 Liguo Zhu
Liguo Zhu Jing Han
Jing Han Li Li
Li Li Ying Wang
Ying Wang Ying Li
Ying Li Shenghong Zhang
Shenghong Zhang
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol. , 27 June 2019
Sec. Inflammation
Volume 10 - 2019 | https://doi.org/10.3389/fimmu.2019.01441
Claudins are a multigene transmembrane protein family comprising at least 27 members. In gastrointestinal tract, claudins are mainly located in the intestinal epithelia; many types of claudins form a network of strands in tight junction plaques within the intercellular space of neighboring epithelial cells and build paracellular selective channels, while others act as signaling proteins and mediates cell behaviors. Claudin dysfunction may contribute to epithelial permeation disorder and multiple intestinal diseases. Over recent years, the importance of claudins in the pathogenesis of inflammatory bowel diseases (IBD) has gained focus and is being investigated. This review analyzes the expression pattern and regulatory mechanism of claudins based on existing evidence and elucidates the fact that claudin dysregulation correlates with increased intestinal permeability, sustained activation of inflammation, epithelial-to-mesenchymal transition (EMT), and tumor progression in IBD as well as consequent colitis-associated colorectal cancer (CAC), possibly shedding new light on further etiologic research and clinical treatments.
Claudins, a multigene transmembrane protein family comprising at least 27 members (1), reportedly contain four transmembrane (TM) helix domains, two extracellular loops (ECLs), a short N-terminus and a C-terminus (2). In gastrointestinal tract, claudins are mainly located in the intestinal epithelia; many species of claudins form a network of strands in tight junction (TJ) plaques within the intercellular space of neighboring epithelial cells and build paracellular selective channels (3–5), while the others act as signaling proteins and modulate cell behaviors (6). Consequently, claudins dysfunction that occurs in enterocytes may contribute to epithelial permeation disorder and multiple intestinal diseases, including inflammatory bowel diseases (IBD) (7, 8). IBD are a series of chronic multifactorial gastrointestinal inflammatory disorders that mainly include Crohn's disease (CD) and Ulcerative Colitis (UC), with rather high non-response rates and recurrence rates in clinical practice. Moreover, evidence regarding the correlation of IBD with colorectal cancers exists, as sustained extensive colitis is a known independent risk factor for colitis-associated colorectal cancer (CAC) (9, 10). However, owing to sophisticated etiology, the dynamics of IBD and consequent tumorigenesis remain obscure. As the roles of claudins have been gradually better understood in recent years, dysregulation of different claudin types may modulate barrier permeability as TJ proteins and impact tumor behaviors as signaling proteins, therefore participating in the pathogenesis of IBD and consequent tumorigenesis (11).
IBD are a series of chronic gastrointestinal inflammatory disorders, among which CD and UC are the most prevalent and well-studied (12). The sophisticated etiology of IBD is described in the following sub-sections, along with the relationship of IBD to claudins (Figure 1).
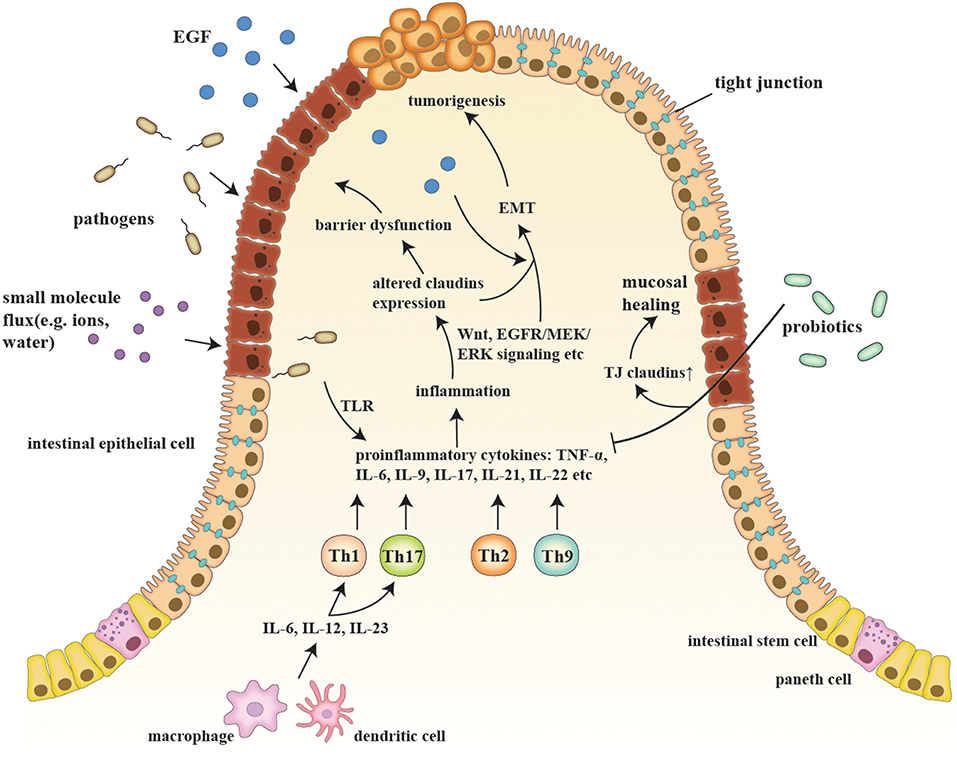
Figure 1. A schematic model of claudins in the pathogenesis of IBD and consequent tumorigenesis. Etiologic factors, such as a pathogenic breach and dysbiosis, engender an overactive inflammatory response by secreting proinflammatory cytokines (e.g., IL-6, IL-9, and IL-23) and generating proinflammatory T cells (e.g., Th1, Th2, Th9, and Th17), which also changes claudin expression via multiple pathways, such as Wnt and MEK/ERK signaling. Claudin dysregulation directly leads to impaired barrier function and luminal bacterial leakage inversely exacerbates inflammation, while EGF influx contributes to tumorigenesis. EMT and tumorigenesis are results of sustained activation of signaling pathways that modulate cell growth and migration, in which claudins act as signaling proteins and modulate aggressive tumor behaviors. Probiotics, on the other hand, recover epithelial claudin expression, and therefore promote mucosal healing.
Over 200 genetic risk loci are associated with IBD in modulating barrier homeostasis, intestinal epithelial renewal, defense against microbes, and innate/adaptive immune regulation, among other things (13, 14). In a study on twins discordant for CD, healthy co-twins exhibited barrier function changes similar to their CD co-twins (e.g., increased small molecule permeability and reduced claudin-5 expression) when compared with non-related controls, indicating that genetic predisposition may be an important etiologic factor in IBD onset (15). Some gene variants may increase IBD predisposition by modulating TJ proteins because they take on the dual role of sealing proteins and signaling proteins. NOD2 mutations engender anti-biotic α-defensin reduction and altered composition of TJ proteins (e.g., claudin-1, claudin-2, occluding, and Zonula Occludens-1), resulting in higher susceptibility to luminal bacterial infection in CD patients (16) and exacerbation of inflammation via interleukin-8 (IL-8)/nuclear factor-κB (NF-κB) activation in epithelia as well as Toll-like receptor-2(TLR2)-dependent interferon-γ (IFN-γ) upregulation in antigen-presenting cells (17, 18). Similarly, SHANK3 mutations downregulate ZO-1 by inhibiting protein kinase Cε (PKCε), thus engendering bacterial paracellular influx and increasing propensity to inflammation in CD patients (19), which is also likely a mechanism of claudins dysregulation in IBD onset that awaits further analysis.
At their core, IBD are a series of autoimmune diseases, and aberrant immune responses may contribute to IBD in sophisticated ways by involving both innate and adaptive immune mechanisms (20). The function of T cells and relevant cytokines is well-studied and considered important in the pathogenesis of IBD. Under physiological conditions, Treg cells and macrophages secrete transforming growth factor-β (TGF-β) and IL-10 to induce immunotolerance (21). However, under pathological conditions such as infections, the upregulated proinflammatory cytokines (e.g., IL-1, IL-6, IL-12, and IL-23) and generation of Th1, Th2, Th9, and Th17 cells, along with the activation of other immune cells (e.g., neutrophils, NK cells), cooperatively construct an elaborate network triggering IBD (21). For example, Th9 subset exacerbates murine experimental colitis, increases bacteria permeability and impairs wound healing by secreting IL-9 and upregulating pore-forming claudin-2 (22). From a viewpoint of clinical applications, except for anti-tumor necrosis factor (TNF)-α agents, the efficacy and safety of other inhibitory agents against the participating immune cells and cytokines, such as anti-integrin and anti-IL-23 agents, still need to be examined through clinical trials (23, 24).
TJ-dependent paracellular passages manage the exchange of paracellular substances between the intestinal lumen and internal environment, thereby playing a role in the balancing of nutrient absorption and waste secretion as well as defense mechanisms against pathogens. In accordance with the well-acknowledged roles of claudins in forming TJs and selective channels, claudins may participate in both types of transepithelial paracellular leakage (25): proinflammatory cytokines-induced small molecule (e.g., ions and mannitol) channel disruption and cell detachment-induced large molecule (e.g., epidermal growth factor, EGF) leakage (26). Thus, as barrier-forming proteins, dysregulated expression and redistribution of claudins may lead to increased intestinal permeability, susceptibility to gut infection and bowel symptoms of IBD patients (27–29).
