 Michelle A. Williams
Michelle A. Williams Amy O'Callaghan
Amy O'Callaghan Sinéad C. Corr
Sinéad C. Corr- Department of Microbiology, School of Genetics and Microbiology, Moyne Institute of Preventative Medicine, Trinity College Dublin, Dublin, Ireland
The IL-1 cytokines are a newly expanded family, with each of its 11 members playing an important role in health and disease. Typically acting as pro- or anti-inflammatory mediators of first-line innate immunity, their production is particularly important in the context of mucosal defenses, through handling breach of the delicate epithelial barrier and mediating a local immune response to invading pathogens. Mucosal immunity is often aberrantly orchestrated in intestinal diseases, such as Inflammatory Bowel Disease (IBD). Various studies have pointed to IL-1 cytokines as being important players in IBD with context-dependent roles, either through promoting auto-inflammatory mechanisms, or alleviating disease through protection against breach of pathogens across the epithelial barrier. This mini-review will succinctly examine the role of IL-1 family members in IBD, with a special focus on the recently described IL-33 as well as IL-18, and will explore the disease models within which these cytokines have been studied. Furthermore, we will examine the evidence of interplay of these cytokines with the gut microbiota, with hopes of summarizing our current knowledge of these family members and their potential for unraveling novel molecular mechanisms of IBD pathology.
Introduction
The IL-1 family of cytokines primarily act on innate immunity to initiate inflammation in the face of local insult, thus playing a particularly important role in the pathophysiology of mucosal diseases. Members of the IL-1 family of cytokines play important, yet context-dependent roles in intestinal homeostasis and inflammation. In this review we will examine two IL-1 family cytokines, IL-33 and IL-18, in the context of mucosal immunity and with a particular focus on the pathogenesis of IBD, a chronic inflammation of the intestinal mucosa. This review will explore their role in host immunity, and describe associations of these cytokines with the host microbiota, a major component in IBD etiology. Together this review will summarize our knowledge of these newly described cytokines, and present an outlook on their individual and complex roles in IBD (Table 1).
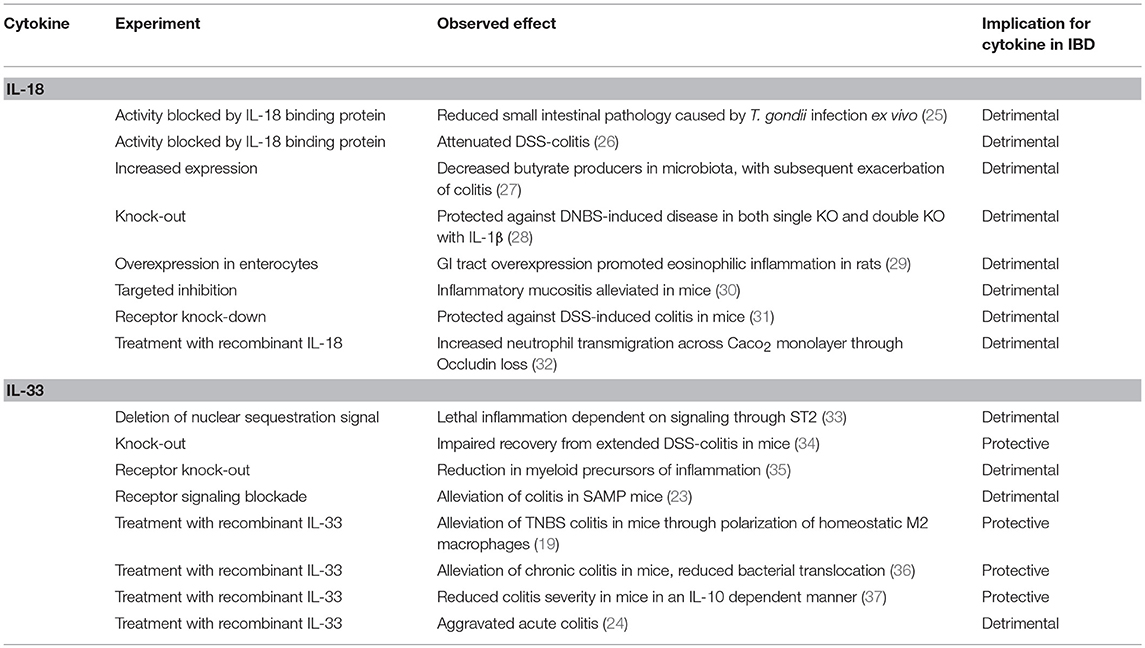
Table 1. Known effects of IL-18 and IL-33 with implications for IBD pathogenesis.
