 Stephanie Menikou
Stephanie Menikou Paul R. Langford
Paul R. Langford
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol. , 12 June 2019
Sec. Inflammation
Volume 10 - 2019 | https://doi.org/10.3389/fimmu.2019.01156
Kawasaki disease (KD) is an inflammatory disease in children associated with vasculitis affecting predominantly the coronary arteries and is now the most common cause of acquired heart disease in children in developed countries. The etiology of KD is unknown but epidemiological studies implicate an infectious agent or toxin, which causes disease in genetically predisposed individuals. The presence of immune complexes (ICs) in the serum of children with KD was established in numerous studies during the 1970s and 80s. More recent genetic studies have identified variation in Fcγ receptors and genes controlling immunoglobulin production associated with KD. In this review we link the genetic findings and IC studies and suggest a key role for their interaction in pathophysiology of the disease.
Kawasaki disease (KD) is a pediatric inflammatory disease associated with a self-limited vasculitis. It was first described in Japan by Dr. Tomisaku Kawasaki in 1967 and has subsequently been recognized as an important childhood disorder worldwide (1). The cause of KD remains unknown, but the seasonal occurrence, prevalence in young children, infrequent relapse, and occurrence of epidemics all point to an infectious agent as the cause. The disease is diagnosed based on the characteristic constellation of fever, lymphadenopathy, rash, mucous membrane changes, and desquamation associated with laboratory evidence of inflammation. In the absence of specific treatment the clinical symptoms usually evolve over 7 to 14 days and resolve gradually, but the fever and severe inflammatory process might persist for 2 to 4 weeks in severe untreated cases (2).
The clinical and epidemiological features of KD strongly suggest an infectious etiology, including the occurrence of epidemics, seasonal variation in incidence and clustering of cases. Although many pathogens have been implicated via superantigen toxins, staphylococcal and streptococcal toxic shock syndromes, other studies have failed to confirm these findings (3–6). A range of other bacteria including Yersinia pseudotuberculosis (7–9), Propionibacterium acnes (10), Mycoplasma pneumoniae (11, 12), Chlamydia pneumoniae (13), Rickettsia species (14), and Coxiella burnetii (15) have been reported. Also, numerous viruses including Epstein Barr virus (16), retroviruses (17, 18), adenovirus (19), and measles virus (20) have been reported to be associated with KD. None of these reported viruses or bacteria have been convincingly replicated.
Intravenous immunoglobulin (IVIG) was shown to be an effective treatment in the 1980s, and the majority of children with KD show a rapid resolution of fever and symptoms following infusion of IVIG (2, 21, 22). IVIG treatment greatly reduces the risk of coronary artery aneurysm (CAA) but 5-20% of KD cases fail to respond, have persistence or reoccurrence of fever and require additional anti-inflammatory treatments (22, 23). IVIG-resistant patients have an increased risk of developing CAA (23–25). For patients that are unresponsive to initial IVIG treatment, a range of alternative therapies to reduce inflammation have been advocated including steroids, and treatment with tumor necrosis factor (TNF) inhibitors. There is growing evidence from recent studies that other anti-inflammatory agents, including cyclosporine A (CyA) and anakinra [an interleukin 1 receptor antagonist (IL1-RA)] may be beneficial in reducing coronary artery damage and at the same time controlling the inflammatory process in KD patients (22, 26–29).
A central feature of KD is the activation of the immune system (2). The immune response in the acute phase of patients with KD involves activation of many different components of the innate and adaptive immune systems (30). There is an intense inflammatory response in the initial weeks of the illness, with elevation in white blood cell (WBC) counts and activation of most classes of WBCs including neutrophil leucocytosis and elevated eosinophil counts in acute and subacute KD patients (31). There is also an intense inflammatory response in the initial weeks of illness with elevation of acute phase proteins such as C-reactive protein (CRP), procalcitonin (PCT) (32), erythrocyte sedimentation rate (ESR) and higher values of alanine aminotransferase and γ glutamyl transferase (GGT) (33). In a study by Katayama et al. (34) peripheral blood CD14+CD16+ monocytes were also increased during the acute phase of KD.
Immune cells including monocytes/macrophages, and T and B lymphocytes produce cytokines such as interleukins (IL) (35). In KD, many cytokines are expressed at significantly higher levels than normal during the acute phase (2). Gene expression profile studies by Hoang et al. (36) identified increased transcript abundance of the genes IL-1R, IL1RN, IL-1RAP, and IL-1R2, supporting the importance of the IL-1 pathway in the pathogenesis of KD (36). Other cytokine genes up-regulated in the blood of KD patients, compared to children with other febrile illnesses and healthy children-adult volunteers, include IL-6 (35), IL-8 (2), and IL-17A (37). A study by Rowley et al. (38) gave another insight into the immunopathogenesis of KD as the authors found significantly increased levels of IgA plasma cells in the trachea, kidney, coronary artery and pancreas of acute phase patients who died; supporting the entrance of a pathological agent through the upper respiratory tract.
Besides the blood changes in neutrophils, monocytes, lymphocytes and cytokines, changes occur in the tissues of acute KD patients as well. Neutrophils are predominant in the peripheral blood of the acute KD patient and have been identified in the arterial wall early in the disease (30, 39, 40). Immunohistochemical studies on CAA from fatal cases who died in the acute and subacute phases of disease showed that invasion of neutrophils into the arterial wall is followed not only by monocytes/macrophages but also by dendritic cells (DCs) and lymphocytes (30, 41). Coronary arteritis in acute KD patients begins with infiltration of a small number of macrophages, lymphocytes and neutrophils in both tunica adventitia and intima and not in the media of the artery. In the subacute phase of illness, the internal elastic lamina is disrupted and there is inflammation of all the layers of coronary artery due to infiltration of lymphocytes, neutrophils and macrophages into the arterial wall (42). The main lymphocyte population identified in the coronary arterial lesions of KD patients were cytotoxic (CD8+) T cells (43, 44). Additional studies confirmed that CD8+ T cells were abnormally activated in the acute phase of KD with an imbalance between their activation and inhibitory actions (43). The results strongly suggest that in the acute phase of KD there is an antigen-driven immune response (41). Yilmaz et al. (45) found that there were increased numbers of mature myeloid DCs with high human leukocyte antigen-antigen D related (HLA-DR) expression in coronary artery lesions of four patients, thus further supporting the antigen-driven immune response as DCs process and transport antigens to the T cell's surface (39, 46).
Antigen-antibody complexes are formed when antibodies are produced against a circulating or tissue antigen. The antigen may be exogenous from an infectious agent, toxin or drug, or may be endogenous as occurs in autoimmune disorders. ICs are formed during many infectious and inflammatory diseases, and may play an important role in immunopathogenesis of infectious and inflammatory processes. They are normally taken up by inflammatory cells through binding of the heavy chain constant region to immunoglobulin Fc receptors (FcRs). Binding of immunoglobulin to some classes of FcRs (FcγRI, FcγRIIA, FcγRIIC, FcγRIIIA, FcγRIIIB) may lead to activation of inflammatory cells while binding to FcγRIIB result in suppression of inflammation. ICs may bind to FcRs and activate a range of cells including monocytes, basophils, eosinophils, lymphocytes and neutrophils (47). The interaction of ICs with neutrophils can lead to phagocytosis, degranulation and respiratory burst generation. In addition, ICs can bind to platelets directly as well by the recognition of FcRs on platelet membranes (47). Platelets are multifunctional cells that contribute to both inflammatory and coagulation functions. Their granules contain a broad range of inflammatory substances including anti-microbial proteins, such as defensins and kinocidins, which may contribute to internalizing pathogens to promote their clearing from host tissues and the bloodstream (48). ICs can have a significant role in immune responses by inducing inflammatory processes and eliminating antigens (49). ICs may also precipitate in tissues, leading to local inflammation or organ damage, complement activation and influx of inflammatory cells (50). They are particularly important in renal diseases such as membranoproliferative glomerulonephritis and systemic lupus erythematous (SLE) nephritis. ICs have been implicated in many autoimmune disease processes including rheumatoid arthritis (RA) and SLE where their deposition in tissues contributes to organ damage (50). They are also found circulating in the blood in patients with malignancies and many infectious diseases such as subacute sclerosing panencephalitis, hepatitis and dengue hemorrhagic fever (50, 51).