Dysbiosis of microbiota may influence mucosal homeostasis, immune response, nutrient uptake, and vitamin production with altered metagenome and perturbed microbial metabolism, finally contributing to IBD (30, 31). For example, adherent-invasive Escherichia coli, a prevalent pathogen in chronic and early recurrent ileal lesions in CD patients (32), interferes with host immune responses by surviving macrophage phagocytosis, inducing neutrophil autophagy, promoting Th17 differentiation, and upregulating proinflammatory cytokines TNF-α and IL-6 (33–35). Probiotics, such as Bacillus subtilis and Bifidobacterium longum, on the other hand, alleviate inflammation and repair barrier function by downregulating proinflammatory cytokines (e.g., IL-17, IL-23, and TNF-α) and upregulating TJ proteins (e.g., claudin-1, occluding, and ZO-1) in colitis mice models (36–38).
Based on existing epidemiologic research, industrialization is regarded as a significant risk factor for IBD because its incidence rate in western countries has increased rapidly since the 1950s and has remained high (39, 40), while the low incidence rate in newly industrialized countries has steadily begun to rise in the twenty-first century (41). Age may also be a risk factor for CD and UC, because the first diagnostic peak appears at 20–30 years in CD and at 30–40 years in UC, along with a possible second peak at 60–70 years (42). Other risk factors, including smoking, appendectomy, dietary habits, antibiotic use, and childhood exposure, may also be relevant to the pathogenesis of IBD (43, 44). Interestingly, second-hand smoking engenders claudin-3 and ZO-2 upregulation in murine large intestines possibly by inhibiting NF-κB signaling and AMP-activated protein kinase (AMPK), but also increases oxidative stress by activating c-Jun N-terminal kinase (JNK) and p38 mitogen-activated protein kinase (MAPK) signaling, indicating the complicated role of smoking in modulating inflammation (45).
The sophisticated pathogenesis of CAC is associated with genomic alterations, inflammation-induced aberrant immune response, and alterations in bowel microbiota, etc. (46). Sequencing analysis has revealed that genomic alterations in CAC are significantly different from sporadic colorectal cancer with regard to a higher frequency of TP53, MYC, and IDH1 mutations, along with lower frequency of APC mutations (47). Moreover, as colitis-associated intestinal barrier leak allows for the paracellular influx of luminal growth factors, EGF triggers sustained activation of Ras/Raf/mitogen-activated ERK kinase (MEK)/extracellular signal-regulated kinase (ERK) signaling, phosphatidylinositol-3-kinase (PI3K)/ protein kinase B (Akt) signaling as well as signal transducer and activator of transcription-3 (STAT3) signaling, along with secretion of proinflammatory cytokines (e.g., IL-17 and IL-23), thus accelerating cell proliferation and engendering tumorigenesis (48–51). Additionally, microbial composition is altered in patients with CAC or sporadic colorectal cancer and varies at different stages of colorectal tumorigenesis (52, 53), possibly owing to infection-associated inflammation, bacterial metabolites, and infection-induced oxidative stress (54). The changes of claudins in CAC and colorectal cancer are summed up in Table 1 (55–76).
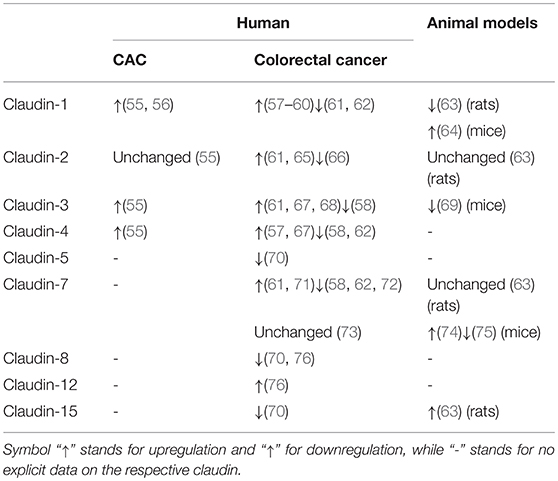
Table 1. Changes of claudins in colitis-associated colorectal cancer (CAC) and colorectal cancer.
Though claudins are widely expressed in various kinds of organs and tissues (77), only a few of them are detectable in the gastrointestinal tract. Multiple studies have confirmed the tissue-specific physiological heterogeneity in normal gastrointestinal epithelium and pathological modification of claudin expression in IBD patients (Table 2) (78–80). Hence, deviation from the physiological expression pattern of claudins probably implies a pathological state. The expression and distribution of claudins may be regarded as an indicator or mediator of mucosal function in concerning diseases that cause intestinal mucosal damage, including IBD.
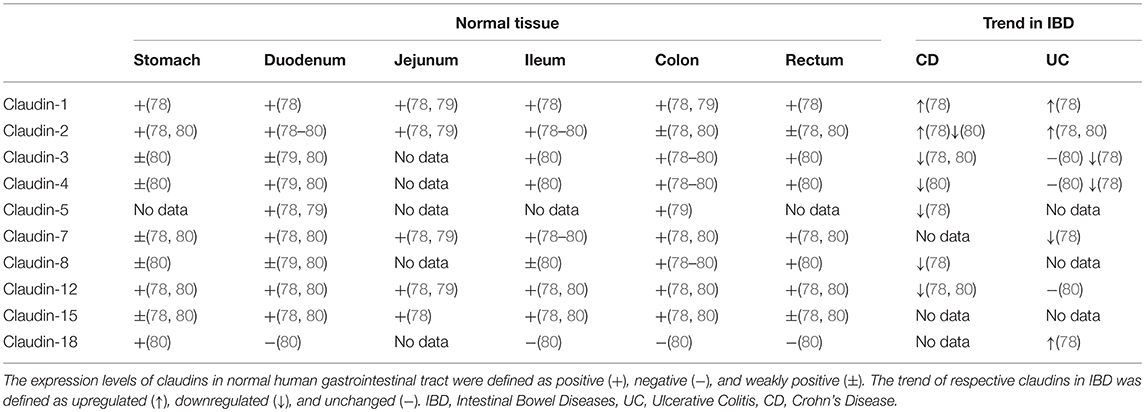
Table 2. Expression patterns of claudins in normal human tissue in comparison with altered expression in IBD.
Different types of claudins have basic structures in common (Figure 2), including four transmembrane helices, two extracellular loops, a short N-terminus and a cytoplasmic C-terminus(2). ECLs and TM mediate the formation of claudin dimers and dimers further assemble themselves into barrier or pores in the paracellular space (81, 82). ECLs are also able to bind C. perfringens enterotoxin and therefore to participate in infection-induced pathogenesis (82). On the other hand, the diverse C-terminus of claudins binds cytoplasmic proteins by PSD-95/Disc-large/ZO-1 (PDZ) domain, which may be the structural foundation for claudins to modulate cell behaviors as signaling proteins (83).
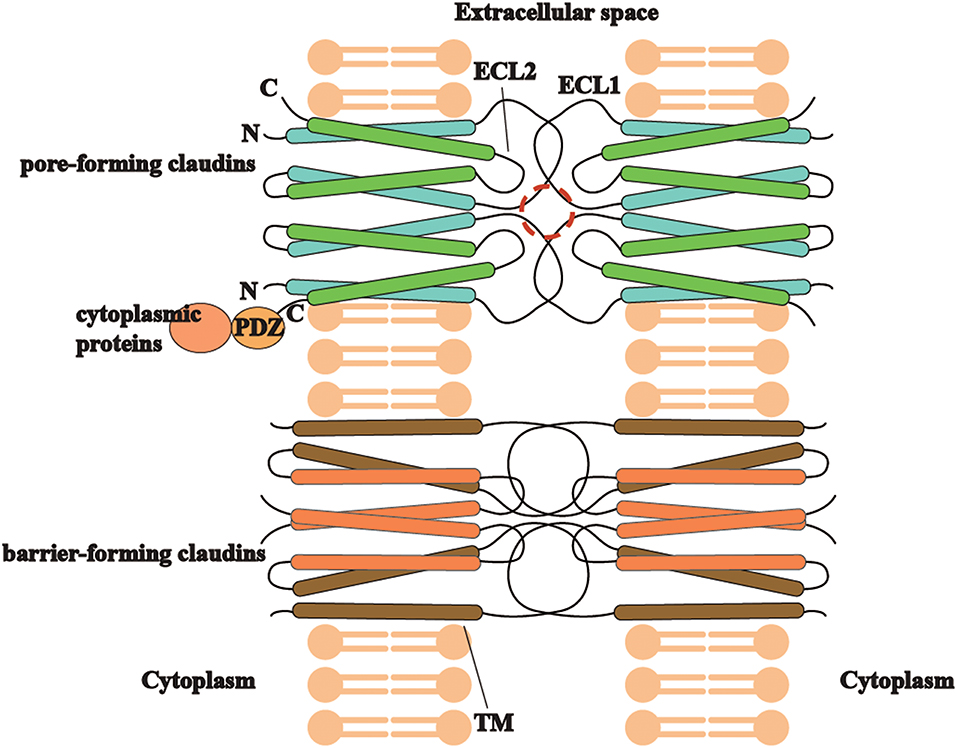
Figure 2. The trait of claudins (pore-forming or barrier-forming) depends on pore-like structures (marked in red circle) and polar amino acid residues of ECLs and occasionally TM segments in claudin dimers interaction. C-terminus of different claudins is diverse and able to bind cytoplasmic proteins by PDZ domain, which may be the structural foundation for claudins to modulate cell behaviors as signaling proteins.