IL-33
IL-33: An Alarmin in Mucosal Immunity
The IL-1 family member IL-33 plays a unique and essential role in mucosal, front-line immunity. Previously known as IL-1F11, IL-33 is a relatively newly described cytokine, with origins tracing back to 2005 (1). It was discovered after the characterization of its cognate receptor, suppressor of tumorigenicity 2 (ST2) (2). IL-33/ST2 signaling not only acts as a front-line herald of tissue damage, but also links innate and adaptive immunity at the host mucosae through potent induction of a type 2 response in T cells, innate lymphoid cells (ILCs) and macrophages (3–5). Despite potentially playing an important role as a mediator of mucosal immunity, and being suggested as a drug target for various disorders, there are currently no IL-33-based therapies for intestinal disease. This presents an interesting opportunity for study of this cytokine and its role in IBD.
The most well-characterized aspect of IL-33 biology is its role as an alarmin: a molecular “fire-alarm” at the barrier tissues of the body, driving inflammatory and fibrotic processes during acute mucosal breach due to cell injury (6). IL-33 is constitutively expressed in epithelial and endothelial cells, and following translation is stored as a full-length, biologically active molecule in the nucleus where it binds to chromatin (7). Following lysis of the cell through destructive mechanisms, IL-33 in the nucleus is immediately available to act as an early signifier of damage, through recruitment of neutrophils, eosinophils, natural killer (NK) cells, and by amplifying a type 2 (Th2, ILC2, M2-like macrophage) response in order to initiate fibrosis and wound healing (8, 9). Interestingly, not only being important for primed release of the cytokine, sequestration of IL-33 in the nucleus allows it to act as a transcriptional regulator, where it can bind to the p65 subunit of NFκB to activate endothelial cells (10). Unlike other members of the IL-1 family, IL-33 does not require processing through an inflammasome in order to achieve biological activity and in fact is inactivated by caspase cleavage (11). However, N-terminal cleavage by neutrophil elastase and cathepsin G proteases, which are found in the microenvironment during inflammation, can increase its potency (12). This again highlights the primary role of IL-33 in orchestrating the response to cellular destruction.
IL-33 in Intestinal Disease
Expression of IL-33 and its receptor ST2 has been well-established in the GI tract, being an important amplifier of innate immunity at the gut mucosa (13). While IL-33 is expressed largely at the mucosae and in myofibroblasts, its receptor is expressed mainly on immune cells, such as ILC2s, Tregs, T helper cells, and CD8+ T cells (14) This allows IL-33/ST2 signaling to act as a bridge between tissue damage and immune system orchestration, which may be a critical component in intestinal immunity. In an experiment whereby the N-terminus of IL-33 was altered such that it could not associate with chromatin, the result was the formation of a whole-body inflammatory response with splenomegaly, increased lymph node infiltration and indeed development of colitis (7). This response was then ablated by knock-down of the ST2 receptor. Not only does this demonstrate the importance of nuclear sequestration of IL-33 but also highlights the potency of IL-33/ST2 signaling in the acute inflammatory response and a potential role in intestinal disease.
IL-33 presents an interesting role in IBD, which is perhaps complicated by the divergent immune pathophysiologies of Crohn's disease (CD) and Ulcerative Colitis (UC). Studies of patient biopsies have shown an increase in IL-33 levels in patients with active IBD, in particular UC, compared with healthy controls (15). Interestingly, UC-associated IL-33 is found in myofibroblasts, which tend to localize at the base of inflamed ulcerations during disease (16). Furthermore, blockade of IL-33/ST2 signaling has been shown to alleviate active disease, suggesting a pathogenic role for the cytokine (17).
In contrast with these findings, IL-33-deficient mice have been shown to be highly susceptible to colitis and colorectal cancer, which would suggest a role as an important protective mediator of intestinal immunity (18). This offers a contradictory and complex role for IL-33 in IBD. This is evidenced by results from various mouse models of disease, including but not limited to the most widely used methods of dextran-sodium sulfate (DSS)-induced and trinitrobenzene sodium (TNBS)-induced colitis. While DSS-induced colitis is largely T cell independent and mediated by chemical damage, TNBS colitis development is dependent on induction of a Th1 response. Given its association with boosting type 2 immunity, IL-33 may act as a “balancing” cytokine in TNBS colitis. This has been shown in a model of TNBS colitis, whereby recombinant IL-33 administration was shown to attenuate disease development through induction of M2-like macrophage polarization (19). Furthermore, IL-33 has been found to ameliorate TNBS colitis, in a manner that was Foxp3 dependent, through promoting a Th2 and Treg response (20). Agreeing with this, IL-33 has been shown to enhance Foxp3+ Treg cell expansion in the intestine via TGF-β (21). Attempting to clarify this, another study employed the use of SAMP colitis mice, which are characterized by development of T cell-driven enteric inflammation. SAMP mice show an early Th1 stage, which is followed by establishment of a chronic Th2-mediated disease (22). UC-like disease in SAMP mice was found to correlate with expression of full-length IL-33 in IECs, and blockade of the IL-33/ST2 signaling pathway was beneficial (23). This demonstrates a somewhat complex role for IL-33 in IBD, wherein its effector role may be determined by the T cell response pattern and intrinsic differences in CD and UC immunology.
