
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol. , 04 June 2019
Sec. Alloimmunity and Transplantation
Volume 10 - 2019 | https://doi.org/10.3389/fimmu.2019.01228
This article is part of the Research Topic Safety, Efficacy and Mechanisms of Action of Mesenchymal Stem Cell Therapies View all 22 articles
 Daniel J. Weiss1Karen English2
Daniel J. Weiss1Karen English2 Anna Krasnodembskaya3
Anna Krasnodembskaya3 Johana M. Isaza-Correa4
Johana M. Isaza-Correa4 Ian J. Hawthorne2
Ian J. Hawthorne2 Bernard P. Mahon4*
Bernard P. Mahon4*Rapid progress is occurring in understanding the mechanisms underlying mesenchymal stromal cell (MSC)-based cell therapies (MSCT). However, the results of clinical trials, while demonstrating safety, have been varied in regard to efficacy. Recent data from different groups have shown profound and significant influences of the host inflammatory environment on MSCs delivered systemically or through organ-specific routes, for example intratracheal, with subsequent actions on potential MSC efficacies. Intriguingly in some models, it appears that dead or dying cells or subcellular particles derived from them, may contribute to therapeutic efficacy, at least in some circumstances. Thus, the broad cellular changes that accompany MSC death, autophagy, pre-apoptotic function, or indeed the host response to these processes may be essential to therapeutic efficacy. In this review, we summarize the existing literature concerning the necrobiology of MSCs and the available evidence that MSCs undergo autophagy, apoptosis, transfer mitochondria, or release subcellular particles with effector function in pathologic or inflammatory in vivo environments. Advances in understanding the role of immune effector cells in cell therapy, especially macrophages, suggest that the reprogramming of immunity associated with MSCT has a weighty influence on therapeutic efficacy. If correct, these data suggest novel approaches to enhancing the beneficial actions of MSCs that will vary with the inflammatory nature of different disease targets and may influence the choice between autologous or allogeneic or even xenogeneic cells as therapeutics.
The efficacy of MSC administration in preclinical inflammatory models is well-documented regardless of the source of MSCs (bone marrow, adipose, placenta, other). The basic biology of MSCs, their mode of action and therapeutic efficacy in clinical studies have been reviewed extensively elsewhere (1–3). However, translation of preclinical efficacy to the clinical setting is proving difficult. A possible reason for this is a lack of understanding of the fate of MSCs when they encounter highly inflammatory microenvironments. Within this inflammatory milieu, MSCs are exposed to insults such as hypoxia and pro-inflammatory cytokines (4). What happens to MSCs during the transient period in which they are at the target site is largely unknown (5). The longstanding working hypothesis has been that viable functional MSCs are critical for efficacy. However, a number of recent studies have suggested that MSC survival in the disease milieu may not be as important as once thought. These studies elegantly demonstrate that apoptotic or dead MSCs can facilitate protection mediated by MSC administration in inflammatory microenvironments in vivo (6–8). However, these studies have opened up a number of questions about the processes involved in the transition from live to dead MSCs. Under what circumstances can dead MSCs substitute for viable cells? What are the limits to use? Can the pre-apoptotic cargo of extracellular vesicles (EVs) produced by MSCs or mitochondria transferred from MSCs to other cells substitute for the MSCs themselves? Is there a role for autophagy or for efferocytosis in MSCT efficacy? Does autophagy influence the soluble factors secreted by MSCs before they die? If we can better understand the fate of MSCs within the diseased microenvironment, perhaps this knowledge would lend itself to development of more optimal MSC-based cell therapies (be that live, autophagic or dead/apoptotic MSCs) and reduce the disparity between pre-clinical models and the clinical setting.
The term “necrobiology” has been used to describe the cellular processes associated with morphological, biochemical, and molecular changes which predispose, precede, and accompany cell death, as well as the consequences and tissue response to cell death (9). The observation that MSC viability and efficacy are not necessarily correlated (6, 7, 10) suggests that the necrobiology of MSCT will be a fruitful and essential area for future study. In this review we focus on key biological processes likely to affect therapeutic efficacy (Figure 1), summarize what is known about the questions above, and for the first time attempt to frame these disparate aspects of research within the concept of necrobiology or the biology of the dying therapeutic cell.
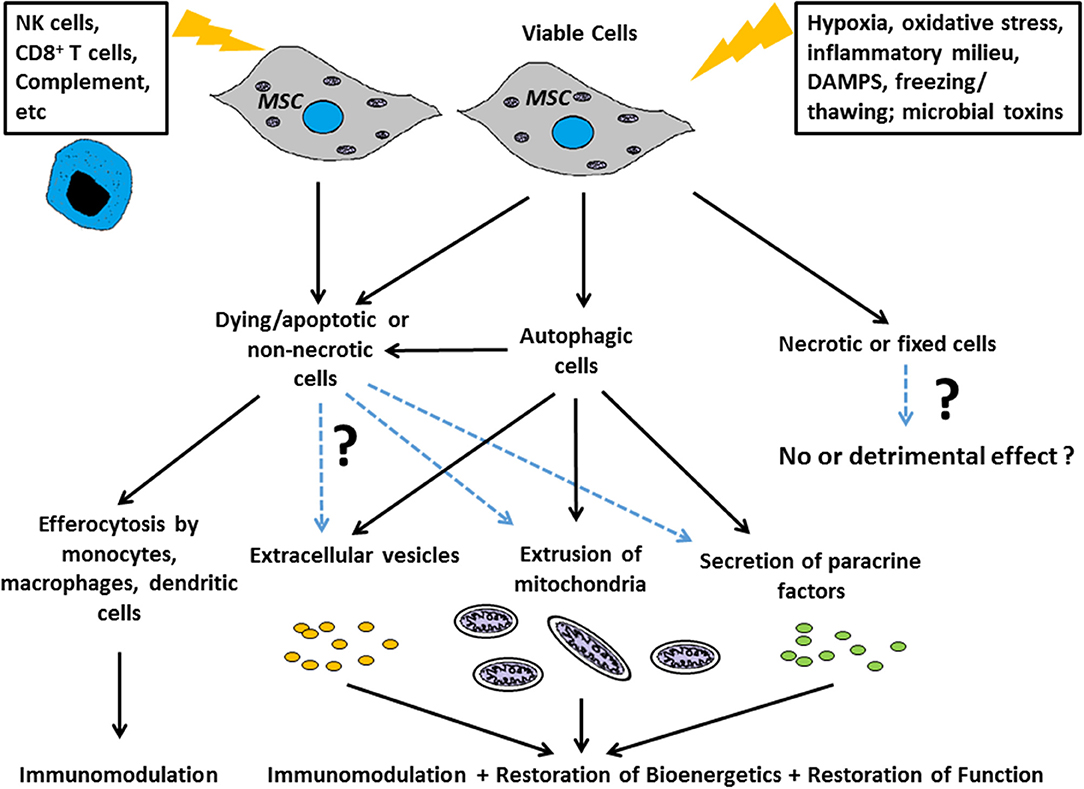
Figure 1. Scheme for how the necrobiology of MSCs influences therapeutic efficacy Putative mechanisms include: as live cells through paracrine mechanisms, and through the cellular processes associated with morphological, biochemical, and molecular changes which predispose, precede, and accompany cell death. These necrobiotic processes include the response to dying and non-necrotic MSCs, the alteration of MSC biology by autophagy, and the delivery of MSC derived mitochondria or EVs to target cells and tissues.
There is relatively little data available in pre-clinical disease models in which apoptotic or dead MSCs were investigated, either as part of a direct investigation of dead/apoptotic cell actions or as part of a control group for live MSC administrations. Using pre-clinical models of respiratory diseases/critical illnesses in mice as representative examples (Table 1), intratracheal administration of apoptotic MSCs in models of acute lung injury or systemic administration of either fixed or heat-killed MSCs in mouse models of asthma and sepsis, respectively, did not mimic the effects of live MSC administration (11–14). Likewise the administration of other cells such as fixed fibroblasts were not beneficial, suggesting a role for MSCs that cannot be replaced by other dead cell types (11, 13). Notably, most of these studies are relatively old and did not exhaustively explore the effects of dead or apoptotic cells on immune or inflammatory cells. Whether this is a phenomenon unique to MSCs is unknown at present as there are few examples of administering other types of cells to the lung that might influence inflammatory or immune pathways. However, there are well documented anti-inflammatory bystander effects when other apoptotic cells are engulfed by macrophages and these have been recently reviewed (15). The extent to which this phenomenon is specific to lung diseases is relatively unexplored and a ripe area for further research.