ICs attracted attention as possible components of the inflammatory process in KD because of their well-established role in experimental and clinical forms of vasculitis. In the serum sickness model of IC vasculitis, following injection of a foreign antigen and production of antibody, ICs form in the circulation in the second and third week following exposure, as antibody production against the foreign antigen occurs, and contributes to tissue damage through activation of platelets and other inflammatory cells (52). As already mentioned, ICs have important roles in many immunological diseases including SLE, RA, and many forms of nephritis and vasculitis. It was therefore not surprising that a role for ICs was considered in KD.
There is considerable evidence that ICs are involved in the pathogenesis of KD. Since the 1970s many studies have identified ICs in the plasma or serum of KD patients as shown in Figure 1. ICs were first reported in patients with KD in 1977 (53). Since then many studies have documented ICs in the acute and early convalescent phase of the disease (47, 53–71). The ICs have been identified by a range of different methods including platelet aggregation tests, C1q binding assay, ELISA and polyethylene glycol (PEG) precipitation.
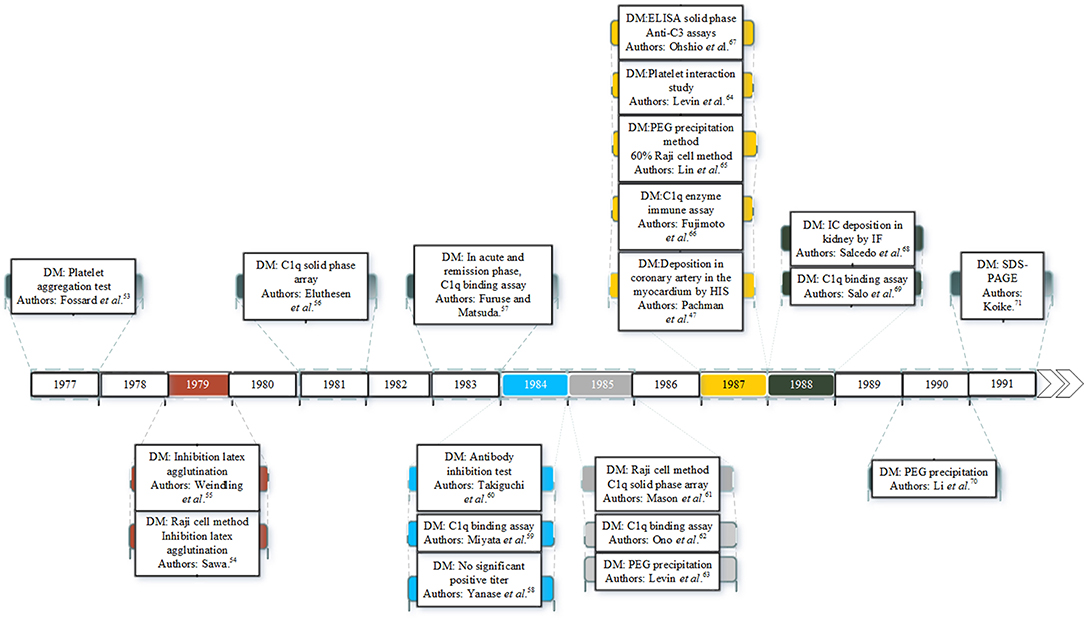
Figure 1. Studies of IC detection in KD patients. DM is the detection method.
Sequential studies of patients in the pre-immunoglobulin therapy era enabled the time course of ICs in the plasma to be identified. ICs appear in the circulation in the first 7 days of the illness, and reach a peak concentration in the second week, before declining slowly by the third and fourth week of the illness (63). The first documented case of ICs in KD in UK was in 1977 in a 22-month-old Japanese boy presenting with increased serum levels of immunoglobulins A and M (IgA, IgM), upper normal levels of immunoglobulin E (IgE), and a strongly positive result on the platelet aggregation test for IC detection (on day 12) (53). Raised concentrations of IgE were also detected in the acute phase of KD patients in both studies of Kusakawa et al. (72) and Furukawa et al. (73). A possible role for IgE was suggested in pathogenesis of vasculitis by increasing vascular permeability (53). In another study, Weindling et al. (55) described a 9 month old Caucasian girl showed normal levels of IgE and complement but increased levels of ICs containing immunoglobulin G. Eluthesen et al. (56) found high levels of circulating ICs in 48 out of 81 early convalescent KD patients (56). Furuse et al. (57), also found high levels of ICs further supporting the possibility of immunopathological involvement in KD.
Several studies have confirmed the presence of IC in the blood in the early phases of KD and an association with severity. These include, higher levels of ICs and immunoglobulin G (IgG) being detected in KD patients with CAA in a study by Miyata et al. (59) and that of Takiguchi et al. (60) who found ICs using the anti-antibody inhibition test in 23% of the patients. Mason et al. (61) detected ICs in the sera of 69% KD patients in all stages of illness but found no correlation between CAA development and the presence of ICs.
In contrast, Ono et al. (62) found that the group of KD patients with CAA had higher levels of ICs compared to the group that did not develop CAA. IgG ICs were present in 47.1% of KD patients in a study by Li et al. (70). The latter authors also found that two of the human IgG subclasses, IgG1 and IgG3, were predominant in ICs and significantly higher in patient sera (70). Levin et al. (63) documented the presence of circulating ICs in the serum of 19 children with KD in the second and third weeks of the illness, with the severity of disease being associated with an increased concentration of ICs (63).
Further studies are necessary to determine whether there is a causal link between ICs and severity of KD. For example, advances in proteomic analytical techniques could be used to characterize the composition of ICs in patients with different degrees of severity of KD and those in children with other illnesses. Such experiments would have the added value of potentially identifying any causative agent of KD.
Levin et al. (63) suggested the hypothesis that KD has three distinct pathophysiological phases: during the first phase (acute) there are high levels of antigen (arising from the unknown pathogen) in the blood and patients exhibit fever, lymphadenopathy and mucocutaneous manifestations (63). In the second phase (days 7–21), ICs are detected in the serum and bind to inhibitory and activating FcRs expressed by macrophages, DCs, B cells, neutrophils and platelets. FcRs may be important as the mediators between ICs and cellular responses. Furthermore, ICs may deposit in the tissues and activate complement, contributing to tissue damage. In the final stage, ICs are cleared and there are persisting antibodies against the causative antigen.
ICs develop when antibodies form complexes with their unknown antigens in the circulation. These complexes interact with platelets causing aggregation and release of vasoactive mediators such as histamine and serotonin, an important part in the initiation of vascular damage (63). Serotonin is an important vasoactive agent stored in the dense granules (74, 75). Levin et al. (63) showed that ICs from KD patients triggered release of serotonin from platelets. Recent studies consider serotonin a critical platelet component implicated in mediating vasodilatation and neutrophil activation (74).
Activated platelets recruit leukocytes and neutrophils to the affected sites following an infection or tissue damage (74). The binding of platelets to neutrophils occurs by CD62P ligand via CD162 which is a P-selectin glycoprotein ligand 1 (48). DCs are also activated by platelets. DCs role is to present antigens to B and T cells (48). It is suggested that naturally occurring autoantibodies or ICs are sufficient to activate DCs (76). This is accomplished by the binding of ICs to FcRs on DCs leading to phagocytosis and antigenic peptide presentation on MHC class I and II molecules that are recognized by T cells via cluster of differentiation 40 (CD40) and CD40 ligand (CD40L) which is also expressed by human platelets. Ultimately, the continuation of antibody production and phagocytosis leads to the elimination of the antigen.
In the third phase (convalescent), the ICs disappear from the circulation and high levels of antibody against the pathogen are present in the blood, (63) coinciding with defervescence of fever and subsidence of mucocutaneous manifestations. Vascular damage that was initiated in the second week of illness can lead to myocardial infarction and CAA that are most commonly detected after this phase of the illness. In summary, the implication of the Levin et al. (63) study was that KD has different phases involving different mechanisms (feverish phase driven by an unknown pathogen and antigen excess, IC vasculitis and convalescent phase after clearance of both antigen and ICs). The proposed IC involvement in KD is illustrated in Figure 2.