In the context of DSS-induced colitis, IL-33 plays a varying role depending on the temporal stage of the disease. For instance, one particular study showed that administration of recombinant IL-33 exacerbated acute colitis, but ameliorated colitis in a chronic model of disease in a manner dependent on amphiregulin-EGFR signaling (24). IL-33 induced neutrophil influx during both stages of disease, which may have contributed to its exacerbating effects on acute colitis through nitric oxide (NO) immunopathology, but was shown by the authors of the study to reduce translocation of pathobionts across the impaired epithelium during chronic disease. This divergence, even through use of the same disease agent, points toward IL-33 expression patterns as an important tool for study of the early and late immune response in IBD, suggesting that despite its role in promoting acute inflammation, it may act to limit chronic inflammation in long-term disease.
IL-18
IL-18 as a Pro-inflammatory Regulator
The role of IL-18 in intestinal disease is largely related to its activity in regulating pro-inflammatory responses. Discovered in 1989 in mouse serum following challenge with bacterial LPS, IL-18 was first identified as a booster of IFNγ activity produced by monocytic cells (38). To compare it with its sister cytokines, IL-18 most similarly resembles IL-1β, in both structure and their association with the inflammasome, a proteolytic complex through which IL-1β and IL-18 precursor molecules become biologically active by caspase-1 cleavage. GI commensals are critically important in regulating intestinal inflammasome assembly, and this has been demonstrated in new-born mammals that feature progressive microbial colonization, accompanied by intestinal barrier formation and immune system maturation. This perhaps further highlights a potential role for aberrant IL-18 in IBD, a disease characterized by an excessive inflammatory response to microbial products. Furthermore, loss-of-function single nucleotide polymorphisms (SNPs) in the IL-18 gene result in an imbalance of the Th1/Th2 response, which promotes host susceptibility to CD (39). Other meta-analysis studies have supported this observation (40).
IL-18 is generally a pro-inflammatory mediator, and its production may be a key etiological factor for patients with IBD (41, 42). Pro-IL-18 is produced by a wide range of cell types, including epithelial cells, myeloid cells and lymphocytes, and following inflammasome activation carries out a wide range of effector functions, including promoting the production of IFNγ, priming of NK cell cytotoxicity (43) and stimulating the differentiation of Th1 cells (44). Despite this, the NLRP6 inflammasome itself is a known regulator of colonic homeostasis, predominantly expressed in intestinal epithelial cells (IECs) with a key role in mucosal renewal, proliferation and secretion (45). Interestingly, deficiency in the NLRP6 inflammasome is detrimental in DSS-induced colitis, in a manner related to insufficient IL-18. Likewise, caspase-1 deficiency is linked to increased DSS severity. These reports would suggest that while IL-18 is a pro-inflammatory mediator, its baseline activity is important for intestinal integrity through unknown mechanisms.
On the other hand, IL-18 has been also shown to contribute to the breakdown of the mucosal barrier, provoking inflammation and amplifying damage elicited to the intestinal epithelium during disease. Clinical studies have correlated increased epithelial secretion of IL-18 with increased severity of IBD (46). This study was supported by Nowarski et al., where deletion of IL-18 receptor (IL-18R) from IECs shielded mice from DSS-induced colitis (31). Transgenic mice deficient in IL-1β, IL-18 or both cytokines protected against TNBS colitis induction in mice (28). Thus, the double knockout increased the protective effects against intestinal inflammation, perhaps due to the inhibition of two converging inflammatory pathways. IL-18 production is elevated in IECs following infection with human immunodeficiency virus (HIV), causing IEC apoptosis through the activation of caspase-1 and caspase-3 (47), two programmed cell death proteases. This study also described how IL-18 disrupts the tight junctions that maintain intestinal epithelium integrity. Collectively, these results suggest that IL-18 overproduction generally contributes to an increase in the permeability of the intestinal monolayer, exacerbating intestinal inflammation. Contribution to inflammation in such a regard presents an opportunity to alleviate IBD symptoms, which is being exploited through development of a monoclonal antibody against IL-18. This is currently in phase one of clinical trials (clinicaltrials.gov ID: NCT01035645).