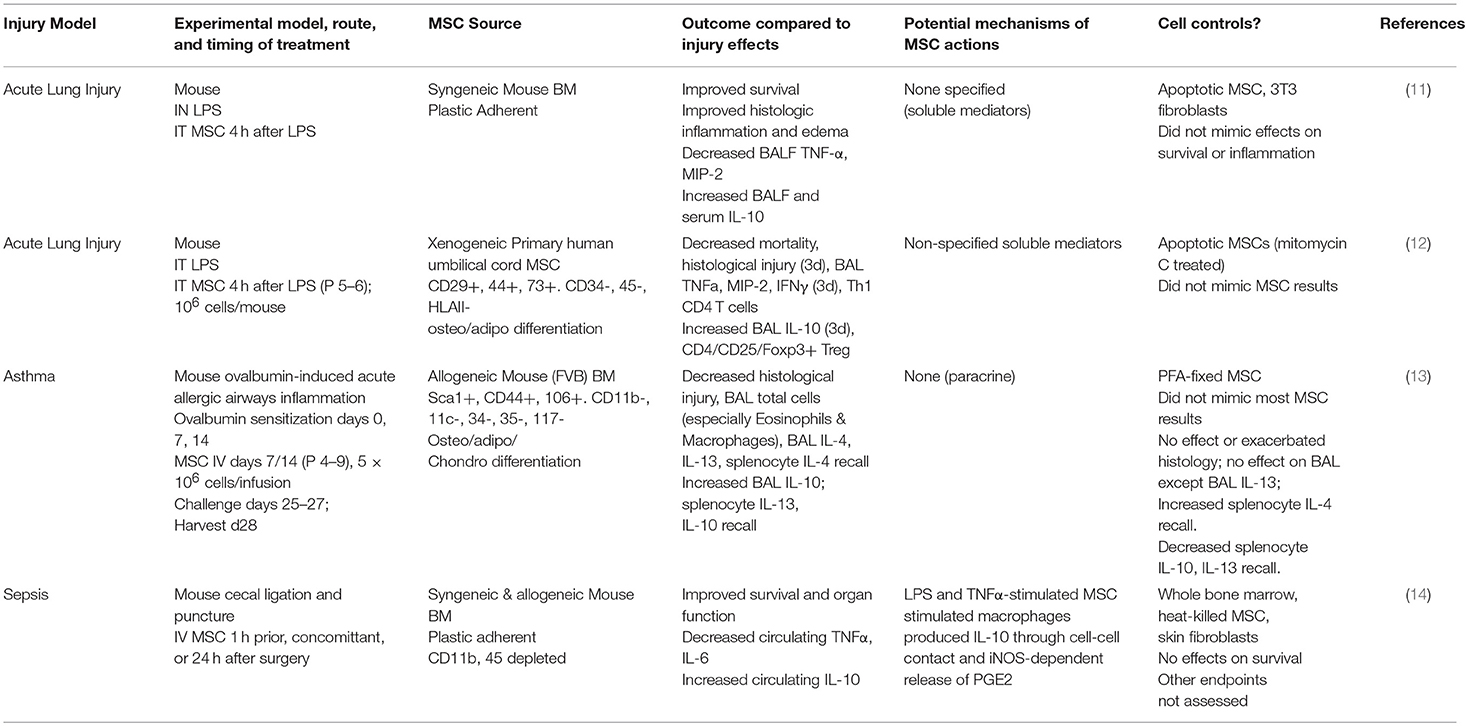
Table 1. Pre-clinical lung injury studies utilizing dead or apoptotic MSCs.
In contrast, more recent studies in pre-clinical models of acute lung injury have suggested that the inflammatory environment in the lung can affect survival and subsequent efficacy of intratracheally-administered MSCs in part through activation of TRL4 signaling pathways (16). MSCs have variable effects in different mouse models of lung injuries with efficacy potentially related to the proteome profile of the BAL fluid in each respective injury (17). Another recent study demonstrated that apoptotic MSCs reduced some inflammatory endpoints in a mouse model of Th2-mediated allergic airway inflammation (7). These effects are not confined to lung disease models, a series of related studies in a rat model of cecal ligation and puncture-induced sepsis demonstrated that administration of rat adipose-derived MSCs, rendered apoptotic by 96 h culture in serum-free media, were more effective than healthy MSCs in improving survival and decreasing lung, kidney and cardiac injuries (18–21) administration of the apoptotic MSCs decreased a number of circulating and organ-specific pro-inflammatory, pro-apoptotic, and oxidative stress markers while increasing anti-apoptotic and anti-oxidant responses. The suggested mechanism(s) were that the apoptotic MSCs were more effective at dampening immune responses to the original injury, however, no specific pathways were delineated. These results suggest a more complex interaction of MSC apoptosis on efficacy in different inflammatory environments such that the inflammatory environment itself directs MSC apoptosis. Unfortunately, other more recent studies of MSC effects in a wide range of pre-clinical lung injury models have generally not included dead or apoptotic cells and thus there is opportunity for more extensive investigation (22–25).
Surprisingly, little is known about how MSCs are killed in different settings. In vitro studies have demonstrated the conditions for NK cell killing of MSCs (26) and this is likely to be an important mechanism for induction of MSC death in vivo, although few studies have examined this in detail. Similarly a role for Complement mediated killing has been proposed (27, 28). Recently, a requirement for cytotoxic CD8+ T cell mediated killing (via apoptotic death) of MSCs has been shown in GvHD (7). However, the mechanisms of MSC killing (e.g., immune-mediated or as a result of exposure to microbial toxins) are likely to influence the type of death induced and the biological consequence. This might be an especially important consideration in designing cell therapeutics for lung diseases or patient subsets where there is a pathogenic microbial burden (e.g., Cystic Fibrosis).
There are even less available data on the effect of apoptotic MSCs in clinical investigations. In a notable recent example, safety but no efficacy was observed in a multi-center double-blinded randomized trial of systemic bone marrow-derived MSCs in patients with ARDS (29). In post-hoc analyses, the unanticipated finding was that up to 85% of the MSCs were non-viable at the time of administration. This suggests that dead MSCs may not have clinical efficacy in ARDS although there are a number of other factors to consider including timing, dose, and route of MSC administration (30). In contrast, a preliminary report of a parallel trial of bone marrow-derived MSC-like cells in ARDS patients demonstrated efficacy in major endpoints of survival, ventilator-free days and ICU stay (31). Notably, the cells utilized were fully viable at the time of administration. Therefore, viable/ live MSCs are not interchangeable therapeutically with apoptotic/dead MSC, but each have potential efficacy in different contexts and presumably by different mechanisms. In combination with the growing experience of dead/apoptotic MSCs in pre-clinical models, these clinical observations raise important and hypothesis-generating mechanistic ideas for further study.
It is now known that non-necrotic cell death can be induced by diverse mechanisms and many of these are linked to the cellular processes that eliminate damaged proteins and organelles, termed autophagy (32, 33). Autophagy is a tightly regulated, complex cascade that controls the efficient delivery and fusion of damaged organelles to the autophagosome (34). Whilst this process supports cell survival and regular cell functioning, it is also associated with at least three forms of cell death- apoptosis, necroptosis, and autosis. Necroptosis is an inflammatory, caspase-independent form of cell death (33) whereas autosis is mediated by the Na+ K+- ATPase pump and is autophagy-gene dependent (35, 36). More broadly, autophagy is activated by microenvironmental and intracellular signals linked to ER stress, hypoxia and immune cell activation (37–39). These signals, related to tissue damage, include damage associated molecular patterns (DAMPs) and MSCs have been shown to sense DAMPs released from dying/stressed cells (40) leading to enhanced pro-reparative and anti-inflammatory effects (40). Thus, the reparative effects of MSCs may be primed or altered by exposure to DAMPs or other stress signals that alter their interactions with other cells. During cell therapy, MSCs become exposed to such signals and autophagy is a common cellular response to such stress. Autophagy influences MSCs' therapeutic effects in at least two contrasting ways- to promote survival of the MSCs, or to induce MSC death through apoptosis, necroptosis, or autosis. The fate of the MSC is thus likely to be dependent on quantitative differences in exposure time to inflammation. Understanding the role of autophagy in MSCs at sites of inflammation could therefore inform therapeutic protocol design in the future.
The role played by autophagy as a survival mechanism to relieve stress and prevent apoptosis has been extensively studied (41). Under starvation conditions (serum deprivation, hypoxia, oxygen/glucose deprivation) or in the presence of reactive oxygen species (ROS), autophagy has been shown to promote MSC survival in vitro (42–44). Importantly, while sufficient levels of ROS are required to activate autophagy, excessive ROS may lead to cell death (45). This has been demonstrated where preconditioning of MSCs to serum deprivation and hypoxic conditions have prolonged survival in ischemic microenvironments through the activation of autophagic processes (46). Moreover, mitophagy in MSCs facilitates interaction with macrophages in conditions of oxidative stress whilst also preventing apoptosis (47). A number of extrinsic factors that modulate autophagy in MSCs have been identified, for example Stromal Cell Derived Factor-1β can promote MSC survival through enhanced autophagy (48). Expression of hypoxia-inducible factor 1α also protects against Oxygen-Glucose deprivation via induction of autophagy and the PI3K/AKT/mTOR signaling pathway (43), while over-expression of CPT1C in human MSCs enhances survival via an increase in autophagic flux (49). In aging mice, knockdown of insulin-like growth factor-1 enhances survival of MSCs through autophagy and prolongs MSC survival in vivo (50). Taken together these studies clearly show that at least in some circumstances autophagy promotes MSC survival under stress.
