
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
HYPOTHESIS AND THEORY article
Front. Immunol. , 06 June 2019
Sec. Inflammation
Volume 10 - 2019 | https://doi.org/10.3389/fimmu.2019.01270
This article is part of the Research Topic Trauma-Induced, DAMP-Mediated Remote Organ Injury and Immunosuppression in the Acutely Ill Patient View all 9 articles
 Patricia Martinez-Quinones1,2*
Patricia Martinez-Quinones1,2* Amel Komic1,2
Amel Komic1,2 Cameron G. McCarthy3
Cameron G. McCarthy3 R. Clinton Webb1
R. Clinton Webb1 Camilla Ferreira Wenceslau3*
Camilla Ferreira Wenceslau3*Despite recent advances in our understanding of the mechanisms underlying systemic inflammatory response syndrome (SIRS) and sepsis, the current therapeutic approach to these critically ill patients is centered around supportive care including fluid resuscitation, vasopressors and source control. The incidence of SIRS and sepsis continues to increase in the United States and patients die due to failure to respond to the traditional therapies of nitric oxide blockade, adrenergic agonists, etc. Bacterial and mitochondrial N-formyl peptides (NFPs) act as damage-associated molecular patterns and activate the innate immune system through formyl peptide receptors (FPR) located in immune and non-immune cells, including the vascular endothelium. The resulting inflammatory response manifests as capillary leak, tissue hypoperfusion and vasoplegia, partially due to endothelium barrier breakdown. Potential strategies to prevent this response include decreasing NFP release, breakdown of NFPs, and blocking NFPs from binding FPR. We propose the use of deformylase, the degrading enzyme for NFPs, as potential therapeutic approach to prevent the deleterious effects of NFPs in SIRS and sepsis.
Systemic inflammatory response syndrome (SIRS) and sepsis are major causes of morbidity and mortality in the United States. Sepsis and its related morbidity and mortality is considered the most expensive condition treated in the United States by the Agency for Healthcare and Research Quality, costing an approximate 20 billion dollars yearly (1–3). Despite advances in critical care, few targeted therapies have been developed for patients with SIRS, sepsis and multi-system organ dysfunction (MSOD) (4). While sepsis has been recently defined as life-threatening organ dysfunction caused by a dysregulated host response to infection (5), SIRS may reflect an appropriate host response, which can be caused by various pathologic insults, including trauma and tissue injury (5, 6). Trauma and tissue injury lead to a sepsis-like clinical picture, “microorganism-free sepsis,” which clinically mimics sepsis, although no microbial pathogen is present.
SIRS and sepsis are complex inflammatory syndromes that involve activation and amplification of the innate immune system through receptor interaction with “danger signals” or “target molecular motifs.” In the case of traumatic tissue injury, this response can be attributed to endogenous molecules known as damage-associated molecular patterns (DAMPs) (7). DAMPs are released when tissue injury occurs and trigger an innate immune response similar to that of pathogen-associated molecular patterns (PAMPs) (8–10). PAMPs are conserved pathogenic structures such as bacterial DNA, bacterial cell wall components and bacterial N-Formyl peptides (fMLP). DAMPs are evolutionarily conserved endogenous molecules not normally found in the circulation such as mitochondrial DNA, mitochondrial N-Formyl peptides (FMITs), basement membrane fragments, histones and heat shock proteins. Innate immune system activation through pattern recognition receptors (PRRs), such as formyl peptide receptors (FPRs) and toll-like receptors (TLRs) (11), leads to the production of high levels of proinflammatory cytokines (tumor necrosis factor (TNF-α), interleukin (IL)-6, interferon gamma (IFN-γ), etc.) causing systemic derangement and cardiovascular collapse (12–14). The sharing of cellular pathways by which DAMPs and PAMPs act may cause the downstream immune responses to SIRS and sepsis to be indistinguishable from one another. This may thus explain the similarity in clinical responses to infective and non-infective challenges.
Increased circulating levels of mitochondrial DAMPs have been associated with increased morbidity and mortality in critically ill adult patients (7, 15, 16). Our laboratory has recently demonstrated that trauma patients with SIRS have increased levels of circulating mitochondrial NFPs compared to control trauma patients without clinically diagnosed SIRS (17). Furthermore, trauma patients who went on to develop sepsis during their hospitalization where found to have even higher levels of mitochondrial NFPs in their plasma, compared to those of SIRS patients (17). In a rat model of hemorrhagic shock, mitochondrial NFPs (N-formyl-Met-Met-Tyr-Ala-Leu-Phe) induced severe hypotension, hyperthermia, lung injury, microvascular thrombosis and vascular leakage (18).
Elevated DAMPs lead to inflammation and end-organ damage in vitro and in vivo (7, 14, 17, 19). In a murine model of acute lung injury with tracheal infusion of mitochondrial NFPs, we showed a concentration-dependent contraction of the trachea, bronchi and bronchioles, which was decreased with FPR-1 antagonist administration (17). Nonetheless, the underlying mechanisms by which NFPs affect non-immune cells and lead to SIRS after traumatic injury are still being investigated. Similarly, targeted degradation of mitochondrial DAMPs in vitro has offered a potential therapeutic alternative for the treatment of these devastating diseases, especially in patients that do not respond to traditional therapies (20).
SIRS and sepsis are different manifestations of an underlying complex pathophysiology with many etiologies. Both SIRS and sepsis can lead to multi-system organ dysfunction and potentially death (21). One of the major characteristics of these conditions is the breakdown of vascular endothelial barrier function (4, 6, 22), which can result in hemodynamic collapse and shock. An increase in vascular permeability (or vascular leakage) leads to progressive subcutaneous and body-cavity edema, clinically referred to as anasarca (4). Whether endothelial barrier dysfunction is a cause or an effect of the disease process underlying SIRS and sepsis has yet to be determined. Nonetheless, understanding the molecular mechanisms causing endothelial barrier breakdown might lead to new pharmacologic approaches for its prevention and ultimately to an innovative treatment.