IL-18 and IL-33 in Host-Microbe Interactions
The current working hypothesis for the etiology of IBD focuses on the loss of host tolerance for the resident microbiota. As such, an exploration of the role of IL-18 and IL-33 should critically examine their known associations with the microbiota, and the ways in which this may alter host pathology during IBD. The role of IL-18 in this regard is well-established in the literature. Indeed, intestinal IL-18 levels progressively increase over the first 5 weeks of post-natal development, harmonizing with microbial colonization (48). Levy et al., demonstrate how gut microbiota bi-products, such as metabolites, influence NLRP6 inflammasome signaling, IL-18 epithelial cell secretion and downstream secretion of anti-microbial peptides (AMPs) (49). This study is supported by Elinav et al., where NLRP6-inflammasome deficient mice had impaired production of IL-18 and concomitant dysbiosis, characterized by an expansion of the bacterial phyla Bacteroidetes (Prevotellaceae) and TM7 (50). This deficiency in NLRP6 and IL-18 results in a failure to produce AMPs, which are essential in mediating pathogenic bacterial clearance. Recently, NLRP6 was shown to prevent the colonization of IBD-inducing bacteria, the mucolytic A. muciniphila, which can induce colitis in both specific pathogen free and germ free (GF) IL10−/− mice. This is mediated by IL-18 (51).
Interestingly, GF mice colonized with a complex human gut-derived microbiota see a transient increase in IL-18 without tissue damage or inflammation, suggesting an ongoing homeostatic response (52). IL-22 is a classic Th1-cell-associated pro-inflammatory cytokine that exacerbates ileitis following infection with protozoa, e.g., Toxoplasma gondii (53). Interestingly, IL-22 knockout mice were associated with reduced production of Th1-promoting IL-18. IL-22 not only influenced the expression of IL-18 messenger RNA (mRNA) in IECs after infection with Toxoplasma gondii or Citrobacter rodentium infection, but was found to promote a homeostatic production of active IL-18 in the ileum (54), which not only is crucial for host defense but also contributes to inflammation, while assisting in microbial clearance. This suggests a double-edged sword role for IL-18, whereby it contributes to inflammation but in doing so, protects the host from pathogenic invasion.
Conversely, very little is currently known about IL-33 crosstalk with the gut microbiota, however some studies have suggested a potential for interaction. One such study in SAMP mice showed that IL-33 is induced in ex-GF mice following transplantation of commensals (55). Furthermore, IL-33 expression has a number of known interactions with the microbial-sensing Toll-like receptors (TLRs), and is indeed induced by pathogen-associated molecular patterns (PAMPs), perhaps as a protective mechanism of the host (56). Further highlighting its potential for enabling host-microbiota crosstalk, IL-33/ST2 interactions can repress the TLR2 pathway through sequestration of the adaptor molecule MYD88 (57).
The susceptibility of IL-33 deficient mice to colitis has been shown to be a result of the ability of IL-33 to promote IgA production from B cells, which notably plays a major role in maintaining microbial homeostasis in the intestine (18). This suggests that IL-33 may indirectly alter the microbiota to protect against colitis through promotion of IgA production, which is already known to be a protective factor in IBD. Supporting this, IL-33 deficient mice developed dysbiosis, characterized by increased levels of mucolytic and colitogenic bacteria, which drastically altered the microbial landscape of the gut making the mice more susceptible to colitis. This effect was found to be dependent on IL-1α release, and colitis susceptibility in IL-33 deficiency could be reversed through either reconstitution of a homeostatic microbiota or by IL-1α ablation. This provides a potential role for IL-33 to quell the colitogenic effects of its more sinister sibling cytokines.
Closing Remarks
In summation, the IL-1 family of cytokines are regulators of mucosal immunity in a manner that appears highly context-dependent. IL-18 itself is a pro-inflammatory mediator capable of exacerbating disease, despite evidence that it can promote homeostasis in some circumstances. The role of IL-33 in IBD is more complex, and perhaps related to disease stage. While both of these cytokines promote early pro-inflammatory responses to effect front-line protection against mucosal breach and pathogenic invasion, their baseline expression is indeed important for the maintenance of overall intestinal integrity. As a result, poorly timed or excessive production of bioactive IL-1 family members may provide a key step in IBD development, with vast potential for therapeutic intervention.
Author Contributions
MW, AO, and SC developed, wrote, and revised the content in this manuscript.