Though it is well-acknowledged that gastrointestinal-specific claudins participate in building intestinal barriers and modulating permeability (79), there is also ample evidence that claudins may act as signaling proteins and participate in inflammation, cell proliferation, differentiation, and tumorigenesis via cellular signaling pathways, including EGFR/MEK/ERK signaling, PI3K/Akt signaling, Wnt/β-catenin signaling and AMPK signaling, etc. (61). Thus, inflammation-induced claudins dysregulation may mediate the interaction between IBD and CAC.
Claudin-1 is located in the junctional areas as well as lateral membranes of crypt epithelial cells and upregulated in entire epithelia during CD and UC (84, 85). Evidence shows that downregulation of claudin-1 contributes to increased intestinal permeability via NF-κB activation and leads to diarrhea in patients with irritable bowel syndrome (86). On the other hand, claudin-1-overexpressing mice exhibit higher susceptibility to experimental colitis, with impaired goblet cell differentiation, deferred epithelial recovery, sustained inflammation and crypt hyperplasia, possibly resulting from matrix metalloproteinases-9(MMP-9)/ERK-induced Notch signaling activation (85). Wnt/β-catenin signaling that participates in inflammation, cell differentiation and proliferation, seems to have a strong correlation with claudin-1, as claudin-1-overexpressing mice exhibit Wnt/β-catenin signaling activation, and caudal homeobox protein-2(Cdx-2)-associated Wnt signaling upregulates claudin-1 inversely in colorectal cancer cell lines (60, 64), along with combined upregulation of claudin-1 and β-catenin in human primary and metastatic colorectal cancer lesions (87). In addition, TNF-α-induced claudin-1 upregulation leads to ERK and SRC signaling activation, thus contributing to EMT and increased tumor invasion, suggesting the underlying role of claudin-1 in triggering colitis and CAC (88).
As claudin-2 is predominantly expressed in TJ region and the apical cytoplasm of surface colonocytes (84), it is considered to increase paracellular permeability by forming cation-selective and water channels on intestinal epithelia (89, 90), along with an overall upregulation in IBD (84, 91) and CAC (92). Contrary to claudin-1, claudin-2-overexpressing mice exhibit higher resistance to experimental colitis by showing decreased cell apoptosis, increased epithelial proliferation and immune tolerance, brought about by downregulation of IL-6-induced NF-κB as well as STAT3 signaling and upregulation of Treg cell population as well as immunoregulatory cytokine TGF-β (93). However, different from the protective role in colitis, claudin-2 is upregulated by Cdx-associated Wnt signaling activation (94) and contributes to tumorigenicity of CAC by promoting cell proliferation via EGFR/ERK signaling in vitro (92).
Claudin-3 is typically located in junctional areas and lateral membranes of healthy colonic surface and crypt epithelia, while its expression in apical regions is lowered in the epithelia of IBD patients (78, 95). A research conducted on murine postnatal intestinal barrier development revealed that intestinal claudin-3 expression peaks during early stages of life and complies to the process of probiotics-induced MyD88-dependent gut maturation, indicating that claudin-3 forms intestinal epithelial barrier on the basis of probiotics-induced TLR activation in early life (96). Evidence from in vitro experiments also demonstrated that claudin-3 is downregulated by proinflammatory cytokines (e.g., IFN-γ, TNF-α, and IL-1β)-induced myosin light chain kinase (MLCK) activation (97, 98).In claudin-3-deficient mice model, colonic epithelial dedifferentiation, barrier dysfunction and rapid tumor progression likely resulted from IL-6/gp130/STAT3 signaling-induced Wnt/β-catenin signaling upregulation, indicating the interaction between inflammation and tumorigenesis mediated by claudin-3 deficiency (69). However, another study on colorectal adenocarcinoma cell line revealed that claudin-3 overexpression promotes tumor malignancy via EGFR/MEK/ERK and PI3K/Akt signaling (68). The aforementioned dual role of claudin-3 in tumorigenesis indicates that the imbalance of claudin-3 may participate in tumorigenesis via multiple pathways.
Claudin-4 is expressed in TJs and lateral membranes of normal colonic surface as well as crypt epithelia, and responsible for physiological chloride reabsorption (95, 99). Downregulation of claudin-4 are mainly observed in junctional areas of inflamed epithelia, probably due to TNF-α/IFN-γ induced MLCK activation (95). AMPK and muscarinic acetylcholine receptors-induced ERK activation is also able to trigger claudin-4 phosphorylation, β-arrestin2/clathrin-mediated redistribution from cellular membranes to cytoplasm, and consequent ubiquitination-induced degradation (100, 101), contributing to increased epithelial paracellular permeability. However, studies on gastrointestinal tumor progression are quite contradictory. Although claudin-4 expression promoted by lncRNA-KRTAP5-AS1 and lncRNA-TUBB2A contributes to aggressive cancer behaviors in gastric cancer (102) and β-catenin expression is synchronous with claudin-4 upregulation in CAC tissues (55), there is also evidence showing that claudin-4 suppression in colorectal cancer tissues correlates with aggressive cancer behaviors (103).
Claudin-5 localizes strictly in junctional areas in normal surface and crypt epithelia, whereas its reduction and redistribution from TJs to sub-junctional membranes are observed in active CD (91). Alteration in the expression of claudin-5 and other sealing claudins leads to decreased number of TJ strands and reduced depth of TJ meshwork, consequently increasing barrier permeability (91). Claudin-5 is upregulated by lamina propria lymphocytes for its function in accelerating intestinal epithelia differentiation via Notch-1 signaling to maintain epithelial homeostasis (104). However, the lymphoepithelial interactions may be a double-edged sword, as PI3K/Akt and MAPK signaling upregulated by lamina propria lymphocytes participate in dysregulated epithelial cell maturation and enhanced intestinal antigen presentation, therefore leading to exacerbation of inflammation (105). On the other hand, as claudin-5 is downregulated by vascular endothelial growth factor receptor-2 (VEGFR2)/PI3K/Akt signaling and IL-8 in endothelial cells, resulting in impaired endothelial integrity and increased vascular permeability (106), the reduction of claudin-5 may play a role in weakened cell adhesion and tumor metastasis in various tumor types, such as ovarian cancer (107), urothelial carcinoma (108), and prostate cancer (109). However, to date, the role of claudin-5 in gastrointestinal tumors remains obscure.
Claudin-7 is found in TJ regions as well as basolateral membranes of colonocytes, whose junctional expression are significantly reduced in active UC (110). Deficiency of claudin-7 mainly causes dysregulated paracellular flux of small organic solutes, such as microflora products, and triggers colonic inflammation in mice models (111). In another study, due to claudin-7 deficiency, upregulation of MMPs (e.g., MMP-3 and MMP-7) turns IL-1β precursor into the active form (112) and triggers NF-κB signaling activation, along with MMPs-induced degradation of claudin-7/claudin-1/integrin-α2 complex, finally engendering colonic inflammation and damaged cell-matrix interaction (113). Further, claudin-7 inhibits ERK and SRC signaling, therefore suppressing EMT and tumor progression in colorectal cancer cell lines (114). However, there is also in vitro evidence that claudin-7 cooperates with epithelial cell adhesion molecule (EpCAM) and generates EpIC, a co-transcription factor that collaborates with β-catenin in cancer initiating cells, contributing to EMT and cancer metastasis (71).
Since claudin-8 acts as sealing claudins in junctional regions of normal epithelial surface as well as crypts(91) and prevents paracellular back-leakage of Na+ in colonic reabsorption(115), its reduction and redistribution from TJs to cytoplasm may lead to TJ structure disruption and increased epithelial permeability in IBD, especially in active CD. In both IBD patients and mice with experimental colitis, claudin-8 is downregulated by hyperactivation of IL-23/miR-223 (116) and IL-9/miR21 (117) pathways, causing inflammation-induced intestinal mucosal damage and retardation of mucosal healing, while IL-9/STAT5 pathway activation may also account for bowel epithelial inflammation (118). On the other hand, as CLDN8 is correlated with cancer progression via Akt and MAPK signaling in prostate cancer (119), the significance of downregulated claudin-8 in colorectal cancer awaits further analysis (76).
The presence of claudin-12 is decreased in the colon epithelia, but increased in the ileum epithelia of CD patients (80). By forming Ca2+ paracellular channels, claudin-12 mediates Vitamin-D dependent Ca2+ permeability in enterocytes (120). Although IL-18/p38 MAPK pathway downregulates claudin-12 and promotes tumor invasion in breast cancer, indicating its possible anti-tumor effect (121), the upregulation of claudin-12 in colorectal cancer needs to be further studied (76).
