 Xiaobo Liu
Xiaobo Liu Christian Gorzelanny2
Christian Gorzelanny2 Stefan W. Schneider
Stefan W. Schneider- 1Department of Dermatology, Medical Faculty Mannheim, University of Heidelberg, Mannheim, Germany
- 2Department of Dermatology and Venereology, University Medical Center Hamburg-Eppendorf, Hamburg, Germany
Systemic lupus erythematosus (SLE), systemic sclerosis (SSc), and small vessel vasculitis are three autoimmune diseases frequently manifested in the skin. They share common pathogenic features, including production of autoantibodies, loss of tolerance to self-antigens, tissue necrosis and fibrosis, vasculopathy and activation of the coagulation system. Platelets occupy a central part within the coagulation cascade and are well-recognized for their hemostatic role. However, recent cumulative evidence implicates their additional and multifaceted immunoregulatory functions. Platelets express immune receptors and they store growth factors, cytokines, and chemokines in their granules enabling a significant contribution to inflammation. A plethora of activating triggers such as damage associated molecular patterns (DAMPs) released from damaged endothelial cells, immune complexes, or complement effector molecules can mediate platelet activation. Activated platelets further foster an inflammatory environment and the crosstalk with the endothelium and leukocytes by the release of immunoactive molecules and microparticles. Further insight into the pathogenic implications of platelet activation will pave the way for new therapeutic strategies targeting autoimmune diseases. In this review, we discuss the inflammatory functions of platelets and their mechanistic contribution to the pathophysiology of SSc, ANCA associated small vessel vasculitis and other autoimmune diseases affecting the skin.
Introduction
Platelets are small circulating cellular fragments that originate from megakaryocytes mainly within the bone marrow (1, 2). Under physiological conditions, platelets have a short lifespan in the circulation as they are eliminated in the spleen and liver after 7–10 days. Under resting conditions, the vascular endothelium continuously prevents platelet adhesion and activation through the release of prostacyclin I2 and nitric oxide (2). Blood vessel damage or detachment of the endothelium upon injury results in the exposure of the pro-coagulant subendothelial matrix and associated perivascular cells which promote platelet activation and blood clotting. However, platelet adhesion and coagulation could also be initiated without the denudation of the endothelial cell layer. Distinct stimulatory agents such as thrombin, histamine, tumor necrosis factor (TNF-α), or CD40 ligand (CD40L, CD154) convert the endothelium into a proinflammatory and procoagulatory surface through the release of von Willebrand factor (VWF) (3–6). Secreted VWF gets immobilized on the luminal site of endothelial cells where it is activated through blood shear flow mediated stretching. These VWF fibers can rapidly interact with GPIb-IX-V on platelets, resulting in the formation of platelet decorated VWF strings (3, 7, 8). Attached platelets translocate GPIIb/IIIb to their surface to stabilize their interaction with VWF. Moreover, these procoagulant platelets expose phosphatidylserine (PS) on their membrane. Together with tissue factor and Factor VII, PS initiates the activation of the coagulation factors X (FX) and II (FII, prothrombin) (9–13). The presence of tissue factor on platelets is controversial discussed. However, more recent studies suggest its expression and its surface exposure upon activation (14, 15). Apart from tissue factor, platelets can enhance hemostasis through the presentation of P-selectin (CD62P) and lysosomal-associated membrane protein 1 and the release of FV, histamine and ADP (2, 10).
Next to their contribution to hemostasis, there is growing body of evidence indicating the action of platelets in inflammation and immune responses (1, 16–18). Moreover, recent findings point toward the significant involvement of platelets in the pathogenesis of autoimmune diseases (7, 19, 20). This review will describe platelet immune functions, and highlight the implication of platelets in the pathogenic mechanisms of autoimmune disorders with frequent but not limited manifestations in the skin. We will in particular focus on systemic lupus erythematosus (SLE), systemic sclerosis (SSc) and antineutrophil cytoplasmic antibody-associated small vessel vasculitis (AAVs).
Inflammatory Functions of Platelet
Upon activation, platelets shed microparticles and they release potent immune modulatory mediators stored in their granules, including proinflammatory cytokines and chemokines (e.g., IL-1ß, TGF-ß, PF4, and PDGF). Platelets are also able to present a number of adhesion (e.g., GPIb-IX-V and P-selectin) and immune receptors (e.g., toll-like or Fc receptors) for prompt responses to the external environment. These receptors enable platelets to interact with activated vascular endothelial cells and immune cells, such as neutrophils, monocytes and lymphocytes. Context dependent, these interactions may tune hemostatic and immune responses, including the activation of the complement system. Figure 1 summarizes various molecules mediating platelet functions in autoimmune diseases.
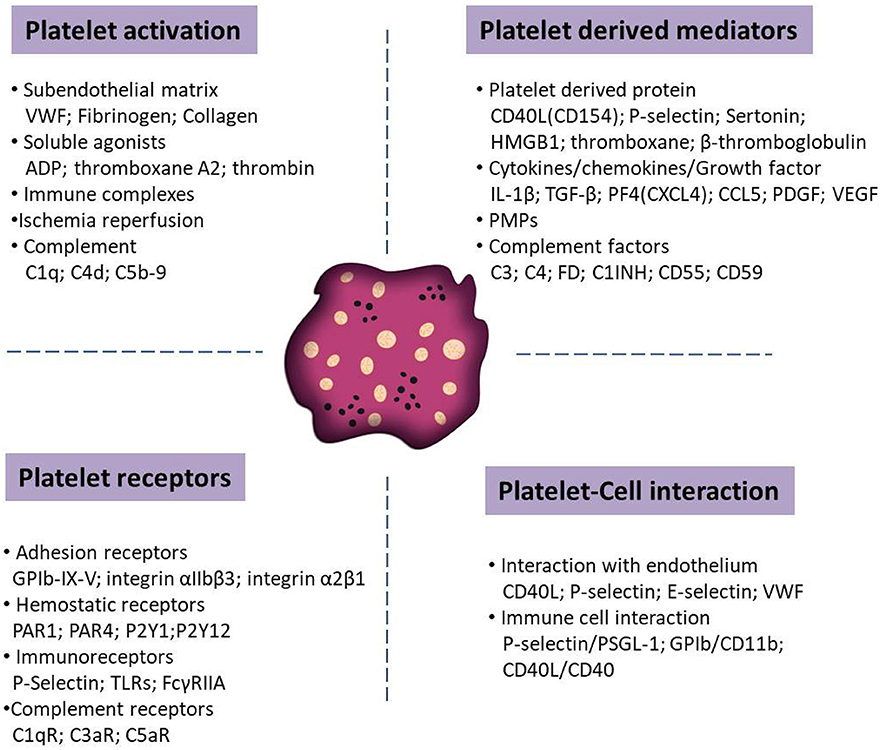
Figure 1. Schematic overview of distinct molecules that tune the function of platelets in autoimmune diseases. Relevant molecules has been categorized into platelet activators, soluble factors released from platelets upon activation, surface receptors that mediate the interaction with other cells and receptors that trigger platelet adhesion and activation.
Platelet Granules and Platelet Derived Microparticles (PMPs)
There are three types of platelet granules: α-granule, dense granule and lysomal granule. Upon platelet stimulation, granules undergo rapid secretion of their contents into the extracellular space.
The most plentiful (40–80 per platelet) and largest platelet granules (200–400 nm) are α-granules (18). They store almost 300 different proteins, including chemokines, cytokines, growth factors, and adhesion receptors (21–23). However, it is of note that the release of these bioactive substances is not random but dependent on the stimulus (20). Recent observations suggest that platelets contain distinct subpopulations of α-granules which facilitate the differential release of specific α-granule components during platelet activation (24, 25). These secretion products do not only contribute to hemostasis and thrombosis but do also play a potential role as immune mediator by amplifying inflammatory responses (16). Several α-granule derived molecules are frequently reported in the context of skin autoimmune diseases, such as complement factors, CD40L, platelet factor 4 (PF4, also known as CXCL4), and P-selectin (19, 20, 26).
The complement is a complex innate immune system for pathogen defense. The dysregulation of the complement mediates excessive inflammation and tissue injury (27–30). Interestingly, platelet α-granules contain a broad spectrum of complement molecules (31–33). For example, platelets store C3, C4, and factor D which are important components of the complement cascades (31, 34). Also complement attenuating factors C1 inhibitor, CD55, and CD59 are secreted upon platelet activation (35).
CD40L is a transmembrane protein of the TNF superfamily. Under quiescent conditions, CD40L is stored in α-granule whereas it gets exposed on the surface during platelet activations. Upon shedding, CD40L could be released as soluble protein (sCD40L) with cytokine-like activities. CD40L interacts with CD40 on dendritic cells (DCs), B cells and T cells, inducing DC maturation (36), T cell activation (37, 38), B cell isotype switching and antibody production (39). This suggests a significant role of the CD40-CD40L axis in regulating the innate and adaptive immune responses (40). Platelet derived CD40L has also been shown to induce tissue factor expression in monocytes (40), which contributes to the activation of the extrinsic coagulation cascade. Moreover, platelet CD40L can bind to CD40 on endothelial cells, inducing the up-regulation of adhesion molecules (E-selectin, VCAM-1, and ICAM-1), chemokines (IL-8 and CCL2) secretion (22, 41), and VWF release (6). Thus, CD40-CD40L signaling further promotes the adhesion and extravasation of leukocytes at the site of platelet activation. Interestingly, constitutive expression of CD40 at the surface of platelets can further mediate platelet CD62P expression and granule release after perception of CD40L (42).
PF4 is one of the most abundant cytokines in α-granules and it is a potent antiangiogenic chemokine (43). In addition, PF4 induces the release of proinflammatory cytokines from leukocytes and promotes neutrophil chemotaxis (44, 45).
α granules contain also P-selectin a glycosylated transmembrane protein (46) recognizing carbohydrate moieties comparabler to C-type lectins. Similar to CD40L, P-selectin is translocated during platelet activation to the surface membrane or secreted into the plasma as soluble P-selectin (sP-selectin). P-selectin is a key adhesion molecule supporting the close interaction with other immune cells (20, 22, 47). P-selectin ligand-1 (PSGL-1) is the major receptor for P-selectin and it is expressed mainly on neutrophils and monocytes (48, 49). The cross-linking between P-selectin and its corresponding ligand PSGL-1 plays a pivotal role in the formation of platelet-leukocyte aggregates (50, 51) and in the upregulation of tissue factor expression on monocyte (52). P-selectin-PSGL-1 stimulated platelets could further activate neutrophils to form neutrophil extracellular traps (NETs) (53). However, the impact of P-selectin to induce NET formation remains controversial. Results reported by Clark et.al and Maugeri et.al indicate that P-selectin is dispensable for NET generation (54, 55). NETs reversely enhance blood coagulation by direct interaction with VWF (56) and through platelet activation (57, 58). In addition, platelets which are tethered on endothelial cells act as a bridge to promote the adhesion of neutrophils on the blood vessel wall through P-selectin (59).
Dense granules, smaller and less abundant than α-granules, store small non-protein molecules, such as ADP, ATP, serotonin (5-HT), and calcium (2, 16). Platelet dense-granule secretion plays a critical role in the amplification of platelets responses and thrombosis (60). As a platelet agonist, serotonin can modulate autocrine and paracrine platelet aggregation through the interaction of serotonin receptor on platelets (61). In line with this, a variety of immunomodulatory functions of serotonin have been reported, including recruitment of neutrophils to the site of inflammation, stimulation of chemokine secretion by monocyte and T cell proliferation (62, 63).
In parallel, activated platelets release microparticles by shedding of the plasma membrane (64). PMPs have diameters ranging from 0.1 to 1 μm, which are marked by the expression of surface CD41 (64, 65). Although various cellular lineages are able to release membrane microparticles, PMPs make up the main source in human circulation (65). Diverse platelet components are presented in PMPs, including transcription factors, cytokines, growth factors, lipid mediators, nucleic acid, lipid mediators, and mitochondria (25, 65, 66). Due to the size of PMPs, PMPs have been shown to selectively infiltrate tissues and deliver these bioactive factors to recipient cells, triggering inflammation and thrombosis (26, 64, 67, 68). For example, PMPs deliver CD40L to B cells, inducing efficient B cell response and antibody production (26, 69). Similarly to activated platelets, PMPs bear negatively charged PS and potentially TF on their surface, which supports coagulation via the activation of FX and prothrombin (70, 71). In addition, PMPs have the ability to directly attach to fibrin and enhance the local production of thrombin which further amplifies the thrombus formation (70, 72).
Platelet Receptors
Platelet has a variety of surface receptors, and the majority of these receptors trigger either platelet activation or platelet adhesion. For example, Platelet glycoprotein complex GPIb-IX-V enables platelet binding to subendothelial and luminal exposed VWF even under high shear stress (73, 74). This interaction is further enforced by collagen or fibrinogen through the platelet receptors GPVI, and integrin α2β1 or integrin αIIbβ3, respectively (2, 73).
Classic hemostatic agonists (thrombin and ADP) not only mediate hemostasis, but are also directly linked to inflammatory receptor mediated signaling pathways. The Gaq-coupled protease-activated receptors PAR1 and PAR4 are the two main thrombin receptors on platelets (75). Signaling through these receptors stimulates the Rho-associated protein kinase and phospholipase Cβ, leading to further downstream protein kinase C (PKC) activation and Ca2+ release (2, 76). ADP is another potent platelet activator and can be secreted from dense granules upon platelet activation. On the membrane of platelets, the two purinergic receptors P2Y1 and P2Y12 are expressed, coupling to Gqα and Giα, respectively (77). Signaling via P2Y1 mediates PKC activation, Ca2+ release into the cytoplasm and induces platelet shape change (78). P2Y12 stimulates phosphoinositide3-kinases activation which triggers platelets granule secretion and aggregation (2, 78). Interestingly, PAR signaling promotes also the release of ADP (79, 80), linking these two pathways and enabling autocrine platelet activation. Clopidogrel, a common used drug to prevent heart disease and stroke, blocks the P2Y12 on platelets explaining its high efficacy.
Platelets could also directly recognize immunoglobulins and immune complexes (IC) through the Fc receptor FcγRIIA (81, 82). IC binding to FcγRIIA induces platelet hypersensitivity to thrombin stimuli (83). In addition, FcγRIIA activation can also support platelets serotonin release (84).