Funding
SC is supported by a Starting Investigator Research Grant from Science Foundation Ireland [SFI] [grant number 11/SIRG/B2099] and a Litwin IBD Pioneer award from the Crohns and Colitis Foundation of America.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Schmitz J, Owyang A, Oldham E, Song Y, Murphy E, McClanahan TK, et al. IL-33, an interleukin-1-like cytokine that signals via the IL-1 receptor-related protein ST2 and induces T helper type 2-associated cytokines. Immunity. (2005) 23:479–90. doi: 10.1016/j.immuni.2005.09.015
2. Tominaga S. A putative protein of a growth specific cDNA from BALB/c-3T3 cells is highly similar to the extracellular portion of mouse interleukin 1 receptor. FEBS Lett. (1989) 258:301–4. doi: 10.1016/0014-5793(89)81679-5
3. Han M, Rajput C, Hong JY, Lei J, Hinde JL, Wu Q, et al. The innate cytokines IL-25, IL-33, and TSLP cooperate in the induction of type 2 innate lymphoid cell expansion and mucous metaplasia in rhinovirus-infected immature mice. J Immunol. (2017) 199:1308–18. doi: 10.4049/jimmunol.1700216
4. He R, Yin H, Yuan B, Liu T, Luo L, Huang P, et al. IL-33 improves wound healing through enhanced M2 macrophage polarization in diabetic mice. Mol Immunol. (2017) 90:42–9. doi: 10.1016/j.molimm.2017.06.249
5. Lohning M, Stroehmann A, Coyle AJ, Grogan JL, Lin S, Gutierrez-Ramos JC, et al. T1/ST2 is preferentially expressed on murine Th2 cells, independent of interleukin 4, interleukin 5, and interleukin 10, and important for Th2 effector function. Proc Natl Acad Sci USA. (1998) 95:6930–5. doi: 10.1073/pnas.95.12.6930
6. Moussion C, Ortega N, Girard JP. The IL-1-like cytokine IL-33 is constitutively expressed in the nucleus of endothelial cells and epithelial cells in vivo: a novel 'alarmin'? PloS ONE. (2008) 3:e3331. doi: 10.1371/journal.pone.0003331
7. Carriere V, Roussel L, Ortega N, Lacorre DA, Americh L, Aguilar L, et al. IL-33, the IL-1-like cytokine ligand for ST2 receptor, is a chromatin-associated nuclear factor in vivo. Proc Natl Acad Sci USA. (2007) 104:282–7. doi: 10.1073/pnas.0606854104
8. Oshio T, Komine M, Tsuda H, Tominaga SI, Saito H, Nakae S, et al. Nuclear expression of IL-33 in epidermal keratinocytes promotes wound healing in mice. J Dermatol Sci. (2017) 85:106–14. doi: 10.1016/j.jdermsci.2016.10.008
9. Rak GD, Osborne LC, Siracusa MC, Kim BS, Wang K, Bayat A, et al. IL-33-dependent group 2 innate lymphoid cells promote cutaneous wound healing. J Investig Dermatol. (2016) 136:487–96. doi: 10.1038/JID.2015.406
10. Choi YS, Park JA, Kim J, Rho SS, Park H, Kim YM, et al. Nuclear IL-33 is a transcriptional regulator of NF-kappaB p65 and induces endothelial cell activation. Biochem Biophys Res Commun. (2012) 421:305–11. doi: 10.1016/j.bbrc.2012.04.005
11. Luthi AU, Cullen SP, McNeela EA, Duriez PJ, Afonina IS, Sheridan C, et al. Suppression of interleukin-33 bioactivity through proteolysis by apoptotic caspases. Immunity. (2009) 31:84–98. doi: 10.1016/j.immuni.2009.05.007
12. Lefrancais E, Roga S, Gautier V, Gonzalez-de-Peredo A, Monsarrat B, Girard JP, et al. IL-33 is processed into mature bioactive forms by neutrophil elastase and cathepsin G. Proc Natl Acad Sci USA. (2012) 109:1673–8. doi: 10.1073/pnas.1115884109
13. Beltran CJ, Nunez LE, Diaz-Jimenez D, Farfan N, Candia E, Heine C, et al. Characterization of the novel ST2/IL-33 system in patients with inflammatory bowel disease. Inflamm Bowel Dis. (2010) 16:1097–107. doi: 10.1002/ibd.21175
14. Griesenauer B, Paczesny S. The ST2/IL-33 axis in immune cells during inflammatory diseases. Front Immunol. (2017) 8:475. doi: 10.3389/fimmu.2017.00475
