cardiomyopathy, caused by a parasitic protozoan Trypanosoma cruzi, represents the third greatest tropical disease burden in the world. Based on a decade of research work, we have developed an effective, experimental subunit vaccine composed of two candidate antigens of T. cruzi. Trypanosoma rangeli is non-pathogenic to mammals and shown to elicit cross-reactive anti-T. cruzi antibodies. In this study, we investigated if adding fixed T. rangeli (fTr) can further enhance the efficacy of the subunit DNA vaccine. We demonstrate that (a) the TcG2 and TcG4 DNA vaccine is simplest in design (so far), (b) the candidate antigens elicit highly effective immunity, and (c) inclusion of fTr is not required to improve the DNA vaccine's efficacy against T. cruzi infection. Future studies will ensure the efficacy of TcG2/TcG4 based prophylactic and therapeutic immunity against diverse parasite strains, parasite doses, and in multiple hosts.
NG provided financial support, conceived the study, and wrote the manuscript. SG, BS-J, and NL designed and performed the experiments, analyzed the data, and confirmed the accuracy of data presentation in the manuscript. JV-C provided T. rangeli, and substantially contributed to the conception of the study. All authors have read and approved the manuscript to be published.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
This work was supported by a grant (R01AI054578) from the National Institute of Allergy and Infectious Diseases of the National Institutes of Health to NG.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2019.01456/full#supplementary-material
Table S1. Splenic and lymph node (LN) cell count post-vaccination.
Table S2. Flow cytometry studies.
Figure S1. Cross-reactivity of antibodies to T. rangeli in vaccinated mice. C57BL/6 female mice were immunized with six different compositions of vaccines as described in Materials and Methods. Mice were vaccinated with dose 1 at day 0, dose 2 at day 21, and euthanized at day 42. Vaccines (per dose) were constituted with pCDNA3.TcG2 and pCDNA3.TcG4 (25 μg each plasmid DNA, intramuscular), 1 × 108 T. rangeli fixed with 0.1 % glutaraldehyde (fTr, subcutaneous), and 5 μg Quil A (QA, subcutaneous). Sera levels of T. rangeli-specific IgG (A), IgG1 (B), IgG2A (C), and IgG2b (D) antibodies were measured by an ELISA. Sera samples from normal and infected mice were used as controls. Data (mean ± SD) are representative of two independent experiments (n = 4 mice per group). Significance is annotated as *none vs. vaccinated or infected vs. vaccinated/infected (*p < 0.05, **p < 0.01, ***p < 0.001).
Figure S2. Antigen presenting capacity of lymph node macrophages (Mφ) in vaccinated mice (± T. cruzi). Mice were vaccinated, infected and euthanized as in Figure S1. Single cell suspensions of lymph node (LN) cells were labeled with fluorochrome-conjugated antibodies, and analyzed by flow cytometry. Bar graphs show ex vivo percentages of Ly6GloCD11b+ LN Mφ that exhibited surface expression of markers of maturation, antigen uptake, and antigen presentation (CD205+, CD209+, MHCI+, MHCII+, CD80+) in vaccinated (A) and vaccinated/infected (B) mice. Lymph node cells from non-vaccinated and non-vaccinated/infected mice were used as controls. Data (mean ± SD) are representative of two independent experiments (n = 3 mice per group per experiment, duplicate observations per mouse). Significance is annotated as *none vs. vaccinated or infected vs. vaccinated/infected (*p < 0.05, **p < 0.01, ***p < 0.001).