Toll-like receptors (TLRs) are another group of immunoreceptors expressed on platelets, which enable platelets to recognize endogenous damage associated molecular patterns (DAMPs) and pathogen associated molecular patterns (85, 86). TLR4 is the most abundantly expressed TLR on platelets and it can detect ligands such as lipopolysaccharide (LPS) and high mobility group protein B1(HMGB1) (87). In this context, Clark et al. reported that LPS induces platelet binding to adherent neutrophils, resulting in neutrophil activation and the formation of NETs (54).
Finally, platelets express several complement receptors (CR) (31). Among them, receptors C3aR and C5aR recognize the strong proinflammatory complement effectors C3a and C5a (88). These two receptors have low expression levels on resting platelets but their expression is increased upon inflammatory stimulation (88, 89). Notably, P-selectin contains nine consensus domains which are common to the structural motif of CRs (90). Therefore, apart from its function as adhesion molecule, P-selectin may also mediate complement effector binding to the surface of platelets to support complement activation (90). Moreover, the surface expression of C1q receptors on platelets has been linked to the initiation of the classical complement pathway activation (91–93).
Platelets Role in Skin Autoimmune Diseases
Systemic Lupus Erythematosus (SLE)
Systemic lupus erythematosus is a chronic autoimmune disease characterized by systemic inflammation in many different organs. SLE is also associated with thrombotic complications and increased cardiovascular morbidity (94, 95). A wide range of research on the pathogenesis of SLE focus on the formation of autoantibodies and autoantibody induced IC, as well as the dysregulation of lymphocyte function and activation of the complement system (95). However, platelets also play an important role in inflammatory activity and immune response. Growing evidence indicates that platelets are activated in SLE patients and contribute to the pathogenesis of SLE (25, 26). Moreover, thrombocytopenia is a common hematologic manifestation in SLE and associated with severe SLE abnormalities such as neurological abnormality and kidney injury (96, 97). As a promoter of complement activation, the presence of antiphospholipid antibodies (aPLs) is often detected in the patients with thrombocytopenia. The mean platelet volume (MPV) is widely used for assessing platelet activation in various inflammatory conditions. However, the current literature on MPV in SLE is contradictory and the usefulness of MPV as a biomarker in SLE still need to be explored (20, 98–101). On the molecular level, the production of thromboxane, P-selection expression and the release of sCD40L, PMPs and β-thromboglobulin have been reported as markers for platelet activation. Their levels are increased in SLE patients and associated with an increased risk of thrombotic events (102–108). The role of platelets in the SLE pathophysiology is depicted in Figure 2.
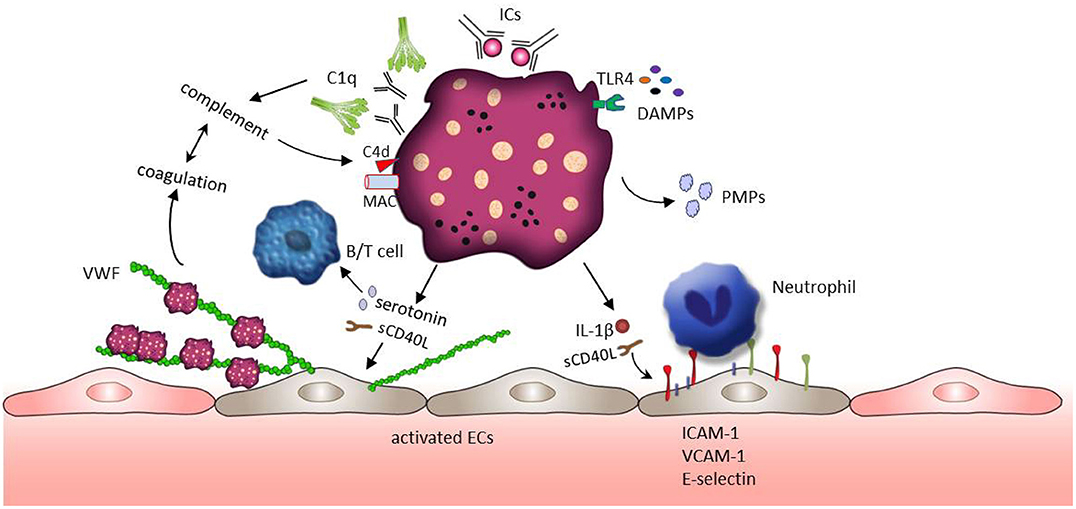
Figure 2. Platelet activation and subsequent effects in SLE. ICs and DAMPs (e.g., HMGB1 or S100A8/9), both enriched in the blood of SLE patients, activate platelets through binding to platelet surface receptors FcγRIIA and TLRs. ICs further induce the complement system activation leading to the deposition of complement fragments (e.g., C1q,C4d, MAC) on the platelet surface which in turn further potentiates platelet activation. Activated platelets release their contents such as IL-1β, sCD40L, and serotonin. These factors mediate the up-regulation of adhesion molecules (e.g., E-selectin, VCAM-1, and ICAM-1) on endothelial cells, promoting the adhesion of immune cells. In addition, sCD40L and serotonin can stimulate the release of VWF from endothelial cells, but also support B cell and T cell activation. VWF mediated trapping and activation of platelets initiate coagulation which may further foster the activity of the complement system building a positive feedback loop.
Several pro-inflammatory mediators are responsible for platelet activation in SLE patients. Among them, DAMPs such as HMGB1, or S100A8/9, both enriched in the blood of SLE patients (109–111), might have the potential to trigger platelet activation through TLR4 signaling. Platelet activation is also mediated by the recognition of SLE-associated ICs through FcγRIIA receptors (25). In addition, TLRs (TLR7, TLR9) exposed at the platelet surface bind to RNA-containing ICs or nucleic acid (ssRNA, dsDNA), contributing to platelets activation (112). ICs are frequently formed by aPLs on the surface of platelets (in almost 40% of the SLE patients) (113, 114). Those platelet-bound ICs could be detected by the complement system mediating the deposition of C4d and the formation of the membrane attack complex (MAC) on the platelets' surface (113, 115). As mentioned above, platelets actively contribute to the complement cascade through the production of several complement factors including C1q, C3, and C4. These complement effectors in turn potentiate the impact of platelets on inflammation (32). For example, MAC promotes platelets to release proinflammatory mediators such as serotonin, thromboxane and β-thromboglobulin stored in their granules (116, 117). Fixation of C4d on the platelet surface supports platelet aggregation and platelet interactions with monocytes and endothelial cells in the context of venous thrombosis (31, 32, 113, 115). Platelets marked with C4d are detected in almost 20% of patients with SLE suggesting C4d positive platelets as a prognostic biomarker (113, 115, 118). The deeply interwoven connection between the complement system and the coagulation (119, 120) has been emphasized by the work of Kölm et al. (121). In their recent study it was demonstrated that VWF bind to C1q and that the C1q-VWF complexes induce platelet adhesion in correlation with the frequently observed thrombotic events in SLE patients (121).
Next to the involvement of the complement system, ICs were shown to mediate the release of serotonin (84, 122, 123), PMPs (108, 124), and IL-1β (106) from platelets. Released serotonin disturbs the endothelial barrier in SLE promoting an increase in vascular permeability (25, 125). Serotonin also has a major effect on T cell activation and proliferation through the interaction with the lymphocyte 5-HT7 receptor (63). Beside serotonin, SLE patients display higher concentrations of PMPs. A study involving 60 SLE patients shows that platelets are the main source of circulating microparticles in SLE (124). Interestingly, those PMPs are C1q+ and can form IC with IgG and IgM (124). Levels of PMPs IgM conjunctions are negatively correlated with SLE severity (124). In contrast, levels of IgG+ PMPs are positively associated with SLE activity (124). IgM autoantibodies bind to apoptotic cells in patients with inactive SLE and may facilitate non-inflammatory removal of PMPs by monocytes or macrophages (124, 126). However, the PMPs-IgG+ stimulate monocytes promoting the expression and release of pro-inflammatory cytokines such as IL-1β, TNF-α, and IFN-α (124). Additionally, IL-1β can also be released from activated platelets. IL-1β induces the expression of NFκB driven inflammatory genes, such as, IL-6 IL-8 and ICAM-1 in endothelial cells which in turn mediates immune cells recruitment and immune-thrombotic complications (106, 127). Endothelial activation could also be directly triggered through platelet derived CD40L (6, 41). This acute activation is characterized by Weibel-Palade body exocytosis, release of VWF multimers and thus the rapid recruitment of further platelets which promotes the sequestration of circulating monocytes by the P-selectin-PSGL-1 interaction (128).
Activated platelets also contribute to the regulation of adaptive immune responses. Platelets are the major source of sCD40L in the circulation (25, 105) and the CD40L signaling through its receptor CD40 on B cells and T cells lead to immunoglobulin IgG and IgM synthesis and the germinal center reaction (38, 39). As reported by several groups, the CD40/CD40L axis promotes DC maturation and IFNα secretion (36). IFNα is a key cytokine in the pathogenesis of SLE indicated by increased levels of the IFNα regulated proteins PRKRA, IFITM1 and CD69 in platelets from SLE patients. The up-regulation of the IFNα system is strongly associated with vascular disease in SLE (20, 129).
Systemic Sclerosis (SSc)
SSc is an autoimmune disease characterized by excessive connective tissue deposition and fibrosis, vasculopathy and a dysregulated immune system (130, 131). Enhanced platelet activation and aggregation can be observed in patients suffering from SSc (131–133). Ischemia reperfusion alternation associated with Raynaud's phenomenon in fingers is frequently the first manifestation of SSc (132). Ischemia-reperfusion injury promotes endothelial cell damage, which results in the release of reactive oxygen species (ROS), DAMPs, the activation of the complement and the exposure of collagen to the blood flow (134–137). This inflammatory environment stimulates platelet recruitment and activation. Similar to SLE, the presence of aPLs related ICs could further promote hypersensitivity of platelets (138). In this context, a specific increase in the number of platelet non-integrin type I collagen receptor was detected in SSc patient's platelets (139). This may explain the accelerated aggregation status of SSc patients' platelets when exposed to collagen. The pathophysiological role of platelet in SSc is shown in Figure 3.
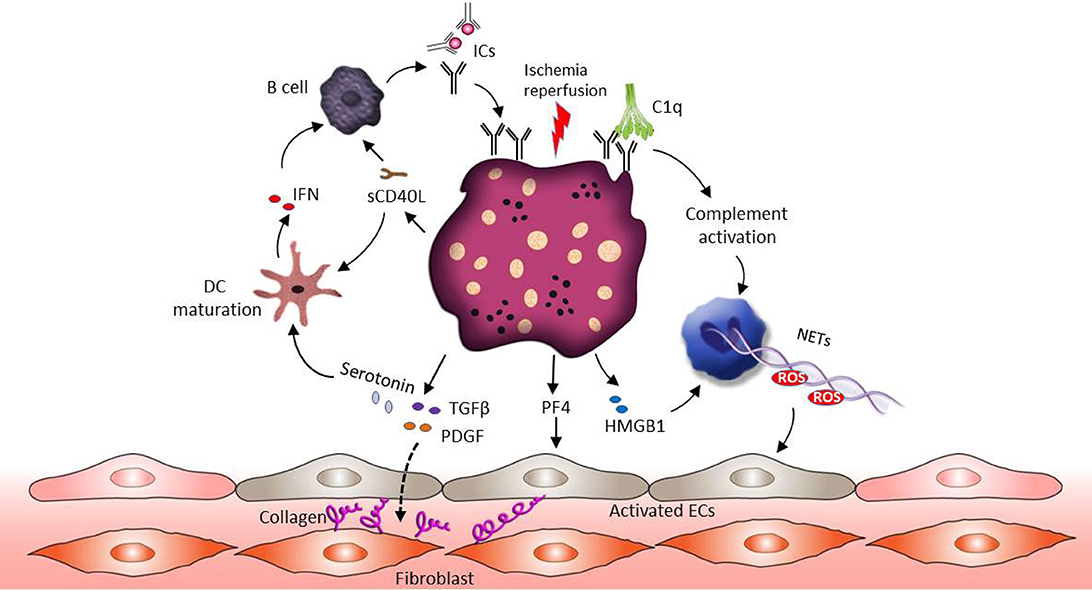
Figure 3. The role of platelets in SSc pathophysiology. Ischemia-reperfusion injury associated endothelial cell damage induces platelet activation. Similar to SLE, the presence of ICs could promote hypersensitivity of platelets and complement activation. Activated platelets release their profibrotic mediators such as TGF-β, serotonin and PDGF, and these factors stimulate connective tissue fibroblasts proliferation and increase collagen production. In addition, both HMGB1 released from platelets and complement activation can support NET formation and ROS release, which further mediates vascular endothelial dysfunction. Apart from NETs, platelets derived PF4 activates endothelial cells and induces SSc associated vascular damage. Activated platelets also release sCD40L to active B cells through CD40 signaling, which leads to B cell auto-antibody production. Moreover, sCD40L and serotonin can promote the maturation of myeloid DCs, followed by IFN production and further B cell activation.
Extensive experimental evidence supports the notion that platelets participate in the fibrotic process mainly by releasing profibrotic mediators (131, 140). Transforming growth factor β (TGF-β) and platelet-derived growth factors (PDGFs) are thought to be the central pathogenic profibrotic mediators. Activation of the TGF-β pro-fibrotic signaling pathway has long been implicated in fibrotic diseases, including SSc (141, 142). Circulating platelets contain high concentrations (about 40–100 times higher compared to other cells) of TGF-β in their α-granules and release it rapidly upon activation (143–145). Several studies showed that TGFβ plays a pivotal role in extracellular matrix remodeling through the control of the collagen synthesis, as well as secretion of fibronectin and thrombospondin-1 (141, 146). Notably, TGF-β stimulates the production of a plethora of secondary mediators from fibroblasts, such as connective tissue growth factor and endothelin-1, modulating the downstream fibrotic signaling activation (141, 147, 148). Similar to TGF-β, activated platelets can also release PDGF, which is able to stimulate connective tissue fibroblasts proliferation and increase collagen production (131, 149). A study revealed that PDGF-A is increased in the dermal interstitial blister fluid of SSc patients (150) and blockade of PDGF receptors by Crenolanib is effective in reducing skin fibrosis in preclinical models of SSc (151). Additionally, Nintedanib, a tyrosine kinase inhibitor, inhibits the PDGF and TGF-β induced activation of SSc fibroblasts and prevents the onset of the disease in different mouse models (152, 153). As mentioned above, platelets are the main source of serotonin in the circulation and the profibrotic role of serotonin has received significant attention recently (154). Serotonin could enhance collagen production in fibroblasts through the 5HT-2B receptor, which is over expressed in SSc patients' skin (154). Platelets in SSc further contribute to skin fibrosis because they interact with dermal microvascular endothelial cell which induces the subsequent secretion of profibrotic mediators such as thymic stromal lymphopoietin (155).