15. Kobori A, Yagi Y, Imaeda H, Ban H, Bamba S, Tsujikawa T, et al. Interleukin-33 expression is specifically enhanced in inflamed mucosa of ulcerative colitis. J Gastroenterol. (2010) 45:999–1007. doi: 10.1007/s00535-010-0245-1
16. Sponheim J, Pollheimer J, Olsen T, Balogh J, Hammarstrom C, Loos T, et al. Inflammatory bowel disease-associated interleukin-33 is preferentially expressed in ulceration-associated myofibroblasts. Am J Pathol. (2010) 177:2804–15. doi: 10.2353/ajpath.2010.100378
17. Sedhom MA, Pichery M, Murdoch JR, Foligne B, Ortega N, Normand S, et al. Neutralisation of the interleukin-33/ST2 pathway ameliorates experimental colitis through enhancement of mucosal healing in mice. Gut. (2013) 62:1714–23. doi: 10.1136/gutjnl-2011-301785
18. Malik A, Sharma D, Zhu Q, Karki R, Guy CS, Vogel P, et al. IL-33 regulates the IgA-microbiota axis to restrain IL-1alpha-dependent colitis and tumorigenesis. J Clin Investig. (2016) 126:4469–81. doi: 10.1172/JCI88625
19. Tu L, Chen J, Xu D, Xie Z, Yu B, Tao Y, et al. IL-33-induced alternatively activated macrophage attenuates the development of TNBS-induced colitis. Oncotarget. (2017) 8:27704–14. doi: 10.18632/oncotarget.15984
20. Duan L, Chen J, Zhang H, Yang H, Zhu P, Xiong A, et al. Interleukin-33 ameliorates experimental colitis through promoting Th2/Foxp3(+) regulatory T-cell responses in mice. Mol Med. (2012) 18:753–61. doi: 10.2119/molmed.2011.00428
21. Schiering C, Krausgruber T, Chomka A, Frohlich A, Adelmann K, Wohlfert EA, et al. The alarmin IL-33 promotes regulatory T-cell function in the intestine. Nature. (2014) 513:564–8. doi: 10.1038/nature13577
22. Mikulski Z, Johnson R, Shaked I, Kim G, Nowyhed H, Goodman W, et al. SAMP1/YitFc mice develop ileitis via loss of CCL21 and defects in dendritic cell migration. Gastroenterology. (2015) 148:783–93.e785. doi: 10.1053/j.gastro.2015.01.027
23. Pastorelli L, Garg RR, Hoang SB, Spina L, Mattioli B, Scarpa M, et al. Epithelial-derived IL-33 and its receptor ST2 are dysregulated in ulcerative colitis and in experimental Th1/Th2 driven enteritis. Proc Natl Acad Sci USA. (2010) 107:8017–22. doi: 10.1073/pnas.0912678107
24. Monticelli LA, Osborne LC, Noti M, Tran SV, Zaiss DM, Artis D. IL-33 promotes an innate immune pathway of intestinal tissue protection dependent on amphiregulin-EGFR interactions. Proc Natl Acad Sci USA. (2015) 112:10762–7. doi: 10.1073/pnas.1509070112
25. Struck D, Frank I, Enders S, Steinhoff U, Schmidt C, Stallmach A, et al. Treatment with interleukin-18 binding protein ameliorates Toxoplasma gondii-induced small intestinal pathology that is induced by bone marrow cell-derived interleukin-18. Eur J Microbiol Immunol. (2012) 2:249–57. doi: 10.1556/EuJMI.2.2012.3.11
26. Sivakumar PV, Westrich GM, Kanaly S, Garka K, Born TL, Derry JM, et al. Interleukin 18 is a primary mediator of the inflammation associated with dextran sulphate sodium induced colitis: blocking interleukin 18 attenuates intestinal damage. Gut. (2002) 50:812–20. doi: 10.1136/gut.50.6.812
27. Tye H, Yu CH, Simms LA, de Zoete MR, Kim ML, Zakrzewski M, et al. NLRP1 restricts butyrate producing commensals to exacerbate inflammatory bowel disease. Nat Commun. (2018) 9:3728. doi: 10.1038/s41467-018-06125-0
28. Impellizzeri D, Siracusa R, Cordaro M, Peritore AF, Gugliandolo E, Mancuso G, et al. Therapeutic potential of dinitrobenzene sulfonic acid (DNBS)-induced colitis in mice by targeting IL-1β and IL-18. Biochem Pharmacol. (2018) 155:150–61. doi: 10.1016/j.bcp.2018.06.029
29. Verma AK, Kandikattu HK, Manohar M, Shukla A, Upparahalli Venkateshaiah S, Zhu X, et al. Intestinal overexpression of IL-18 promotes eosinophils-mediated allergic disorders. Immunology. (2019). doi: 10.1111/imm.13051. [Epub ahead of print].