An increase in vascular endothelium permeability, secondary to endothelial barrier dysfunction, has been previously associated with pro-inflammatory factors such as reactive oxygen species, TNF-α, IL-1, IL-2, and IL-6 (23), known to be elevated in SIRS and sepsis. However, pharmacological interventions that inhibit these molecules have not been successful at preventing or reversing endothelial damage (22). Further, inhibition of TLR-4 with the antagonists E5564 and TAK-242 showed no effects on 28-days mortality reduction in sepsis (24, 25). Similarly, polyclonal intravenous immune globulin administration has shown variable results; however, randomized trials showed no benefits when compared to placebo (26–28). Additionally, use of a recombinant, non-glycosylated human IL-1 receptor antagonist also showed no improvement in patients with severe sepsis and septic shock (29, 30).
Due to the lack of understanding of the molecular mechanisms underlying endothelial barrier dysfunction, therapies targeting vascular leakage in SIRS and sepsis are not currently available. Our aim is to better understand the underlying mechanisms of how bacterial and mitochondrial NFPs lead to vascular leakage, and to devise strategies which may specifically target NFP pathways. With this knowledge we can devise potential strategies which may target NFPs, breakdown of circulating NFPs and/or preventing NFPs from binding its target receptor, FPR-1.
The pro-inflammatory nature of NFPs and their critical role in initiating pathogenic and sterile inflammatory responses makes them an appealing therapeutic target. While activation of the innate immune system is necessary for clearance of the offending bacterial organism or injured tissue, it is unknown how much NFP is needed to potentiate the inflammatory response and alter this response from adaptive to maladaptive. Bacterial NFPs all contain a conserved secondary structure, allowing for a large pool of pathogens to activate FPR-1 with similar affinity and elicit a similar response (31). FPR-1 activation by fMLP (a bacterial NFP) triggers neutrophil chemotaxis, diapedesis, and degranulation (32–34) and neutrophils deficient in FPR-1 display impaired chemotaxis (35). As mentioned above, we have previously shown that fMLP induce vascular leakage and exacerbate vasodilatation in rat mesenteric resistance arteries, and that Cyclosporin-H (CsH), an FPR-1 antagonist, inhibited this response (14).
FPR-1 has differential expression in various immune cells (e.g., dendritic cells, neutrophils, mast cells) and non-immune cells (e.g., somatic cells of the cardiovascular system, including the endothelium) (33). FPR-1 detects evolutionarily conserved molecules found in bacteria and recognizes the bacterial origin of mitochondria (7, 14, 36). FMIT exposure to vessels also induces FPR-1-mediated vascular relaxation that is inhibited by CsH (14).
FPR belongs to G-protein coupled receptor (GPCR) family and important components of the innate immune system (4). FPRs were first discovered in neutrophils and are now known to be comprised of 3 members in humans (FPR-1, FPR-2, and FPR-3) and 8 in mice (37, 38). Each member of the FPR family has differential expression and binding affinities, with FPR-2 and FPR-3 having lower affinity and a greater number of ligands (39, 40). FPR-1 activation by NFPs triggers immune reactions in neutrophils, monocytes and macrophages (41). FPR-1 is a high affinity binding site for the NFP sequence fMLP, with the ability to recognize even small picomolar concentrations (42). FPR-1 activation in phagocytic cells triggers degranulation, pro-inflammatory cytokine and chemokine production and reactive oxygen species generation (41). Contrary to FPR-1, FPR-2 can bind a variety of ligands, although with lower affinity, including select bacterial NFPs and non-formylated ligands like annexin-1, resolvin D1 and lipoxin A4, among others (33, 42, 43). FPR-2 has been observed to prevent excessive inflammatory responses in animal models of meningitis and Alzheimer's disease (44, 45). FPR-3 function remains unclear, as it is not present in hematopoietic cells and its ligands do not overlap with FPR-1 and FPR-2 (46, 47). To our knowledge there are no data to suggest FPR-3 involvement in endothelium integrity.
FPR-1 is also expressed in non-immune cells, suggesting that FPR-1 serves other functions besides sensing targeted molecular motifs (48). FPR-1 is essential for vascular homeostasis, as shown in our recent work where FPR-1 was found to be fundamental for myogenic vascular contraction under physiological conditions (49). FPR-1 has been implicated in cell growth and proliferation in tumorigenesis (50, 51). FPR-1 activation in immune and non-immune cells triggers intracellular signal transduction pathways responsible for transcriptional regulation, cytoskeletal reorganization, superoxide production, and exocytosis of granules (34, 52, 53). Activation of FPR-1 signaling contributes to the physiological defense against danger signals and makes FPR-1 an attractive therapeutic target.
The identification of selective FPR antagonists has allowed for the continued discovery of this receptor family interactions and potential implications in disease. Interestingly, some pathogens produce FPR antagonists. For instance, the pertussis toxin from Bordetella pertussis is a potent inhibitor of GPCR-mediated leukocyte chemotaxis by inactivating the Gαi protein of FPR (42). The most widely used FPR-1 antagonist Cyclosporin H (CsH) is a high affinity inverse agonist, selective for FPR-1, which “locks” FPR-1 into an inactive conformation (54, 55). Due to the intrinsic relationship between FPR-1, the actin cytoskeleton and transcription regulation, antagonizing this receptor is a problematic approach (37). In a model of pneumococcal meningitis, FPR-1 deficient mice were found to have increased bacterial burden, increased neutrophil infiltration and elevated mortality rates (44). Oldekamp and colleagues also showed that FPR-1 deficient microglial cells have attenuated cell viability after bacterial exposure to S. pneumoniae and N. meningitidis (44). FPR-1 deficient mice also have increased susceptibility to Listeria monocytogenes as evidenced by increased bacterial load in the spleen and liver (56). Increased knowledge of the direct importance of FPR-1 in physiological and pathophysiological conditions is still needed. Challenges exist in targeting FPR-1 directly because of its intrinsic functional properties and its ability to mediate both pro-inflammatory and anti-inflammatory effects depending on the activating ligand (57). Furthermore, since its absence leads to an enhanced inflammatory response, other approaches to targeting FPR-1 signaling must be considered for potential therapeutic applications.