Claudin-15 modulates small intestinal Na+ permeability by forming Na+ channels (122), whose deficiency results in Na+-dependent glucose/amino acid/fat absorption defect in mice models (123, 124). Interestingly, claudin-15-deficient mice develop dilated and lengthened upper small intestines without signs of epithelial neoplasia, suggesting that claudin-15 may regulate intestinal epithelial cell proliferation and organ size, while the underlying mechanism remains to be discovered (125). Owing to a scarcity of research, the significance of reduced claudin-15 expression in colorectal adenocarcinoma is still undefined (70).
CLDN18 gene produces lung-specific and stomach-specific claudin-18 (126). Stomach-specific claudin-18 (claudin-18.2) forms TJ strands in gastric epithelial cells and limits paracellular H+ efflux; its deficiency may trigger gastritis by upregulating IL-1β, TNF-α, and cyclooxygenase-2 (COX-2)/prostaglandin-E pathway (127). Intriguingly, claudin-18.2-deficient mice develop gastric mucosal neoplasia, which is associated with the activation of multiple pathways, including CD44, ephrin (EFN)/ ephrin receptor (EPH) and yes-associated protein-1 (YAP1)/HIPPO signaling (128). However, the existence of claudin-18.2 in primary and metastatic lesions of gastric cancer, along with its significant ectopic expression in various types of cancers (e.g., pancreatic adenocarcinomas, esophageal tumors, and bile duct cancers), indicates the underlying role of claudin-18.2 as a pan-cancer therapeutic target (129, 130). On the other hand, though lung-specific claudin-18 (claudin-18.1) is expressed in the colonic epithelia of UC patients and experimental colitis mice models, its significance awaits further analysis (131).
Though claudins participate in the pathogenesis of IBD and consequent tumorigenesis, actual prognostic or therapeutic application of claudins in IBD remains scarce. As for the potential role of claudins in colorectal cancer, anti-claudin-1 antibodies specifically bind claudin-1 on the membrane of tumor cells and inhibit tumor progression in vivo and in vitro, indicating that claudin-1 may be a therapeutic target for colorectal cancer, especially for subtypes with KRAS mutations and Wnt signaling activation (132). However, even though claudin-1 is upregulated in human colon cancer tissues when compared with adjacent normal epithelia, claudin-1 reduction is strongly correlated with poor prognosis, including high recurrence rate and poor survival, in tumor-node-metastasis (TNM) stage II colon cancer patients (133). On the other hand, in a phase IIb study on patients with claudin-18.2 positive gastric cancer or gastroesophageal junction adenocarcinoma, the combination of anti-claudin-18.2 antibodies claudiximab and first line chemotherapy improves response rate, progression-free survival, and overall survival in comparison with chemotherapy alone, suggesting the potential of claudin-18.2 as a therapeutic target for claudin-18.2-positive gastric cancer (134) and other cancer types with claudin-18.2 ectopic expression (e.g., pancreatic cancer) (129, 135).
Previous studies have shown that claudins modulate barrier function, inflammation and tumorigenesis in gastrointestinal tract, while their roles in genetic propensity, immune response apart from T cells activation and microbial dysbiosis still need to be further studied. Also, for several gastrointestinal claudins (e.g., claudin−12,−15, and−18), where does the dysregulation exactly occur among various types of gastrointestinal cells and the relevant modulating mechanisms remain obscure. Though claudins dysregulation predominantly occurs in epithelial cells, whether it influences other cell types (e.g., intestinal stem cells and goblet cells) awaits further analysis. In addition, the sophisticated modulating mechanism of claudins makes it difficult to determine which signaling pathway is primary in IBD and CAC, especially for different cancer subtypes. Therefore, conclusions from different signaling studies may be contradictory, which has become an obstacle for claudins to be therapeutic targets for IBD and CAC. The potential of claudins in promoting mucosal healing by restoring TJ structures as TJ-forming proteins and regulating inflammation as well as tumor behaviors as signaling proteins awaits further analysis.
LZ and JH searched for and analyzed referenced articles. LZ and JH wrote the manuscript. LZ, JH, LL, YW, and YL summarized relevant information and designed the tables and figures in the review. SZ designed the study and revised the review. All authors agreed to be responsible for the content of the review.
The study was funded by grants from the National Natural Science Foundation of China (#81870374, #81670498), Pearl River S&T Nova Program of Guangzhou (#201610010126), Guangdong Science and Technology (#2017A030306021), and Science and Technology Innovation Young Talents of Guangdong Special Support Plan (#2016TQ03R296).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
IBD, inflammatory bowel diseases; TJ, tight junction; CD, Crohn's Disease; UC, Ulcerative Colitis; CAC, colitis-associated colorectal cancer; TM, transmembrane; ECL, extracellular loop; EMT, epithelial-to-mesenchymal transition; ZO, Zonula Occludens; IL, interleukin; NF-κB, nuclear factor-κB; TLR, Toll-like receptor; IFN, interferon; PKC, protein kinase C; TGF-β, transforming growth factor-β; TNF, tumor necrosis factor; EGF, epidermal growth factor; AMPK, AMP-activated protein kinase; JNK, c-Jun N-terminal kinase; MAPK, mitogen-activated protein kinase; MEK, mitogen-activated ERK kinase; ERK, extracellular signal-regulated kinase; PI3K, phosphatidylinositol-3-kinase; Akt, protein kinase B; STAT, signal transducer and activator of transcription; PDZ, PSD-95/Disc-large/ZO-1; MMP, matrix metalloproteinases; Cdx, caudal homeobox protein; MLCK, myosin light chain kinase; VEGFR, vascular endothelial growth factor receptor; EpCAM, epithelial cell adhesion molecule; COX, cyclooxygenase; EFN, ephrin; EPH, ephrin receptor; YAP, yes-associated protein; TNM, tumor-node-metastasis.
1. Mineta K, Yamamoto Y, Yamazaki Y, Tanaka H, Tada Y, Saito K, et al. Predicted expansion of the claudin multigene family. FEBS Lett. (2011) 585:606–12. doi: 10.1016/j.febslet.2011.01.028
2. Suzuki H, Nishizawa T, Tani K, Yamazaki Y, Tamura A, Ishitani R, et al. Crystal structure of a claudin provides insight into the architecture of tight junctions. Science. (2014) 344:304–7. doi: 10.1126/science.1248571
3. Schneeberger EE, Lynch RD. The tight junction: a multifunctional complex. Am J Physiol Cell Physiol. (2004) 286:C1213–28. doi: 10.1152/ajpcell.00558.2003
4. Tsukita S, Furuse M. The structure and function of claudins, cell adhesion molecules at tight junctions. Ann N Y Acad Sci. (2000) 915:129–35. doi: 10.1111/j.1749-6632.2000.tb05235.x
5. Ahmad R, Sorrell MF, Batra SK, Dhawan P, Singh AB. Gut permeability and mucosal inflammation: bad, good or context dependent. Mucosal Immunol. (2017) 10:307–17. doi: 10.1038/mi.2016.128
6. Singh AB, Uppada SB, Dhawan P. Claudin proteins, outside-in signaling, and carcinogenesis. Pflugers Arch. (2017) 469:69–75. doi: 10.1007/s00424-016-1919-1
7. Bertiaux-Vandaele N, Youmba SB, Belmonte L, Lecleire S, Antonietti M, Gourcerol G, et al. The expression and the cellular distribution of the tight junction proteins are altered in irritable bowel syndrome patients with differences according to the disease subtype. Am J Gastroenterol. (2011) 106:2165–73. doi: 10.1038/ajg.2011.257