30. Lima-Junior RC, Freitas HC, Wong DV, Wanderley CW, Nunes LG, Leite LL, et al. Targeted inhibition of IL-18 attenuates irinotecan-induced intestinal mucositis in mice. Br J Pharmacol. (2014) 171:2335–50. doi: 10.1111/bph.12584
31. Nowarski R, Jackson R, Gagliani N, de Zoete MR, Palm NW, Bailis W, et al. Epithelial IL-18 equilibrium controls barrier function in colitis. Cell. (2015) 163:1444–56. doi: 10.1016/j.cell.2015.10.072
32. Lapointe TK, Buret AG. Interleukin-18 facilitates neutrophil transmigration via myosin light chain kinase-dependent disruption of occludin, without altering epithelial permeability. Am J Physiol Gastrointest Liver Physiol. (2012) 302:G343–51. doi: 10.1152/ajpgi.00202.2011
33. Bessa J, Meyer CA, de Vera Mudry MC, Schlicht S, Smith SH, Iglesias A, et al. Altered subcellular localization of IL-33 leads to non-resolving lethal inflammation. J Autoimmun. (2014) 55:33–41. doi: 10.1016/j.jaut.2014.02.012
34. Lopetuso LR, De Salvo C, Pastorelli L, Rana N, Senkfor HN, Petito V, et al. IL-33 promotes recovery from acute colitis by inducing miR-320 to stimulate epithelial restitution and repair. Proc Natl Acad Sci USA. (2018) 115:E9362–70. doi: 10.1073/pnas.1803613115
35. Lei-Leston AC, Murphy AG, Maloy KJ. Epithelial cell inflammasomes in intestinal immunity and inflammation. Front Immunol. (2017) 8:1168. doi: 10.3389/fimmu.2017.01168
36. Grobeta P, Doser K, Falk W, Obermeier F, Hofmann C. IL-33 attenuates development and perpetuation of chronic intestinal inflammation. Inflamm Bowel Dis. (2012) 18:1900–9. doi: 10.1002/ibd.22900
37. Sattler S, Ling GS, Xu D, Hussaarts L, Romaine A, Zhao H, et al. IL-10-producing regulatory B cells induced by IL-33 (Breg(IL-33)) effectively attenuate mucosal inflammatory responses in the gut. J Autoimmun. (2014) 50:107–22. doi: 10.1016/j.jaut.2014.01.032
38. Okamura H, Nagata K, Komatsu T, Tanimoto T, Nukata Y, Tanabe F, et al. A novel costimulatory factor for gamma interferon induction found in the livers of mice causes endotoxic shock. Infect Immunity. (1995) 63:3966–72.
39. Pan HF, Leng RX, Ye DQ. Lack of association of interleukin-18 gene promoter−607 A/C polymorphism with susceptibility to autoimmune diseases: a meta-analysis. Lupus. (2011) 20:945–51. doi: 10.1177/0961203311400114
40. Gao SJ, Zhang L, Lu W, Wang L, Chen L, Zhu Z, et al. Interleukin-18 genetic polymorphisms contribute differentially to the susceptibility to Crohn's disease. World J Gastroenterol. (2015) 21:8711–22. doi: 10.3748/wjg.v21.i28.8711
41. Loher F, Bauer C, Landauer N, Schmall K, Siegmund B, Lehr HA, et al. The interleukin-1 beta-converting enzyme inhibitor pralnacasan reduces dextran sulfate sodium-induced murine colitis and T helper 1 T-cell activation. J Pharmacol Exp Ther. (2004) 308:583–90. doi: 10.1124/jpet.103.057059
42. Siegmund B, Fantuzzi G, Rieder F, Gamboni-Robertson F, Lehr HA, Hartmann G, et al. Neutralization of interleukin-18 reduces severity in murine colitis and intestinal IFN-gamma and TNF-alpha production. Am J Physiol Regul Integr Comp Physiol. (2001) 281:R1264–1273. doi: 10.1152/ajpregu.2001.281.4.R1264
43. Chaix J, Tessmer MS, Hoebe K, Fuséri N, Ryffel B, Dalod M, et al. Cutting edge: priming of NK cells by IL-18. J Immunol. (2008) 181:1627–31. doi: 10.4049/jimmunol.181.3.1627
44. Matsumoto S, Tsuji-Takayama K, Aizawa Y, Koide K, Takeuchi M, Ohta T, et al. Interleukin-18 activates NF-kappaB in murine T helper type 1 cells. Biochem Biophys Res Commun. (1997) 234:454–7. doi: 10.1006/bbrc.1997.6665