In addition to factors influencing the autophagic pathway in MSCs, autophagy may also lead to the production of soluble factors important for MSC's therapeutic efficacy. Vascular endothelial growth factor (VEGF) plays a key role in MSCs promotion of wound healing (51, 52), a recent study has identified that increased VEGF secretion from autophagic MSCs promoted vascularization in cutaneous wounds and improved healing (52). The induction of autophagy in MSCs may also alter their immunomodulatory function. Autophagic human bone marrow-derived MSCs can regulate CD4+ T helper cell proliferation via TGF-β1 signaling (53). Activation of autophagy by rapamycin in a co-culture system enhanced MSC's ability to suppress CD4+ T helper cell proliferation, whilst 3-methyladenine (3-MA), an autophagic inhibitor, reduced it. These data indicate a role for autophagy in MSCs' immunomodulatory functions of the adaptive immune response, and therefore suggest that the autophagic status of the MSCs will influence therapeutic efficacy under inflammatory conditions. However, the precise limits of this effect are unknown and there are clearly redundant and parallel mechanisms operating. For example, Chinnadurai et al. showed that while interferon gamma (IFN-γ) stimulation of MSCs upregulated the expression of autophagy genes, inhibition of autophagy via 3-MA did not affect MSCs' immunomodulatory potential (54). Furthermore, in some studies, autophagy was shown to have adverse effects on MSCs' immunomodulatory capacity. When rodent MSCs were stimulated with tumor necrosis factor (TNF) and IFN-γ, autophagy reduced MSCs' immunomodulatory effects whereas inhibition through the knockdown of Bcn1 was beneficial (55). It is difficult to compare studies that inhibit autophagy when diverse inducers and inhibitors have been used, or when different inhibitor concentrations and time points have been studied. Nevertheless, these differences are important, autophagy and indeed apoptosis are time dependent processes, and it is reasonable to assume that the activity and function of the MSCs transitioning through these processes will vary with each disease and over time. The implication for developing therapies is that future preclinical approaches will have to account more comprehensively for temporal and dose effects to be informative, but such information could well-shorten therapeutic development times if it leads to improved understanding of delivery route and dosage.
Autophagy can alter biological function following starvation or inflammation (56), and in contrast to the above, can promote autophagic cell death or autosis rather than survival. This switch in roles for autophagy is thought to be dependent on the strength of the signals present, time of treatment and availability of ATP (57). At present our understanding of the type of death induced by autophagy, tends to reflect the greater understanding of apoptosis compared to necroptosis and autosis (58). Nevertheless, autophagy-induced apoptosis has been reported as an alternative fate of MSCs exposed to an inflammatory microenvironment (59). Dang et al. demonstrated that autophagy may cause cell death in a sepsis model of inflammation. These data suggest that the cytokine cocktail presented to the MSCs from the microenvironment causes autophagy to trigger death instead of promoting cell survival. This was mediated via the interaction with the ROS/ERK pathway resulting in the downregulation of Bcl-2. Inhibition of autophagy in MSCs led to increased production of prostaglandin E2 (55) and enhanced immunoregulation in pre-clinical models of EAE (55) and sepsis (59). Dang et al. (55) also reported that the induction of apoptosis reduced the therapeutic effect of MSCs, however, it has recently been demonstrated in a GvHD model that apoptotic MSCs are still immunosuppressive (7). Galleu et al. recorded that apoptotic MSCs (apoMSC) could reduce effector cell number in the lung and spleen of GvHD mice (7). Importantly, phagocytes producing indolamine 2,3-dioxygenase were required for the protection associated with apoMSC when administered intraperitoneally but not intravenously (60). These and other studies from the Hoogduijn group (5, 6) suggest that the therapeutic effects of both live and apoMSC are dependent on interactions with specific phagocytic cell populations. These observations also highlight the important interaction between MSCs and macrophages and the contribution of innate immune modulation to therapeutic efficacy (61). Given the contrasting data surrounding the effects on efficacy of autophagy in MSCs it is clear that further study is needed, especially of dose and temporal responses. Nevertheless, it is possible to state that the inflammatory environment plays an important role in the MSC fate of survival or death, that autophagic processes are involved in this fate decision, and that subsequent interaction of MSCs with innate cells such as monocytes/macrophages influence therapeutic efficacy. From the above studies, it seems likely that whereas pro-survival processes are likely to be linked in part to therapeutic effects through MSCs' production of paracrine factors (e.g., VEGF, etc.), necrobiological-related efficacy is more likely to operate through the interaction between MSCs and the innate immune cells such as monocytes/macrophages (6).
Cell death, oxidative stress, and autophagy are all linked to mitochondrial function (62), so it understandable that the mitochondrion has a role in MSC efficacy. More surprising have been the now well-documented reports that reprogramming of host cells by MSCs is significantly mediated by their ability to transfer functionally active mitochondria to somatic recipient cells. Mitochondrial transfer has been found to play a critical role in therapeutic effect of MSCs in the pre-clinical models of multiple diseases including brain injury, cardiac myopathies, muscle sepsis, and acute (ARDS) (63, 64) and chronic respiratory disorders (asthma and COPD) (65, 66). Mitochondria are transferred between cells via tunneling nanotubules (TNTs), cell fusion, and can also be contained in secreted extracellular vesicles (EV) (67). These mitochondria are functionally active and their transfer results in the enhancement of oxidative phosphorylation coupled with alleviation of oxidative stress in recipient cells leading to restoration of impaired functional activity (e.g., surfactant secretion, phagocytosis and wound healing) and cytoprotective effects. The consequences and mechanisms of mitochondrial transfer have been comprehensively reviewed previously (67–69). As mitochondrial dysfunction contributes to pathophysiology of various diseases, strategies aiming to protect mitochondria from injury or to increase biogenesis are being increasingly explored as promising therapeutic opportunities. Replacement of damaged mitochondria through donation from MSCs is a faster and physiologically more economical route for the recipient cell at the site of injury as compared to the mitochondrial biogenesis and therefore, appears to be an efficient means for disease attenuation (70).
In addition to protective effects due to improved bioenergetics, there is evidence of the involvement of mitochondrial transfer in cellular rejuvenation and transcriptional reprogramming (71). Studies by Acquistapace et al. demonstrated a key beneficial role of MSC mitochondria for reprogramming of post-mitotic murine cardiomyocytes toward proliferating cardiac progenitor-like cells through spontaneous cell fusion (72).
Although the precise mechanisms regulating mitochondrial extrusion from MSCs as well as their uptake by recipient cells remain to be investigated, it is clear that the injury microenvironment will have an impact on the rate and efficiency of this process. Thus, we have recently demonstrated that hypercapnia, a condition often associated with low tidal volume ventilation in ARDS, induces mitochondrial dysfunction and although the rate of mitochondrial transfer from MSCs to recipient cells is not changed, these dysfunctional mitochondria are not able to improve recipient cell bioenergetics and promote capacity of the lung epithelial cells to wound closure. This is in good agreement with the finding of Paliwal et al. demonstrating that mitochondria from MSCs with higher mitochondrial respiration capacities are more effective in suppression of mtROS in stressed recipient cells (73). Li et al. have demonstrated that pre-treatment with anti-oxidants such as N-acetyl-L-cysteine and L-ascorbic acid 2-phosphate enhanced mitochondrial transfer from the anti-oxidant treated population of the bone marrow derived MSCs to the untreated population of MSCs injured by H2O2 (74).
A key study by Mahrouf-Yorgov et al. reports that mitochondria released from dying cells at the site of injury are an important environmental cue that controls the cytoprotective function of MSCs and regulates their capacity for mitochondrial transfer (75). It was shown that upon oxidative stress, somatic cells (cardiomyocytes and endothelial cells) release mitochondria which are engulfed and degraded by MSCs, leading to induction of heme oxygenase-1 (HO-1) and stimulation of autophagy and mitochondrial biogenesis. As a result, the ability of MSCs to donate their mitochondria to injured cells to alleviate oxidative stress injury was enhanced (75). Reactive oxygen species and inflammatory cytokines (e.g., TNF-α) have also been postulated to play a role in the regulation of mitochondrial donation (67–69, 76). Our unpublished data suggest that mitochondrial transfer from MSCs to lung epithelial cells is enhanced in inflammatory environments. Taken together these data strongly suggest that mitochondrial transfer and cell death are related and relevant to clinical efficacy.