8. Su L, Shen L, Clayburgh DR, Nalle SC, Sullivan EA, Meddings JB, et al. Targeted epithelial tight junction dysfunction causes immune activation and contributes to development of experimental colitis. Gastroenterology. (2009) 136:551–63. doi: 10.1053/j.gastro.2008.10.081
9. Rubin DT, Huo D, Kinnucan JA, Sedrak MS, McCullom NE, Bunnag AP, et al. Inflammation is an independent risk factor for colonic neoplasia in patients with ulcerative colitis: a case-control study. Clin Gastroenterol Hepatol. (2013) 11:1601–8. doi: 10.1016/j.cgh.2013.06.023
10. Beaugerie L, Svrcek M, Seksik P, Bouvier AM, Simon T, Allez M, et al. Risk of colorectal high-grade dysplasia and cancer in a prospective observational cohort of patients with inflammatory bowel disease. Gastroenterology. (2013) 145:166–75. doi: 10.1053/j.gastro.2013.03.044
11. Takehara M, Nishimura T, Mima S, Hoshino T, Mizushima T. Effect of claudin expression on paracellular permeability, migration and invasion of colonic cancer cells. Biol Pharm Bull. (2009) 32:825–31. doi: 10.1248/bpb.32.825
12. Xavier RJ, Podolsky DK. Unravelling the pathogenesis of inflammatory bowel disease. Nature. (2007) 448:427–34. doi: 10.1038/nature06005
13. Uniken VW, Voskuil MD, Dijkstra G, Weersma RK, Festen EA. The genetic background of inflammatory bowel disease: from correlation to causality. J Pathol. (2017) 241:146–58. doi: 10.1002/path.4817
14. Khor B, Gardet A, Xavier RJ. Genetics and pathogenesis of inflammatory bowel disease. Nature. (2011) 474:307–17. doi: 10.1038/nature10209
15. Keita ÅV, Lindqvist CM, Öst Å, Magana CDL, Schoultz I, Halfvarson J. Gut barrier dysfunction- a primary defect in twins with Crohn's disease predominantly caused by genetic predisposition. J Crohns Colitis. (2018) 12:1200–9. doi: 10.1093/ecco-jcc/jjy045
16. Wehkamp J, Harder J, Weichenthal M, Schwab M, Schaffeler E, Schlee M, et al. NOD2 (CARD15) mutations in Crohn's disease are associated with diminished mucosal alpha-defensin expression. Gut. (2004) 53:1658–64. doi: 10.1136/gut.2003.032805
17. Watanabe T, Kitani A, Murray PJ, Wakatsuki Y, Fuss IJ, Strober W. Nucleotide binding oligomerization domain 2 deficiency leads to dysregulated TLR2 signaling and induction of antigen-specific colitis. Immunity. (2006) 25:473–85. doi: 10.1016/j.immuni.2006.06.018
18. Kosovac K, Brenmoehl J, Holler E, Falk W, Schoelmerich J, Hausmann M, et al. Association of the NOD2 genotype with bacterial translocation via altered cell-cell contacts in Crohn's disease patients. Inflamm Bowel Dis. (2010) 16:1311–21. doi: 10.1002/ibd.21223
19. Wei SC, Yang-Yen HF, Tsao PN, Weng MT, Tung CC, Yu LCH, et al. SHANK3 regulates intestinal barrier function through modulating ZO-1 expression through the PKCε-dependent pathway. Inflamm Bowel Dis. (2017) 23:1730–40. doi: 10.1097/MIB.0000000000001250
20. Park JH, Peyrin-Biroulet L, Eisenhut M, Shin JI. IBD immunopathogenesis: a comprehensive review of inflammatory molecules. Autoimmun Rev. (2017) 16:416–26. doi: 10.1016/j.autrev.2017.02.013
21. de Souza HS, Fiocchi C. Immunopathogenesis of IBD: current state of the art. Nat Rev Gastroenterol Hepatol. (2016) 13:13–27. doi: 10.1038/nrgastro.2015.186
22. Gerlach K, Hwang Y, Nikolaev A, Atreya R, Dornhoff H, Steiner S, et al. TH9 cells that express the transcription factor PU.1 drive T cell-mediated colitis via IL-9 receptor signaling in intestinal epithelial cells. Nat Immunol. (2014) 15:676–86. doi: 10.1038/ni.2920
23. Colombel J, Sands BE, Rutgeerts P, Sandborn W, Danese S, D'Haens G, et al. The safety of vedolizumab for ulcerative colitis and Crohn's disease. Gut. (2017) 66:839–51. doi: 10.1136/gutjnl-2015-311079
24. Feagan BGP, Sandborn WJM, D'Haens GP, Panés JP, Kaser AP, Ferrante MP, et al. Induction therapy with the selective interleukin-23 inhibitor risankizumab in patients with moderate-to-severe Crohn's disease: a randomised, double-blind, placebo-controlled phase 2 study. Lancet. (2017) 389:1699–709. doi: 10.1016/S0140-6736(17)30570-6
25. Mullin JM, Marano CW, Laughlin KV, Nuciglio M, Stevenson BR, Soler P. Different size limitations for increased transepithelial paracellular solute flux across phorbol ester and tumor necrosis factor-treated epithelial cell sheets. J Cell Physiol. (1997) 171:226–33.
26. DiGuilio KM, Mercogliano CM, Born J, Ferraro B, To J, Mixson B, et al. Sieving characteristics of cytokine- and peroxide-induced epithelial barrier leak: Inhibition by berberine. World J Gastrointest Pathophysiol. (2016) 7:223–34. doi: 10.4291/wjgp.v7.i2.223
27. Landy J, Ronde E, English N, Clark SK, Hart AL, Knight SC, et al. Tight junctions in inflammatory bowel diseases and inflammatory bowel disease associated colorectal cancer. World J Gastroenterol. (2016) 22:3117–26. doi: 10.3748/wjg.v22.i11.3117
28. Teshima CW, Dieleman LA, Meddings JB. Abnormal intestinal permeability in Crohn's disease pathogenesis. Ann N Y Acad Sci. (2012) 1258:159–65. doi: 10.1111/j.1749-6632.2012.06612.x
29. Chang J, Leong RW, Wasinger VC, Ip M, Yang M, Phan TG. Impaired intestinal permeability contributes to ongoing bowel symptoms in patients with inflammatory bowel disease and mucosal healing. Gastroenterology. (2017) 153:723–31. doi: 10.1053/j.gastro.2017.05.056
30. Sartor RB, Wu GD. Roles for Intestinal bacteria, viruses, and fungi in pathogenesis of inflammatory bowel diseases and therapeutic approaches. Gastroenterology. (2017) 152:327–39. doi: 10.1053/j.gastro.2016.10.012
31. Morgan XC, Tickle TL, Sokol H, Gevers D, Devaney KL, Ward DV, et al. Dysfunction of the intestinal microbiome in inflammatory bowel disease and treatment. Genome Biol. (2012) 13:R79. doi: 10.1186/gb-2012-13-9-r79
32. Darfeuille-Michaud A, Boudeau J, Bulois P, Neut C, Glasser AL, Barnich N, et al. High prevalence of adherent-invasive Escherichia coli associated with ileal mucosa in Crohn's disease. Gastroenterology. (2004) 127:412–21. doi: 10.1053/j.gastro.2004.04.061
33. Chargui A, Cesaro A, Mimouna S, Fareh M, Brest P, Naquet P, et al. Subversion of autophagy in adherent invasive Escherichia coli-infected neutrophils induces inflammation and cell death. PLoS ONE. (2012) 7:e51727. doi: 10.1371/journal.pone.0051727
34. Vazeille E, Buisson A, Bringer MA, Goutte M, Ouchchane L, Hugot JP, et al. Monocyte-derived macrophages from Crohn's Disease patients are impaired in the ability to control intracellular adherent-invasive Escherichia coli and exhibit disordered cytokine secretion profile. J Crohns Colitis. (2015) 9:410–20. doi: 10.1093/ecco-jcc/jjv053
35. Viladomiu M, Kivolowitz C, Abdulhamid A, Dogan B, Victorio D, Castellanos JG, et al. IgA-coated E. coli enriched in Crohn's disease spondyloarthritis promote TH17-dependent inflammation. Sci Transl Med. (2017) 9:eaaf9655. doi: 10.1126/scitranslmed.aaf9655
36. Gong Y, Li H, Li Y. Effects of Bacillus subtilis on epithelial tight junctions of mice with inflammatory Bowel Disease. J Interferon Cytokine Res. (2016) 36:75–85. doi: 10.1089/jir.2015.0030
37. Chen X, Fu Y, Wang L, Qian W, Zheng F, Hou X. Bifidobacterium longum and VSL#3® amelioration of TNBS-induced colitis associated with reduced HMGB1 and epithelial barrier impairment. Dev Comp Immunol. (2019) 92:77–86. doi: 10.1016/j.dci.2018.09.006
38. Isidro RA, Lopez A, Cruz ML, Gonzalez Torres MI, Chompre G, Isidro AA, et al. The Probiotic VSL#3 modulates colonic macrophages, inflammation, and microflora in acute trinitrobenzene sulfonic acid colitis. J Histochem Cytochem. (2017) 65:445–61. doi: 10.1369/0022155417718542
39. Ng SC, Shi HY, Hamidi N, Underwood FE, Tang W, Benchimol EI, et al. Worldwide incidence and prevalence of inflammatory bowel disease in the 21st century: a systematic review of population-based studies. Lancet. (2018) 390:2769–78. doi: 10.1016/S0140-6736(17)32448-0