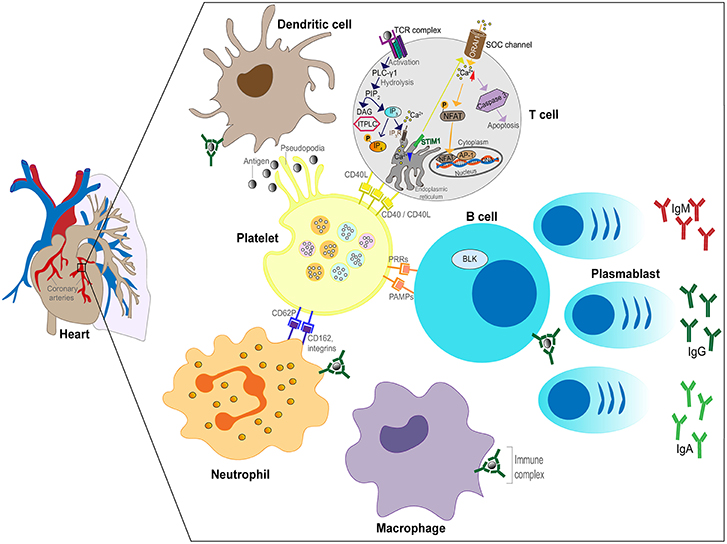
Figure 2. Schematic of the role of ICs in the inflammatory process in KD. Plasmablasts produce antibodies (IgG, IgM, or IgA) against the unknown pathogen resulting in IC formation. ICs bind to FcRs expressed by neutrophils, B cells, DCs and macrophages leading to activation of the cells. Neutrophils invade the arterial wall followed by macrophages, DCs and T cells. ICs activate platelets releasing soluble mediators and binding to neutrophils. Phagocytosis of ICs by macrophages leads to antigenic peptide presentation and T cell activation.
Although there are many studies demonstrating ICs in KD patients, a few have not supported an important role for ICs. In the study of Mason et al. (77) no correlation was found between the symptoms of KD (in particular fever and CAA) and the presence of ICs. In another study by Falcini et al. (78), 66% of KD patients had circulating ICs, with lower levels in convalescent compared to acute phase sera, but there was no significant relationship between ICs and the occurrence of aneurysm or any other clinical symptoms.
It should again be noted that most of the literature on ICs in KD dates from the 1980s, and most of the reports give insufficient clinical details to accurately determine the stage of the disease at which blood was tested for ICs. Furthermore, a range of different assays were used to detect ICs, some based on interaction with specific cells (such as platelets or Raji cells), and others based on the complement binding property of ICs. The fact that not all studies have confirmed ICs in all KD patients may reflect the differences in timing of the test in relationship to disease onset, or the differences in methodology for IC detection. Since the introduction of treatment with IVIG, studies on the “natural” pathophysiology have become difficult, as the IVIG may work in KD through an effect on the ICs or interaction with their receptors. Subsequent studies in the field were focused on macrophages, B cells, cytokines and T cells, and did not address a causal link between ICs and KD severity. It should be noted that these attempts to link severity (usually based on presence or not of CAA) and ICs were undertaken in the early days of ultrasound detection of CAA, and before the introduction of Z scores to quantify the size of CAA. To fully address the question of a relationship between presence of ICs and disease severity, more detailed studies using, in addition to quantitative documentation of coronary artery dimensions by echocardiography, other markers of disease severity such as height or duration of inflammatory markers such as CRP, IL-6, white cell or platelet count as well as response to IVIG would be needed. Furthermore, while the ICs may themselves contribute to disease pathogenesis, the nature and load of antigen may also be important contributors to disease severity. Severity of the initial febrile illness may be caused by a direct effect of the infecting agent or its antigens or toxins. The production of antibodies (the rate and nature of which may be genetically determined) and formation of ICs may be a second factor determining severity, and how ICs interact with cellular receptors such as the Fc receptors on immune cells or other tissues (which also depends on genetic factors) may play an additional role.
A variety of methods are used for IC isolation. Some of these methods are based on the attachment of ICs to specific cellular receptors such as Complement component 3 (C3) receptors on Raji cells, platelets or FcRs on macrophages or C1q (50). However, these techniques identify elements that are associated with but do not directly identify ICs themselves (79). Morrow et al. (80) found that PEG precipitation can be used to isolate ICs, and that there was no correlation between serum IgM and IgG levels and PEG-precipitated IgM and IgG levels in ICs in disease sera. Other studies supported Morrow's findings, and that lower concentrations of PEG resulted in higher yields of ICs with minimal free IgG or other proteins being precipitated (79). PEG appears to precipitate antibody preferentially when in complexes with antigens. PEG causes precipitation of proteins and ICs by steric exclusion in which larger proteins are precipitated only from the regions of the sample that are surrounded by linear chains of PEG (81, 82). Steric exclusion increases concentration and when the solubility is exceeded, precipitation of proteins occurs (82).
Several other techniques have been used to detect ICs including platelet aggregation tests for circulating ICs (83, 84), gel diffusion methods which measure bound exogenous C1q to ICs and aggregated y-globulin (85), anti-complementary activity by binding to Complement 1 (C1) (86, 87), gel filtration using Sepharose 6B (88), or using radiolabeled C1q that reacts with sera containing ICs in combination with PEG precipitation for removal of free C1q (89). Additional studies used labeled macrophages and aggregated IgG to study IC inhibition of phagocytosis (79). Other techniques for IC quantification use binding to C1q followed by addition of a radiolabelled anti-human IgG to determine the amount of ICs (90), or using purified bovine conglutinin (instead of C1q) to detect ICs with minimal monomeric IgG influence (91).
As indicated above, many different methods have been used to detect ICs. However, those involving PEG precipitation are considered the most reliable due to their simplicity, involving only comparatively few technical steps, that they do not induce IC formation (92), and there is low propensity for denaturation of proteins present, facilitating the search for new antigens within ICs. It should be noted that, dependent on the concentration used, PEG-precipitated sera may contain free immunoglobulins and C3 but also other proteins such as albumin and fibronectin (92, 93), which can be advantageous when such proteins are also under investigation. The last decade has seen an explosion in mass spectrometric-based proteomic techniques (94, 95), and we believe that their use with PEG-precipitated ICs from KD patients has great potential to identify the cause of the disease, complementing genetic approaches (as reviewed below).
Although all ethnic groups are affected by KD, the high incidence in Asian countries, and in those of Asian ethnicity that migrate to other countries (96), and the higher frequencies among siblings and twins, suggests a genetic contribution to the risk of KD (97, 98).
Candidate gene case control studies have identified a large number of associations with KD susceptibility. These include polymorphisms in cytokines such as IL1-RA (99), IL-4 (100), IL-10, TGFB2 and TGFBR2 (genes of the TGF-β pathway) (101) and chemokines including Chemokine (C-X-C motif) receptor 1 and 2 (102) and Chemokine (C-C) receptor 2, 3 and 5 (102), matrix metallopeptidases such as matrix metallopeptidase 3 (MMP3) (103), and HLA genes (B35, B37 and Bw44) (104). Polymorphisms in HLA, transforming growth factor beta (TGF-β) and calcium signaling pathways account for a proportion of the genetic risk (30). Genes associated with increased risk of CAA have also been identified in association studies including a polymorphism in the IL-10 promoter (105), a functional polymorphism in the MMP-13 gene (103), a polymorphism of the inflammatory gene CRP (106) and the TNF-α gene (106).
Genome wide association studies (GWAS) have identified a number of validated associations including a functional single nucleotide polymorphism (SNP) in the ITPKC gene, encoding inositol 1,4,5-trisphosphate 3-kinase C, whose role is to negatively regulate T cell activation through the calcium signaling pathway (Figure 2) (107). Functional polymorphisms in ITPKC can result in increased T cell activation, increased expression of cytokines, and these polymorphisms were found to be significantly associated with increased susceptibility to KD and an increased risk of coronary artery lesions in US and Japanese children (107).
Further genetic associations with KD susceptibility published to date suggest an important role for ICs in KD. Firstly, the finding from GWAS that polymorphisms in the IgG receptor gene FCGR2A, identified as a susceptibility locus for KD in European and Asian populations, is an important clue to the role of ICs (108). Immune cells (DCs, neutrophils, monocytes/macrophages) express on their surface FCGR2A and when this receptor is ligated to ICs it transduces activation signals in the cells (Figure 2) (109). These signals lead to either activation of inflammation or in some cases immune suppression. Therefore, polymorphisms in FCGR2A suggest an important role for IgG receptors in the immunopathogenesis of KD as individuals who carry the functional polymorphism may respond to ICs differently from the majority of the population with a persistent or excessive inflammatory response.