45. Wlodarska M, Thaiss CA, Nowarski R, Henao-Mejia J, Zhang JP, Brown EM, et al. NLRP6 inflammasome orchestrates the colonic host-microbial interface by regulating goblet cell mucus secretion. Cell. (2014) 156:1045–59. doi: 10.1016/j.cell.2014.01.026
46. Pizarro TT, Michie MH, Bentz M, Woraratanadharm J, Smith MF, Foley E, et al. IL-18, a novel immunoregulatory cytokine, is up-regulated in Crohn's disease: expression and localization in intestinal mucosal cells. J Immunol. (1999) 162:6829–35. doi: 10.1016/S0016-5085(98)84319-9
47. Allam O, Samarani S, Mehraj V, Jenabian MA, Tremblay C, Routy JP, et al. HIV induces production of IL-18 from intestinal epithelial cells that increases intestinal permeability and microbial translocation. PLoS ONE. (2018) 13:e0194185. doi: 10.1371/journal.pone.0194185
48. Kempster SL, Belteki G, Forhead AJ, Fowden AL, Catalano RD, Lam BY, et al. Developmental control of the Nlrp6 inflammasome and a substrate, IL-18, in mammalian intestine. Am J Physiol Gastrointest Liver Physiol. (2011) 300:G253–63. doi: 10.1152/ajpgi.00397.2010
49. Levy M, Thaiss CA, Zeevi D, Dohnalová L, Zilberman-Schapira G, Mahdi JA, et al. Microbiota-modulated metabolites shape the intestinal microenvironment by regulating NLRP6 inflammasome signaling. Cell. (2015) 163:1428–43. doi: 10.1016/j.cell.2015.10.048
50. Elinav E, Strowig T, Kau AL, Henao-Mejia J, Thaiss CA, Booth CJ, et al. NLRP6 inflammasome regulates colonic microbial ecology and risk for colitis. Cell. (2011) 145:745–57. doi: 10.1016/j.cell.2011.04.022
51. Seregin SS, Golovchenko N, Schaf B, Chen J, Pudlo NA, Mitchell J, et al. NLRP6 protects Il10 -/- mice from colitis by limiting colonization of Akkermansia muciniphila. Cell Rep. (2017) 19:733–45. doi: 10.1016/j.celrep.2017.03.080
52. Hayes CL, Dong J, Galipeau HJ, Jury J, McCarville J, Huang X, et al. Commensal microbiota induces colonic barrier structure and functions that contribute to homeostasis. Sci Rep. (2018) 8:14184. doi: 10.1038/s41598-018-32366-6
53. Munoz M, Liesenfeld O, Heimesaat MM. Immunology of Toxoplasma gondii. Immunol Rev. (2011) 240:269–85. doi: 10.1111/j.1600-065X.2010.00992.x
54. Muñoz M, Eidenschenk C, Ota N, Wong K, Lohmann U, Kühl AA, et al. Interleukin-22 induces interleukin-18 expression from epithelial cells during intestinal infection. Immunity. (2015) 42:321–31. doi: 10.1016/j.immuni.2015.01.011
55. De Salvo C, Wang XM, Pastorelli L, Mattioli B, Omenetti S, Buela KA, et al. IL-33 Drives eosinophil infiltration and pathogenic type 2 helper T-cell immune responses leading to chronic experimental ileitis. Am J Pathol. (2016) 186:885–98. doi: 10.1016/j.ajpath.2015.11.028
56. Natarajan C, Yao SY, Sriram S. TLR3 agonist poly-ic induces IL-33 and promotes myelin repair. PloS ONE. (2016) 11:e0152163. doi: 10.1371/journal.pone.0152163
Keywords: IBD, IL-18, IL-33, colitis, microbiota
Citation: Williams MA, O'Callaghan A and Corr SC (2019) IL-33 and IL-18 in Inflammatory Bowel Disease Etiology and Microbial Interactions. Front. Immunol. 10:1091. doi: 10.3389/fimmu.2019.01091
Received: 29 January 2019; Accepted: 29 April 2019;
Published: 14 May 2019.
Edited by:
Elizabeth Brint, University College Cork, IrelandReviewed by:
Vanessa Pinho, Federal University of Minas Gerais, BrazilClaudia Nold, Hudson Institute of Medical Research, Australia
Copyright © 2019 Williams, O'Callaghan and Corr. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Sinéad C. Corr, corrsc@tcd.ie