The involvement of the endothelium in patients suffering from SSc is mirrored by an increased risk of vascular disease such as Raynaud's phenomenon, pulmonary arterial hypertension, ischemic ulcer due to vascular damage, renal and cardiac disease (131, 156). Several mechanisms have been suggested to explain the role of platelets in SSc associated vasculopathy. Experimental data have shown that platelets derived PF4 could enhance the expression of thrombospondin-1 and promote endothelin-1 secretion from human endothelial cells, resulting in an inflammatory phenotype of endothelial cells and SSc associated vascular damage (157). Similarly to SLE, the existence of PMP is abundant in the blood of SSC patients, especially HMGB1-associated PMPs (158–160). HMGB1 released from activated platelets in SSc patients, sustains autophagy associated activation of neutrophils in SSc and commits them to generate NETs, leading to vascular endothelium dysfunction (158). Recent studies suggest that antiangiogenic factors such as VEGF165b, together with proinflammatory (CD40L) and profibrotic (TGF-β) factors secreted by platelets, can contribute to the progression of peripheral microvascular damage and defective vascular repair in SSc (161).
It is becoming increasingly clear that platelets act as key regulators of the immune response participating in the pathogenesis of SSc. A large array of proinflammatory mediators are either synthesized within platelets or stored in the granules and released upon activation. Platelets derived CCL5, PF4, CXCL5, and leukotriene-B4 hold important leukocyte chemoattractant properties, recruiting neutrophils, monocytes, and fibroblasts to the site of inflammation, which in turn amplify local inflammatory reactions (132, 162). What is more, it has been shown that PF4-activated monocytes trigger ROS production and the release of the procoagulant VWF in endothelial cells (163). Also P-selectin translocates to the platelet membrane and forms an adhesive bridge between endothelial cells and neutrophils, monocytes and T cells, thereby facilitating the formation of heterotypic platelet-leukocyte aggregates on the surface of blood vessel (162). Activated platelets express and release sCD40L which accounts for almost 90% of circulating sCD40L (164). The interaction between sCD40L and CD40 on B cells leads to increased immumoglobulins production and B cell proliferation, highlighting the remarkable role of platelets in the regulation of adaptive immune response (39). Another important SSc related immune mediator is serotonin (63). Apart from the role in the regulation of collagen production, serotonin also enhances T cell activation and proliferation through 5-HT7 receptor signaling (165). Some studies also suggest the immunomodulatory properties of serotonin through the maturation of myeloid DCs (166) and the regulation of the production of proinflammatory mediators such as IL-6 and TNF-α from monocyte (167).
Antineutrophil Cytoplasmic Antibody (ANCA)—Associated Small Vessel Vasculitis
Vasculitis represents a group of complex diseases with the pathology of blood vessel wall inflammation (168, 169). Based on the size of vessels involved, vasculitis is categorized by large, medium, and small vessel vasculitis (170). The pathogenesis of vasculitis remains incompletely understood and few studies reported about the role of platelet in large vessel vasculitis. Our review will focus on ANCA-associated small vessel vasculitis (AAVs), which are characterized by the inflammatory cell infiltration of small sized vessel walls in multiple organ systems (171). AAVs also display a broad variety of cutaneous manifestations, including tissue necrosis, and vascular destruction (172). The neutrophil derived proteins myeloperoxidase and proteinase 3 are the main target antigens of ANCA (173). Platelets are first responders during vascular injury and they are inflammatory effector cells closely correlated with the activity of vasculitis (7, 174). In line with that PMPs containing proinflammatory cytokines are increased in AAVs patients and supposed to trigger an acute inflammatory state in AAVs (175). Figure 4 summarizes the platelet activation and subsequent effects in AAVs.
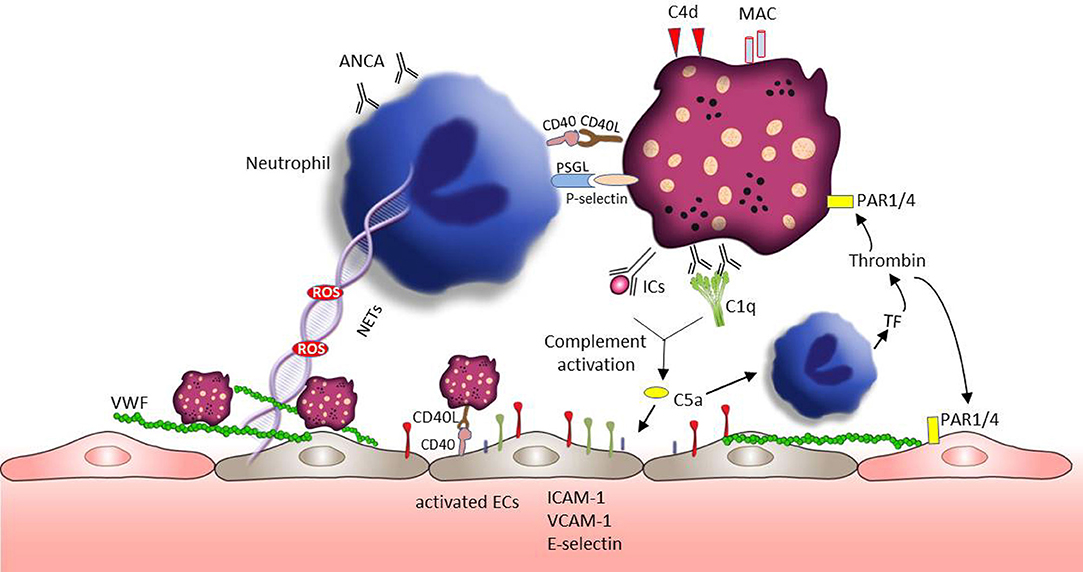
Figure 4. Platelets involvement in the pathogenesis of AAVs. Neutrophils play a crucial role in the development of AAVs. Platelet receptors CD40L and P-selectin enable the interaction with neutrophils promoting ROS generation and NET formation. ROS and NETs are toxic to the endothelium and can lead to VWF release and vascular injury. VWF is well-known to mediate platelets adhesion and aggregation. Similar to neutrophils, sCD40L-CD40 can also mediate platelet -endothelial cell interaction, which can enhance the expression of endothelial cell adhesion proteins (e.g., E-selectin, VCAM-1, and ICAM-1). C5a, a chemoattractant anaphylatoxin, can directly attract and activate neutrophils promoting the exposure of tissue factor. Tissue factor initiates the plasmatic coagulation which culminates in the generation of thrombin. Thrombin is able to further activate platelets and endothelial cells through PAR signaling pathway, followed by platelet degranulation and VWF release. In addition, C5a can also directly active endothelial cells accelerating vascular injury.
CD40 and CD40L are expressed on endothelial cells, platelets and epithelial cells and their signaling mediates several processes of vascular inflammation. sCD40L-CD40 mediated platelet-endothelial cell interaction, induces cytokine and growth factors production (such as IL-1β, TNF-α, IL-2, VEGF) in endothelial cells and enhance the expression of endothelial cell adhesion proteins (such as ICAM-1, VCAM-1, P-selectin) (40, 41). This further leads to the recruitment of neutrophils and lymphocytes to the site of injury. sCD40L also enhances endothelial expression of tissue factor (176) and contributes to the endothelial dysfunction through ROS generation (177, 178). Neutrophils are strong mediators of the pathogenesis of AAVs. sCD40L can also directly activate neutrophils, mediating ROS release (179) and macrophage 1 antigen (CD11b/CD18) expression (180). Notably, activated platelets can stimulate NET formation by the release of sP-selectin (53). In turn, NET fibers could bind platelets supporting their aggregation (181). Additionally, NET components, such as histones, can further stimulate platelet and endothelial cell activation (181–183). Blocking of neutrophil PSGL-1 completely inhibits the activated platelet mediated NET formation (53). Similar to SLE and SSc, the development of vasculitis is at least partially related to the activity of the complement system (184). The activation of the complement alternative pathway both in the fluid phase and on the surface of platelets leads to MAC deposition on platelets and generation of the inflammatory factors C3a and C5a (185). Both complement effector molecules elicit the expression of cellular adhesion molecules such E-selectin, ICAM-1, and VCAM-1 on endothelial cells and the production of cytokines/chemokines and related receptors (such as VEGFC-R, IL-6R, IL-18R) (186). Because of the presence of C3aR and C5aR on platelets surface, C3a and C5a can also promote the activation of platelets, supporting coagulation and inflammation in AAVs (32, 185). C5a may also trigger the release of tissue factor expressing microparticles and ROS from ANCA sensitized neutrophils (187). Tissue factor, as a pivotal part of the coagulation cascade, catalyzes the generation of thrombin, indicating again the close connection between the complement and the coagulation system. Thrombin activates platelets promoting platelet degranulation and the release of P-selectin. In addition, thrombin can also induce the release of VWF from the endothelium mediating platelet adhesion and aggregation amplifying the cross-talk between coagulation and the innate immune system.
Other Skin Autoimmune Diseases
A limited number of studies have reported on the potential involvement of platelets in other skin autoimmune diseases such as chronic urticarial (CU), vitiligo, bullous pemphigoid (BP), and psoriasis.
In CU patients the assessment of MPV as a supposed marker for platelet activation provided conflicting results (188–190). Contradiction might have been due to the variations in patients' selection, the stage of the disease and patients' disorders (such as obesity and diabetes) during the laboratory analysis (20, 191).
Some studies investigated the intradermal injection of platelet rich plasma (PRP) into vitiligo patients (192). PRP is an autologous blood-derived product with enriched platelets and high concentration of growth factors secreted from platelet α –granules (193, 194). These factors can stimulate melanocyte migration and promote keratinocytes proliferation. Several research groups reported the combination of PRP treatment with narrowband–ultraviolet B phototherapy or exposure to fractional CO2 laser light results in a significant improvement in repigmentation, improving patient compliance (194–196). However, the limitations of those studies are the small sample size and short follow-up periods.
BP is a common autoimmune bullous disease, characterized by autoantibodies directed against hemidesmosomal proteins (BP180 and BP320) of the skin followed by subepidermal blistering (197). The pathogenesis of BP is not fully understood, and the majority of research that has previously address the pathogenesis of BP focused on the immune response caused by autoantibodies. Research about the role of platelet in BP is scarce. A study found significantly higher number of eosinophil and MPV values in BP patients, pointing to a disease related platelet activation (198). Another study discovered increased levels of sP-selectin in BP patients suggesting also a potential platelet activation (199). However, further studies are required to better understand the contribution of platelets on BP.
Psoriasis is a chronic autoimmune-mediated skin disease characterized by red scaly plaques. Some previous studies showed a close association between platelets activation and psoriasis activity. In vitro, platelets from psoriasis patients are more sensitive to thrombin and ADP (200, 201). In line with this increased sensitivity, plasma levels of platelet activation markers such as ß-thromboglobulin, PF4, PMPs and sP-selectin were also elevated in psoriasis patients (202, 203). Another recent study evaluated 320 patients with psoriasis vulgaris and found that the mass of platelets is increased in the affected patients (204). Interestingly, platelet activation was also attributed to the increased endogenous antimicrobial cathelicidin LL37 in psoriasis patients (205). In platelet, LL37 can induce enhanced fibrinogen binding, p-selectin exposure and Ca2+ mobilization (206). In a murine psoriasis model, the P-selectin/PSGL-1 interaction was shown to promote the formation of platelet-leukocyte aggregates and to favor leukocyte rolling in murine skin microvasculature (207). Similarly, Teague et al. showed a disease severity dependent interaction between low density of neutrophils and platelets in psoriasis patients (208).
Platelets as Potential Biomarker for Monitoring and Diagnosis of Skin Autoimmune Diseases
Platelet derived components and platelet indices can potentially be used to monitor and diagnose autoimmune diseases. In SLE, the serum or plasma levels of platelet associated molecules, such as HMGB1, S100A8/A9, sCD40L, and CCL5 as well as platelet derived PMPs harboring IgGs have been shown to correlate with the SLE disease activity index (SLEDAI) score (105, 110, 124, 209, 210). Lood et al. have recently correlated platelet derived S100A8/A9 with cardiovascular complications in SLE patients (109). Apart from platelet released molecules also the presentation of P-selectin at the surface of platelets has been associated with the severity of SLE (106). Next to an active P-selectin exposure, also the deposition of C4d on the platelet surface has been correlated to the occurrence of aPL-related venous thrombosis and to the SLEDAI score (115, 211). Notably, in a cross-sectional study of 105 patients with SLE, the authors reported that compared with healthy individuals, platelet C4d was 100% specific for SLE patients (115). As further demonstrated in a retrospective cohort study, platelet C4d is significantly associated with all-cause mortality and ischemic stroke in SLE patients (211). Therefore, C4d bound platelet can be used as a reliably biomarker for SLE and a predictor for thrombotic events in SLE patients.
In SSc, the levels of platelet released mediator such as serotonin, sCD40L, sP-selectin, HMGB1, and PDGF have been connected to the severity of the SSc related fibrosis or vasculopathy (151, 154, 212–214). Moreover, increased numbers of PMPs have also been observed in SSc patients (158). These few reports suggest the potential usage of platelets and platelet associated compounds as prognostic biomarker, however further validating research is required.