Another genetic association suggesting the importance of ICs in KD is the association with polymorphisms in CD40 and CD40L. CD40 is expressed on the endothelial cells, epithelial, B-cells, DCs, monocytes, macrophages, fibroblasts, myofibroblasts and it engages with CD40L (110). This ligand is a type II integral membrane protein of the TNF family that is expressed primarily on the surface of activated CD4+ T cells, B cells and platelets transducing signals that result in endothelial cell damage, various inflammatory and immune responses (111). CD40/CD40L engagement on the surface of DCs enables cross-presentation of antigens and promotes cytokine production to effectively stimulate activation and differentiation of T cells (110). Also, their interaction on B cells promotes early immunoglobulin production, isotype switching and enhancing affinity for antigen by somatic hypermutation of immunoglobulin genes (110). Acute KD patients had significantly higher expression levels of CD40L when compared to febrile controls, and these expression levels were higher in KD patients with CAA (111, 112). Gene polymorphisms in CD40 (113, 114)/CD40L (115) are associated with KD susceptibility and CAA formation. These genetic polymorphisms and differences in expression levels of CD40/CD40L might affect immunoglobulin production rate, or somatic hypermutation of immunoglobulin that would enhance affinity for antigen or lead to different isotype switching. Hence, individuals who are genetically determined to produce a different class or amount of immunoglobulin antibodies may produce different types of ICs.
Besides FCGR2A and CD40/CD40L, the association of the BLK gene with susceptibility to KD further supports the hypothesis that a genetically determined difference in rate or type of antibody produced in response to an infection is involved in KD. BLK is a gene encoding B-lymphoid tyrosine kinase, expressed primarily in the B cell lineage and is part of one of the members of the Src family of kinases downstream of the B cell receptor. B cell receptor signaling is important for secretion of antibodies and B cell activation. An important example of B cell negative regulation, is through the CD40/CD40L pathway to repress T cell proliferation (116). GWAS showed that the most significant association of the BLK gene with KD was in individuals carrying the risk T allele of rs273640 in the promoter region and the first intron of the BLK gene (8p23.1) (117). During the acute phase of KD, this risk allele is associated with lower expression of BLK in peripheral blood B cells altering B cell function (116). One consequence might be impaired or abnormal antibody production. Another genetic variant in BLK region, rs2254546 at an intergenic region between FAM167A and BLK genes, was significantly associated with KD in the Japanese population (113).
Thus, both CD40/CD40L and BLK genetic variant associations may result in KD patients having a different immunoglobulin production speed, quantity or immunoglobulin isotype classes and therefore differing from healthy children in types of ICs formed. Also, the differences in FcγRs due to functional polymorphisms in the IgG receptor gene FCGR2A, contribute to an aberrant response in which ICs are central players.
In view of the evidence of aberration in KD patients in genes involved in the production of immunoglobulin and class switching, or clearance of ICs, the dramatic effect of IVIG may be due to several possible effects on IC-mediated inflammation. IVIG administration results in rapid resolution of symptoms and reduction in rash, fever and conjunctival injection in the majority of patients. There are several possible mechanisms by which IVIG might induce such a rapid resolution of inflammation in KD. Antibodies in IVIG might neutralize the unknown toxin or antigen that triggers the inflammatory process. However, an alternative explanation is that antibodies in IVIG might compete with endogenous antibody, to alter the size or composition of the ICs. Alternatively, the immunoglobulin might compete with the ICs for binding to FcγRs, thus reducing the aberrant cellular activation occurring in KD. In a more generic view, IVIG treatment may depend on the F(ab′)2 portion of immunoglobulins which mediates inhibition of the antibodies, cytokines and receptors. Alternatively, it may depend on the Fc portion which is associated with inhibition of interaction with complement, regulation of DCs, B cells and FcγR binding and activation. Attempts to define the mechanisms of IVIG action in other diseases may be relevant to KD. Debre et al. (118) found that in acute immune thrombocytopenic purpura (ITP), purified Fc fragments alone were an effective treatment.
Nagelkerke et al. (119) found that IVIG had an inhibiting effect on FcγRs from monocyte-derived macrophages and this blocking was most effective in the presence of dimeric and multimeric IgGs. Preference toward dimeric/multimeric IgGs could be linked to the fact that most FcγRs can only interact with antibodies in the form of ICs. Abe et al. (120) found monocytes and macrophages were suppressed following IVIG infusion due to down-regulation of FcγRIA and FcγR3A. These studies suggest that IVIG may compete with ICs for binding to the FcγRs, preventing activation of these cells by ICs.
IVIG may also act through DCs which are involved in antigen capture and presentation of cell-bound antigens to lymphocytes, initiating T cell responses. IVIG modulates DC functions and is associated with induction of regulatory T cells (Treg) via cyclooxygenase (COX)-2-depedent prostaglandin E2 (PGE2) (121). This effect was Fab' dependent, and affected by sialylation of immunoglobulin. The importance of Treg's is highlighted by the observation that Tregs stimulated via Fc secrete IL-10 (122), and IL-10 levels were significantly higher in KD patients with CAA and in IVIG non-responders before IVIG treatment (123). IC bind to Fc receptors enabling priming of CD8 cytotoxic T cell responses and expansion of Treg cells. ICs are more active in triggering DC activation than antigen alone and trigger stronger immune responses (109). We hypothesize that IVIG infusion in KD inhibits binding of ICs to the DCs, reducing immunological responses.
There is also evidence that IVIG affects Treg secretion of anti-inflammatory cytokines such as TGF-β and IL-10 (124). Immunoglobulin can induce Treg production and function by promoting STAT5 expression. CD8+ T cells are also abnormally activated during acute KD, and the greater the degree of activation the higher the chance of IVIG resistance (43).
Neutrophil activation is also affected by IVIG. Sialic acid-binding immunoglobulin-like lectin 9 (Siglec) is a surface molecule expressed on neutrophils. IVIG contains antibodies against Siglecs that induce cell death (125), which requires only the Fab' portion of the IgG (126). Thus, although neutrophils express Fc receptors, IVIG may affect neutrophil viability through a number of mechanisms in addition to competing with ICs for bind to the FcγRs.
In summary multiple pathways are targeted by IVIG to reduce fever, tissue inflammation, and prevent CAA. However, further studies are required to understand in depth the mechanisms of action of IVIG that could lead to more effective and targeted therapeutic approaches in KD.
The 50-year-old mystery as to the cause of KD remains to be solved. We still do not know whether it is caused by an infectious organism, a toxin, a chemical substance or other agent. ICs seem to be important in the etiopathogenesis of KD and may provide a clue to the triggering antigen.
The large number of studies which have documented ICs in the plasma or serum of patients with KD suggests that they play an immunopathogenic role in the disease. The fact that many of the genes now established as being associated with KD are those controlling B cells (BLK), immunoglobulin class switching (CD40/CD40L) or recognition of ICs' (FCGR2) support a role for the antibody response and clearance of antibody in KD. As the ICs are likely to contain antigens which have initiated the immune response, identification of the antigen within the ICs may be an important step in identifying the causative agent of KD. Future studies, using modern proteomic methods to isolate ICs in KD, and characterization of the antigen(s) may lead to identification of the causative agent as well as improved understanding of the inflammatory process.
SM, PL, and ML: conceptualization. SM: writing—original draft. SM, PL, and ML: writing—reviewing & editing.
ML and SM have received a research grant from COSMIC (Children of St. Mary's Intensive Care) (R&D 21) for work related to this manuscript.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We thank Mr. Andreas Menikou for his constructive feedback on the figures, Dr. Victoria Wright (Imperial College London), Dr. Jethro Herberg (Imperial College London), and Dr. Shea Hamilton (Imperial College London) for their constructive discussion.
1. Kawasaki T. [Acute febrile mucocutaneous syndrome with lymphoid involvement with specific desquamation of the fingers and toes in children]. Arerugi. (1967) 16:178–222.