40. Molodecky NA, Soon IS, Rabi DM, Ghali WA, Ferris M, Chernoff G, et al. Increasing incidence and prevalence of the inflammatory bowel diseases with time, based on systematic review. Gastroenterology. (2012) 142:46–54. doi: 10.1053/j.gastro.2011.10.001
41. Kaplan GG. The global burden of IBD: from 2015 to 2025. Nat Rev Gastroenterol Hepatol. (2015) 12:720–7. doi: 10.1038/nrgastro.2015.150
42. Cosnes J, Gower Rousseau C, Seksik P, Cortot A. Epidemiology and natural history of inflammatory bowel diseases. Gastroenterology. (2011) 140:1785–94. doi: 10.1053/j.gastro.2011.01.055
43. Ng SC, Tang W, Leong RW, Chen M, Ko Y, Studd C, et al. Environmental risk factors in inflammatory bowel disease: a population-based case-control study in Asia-Pacific. Gut. (2015) 64:1063–71. doi: 10.1136/gutjnl-2014-307410
44. Ng SC, Bernstein CN, Vatn MH, Lakatos PL, Loftus EV, Tysk C, et al. Geographical variability and environmental risk factors in inflammatory bowel disease. Gut. (2013) 62:630–49. doi: 10.1136/gutjnl-2012-303661
45. Wang H, Zhao JX, Hu N, Ren J, Du M, Zhu MJ. Side-stream smoking reduces intestinal inflammation and increases expression of tight junction proteins. World J Gastroenterol. (2012) 18:2180–7. doi: 10.3748/wjg.v18.i18.2180
46. Kim ER, Chang DK. Colorectal cancer in inflammatory bowel disease: the risk, pathogenesis, prevention and diagnosis. World J Gastroenterol. (2014) 20:9872–81. doi: 10.3748/wjg.v20.i29.9872
47. Yaeger R, Shah MA, Miller VA, Kelsen JR, Wang K, Heins ZJ, et al. Genomic alterations observed in colitis-associated cancers are distinct from those found in sporadic colorectal cancers and vary by type of inflammatory bowel disease. Gastroenterology. (2016) 151:278–87. doi: 10.1053/j.gastro.2016.04.001
48. Mullin JM. Epithelial barriers, compartmentation, and cancer. Sci STKE. (2004) 216:pe2. doi: 10.1126/stke.2162004pe2
49. Zeng F, Harris RC. Epidermal growth factor, from gene organization to bedside. Semin Cell Dev Biol. (2014) 28:2–11. doi: 10.1016/j.semcdb.2014.01.011
50. Wu S, Rhee KJ, Albesiano E, Rabizadeh S, Wu X, Yen HR, et al. A human colonic commensal promotes colon tumorigenesis via activation of T helper type 17 T cell responses. Nat Med. (2009) 15:1016–22. doi: 10.1038/nm.2015
51. Grivennikov SI, Karin M. Dangerous liaisons: STAT3 and NF-κB collaboration and crosstalk in cancer. Cytokine Growth Factor Rev. (2010) 21:11–9. doi: 10.1016/j.cytogfr.2009.11.005
52. Kang M, Martin A. Microbiome and colorectal cancer: Unraveling host-microbiota interactions in colitis-associated colorectal cancer development. Semin Immunol. (2017) 32:3–13. doi: 10.1016/j.smim.2017.04.003
53. Nakatsu G, Li X, Zhou H, Sheng J, Wong SH, Wu WK, et al. Gut mucosal microbiome across stages of colorectal carcinogenesis. Nat Commun. (2015) 6:8727. doi: 10.1038/ncomms9727
54. Gagnière J, Raisch J, Veziant J, Barnich N, Bonnet R, Buc E, et al. Gut microbiota imbalance and colorectal cancer. World J Gastroenterol. (2016) 22:501–18. doi: 10.3748/wjg.v22.i2.501
55. Mees ST, Mennigen R, Spieker T, Rijcken E, Senninger N, Haier J, et al. Expression of tight and adherens junction proteins in ulcerative colitis associated colorectal carcinoma: upregulation of claudin-1, claudin-3, claudin-4, and beta-catenin. Int J Colorectal Dis. (2009) 24:361–8. doi: 10.1007/s00384-009-0653-y
56. Kinugasa T, Yoshida T, Shirouzu K, Akagi Y. Increased expression of claudin-1 contributes to tumorigenesis in ulcerative colitis-associated colorectal cancer. Gastroenterology. (2011) 140:S352. doi: 10.1016/S0016-5085(11)61434-0
57. Wang X, Tully O, Ngo B, Zitin M, Mullin JM. Epithelial tight junctional changes in colorectal cancer tissues. Sci World J. (2011) 11:826–41. doi: 10.1100/tsw.2011.86
58. Tang W, Dou T, Zhong M, Wu Z. Dysregulation of Claudin family genes in colorectal cancer in a Chinese population. Biofactors. (2011) 37:65–73. doi: 10.1002/biof.138
59. Singh AB, Sharma A, Smith JJ, Krishnan M, Chen X, Eschrich S, et al. Claudin-1 up-regulates the repressor ZEB-1 to inhibit E-cadherin expression in colon cancer cells. Gastroenterology. (2011) 141:2140–53. doi: 10.1053/j.gastro.2011.08.038
60. Bhat AA, Sharma A, Pope J, Krishnan M, Washington MK, Singh AB, et al. Caudal homeobox protein Cdx-2 cooperates with Wnt pathway to regulate claudin-1 expression in colon cancer cells. PLoS ONE. (2012) 7:e37174. doi: 10.1371/journal.pone.0037174
61. Zeisel MB, Dhawan P, Baumert TF. Tight junction proteins in gastrointestinal and liver disease. Gut. (2019) 68:547–61. doi: 10.1136/gutjnl-2018-316906
62. Hahn-Stromberg V, Askari S, Ahmad A, Befekadu R, Nilsson TK. Expression of claudin 1, claudin 4, and claudin 7 in colorectal cancer and its relation with CLDN DNA methylation patterns. Tumour Biol. (2017) 39:1010428317697569. doi: 10.1177/1010428317697569
63. Arimura Y, Nagaishi K, Hosokawa M. Dynamics of claudins expression in colitis and colitis-associated cancer in rat. Methods Mol Biol. (2011) 762:409–25. doi: 10.1007/978-1-61779-185-7_29
64. Pope JL, Ahmad R, Bhat AA, Washington MK, Singh AB, Dhawan P. Claudin-1 overexpression in intestinal epithelial cells enhances susceptibility to adenamatous polyposis coli-mediated colon tumorigenesis. Mol Cancer. (2014) 13:167. doi: 10.1186/1476-4598-13-167
65. Paquet-Fifield S, Koh SL, Cheng L, Beyit LM, Shembrey C, Mølck C, et al. Tight junction protein claudin-2 promotes self-renewal of human colorectal cancer stem-like cells. Cancer Res. (2018) 78:2925–38. doi: 10.1158/0008-5472.CAN-17-1869
66. Hahn-Stromberg V, Edvardsson H, Bodin L, Franzen L. Tumor volume of colon carcinoma is related to the invasive pattern but not to the expression of cell adhesion proteins. APMIS. (2009) 117:205–11. doi: 10.1111/j.1600-0463.2008.00011.x
67. de Oliveira SS, de Oliveira IM, De Souza W, Morgado-Diaz JA. Claudins upregulation in human colorectal cancer. FEBS Lett. (2005) 579:6179–85. doi: 10.1016/j.febslet.2005.09.091
68. de Souza WF, Fortunato-Miranda N, Robbs BK, et al. Claudin-3 overexpression increases the malignant potential of colorectal cancer cells: roles of ERK1/2 and PI3K-Akt as modulators of EGFR signaling. PLoS ONE. (2013) 8:e74994. doi: 10.1371/journal.pone.0074994
69. Ahmad R, Kumar B, Chen Z, et al. Loss of claudin-3 expression induces IL6/gp130/Stat3 signaling to promote colon cancer malignancy by hyperactivating Wnt/beta-catenin signaling. Oncogene. (2017) 36:6592–604. doi: 10.1038/onc.2017.259
70. Bujko M, Kober P, Mikula M, Ligaj M, Ostrowski J, Siedlecki JA. Expression changes of cell-cell adhesion-related genes in colorectal tumors. Oncol Lett. (2015) 9:2463–70. doi: 10.3892/ol.2015.3107
71. Philip R, Heiler S, Mu W, Buchler MW, Zoller M, Thuma F. Claudin-7 promotes the epithelial-mesenchymal transition in human colorectal cancer. Oncotarget. (2015) 6:2046–63. doi: 10.18632/oncotarget.2858
72. Wang K, Li T, Xu C, Ding Y, Li W, Ding L. Claudin-7 downregulation induces metastasis and invasion in colorectal cancer via the promotion of epithelial-mesenchymal transition. Biochem Biophys Res Commun. (2019) 508:797–804. doi: 10.1016/j.bbrc.2018.10.049
73. Oshima T, Kunisaki C, Yoshihara K, Yamada R, Yamamoto N, Sato T, et al. Reduced expression of the claudin-7 gene correlates with venous invasion and liver metastasis in colorectal cancer. Oncol Rep. (2008) 19:953–9. doi: 10.3892/or.19.4.953
74. Darido C, Buchert M, Pannequin J, Bastide P, Zalzali H, Mantamadiotis T, et al. Defective claudin-7 regulation by Tcf-4 and Sox-9 disrupts the polarity and increases the tumorigenicity of colorectal cancer cells. Cancer Res. (2008) 68:4258–68. doi: 10.1158/0008-5472.CAN-07-5805