Tomasson et al. reported that sP-selectin and sCD40L released by platelets are positively associated with vasculitic activities (215). Platelet counts are increased in the active stage of AAVs, which can be used to distinguish active disease from acute systemic infections (176). In line with SLE, SSC, and AAV, elevated sP-selectin levels were also previously detected by several groups in patients with CU, especially in autologous plasma skin test positive patients and those which are aspirin-intolerant (216). In addition, the sP-selectin levels correlated positively with the urticarial severity score (217), indicating platelet activation as a possible indicator of the CU disease activity. In psoriasis, several studies reported that the plasma PMPs and P-selectin levels were significantly correlated with the psoriasis area and the severity index (PASI) score (203, 218). Recently, Raghavan et al. proposed a strong correlation between the PASI score and MPV values and platelet counts (219). However, further research is required to confirm the usability of platelet related parameters for the diagnosis and the monitoring of AAV, CU, and psoriasis.
Platelets as Therapeutic Targets in Skin Autoimmune Diseases and Future Perspectives
A number of small molecule inhibitors and monoclonal antibodies have been developed to target platelet activation in skin autoimmune diseases. For example, hydroxychloroquine (HCQ) is an antimalarial compound that provides an effective treatment (220) in all types of Lupus erythematodes. One beneficial mechanism of HCQ therapy is the inhibition of platelet aggregation and degranulation (221, 222). HCQ mediated platelet inhibition is also effective for the treatment of SSc (223). Another therapeutic option to block platelet activation is the use of the P2Y12 receptor inhibitor clopidogrel (224). Several studies reported that in lupus-prone mice clopidogrel treatment inhibits the release of platelet derived sCD40L and P-selectin (36, 225). In SSc mice models, clopidogrel has been shown to decrease fibrosis (154). However, one study reported that a standard dose of clopidogrel (75 mg) inhibits platelet activation in SSc patients but had no effect on plasma serotonin levels. Clopidogrel treatment even led to a significant increase in soluble VCAM-1, indicating endothelial dysfunction as adverse effect (226). Additionally, fibrosis has been successfully managed in an SSc murine model using the serotonin receptor inhibitors terguride, cyproheptadine, and SB 204741 (154, 227). Dapirolizumab is an anti-CD40L Fab antibody fragment and currently evaluated in a phase 2 clinical trial (228). The treatment exhibit an excellent tolerance profile and convincing clinical efficacy in SLE patients. Notably, no thromboembolic adverse events occurred in the phase 1 clinical trial (228). CD40L signaling is also inhibited by Statins, because this class of small molecular compounds is able to reduce the CD40L content in platelets and thus sCD40L levels (229, 230). Therefore, treatment of SLE patients with statins can decrease platelets thrombotic activities (231). Platelet inhibition as therapy of AAV has to our knowledge never been investigated. Figure 5 summarizes current treatment options targeting platelets in skin autoimmune diseases.
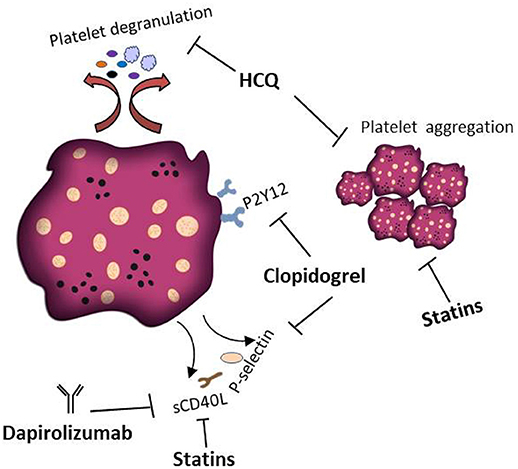
Figure 5. Platelets as therapeutic targets in skin autoimmune disease. Several treatments that interfere with the pathophysiological activity of platelets in skin autoimmune diseases have been proposed. HCQ inhibits platelet aggregation and degranulation. Clopidogrel is a P2Y12 receptor inhibitor preventing the release of platelet derived sCD40L and P-selectin. Dapirolizumab is a monoclonal antibody specifically blocking sCD40L functions. Statins are supposed to affect the thrombotic activities of platelets and to decrease the plasma levels of sCD40L.
As future perspectives, several potential treatment options targeting platelet components or preventing platelet activation might be worth to be investigated. For instance, because P-selectin plays an important role in the pathogenesis of autoimmune diseases, Inclacumab, a novel monoclonal antibody against P-selectin (232), might be considered for the treatment of skin autoimmune diseases in future. As discussed in the previous section, C4d deposition on platelets was found to be highly specific for the diagnosis of SLE, and C4d positive platelets are regarded as a risk factor for thrombotic event in SLE patients (115). In line with this, inhibition of the complement system by eculizumab, a C5 neutralizing monoclonal antibody, prevented thrombotic microangiopathy in SLE (233). Therefore, it would be highly interesting to further investigate the treatment effects of C4 neutralizing antibodies or to develop specific compounds preventing the deposition of C4d on the surface of platelets.
Platelets activation is controlled by a plethora of distinct pathways and a better understanding of these signaling routes may open new therapeutic perspectives. Such as, collagen triggers the phosphorylation of Bruton's Tyrosine Kinase (BTK), and signal transducer and activator of transcription 3 (STAT3) can modulate platelets aggregation and activation (234, 235). BTK inhibitor RN486 has been reported to abrogate the generation of PMPs (234) and the STAT3 inhibitor SC99 was effective to inhibit thrombin-induced P-selectin expression and platelet activation (235). In addition, activated platelets are the main source of sCD40L and blockade of the CD40–CD40L interaction by monoclonal antibodies appears to be a promising treatment of autoimmune diseases. Apart from anti-CD40L antibodies, a CD40-targeting peptide was designed recently (236). This peptide mimics the CD40L domain critical for the interaction with CD40 and it has been shown to effectively block CD40-CD40L signaling (236). In a comparable approach, Chen et al. identified small molecule compounds that can interfere with the CD40–CD40L protein-protein interaction (237). To which extend those drugs are applicable to treat autoimmune diseases remains elusive. However, the development of platelet inhibiting compounds is an emerging field of research and it envisions the discovery of novel compounds efficient for the treatment of skin autoimmune diseases.
Conclusions
Platelets are the main participants of hemostasis and have multiple immunoregulating functions linking coagulation and inflammation. It is becoming apparent that platelet activation might be a biomarker of skin autoimmune diseases activity. Multiple compounds in circulation, such as ICs, DAMPs, VWF and collagen, can stimulate continuous platelet activation. Platelet activation appeared to be further potentiated by the deposition of complement factors at their surface. However, more details about the crosstalk between platelets and complement still needs to be investigated. Upon activation, platelets release a variety of pro-inflammatory mediators such as sCD40L, P-selectin, serotonin, PDGFs, and TGF-β. These released factors contribute to the immune response, ultimately resulting in chronic inflammation and local tissue damage. Activated platelets modulate the function of the innate and adaptive immune system by secreting immune mediators or through direct cellular interactions with immune cells, such as DC, neutrophils, B and T cells. However, the interplay between platelets with other immune cells such as mast cells and monocytes requires further clarification. It would be also worth to investigate novel platelet derived biomarkers to predict and assess the status of the respective disease. There are still some controversies on the assessment of the commonly measured MPV in autoimmune diseases. Contradictive results suggest that only MPV measurements are not sufficient to evaluate platelet function. Platelet activation is a variable and dynamic process and research addressing the role of platelets in autoimmune disease should be not limited to distinct platelet activation processes or distinct factors released by platelets. Right now, targeting platelets using anti-CD40L monoclonal antibodies and ADP receptor inhibitors has been reported to reduce autoimmune reactions in several clinical trials addressing autoimmune diseases. A better understanding of the role of platelets in autoimmune diseases will reveal new therapeutic options in the future.
Author Contributions
XL, CG, and SS contributed to reviewing the current literature and writing of the manuscript.
Funding
Financial support was obtained from the Heike und Wolfgang Mühlbauer Stiftung, Hamburg, Germany.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors thank Birgit Schneider for the design of the artwork.
References
1. Semple JW, Italiano JE Jr, Freedman J. Platelets and the immune continuum. Nat Rev Immunol. (2011) 11:264–74. doi: 10.1038/nri2956
2. Van Der Meijden PEJ, Heemskerk JWM. Platelet biology and functions: new concepts and clinical perspectives. Nat Rev Cardiol. (2019) 16:166–79. doi: 10.1038/s41569-018-0110-0
3. Huck V, Schneider MF, Gorzelanny C, Schneider SW. The various states of von Willebrand factor and their function in physiology and pathophysiology. Thromb Haemost. (2014) 111:598–609. doi: 10.1160/TH13-09-0800
4. Pappelbaum KI, Gorzelanny C, Grassle S, Suckau J, Laschke MW, Bischoff M, et al. Ultralarge von Willebrand factor fibers mediate luminal Staphylococcus aureus adhesion to an intact endothelial cell layer under shear stress. Circulation. (2013) 128:50–9. doi: 10.1161/CIRCULATIONAHA.113.002008
5. Bauer AT, Suckau J, Frank K, Desch A, Goertz L, Wagner AH, et al. von Willebrand factor fibers promote cancer-associated platelet aggregation in malignant melanoma of mice and humans. Blood. (2015) 125:3153–63. doi: 10.1182/blood-2014-08-595686
6. Moller K, Adolph O, Grunow J, Elrod J, Popa M, Ghosh S, et al. Mechanism and functional impact of CD40 ligand-induced von Willebrand factor release from endothelial cells. Thromb Haemost. (2015) 113:1095–108. doi: 10.1160/TH14-04-0336
7. Rondina MT, Weyrich AS, Zimmerman GA. Platelets as cellular effectors of inflammation in vascular diseases. Circ Res. (2013) 112:1506–19. doi: 10.1161/CIRCRESAHA.113.300512
8. Kalagara T, Moutsis T, Yang Y, Pappelbaum KI, Farken A, Cladder-Micus L, et al. The endothelial glycocalyx anchors von Willebrand factor fibers to the vascular endothelium. Blood Adv. (2018) 2:2347–57. doi: 10.1182/bloodadvances.2017013995
9. Jackson SP, Schoenwaelder SM. Procoagulant platelets: are they necrotic? Blood. (2010) 116:2011–8. doi: 10.1182/blood-2010-01-261669
10. Heemskerk JW, Mattheij NJ, Cosemans JM. Platelet-based coagulation: different populations, different functions. J Thromb Haemost. (2013) 11:2–16. doi: 10.1111/jth.12045
11. Heemskerk JW, Bevers EM, Lindhout T. Platelet activation and blood coagulation. Thromb Haemost. (2002) 88:186–93. doi: 10.1055/s-0037-1613209
12. Monroe DM, Hoffman M, Roberts HR. Platelets and thrombin generation. Arterioscler Thromb Vasc Biol. (2002) 22:1381–9. doi: 10.1161/01.ATV.0000031340.68494.34
13. Swieringa F, Spronk HMH, Heemskerk JWM, Van Der Meijden PEJ. Integrating platelet and coagulation activation in fibrin clot formation. Res Pract Thromb Haemost. (2018) 2:450–60. doi: 10.1002/rth2.12107
14. Cimmino G, Cirillo P. Tissue factor: newer concepts in thrombosis and its role beyond thrombosis and hemostasis. Cardiovasc Diagn Ther. (2018) 8:581–93. doi: 10.21037/cdt.2018.10.14
15. Capozzi A, Manganelli V, Riitano G, Recalchi S, Truglia S, Alessandri C, et al. Tissue factor over-expression in platelets of patients with anti-phospholipid syndrome: induction role of anti-beta2-GPI antibodies. Clin Exp Immunol. (2019) 196:59–66. doi: 10.1111/cei.13248
16. Duerschmied D, Bode C, Ahrens I. Immune functions of platelets. Thromb Haemost. (2014) 112:678–91. doi: 10.1160/TH14-02-0146
17. Rondina MT, Garraud O. Emerging evidence for platelets as immune and inflammatory effector cells. Front Immunol. (2014) 5:653. doi: 10.3389/fimmu.2014.00653
18. Morrell CN, Aggrey AA, Chapman LM, Modjeski KL. Emerging roles for platelets as immune and inflammatory cells. Blood. (2014) 123:2759–67. doi: 10.1182/blood-2013-11-462432
19. Habets KL, Huizinga TW, Toes RE. Platelets and autoimmunity. Eur J Clin Invest. (2013) 43:746–57. doi: 10.1111/eci.12101