The processes of MSC mitochondrial transfer and autophagy are intrinsically interdependent. Phinney et al. have demonstrated that MSCs extrude their mitochondria in EVs which express autophagosomal markers, suggesting that this phenomenon is a result of incomplete autophagy (47). Physiologically, MSCs reside in the low oxygen environment of the bone marrow stem cell niche and the authors observed that conventional culture of MSCs in normoxia- (21% oxygen) induced oxidative stress, thereby promoting MSC mitophagy, however instead of degradation, mitochondria were directed outside of the cells (47). Ghanta et al. then demonstrated the importance of autophagy in maintaining healthy mitochondrial function and promoting survival in MSCs during oxidative stress (45).
In the view of accumulating evidence that after in vivo administration, MSCs undergo apoptosis and fragmentation and subsequent elimination by phagocytes (6, 7, 77), it is plausible to hypothesize that mitochondria could be released during fragmentation and taken up by surrounding somatic cells and particularly macrophages. We have previously demonstrated that mitochondrial transfer from healthy MSCs through extracellular vesicles results in macrophage metabolic reprogramming toward M2-like phenotype with enhanced phagocytic activity (63, 64) however whether or not mitochondria released from dying MSCs retain the same properties and exert similar effects remains to be determined.
If there is a cell-specific therapeutic benefit from using MSCs as opposed to any apoptotic cell, then cell-specific characteristics and mechanisms need further exploration. Apoptotic cells maintain some biological activities as they begin the orderly process of disassembly and death. In particular apoptotic cells can produce a range of EVs and apoptotic bodies that can influence their microenvironment (78, 79). There has been an explosion in the literature describing how exosomes and other EVs can act as biological modulators. Healthy, viable MSCs are well-characterized producers of a wide range of EVs with different cargos. These can include microvesicles, including those bearing mitochondria (see above), as well as exosomes (66) that are now recognized as powerful mediators of intercellular communication locally and systemically. Exosomes, and presumably their cargo, can activate or suppress aspects of immunity such as cytokine secretion, immune cell differentiation and polarization, and T cell activation (47, 80–82). In addition, processes such as angiogenesis, proliferation, oncogenesis, and microenvironmental conditioning can all be affected by exosomes. MSC derived exosomes (even in the absence of their viable MSC producer) can thus have detectable therapeutic influences in human systems (Table 2). The influences of exosomes are largely defined by their cargo, which can include cytosolic and membrane proteins, mRNA and non-coding RNA including miRNA (miR), and the nature of the EV cargo of MSCs is influenced by the extracellular environment (Table 3). Several studies have linked treatment with MSC-derived exosomes to improvement in models of liver, kidney, heart, skin, lung and other diseases (90–93). The influence of MSC-derived exosomes on lung injury is especially important to studies of clinical efficacy given that the lung is a major site of MSC accumulation in the early period after delivery (94, 95). MSC-derived exosomes regulate vascular remodeling and reduce hypoxic pulmonary hypertension in rodent models. These exosomes reduced the activation of the hypoxic transcription factor STAT3 and the expression of the miRNA-17 superfamily but restored miRNA-204 in lung (normally reduced in human pulmonary hypertension) (83). In an acute respiratory distress syndrome (ARDS) model, alveolar macrophages treated with MSC-derived CD44+ EVs also reduced lung injury (64). As discussed above, EVs promoted mitochondrial transfer to the macrophages increasing their phagocytic capacity and inducing an anti-inflammatory response. These findings suggest that intravenously delivered MSC therapies that see an accumulation of viable pre-apoptotic MSCs in the lung vasculature, have the potential to produce EV with extensive biological effects.
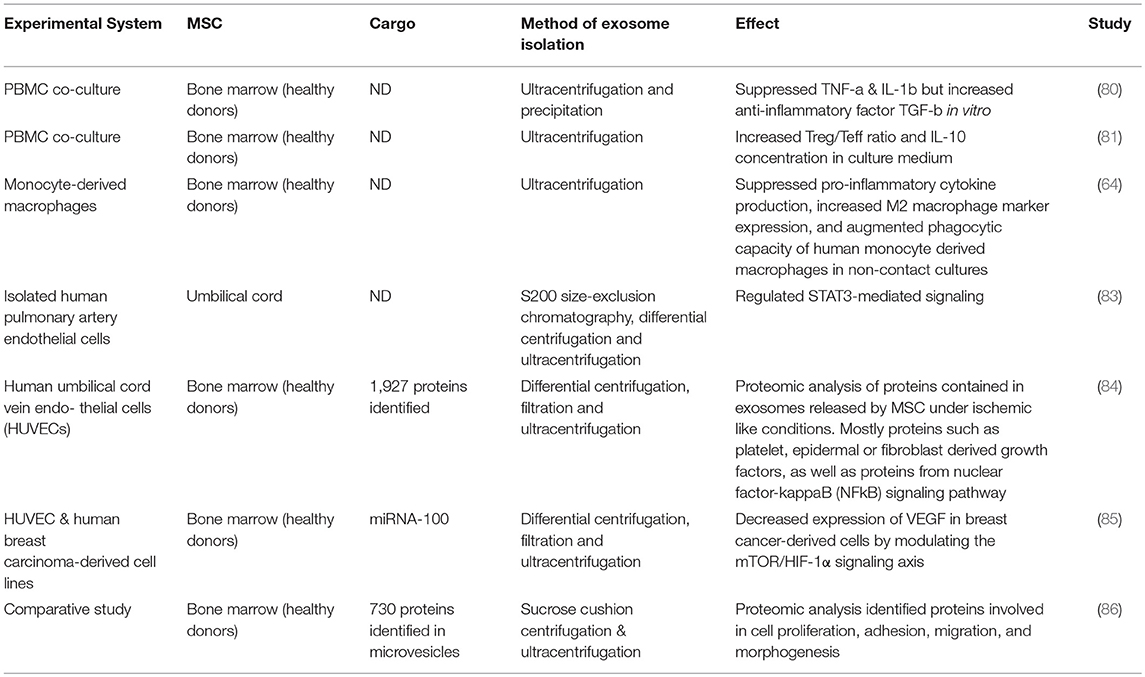
Table 2. Selected studies of MSC-derived exosomes in human models.
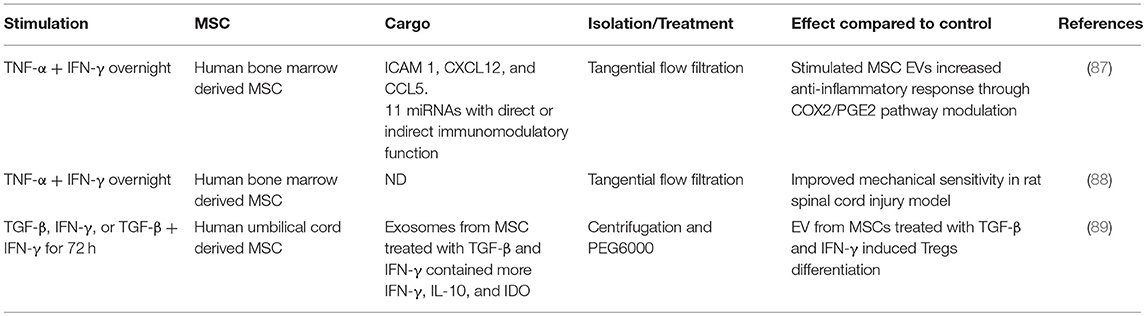
Table 3. Studies showing the influence of inflammatory environment on human MSC-derived exosome cargo.
Perhaps the most profound influence of MSC-derived exosomes and EVs is the regulation of innate immune responses. Phinney et al. showed that MSC-derived exosomes with miRNA cargo inhibited macrophage activation by modulating Toll-like receptor signaling (47, 96). Macrophages treated with MSC-derived exosomes activated NF-kB and changed the expression of 50 of the 84 TLR-associated proteins evaluated, including IL-1β, COX2, IL-10, CCL2, TNF, MyD88, TLR 1,4,5,7,8 and 9, IRAK1, and TRAF6 (47). The breadth of biological processes downstream of these factors is very extensive and hints at the potential scale of effects that might be influenced by exosomes produced in the early, pre-apoptotic, phase of MSC therapy. In adaptive immunity (and hence of relevance to cell therapy for autoimmune disease and transplantation), EVs from bone marrow-derived MSCs increased production of immunosuppressive IL-10 and the proliferation of regulatory T cells in peripheral blood mononuclear cell cultures stimulated with anti-CD3/CD28 beads (81). In this research, treatment with MSC-EVs alone resulted in apoptosis of T cell populations. Interestingly, in other studies, exosomes secreted by HIF-1α-overexpressing donor MSCs were enriched for the Notch ligand Jagged-1 (97). Subcutaneous injection of these exosomes in a Matrigel plug assay induced angiogenesis (97). This builds on earlier work showing that Jagged-1 was an important contact dependent signal by which MSCs induced tolerogenic dendritic cells (DC) (98). Given that DCs have an antigen acquisition sentinel function throughout the body, and that these are key cells in shaping adaptive immunity, exosomal cargos of Notch ligands might prove an important modulator of immunity in multiple cell therapies. The immunomodulatory properties of MSC-derived subcellular particles indicate their potential as a novel cell-free therapy for treatment of immunological disorders, especially through interaction with antigen presenting cells (61). This is borne out by a recent study showing differential effects of membrane derived particles from MSCs either untreated or pre-treated with IFN-γ. Whilst both particle types decreased the frequency of CD14+ CD16+ inflammatory monocytes, the particles derived from IFN-γ treated cells also promoted anti-inflammatory PD-L1 expressing monocytes (10). This provides a mechanistic basis for earlier work showing that IFN-γ does not break but enhances the immunosuppressive capacities of MSCs and MSC-like cells (26, 99, 100). In the context of necrobiology, these data indicate that pre-apoptotic MSCs used as therapies in inflammatory microenvironments could be responsible for a switch toward an anti-inflammatory response through subcellular particles through their intra-vesicular or surface cargo (10).