When FPR-1 is activated, it mediates chemotaxis (58), signals intracellular cascades (59), induces cell cytoskeleton rearrangement (48), and may act as a mechanosensor (49, 60). In phagocytic cells, FPR-1 blockade in these cells impairs their function, prevents their migration to sites of infection and decreases bacterial clearance (35). FPR-1 activation in non-immune cells may occur through neutrophil-dependent and/or neutrophil-independent pathways (19). In accordance with prior studies, we showed that FPR-1 is not only present in immune cells but also on vascular endothelial and vascular smooth muscle cells (18, 48, 61). FPR-1 expression in somatic vascular cells is consistent with the theory that each tissue and cell type can tailor its own immune response. FPR-1 mediated generation of pro-inflammatory cytokines, chemokines, and adhesion molecules has been extensively studied in immune cells, specifically neutrophils. However, whether or not the same signaling pathways and downstream effects of FPR-1 activation occur in vascular endothelial cells has yet to be determined.
In the pathophysiologic states of SIRS and sepsis, disturbances in the microcirculation are associated with worse clinical outcomes, and this occurs independent of macrohemodynamic changes. Factors proposed to contribute to this capillary leakage, and disturbances in blood flow, include platelet aggregation, endothelial cell injury, and increased microvascular permeability with accompanying interstitial edema, among others (62). Microvascular injury compromises capillary blood flow, leading to capillary flow cessation and potentially causing further hypoxic tissue injury. Endothelial cell activation, independent of leukocyte activation, triggers a localized inflammatory response and exacerbates microvascular leakage (62).
Vascular endothelium is one of the tissues most affected by sepsis and traumatic injury, either by trauma itself and/or the inflammatory reaction after trauma. An intact vascular endothelium is necessary to maintain barrier function, osmotic balance, solute transport, and to prevent pathogens and molecules from reaching the sub-endothelial connective tissue (22). The endothelial cell lining of the vasculature constitutes a semi-permeable barrier between the intravascular and the interstitial space. Under physiological conditions, activation of FPR-1 in vascular endothelial cells is necessary to allow neutrophil and monocyte migration to sites of inflammation, allowing for reduction of bacterial burden (35). However, an exacerbated inflammatory response may lead to endothelial cell apoptosis and necrosis, detachment and loss of endothelium barrier function (63). Increase in endothelium permeability allows for immune cell infiltration, interstitial edema, and potentiation of end-organ damage. Breakdown of the endothelium lining may hinder bacterial clearance as an intact endothelial cytoskeleton is necessary for paracellular transport and leukocyte transmigration (64). Disruption of this barrier integrity manifests as hyper-permeability which is associated with many systemic diseases, including SIRS and sepsis.
The integrity of vascular endothelium is influenced by an intact endothelial cytoskeleton. FPR-1 activation leads to cytoskeletal rearrangement resulting in endothelial cell contraction via actin-myosin interaction and actin polymerization (48). However, the intracellular molecular mechanisms by which NFPs, from bacteria and mitochondria, lead to vascular injury and endothelial barrier breakdown remain incompletely understood. Our working hypothesis is that NFPs, whether exogenous or endogenous, lead to increased vascular endothelial cell permeability through FPR-1 activation, causing downstream actin cytoskeletal rearrangement and endothelial contraction.
There are still major gaps in our understanding of the underlying pathophysiology of trauma-induced SIRS and sepsis. Particularly how DAMPs and PAMPs interaction with PRRs give rise to the multiple cytokines and chemokines produced during SIRS and sepsis, and the subsequent physiological consequences. The inability to treat or prevent trauma-induced SIRS and microbial sepsis may be due to our limited understanding of the underlying molecular mechanisms causing endothelial dysfunction and vascular leakage (48). Given that loss of FPR-1 function could affect appropriate innate immune system response, it should be important to identify means for restoring or bypassing deficiencies in FPR-1 signaling.
FPR-1 antagonists have the potential to inhibit the functional intrinsic properties of this receptor in endothelial cells. However, since FPR-1 ligands involved in eliciting innate immune response can be discriminated, this may offer an opportunity to prevent deleterious downstream FPR-1 signaling. Targeting DAMPs and PAMPs and their receptors is a promising therapeutic strategy for the management of inflammatory pathologies (65). For instance, cell-free mitochondrial DNA is currently being studied as a therapeutic target in myocardial infarction (66) with the use of Endonuclease III, an enzyme that digests DNA. Additionally DNase, another family of enzymes that digests DNA (67), has shown the potential to degrade mitochondrial DNA in in vitro studies (68). DNase has been shown to be elevated in the systemic circulation following traumatic injury (69) and may have potential targeting against mitochondrial DNA. However, no enzymes with the potential to digest NFPs have been identified to be present in the systemic circulation.
Peptide deformylase, a metalloenzyme, has the inherent activity to degrade NFPs before they bind FPR-1, and may in fact serve as a potential pharmacologic agent for the treatment of trauma-induced SIRS and sepsis. Deformylase removes the formyl group at the N terminus of nascent polypeptides in bacteria and mitochondria (70). This enzyme, acting as a monomer, binds a metal ion and catalyzes the reaction: N-formyl-L-methionine + H2O = formate + methionyl peptide (71). Deformylase is essential in prokaryotes and it was previously thought that deformylase was unique to bacteria, hence it has been a target for the creation of antibacterial agents against its activity (70, 72). Deformylase was previously targeted by antibacterial agents after the finding that its inhibition in E. coli was bactericidal (73–75). These agents were found to target both bacterial and human mitochondrial deformylase, as it was later discovered that the three-dimensional structure of deformylase is evolutionarily conserved (76). This led to the identification of deformylase homologs in eukaryotes, including human mitochondria (72, 77). To date, its function in human mitochondria is not well-defined. What is known, is that human mitochondrial deformylase is necessary for translation initiation of respiratory complexes; therefore its inhibition disrupts mitochondrial function (72). Some peptide deformylase inhibitors with activity against bacterial peptide deformylase have been isolated and studied in Phase I clinical trials with variable but non-clinically significant adverse effects (70, 72, 78). The deformylase inhibitors BB83698 and LBM415 studied in phase I clinical trials in humans were ultimately found to have poor selectivity (78). The first deformylase inhibitor to be studied in a clinical trial was LBM415 (Novartis Pharmaceuticals), its oral administration led to the unexpected finding of methemoglobinemia in study participants; these results were later confirmed with in vitro and in vivo animal studies (79). Deformylase inhibitors impair not only bacterial deformylase but also human mitochondrial deformylase and prevent mitochondrial translation and oxidative phosphorylation (80, 81).