20. Łukasik ZM, Makowski M, Makowska JS. From blood coagulation to innate and adaptive immunity: the role of platelets in the physiology and pathology of autoimmune disorders. Rheumatol Int. (2018) 38:959–74. doi: 10.1007/s00296-018-4001-9
21. Maynard DM, Heijnen HF, Horne MK, White JG, Gahl WA. Proteomic analysis of platelet alpha-granules using mass spectrometry. J Thromb Haemost. (2007) 5:1945–55. doi: 10.1111/j.1538-7836.2007.02690.x
22. Thomas MR, Storey RF. The role of platelets in inflammation. Thromb Haemost. (2015) 114:449–58. doi: 10.1160/TH14-12-1067
23. Herter JM, Rossaint J, Zarbock A. Platelets in inflammation and immunity. J Thromb Haemost. (2014) 12:1764–75. doi: 10.1111/jth.12730
24. Italiano JE Jr, Battinelli EM. Selective sorting of alpha-granule proteins. J Thromb Haemost. (2009) 7 (Suppl. 1):173–6. doi: 10.1111/j.1538-7836.2009.03387.x
25. Linge P, Fortin PR, Lood C, Bengtsson AA, Boilard E. The non-haemostatic role of platelets in systemic lupus erythematosus. Nat Rev Rheumatol. (2018) 14:195–213. doi: 10.1038/nrrheum.2018.38
26. Boilard E, Blanco P, Nigrovic PA. Platelets: active players in the pathogenesis of arthritis and SLE. Nat Rev Rheumatol. (2012) 8:534–42. doi: 10.1038/nrrheum.2012.118
27. Ballanti E, Perricone C, Greco E, Ballanti M, Di Muzio G, Chimenti MS, et al. Complement and autoimmunity. Immunol Res. (2013) 56:477–91. doi: 10.1007/s12026-013-8422-y
28. Vignesh P, Rawat A, Sharma M, Singh S. Complement in autoimmune diseases. Clin Chim Acta. (2017) 465:123–30. doi: 10.1016/j.cca.2016.12.017
29. Panelius J, Meri S. Complement system in dermatological diseases - fire under the skin. Front Med. (2015) 2:3. doi: 10.3389/fmed.2015.00003
30. Trouw LA, Pickering MC, Blom AM. The complement system as a potential therapeutic target in rheumatic disease. Nat Rev Rheumatol. (2017) 13:538–47. doi: 10.1038/nrrheum.2017.125
31. Speth C, Rambach G, Wurzner R, Lass-Florl C, Kozarcanin H, Hamad OA, et al. Complement and platelets: mutual interference in the immune network. Mol Immunol. (2015) 67:108–18. doi: 10.1016/j.molimm.2015.03.244
32. Peerschke EI, Yin W, Ghebrehiwet B. Complement activation on platelets: implications for vascular inflammation and thrombosis. Mol Immunol. (2010) 47:2170–5. doi: 10.1016/j.molimm.2010.05.009
33. Peerschke EI, Yin W, Ghebrehiwet B. Platelet mediated complement activation. Adv Exp Med Biol. (2008) 632:81–91. doi: 10.1007/978-0-387-78952-1_7
34. Kenney DM, Davis AE III. Association of alternative complement pathway components with human blood platelets: secretion and localization of factor D and beta 1H Globulin. Clin Immunol Immunopathol. (1981) 21:351–63. doi: 10.1016/0090-1229(81)90224-5
35. Yin W, Ghebrehiwet B, Peerschke EI. Expression of complement components and inhibitors on platelet microparticles. Platelets. (2008) 19:225–33. doi: 10.1080/09537100701777311
36. Duffau P, Seneschal J, Nicco C, Richez C, Lazaro E, Douchet I, et al. Platelet CD154 potentiates interferon-alpha secretion by plasmacytoid dendritic cells in systemic lupus erythematosus. Sci Transl Med. (2010) 2:47ra63. doi: 10.1126/scitranslmed.3001001
37. Elzey BD, Schmidt NW, Crist SA, Kresowik TP, Harty JT, Nieswandt B, et al. Platelet-derived CD154 enables T-cell priming and protection against Listeria monocytogenes challenge. Blood. (2008) 111:3684–91. doi: 10.1182/blood-2007-05-091728
38. Elzey BD, Grant JF, Sinn HW, Nieswandt B, Waldschmidt TJ, Ratliff TL. Cooperation between platelet-derived CD154 and CD4+ T cells for enhanced germinal center formation. J Leukoc Biol. (2005) 78:80–4. doi: 10.1189/jlb.1104669
39. Cognasse F, Hamzeh-Cognasse H, Lafarge S, Chavarin P, Cogne M, Richard Y, et al. Human platelets can activate peripheral blood B cells and increase production of immunoglobulins. Exp Hematol. (2007) 35:1376–87. doi: 10.1016/j.exphem.2007.05.021
40. Lievens D, Eijgelaar WJ, Biessen EA, Daemen MJ, Lutgens E. The multi-functionality of CD40L and its receptor CD40 in atherosclerosis. Thromb Haemost. (2009) 102:206–14. doi: 10.1160/TH09-01-0029
41. Henn V, Slupsky JR, Grafe M, Anagnostopoulos I, Forster R, Muller-Berghaus G, et al. CD40 ligand on activated platelets triggers an inflammatory reaction of endothelial cells. Nature. (1998) 391:591–4. doi: 10.1038/35393
42. Inwald DP, Mcdowall A, Peters MJ, Callard RE, Klein NJ. CD40 is constitutively expressed on platelets and provides a novel mechanism for platelet activation. Circ Res. (2003) 92:1041–8. doi: 10.1161/01.RES.0000070111.98158.6C
43. Struyf S, Burdick MD, Proost P, Van Damme J, Strieter RM. Platelets release CXCL4L1, a nonallelic variant of the chemokine platelet factor-4/CXCL4 and potent inhibitor of angiogenesis. Circ Res. (2004) 95:855–7. doi: 10.1161/01.RES.0000146674.38319.07
44. Fleischer J, Grage-Griebenow E, Kasper B, Heine H, Ernst M, Brandt E, et al. Platelet factor 4 inhibits proliferation and cytokine release of activated human T cells. J Immunol. (2002) 169:770–7. doi: 10.4049/jimmunol.169.2.770
45. Deuel TF, Senior RM, Chang D, Griffin GL, Heinrikson RL, Kaiser ET. Platelet factor 4 is chemotactic for neutrophils and monocytes. Proc Natl Acad Sci USA. (1981) 78:4584–7. doi: 10.1073/pnas.78.7.4584
46. Lewandrowski U, Moebius J, Walter U, Sickmann A. Elucidation of N-glycosylation sites on human platelet proteins: a glycoproteomic approach. Mol Cell Proteomics. (2006) 5:226–33. doi: 10.1074/mcp.M500324-MCP200
47. Buttrum SM, Hatton R, Nash GB. Selectin-mediated rolling of neutrophils on immobilized platelets. Blood. (1993) 82:1165–74.
48. Spertini O, Cordey AS, Monai N, Giuffre L, Schapira M. P-selectin glycoprotein ligand 1 is a ligand for L-selectin on neutrophils, monocytes, and CD34+ hematopoietic progenitor cells. J Cell Biol. (1996) 135:523–31. doi: 10.1083/jcb.135.2.523
49. Lam FW, Vijayan KV, Rumbaut RE. Platelets and their interactions with other immune cells. Compr Physiol. (2015) 5:1265–80. doi: 10.1002/cphy.c140074
50. Sreeramkumar V, Adrover JM, Ballesteros I, Cuartero MI, Rossaint J, Bilbao I, et al. Neutrophils scan for activated platelets to initiate inflammation. Science. (2014) 346:1234–8. doi: 10.1126/science.1256478
51. Evangelista V, Manarini S, Sideri R, Rotondo S, Martelli N, Piccoli A, et al. Platelet/polymorphonuclear leukocyte interaction: P-selectin triggers protein-tyrosine phosphorylation-dependent CD11b/CD18 adhesion: role of PSGL-1 as a signaling molecule. Blood. (1999) 93:876–85.
52. Celi A, Pellegrini G, Lorenzet R, De Blasi A, Ready N, Furie BC, et al. P-selectin induces the expression of tissue factor on monocytes. Proc Natl Acad Sci USA. (1994) 91:8767–71. doi: 10.1073/pnas.91.19.8767
53. Etulain J, Martinod K, Wong SL, Cifuni SM, Schattner M, Wagner DD. P-selectin promotes neutrophil extracellular trap formation in mice. Blood. (2015) 126:242–6. doi: 10.1182/blood-2015-01-624023
54. Clark SR, Ma AC, Tavener SA, Mcdonald B, Goodarzi Z, Kelly MM, et al. Platelet TLR4 activates neutrophil extracellular traps to ensnare bacteria in septic blood. Nat Med. (2007) 13:463–9. doi: 10.1038/nm1565
55. Maugeri N, Campana L, Gavina M, Covino C, De Metrio M, Panciroli C, et al. Activated platelets present high mobility group box 1 to neutrophils, inducing autophagy and promoting the extrusion of neutrophil extracellular traps. J Thromb Haemost. (2014) 12:2074–88. doi: 10.1111/jth.12710
56. Grassle S, Huck V, Pappelbaum KI, Gorzelanny C, Aponte-Santamaria C, Baldauf C, et al. von Willebrand factor directly interacts with DNA from neutrophil extracellular traps. Arterioscler Thromb Vasc Biol. (2014) 34:1382–9. doi: 10.1161/ATVBAHA.113.303016
57. Fuchs TA, Brill A, Duerschmied D, Schatzberg D, Monestier M, Myers DD Jr, et al. Extracellular DNA traps promote thrombosis. Proc Natl Acad Sci USA. (2010) 107:15880–5. doi: 10.1073/pnas.1005743107
58. Mcdonald B, Davis RP, Kim SJ, Tse M, Esmon CT, Kolaczkowska E, et al. Platelets and neutrophil extracellular traps collaborate to promote intravascular coagulation during sepsis in mice. Blood. (2017) 129:1357–67. doi: 10.1182/blood-2016-09-741298
59. Mine S, Fujisaki T, Suematsu M, Tanaka Y. Activated platelets and endothelial cell interaction with neutrophils under flow conditions. Intern Med. (2001) 40:1085–92. doi: 10.2169/internalmedicine.40.1085
60. Golebiewska EM, Poole AW. Platelet secretion: From haemostasis to wound healing and beyond. Blood Rev. (2015) 29:153–62. doi: 10.1016/j.blre.2014.10.003
61. Angiolillo DJ, Ueno M, Goto S. Basic principles of platelet biology and clinical implications. Circ J. (2010) 74:597–607. doi: 10.1253/circj.CJ-09-0982
62. Shajib MS, Khan WI. The role of serotonin and its receptors in activation of immune responses and inflammation. Acta Physiol. (2015) 213:561–74. doi: 10.1111/apha.12430
63. Herr N, Bode C, Duerschmied D. The effects of serotonin in immune cells. Front Cardiovasc Med. (2017) 4:48. doi: 10.3389/fcvm.2017.00048
64. Melki I, Tessandier N, Zufferey A, Boilard E. Platelet microvesicles in health and disease. Platelets. (2017) 28:214–21. doi: 10.1080/09537104.2016.1265924
65. Arraud N, Linares R, Tan S, Gounou C, Pasquet JM, Mornet S, et al. Extracellular vesicles from blood plasma: determination of their morphology, size, phenotype and concentration. J Thromb Haemost. (2014) 12:614–27. doi: 10.1111/jth.12554
66. Garcia BA, Smalley DM, Cho H, Shabanowitz J, Ley K, Hunt DF. The platelet microparticle proteome. J Proteome Res. (2005) 4:1516–21. doi: 10.1021/pr0500760
67. Diehl P, Fricke A, Sander L, Stamm J, Bassler N, Htun N, et al. Microparticles: major transport vehicles for distinct microRNAs in circulation. Cardiovasc Res. (2012) 93:633–44. doi: 10.1093/cvr/cvs007
68. Mause SF, Von Hundelshausen P, Zernecke A, Koenen RR, Weber C. Platelet microparticles: a transcellular delivery system for RANTES promoting monocyte recruitment on endothelium. Arterioscler Thromb Vasc Biol. (2005) 25:1512–8. doi: 10.1161/01.ATV.0000170133.43608.37
69. Sprague DL, Elzey BD, Crist SA, Waldschmidt TJ, Jensen RJ, Ratliff TL. Platelet-mediated modulation of adaptive immunity: unique delivery of CD154 signal by platelet-derived membrane vesicles. Blood. (2008) 111:5028–36. doi: 10.1182/blood-2007-06-097410
70. Boilard E, Duchez AC, Brisson A. The diversity of platelet microparticles. Curr Opin Hematol. (2015) 22:437–44. doi: 10.1097/MOH.0000000000000166
71. Wang ZT, Wang Z, Hu YW. Possible roles of platelet-derived microparticles in atherosclerosis. Atherosclerosis. (2016) 248:10–6. doi: 10.1016/j.atherosclerosis.2016.03.004
72. Berckmans RJ, Nieuwland R, Boing AN, Romijn FP, Hack CE, Sturk A. Cell-derived microparticles circulate in healthy humans and support low grade thrombin generation. Thromb Haemost. (2001) 85:639–46. doi: 10.1055/s-0037-1615646
73. Koupenova M, Kehrel BE, Corkrey HA, Freedman JE. Thrombosis and platelets: an update. Eur Heart J. (2017) 38:785–91. doi: 10.1093/eurheartj/ehw550
74. Schneider SW, Nuschele S, Wixforth A, Gorzelanny C, Alexander-Katz A, Netz RR, et al. Shear-induced unfolding triggers adhesion of von Willebrand factor fibers. Proc Natl Acad Sci USA. (2007) 104:7899–903. doi: 10.1073/pnas.0608422104
75. Sveshnikova AN, Balatskiy AV, Demianova AS, Shepelyuk TO, Shakhidzhanov SS, Balatskaya MN, et al. Systems biology insights into the meaning of the platelet's dual-receptor thrombin signaling. J Thromb Haemost. (2016) 14:2045–57. doi: 10.1111/jth.13442
76. Coughlin SR. Thrombin signalling and protease-activated receptors. Nature. (2000) 407:258–64. doi: 10.1038/35025229
77. Hechler B, Gachet C. Purinergic receptors in thrombosis and inflammation. Arterioscler Thromb Vasc Biol. (2015) 35:2307–15. doi: 10.1161/ATVBAHA.115.303395
78. Murugappa S, Kunapuli SP. The role of ADP receptors in platelet function. Front Biosci. (2006) 11:1977–86. doi: 10.2741/1939
79. Versteeg HH, Heemskerk JW, Levi M, Reitsma PH. New fundamentals in hemostasis. Physiol Rev. (2013) 93:327–58. doi: 10.1152/physrev.00016.2011
80. Vieira-De-Abreu A, Campbell RA, Weyrich AS, Zimmerman GA. Platelets: versatile effector cells in hemostasis, inflammation, and the immune continuum. Semin Immunopathol. (2012) 34:5–30. doi: 10.1007/s00281-011-0286-4
81. Qiao J, Al-Tamimi M, Baker RI, Andrews RK, Gardiner EE. The platelet Fc receptor, FcgammaRIIa. Immunol Rev. (2015) 268:241–52. doi: 10.1111/imr.12370
82. Worth RG, Chien CD, Chien P, Reilly MP, Mckenzie SE, Schreiber AD. Platelet FcgammaRIIA binds and internalizes IgG-containing complexes. Exp Hematol. (2006) 34:1490–5. doi: 10.1016/j.exphem.2006.06.015