The second aspect of MSC necrobiology that could affect therapeutic efficacy is the very recent observation from tumor biology that apoptotic cancer cells produce EVs with the characteristics of exosomes (78, 101). Apoptotic cell-derived extracellular vesicles (apoEVs) appear to be enriched with snRNA and spliceosomal proteins that can alter mRNA splicing in recipient cells (101). This finding is consistent with other studies showing that EVs produced during apoptosis are not simply debris but have important immune regulatory roles in autoimmunity, infection and cancer (78, 79). Thus, apoEVs including those with exosome characteristics are the conduit of intercellular communication in physiologic and pathologic contexts. In this regard, it is important to note that there has yet to be a comprehensive description of exosomes produced by apoptotic MSCs. We do not know the degree to which apoptotic MSCs produce apoEVs, nor how this is affected by MSC history, stimulation, source, or apoptotic stage. Nevertheless, it is clear that MSC-derived subcellular particles' contents are not static but vary by tissue origin, MSC activity, and the cellular environment of the MSCs (96). It remains reasonable to assume that apoptotic MSCs produce apoEVs with potential to modify target cells.
Overall, the important implication is that MSCs (whether viable or non-viable) delivered to a patient are likely to be accompanied by or result in EV with diverse cargo produced prior to or after therapeutic deployment. However, it is also worth remembering that broader animal studies of MSCs that do not consider exosome function, could be unwittingly measuring a confounding effect of bovine exosomes present in the serum constituents of culture medium. This is usually well-controlled for in studies designed to discover exosome effects, but less often in studies of the MSC function itself. The range of such effects are extensive and could be influencing multiple disease models (Table 2). Nonetheless, the beneficial effects of exosomes derived from various sources has led to over 100 human phase I/II clinical trials, although to date there are very few reports of trials involving human MSC derived exosomes (www.clinicaltrials.gov). Those that have been registered target pancreatic cancer, macular holes, cerebrovascular disorders and diabetes, but most seem to be in the recruitment phase at present.
A common aspect of all the above aspects of MSC necrobiology is the significant role played by innate immune cells to counter the pathologic processes. Thus, efferocytosis or the processes linked to removing apoptotic MSCs are likely to contribute to the therapeutic benefit in studies where MSC viability is not essential (2, 102). This is likely to extend beyond simple uptake of apoptotic MSCs by macrophages, dendritic or other cells (61), and extend to the range of EVs, mitochondria, and other signals produced by dying MSCs and which profoundly alter the tissue microenvironment and innate immune cells (8, 102). The importance of these processes in the regular homeostatic function of endogenous MSCs is not known. Nevertheless, in the context of cell therapy, the efficacy of MSC can be attributed to either live/viable or dying/dead MSCs in different disease contexts, and these benefits are attributable to downstream effects linked to: a) the biological activity to (or evoked by) the intended therapeutic component (the viable MSC itself or its derivatives) and/or b) the recipient's response to MSCs that are in the process of dying (Figure 1). Without this understanding, and a greater appreciation of the complex necrobiology of MSCs, we are unlikely to understand the mechanisms of cell therapy action or rationally design improvements. Thus, the necrobiology of the mesenchymal stromal cell is likely to be a fruitful area for improving the efficacy or removing confounding influences on cell therapy.
DW has performed literature research, designed the review layout, wrote, and revised the review. KE has performed literature research, designed the review layout, wrote, and revised the review. AK has performed literature research, designed the review layout, wrote, and revised the review. JI-C has performed the literature research and contributed to the section on extracellular vesicles. IH has performed the literature research and contributed to the section on autophagy. BM has performed literature research, designed the review layout, wrote, and revised the review. All authors agree to be accountable for the content of the work.
DW is funded by the National Institutes of Health (HL127144-01, EB024329), the Cystic Fibrosis Foundation, and the Medical Technology Consortium. KE is supported by Science Foundation Ireland (SFI) under Grant Number 13/SIRG/2172 through a Starting Investigator Research Grant award and the Irish Research Council Laureate award IRCLA/2017/288. AK is supported by UK Medical Research Council Research Awards (MRC MR/R025096/1 and MR/S009426/1).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
1. English K. Mechanisms of mesenchymal stromal cell immunomodulation. Immunol Cell Biol. (2013) 91:19–26. doi: 10.1038/icb.2012.56
2. Galipeau J, Sensebe L. Mesenchymal stromal cells: clinical challenges and therapeutic opportunities. Cell Stem Cell. (2018) 22:824–33. doi: 10.1016/j.stem.2018.05.004
3. Weiss DJ. Concise review: current status of stem cells and regenerative medicine in lung biology and diseases. Stem Cells. (2014) 32:16–25. doi: 10.1002/stem.1506
4. Greco SJ, Rameshwar P. Microenvironmental considerations in the application of human mesenchymal stem cells in regenerative therapies. Biologics Targets Ther. (2008) 2:699–705. doi: 10.2147/BTT.S2765
5. Eggenhofer E, Benseler V, Kroemer A, Popp FC, Geissler EK, Schlitt HJ, et al. Mesenchymal stem cells are short-lived and do not migrate beyond the lungs after intravenous infusion. Front Immunol. (2012) 3:297. doi: 10.3389/fimmu.2012.00297
6. de Witte SFH, Luk F, Sierra Parraga JM, Gargesha M, Merino A, Korevaar SS, et al. Immunomodulation by therapeutic mesenchymal stromal cells (MSC) is triggered through phagocytosis of MSC by monocytic cells. Stem Cells. (2018) 36:602–15. doi: 10.1002/stem.2779
7. Galleu A, Riffo-Vasquez Y, Trento C, Lomas C, Dolcetti L, Cheung TS, et al. Apoptosis in mesenchymal stromal cells induces in vivo recipient-mediated immunomodulation. Sci Transl Med. (2017) 9:eaam7828. doi: 10.1126/scitranslmed.aam7828
8. Luk F, de Witte SF, Korevaar SS, Roemeling-van Rhijn M, Franquesa M, Strini T, et al. Inactivated mesenchymal stem cells maintain immunomodulatory capacity. Stem Cells Dev. (2016) 25:1342–54. doi: 10.1089/scd.2016.0068
9. Darzynkiewicz Z, Juan G, Li X, Gorczyca W, Murakami T, Traganos F. Cytometry in cell necrobiology: analysis of apoptosis and accidental cell death (necrosis). Cytometry. (1997) 27:1–20.