The proteolytic effects of deformylase make it an attractive target for drug development. To date peptide deformylase itself has not been studied as a therapeutic option for the management of SIRS or sepsis. Our group has investigated the links between trauma, vascular collapse and sepsis (Figure 1), and our results suggest that NFPs and FPR-1 may serve as that link (14, 17, 18, 48). We have recently found that deformylase is a potent treatment for sepsis in a murine cecal ligation and puncture model of intraperitoneal sepsis and an in vitro cell culture model of SIRS (unpublished) (US Provisional Patent Application 62/790, 185 “Methods and Compositions of Treating Sepsis and Systemic Inflammatory Response Syndrome”).
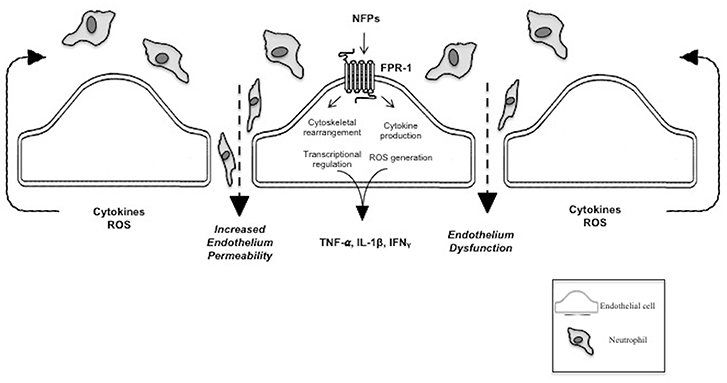
Figure 1. N-Formyl peptide mediated pathophysiology of SIRS and sepsis. This figure shows the pathogenic effect of NFPs on the vascular endothelium via FPR-1 activation. FPR-1 activation results in dysfunction of the vascular barrier allowing infiltration of immune cells and molecules into the interstitial and extravascular space. NFPs, bacterial and mitochondrial N-Formyl peptides; FPR-1, formyl peptide receptor-1; TNF-α, tumor necrosis factor alpha; ILF-1β, interleukin-1 beta; IFNγ, Interferon gamma; ROS, reactive oxygen species.
Based on previous findings, it is reasonable to speculate that administration of deformylase, as the degrading enzyme for both bacterial and mitochondrial NFPs, may serve a therapeutic role in preventing FPR-1 activation and its subsequent endothelium barrier dysfunction and vascular leakage (Figure 2). To assess the potential therapeutic strategy of NFP degradation in SIRS and sepsis, we still need to understand the settings in which activation or inhibition of FPR-1 is beneficial or detrimental to injury repair and pathogenic clearance. Furthermore, the quantity of NFPs needed to potentiate an inflammatory response and tilt the balance from an adaptive response to a maladaptive response is unknown.
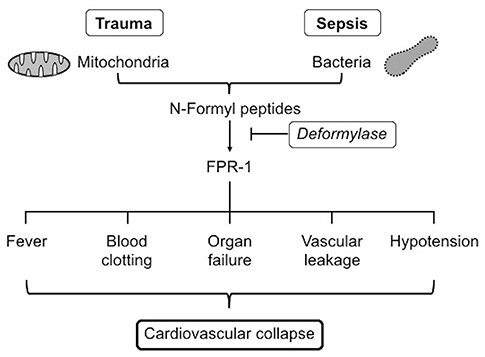
Figure 2. FPR-1, formyl peptide receptor-1.
Despite advances in the care of critically ill patients, further improvements in the field are limited by a lack of knowledge of the underlying pathophysiology in SIRS and sepsis. Our current therapeutic approach is centered around supportive care including fluid resuscitation, vasopressors and source control with antibiotics or surgical intervention. The endothelium plays a central role in initiating, promoting and exacerbating the overwhelming inflammatory response. The molecular mechanisms involved in endothelium barrier dysfunction, vascular leakage, and cardiovascular collapse are still being discovered. Bacterial and mitochondrial NFPs, and their activation of FPR-1 in immune and non-immune cells, may serve as a link to the underlying pathophysiology of trauma-induced SIRS and sepsis. The potent pro-inflammatory nature of NFPs and their role in initiating sterile and infective inflammation make them an attractive therapeutic target. The development of therapeutic agents to neutralize the inflammatory effects of NFPs promises to dramatically improve trauma management. The potential use of deformylase itself holds translational value for further pre-clinical and clinical testing considering that very few, if any, targeted therapies are currently available for the treatment of trauma-induced SIRS and sepsis.
Much is still yet to be determined about the consequences of NFPs and FPR-1 receptor activation, especially in vascular somatic cells. Major barriers still exist in the search for immune-based interventions in trauma and sepsis: (1) understanding the underlying molecular mechanisms triggering SIRS, (2) identification of potential biomarkers that could serve as therapeutic targets, (3) identification of meaningful pre-clinical and clinical endpoints other than death, and (4) clinically, establishment of early intervention and logistics for consent waived trials. Our current work and hypotheses may have implications in the management of SIRS and sepsis patients, especially in those who are non-responders to traditional vasopressors. FPR-1-NFP interaction may serve as the missing link between host-derived danger signals, inflammation and vascular dysfunction in SIRS and sepsis.