83. Berlacher MD, Vieth JA, Heflin BC, Gay SR, Antczak AJ, Tasma BE, et al. FcgammaRIIa ligation induces platelet hypersensitivity to thrombotic stimuli. Am J Pathol. (2013) 182:244–54. doi: 10.1016/j.ajpath.2012.09.005
84. Cloutier N, Allaeys I, Marcoux G, Machlus KR, Mailhot B, Zufferey A, et al. Platelets release pathogenic serotonin and return to circulation after immune complex-mediated sequestration. Proc Natl Acad Sci USA. (2018) 115:E1550–9. doi: 10.1073/pnas.1720553115
85. Cognasse F, Nguyen KA, Damien P, Mcnicol A, Pozzetto B, Hamzeh-Cognasse H, et al. The inflammatory role of platelets via their TLRs and siglec receptors. Front Immunol. (2015) 6:83. doi: 10.3389/fimmu.2015.00083
86. Lp DA, Schattner M. Platelet toll-like receptors in thromboinflammation. Front Biosci. (2017) 22:1867–83. doi: 10.2741/4576
87. Vallance TM, Zeuner M-T, Williams HF, Widera D, Vaiyapuri S. Toll-Like receptor 4 signalling and its impact on platelet function, thrombosis, and haemostasis. Mediators Inflamm. (2017) 2017:13. doi: 10.1155/2017/9605894
88. Patzelt J, Mueller KA, Breuning S, Karathanos A, Schleicher R, Seizer P, et al. Expression of anaphylatoxin receptors on platelets in patients with coronary heart disease. Atherosclerosis. (2015) 238:289–95. doi: 10.1016/j.atherosclerosis.2014.12.002
89. Martel C, Cointe S, Maurice P, Matar S, Ghitescu M, Theroux P, et al. Requirements for membrane attack complex formation and anaphylatoxins binding to collagen-activated platelets. PLoS ONE. (2011) 6:e18812. doi: 10.1371/journal.pone.0018812
90. Del Conde I, Cruz MA, Zhang H, Lopez JA, Afshar-Kharghan V. Platelet activation leads to activation and propagation of the complement system. J Exp Med. (2005) 201:871–9. doi: 10.1084/jem.20041497
91. Peerschke EI, Yin W, Grigg SE, Ghebrehiwet B. Blood platelets activate the classical pathway of human complement. J Thromb Haemost. (2006) 4:2035–42. doi: 10.1111/j.1538-7836.2006.02065.x
92. Peerschke EI, Murphy TK, Ghebrehiwet B. Activation-dependent surface expression of gC1qR/p33 on human blood platelets. Thromb Haemost. (2003) 89:331–9. doi: 10.1055/s-0037-1613450
93. Peerschke EI, Ghebrehiwet B. Human blood platelet gC1qR/p33. Immunol Rev. (2001) 180:56–64. doi: 10.1034/j.1600-065X.2001.1800105.x
94. Bengtsson AA, Ronnblom L. Systemic lupus erythematosus: still a challenge for physicians. J Intern Med. (2017) 281:52–64. doi: 10.1111/joim.12529
95. Kaul A, Gordon C, Crow MK, Touma Z, Urowitz MB, Van Vollenhoven R, et al. Systemic lupus erythematosus. Nat Rev Disease Primers. (2016) 2:16039. doi: 10.1038/nrdp.2016.39
96. Jung JH, Soh MS, Ahn YH, Um YJ, Jung JY, Suh CH, et al. Thrombocytopenia in systemic lupus erythematosus: clinical manifestations, treatment, and prognosis in 230 patients. Medicine. (2016) 95:e2818. doi: 10.1097/MD.0000000000002818
97. Zhao H, Li S, Yang R. Thrombocytopenia in patients with systemic lupus erythematosus: significant in the clinical implication and prognosis. Platelets. (2010) 21:380–5. doi: 10.3109/09537101003735564
98. Khan A, Haider I, Ayub M, Khan S. Mean Platelet Volume (MPV) as an indicator of disease activity and severity in lupus. F1000Res. (2017) 6:126. doi: 10.12688/f1000research.10763.2
99. Chen SY, Du J, Lu XN, Xu JH. Platelet distribution width as a novel indicator of disease activity in systemic lupus erythematosus. J Res Med Sci. (2018) 23:48. doi: 10.4103/jrms.JRMS_1038_16
100. Zhao CN, Mao YM, Wang P, Guan SY, Sam NB, Li XM, et al. Lack of association between mean platelet volume and disease activity in systemic lupus erythematosus patients: a systematic review and meta-analysis. Rheumatol Int. (2018) 38:1635–41. doi: 10.1007/s00296-018-4065-6
101. Bai M, Xing L, Feng J, Cui C, Huang L, Liang G. Mean platelet volume could reflect disease activity of adult patients with systemic lupus erythematosus. Clin Lab. (2016) 62:1317–22. doi: 10.7754/Clin.Lab.2015.151134
102. Kim KJ, Baek IW, Yoon CH, Kim WU, Cho CS. Elevated levels of soluble CD40 ligand are associated with antiphospholipid antibodies in patients with systemic lupus erythematosus. Clin Exp Rheumatol. (2017) 35:823–30.
103. Ferro D, Basili S, Roccaforte S, Di Franco M, Cipollone F, Ciabattoni G, et al. Determinants of enhanced thromboxane biosynthesis in patients with systemic lupus erythematosus. Arthritis Rheum. (1999) 42:2689–97.
104. Nagahama M, Nomura S, Ozaki Y, Yoshimura C, Kagawa H, Fukuhara S. Platelet activation markers and soluble adhesion molecules in patients with systemic lupus erythematosus. Autoimmunity. (2001) 33:85–94. doi: 10.3109/08916930108995993
105. Goules A, Tzioufas AG, Manousakis MN, Kirou KA, Crow MK, Routsias JG. Elevated levels of soluble CD40 ligand (sCD40L) in serum of patients with systemic autoimmune diseases. J Autoimmun. (2006) 26:165–71. doi: 10.1016/j.jaut.2006.02.002
106. Nhek S, Clancy R, Lee KA, Allen NM, Barrett TJ, Marcantoni E, et al. Activated platelets induce endothelial cell activation via an interleukin-1β pathway in systemic lupus erythematosus. Arterioscler Thromb Vasc Biol. (2017) 37:707–16. doi: 10.1161/ATVBAHA.116.308126
107. Ekdahl KN, Ronnblom L, Sturfelt G, Nilsson B. Increased phosphate content in complement component C3, fibrinogen, vitronectin, and other plasma proteins in systemic lupus erythematosus: covariation with platelet activation and possible association with thrombosis. Arthritis Rheum. (1997) 40:2178–86. doi: 10.1002/art.1780401212
108. Scherlinger M, Sisirak V, Richez C, Lazaro E, Duffau P, Blanco P. New insights on platelets and platelet-derived microparticles in systemic lupus erythematosus. Curr Rheumatol Rep. (2017) 19:48. doi: 10.1007/s11926-017-0678-0
109. Lood C, Tyden H, Gullstrand B, Jonsen A, Kallberg E, Morgelin M, et al. Platelet-Derived S100A8/A9 and cardiovascular disease in systemic lupus erythematosus. Arthritis Rheumatol. (2016) 68:1970–80. doi: 10.1002/art.39656
110. Ma CY, Jiao YL, Zhang J, Yang QR, Zhang ZF, Shen YJ, et al. Elevated plasma level of HMGB1 is associated with disease activity and combined alterations with IFN-alpha and TNF-alpha in systemic lupus erythematosus. Rheumatol Int. (2012) 32:395–402. doi: 10.1007/s00296-010-1636-6
111. Li J, Xie H, Wen T, Liu H, Zhu W, Chen X. Expression of high mobility group box chromosomal protein 1 and its modulating effects on downstream cytokines in systemic lupus erythematosus. J Rheumatol. (2010) 37:766–75. doi: 10.3899/jrheum.090663
112. Lafyatis R, Marshak-Rothstein A. Toll-like receptors and innate immune responses in systemic lupus erythematosus. Arthritis Res Ther. (2007) 9:222. doi: 10.1186/ar2321
113. Lood C, Tyden H, Gullstrand B, Sturfelt G, Jonsen A, Truedsson L, et al. Platelet activation and anti-phospholipid antibodies collaborate in the activation of the complement system on platelets in systemic lupus erythematosus. PLoS ONE. (2014) 9:e99386. doi: 10.1371/journal.pone.0099386
114. Willis R, Harris EN, Pierangeli SS. Pathogenesis of the antiphospholipid syndrome. Semin Thromb Hemost. (2012) 38:305–21. doi: 10.1055/s-0032-1311827
115. Navratil JS, Manzi S, Kao AH, Krishnaswami S, Liu CC, Ruffing MJ, et al. Platelet C4d is highly specific for systemic lupus erythematosus. Arthritis Rheum. (2006) 54:670–4. doi: 10.1002/art.21627
116. Wiedmer T, Sims PJ. Effect of complement proteins C5b-9 on blood platelets. Evidence for reversible depolarization of membrane potential. J Biol Chem. (1985) 260:8014–9.
117. Wiedmer T, Esmon CT, Sims PJ. Complement proteins C5b-9 stimulate procoagulant activity through platelet prothrombinase. Blood. (1986) 68:875–80.
118. Peerschke EI, Yin W, Alpert DR, Roubey RA, Salmon JE, Ghebrehiwet B. Serum complement activation on heterologous platelets is associated with arterial thrombosis in patients with systemic lupus erythematosus and antiphospholipid antibodies. Lupus. (2009) 18:530–8. doi: 10.1177/0961203308099974
119. Liang Y, Xie SB, Wu CH, Hu Y, Zhang Q, Li S, et al. Coagulation cascade and complement system in systemic lupus erythematosus. Oncotarget. (2018) 9:14862–81. doi: 10.18632/oncotarget.23206
120. Markiewski MM, Nilsson B, Ekdahl KN, Mollnes TE, Lambris JD. Complement and coagulation: strangers or partners in crime? Trends Immunol. (2007) 28:184–92. doi: 10.1016/j.it.2007.02.006
121. Kölm R, Schaller M, Roumenina LT, Niemiec I, Kremer Hovinga JA, Khanicheh E, et al. Von Willebrand Factor Interacts with Surface-Bound C1q and Induces Platelet Rolling. J Immunol. (2016) 197:3669–79. doi: 10.4049/jimmunol.1501876
122. Meyerhoff J, Dorsch CA. Decreased platelet serotonin levels in systemic lupus erythematosus. Arthritis Rheum. (1981) 24:1495–500. doi: 10.1002/art.1780241207
123. Lood C, Tyden H, Gullstrand B, Klint C, Wenglen C, Nielsen CT, et al. Type I interferon-mediated skewing of the serotonin synthesis is associated with severe disease in systemic lupus erythematosus. PLoS ONE. (2015) 10:e0125109. doi: 10.1371/journal.pone.0125109
124. Burbano C, Villar-Vesga J, Orejuela J, Munoz C, Vanegas A, Vasquez G, et al. Potential involvement of platelet-derived microparticles and microparticles forming immune complexes during monocyte activation in patients with systemic lupus erythematosus. Front Immunol. (2018) 9:322. doi: 10.3389/fimmu.2018.00322
125. Vanhoutte PM. Platelet-derived serotonin, the endothelium, and cardiovascular disease. J Cardiovasc Pharmacol. (1991) 17 (Suppl. 5):S6–12. doi: 10.1097/00005344-199100175-00003
126. Litvack ML, Post M, Palaniyar N. IgM promotes the clearance of small particles and apoptotic microparticles by macrophages. PLoS ONE. (2011) 6:e17223. doi: 10.1371/journal.pone.0017223
127. Boilard E. Platelet-derived Interleukin-1beta fuels the fire in blood vessels in systemic lupus erythematosus. Arterioscler Thromb Vasc Biol. (2017) 37:607–8. doi: 10.1161/ATVBAHA.117.309108
128. Furie B, Furie BC. The molecular basis of platelet and endothelial cell interaction with neutrophils and monocytes: role of P-selectin and the P-selectin ligand, PSGL-1. Thromb Haemost. (1995) 74:224–7. doi: 10.1055/s-0038-1642681
129. Lood C, Amisten S, Gullstrand B, Jonsen A, Allhorn M, Truedsson L, et al. Platelet transcriptional profile and protein expression in patients with systemic lupus erythematosus: up-regulation of the type I interferon system is strongly associated with vascular disease. Blood. (2010) 116:1951–7. doi: 10.1182/blood-2010-03-274605
130. Pattanaik D, Brown M, Postlethwaite BC, Postlethwaite AE. Pathogenesis of systemic sclerosis. Front Immunol. (2015) 6:272. doi: 10.3389/fimmu.2015.00272
131. Ntelis K, Solomou EE, Sakkas L, Liossis SN, Daoussis D. The role of platelets in autoimmunity, vasculopathy, and fibrosis: implications for systemic sclerosis. Semin Arthritis Rheum. (2017) 47:409–17. doi: 10.1016/j.semarthrit.2017.05.004
132. Pauling JD, O'donnell VB, Mchugh NJ. The contribution of platelets to the pathogenesis of Raynaud's phenomenon and systemic sclerosis. Platelets. (2013) 24:503–15. doi: 10.3109/09537104.2012.719090
133. Ramirez GA, Franchini S, Rovere-Querini P, Sabbadini MG, Manfredi AA, Maugeri N. The role of platelets in the pathogenesis of systemic sclerosis. Front Immunol. (2012) 3:160. doi: 10.3389/fimmu.2012.00160
134. Widgerow AD. Ischemia-reperfusion injury: influencing the microcirculatory and cellular environment. Ann Plast Surg. (2014) 72:253–60. doi: 10.1097/SAP.0b013e31825c089c
135. Jansen MP, Emal D, Teske GJ, Dessing MC, Florquin S, Roelofs JJ. Release of extracellular DNA influences renal ischemia reperfusion injury by platelet activation and formation of neutrophil extracellular traps. Kidney Int. (2017) 91:352–64. doi: 10.1016/j.kint.2016.08.006
136. Scherlinger M, Guillotin V, Truchetet ME, Contin-Bordes C, Sisirak V, Duffau P, et al. Systemic lupus erythematosus and systemic sclerosis: all roads lead to platelets. Autoimmun Rev. (2018) 17:625–35. doi: 10.1016/j.autrev.2018.01.012
137. Gualtierotti R, Ingegnoli F, Griffini S, Grovetti E, Borghi MO, Bucciarelli P, et al. Detection of early endothelial damage in patients with Raynaud's phenomenon. Microvasc Res. (2017) 113:22–8. doi: 10.1016/j.mvr.2017.04.004
138. Morrisroe KB, Stevens W, Nandurkar H, Prior D, Thakkar V, Roddy J, et al. The association of antiphospholipid antibodies with cardiopulmonary manifestations of systemic sclerosis. Clin Exp Rheumatol. (2014) 32:S133–17.