1. Lopez M, Tanowitz HB, Garg NJ. Pathogenesis of chronic Chagas disease: macrophages, mitochondria, and oxidative Stress. Curr Clin Microbiol Rep. (2018) 5:45–54. doi: 10.1007/s40588-018-0081-2
2. Tanowitz HB, Machado FS, Spray DC, Friedman JM, Weiss OS, Lora JN, et al. Developments in the management of Chagas cardiomyopathy. Expert Rev Cardiovasc Ther. (2015). 13:1393–409. doi: 10.1586/14779072.2015.1103648
3. El-Sayed NM, Myler PJ, Bartholomeu DC, Nilsson D, Aggarwal G, Tran AN, et al. The genome sequence of Trypanosoma cruzi, etiologic agent of Chagas disease. Science. (2005) 309:409–15. doi: 10.1126/science.1112631
4. Bhatia V, Sinha M, Luxon B, Garg NJ. Utility of Trypanosoma cruzi sequence database for the identification of potential vaccine candidates: in silico and in vitro screening. Infect Immun. (2004) 72:6245–54. doi: 10.1128/IAI.72.11.6245-6254.2004
5. Bhatia V, Garg NJ. Previously unrecognized vaccine candidates control Trypanosoma cruzi infection and immunopathology in mice. Clin Vaccine Immunol. (2008) 15:1158–64. doi: 10.1128/CVI.00144-08
6. Aparicio-Burgos JE, Ochoa-Garcia L, Zepeda-Escobar JA, Gupta S, Dhiman M, et al. Testing the efficacy of a multi-component DNA-prime/DNA-boost vaccine against Trypanosoma cruzi infection in dogs. PLoS Negl Trop Dis. (2011) 5:e1050. doi: 10.1371/journal.pntd.0001050
7. Gupta S, Wan X-X, Zago MP, Martinez VCS, Silva TS, et al. Antigenicity and diagnostic potential of vaccine candidates in human Chagas disease. Plos NTD. (2013) 7:e2018. doi: 10.1371/journal.pntd.0002018
8. Gupta S, Garg NJ. Prophylactic efficacy of TcVac2 against Trypanosoma cruzi in mice. PLoS Negl Trop Dis. (2010) 4:e797. doi: 10.1371/journal.pntd.0000797
9. Gupta S, Garg NJ. Delivery of antigenic candidates by a DNA/MVA heterologous approach elicits effector CD8+T cell mediated immunity against Trypanosoma cruzi. Vaccine. (2012) 12:1464–78. doi: 10.1016/j.vaccine.2012.10.018
10. Gupta S, Garg NJ. TcVac3 induced control of Trypanosoma cruzi infection and chronic myocarditis in mice. PLOS ONE. (2013) 8:e59434. doi: 10.1371/journal.pone.0059434
11. Gupta S, Garg NJ. A two-component DNA-prime/protein-boost vaccination strategy for eliciting long-term, protective T Cell immunity against Trypanosoma cruzi. PLoS Pathog. (2015) 11:e1004828. doi: 10.1371/journal.ppat.1004828
12. Aparicio-Burgos JE, Zepeda-Escobar JA, de Oca-Jimenez RM, Estrada-Franco JG, Barbabosa-Pliego A, Ochoa-García L, et al. Immune protection against Trypanosoma cruzi induced by TcVac4 in a canine model. PLoS Negl Trop Dis. (2015) 9:e0003625. doi: 10.1371/journal.pntd.0003625
13. Dumonteil E. DNA vaccines against protozoan parasites: advances and challenges. J Biomed Biotechnol. (2007) 2007:90520. doi: 10.1155/2007/90520
14. Vázquez-Chagoyán JC, Gupta S, Garg NJ. Vaccine development against Trypanosoma cruzi and Chagas disease. Adv Parasitol. (2011) 75:121–46. doi: 10.1016/B978-0-12-385863-4.00006-X
15. Dos Santos Virgilio F, Pontes C, Dominguez MR, Ersching J, Rodrigues MM, Vasconcelos JR, et al. CD8(+) T cell-mediated immunity during Trypanosoma cruzi infection: a path for vaccine development? Mediators Inflamm. (2014) 2014:243786. doi: 10.1155/2014/243786
16. Gupta S, Smith C, Auclair S, Delgadillo Ade J, Garg NJ. Therapeutic efficacy of a subunit vaccine in controlling chronic Trypanosoma cruzi infection and Chagas disease Is enhanced by glutathione peroxidase over-expression. PLoS ONE. (2015) 10:e0130562. doi: 10.1371/journal.pone.0130562
17. Vermont CL, van Dijken HH, van Limpt CJ, de Groot R, van Alphen L, et al. Antibody avidity and immunoglobulin G isotype distribution following immunization with a monovalent meningococcal B outer membrane vesicle vaccine. Infect Immun. (2002) 70:584–90. doi: 10.1128/IAI.70.2.584-590.2002
18. Gupta S, Silva TS, Osizugbo JE, Tucker L, Garg NJ. Serum mediated activation of macrophages reflects Tcvac2 vaccine efficacy against Chagas disease. Infect Immun. (2014) 82:1382–9. doi: 10.1128/IAI.01186-13
19. Padilla AM, Bustamante JM, Tarleton RL. CD8+ T cells in Trypanosoma cruzi infection. Curr Opin Immunol. (2009) 21:385–90. doi: 10.1016/j.coi.2009.07.006
20. Zak DE, Aderem A. Overcoming limitations in the systems vaccinology approach: a pathway for accelerated HIV vaccine development. Curr Opin HIV AIDS. (2012) 7:58–63. doi: 10.1097/COH.0b013e32834ddd31
21. Garg NJ, Tarleton RL Elicitation of protective cellular and humoral immune responses to Trypanosoma cruzi infection using DNA vaccines can be augmented with cytokines. In: Proceedings 10th International Congress of Immunology. New Delhi (1998). p. 1421–6.