2. Burns JC, Glodé MP. Kawasaki syndrome. Lancet. (2004) 364:533–44. doi: 10.1016/S0140-6736(04)16814-1
3. Xu SX, McCormick JK. Staphylococcal superantigens in colonization and disease. Front Cell Infect Microbiol. (2012) 2:52. doi: 10.3389/fcimb.2012.00052
4. Curtis N, Chan B, Levin M. Toxic shock syndrome toxin-secreting Staphylococcus aureus in Kawasaki syndrome. Lancet. (1994) 343:299. doi: 10.1016/S0140-6736(94)91149-5
5. Morita A, Imada Y, Igarashi H, Yutsudo T. Serologic evidence that streptococcal superantigens are not involved in the pathogenesis of Kawasaki disease. Microbiol Immunol. (1997) 41:895–900. doi: 10.1111/j.1348-0421.1997.tb01947.x
6. Gupta-Malhotra M, Viteri-Jackson A, Thomas W, Zabriskie JB. Antibodies to highly conserved peptide sequence of staphylococcal and streptococcal superantigens in Kawasaki disease. Exp Mol Pathol. (2004) 76:117–21. doi: 10.1016/j.yexmp.2003.12.003
7. Tahara M, Baba K, Waki K, Arakaki Y. Analysis of Kawasaki disease showing elevated antibody titres of Yersinia pseudotuberculosis. Acta Paediatr. (2006) 95:1661–4. doi: 10.1080/08035250600750080
8. Usui D, Ishii Y, Akaike H, Isumi H, Komura H, Kawasaki K, et al. [Yersinia pseudotuberculosis type 4a infection meeting the diagnostic criteria for Kawasaki disease complicated by disseminated intravascular coagulation]. Kansenshogaku zasshi J Jap Assoc Infect Dis. (2005) 79:895–9. doi: 10.11150/kansenshogakuzasshi1970.79.895
9. Chou CT, Chang JS, Ooi SE, Huo AP, Chang SJ, Chang HN, et al. Serum anti-Yersinia antibody in Chinese patients with Kawasaki disease. Arch Med Res. (2005) 36:14–8. doi: 10.1016/j.arcmed.2004.09.004
10. Tomita S, Kato H, Fujimoto T, Inoue O, Koga Y, Kuriya N. Cytopathogenic protein in filtrates from cultures of Propionibacterium acnes isolated from patients with Kawasaki disease. Br Med J. (1987) 295:1229–32. doi: 10.1136/bmj.295.6608.1229
11. Ebrahim M, Gabay M, Rivas-Chacon RF. Evidence of acute mycoplasma infection in a patient with incomplete and atypical kawasaki disease: a case report. Case Rep Med. (2011) 2011:606920. doi: 10.1155/2011/606920
12. Lee MN, Cha JH, Ahn HM, Yoo JH, Kim HS, Sohn S, et al. Mycoplasma pneumoniae infection in patients with Kawasaki disease. Kor J Pediatr. (2011) 54:123–7. doi: 10.3345/kjp.2011.54.3.123
13. Normann E, Naas J, Gnarpe J, Backman H, Gnarpe H. Demonstration of Chlamydia pneumoniae in cardiovascular tissues from children with Kawasaki disease. Pediatr Infect Dis J. (1999) 18:72–3. doi: 10.1097/00006454-199901000-00020
14. Carter RF, Haynes ME, Morton J. Rickettsia-like bodies and splenitis in Kawasaki disease. Lancet. (1976) 2:1254–5. doi: 10.1016/S0140-6736(76)91192-2
15. Weir WR, Bouchet VA, Mitford E, Taylor RF, Smith H. Kawasaki disease in European adult associated with serological response to Coxiella burnetii. Lancet. (1985) 2:504. doi: 10.1016/S0140-6736(85)90440-4
16. Muso E, Fujiwara H, Yoshida H, Hosokawa R, Yashiro M, Hongo Y, et al. Epstein-Barr virus genome-positive tubulointerstitial nephritis associated with Kawasaki disease-like coronary aneurysms. Clin Nephrol. (1993) 40:7–15.
17. Burns JC, Geha RS, Schneeberger EE, Newburger JW, Rosen FS, Glezen LS, et al. Polymerase activity in lymphocyte culture supernatants from patients with Kawasaki disease. Nature. (1986) 323:814–6. doi: 10.1038/323814a0
18. Lin CY, Chen IC, Cheng TI, Liu WT, Hwang B, Chiang BN. Virus-like particles with reverse transcriptase activity associated with Kawasaki disease. J Med Virol. (1992) 38:175–82. doi: 10.1002/jmv.1890380305
19. Okano M, Thiele GM, Sakiyama Y, Matsumoto S, Purtilo DT. Adenovirus infection in patients with Kawasaki disease. J Med Virol. (1990) 32:53–7. doi: 10.1002/jmv.1890320109
20. Schulz TF, Hoad JG, Whitby D, Tizard EJ, Dillon MJ, Weiss RA. A measles virus isolate from a child with Kawasaki disease: sequence comparison with contemporaneous isolates from 'classical' cases. J Gen Virol. (1992) 73 (Pt 6):1581–6. doi: 10.1099/0022-1317-73-6-1581
21. Newburger JW, Takahashi M, Burns JC, Beiser AS, Chung KJ, Duffy CE, et al. The treatment of Kawasaki syndrome with intravenous gamma globulin. N Engl J Med. (1986) 315:341–7. doi: 10.1056/NEJM198608073150601
22. Campbell AJ, Burns JC. Adjunctive therapies for Kawasaki disease. J Infect. (2016) 72(Suppl):S1–5. doi: 10.1016/j.jinf.2016.04.015
23. Burns JC, Capparelli EV, Brown JA, Newburger JW, Glode MP. Intravenous gamma-globulin treatment and retreatment in Kawasaki disease. US/Canadian Kawasaki Syndrome Study Group. Pediatr Infect Dis J. (1998) 17:1144–8. doi: 10.1097/00006454-199812000-00009
24. Tremoulet AH, Best BM, Song S, Wang S, Corinaldesi E, Eichenfield JR, et al. Resistance to intravenous immunoglobulin in children with Kawasaki disease. J Pediatr. (2008) 153:117–21. doi: 10.1016/j.jpeds.2007.12.021
25. Sittiwangkul R, Pongprot Y, Silvilairat S, Phornphutkul C. Management and outcome of intravenous gammaglobulin-resistant Kawasaki disease. Singapore Med J. (2006) 47:780–4.
26. Levin M, Burns JC, Gordon JB. Warfarin plus aspirin or aspirin alone for patients with giant coronary artery aneurysms secondary to Kawasaki disease? Cardiology. (2014) 129:174–7. doi: 10.1159/000366052
27. Cohen S, Tacke CE, Straver B, Meijer N, Kuipers IM, Kuijpers TW. A child with severe relapsing Kawasaki disease rescued by IL-1 receptor blockade and extracorporeal membrane oxygenation. Ann Rheum Dis. (2012) 71:2059–61. doi: 10.1136/annrheumdis-2012-201658
28. Shafferman A, Birmingham JD, Cron RQ. High dose Anakinra for treatment of severe neonatal Kawasaki disease: a case report. Pediatr Rheumatol Online J. (2014) 12:26. doi: 10.1186/1546-0096-12-26
29. Tremoulet AH, Pancoast P, Franco A, Bujold M, Shimizu C, Onouchi Y, et al. Calcineurin inhibitor treatment of intravenous immunoglobulin-resistant Kawasaki disease. J Pediatr. (2012) 161:506–12 e1. doi: 10.1016/j.jpeds.2012.02.048
30. Newburger JW, Takahashi M, Burns JC. Kawasaki disease. J Am Coll Cardiol. (2016) 67:1738–49. doi: 10.1016/j.jacc.2015.12.073
31. Tremoulet AH, Devera G, Best BM, Jimenez-Fernandez S, Sun X, Jain S, et al. Increased incidence and severity of Kawasaki disease among Filipino-Americans in San Diego county. Pediatr Infect Dis J. (2011) 30:909–11. doi: 10.1097/INF.0b013e31821e52c6
32. Lee NH, Choi HJ, Kim YH. Clinical usefulness of serum procalcitonin level in distinguishing between Kawasaki disease and other infections in febrile children. Korean J Pediatr. (2017) 60:112–7. doi: 10.3345/kjp.2017.60.4.112
33. Ting EC, Capparelli EV, Billman GF, Lavine JE, Matsubara T, Burns JC. Elevated gamma-glutamyltransferase concentrations in patients with acute Kawasaki disease. Pediatr Infect Dis J. (1998) 17:431–2. doi: 10.1097/00006454-199805000-00020
34. Katayama K, Matsubara T, Fujiwara M, Koga M, Furukawa S. CD14+CD16+ monocyte subpopulation in Kawasaki disease. Clin Exp Immunol. (2000) 121:566–70. doi: 10.1046/j.1365-2249.2000.01321.x
35. Ueno Y, Takano N, Kanegane H, Yokoi T, Yachie A, Miyawaki T, et al. The acute phase nature of interleukin 6: studies in Kawasaki disease and other febrile illnesses. Clin Exp Immunol. (1989) 76:337–42.