139. Chiang TM, Takayama H, Postlethwaite AE. Increase in platelet non-integrin type I collagen receptor in patients with systemic sclerosis. Thromb Res. (2006) 117:299–306. doi: 10.1016/j.thromres.2005.03.003
140. Laurent P, Sisirak V, Lazaro E, Richez C, Duffau P, Blanco P, et al. Innate immunity in systemic sclerosis fibrosis: recent advances. Front Immunol. (2018) 9:1702. doi: 10.3389/fimmu.2018.01702
141. Lafyatis R. Transforming growth factor beta–at the centre of systemic sclerosis. Nat Rev Rheumatol. (2014) 10:706–19. doi: 10.1038/nrrheum.2014.137
142. Sacchetti C, Bai Y, Stanford SM, Di Benedetto P, Cipriani P, Santelli E, et al. PTP4A1 promotes TGFbeta signaling and fibrosis in systemic sclerosis. Nat Commun. (2017) 8:1060. doi: 10.1038/s41467-017-01168-1
143. Blakytny R, Ludlow A, Martin GE, Ireland G, Lund LR, Ferguson MW, et al. Latent TGF-beta1 activation by platelets. J Cell Physiol. (2004) 199:67–76. doi: 10.1002/jcp.10454
144. Assoian RK, Komoriya A, Meyers CA, Miller DM, Sporn MB. Transforming growth factor-beta in human platelets. Identification of a major storage site, purification, and characterization. J Biol Chem. (1983) 258:7155–60.
145. Meyer A, Wang W, Qu J, Croft L, Degen JL, Coller BS, et al. Platelet TGF-β1 contributions to plasma TGF-β1, cardiac fibrosis, and systolic dysfunction in a mouse model of pressure overload. Blood. (2012) 119:1064–74. doi: 10.1182/blood-2011-09-377648
146. Roberts AB, Heine UI, Flanders KC, Sporn MB. Transforming growth factor-beta. Major role in regulation of extracellular matrix. Ann N Y Acad Sci. (1990) 580:225–32. doi: 10.1111/j.1749-6632.1990.tb17931.x
147. Chen Y, Shi-Wen X, Eastwood M, Black CM, Denton CP, Leask A, et al. Contribution of activin receptor–like kinase 5 (transforming growth factor β receptor type I) signaling to the fibrotic phenotype of scleroderma fibroblasts. Arthritis Rheum. (2006) 54:1309–16. doi: 10.1002/art.21725
148. Leask A. The role of endothelin-1 signaling in the fibrosis observed in systemic sclerosis. Pharmacol Res. (2011) 63:502–3. doi: 10.1016/j.phrs.2011.01.011
149. Bonner JC. Regulation of PDGF and its receptors in fibrotic diseases. Cytokine Growth Factor Rev. (2004) 15:255–73. doi: 10.1016/j.cytogfr.2004.03.006
150. Clark KE, Lopez H, Abdi BA, Guerra SG, Shiwen X, Khan K, et al. Multiplex cytokine analysis of dermal interstitial blister fluid defines local disease mechanisms in systemic sclerosis. Arthritis Res Ther. (2015) 17:73. doi: 10.1186/s13075-015-0575-8
151. Makino K, Makino T, Stawski L, Mantero JC, Lafyatis R, Simms R, et al. Blockade of PDGF receptors by crenolanib has therapeutic effect in patient fibroblasts and in preclinical models of systemic sclerosis. J Invest Dermatol. (2017) 137:1671–81. doi: 10.1016/j.jid.2017.03.032
152. Huang J, Maier C, Zhang Y, Soare A, Dees C, Beyer C, et al. Nintedanib inhibits macrophage activation and ameliorates vascular and fibrotic manifestations in the Fra2 mouse model of systemic sclerosis. Ann Rheum Dis. (2017) 76:1941–8. doi: 10.1136/annrheumdis-2016-210823
153. Huang J, Beyer C, Palumbo-Zerr K, Zhang Y, Ramming A, Distler A, et al. Nintedanib inhibits fibroblast activation and ameliorates fibrosis in preclinical models of systemic sclerosis. Ann Rheum Dis. (2016) 75:883–90. doi: 10.1136/annrheumdis-2014-207109
154. Dees C, Akhmetshina A, Zerr P, Reich N, Palumbo K, Horn A, et al. Platelet-derived serotonin links vascular disease and tissue fibrosis. J Exp Med. (2011) 208:961–72. doi: 10.1084/jem.20101629
155. Truchetet ME, Demoures B, Eduardo Guimaraes J, Bertrand A, Laurent P, Jolivel V, et al. Platelets induce thymic stromal lymphopoietin production by endothelial cells: contribution to fibrosis in human systemic sclerosis. Arthritis Rheumatol. (2016) 68:2784–94. doi: 10.1002/art.39817
156. Pattanaik D, Brown M, Postlethwaite AE. Vascular involvement in systemic sclerosis (scleroderma). J Inflamm Res. (2011) 4:105–25. doi: 10.2147/JIR.S18145
157. Van Bon L, Affandi AJ, Broen J, Christmann RB, Marijnissen RJ, Stawski L, et al. Proteome-wide analysis and CXCL4 as a biomarker in systemic sclerosis. N Engl J Med. (2014) 370:433–43. doi: 10.1056/NEJMoa1114576
158. Maugeri N, Capobianco A, Rovere-Querini P, Ramirez GA, Tombetti E, Valle PD, et al. Patelet microparticles sustain autophagy-associated activation of ne1utrophils in systemic sclerosis. Sci Translational Med. (2018) 10:eaao3089. doi: 10.1126/scitranslmed.aao3089
159. Maugeri N, Rovere-Querini P, Baldini M, Baldissera E, Sabbadini MG, Bianchi ME, et al. Oxidative stress elicits platelet/leukocyte inflammatory interactions via HMGB1: a candidate for microvessel injury in sytemic sclerosis. Antioxid Redox Signal. (2014) 20:1060–74. doi: 10.1089/ars.2013.5298
160. Maugeri N, Franchini S, Campana L, Baldini M, Ramirez GA, Sabbadini MG, et al. Circulating platelets as a source of the damage-associated molecular pattern HMGB1 in patients with systemic sclerosis. Autoimmunity. (2012) 45:584–7. doi: 10.3109/08916934.2012.719946
161. Hirigoyen D, Burgos PI, Mezzano V, Duran J, Barrientos M, Saez CG, et al. Inhibition of angiogenesis by platelets in systemic sclerosis patients. Arthritis Res Ther. (2015) 17:332. doi: 10.1186/s13075-015-0848-2
162. Ed Rainger G, Chimen M, Harrison MJ, Yates CM, Harrison P, Watson SP, et al. The role of platelets in the recruitment of leukocytes during vascular disease. Platelets. (2015) 26:507–20. doi: 10.3109/09537104.2015.1064881
163. Woller G, Brandt E, Mittelstadt J, Rybakowski C, Petersen F. Platelet factor 4/CXCL4-stimulated human monocytes induce apoptosis in endothelial cells by the release of oxygen radicals. J Leukoc Biol. (2008) 83:936–45. doi: 10.1189/jlb.0907592
164. Aloui C, Prigent A, Tariket S, Sut C, Fagan J, Cognasse F, et al. Levels of human platelet-derived soluble CD40 ligand depend on haplotypes of CD40LG-CD40-ITGA2. Sci Rep. (2016) 6:24715. doi: 10.1038/srep24715
165. Leon-Ponte M, Ahern GP, O'connell PJ. Serotonin provides an accessory signal to enhance T-cell activation by signaling through the 5-HT7 receptor. Blood. (2007) 109:3139–46. doi: 10.1182/blood-2006-10-052787
166. Li N, Ghia JE, Wang H, Mcclemens J, Cote F, Suehiro Y, et al. Serotonin activates dendritic cell function in the context of gut inflammation. Am J Pathol. (2011) 178:662–71. doi: 10.1016/j.ajpath.2010.10.028
167. Durk T, Panther E, Muller T, Sorichter S, Ferrari D, Pizzirani C, et al. 5-Hydroxytryptamine modulates cytokine and chemokine production in LPS-primed human monocytes via stimulation of different 5-HTR subtypes. Int Immunol. (2005) 17:599–606. doi: 10.1093/intimm/dxh242
168. Loricera J, Blanco R. Vasculitis syndromes: A closer look at skin involvement in vasculitis. Nat Rev Rheumatol. (2018) 14:186–7. doi: 10.1038/nrrheum.2018.26
169. Renauer P, Coit P, Sawalha AH. Epigenetics and vasculitis: a comprehensive review. Clin Rev Allergy Immunol. (2016) 50:357–66. doi: 10.1007/s12016-015-8495-6
170. Jennette JC. Overview of the 2012 revised International chapel hill consensus conference nomenclature of vasculitides. Clin Exp Nephrol. (2013) 17:603–6. doi: 10.1007/s10157-013-0869-6
171. Nakazawa D, Masuda S, Tomaru U, Ishizu A. Pathogenesis and therapeutic interventions for ANCA-associated vasculitis. Nat Rev Rheumatol. (2019) 15:91–101. doi: 10.1038/s41584-018-0145-y
172. Marzano AV, Raimondo MG, Berti E, Meroni PL, Ingegnoli F. Cutaneous manifestations of ANCA-associated small vessels vasculitis. Clin Rev Allergy Immunol. (2017) 53:428–38. doi: 10.1007/s12016-017-8616-5
173. Jennette JC, Falk RJ. Pathogenesis of antineutrophil cytoplasmic autoantibody-mediated disease. Nat Rev Rheumatol. (2014) 10:463. doi: 10.1038/nrrheum.2014.103
174. Meijer-Jorna LB, Mekkes JR, Van Der Wal AC. Platelet involvement in cutaneous small vessel vasculitis. J Cutan Pathol. (2002) 29:176–80. doi: 10.1034/j.1600-0560.2002.290309.x
175. Miao D, Ma TT, Chen M, Zhao MH. Platelets release proinflammatory microparticles in anti-neutrophil cytoplasmic antibody-associated vasculitis. Rheumatology. (2019). doi: 10.1093/rheumatology/kez044. [Epub ahead of print].