10. Goncalves FDC, Luk F, Korevaar SS, Bouzid R, Paz AH, López-Iglesias C, et al. Membrane particles generated from mesenchymal stromal cells modulate immune responses by selective targeting of pro-inflammatory monocytes. Sci Rep. (2017) 7:12100. doi: 10.1038/s41598-017-12121-z
11. Gupta N, Su X, Popov B, Lee JW, Serikov V, Matthay MA. Intrapulmonary delivery of bone marrow-derived mesenchymal stem cells improves survival and attenuates endotoxin-induced acute lung injury in mice. J Immunol. (2007) 179:1855–63. doi: 10.4049/jimmunol.179.3.1855
12. Sun J, Han ZB, Liao W, Yang SG, Yang Z, Yu J, et al. Intrapulmonary delivery of human umbilical cord mesenchymal stem cells attenuates acute lung injury by expanding CD4+CD25+ Forkhead Boxp3 (FOXP3)+ regulatory T cells and balancing anti- and pro-inflammatory factors. Cell Physiol Biochem. (2011) 27:587–96. doi: 10.1159/000329980
13. Kavanagh H, Mahon BP. Allogeneic mesenchymal stem cells prevent allergic airway inflammation by inducing murine regulatory T cells. Allergy. (2011) 66:523–31. doi: 10.1111/j.1398-9995.2010.02509.x
14. Nemeth K, Leelahavanichkul A, Yuen PS, Mayer B, Parmelee A, Doi K, et al. Bone marrow stromal cells attenuate sepsis via prostaglandin E(2)-dependent reprogramming of host macrophages to increase their interleukin-10 production. Nat Med. (2009) 15:42–9. doi: 10.1038/nm.1905
15. Saas P, Daguindau E, Perruche S. Concise review: apoptotic cell-based therapies-rationale, preclinical results and future clinical developments. Stem Cells. (2016) 34:1464–73. doi: 10.1002/stem.2361
16. Gupta N, Sinha R, Krasnodembskaya A, Xu X, Nizet V, Matthay MA, et al. The TLR4-PAR1 axis regulates bone marrow mesenchymal stromal cell survival and therapeutic capacity in experimental bacterial pneumonia. Stem Cells. (2018) 36:796–806. doi: 10.1002/stem.2796
17. Islam D, Huang Y, Fanelli V, Delsedime L, Wu S, Khang J, et al. Identification and modulation of microenvironment is crucial for effective MSC therapy in acute lung injury. Am J Respir Crit Care Med. (2018) 199:1214–24. doi: 10.1164/rccm.201802-0356OC
18. Chang CL, Leu S, Sung HC, Zhen YY, Cho CL, Chen A, et al. Impact of apoptotic adipose-derived mesenchymal stem cells on attenuating organ damage and reducing mortality in rat sepsis syndrome induced by cecal puncture and ligation. J Transl Med. (2012) 10:244. doi: 10.1186/1479-5876-10-244
19. Chen HH, Chang CL, Lin KC, Sung PH, Chai HT, Zhen YY, et al. Melatonin augments apoptotic adipose-derived mesenchymal stem cell treatment against sepsis-induced acute lung injury. Am J Transl Res. (2014) 6:439–58.
20. Chen HH, Lin KC, Wallace CG, Chen YT, Yang CC, Leu S, et al. Additional benefit of combined therapy with melatonin and apoptotic adipose-derived mesenchymal stem cell against sepsis-induced kidney injury. J Pineal Res. (2014) 57:16–32. doi: 10.1111/jpi.12140
21. Sung PH, Chang CL, Tsai TH, Chang LT, Leu S, Chen YL, et al. Apoptotic adipose-derived mesenchymal stem cell therapy protects against lung and kidney injury in sepsis syndrome caused by cecal ligation puncture in rats. Stem Cell Res Ther. (2013) 4:155. doi: 10.1186/scrt385
22. Augustine S, Avey MT, Harrison B, Locke T, Ghannad M, Moher D, et al. Mesenchymal stromal cell therapy in bronchopulmonary dysplasia: systematic review and meta-analysis of preclinical studies. Stem Cells Transl Med. (2017) 6:2079–93. doi: 10.1002/sctm.17-0126
23. Mei SH, Dos Santos CC, Stewart DJ. Advances in stem cell and cell-based gene therapy approaches for experimental acute lung injury: a review of preclinical studies. Hum Gene Ther. (2016) 27:802–12. doi: 10.1089/hum.2016.063
24. Serrano-Mollar A. Cell therapy in idiopathic pulmonary fibrosis(dagger). Med Sci. (2018) 6:64. doi: 10.3390/medsci6030064
25. Zhang LB, He M. Effect of mesenchymal stromal (stem) cell (MSC) transplantation in asthmatic animal models: a systematic review and meta-analysis. Pulmonary Pharmacol Therapeut. (2019) 54:39–52. doi: 10.1016/j.pupt.2018.11.007
26. Noone C, Kihm A, English K, O'Dea S, Mahon BP. IFN-gamma stimulated human umbilical-tissue-derived cells potently suppress NK activation and resist NK-mediated cytotoxicity in vitro. Stem Cells Dev. (2013) 22:3003–14. doi: 10.1089/scd.2013.0028
27. Li Y, Fung J, Lin F. Local inhibition of complement improves mesenchymal stem cell viability and function after administration. Mol Ther. (2016) 24:1665–74. doi: 10.1038/mt.2016.142
28. Li Y, Lin F. Mesenchymal stem cells are injured by complement after their contact with serum. Blood. (2012) 120:3436–43. doi: 10.1182/blood-2012-03-420612
29. Matthay MA, Calfee CS, Zhuo H, Thompson BT, Wilson JG, Levitt JE, et al. Treatment with allogeneic mesenchymal stromal cells for moderate to severe acute respiratory distress syndrome (START study): a randomised phase 2a safety trial. Lancet Respiratory Med. (2019) 7:154–62. doi: 10.1016/S2213-2600(18)30418-1
30. Weiss DJ. Cell-based therapies for acute respiratory distress syndrome. Lancet Respiratory Med. (2019) 7:105–6. doi: 10.1016/S2213-2600(18)30477-6
31. Bellingan G, Jacono F, Bannard-Smith J, Brealey D, Meyer N, Thickett D, et al. Ting, primary analysis of a phase 1/2 study to assess MultiStem® cell therapy, a regenerative Advanced Therapy Medicinal Product (ATMP), in Acute Respiratory Distress Syndrome (MUST-ARDS). B14. Late Break Clin Trials. (2019) 199:A7353. doi: 10.1164/ajrccm-conference.2019.199.1_MeetingAbstracts.A7353
32. Dunn WA Jr. Autophagy and related mechanisms of lysosome-mediated protein degradation. Trends Cell Biol. (1994) 4:139–43. doi: 10.1016/0962-8924(94)90069-8
33. Goodall ML, Fitzwalter BE, Zahedi S, Wu M, Rodriguez D, Mulcahy-Levy JM, et al. The autophagy machinery controls cell death switching between apoptosis and necroptosis. Dev Cell. (2016) 37:337–49. doi: 10.1016/j.devcel.2016.04.018
34. Kirisako T, Baba M, Ishihara N, Miyazawa K, Ohsumi M, Yoshimori T, et al. Formation process of autophagosome is traced with Apg8/Aut7p in yeast. J Cell Biol. (1999) 147:435–46. doi: 10.1083/jcb.147.2.435
35. Liu Y, Levine B. Autosis and autophagic cell death: the dark side of autophagy. Cell Death Differentiation. (2014) 22:367. doi: 10.1038/cdd.2014.143
36. Liu Y, Shoji-Kawata S, Sumpter RM Jr, Wei Y, Ginet V, Zhang L, et al. Autosis is a Na+,K+-ATPase-regulated form of cell death triggered by autophagy-inducing peptides, starvation, and hypoxia-ischemia. Proc Natl Acad Sci USA. (2013) 110:20364–71. doi: 10.1073/pnas.1319661110
37. Fang Y, Tan J, Zhang Q. Signaling pathways and mechanisms of hypoxia-induced autophagy in the animal cells. Cell Biol Int. (2015) 39:891–8. doi: 10.1002/cbin.10463
38. Fougeray S, Pallet N. Mechanisms and biological functions of autophagy in diseased and ageing kidneys. Nat Rev Nephrol. (2015) 11:34–45. doi: 10.1038/nrneph.2014.201
39. Kouroku Y, Fujita E, Tanida I, Ueno T, Isoai A, Kumagai H, et al. ER stress (PERK/eIF2alpha phosphorylation) mediates the polyglutamine-induced LC3 conversion, an essential step for autophagy formation. Cell Death Differentiation. (2007) 14:230–9. doi: 10.1038/sj.cdd.4401984
40. Auletta JJ, Deans RJ, Bartholomew AM. Emerging roles for multipotent, bone marrow-derived stromal cells in host defense. Blood. (2012) 119:1801–9. doi: 10.1182/blood-2011-10-384354
41. Sommermann TG, Mack HI, Cahir-McFarland E. Autophagy prolongs survival after NFkappaB inhibition in B-cell lymphomas. Autophagy. (2012) 8:265–7. doi: 10.4161/auto.8.2.18763
42. Liu J, Hao H, Huang H, Tong C, Ti D, Dong L, et al. Hypoxia regulates the therapeutic potential of mesenchymal stem cells through enhanced autophagy. Int J Lower Extremity Wounds. (2015) 14:63–72. doi: 10.1177/1534734615573660
43. Lv B, Hua T, Li F, Han J, Fang J, Xu L, et al. Hypoxia-inducible factor 1 alpha protects mesenchymal stem cells against oxygen-glucose deprivation-induced injury via autophagy induction and PI3K/AKT/mTOR signaling pathway. Am J Transl Res. (2017) 9:2492–9.