PM-Q, AK, and CGM co-wrote the manuscript. RCW critically reviewed the manuscript and contributed to Figures 1, 2. CFW co-wrote the manuscript and supervised the project. All authors agree to be accountable for the content of the work.
This study was performed with the financial support of NIH Grant 1R00GM118885-01 (to CFW) and PO1HL134604 (to RCW), and AHA Grant 18POST3406003 (to CGM).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The handling editor is currently co-organizing a Research Topic with one of the authors CFW, and confirms the absence of any other collaboration.
1. Angus DC, Linde-Zwirble WT, Lidicker J, Clermont G, Carcillo J, Pinsky MR. Epidemiology of severe sepsis in the United States: analysis of incidence, outcome, and associated costs of care. Crit Care Med. (2001) 29:1303–10. doi: 10.1097/00003246-200107000-00002
2. Torio CM, Moore BJ. National inpatient hospital costs: the most expensive conditions by Payer, 2013: statistical brief #204. In: Healthcare Cost and Utilization Project (HCUP) Statistical Briefs. Rockville, MD: Agency for Healthcare Research and Quality (US) (2006).
3. Torio C, Moore B. National Inpatient Hospital Costs: The Most Expensive Conditions by Payer, 2013. Agency for Healthcare Research and Quality (2016). Avilable online at: http://www.hcup-us.ahrq.gov/reports/statbriefs/sb204-Most-Expensive-Hospital-Conditions.pdf (accessed April 6, 2019).
4. Lee WL, Slutsky AS. Sepsis and endothelial permeability. N Engl J Med. (2010) 363:689–91. doi: 10.1056/NEJMcibr1007320
5. Singer M, Deutschman CS, Seymour CW, Shankar-Hari M, Annane D, Bauer M, et al. The third international consensus definitions for sepsis and septic shock (sepsis-3). JAMA. (2016) 315:801–10. doi: 10.1001/jama.2016.0287
6. Rangel-Frausto MS, Pittet D, Costigan M, Hwang T, Davis CS, Wenzel RP. The natural history of the systemic inflammatory response syndrome (SIRS). A prospective study. JAMA. (1995) 273:117–23. doi: 10.1001/jama.1995.03520260039030
7. Zhang Q, Raoof M, Chen Y, Sumi Y, Sursal T, Junger W, et al. Circulating mitochondrial DAMPs cause inflammatory responses to injury. Nature. (2010) 464:104–7. doi: 10.1038/nature08780
8. Matzinger P. Tolerance, danger, and the extended family. Annu Rev Immunol. (1994) 12:991–1045. doi: 10.1146/annurev.iy.12.040194.005015
9. Matzinger P. An innate sense of danger. Semin Immunol. (1998) 10:399–415. doi: 10.1006/smim.1998.0143
10. Matzinger P. The danger model: a renewed sense of self. Science. (2002) 296:301–5. doi: 10.1126/science.1071059
11. Tang BM, Huang SJ, McLean AS. Genome-wide transcription profiling of human sepsis: a systematic review. Crit Care. (2010) 14:R237. doi: 10.1186/cc9392
12. Okusawa S, Gelfand JA, Ikejima T, Connolly RJ, Dinarello CA. Interleukin 1 induces a shock-like state in rabbits. Synergism with tumor necrosis factor and the effect of cyclooxygenase inhibition. J Clin Invest. (1988) 81:1162–72. doi: 10.1172/JCI113431
13. Lakhani SA, Bogue CW. Toll-like receptor signaling in sepsis. Curr Opin Pediatr. (2003) 15:278–82. doi: 10.1097/00008480-200306000-00009
14. Wenceslau CF, McCarthy CG, Goulopoulou S, Szasz T, NeSmith EG. Mitochondrial-derived N-formyl peptides: novel links between trauma, vascular collapse and sepsis. Med Hypotheses. (2013) 81:532–5. doi: 10.1016/j.mehy.2013.06.026
15. Simmons JD, Lee YL, Mulekar S, Kuck JL, Brevard SB, Gonzalez RP, et al. Elevated levels of plasma mitochondrial DNA DAMPs are linked to clinical outcome in severely injured human subjects. Ann Surg. (2013) 258:591–6. discussion: 596–8. doi: 10.1097/SLA.0b013e3182a4ea46
16. Krychtiuk KA, Ruhittel S, Hohensinner PJ, Koller L, Kaun C, Lenz M, et al. Mitochondrial DNA and Toll-like receptor-9 are associated with mortality in critically Ill patients. Crit Care Med. (2015) 43:2633–41. doi: 10.1097/CCM.0000000000001311
17. Wenceslau CF, Szasz T, McCarthy CG, Baban B, NeSmith E, Webb RC. Mitochondrial N-formyl peptides cause airway contraction and lung neutrophil infiltration via formyl peptide receptor activation. Pulm Pharmacol Ther. (2016) 37:49–56. doi: 10.1016/j.pupt.2016.02.005
18. Wenceslau CF, McCarthy CG, Szasz T, Goulopoulou S, Webb RC. Mitochondrial N-formyl peptides induce cardiovascular collapse and sepsis-like syndrome. Am J Physiol Heart Circ Physiol. (2015) 308:H768–77. doi: 10.1152/ajpheart.00779.2014
19. Sun S, Sursal T, Adibnia Y, Zhao C, Zheng Y, Li H, et al. Mitochondrial DAMPs increase endothelial permeability through neutrophil dependent and independent pathways. PLoS ONE. (2013) 8:e59989. doi: 10.1371/journal.pone.0059989