36. Hoang LT, Shimizu C, Ling L, Naim AN, Khor CC, Tremoulet AH, et al. Global gene expression profiling identifies new therapeutic targets in acute Kawasaki disease. Genome Med. (2014) 6:541. doi: 10.1186/s13073-014-0102-6
37. Guo MM, Tseng WN, Ko CH, Pan HM, Hsieh KS, Kuo HC. Th17- and Treg-related cytokine and mRNA expression are associated with acute and resolving Kawasaki disease. Allergy. (2015) 70:310–8. doi: 10.1111/all.12558
38. Rowley AH, Shulman ST, Mask CA, Finn LS, Terai M, Baker SC, et al. IgA plasma cell infiltration of proximal respiratory tract, pancreas, kidney, and coronary artery in acute Kawasaki disease. J Infect Dis. (2000) 182:1183–91. doi: 10.1086/315832
39. Rowley AH, Shulman ST. Pathogenesis and management of Kawasaki disease. Expert Rev Anti Infect Ther. (2010) 8:197–203. doi: 10.1586/eri.09.109
40. Takahashi K, Oharaseki T, Naoe S, Wakayama M, Yokouchi Y. Neutrophilic involvement in the damage to coronary arteries in acute stage of Kawasaki disease. Pediatr Int. (2005) 47:305–10. doi: 10.1111/j.1442-200x.2005.02049.x
41. Brown TJ, Crawford SE, Cornwall ML, Garcia F, Shulman ST, Rowley AH. CD8 T lymphocytes and macrophages infiltrate coronary artery aneurysms in acute Kawasaki disease. J Infect Dis. (2001) 184:940–3. doi: 10.1086/323155
42. Takahashi K, Oharaseki T, Yokouchi Y. Histopathological aspects of cardiovascular lesions in Kawasaki disease. Int J Rheumat Dis. (2018) 21:31–5. doi: 10.1111/1756-185X.13207
43. Ye Q, Gong FQ, Shang SQ, Hu J. Intravenous immunoglobulin treatment responsiveness depends on the degree of CD8+ T cell activation in Kawasaki disease. Clin Immunol. (2016) 171:25–31. doi: 10.1016/j.clim.2016.08.012
44. Janeway CJ, Travers P, Walport M, Shlomchik MJ. Immunobiology: The Immune System in Health and Disease. 5th ed. New York, NY: Garland Science (2001).
45. Yilmaz A, Rowley A, Schulte DJ, Doherty TM, Schroder NW, Fishbein MC, et al. Activated myeloid dendritic cells accumulate and co-localize with CD3+ T cells in coronary artery lesions in patients with Kawasaki disease. Exp Mol Pathol. (2007) 83:93–103. doi: 10.1016/j.yexmp.2007.01.007
46. Liu YJ. Dendritic cell subsets and lineages, and their functions in innate and adaptive immunity. Cell. (2001) 106:259–62. doi: 10.1016/S0092-8674(01)00456-1
47. Pachman LM, Herold BC, Davis AT, Hang LM, Schaller JG, Beckwith B, et al. Immune complexes in Kawasaki syndrome: a review. Prog Clin Biol Res. (1987) 250:193–207.
48. Yeaman MR. Platelets: at the nexus of antimicrobial defence. Nat Rev Microbiol. (2014) 12:426–37. doi: 10.1038/nrmicro3269
49. Ronnelid J, Tejde A, Mathsson L, Nilsson-Ekdahl K, Nilsson B. Immune complexes from SLE sera induce IL10 production from normal peripheral blood mononuclear cells by an FcgammaRII dependent mechanism: implications for a possible vicious cycle maintaining B cell hyperactivity in SLE. Ann Rheum Dis. (2003) 62:37–42. doi: 10.1136/ard.62.1.37
50. Chia D, Barnett EV, Yamagata J, Knutson D, Restivo C, Furst D. Quantitation and characterization of soluble immune complexes precipitated from sera by polyethylene glycol (PEG). Clin Exp Immunol. (1979) 37:399–407.
51. Theofilopoulos AN, Wilson CB, Dixon FJ. The Raji cell radioimmune assay for detecting immune complexes in human sera. J Clin Invest. (1976) 57:169–82. doi: 10.1172/JCI108257
52. Mannik M. Experimental models for immune complex-mediated vascular inflammation. Acta Med Scan Suppl. (1987) 715:145–55. doi: 10.1111/j.0954-6820.1987.tb09915.x
53. Fossard C, Thompson RA. Mucocutaneous lymph-node syndrome (Kawasaki disease): probable soluble-complex disorder. Br Med J. (1977) 1:883. doi: 10.1136/bmj.1.6065.883
54. Sawa F. Circulating Immune complexes in MCLS. Pediatr Int. (1979) 21:82-. doi: 10.1111/j.1442-200X.1979.tb00210.x
55. Weindling AM, Levinsky RJ, Marshall WC, Hood J. Circulating immune complexes in mucocutaneous lymph-node syndrome (Kawasaki disease). Arch Dis Child. (1979) 54:241–2. doi: 10.1136/adc.54.3.241
56. Eluthesen K, Marchette NJ, Melish ME, Hicks RM. Circulating immune complexes in Kawasaki's disease: detection by Clq binding assay. In: Proceedings of 21st Interscience Conference on Antimicrobial Agents and Chemotherapy, Chicago, IL (1981).
57. Furuse A, Matsuda I. Circulating immune complex in the mucocutaneous lymph node syndrome. Eur J Pediatr. (1983) 141:50–1. doi: 10.1007/BF00445669
58. Yanase Y, Kawasaki T, Yoshinoya S, Aikawa T, Hashimoto Y, Mitamura T, et al. [A study of immune complexes in Kawasaki disease]. Arerugi. (1984) 33:59–65.
59. Miyata K, Kawakami K, Onimaru T, Baba Y, Ono S, Hokonohara M, et al. Circulating immune complexes and granulocytes chemotaxis in Kawasaki disease. Jpn Circ J. (1984) 48:1350–3. doi: 10.1253/jcj.48.1350
60. Takiguchi M, Tamura T, Goto M, Kusakawa S, Milgrom F, Kano K. Immunological studies on Kawasaki disease. I. Appearance of Hanganutziu-Deicher antibodies. Clin Exp Immunol. (1984) 56:345–52.
61. Mason WH, Jordan SC, Sakai R, Takahashi M, Bernstein B. Circulating immune complexes in Kawasaki syndrome. Pediatr Infect Dis. (1985) 4:48–51. doi: 10.1097/00006454-198501000-00012
62. Ono S, Onimaru T, Kawakami K, Hokonohara M, Miyata K. Impaired granulocyte chemotaxis and increased circulating immune complexes in Kawasaki disease. J Pediatr. (1985) 106:567–70. doi: 10.1016/S0022-3476(85)80073-1
63. Levin M, Holland PC, Nokes TJ, Novelli V, Mola M, Levinsky RJ, et al. Platelet immune complex interaction in pathogenesis of Kawasaki disease and childhood polyarteritis. Br Med J. (1985) 290:1456–60. doi: 10.1136/bmj.290.6480.1456
64. Levin M, Holland PC, Novelli V. Platelet immune complex interaction in the pathogenesis of Kawasaki disease. Prog Clin Biol Res. (1987) 250:227–37.
65. Lin CY, Hwang B. Serial immunologic studies in patients with mucocutaneous lymph node syndrome (Kawasaki disease). Ann Allergy. (1987) 59:291–7.
66. Fujimoto T, Kato H, Inoue O, Tomita S, Koga Y. Immune complex study of biopsy specimens from Kawasaki disease patients. Prog Clin Biol Res. (1987) 250:209–17.
67. Ohshio G, Furukawa F, Khine M, Yoshioka H, Kudo H, Hamashima Y. High levels of IgA-containing circulating immune complex and secretory IgA in Kawasaki disease. Microbiol Immunol. (1987) 31:891–8. doi: 10.1111/j.1348-0421.1987.tb03150.x
68. Salcedo JR, Greenberg L, Kapur S. Renal histology of mucocutaneous lymph node syndrome (Kawasaki disease). Clin Nephrol. (1988) 29:47–51.