22. Vallejo GA, Guhl F, Schaub GA. Triatominae-Trypanosoma cruzi/T. rangeli: vector-parasite interactions Acta Trop. (2009) 110:137–47. doi: 10.1016/j.actatropica.2008.10.001
23. Bhatia V, Garg NJ. Current status and future prospects for a vaccine against American trypanosomiasis. Expert Rev Vaccines. (2005) 4:867–80. doi: 10.1586/14760584.4.6.867
24. Garg NJ, Tarleton RL. Genetic immunization elicits antigen-specific protective immune responses and decreases disease severity in Trypanosoma cruzi infection. Infect Immun. (2002) 70:5547–55. doi: 10.1128/IAI.70.10.5547-5555.2002
25. Bhatia V, Wen J-J, Zacks MA, Garg NJ American trypanosomiasis and perspectives on vaccine development. In: Stanberry LR, Barrett AD, editors. Vaccines for Biodefense and Emerging and Neglected Diseases. New York, NY: Academic Press (2009). p. 1407–34. doi: 10.1016/B978-0-12-369408-9.00069-X
26. Stevens JR, Teixeira MM, Bingle LE, Gibson WC. The taxonomic position and evolutionary relationships of Trypanosoma rangeli. Int J Parasitol. (1999) 29:749–57.
27. Stoco PH, Wagner G, Talavera-Lopez C, Gerber A, Zaha A, Thompson CE, et al. Genome of the avirulent human-infective trypanosome–Trypanosoma rangeli. PLoS Negl Trop Dis. (2014) 8:e3176. doi: 10.1371/journal.pntd.0003176
28. Vallejo GA, Guhl F, Carranza JC, Triana O, Perez G, Ortiz PA, et al. Trypanosoma rangeli parasite-vector-vertebrate interactions and their relationship to the systematics and epidemiology of American trypanosomiasis. Biomedica. (2007) 27(Suppl. 1):110–8.
29. Vasquez AM, Samudio FE, Saldana A, Paz HM, Calzada JE. Eco-epidemiological aspects of Trypanosoma cruzi, Trypanosoma rangeli and their vector (Rhodnius pallescens) in Panama. Rev Inst Med. (2004) 46:217–22. doi: 10.1590/S0036-46652004000400008
30. Basso B, Marini V. Experimental Chagas disease. Innate immune response in Balb/c mice previously vaccinated with Trypanosoma rangeli I The macrophage shows immunological memory: Reality or fiction? Immunobiology. (2014) 219:275–84. doi: 10.1016/j.imbio.2013.10.012
31. Basso B, Moretti E, Fretes R. Vaccination with Trypanosoma rangeli induces resistance of guinea pigs to virulent Trypanosoma cruzi. Vet Immunol Immunopathol. (2014) 157:119–23. doi: 10.1016/j.vetimm.2013.10.011
32. Basso B, Marini V. Experimental Chagas disease in Balb/c mice previously vaccinated with T. rangeli II The innate immune response shows immunological memory: reality or fiction? Immunobiology. (2015) 220:428–36. doi: 10.1016/j.imbio.2014.10.003
33. Haniffa M, Bigley V, Collin M. Human mononuclear phagocyte system reunited. Semin Cell Dev Biol. (2015) 41:59–69. doi: 10.1016/j.semcdb.2015.05.004
34. Koo SJ, Chowdhury IH, Szczesny B, Wan X, Garg NJ. Macrophages promote oxidative metabolism to drive nitric oxide generation in response to Trypanosoma cruzi. Infect Immun. (2016) 84:3527–41. doi: 10.1128/IAI.00809-16
35. Koo SJ, Garg NJ. Metabolic programming of macrophage functions and pathogens control. Redox Biol. (2019) 24:101198. doi: 10.1016/j.redox.2019.101198