20. Aswani A, Manson J, Itagaki K, Chiazza F, Collino M, Wupeng WL, et al. Scavenging circulating mitochondrial DNA as a potential therapeutic option for multiple organ dysfunction in trauma hemorrhage. Front Immunol. (2018) 9:891. doi: 10.3389/fimmu.2018.00891
21. Fang H, Jiang W, Cheng J, Lu Y, Liu A, Kan L, et al. Balancing innate immunity and inflammatory state via modulation of neutrophil function: a novel strategy to fight sepsis. J Immunol Res. (2015) 2015:187048. doi: 10.1155/2015/187048
22. Ince C, Mayeux PR, Nguyen T, Gomez H, Kellum JA, Ospina-Tascon GA, et al. The endothelium in sepsis. Shock. (2016) 45:259–70. doi: 10.1097/SHK.0000000000000473
23. Peters K, Unger R, Brunner J, Kirkpatrick CJ. Molecular basis of endothelial dysfunction in sepsis. Cardiovasc Res. (2003) 60:49–57. doi: 10.1016/S0008-6363(03)00397-3
24. Rice TW, Wheeler AP, Bernard GR, Vincent JL, Angus DC, Aikawa N, et al. A randomized, double-blind, placebo-controlled trial of TAK-242 for the treatment of severe sepsis. Crit Care Med. (2010) 38:1685–94. doi: 10.1097/CCM.0b013e3181e7c5c9
25. Opal SM, Laterre PF, Francois B, LaRosa SP, Angus DC, Mira JP, et al. Effect of eritoran, an antagonist of MD2-TLR4, on mortality in patients with severe sepsis: the ACCESS randomized trial. JAMA. (2013) 309:1154–62. doi: 10.1001/jama.2013.2194
26. Laupland KB, Kirkpatrick AW, Delaney A. Polyclonal intravenous immunoglobulin for the treatment of severe sepsis and septic shock in critically ill adults: a systematic review and meta-analysis. Crit Care Med. (2007) 35:2686–92. doi: 10.1097/00003246-200712000-00002
27. Werdan K, Pilz G, Bujdoso O, Fraunberger P, Neeser G, Schmieder RE, et al. Score-based immunoglobulin G therapy of patients with sepsis: the SBITS study. Crit Care Med. (2007) 35:2693–701. doi: 10.1097/00003246-200712000-00003
28. Alejandria MM, Lansang MAD, Dans LF, Mantaring JB III. Intravenous immunoglobulin for treating sepsis, severe sepsis and septic shock. Cochrane Database Syst Rev. (2013) CD001090. doi: 10.1002/14651858.CD001090.pub2
29. Fisher CJ Jr., Dhainaut JF, Opal SM, Pribble JP, Balk RA, Slotman GJ, et al. Recombinant human interleukin 1 receptor antagonist in the treatment of patients with sepsis syndrome. Results from a randomized, double-blind, placebo-controlled trial. Phase III rhIL-1ra Sepsis Syndrome Study Group. JAMA. (1994) 271:1836–43. doi: 10.1001/jama.1994.03510470040032
30. Opal SM, Fisher CJ Jr., Dhainaut JF, Vincent JL, Brase R, Lowry SF, et al. Confirmatory interleukin-1 receptor antagonist trial in severe sepsis: a phase III, randomized, double-blind, placebo-controlled, multicenter trial. The Interleukin-1 Receptor Antagonist Sepsis Investigator Group. Crit. Care Med. (1997) 25:1115–24. doi: 10.1097/00003246-199707000-00010
31. Bufe B, Schumann T, Kappl R, Bogeski I, Kummerow C, Podgorska M, et al. Recognition of bacterial signal peptides by mammalian formyl peptide receptors: a new mechanism for sensing pathogens. J Biol Chem. (2015) 290:7369–87. doi: 10.1074/jbc.M114.626747
32. Schiffmann E, Corcoran BA, Wahl SM. N-formylmethionyl peptides as chemoattractants for leucocytes. Proc Natl Acad Sci USA. (1975) 72:1059–62. doi: 10.1073/pnas.72.3.1059
33. Le Y, Murphy PM, Wang JM. Formyl-peptide receptors revisited. Trends Immunol. (2002) 23:541–8. doi: 10.1016/S1471-4906(02)02316-5
34. Liu M, Chen K, Yoshimura T, Liu Y, Gong W, Wang A, et al. Formylpeptide receptors are critical for rapid neutrophil mobilization in host defense against Listeria monocytogenes. Sci Rep. (2012) 2:786. doi: 10.1038/srep00786
35. McDonald B, Pittman K, Menezes GB, Hirota SA, Slaba I, Waterhouse CC, et al. Intravascular danger signals guide neutrophils to sites of sterile inflammation. Science. (2010) 330:362–6. doi: 10.1126/science.1195491
36. Raoof M, Zhang Q, Itagaki K, Hauser CJ. Mitochondrial peptides are potent immune activators that activate human neutrophils via FPR-1. J Trauma. (2010) 68:1328–32. discussion: 1332–4. doi: 10.1097/TA.0b013e3181dcd28d
37. Schepetkin IA, Khlebnikov AI, Kirpotina LN, Quinn MT. Antagonism of human formyl peptide receptor 1 with natural compounds and their synthetic derivatives. Int Immunopharmacol. (2016) 37:43–58. doi: 10.1016/j.intimp.2015.08.036
38. Yang SC, Hwang TL. The potential impacts of formyl peptide receptor 1 in inflammatory diseases. Front Biosci (Elite Ed). (2016) 8:436–49. doi: 10.2741/e778
39. Gao JL, Murphy PM. Species and subtype variants of the N-formyl peptide chemotactic receptor reveal multiple important functional domains. J Biol Chem. (1993) 268:25395–401.