69. Salo E, Kekomaki R, Pelkonen P, Ruuskanen O, Viander M, Wagner O. Kawasaki disease: monitoring of circulating immune complexes. Eur J Pediatr. (1988) 147:377–80. doi: 10.1007/BF00496414
70. Li CR, Yang XQ, Shen J, Li YB, Jiang LP. Immunoglobulin G subclasses in serum and circulating immune complexes in patients with Kawasaki syndrome. Pediatr Infect Dis J. (1990) 9:544–7. doi: 10.1097/00006454-199008000-00003
71. Koike R. The effect of immunoglobulin on immune complexes in patients with Kawasaki disease (MCLS). Acta Paediatr Jpn. (1991) 33:300–9. doi: 10.1111/j.1442-200X.1991.tb01558.x
72. Kusakawa S, Heiner DC. Elevated levels of immunoglobulin E in the acute febrile mucocutaneous lymph node syndrome. Pediatr Res. (1976) 10:108–11. doi: 10.1203/00006450-197602000-00007
73. Furukawa S, Matsubara T, Motohashi T, Sasai K, Nakachi S, Umezawa Y, et al. Increased expression of Fc epsilon R2/CD23 on peripheral blood B lymphocytes and serum IgE levels in Kawasaki disease. Int Arch Allergy Appl Immunol. (1991) 95:7–12. doi: 10.1159/000235446
74. Cloutier N, Allaeys I, Marcoux G, Machlus KR, Mailhot B, Zufferey A, et al. Platelets release pathogenic serotonin and return to circulation after immune complex-mediated sequestration. Proc Natl Acad Sci USA. (2018) 115:E1550–e9. doi: 10.1073/pnas.1720553115
75. Duerschmied D, Suidan GL, Demers M, Herr N, Carbo C, Brill A, et al. Platelet serotonin promotes the recruitment of neutrophils to sites of acute inflammation in mice. Blood. (2013) 121:1008–15. doi: 10.1182/blood-2012-06-437392
76. Dhodapkar KM, Kaufman JL, Ehlers M, Banerjee DK, Bonvini E, Koenig S, et al. Selective blockade of inhibitory Fcgamma receptor enables human dendritic cell maturation with IL-12p70 production and immunity to antibody-coated tumor cells. Proc Natl Acad Sci USA. (2005) 102:2910–5. doi: 10.1073/pnas.0500014102
77. Mason W, Jordan S, Sakai R, Takahashi M. Lack of effect of gamma-globulin infusion on circulating immune complexes in patients with Kawasaki syndrome. Pediatr Infect Dis J. (1988) 7:94–9. doi: 10.1097/00006454-198802000-00006
78. Falcini F, Trapani S, Turchini S, Farsi A, Ermini M, Keser G, et al. Immunological findings in Kawasaki disease: an evaluation in a cohort of Italian children. Clin Exp Rheumatol. (1997) 15:685–9.
79. Digeon M, Laver M, Riza J, Bach JF. Detection of circulating immune complexes in human sera by simplified assays with polyethylene glycol. J Immunol Methods. (1977) 16:165–83. doi: 10.1016/0022-1759(77)90051-5
80. Morrow WJ, Wharton M, Stricker RB, Levy JA. Circulating immune complexes in patients with acquired immune deficiency syndrome contain the AIDS-associated retrovirus. Clin Immunol Immunopathol. (1986) 40:515–24. doi: 10.1016/0090-1229(86)90196-0
81. Coppock JS, Scott DL, Carter SD, Moreland TM, Robinson MW. Fibronectin in polyethylene glycol precipitates: evidence for a role in immune complexes. Rheumatol Int. (1986) 6:79–83. doi: 10.1007/BF00541509
82. Sharma VK, Kalonia DS. Polyethylene glycol-induced precipitation of interferon Alpha-2a followed by vacuum drying: development of a novel process for obtaining a dry, stable powder. AAPS PharmSci. (2004) 6:E4. doi: 10.1208/ps060104
83. Myllyla G, Vaheri A, Penttinen K. Detection and characterization of immune complexes by the platelet aggregation test. II. Circulating complexes. Clin Exp Immunol. (1971) 8:399–408.
84. Biberfeld G, Norberg R. Circulating immune complexes in Mycoplasma pneumoniae infection. J Immunol. (1974) 112:413–5.
85. Agnello V, Winchester RJ, Kunkel HG. Precipitin reactions of the C1q component of complement with aggregated gamma-globulin and immune complexes in gel diffusion. Immunology. (1970) 19:909–19.
86. Mowbray JF, Hoffbrand AV, Holborow EJ, Seah PP, Fry L. Circulating immune complexes in dermatitis herpetiformis. Lancet. (1973) 1:400–2. doi: 10.1016/S0140-6736(73)90253-5
87. Johnson A, Mowbray JF, Porter KA. Detection of circulating immune complexes in pathological human sera. Lancet. (1975) 305:762–5. doi: 10.1016/S0140-6736(75)92433-2
88. Jewell DP, MacLennan IC. Circulating immune complexes in inflammatory bowel disease. Clin Exp Immunol. (1973) 14:219–26.
89. Nydegger UE, Lambert PH, Gerber H, Miescher PA. Circulating immune complexes in the serum in systemic lupus erythematosus and in carriers of hepatitis B antigen. Quantitation by binding to radiolabeled C1q. J Clin Invest. (1974) 54:297–309. doi: 10.1172/JCI107765
90. Hay FC, Nineham LJ, Roitt IM. Routine assay for the detection of immune complexes of known immunoglobulin class using solid phase C1q. Clin Exp Immunol. (1976) 24:396–400.
91. Eisenberg RA, Theofilopoulos AN, Dixon FJ. Use of bovine conglutinin for the assay of immune complexes. J Immunol. (1977) 118:1428–34.
92. Mathsson L, Lampa J, Mullazehi M, Ronnelid J. Immune complexes from rheumatoid arthritis synovial fluid induce FcgammaRIIa dependent and rheumatoid factor correlated production of tumour necrosis factor-alpha by peripheral blood mononuclear cells. Arthr Res Ther. (2006) 8:R64. doi: 10.1186/ar1926
93. Crowley-Nowick PA, Campbell E, Schrohenloher RE, Mestecky J, Mestecky J, Jackson S. Polyethylene glycol precipitates of serum contain a large proportion of uncomplexed immunoglobulins and C3. Immunol Invest. (1996) 25:91–101. doi: 10.3109/08820139609059293
94. Candia J, Cheung F, Kotliarov Y, Fantoni G, Sellers B, Griesman T, et al. Assessment of Variability in the SOMAscan Assay. Sci Rep. (2017) 7:14248. doi: 10.1038/s41598-017-14755-5
95. Zolg DP, Wilhelm M, Schmidt T, Medard G, Zerweck J, Knaute T, et al. Proteometools: systematic characterization of 21 post-translational protein modifications by liquid chromatography tandem mass spectrometry (LC-MS/MS) using synthetic peptides. Mol Cell Proteom. (2018) 17:1850–63. doi: 10.1074/mcp.TIR118.000783
96. Holman RC, Christensen KY, Belay ED, Steiner CA, Effler PV, Miyamura J, et al. Racial/ethnic differences in the incidence of Kawasaki syndrome among children in Hawaii. Hawaii Med J. (2010) 69:194–7.
97. Eleftheriou D, Levin M, Shingadia D, Tulloh R, Klein NJ, Brogan PA. Management of Kawasaki disease. Arch Dis Child. (2014) 99:74–83. doi: 10.1136/archdischild-2012-302841
98. Fujita Y, Nakamura Y, Sakata K, Hara N, Kobayashi M, Nagai M, et al. Kawasaki disease in families. Pediatrics. (1989) 84:666–9.