75. Wang K, Xu C, Li W, Ding L. Emerging clinical significance of claudin-7 in colorectal cancer: a review. Cancer Manag Res. (2018) 10:3741–52. doi: 10.2147/CMAR.S175383
76. Grone J, Weber B, Staub E, Heinze M, Klaman I, Pilarsky C, et al. Differential expression of genes encoding tight junction proteins in colorectal cancer: frequent dysregulation of claudin-1,−8 and−12. Int J Colorectal Dis. (2007) 22:651–9. doi: 10.1007/s00384-006-0197-3
77. Elkouby-Naor L, Ben-Yosef T. Functions of claudin tight junction proteins and their complex interactions in various physiological systems. Int Rev Cell Mol Biol. (2010) 279:1–32. doi: 10.1016/S1937-6448(10)79001-8
78. Garcia-Hernandez V, Quiros M, Nusrat A. Intestinal epithelial claudins: expression and regulation in homeostasis and inflammation. Ann N Y Acad Sci. (2017) 1397:66–79. doi: 10.1111/nyas.13360
79. Amasheh S, Fromm M, Günzel D. Claudins of intestine and nephron - a correlation of molecular tight junction structure and barrier function. Acta Physiol. (2011) 201:133–40. doi: 10.1111/j.1748-1716.2010.02148.x
80. Lameris AL, Huybers S, Kaukinen K, Mäkelä TH, Bindels RJ, Hoenderop JG, et al. Expression profiling of claudins in the human gastrointestinal tract in health and during inflammatory bowel disease. Scand J Gastroenterol. (2013) 48:58–69. doi: 10.3109/00365521.2012.741616
81. Irudayanathan FJ, Wang N, Wang X, Nangia S. Architecture of the paracellular channels formed by claudins of the blood-brain barrier tight junctions. Ann N Y Acad Sci. (2017) 1405:131–46. doi: 10.1111/nyas.13378
82. Irudayanathan FJ, Wang X, Wang N, Willsey SR, Seddon IA, Nangia S. Self-assembly simulations of classic claudins-insights into the pore structure, selectivity, and higher order complexes. J Phys Chem B. (2018) 122:7463–74. doi: 10.1021/acs.jpcb.8b03842
83. Van Itallie CM, Anderson JM. Claudins and epithelial paracellular transport. Annu Rev Physiol. (2006) 68:403–29. doi: 10.1146/annurev.physiol.68.040104.131404
84. Weber CR, Nalle SC, Tretiakova M, Rubin DT, Turner JR. Claudin-1 and claudin-2 expression is elevated in inflammatory bowel disease and may contribute to early neoplastic transformation. Lab Invest. (2008) 88:1110–20. doi: 10.1038/labinvest.2008.78
85. Pope JL, Bhat AA, Sharma A, Ahmad R, Krishnan M, Washington MK, et al. Claudin-1 regulates intestinal epithelial homeostasis through the modulation of Notch-signalling. Gut. (2014) 63:622–34. doi: 10.1136/gutjnl-2012-304241
86. Zhou Q, Costinean S, Croce CM, Brasier AR, Merwat S, Larson SA, et al. MicroRNA 29 targets nuclear factor-κB-repressing factor and claudin 1 to increase intestinal permeability. Gastroenterology. (2015) 148:158–69. doi: 10.1053/j.gastro.2014.09.037
87. Kinugasa T, Akagi Y, Ochi T, Tanaka N, Kawahara A, Ishibashi Y, et al. Increased claudin-1 protein expression in hepatic metastatic lesions of colorectal cancer. Anticancer Res. (2012) 32:2309–14. doi: 10.1016/j.semcancer.2012.03.005
88. Bhat AA, Ahmad R, Uppada SB, Singh AB, Dhawan P. Claudin-1 promotes TNF-α-induced epithelial-mesenchymal transition and migration in colorectal adenocarcinoma cells. Exp Cell Res. (2016) 349:119–27. doi: 10.1016/j.yexcr.2016.10.005
89. Amasheh S, Meiri N, Gitter AH, Schoneberg T, Mankertz J, Schulzke JD, et al. Claudin-2 expression induces cation-selective channels in tight junctions of epithelial cells. J Cell Sci. (2002) 115:4969–76. doi: 10.1242/jcs.00165
90. Rosenthal R, Milatz S, Krug SM, Oelrich B, Schulzke JD, Amasheh S, et al. Claudin-2, a component of the tight junction, forms a paracellular water channel. J Cell Sci. (2010) 123:1913–21. doi: 10.1242/jcs.060665
91. Zeissig S, Bürgel N, Günzel D, Richter J, Mankertz J, Wahnschaffe U, et al. Changes in expression and distribution of claudin 2, 5 and 8 lead to discontinuous tight junctions and barrier dysfunction in active Crohn's disease. Gut. (2007) 56:61–72. doi: 10.1136/gut.2006.094375
92. Dhawan P, Ahmad R, Chaturvedi R, Smith JJ, Midha R, Mittal MK, et al. Claudin-2 expression increases tumorigenicity of colon cancer cells: role of epidermal growth factor receptor activation. Oncogene. (2011) 30:3234–47. doi: 10.1038/onc.2011.43
93. Ahmad R, Chaturvedi R, Olivares-Villagómez D, Habib T, Asim M, Shivesh P, et al. Targeted colonic claudin-2 expression renders resistance to epithelial injury, induces immune suppression and protects from colitis. Mucosal Immunol. (2014) 7:1340–53. doi: 10.1038/mi.2014.21
94. Mankertz J, Hillenbrand B, Tavalali S, Huber O, Fromm M, Schulzke JD, et al. Functional crosstalk between Wnt signaling and Cdx-related transcriptional activation in the regulation of the claudin-2 promoter activity. Biochem Biophys Res Commun. (2004) 314:1001–7. doi: 10.1016/j.bbrc.2003.12.185
95. Prasad S, Mingrino R, Kaukinen K, Hayes KL, Powell RM, MacDonald TT, et al. Inflammatory processes have differential effects on claudins 2, 3 and 4 in colonic epithelial cells. Lab Invest. (2005) 85:1139–62. doi: 10.1038/labinvest.3700316
96. Patel RM, Myers LS, Kurundkar AR, Maheshwari A, Nusrat A, Lin PW. Probiotic bacteria induce maturation of intestinal claudin 3 expression and barrier function. Am J Pathol. (2012) 180:626–35. doi: 10.1016/j.ajpath.2011.10.025
97. Wang F, Graham WV, Wang Y, Witkowski ED, Schwarz BT, Turner JR. Interferon-gamma and tumor necrosis factor-alpha synergize to induce intestinal epithelial barrier dysfunction by up-regulating myosin light chain kinase expression. Am J Pathol. (2005) 166:409–19. doi: 10.1016/S0002-9440(10)62264-X
98. Haines RJ, Beard RS, Chen L, Eitnier RA, Wu MH. Interleukin-1β mediates β-catenin-driven downregulation of claudin-3 and barrier dysfunction in caco2 cells. Digest Dis Sci. (2016) 61:2252–61. doi: 10.1007/s10620-016-4145-y
99. Hou J, Renigunta A, Yang J, Waldegger S. Claudin-4 forms paracellular chloride channel in the kidney and requires claudin-8 for tight junction localization. Proc Natl Acad Sci USA. (2010) 107:18010–5. doi: 10.1073/pnas.1009399107
100. Xiang RL, Mei M, Cong X, Li J, Zhang Y, Ding C, et al. Claudin-4 is required for AMPK-modulated paracellular permeability in submandibular gland cells. J Mol Cell Biol. (2014) 6:486–97. doi: 10.1093/jmcb/mju048
101. Cong X, Zhang Y, Li J, Mei M, Ding C, Xiang RL, et al. Claudin-4 is required for modulation of paracellular permeability by muscarinic acetylcholine receptor in epithelial cells. J Cell Sci. (2015) 128:2271–86. doi: 10.1242/jcs.165878
102. Song YX, Sun JX, Zhao JH, Yang YC, Shi JX, Wu ZH, et al. Non-coding RNAs participate in the regulatory network of CLDN4 via ceRNA mediated miRNA evasion. Nat Commun. (2017) 8:289. doi: 10.1038/s41467-017-00304-1
103. Süren D, Yildirim M, Kaya V, Alikanoglu AS, Bülbüller N, Yildiz M, et al. Loss of tight junction proteins (Claudin 1, 4, and 7) correlates with aggressive behavior in colorectal carcinoma. Med Sci Monit. (2014) 20:1255–62. doi: 10.12659/MSM.890598
104. Dahan S, Rabinowitz KM, Martin AP, Berin MC, Unkeless JC, Mayer L. Notch-1 signaling regulates intestinal epithelial barrier function, through interaction with CD4+ T cells, in mice and humans. Gastroenterology. (2011) 140:550–9. doi: 10.1053/j.gastro.2010.10.057
105. Dahan S, Roda G, Pinn D, Roth Walter F, Kamalu O, Marctin AP, et al. Epithelial: lamina propria lymphocyte interactions promote epithelial cell differentiation. Gastroenterology. (2008) 134:192–203. doi: 10.1053/j.gastro.2007.10.022
106. Laakkonen JP, Lappalainen JP, Theelen TL, Toivanen PI, Nieminen T, Jauhiainen S, et al. Differential regulation of angiogenic cellular processes and claudin-5 by histamine and VEGF via PI3K-signaling, transcription factor SNAI2 and interleukin-8. Angiogenesis. (2017) 20:109–24. doi: 10.1007/s10456-016-9532-7