40. Murphy PM, Tiffany HL, McDermott D, Ahuja SK. Sequence and organization of the human N-formyl peptide receptor-encoding gene. Gene. (1993) 133:285–90. doi: 10.1016/0378-1119(93)90653-K
41. Migeotte I, Communi D, Parmentier M. Formyl peptide receptors: a promiscuous subfamily of G protein-coupled receptors controlling immune responses. Cytokine Growth Factor Rev. (2006) 17:501–19. doi: 10.1016/j.cytogfr.2006.09.009
42. Ye RD, Boulay F, Wang JM, Dahlgren C, Gerard C, Parmentier M, et al. International Union of basic and clinical pharmacology. LXXIII Nomenclature for the formyl peptide receptor (FPR) family. Pharmacol Rev. (2009) 61:119–61. doi: 10.1124/pr.109.001578
43. Stenfeldt AL, Karlsson J, Wenneras C, Bylund J, Fu H, Dahlgren C. The non-steroidal anti-inflammatory drug piroxicam blocks ligand binding to the formyl peptide receptor but not the formyl peptide receptor like 1. Biochem Pharmacol. (2007) 74:1050–6. doi: 10.1016/j.bcp.2007.06.049
44. Oldekamp S, Pscheidl S, Kress E, Soehnlein O, Jansen S, Pufe T, et al. Lack of formyl peptide receptor 1 and 2 leads to more severe inflammation and higher mortality in mice with of pneumococcal meningitis. Immunology. (2014) 143:447–61. doi: 10.1111/imm.12324
45. Kantarci A, Aytan N, Palaska I, Stephens D, Crabtree L, Benincasa C, et al. Combined administration of resolvin E1 and lipoxin A4 resolves inflammation in a murine model of Alzheimer's disease. Exp Neurol. (2018) 300:111–20. doi: 10.1016/j.expneurol.2017.11.005
46. Devosse T, Guillabert A, D'Haene N, Berton A, De Nadai P, Noel S, et al. Formyl peptide receptor-like 2 is expressed and functional in plasmacytoid dendritic cells, tissue-specific macrophage subpopulations, and eosinophils. J Immunol. (2009) 182:4974–84. doi: 10.4049/jimmunol.0803128
47. Bufe B, Schumann T, Zufall F. Formyl peptide receptors from immune and vomeronasal system exhibit distinct agonist properties. J Biol Chem. (2012) 287:33644–55. doi: 10.1074/jbc.M112.375774
48. Wenceslau CF, McCarthy CG, Webb RC. Formyl peptide receptor activation elicits endothelial cell contraction and vascular leakage. Front Immunol. (2016) 7:297. doi: 10.3389/fimmu.2016.00297
49. Wenceslau CF, McCarthy CG, Szasz T, Calmasini FB, Mamenko M, Webb RC. Formyl peptide receptor-1 activation exerts a critical role for the dynamic plasticity of arteries via actin polymerization. Pharmacol Res. (2019) 141:276–90. doi: 10.1016/j.phrs.2019.01.015
50. Huang J, Chen K, Huang J, Gong W, Dunlop NM, Howard OM, et al. Regulation of the leucocyte chemoattractant receptor FPR in glioblastoma cells by cell differentiation. Carcinogenesis. (2009) 30:348–55. doi: 10.1093/carcin/bgn266
51. Cheng TY, Wu MS, Lin JT, Lin MT, Shun CT, Hua KT, et al. Formyl peptide receptor 1 expression is associated with tumor progression and survival in gastric cancer. Anticancer Res. (2014) 34:2223–9.
52. Prossnitz ER, Ye RD. The N-formyl peptide receptor: a model for the study of chemoattractant receptor structure and function. Pharmacol Ther. (1997) 74:73–102. doi: 10.1016/S0163-7258(96)00203-3
53. Dorward DA, Lucas CD, Chapman GB, Haslett C, Dhaliwal K, Rossi AG. The role of formylated peptides and formyl peptide receptor 1 in governing neutrophil function during acute inflammation. Am J Pathol. (2015) 185:1172–84. doi: 10.1016/j.ajpath.2015.01.020
54. Loor F, Tiberghien F, Wenandy T, Didier A, Traber R. Cyclosporins: structure-activity relationships for the inhibition of the human FPR1 formylpeptide receptor. J Med Chem. (2002) 45:4613–28. doi: 10.1021/jm010987v
55. Stenfeldt AL, Karlsson J, Wenneras C, Bylund J, Fu H, Dahlgren C. Cyclosporin H, Boc-MLF and Boc-FLFLF are antagonists that preferentially inhibit activity triggered through the formyl peptide receptor. Inflammation. (2007) 30:224–9. doi: 10.1007/s10753-007-9040-4
56. Gao JL, Lee EJ, Murphy PM. Impaired antibacterial host defense in mice lacking the N-formylpeptide receptor. J Exp Med. (1999) 189:657–62. doi: 10.1084/jem.189.4.657
57. Dahlgren C, Gabl M, Holdfeldt A, Winther M, Forsman H. Basic characteristics of the neutrophil receptors that recognize formylated peptides, a danger-associated molecular pattern generated by bacteria and mitochondria. Biochem Pharmacol. (2016) 114:22–39. doi: 10.1016/j.bcp.2016.04.014
58. Korff S, Loughran P, Cai C, Lee YS, Scott M, Billiar TR. Eritoran attenuates tissue damage and inflammation in hemorrhagic shock/trauma. J Surg Res. (2013) 184:e17–25. doi: 10.1016/j.jss.2013.03.023
59. Glogauer M, Hartwig J, Stossel T. Two pathways through Cdc42 couple the N-formyl receptor to actin nucleation in permeabilized human neutrophils. J Cell Biol. (2000) 150:785–96. doi: 10.1083/jcb.150.4.785
60. Makino A, Prossnitz ER, Bunemann M, Wang JM, Yao W, Schmid-Schonbein GW. G protein-coupled receptors serve as mechanosensors for fluid shear stress in neutrophils. Am J Physiol Cell Physiol. (2006) 290:C1633–9. doi: 10.1152/ajpcell.00576.2005
61. Heo SC, Kwon YW, Jang IH, Jeong GO, Yoon JW, Kim CD, et al. WKYMVm-induced activation of formyl peptide receptor 2 stimulates ischemic neovasculogenesis by promoting homing of endothelial colony-forming cells. Stem Cells. (2014) 32:779–90. doi: 10.1002/stem.1578
62. Wenceslau CF, McCarthy CG, Szasz T, Spitler K, Goulopoulou S, Webb RC, et al. Mitochondrial damage-associated molecular patterns and vascular function. Eur Heart J. (2014) 35:1172–7. doi: 10.1093/eurheartj/ehu047
63. Legrand M, Klijn E, Payen D, Ince C. The response of the host microcirculation to bacterial sepsis: does the pathogen matter? J Mol Med. (2010) 88:127–33. doi: 10.1007/s00109-009-0585-6
64. Hickey MJ, Kwan RY, Awad MM, Kennedy CL, Young LF, Hall P, et al. Molecular and cellular basis of microvascular perfusion deficits induced by Clostridium perfringens and Clostridium septicum. PLoS Pathog. (2008) 4:e1000045. doi: 10.1371/journal.ppat.1000045
65. Piccinini AM, Midwood KS. DAMPening inflammation by modulating TLR signaling. Mediators Inflamm. (2010) 2010:672395. doi: 10.1155/2010/672395
66. Yang XM, Cui L, White J, Kuck J, Ruchko MV, Wilson GL, et al. Mitochondrially targeted Endonuclease III has a powerful anti-infarct effect in an in vivo rat model of myocardial ischemia/reperfusion. Basic Res Cardiol. (2015) 110:3. doi: 10.1007/s00395-014-0459-0
67. Love JD, Hewitt RR. The relationship between human serum and human pancreatic DNase I. J Biol Chem. (1979) 254:12588–94.