99. Wu SF, Chang JS, Wan L, Tsai CH, Tsai FJ. Association of IL-1Ra gene polymorphism, but no association of IL-1beta and IL-4 gene polymorphisms, with Kawasaki disease. J Clin Lab Anal. (2005) 19:99–102. doi: 10.1002/jcla.20059
100. Burns JC, Shimizu C, Shike H, Newburger JW, Sundel RP, Baker AL, et al. Family-based association analysis implicates IL-4 in susceptibility to Kawasaki disease. Genes Immun. (2005) 6:438–44. doi: 10.1038/sj.gene.6364225
101. Shimizu C, Jain S, Davila S, Hibberd ML, Lin KO, Molkara D, et al. Transforming growth factor-beta signaling pathway in patients with Kawasaki disease. Circ Cardiovasc Genet. (2011) 4:16–25. doi: 10.1161/CIRCGENETICS.110.940858
102. Breunis WB, Biezeveld MH, Geissler J, Kuipers IM, Lam J, Ottenkamp J, et al. Polymorphisms in chemokine receptor genes and susceptibility to Kawasaki disease. Clin Exp Immunol. (2007) 150:83–90. doi: 10.1111/j.1365-2249.2007.03457.x
103. Ikeda K, Ihara K, Yamaguchi K, Muneuchi J, Ohno T, Mizuno Y, et al. Genetic analysis of MMP gene polymorphisms in patients with Kawasaki disease. Pediatr Res. (2008) 63:182–5. doi: 10.1203/PDR.0b013e31815ef224
104. Kim KY, Kim DS. Recent Advances in Kawasaki Disease. Yonsei Med J. (2016) 57:15–21. doi: 10.3349/ymj.2016.57.1.15
105. Jin HS, Kim HB, Kim BS, Lee JK, Seo EJ, Yoo HW, et al. The IL-10 (-627 A/C) promoter polymorphism may be associated with coronary aneurysms and low serum albumin in Korean children with Kawasaki disease. Pediatr Res. (2007) 61 (5 Pt 1):584–7. doi: 10.1203/pdr.0b013e3180459fb5
106. Cheung YF, Huang GY, Chen SB, Liu XQ, Xi L, Liang XC, et al. Inflammatory gene polymorphisms and susceptibility to Kawasaki disease and its arterial sequelae. Pediatrics. (2008) 122:e608–14. doi: 10.1542/peds.2008-0646
107. Onouchi Y, Gunji T, Burns JC, Shimizu C, Newburger JW, Yashiro M, et al. ITPKC functional polymorphism associated with Kawasaki disease susceptibility and formation of coronary artery aneurysms. Nat Genet. (2008) 40:35–42. doi: 10.1038/ng.2007.59
108. Khor CC, Davila S, Breunis WB, Lee YC, Shimizu C, Wright VJ, et al. Genome-wide association study identifies FCGR2A as a susceptibility locus for Kawasaki disease. Nat Genet. (2011) 43:1241–6. doi: 10.1038/ng.981
109. Nimmerjahn F, Ravetch JV. Fcgamma receptors as regulators of immune responses. Nat Rev Immunol. (2008) 8:34–47. doi: 10.1038/nri2206
110. Elgueta R, Benson MJ, de Vries VC, Wasiuk A, Guo Y, Noelle RJ. Molecular mechanism and function of CD40/CD40L engagement in the immune system. Immunol Rev. (2009) 229:152–72. doi: 10.1111/j.1600-065X.2009.00782.x
111. Wang CL, Wu YT, Liu CA, Lin MW, Lee CJ, Huang LT, et al. Expression of CD40 ligand on CD4+ T-cells and platelets correlated to the coronary artery lesion and disease progress in Kawasaki disease. Pediatrics. (2003) 111:E140–7. doi: 10.1542/peds.111.2.e140
112. Kuo HC, Wang CL, Yang KD, Lo MH, Hsieh KS, Li SC, et al. Plasma prostaglandin E2 levels correlated with the prevention of intravenous immunoglobulin resistance and coronary artery lesions formation via CD40L in Kawasaki Disease. PLOS ONE. (2016) 11:e0161265. doi: 10.1371/journal.pone.0161265
113. Onouchi Y, Ozaki K, Burns JC, Shimizu C, Terai M, Hamada H, et al. A genome-wide association study identifies three new risk loci for Kawasaki disease. Nat Genet. (2012) 44:517–21. doi: 10.1038/ng.2220
114. Kuo HC, Chao MC, Hsu YW, Lin YC, Huang YH, Yu HR, et al. CD40 Gene polymorphisms associated with susceptibility and coronary artery lesions of Kawasaki disease in the Taiwanese population. Sci World J. (2012) 2012:520865. doi: 10.1100/2012/520865
115. Onouchi Y, Onoue S, Tamari M, Wakui K, Fukushima Y, Yashiro M, et al. CD40 ligand gene and Kawasaki disease. Eur J Hum Genet. (2004) 12:1062–8. doi: 10.1038/sj.ejhg.5201266
116. Chang CJ, Kuo HC, Chang JS, Lee JK, Tsai FJ, Khor CC, et al. Replication and meta-analysis of GWAS identified susceptibility loci in Kawasaki disease confirm the importance of B lymphoid tyrosine kinase (BLK) in disease susceptibility. PLOS ONE. (2013) 8:e72037. doi: 10.1371/journal.pone.0072037
117. Lou J, Zhong R, Shen N, Lu XZ, Ke JT, Duan JY, et al. Systematic confirmation study of GWAS-identified genetic variants for Kawasaki disease in a Chinese population. Sci Rep. (2015) 5:8194. doi: 10.1038/srep08194
118. Debre M, Bonnet MC, Fridman WH, Carosella E, Philippe N, Reinert P, et al. Infusion of Fc gamma fragments for treatment of children with acute immune thrombocytopenic purpura. Lancet. (1993) 342:945–9. doi: 10.1016/0140-6736(93)92000-J
119. Nagelkerke SQ, Dekkers G, Kustiawan I, van de Bovenkamp FS, Geissler J, Plomp R, et al. Inhibition of FcgammaR-mediated phagocytosis by IVIg is independent of IgG-Fc sialylation and FcgammaRIIb in human macrophages. Blood. (2014) 124:3709–18. doi: 10.1182/blood-2014-05-576835
120. Abe J, Jibiki T, Noma S, Nakajima T, Saito H, Terai M. Gene expression profiling of the effect of high-dose intravenous Ig in patients with Kawasaki disease. J Immunol. (2005) 174:5837–45. doi: 10.4049/jimmunol.174.9.5837
121. Trinath J, Hegde P, Sharma M, Maddur MS, Rabin M, Vallat JM, et al. Intravenous immunoglobulin expands regulatory T cells via induction of cyclooxygenase-2-dependent prostaglandin E2 in human dendritic cells. Blood. (2013) 122:1419–27. doi: 10.1182/blood-2012-11-468264
122. Burns JC, Song Y, Bujold M, Shimizu C, Kanegaye JT, Tremoulet AH, et al. Immune-monitoring in Kawasaki disease patients treated with infliximab and intravenous immunoglobulin. Clin Exp Immunol. (2013) 174:337–44. doi: 10.1111/cei.12182
123. Wang Y, Wang W, Gong F, Fu S, Zhang Q, Hu J, et al. Evaluation of intravenous immunoglobulin resistance and coronary artery lesions in relation to Th1/Th2 cytokine profiles in patients with Kawasaki disease. Arthr Rheumat. (2013) 65:805–14. doi: 10.1002/art.37815
124. Li J, Chen T, Yuan C, Zhao G, Xu M, Li X, et al. Effect of intravenous immunoglobulin on the function of Treg cells derived from immunosuppressed mice with Pseudomonas aeruginosa pneumonia. PLOS ONE. (2017) 12:e0176843. doi: 10.1371/journal.pone.0176843
125. von Gunten S, Schaub A, Vogel M, Stadler BM, Miescher S, Simon HU. Immunologic and functional evidence for anti-Siglec-9 autoantibodies in intravenous immunoglobulin preparations. Blood. (2006) 108:4255–9. doi: 10.1182/blood-2006-05-021568
Keywords: Kawasaki disease, immune complexes, immunopathology, IVIG, pathogens
Citation: Menikou S, Langford PR and Levin M (2019) Kawasaki Disease: The Role of Immune Complexes Revisited. Front. Immunol. 10:1156. doi: 10.3389/fimmu.2019.01156
Received: 19 October 2018; Accepted: 07 May 2019;
Published: 12 June 2019.
Edited by:
Soohyun Kim, Konkuk University, South KoreaReviewed by:
James Harris, Monash University, AustraliaCopyright © 2019 Menikou, Langford and Levin. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Stephanie Menikou, cy5tZW5pa291QGltcGVyaWFsLmFjLnVr
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.