36. Koo SJ, Szczesny B, Wan X, Putluri N, Garg NJ. Pentose phosphate shunt modulates reactive oxygen species and nitric oxide production controlling Trypanosoma cruzi in macrophages. Front Immunol. (2018) 9:202. doi: 10.3389/fimmu.2018.00202
37. Chan LC, Rossetti M, Miller LS, Filler SG, Johnson CW, Lee HK, et al. Protective immunity in recurrent Staphylococcus aureus infection reflects localized immune signatures and macrophage-conferred memory. Proc Natl Acad Sci USA. (2018) 115:E11111–19. doi: 10.1073/pnas.1808353115
38. Arts RJW, Moorlag S, Novakovic B, Li Y, Wang SY, Oosting M, et al. BCG vaccination protects against experimental viral infection in humans through the induction of cytokines associated with trained immunity. Cell Host Microbe. (2018) 23: 89–100 e105. doi: 10.1016/j.chom.2017.12.010
39. Arts RJW, Carvalho A, La Rocca C, Palma C, Rodrigues F, Silvestre R, et al. Immunometabolic pathways in BCG-induced trained immunity. Cell Rep. (2016) 17:2562–71. doi: 10.1016/j.celrep.2016.11.011
40. Gruenbacher G, Thurnher M. Mevalonate metabolism in cancer stemness and trained immunity. Front Oncol. (2018) 8:394. doi: 10.3389/fonc.2018.00394
41. Bonney KM, Luthringer DJ, Kim SA, Garg NJ, Engman DM. Pathology and pathogenesis of chagas heart disease. Annu Rev Pathol Mechan Dis. (2019) 14:421–47. doi: 10.1146/annurev-pathol-020117-043711
42. Lander N, Li ZH, Niyogi S, Docampo R. CRISPR/Cas9-Induced disruption of paraflagellar rod protein 1 and 2 genes in Trypanosoma cruzi reveals their role in flagellar attachment. MBio. (2015) 6:e01012. doi: 10.1128/mBio.01012-15
43. Peng D, Kurup SP, Yao PY, Minning TA, Tarleton RL. CRISPR-Cas9-mediated single-gene and gene family disruption in Trypanosoma cruzi. MBio. (2014) 6:e02097–02014. doi: 10.1128/mBio.02097-14
44. Sanchez-Valdez FJ, Perez Brandan C, Ferreira A, Basombrio MA. Gene-deleted live-attenuated Trypanosoma cruzi parasites as vaccines to protect against Chagas disease. Expert Rev Vaccines. (2015) 14:681–97. doi: 10.1586/14760584.2015.989989
Keywords: Trypanosoma cruzi, Chagas disease, recombinant DNA vaccine, T. rangeli, immune efficacy
Citation: Gupta S, Salgado-Jiménez B, Lokugamage N, Vázquez-Chagoyán JC and Garg NJ (2019) TcG2/TcG4 DNA Vaccine Induces Th1 Immunity Against Acute Trypanosoma cruzi Infection: Adjuvant and Antigenic Effects of Heterologous T. rangeli Booster Immunization. Front. Immunol. 10:1456. doi: 10.3389/fimmu.2019.01456
Received: 04 March 2019; Accepted: 10 June 2019;
Published: 26 June 2019.
Edited by:
Pedro A. Reche, Complutense University of Madrid, SpainReviewed by:
Julio Alonso Padilla, Instituto Salud Global Barcelona (ISGlobal), SpainCopyright © 2019 Gupta, Salgado-Jiménez, Lokugamage, Vázquez-Chagoyán and Garg. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Nisha Jain Garg, bmlnYXJnQHV0bWIuZWR1
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.