176. Bavendiek U, Libby P, Kilbride M, Reynolds R, Mackman N, Schonbeck U. Induction of tissue factor expression in human endothelial cells by CD40 ligand is mediated via activator protein 1, nuclear factor kappa B, and Egr-1. J Biol Chem. (2002) 277:25032–9. doi: 10.1074/jbc.M204003200
177. Urbich C, Dernbach E, Aicher A, Zeiher AM, Dimmeler S. CD40 ligand inhibits endothelial cell migration by increasing production of endothelial reactive oxygen species. Circulation. (2002) 106:981–6. doi: 10.1161/01.CIR.0000027107.54614.1A
178. Rizvi M, Pathak D, Freedman JE, Chakrabarti S. CD40-CD40 ligand interactions in oxidative stress, inflammation and vascular disease. Trends Mol Med. (2008) 14:530–8. doi: 10.1016/j.molmed.2008.09.006
179. Vanichakarn P, Blair P, Wu C, Freedman JE, Chakrabarti S. Neutrophil CD40 enhances platelet-mediated inflammation. Thromb Res. (2008) 122:346–58. doi: 10.1016/j.thromres.2007.12.019
180. Li G, Sanders JM, Bevard MH, Sun Z, Chumley JW, Galkina EV, et al. CD40 ligand promotes Mac-1 expression, leukocyte recruitment, and neointima formation after vascular injury. Am J Pathol. (2008) 172:1141–52. doi: 10.2353/ajpath.2008.070633
181. Papayannopoulos V. Neutrophil extracellular traps in immunity and disease. Nat Rev Immunol. (2017) 18:134. doi: 10.1038/nri.2017.105
182. Andrews RK, Arthur JF, Gardiner EE. Neutrophil extracellular traps (NETs) and the role of platelets in infection. Thromb Haemost. (2014) 112:659–65. doi: 10.1160/TH14-05-0455
183. Zawrotniak M, Rapala-Kozik M. Neutrophil extracellular traps (NETs) - formation and implications. Acta Biochim Pol. (2013) 60:277–84. doi: 10.18388/abp.2013_1983
184. Chen M, Jayne DRW, Zhao M-H. Complement in ANCA-associated vasculitis: mechanisms and implications for management. Nat Rev Nephrol. (2017) 13:359. doi: 10.1038/nrneph.2017.37
185. Miao D, Li DY, Chen M, Zhao MH. Platelets are activated in ANCA-associated vasculitis via thrombin-PARs pathway and can activate the alternative complement pathway. Arthritis Res Ther. (2017) 19:252. doi: 10.1186/s13075-017-1458-y
186. Albrecht EA, Chinnaiyan AM, Varambally S, Kumar-Sinha C, Barrette TR, Sarma JV, et al. C5a-induced gene expression in human umbilical vein endothelial cells. Am J Pathol. (2004) 164:849–59. doi: 10.1016/S0002-9440(10)63173-2
187. Huang YM, Wang H, Wang C, Chen M, Zhao MH. Promotion of hypercoagulability in antineutrophil cytoplasmic antibody-associated vasculitis by C5a-induced tissue factor-expressing microparticles and neutrophil extracellular traps. Arthritis Rheumatol. (2015) 67:2780–90. doi: 10.1002/art.39239
188. Isiksacan N, Koser M, Cemsitoglu F, Kucuksezer UC, Gurdol F. Platelet and other hemostatic characteristics in patients with chronic urticaria. Angiology. (2015) 66:387–91. doi: 10.1177/0003319714552693
189. Magen E, Mishal J, Zeldin Y, Feldman V, Kidon M, Schlesinger M, et al. Increased mean platelet volume and C-reactive protein levels in patients with chronic urticaria with a positive autologous serum skin test. Am J Med Sci. (2010) 339:504–8. doi: 10.1097/MAJ.0b013e3181db6ed5
190. Vena GA, Cassano N, Marzano AV, Asero R. The role of platelets in chronic urticaria. Int Arch Allergy Immunol. (2016) 169:71–9. doi: 10.1159/000444085
191. Beyan C, Kaptan K, Ifran A. Platelet count, mean platelet volume, platelet distribution width, and plateletcrit do not correlate with optical platelet aggregation responses in healthy volunteers. J Thromb Thrombolysis. (2006) 22:161–4. doi: 10.1007/s11239-006-9014-7
192. Zhang M, Park G, Zhou B, Luo D. Applications and efficacy of platelet-rich plasma in dermatology: a clinical review. J Cosmet Dermatol. (2018) 17:660–5. doi: 10.1111/jocd.12673
193. Marx RE. Platelet-rich plasma: evidence to support its use. J Oral Maxillofac Surg. (2004) 62:489–96. doi: 10.1016/j.joms.2003.12.003
194. Ibrahim ZA, El-Ashmawy AA, El-Tatawy RA, Sallam FA. The effect of platelet-rich plasma on the outcome of short-term narrowband-ultraviolet B phototherapy in the treatment of vitiligo: a pilot study. J Cosmet Dermatol. (2016) 15:108–16. doi: 10.1111/jocd.12194
195. Abdelghani R, Ahmed NA, Darwish HM. Combined treatment with fractional carbon dioxide laser, autologous platelet-rich plasma, and narrow band ultraviolet B for vitiligo in different body sites: a prospective, randomized comparative trial. J Cosmet Dermatol. (2018) 17:365–72. doi: 10.1111/jocd.12397
196. Kadry M, Tawfik A, Abdallah N, Badawi A, Shokeir H. Platelet-rich plasma versus combined fractional carbon dioxide laser with platelet-rich plasma in the treatment of vitiligo: a comparative study. Clin Cosmet Investig Dermatol. (2018) 11:551–9. doi: 10.2147/CCID.S178817
197. Bagci IS, Horvath ON, Ruzicka T, Sardy M. Bullous pemphigoid. Autoimmun Rev. (2017) 16:445–55. doi: 10.1016/j.autrev.2017.03.010
198. Rifaioglu EN, Sen BB, Ekiz O, Dogramaci AC. Mean platelet volume and eosinophilia relationship in patients with bullous pemphigoid. Platelets. (2014) 25:264–7. doi: 10.3109/09537104.2013.784735
199. Bieber K, Ernst AL, Tukaj S, Holtsche MM, Schmidt E, Zillikens D, et al. Analysis of serum markers of cellular immune activation in patients with bullous pemphigoid. Exp Dermatol. (2017) 26:1248–52. doi: 10.1111/exd.13382
200. Tamagawa-Mineoka R. Important roles of platelets as immune cells in the skin. J Dermatol Sci. (2015) 77:93–101. doi: 10.1016/j.jdermsci.2014.10.003
201. Berrettini M, Parise P, Constantini V, Grasselli S, Nenci GG. Platelet activation in psoriasis. Thromb Haemost. (1985) 53:195–7. doi: 10.1055/s-0038-1661271
202. Tamagawa-Mineoka R, Katoh N, Ueda E, Masuda K, Kishimoto S. Elevated platelet activation in patients with atopic dermatitis and psoriasis: increased plasma levels of beta-thromboglobulin and platelet factor 4. Allergol Int. (2008) 57:391–6. doi: 10.2332/allergolint.O-08-537
203. Tamagawa-Mineoka R, Katoh N, Kishimoto S. Platelet activation in patients with psoriasis: increased plasma levels of platelet-derived microparticles and soluble P-selectin. J Am Acad Dermatol. (2010) 62:621–6. doi: 10.1016/j.jaad.2009.06.053
204. Unal M. Platelet mass index is increased in psoriasis. A possible link between psoriasis and atherosclerosis. Arch Med Sci Atheroscler Dis. (2016) 1:e145–9. doi: 10.5114/amsad.2016.64444
205. Ong PY, Ohtake T, Brandt C, Strickland I, Boguniewicz M, Ganz T, et al. Endogenous antimicrobial peptides and skin infections in atopic dermatitis. N Engl J Med. (2002) 347:1151–60. doi: 10.1056/NEJMoa021481
206. Salamah MF, Ravishankar D, Kodji X, Moraes LA, Williams HF, Vallance TM, et al. The endogenous antimicrobial cathelicidin LL37 induces platelet activation and augments thrombus formation. Blood Adv. (2018) 2:2973–85. doi: 10.1182/bloodadvances.2018021758
207. Ludwig RJ, Schultz JE, Boehncke WH, Podda M, Tandi C, Krombach F, et al. Activated, not resting, platelets increase leukocyte rolling in murine skin utilizing a distinct set of adhesion molecules. J Invest Dermatol. (2004) 122:830–6. doi: 10.1111/j.0022-202X.2004.22318.x
208. Teague HL, Varghese NJ, Tsoi LC, Dey AK, Garshick MS, Silverman JI, et al. Neutrophil subsets, platelets, and vascular disease in psoriasis. JACC Basic Transl Sci. (2019) 4:1–14. doi: 10.1016/j.jacbts.2018.10.008
209. Soyfoo MS, Roth J, Vogl T, Pochet R, Decaux G. Phagocyte-specific S100A8/A9 protein levels during disease exacerbations and infections in systemic lupus erythematosus. J Rheumatol. (2009) 36:2190–4. doi: 10.3899/jrheum.081302
210. Patsouras MD, Sikara MP, Grika EP, Moutsopoulos HM, Tzioufas AG, Vlachoyiannopoulos PG. Elevated expression of platelet-derived chemokines in patients with antiphospholipid syndrome. J Autoimmun. (2015) 65:30–7. doi: 10.1016/j.jaut.2015.08.001
211. Kao AH, Mcburney CA, Sattar A, Lertratanakul A, Wilson NL, Rutman S, et al. Relation of platelet C4d with all-cause mortality and ischemic stroke in patients with systemic lupus erythematosus. Transl Stroke Res. (2014) 5:510–8. doi: 10.1007/s12975-013-0295-9
212. Komura K, Sato S, Hasegawa M, Fujimoto M, Takehara K. Elevated circulating CD40L concentrations in patients with systemic sclerosis. J Rheumatol. (2004) 31:514–19.
213. Iversen LV, Ostergaard O, Ullman S, Nielsen CT, Halberg P, Karlsmark T, et al. Circulating microparticles and plasma levels of soluble E- and P-selectins in patients with systemic sclerosis. Scand J Rheumatol. (2013) 42:473–82. doi: 10.3109/03009742.2013.796403
214. Yoshizaki A, Komura K, Iwata Y, Ogawa F, Hara T, Muroi E, et al. Clinical significance of serum HMGB-1 and sRAGE levels in systemic sclerosis: association with disease severity. J Clin Immunol. (2009) 29:180–9. doi: 10.1007/s10875-008-9252-x
215. Tomasson G, Lavalley M, Tanriverdi K, Finkielman JD, Davis JC Jr, Hoffman GS, et al. Relationship between markers of platelet activation and inflammation with disease activity in Wegener's granulomatosis. J Rheumatol. (2011) 38:1048–54. doi: 10.3899/jrheum.100735
216. Palikhe S, Palikhe NS, Kim SH, Yoo HS, Shin YS, Park HS. Elevated platelet activation in patients with chronic urticaria: a comparison between aspirin-intolerant and aspirin-tolerant groups. Ann Allergy Asthma Immunol. (2014) 113:276–81. doi: 10.1016/j.anai.2014.06.011
217. Chandrashekar L, Rajappa M, Sundar I, Munisamy M, Ananthanarayanan PH, Thappa DM, et al. Platelet activation in chronic urticaria and its correlation with disease severity. Platelets. (2014) 25:162–5. doi: 10.3109/09537104.2013.786822
218. Papadavid E, Diamanti K, Spathis A, Varoudi M, Andreadou I, Gravanis K, et al. Increased levels of circulating platelet-derived microparticles in psoriasis: possible implications for the associated cardiovascular risk. World J Cardiol. (2016) 8:667–75. doi: 10.4330/wjc.v8.i11.667
219. Raghavan V, Radha RKN, Rao RK, Kuberan A. A correlative study between platelet count, mean platelet volume and red cell distribution width with the disease severity index in psoriasis patients. J Clin Diagn Res. (2017) 11:EC13–6. doi: 10.7860/JCDR/2017/31172.10639
220. Ponticelli C, Moroni G. Hydroxychloroquine in systemic lupus erythematosus (SLE). Expert Opin Drug Saf. (2017) 16:411–9. doi: 10.1080/14740338.2017.1269168
221. Johansson E, Forsberg K, Johnsson H. Clinical and experimental evaluation of the thromboprophylactic effect of hydroxychloroquine sulfate after total hip replacement. Haemostasis. (1981) 10:89–96. doi: 10.1159/000214391
222. Prowse C, Pepper D, Dawes J. Prevention of the platelet alpha-granule release reaction by membrane-active drugs. Thromb Res. (1982) 25:219–27. doi: 10.1016/0049-3848(82)90241-9
223. Bruni C, Praino E, Guiducci S, Bellando-Randone S, Furst DE, Matucci-Cerinic M. Hydroxychloroquine and joint involvement in systemic sclerosis: Preliminary beneficial results from a retrospective case-control series of an EUSTAR center. Joint Bone Spine. (2017) 84:747–8. doi: 10.1016/j.jbspin.2016.10.002
224. Wang L, Erling P, Bengtsson AA, Truedsson L, Sturfelt G, Erlinge D. Transcriptional down-regulation of the platelet ADP receptor P2Y(12) and clusterin in patients with systemic lupus erythematosus. J Thromb Haemost. (2004) 2:1436–42. doi: 10.1111/j.1538-7836.2004.00854.x
225. Thomas MR, Storey RF. Effect of P2Y12 inhibitors on inflammation and immunity. Thromb Haemost. (2015) 114:490–7. doi: 10.1160/TH14-12-1068
226. Ntelis K, Gkizas V, Filippopoulou A, Davlouros P, Alexopoulos D, Andonopoulos AP, et al. Clopidogrel treatment may associate with worsening of endothelial function and development of new digital ulcers in patients with systemic sclerosis: results from an open label, proof of concept study. BMC Musculoskelet Disord. (2016) 17:213. doi: 10.1186/s12891-016-1072-1
227. Iwamoto N, Distler O. Molecular targets for therapy in systemic sclerosis. Fibrogenesis Tissue Repair. (2012) 5:S19. doi: 10.1186/1755-1536-5-S1-S19
228. Chamberlain C, Colman PJ, Ranger AM, Burkly LC, Johnston GI, Otoul C, et al. Repeated administration of dapirolizumab pegol in a randomised phase I study is well tolerated and accompanied by improvements in several composite measures of systemic lupus erythematosus disease activity and changes in whole blood transcriptomic profiles. Ann Rheum Dis. (2017) 76:1837–44. doi: 10.1136/annrheumdis-2017-211388
229. Cipollone F, Mezzetti A, Porreca E, Di Febbo C, Nutini M, Fazia M, et al. Association between enhanced soluble CD40L and prothrombotic state in hypercholesterolemia: effects of statin therapy. Circulation. (2002) 106:399–402. doi: 10.1161/01.CIR.0000025419.95769.F0
230. Sanguigni V, Pignatelli P, Lenti L, Ferro D, Bellia A, Carnevale R, et al. Short-term treatment with atorvastatin reduces platelet CD40 ligand and thrombin generation in hypercholesterolemic patients. Circulation. (2005) 111:412–9. doi: 10.1161/01.CIR.0000153810.81187.7D
231. Watanabe T, Oku K, Amengual O, Hisada R, Ohmura K, Nakagawa I, et al. Effects of statins on thrombosis development in patients with systemic lupus erythematosus and antiphospholipid antibodies. Lupus. (2018) 27:225–34. doi: 10.1177/0961203317716787
232. Kling D, Stucki C, Kronenberg S, Tuerck D, Rheaume E, Tardif JC, et al. Pharmacological control of platelet-leukocyte interactions by the human anti-P-selectin antibody inclacumab–preclinical and clinical studies. Thromb Res. (2013) 131:401–10. doi: 10.1016/j.thromres.2013.02.020
233. De Holanda MI, Porto LC, Wagner T, Christiani LF, Palma LMP. Use of eculizumab in a systemic lupus erythemathosus patient presenting thrombotic microangiopathy and heterozygous deletion in CFHR1-CFHR3. A case report and systematic review. Clin Rheumatol. (2017) 36:2859–67. doi: 10.1007/s10067-017-3823-2
234. Hsu J, Gu Y, Tan SL, Narula S, Demartino JA, Liao C. Bruton's Tyrosine Kinase mediates platelet receptor-induced generation of microparticles: a potential mechanism for amplification of inflammatory responses in rheumatoid arthritis synovial joints. Immunol Lett. (2013) 150:97–104. doi: 10.1016/j.imlet.2012.12.007
235. Xu Z, Xu Y-J, Hao Y-N, Ren L-J, Zhang Z-B, Xu X, et al. A novel STAT3 inhibitor negatively modulates platelet activation and aggregation. Acta Pharmacol Sin. (2017) 38:651. doi: 10.1038/aps.2016.155
236. Vaitaitis GM, Olmstead MH, Waid DM, Carter JR, Wagner DH Jr. A CD40-targeted peptide controls and reverses type 1 diabetes in NOD mice. Diabetologia. (2014) 57:2366–73. doi: 10.1007/s00125-014-3342-5
Keywords: platelets, autoimmune disease, SLE, SSc, vasculitis, neutrophil, endothelial cell, complement
Citation: Liu X, Gorzelanny C and Schneider SW (2019) Platelets in Skin Autoimmune Diseases. Front. Immunol. 10:1453. doi: 10.3389/fimmu.2019.01453
Received: 31 March 2019; Accepted: 10 June 2019;
Published: 04 July 2019.
Edited by:
Ralf J. Ludwig, Universität zu Lübeck, GermanyReviewed by:
Angelo A. Manfredi, Vita-Salute San Raffaele University, ItalyNorma Maugeri, Vita-Salute San Raffaele University, Italy
Unni Samavedam, University of Cincinnati, United States
Copyright © 2019 Liu, Gorzelanny and Schneider. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Stefan W. Schneider, c3Quc2NobmVpZGVyQHVrZS5kZQ==