44. Zhang Q, Yang YJ, Wang H, Dong QT, Wang TJ, Qian HY, et al. Autophagy activation: a novel mechanism of atorvastatin to protect mesenchymal stem cells from hypoxia and serum deprivation via AMP-activated protein kinase/mammalian target of rapamycin pathway. Stem Cells Dev. (2012) 21:1321–32. doi: 10.1089/scd.2011.0684
45. Ghanta S, Tsoyi K, Liu X, Nakahira K, Ith B, Coronata AA, et al. Mesenchymal stromal cells deficient in autophagy proteins are susceptible to oxidative injury and mitochondrial dysfunction. Am J Respir Cell Mol Biol. (2017) 56:300–9. doi: 10.1165/rcmb.2016-0061OC
46. Moya A, Larochette N, Paquet J, Deschepper M, Bensidhoum M, Izzo V, et al. Quiescence preconditioned human multipotent stromal cells adopt a metabolic profile favorable for enhanced survival under ischemia. Stem Cells. (2017) 35:181–96. doi: 10.1002/stem.2493
47. Phinney DG, Di Giuseppe M, Njah J, Sala E, Shiva S, St. Croix CM, et al. Mesenchymal stem cells use extracellular vesicles to outsource mitophagy and shuttle microRNAs. Nat Commun. (2015) 6:8472. doi: 10.1038/ncomms9472
48. Herberg S, Shi X, Johnson MH, Hamrick MW, Isales CM, Hill WD. Stromal cell-derived factor-1beta mediates cell survival through enhancing autophagy in bone marrow-derived mesenchymal stem cells. PLoS ONE. (2013) 8:e58207. doi: 10.1371/journal.pone.0058207
49. Roa-Mansergas X, Fado R, Atari M, Mir JF, Muley H, Serra D, et al. CPT1C promotes human mesenchymal stem cells survival under glucose deprivation through the modulation of autophagy. Sci Rep. (2018) 8:6997. doi: 10.1038/s41598-018-25485-7
50. Yang M, Wen T, Chen H, Deng J, Yang C, Zhang Z. Knockdown of insulin-like growth factor 1 exerts a protective effect on hypoxic injury of aged BM-MSCs: role of autophagy. Stem Cell Res Ther. (2018) 9:284. doi: 10.1186/s13287-018-1028-5
51. Wu Y, Chen L, Scott PG, Tredget EE. Mesenchymal stem cells enhance wound healing through differentiation and angiogenesis. Stem Cells. (2007) 25:2648–59. doi: 10.1634/stemcells.2007-0226
52. An Y, Liu WJ, Xue P, Ma Y, Zhang LQ, Zhu B, et al. Autophagy promotes MSC-mediated vascularization in cutaneous wound healing via regulation of VEGF secretion. Cell Death Dis. (2018) 9:58. doi: 10.1038/s41419-017-0082-8
53. Gao L, Cen S, Wang P, Xie Z, Liu Z, Deng W, et al. Autophagy improves the immunosuppression of CD4+ T cells by mesenchymal stem cells through transforming growth factor-beta1. Stem Cells Transl Med. (2016) 5:1496–505. doi: 10.5966/sctm.2015-0420
54. Chinnadurai R, Copland IB, Ng S, Garcia M, Prasad M, Arafat D, et al. Mesenchymal stromal cells derived from Crohn's patients deploy indoleamine 2,3-dioxygenase-mediated immune suppression, independent of autophagy. Mol Ther. (2015) 23:1248–61. doi: 10.1038/mt.2015.67
55. Dang S, Xu H, Xu C, Cai W, Li Q, Cheng Y, et al. Autophagy regulates the therapeutic potential of mesenchymal stem cells in experimental autoimmune encephalomyelitis. Autophagy. (2014) 10:1301–15. doi: 10.4161/auto.28771
56. Marino G, Niso-Santano M, Baehrecke EH, Kroemer G. Self-consumption: the interplay of autophagy and apoptosis. Nat Rev Mol Cell Biol. (2014) 15:81–94. doi: 10.1038/nrm3735
57. Munoz-Pinedo C, Martin SJ. Autosis: a new addition to the cell death Tower of Babel. Cell Death Dis. (2014) 5:e1319. doi: 10.1038/cddis.2014.246
58. Kroemer G, Levine B. Autophagic cell death: the story of a misnomer. Nat Rev Mol Cell Biol. (2008) 9:1004–10. doi: 10.1038/nrm2529
59. Dang S, Yu ZM, Zhang CY, Zheng J, Li KL, Wu Y, et al. Autophagy promotes apoptosis of mesenchymal stem cells under inflammatory microenvironment. Stem Cell Res Ther. (2015) 6:247. doi: 10.1186/s13287-015-0245-4
60. Cheung TS, Galleu A, von Bonin M, Bornhauser M, Dazzi F. Apoptotic mesenchymal stromal cells induce prostaglandin E2 in monocytes: implications for the monitoring of mesenchymal stromal cells activity. Haematologica. (2019). doi: 10.3324/haematol.2018.214767. [Epub ahead of print].
61. Carty F, Mahon BP, English K. The influence of macrophages on mesenchymal stromal cell therapy: passive or aggressive agents? Clin Exp Immunol. (2017) 188:1–11. doi: 10.1111/cei.12929
62. Scherz-Shouval R, Elazar Z. ROS, mitochondria and the regulation of autophagy. Trends Cell Biol. (2007) 17:422–7. doi: 10.1016/j.tcb.2007.07.009
63. Jackson MV, Morrison TJ, Doherty DF, McAuley DF, Matthay MA, Kissenpfennig A, et al. Mitochondrial transfer via tunneling nanotubes is an important mechanism by which mesenchymal stem cells enhance macrophage phagocytosis in the in vitro and in vivo models of ARDS. Stem Cells. (2016) 34:2210–23. doi: 10.1002/stem.2372
64. Morrison TJ, Jackson MV, Cunningham EK, Kissenpfennig A, McAuley DF, O'Kane CM, et al. Mesenchymal stromal cells modulate macrophages in clinically relevant lung injury models by extracellular vesicle mitochondrial transfer. Am J Respir Crit Care Med. (2017) 196:1275–86. doi: 10.1164/rccm.201701-0170OC
65. Ahmad T, Mukherjee S, Pattnaik B, Kumar M, Singh S, Kumar M, et al. Miro1 regulates intercellular mitochondrial transport & enhances mesenchymal stem cell rescue efficacy. EMBO J. (2014) 33:994–1010. doi: 10.1002/embj.201386030
66. Li X, Zhang Y, Yeung SC, Liang Y, Liang X, Ding Y, et al. Mitochondrial transfer of induced pluripotent stem cell-derived mesenchymal stem cells to airway epithelial cells attenuates cigarette smoke-induced damage. Am J Respir Cell Mol Biol. (2014) 51:455–65. doi: 10.1165/rcmb.2013-0529OC
67. Torralba D, Baixauli F, Sanchez-Madrid F. Mitochondria know no boundaries: mechanisms and functions of intercellular mitochondrial transfer. Front Cell Dev Biol. (2016) 4:107. doi: 10.3389/fcell.2016.00107
68. Murray LMA, Krasnodembskaya AD. Concise review: intercellular communication via organelle transfer in the biology and therapeutic applications of stem cells. Stem Cells. (2019) 37:14–25. doi: 10.1002/stem.2922
69. Paliwal S, Chaudhuri R, Agrawal A, Mohanty S. Regenerative abilities of mesenchymal stem cells through mitochondrial transfer. J Biomed Sci. (2018) 25:31. doi: 10.1186/s12929-018-0429-1
70. Agrawal A, Mabalirajan U. Rejuvenating cellular respiration for optimizing respiratory function: targeting mitochondria. Am J Physiol Lung Cell Mol Physiol. (2016) 310:L103–L113. doi: 10.1152/ajplung.00320.2015
71. Fergie N, Todd N, McClements L, McAuley D, O'Kane C, Krasnodembskaya A. Hypercapnic acidosis induces mitochondrial dysfunction and impairs the ability of mesenchymal stem cells to promote distal lung epithelial repair. FASEB J. (2019) 33:5585–98. doi: 10.1096/fj.201802056R
72. Acquistapace A, Bru T, Lesault PF, Figeac F, Coudert AE, le Coz O, et al. Human mesenchymal stem cells reprogram adult cardiomyocytes toward a progenitor-like state through partial cell fusion and mitochondria transfer. Stem Cells. (2011) 29:812–24. doi: 10.1002/stem.632
73. Paliwal S, Chaudhuri R, Agrawal A, Mohanty S. Human tissue-specific MSCs demonstrate differential mitochondria transfer abilities that may determine their regenerative abilities. Stem Cell Res Ther. (2018) 9:298. doi: 10.1186/s13287-018-1012-0
74. Li CJ, Chen PK, Sun LY, Pang CY. Enhancement of mitochondrial transfer by antioxidants in human mesenchymal stem cells. Oxid Med Cell Longevity. (2017) 2017:8510805. doi: 10.1155/2017/8510805