107. Bekes I, Friedl TW, Köhler T, Möbus V, Janni W, Wöckel A, et al. Does VEGF facilitate local tumor growth and spread into the abdominal cavity by suppressing endothelial cell adhesion, thus increasing vascular peritoneal permeability followed by ascites production in ovarian cancer? Mol Cancer. (2016) 15:13. doi: 10.1186/s12943-016-0497-3
108. Chung YH, Li SC, Kao YH, Luo HL, Cheng YT, Lin PR, et al. MiR-30a-5p Inhibits epithelial-to-mesenchymal transition and upregulates expression of tight junction protein claudin-5 in human upper tract urothelial carcinoma cells. Int J Mol Sci. (2017) 18:E1826. doi: 10.3390/ijms18081826
109. Gao F, Alwhaibi A, Artham S, Verma A, Somanath PR. Endothelial Akt1 loss promotes prostate cancer metastasis via beta-catenin-regulated tight-junction protein turnover. Br J Cancer. (2018) 118:1464–75. doi: 10.1038/s41416-018-0110-1
110. Oshima T, Miwa H, Joh T. Changes in the expression of claudins in active ulcerative colitis. J Gastroenterol Hepatol. (2008) 23 (Suppl. 2):S146–50. doi: 10.1111/j.1440-1746.2008.05405.x
111. Tanaka H, Takechi M, Kiyonari H, Shioi G, Tamura A, Tsukita S. Intestinal deletion of Claudin-7 enhances paracellular organic solute flux and initiates colonic inflammation in mice. Gut. (2015) 64:1529–38. doi: 10.1136/gutjnl-2014-308419
112. Manicone AM, McGuire JK. Matrix metalloproteinases as modulators of inflammation. Semin Cell Dev Biol. (2008) 19:34–41. doi: 10.1016/j.semcdb.2007.07.003
113. Ding L, Lu Z, Foreman O, Tatum R, Lu Q, Renegar R, et al. Inflammation and disruption of the mucosal architecture in claudin-7-deficient mice. Gastroenterology. (2012) 142:305–15. doi: 10.1053/j.gastro.2011.10.025
114. Bhat AA, Pope JL, Smith JJ, Ahmad R, Chen X, Washington MK, et al. Claudin-7 expression induces mesenchymal to epithelial transformation (MET) to inhibit colon tumorigenesis. Oncogene. (2015) 34:4570–80. doi: 10.1038/onc.2014.385
115. Amasheh S, Milatz S, Krug SM, Bergs M, Amasheh M, Schulzke JD, et al. Na+ absorption defends from paracellular back-leakage by claudin-8 upregulation. Biochem Biophys Res Commun. (2009) 378:45–50. doi: 10.1016/j.bbrc.2008.10.164
116. Wang H, Chao K, Ng SC, Bai AH, Yu Q, Yu J, et al. Pro-inflammatory miR-223 mediates the cross-talk between the IL23 pathway and the intestinal barrier in inflammatory bowel disease. Genome Biol. (2016) 17:58. doi: 10.1186/s13059-016-0901-8
117. Li L, Huang S, Wang H, Chao K, Ding L, Feng R, et al. Cytokine IL9 triggers the pathogenesis of inflammatory bowel disease through the miR21-CLDN8 pathway. Inflamm Bowel Dis. (2018) 24:2211–23. doi: 10.1093/ibd/izy187
118. Nalleweg N, Chiriac MT, Podstawa E, Lehmann C, Rau TT, Atreya R, et al. IL-9 and its receptor are predominantly involved in the pathogenesis of UC. Gut. (2015) 64:743–55. doi: 10.1136/gutjnl-2013-305947
119. Ashikari D, Takayama KI, Obinata D, Takahashi S, Inoue S. CLDN8, an androgen-regulated gene, promotes prostate cancer cell proliferation and migration. Cancer Sci. (2017) 108:1386–93. doi: 10.1111/cas.13269
120. Fujita H, Sugimoto K, Inatomi S, Maeda T, Osanai M, Uchiyama Y, et al. Tight junction proteins claudin-2 and−12 are critical for vitamin D-dependent Ca2+ absorption between enterocytes. Mol Biol Cell. (2008) 19:1912–21. doi: 10.1091/mbc.E07-09-0973
121. Yang Y, Cheon S, Jung MK, Song SB, Kim D, Kim HJ, et al. Interleukin-18 enhances breast cancer cell migration via down-regulation of claudin-12 and induction of the p38 MAPK pathway. Biochem Biophys Res Commun. (2015) 459:379–86. doi: 10.1016/j.bbrc.2015.02.108
122. Alberini G, Benfenati F, Maragliano L. A refined model of claudin-15 tight junction paracellular architecture by molecular dynamics simulations. PLoS ONE. (2017) 12:e0184190. doi: 10.1371/journal.pone.0184190
123. Tamura A, Hayashi H, Imasato M, Yamazaki Y, Hagiwara A, Wada M, et al. Loss of claudin-15, but not claudin-2, causes Na+ deficiency and glucose malabsorption in mouse small intestine. Gastroenterology. (2011) 140:913–23. doi: 10.1053/j.gastro.2010.08.006
124. Wada M, Tamura A, Takahashi N, Tsukita S. Loss of claudins 2 and 15 from mice causes defects in paracellular Na+ flow and nutrient transport in gut and leads to death from malnutrition. Gastroenterology. (2013) 144:369–80. doi: 10.1053/j.gastro.2012.10.035
125. Tamura A, Kitano Y, Hata M, Katsuno T, Moriwaki K, Sasaki H, et al. Megaintestine in claudin-15-deficient mice. Gastroenterology. (2008) 134:523–34. doi: 10.1053/j.gastro.2007.11.040
126. Niimi T, Nagashima K, Ward JM, Minoo P, Zimonjic DB, Popescu NC, et al. claudin-18, a novel downstream target gene for the T/EBP/NKX2.1 homeodomain transcription factor, encodes lung- and stomach-specific isoforms through alternative splicing. Mol Cell Biol. (2001) 21:7380–90. doi: 10.1128/MCB.21.21.7380-7390.2001
127. Hayashi D, Tamura A, Tanaka H, Yamazaki Y, Watanabe S, Suzuki K, et al. Deficiency of claudin-18 causes paracellular H+ leakage, up-regulation of interleukin-1β, and atrophic gastritis in mice. Gastroenterology. (2012) 142:292–304. doi: 10.1053/j.gastro.2011.10.040
128. Hagen SJ, Ang LH, Zheng Y, Karahan SN, Wu J, Wang YE, et al. Loss of tight junction protein claudin 18 promotes progressive neoplasia development in mouse stomach. Gastroenterology. (2018) 155:1852–67. doi: 10.1053/j.gastro.2018.08.041
129. Sahin U, Koslowski M, Dhaene K, Usener D, Brandenburg G, Seitz G, et al. Claudin-18 splice variant 2 is a pan-cancer target suitable for therapeutic antibody development. Clin Cancer Res. (2008) 14:7624–34. doi: 10.1158/1078-0432.CCR-08-1547
130. Takasawa K, Takasawa A, Osanai M, Aoyama T, Ono Y, Kono T, et al. Claudin-18 coupled with EGFR/ERK signaling contributes to the malignant potentials of bile duct cancer. Cancer Lett. (2017) 403:66–73. doi: 10.1016/j.canlet.2017.05.033
131. Zwiers A, Fuss IJ, Leijen S, Mulder CJ, Kraal G, Bouma G. Increased expression of the tight junction molecule claudin-18 A1 in both experimental colitis and ulcerative colitis. Inflamm Bowel Dis. (2008) 14:1652–59. doi: 10.1002/ibd.20695
132. Cherradi S, Ayrolles-Torro A, Vezzo-Vié N, Gueguinou N, Denis V, Combes E, et al. Antibody targeting of claudin-1 as a potential colorectal cancer therapy. J Exp Clin Cancer Res. (2017) 36:89. doi: 10.1186/s13046-017-0558-5
133. Resnick MB, Konkin T, Routhier J, Sabo E, Pricolo VE. Claudin-1 is a strong prognostic indicator in stage II colonic cancer: a tissue microarray study. Mod Pathol. (2005) 18:511–8. doi: 10.1038/modpathol.3800301
134. Singh P, Toom S, Huang Y. Anti-claudin 18.2 antibody as new targeted therapy for advanced gastric cancer. J Hematol Oncol. (2017) 10:105. doi: 10.1186/s13045-017-0473-4
Keywords: inflammatory bowel diseases, claudin, intestinal permeability, colitis-associated colorectal cancer, protein family
Citation: Zhu L, Han J, Li L, Wang Y, Li Y and Zhang S (2019) Claudin Family Participates in the Pathogenesis of Inflammatory Bowel Diseases and Colitis-Associated Colorectal Cancer. Front. Immunol. 10:1441. doi: 10.3389/fimmu.2019.01441
Received: 22 January 2019; Accepted: 07 June 2019;
Published: 27 June 2019.
Edited by:
Kai Fang, University of California, Los Angeles, United StatesReviewed by:
James Michael Mullin, Lankenau Institute for Medical Research, United StatesCopyright © 2019 Zhu, Han, Li, Wang, Li and Zhang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Shenghong Zhang, c2hlbmdob25nemhhbmdAMTYzLmNvbQ==
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.