68. Puyo CA, Peruzzi D, Earhart A, Roller E, Karanikolas M, Kollef MH, et al. Endotracheal tube-induced sore throat pain and inflammation is coupled to the release of mitochondrial DNA. Mol Pain. (2017) 13:1744806917731696. doi: 10.1177/1744806917731696
69. Meng W, Paunel-Gorgulu A, Flohe S, Witte I, Schadel-Hopfner M, Windolf J, et al. Deoxyribonuclease is a potential counter regulator of aberrant neutrophil extracellular traps formation after major trauma. Mediators Inflamm. (2012) 2012:149560. doi: 10.1155/2012/149560
70. Sangshetti JN, Khan FA, Shinde DB. Peptide deformylase: a new target in antibacterial, antimalarial and anticancer drug discovery. Curr Med Chem. (2015) 22:214–36. doi: 10.2174/0929867321666140826115734
71. Lucchini G, Bianchetti R. Initiation of protein synthesis in isolated mitochondria and chloroplasts. Biochim Biophys Acta. (1980) 608:54–61. doi: 10.1016/0005-2787(80)90133-1
72. Olaleye OA, Bishai WR, Liu JO. Targeting the role of N-terminal methionine processing enzymes in Mycobacterium tuberculosis. Tuberculosis (Edinb). (2009) 89(Suppl 1):S55–9. doi: 10.1016/S1472-9792(09)70013-7
73. Mazel D, Pochet S, Marliere P. Genetic characterization of polypeptide deformylase, a distinctive enzyme of eubacterial translation. EMBO J. (1994) 13:914–23. doi: 10.1002/j.1460-2075.1994.tb06335.x
74. Leeds JA, Dean CR. Peptide deformylase as an antibacterial target: a critical assessment. Curr Opin Pharmacol. (2006) 6:445–52. doi: 10.1016/j.coph.2006.06.003
75. Pichota A, Duraiswamy J, Yin Z, Keller TH, Alam J, Liung S, et al. Peptide deformylase inhibitors of Mycobacterium tuberculosis: synthesis, structural investigations, and biological results. Bioorg Med Chem Lett. (2008) 18:6568–72. doi: 10.1016/j.bmcl.2008.10.040
76. Guilloteau JP, Mathieu M, Giglione C, Blanc V, Dupuy A, Chevrier M, et al. The crystal structures of four peptide deformylases bound to the antibiotic actinonin reveal two distinct types: a platform for the structure-based design of antibacterial agents. J Mol Biol. (2002) 320:951–62. doi: 10.1016/S0022-2836(02)00549-1
77. Giglione C, Pierre M, Meinnel T. Peptide deformylase as a target for new generation, broad spectrum antimicrobial agents. Mol Microbiol. (2000) 36:1197–205. doi: 10.1046/j.1365-2958.2000.01908.x
78. Ramanathan-Girish S, McColm J, Clements JM, Taupin P, Barrowcliffe S, Hevizi J, et al. Pharmacokinetics in animals and humans of a first-in-class peptide deformylase inhibitor. Antimicrob Agents Chemother. (2004) 48:4835–42. doi: 10.1128/AAC.48.12.4835-4842.2004
79. Rolan P, Sun H, Macleod C, Bracken K, Evans TG. Pharmacokinetics and unexpected safety issues of LBM415, a novel oral peptide deformylase inhibitor. Clin Pharmacol Ther. (2011) 90:256–62. doi: 10.1038/clpt.2011.101
80. Escobar-Alvarez S, Gardner J, Sheth A, Manfredi G, Yang G, Ouerfelli O, et al. Inhibition of human peptide deformylase disrupts mitochondrial function. Mol Cell Biol. (2010) 30:5099–109. doi: 10.1128/MCB.00469-10
Keywords: sepsis, SIRS, trauma, DAMPs, formyl peptide receptor-1, deformylase, endothelium
Citation: Martinez-Quinones P, Komic A, McCarthy CG, Webb RC and Wenceslau CF (2019) Targeting Endothelial Barrier Dysfunction Caused by Circulating Bacterial and Mitochondrial N-Formyl Peptides With Deformylase. Front. Immunol. 10:1270. doi: 10.3389/fimmu.2019.01270
Received: 05 October 2018; Accepted: 17 May 2019;
Published: 06 June 2019.
Edited by:
Julien Pottecher, Hôpitaux Universitaires de Strasbourg, FranceReviewed by:
Cheng Xue Helena Qin, Baker Heart and Diabetes Institute, AustraliaCopyright © 2019 Martinez-Quinones, Komic, McCarthy, Webb and Wenceslau. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Camilla Ferreira Wenceslau, Q2FtaWxsYS5XZW5jZXNsYXVAdXRvbGVkby5lZHU=; Patricia Martinez-Quinones, cG1hcnRpbmV6cXVpbm9uQGF1Z3VzdGEuZWR1
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.