75. Mahrouf-Yorgov M, Augeul L, Da Silva CC, Jourdan M, Rigolet M, Manin S, et al. Mesenchymal stem cells sense mitochondria released from damaged cells as danger signals to activate their rescue properties. Cell Death Differentiation. (2017) 24:1224–38. doi: 10.1038/cdd.2017.51
76. Zhang Y, Yu Z, Jiang D, Liang X, Liao S, Zhang Z, et al. iPSC-MSCs with high intrinsic MIRO1 and sensitivity to TNF-alpha yield efficacious mitochondrial transfer to rescue anthracycline-induced cardiomyopathy. Stem Cell Rep. (2016) 7:749–63. doi: 10.1016/j.stemcr.2016.08.009
77. Braza F, Dirou S, Forest V, Sauzeau V, Hassoun D, Chesne J, et al. Mesenchymal stem cells induce suppressive macrophages through phagocytosis in a mouse model of asthma. Stem Cells. (2016) 34:1836–45. doi: 10.1002/stem.2344
78. Caruso S, Poon IKH. Apoptotic cell-derived extracellular vesicles: more than just debris. Front Immunol. (2018) 9:1486. doi: 10.3389/fimmu.2018.01486
79. Lynch C, Panagopoulou M, Gregory CD. Extracellular vesicles arising from apoptotic cells in tumors: roles in cancer pathogenesis and potential clinical applications. Front Immunol. (2017) 8:1174. doi: 10.3389/fimmu.2017.01174
80. Chen W, Huang Y, Han J, Yu L, Li Y, Lu Z, et al. Immunomodulatory effects of mesenchymal stromal cells-derived exosome. Immunol Res. (2016) 64:831–40. doi: 10.1007/s12026-016-8798-6
81. Del Fattore A, Luciano R, Pascucci L, Goffredo BM, Giorda E, Scapaticci M, et al. Immunoregulatory effects of mesenchymal stem cell-derived extracellular vesicles on T lymphocytes. Cell Transplant. (2015) 24:2615–27. doi: 10.3727/096368915X687543
82. Del Fattore A, Luciano R, Saracino R, Battafarano G, Rizzo C, Pascucci L, et al. Differential effects of extracellular vesicles secreted by mesenchymal stem cells from different sources on glioblastoma cells. Expert Opin Biol Ther. (2015) 15:495–504. doi: 10.1517/14712598.2015.997706
83. Lee C, Mitsialis SA, Aslam M, Vitali SH, Vergadi E, Konstantinou G, et al. Exosomes mediate the cytoprotective action of mesenchymal stromal cells on hypoxia-induced pulmonary hypertension. Circulation. (2012) 126:2601–11. doi: 10.1161/CIRCULATIONAHA.112.114173
84. Anderson JD, Johansson HJ, Graham CS, Vesterlund M, Pham MT, Bramlett CS, et al. Comprehensive proteomic analysis of mesenchymal stem cell exosomes reveals modulation of angiogenesis via nuclear factor-kappaB signaling. Stem Cells. (2016) 34:601–13. doi: 10.1002/stem.2298
85. Pakravan K, Babashah S, Sadeghizadeh M, Mowla SJ, Mossahebi-Mohammadi M, Ataei F, et al. MicroRNA-100 shuttled by mesenchymal stem cell-derived exosomes suppresses in vitro angiogenesis through modulating the mTOR/HIF-1alpha/VEGF signaling axis in breast cancer cells. Cell Oncol. (2017) 40:457–70. doi: 10.1007/s13402-017-0335-7
86. Kim HS, Choi DY, Yun SJ, Choi SM, Kang JW, Jung JW, et al. Proteomic analysis of microvesicles derived from human mesenchymal stem cells. J Proteome Res. (2012) 11:839–49. doi: 10.1021/pr200682z
87. Harting MT, Srivastava AK, Zhaorigetu S, Bair H, Prabhakara KS, Toledano Furman NE, et al. Inflammation-stimulated mesenchymal stromal cell-derived extracellular vesicles attenuate inflammation. Stem Cells. (2018) 36:79–90. doi: 10.1002/stem.2730
88. Ruppert KA, Nguyen TT, Prabhakara KS, Toledano Furman NE, Srivastava AK, Harting MT, et al. Human mesenchymal stromal cell-derived extracellular vesicles modify microglial response and improve clinical outcomes in experimental spinal cord injury. Sci Rep. (2018) 8:480. doi: 10.1038/s41598-017-18867-w
89. Zhang Q, Fu L, Liang Y, Guo Z, Wang L, Ma C, et al. Exosomes originating from MSCs stimulated with TGF-beta and IFN-gamma promote Treg differentiation. J Cell Physiol. (2018) 233:6832–40. doi: 10.1002/jcp.26436
90. Collino F, Pomatto M, Bruno S, Lindoso RS, Tapparo M, Sicheng W, et al. Exosome and microvesicle-enriched fractions isolated from mesenchymal stem cells by gradient separation showed different molecular signatures and functions on renal tubular epithelial cells. Stem Cell Rev. (2017) 13:226–43. doi: 10.1007/s12015-016-9713-1
91. Lai RC, Arslan F, Lee MM, Sze NS, Choo A, Chen TS, et al. Exosome secreted by MSC reduces myocardial ischemia/reperfusion injury. Stem Cell Res. (2010) 4:214–22. doi: 10.1016/j.scr.2009.12.003
92. Lou G, Chen Z, Zheng M, Liu Y. Mesenchymal stem cell-derived exosomes as a new therapeutic strategy for liver diseases. Exp Mol Med. (2017) 49:e346. doi: 10.1038/emm.2017.63
93. Wu P, Zhang B, Shi H, Qian H, Xu W. MSC-exosome: a novel cell-free therapy for cutaneous regeneration. Cytotherapy. (2018) 20:291–301. doi: 10.1016/j.jcyt.2017.11.002
94. Cahill EF, Kennelly H, Carty F, Mahon BP, English K. Hepatocyte growth factor is required for mesenchymal stromal cell protection against bleomycin-induced pulmonary fibrosis. Stem Cells Transl Med. (2016) 5:1307–18. doi: 10.5966/sctm.2015-0337
95. Kennelly H, Mahon BP, English K. Human mesenchymal stromal cells exert HGF dependent cytoprotective effects in a human relevant pre-clinical model of COPD. Sci Rep. (2016) 6:38207. doi: 10.1038/srep38207
96. Phinney DG, Pittenger MF. Concise review: MSC-derived exosomes for cell-free therapy. Stem Cells. (2017) 35:851–8. doi: 10.1002/stem.2575
97. Gonzalez-King H, Garcia NA, Ontoria-Oviedo I, Ciria M, Montero JA, Sepulveda P. Hypoxia inducible factor-1alpha potentiates jagged 1-mediated angiogenesis by mesenchymal stem cell-derived exosomes. Stem Cells. (2017) 35:1747–59. doi: 10.1002/stem.2618
98. Cahill EF, Tobin LM, Carty F, Mahon BP, English K. Jagged-1 is required for the expansion of CD4+ CD25+ FoxP3+ regulatory T cells and tolerogenic dendritic cells by murine mesenchymal stromal cells. Stem Cell Res Ther. (2015) 6:19. doi: 10.1186/s13287-015-0021-5
99. English K, Barry FP, Field-Corbett CP, Mahon BP. IFN-gamma and TNF-alpha differentially regulate immunomodulation by murine mesenchymal stem cells. Immunol Lett. (2007) 110:91–100. doi: 10.1016/j.imlet.2007.04.001
100. Ryan JM, Barry F, Murphy JM, Mahon BP. Interferon-gamma does not break, but promotes the immunosuppressive capacity of adult human mesenchymal stem cells. Clin Exp Immunol. (2007) 149:353–63. doi: 10.1111/j.1365-2249.2007.03422.x
101. Pavlyukov MS, Yu H, Bastola S, Minata M, Shender VO, Lee Y, et al. Apoptotic cell-derived extracellular vesicles promote malignancy of glioblastoma via intercellular transfer of splicing factors. Cancer Cell. (2018) 34:119–35.e10. doi: 10.1016/j.ccell.2018.05.012
Keywords: mesenchymal stromal cell, cell therapy, apoptosis, autophagy, mitochondria, extracellular vesicles, efficacy
Citation: Weiss DJ, English K, Krasnodembskaya A, Isaza-Correa JM, Hawthorne IJ and Mahon BP (2019) The Necrobiology of Mesenchymal Stromal Cells Affects Therapeutic Efficacy. Front. Immunol. 10:1228. doi: 10.3389/fimmu.2019.01228
Received: 08 March 2019; Accepted: 14 May 2019;
Published: 04 June 2019.
Edited by:
Martin Johannes Hoogduijn, Erasmus University Rotterdam, NetherlandsReviewed by:
Armand Keating, University of Toronto, CanadaCopyright © 2019 Weiss, English, Krasnodembskaya, Isaza-Correa, Hawthorne and Mahon. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Bernard P. Mahon, QmVybmFyZC5NYWhvbkBtdS5pZQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.