 Heidi Harjunpää
Heidi Harjunpää Marc Llort Asens
Marc Llort Asens Carla Guenther
Carla Guenther Susanna C. Fagerholm
Susanna C. Fagerholm- Research Program of Molecular and Integrative Biosciences, Faculty of Bio- and Environmental Sciences, University of Helsinki, Helsinki, Finland
The immune system and cancer have a complex relationship with the immune system playing a dual role in tumor development. The effector cells of the immune system can recognize and kill malignant cells while immune system-mediated inflammation can also promote tumor growth and regulatory cells suppress the anti-tumor responses. In the center of all anti-tumor responses is the ability of the immune cells to migrate to the tumor site and to interact with each other and with the malignant cells. Cell adhesion molecules including receptors of the immunoglobulin superfamily and integrins are of crucial importance in mediating these processes. Particularly integrins play a vital role in regulating all aspects of immune cell function including immune cell trafficking into tissues, effector cell activation and proliferation and the formation of the immunological synapse between immune cells or between immune cell and the target cell both during homeostasis and during inflammation and cancer. In this review we discuss the molecular mechanisms regulating integrin function and the role of integrins and other cell adhesion molecules in immune responses and in the tumor microenvironment. We also describe how malignant cells can utilize cell adhesion molecules to promote tumor growth and metastases and how these molecules could be targeted in cancer immunotherapy.
Introduction
Cancer and the Immune System
The immune system and cancer cells share complex interactions during tumor development. Indeed, all types of immune cells can be found in different tumors (1–3). These include lymphocytes such as T and B cells and natural killer (NK) cells, myeloid cells such as dendritic cells (DCs) and macrophages and granulocytes such as neutrophils, eosinophils, and mast cells. The immune contexture, or the frequency, location and functional orientation of different immune cell subsets, varies substantially between tumor types and also between individuals with seemingly identical cancers (2). Interestingly, correlations between immune contexture in the tumor microenvironment and clinical outcomes have been examined in various malignancies. In general, a strong infiltration of memory CD8+ T cells and T helper 1 (Th1) cells correlates with favorable prognosis while strong T helper 2 (Th2) or T helper 17 (Th17) orientation is associated with poor prognosis in terms of overall survival (2, 3). In addition, high infiltration of regulatory cells such as regulatory T cells (Tregs) and myeloid-derived suppressor cells (MDSCs) in tumors often correlates with decreased survival (4–6). Understanding the role of the immune system for tumor development has been the central focus of tumor immunology since its inception. It has become evident that immune cells are able to recognize and kill malignant cells and thus suppress tumor growth in a process known as cancer immunosurveillance (7). In addition to directly targeting the cancer cells, the immune system prevents viral infections and thus the growth of virus-induced tumors and inhibits tumor-promoting inflammation by eradicating pathogens and by clearing existing inflammation. However, it is now known that the immune system can also promote tumor growth by maintaining chronic inflammation, by shaping tumor immunogenicity and by suppressing anti-tumor immunity, and thus a concept called cancer immunoediting is currently preferred to cancer immunosurveillance (7).
Cancer immunoediting is a process in which both innate and adaptive immune systems work together to suppress/control and promote/shape tumor growth (8). In order for this process to take place, the DCs need to take up tumor antigens, migrate to lymph nodes, present tumor antigens to T cells in the lymph nodes and then activated T cells need to expand and traffic into the tumor site where they interact with the malignant cells. These events can eventually lead to tumor cell killing or immune cell exhaustion. Cell adhesion molecules such as integrins and receptors of the immunoglobulin superfamily play a crucial role in all these events.
Cell Adhesion Molecules (CAMs)
Adhesion molecules are generally divided into five groups: integrins (discussed in greater detail below), selectins, cadherins, members of the immunoglobulin superfamily (IgSF) including nectins and others such as mucins (9). In addition to the conventional adhesion molecules, certain enzymes such as vascular adhesion protein 1 (VAP-1) also play a role in cell adhesion (10).
Apart from structural differences, cell adhesion molecules also bind to different ligands. Integrins typically bind to the extracellular matrix, while selectins, cadherins, and IgSF members are associated with cell-cell adhesion (9). However, immune cell integrins also bind to soluble ligands and ligands on other cells. The cell-cell adhesion mediating molecules can further be divided by their ligands as selectins bind carbohydrates in a calcium dependent manner (11), cadherins mediate preferably homophilic bonds in a calcium-dependent manner (12) and the IgSF subfamily nectins mediate homophilic and heterophilic bonds (9).
Selectins are further divided into P-, E- and L-selectins originally based on which cell types they were found in: platelets, endothelial cells and leukocytes (however, P-selectin is also expressed on endothelial cells) (13). Selectins differ in kinetics of expression, as P-selectins are expressed within minutes and E-selectins within hours (13). Selectins are especially important for leukocyte trafficking, migration of lymphocytes to peripheral lymph nodes and to the skin. Their most prominent function is associated with the initial stage of the rolling cell adhesion cascade in which selectin binding enables rolling (14). Selectin binding also initiates the subsequent integrin dependent step of slow rolling and cell arrest as selectin binding together with chemokine receptor activation initiates inside out signaling leading to integrin activation (see later sections) (13, 15). L-selectins in particular display a force dependent binding e.g., L-selectin forms catch bonds with its ligands (bonds that strengthen under force). Catch bonds dominate until the applied force reaches a force threshold upon which slip bonds are formed (bonds that weaken under force) (13, 14). In other selectins this occurs to a lesser extent (13, 14).
Proper functionality of selectins is carbohydrate-dependent as is demonstrated in a disorder called leukocyte adhesion deficiency II (LAD II). LAD II is caused by a mutation in a fucose transporter protein leading to fucose not being incorporated into selectin ligands, which ultimately leads to disruption of selectin-mediated leukocyte adhesion (13). Many selectin ligands, including P-selectin glycoprotein ligand 1 (PSGL-1) which is the main ligand for all three selectins (13), mucosal addressin cell adhesion molecule-1 (MAdCAM-1) and peripheral node addressin (PNAd) belong to a glycoprotein family called mucins which function as major components of the mucous protecting epithelial cells lining the digestive, respiratory and urogenital tracts (16). Interestingly, over-expression of mucins, MUC-1 in particular, have been detected in many human malignancies and seem to promote cancer cell growth and survival (17).
Cadherins are associated with cell-cell adhesive bonds in solid tissues (12). Molecules of this family feature cadherin repeat sequences which are stabilized by calcium ions. The essential role of calcium for cadherin adhesive function is also reflected in the protein family name which is an abbreviation of “calcium-dependent adherent proteins” (12). Cadherins in turn can be subdivided into classical cadherins (type I and II), protocadherins and atypical cadherins (9, 12).
The IgSF is one of the largest and most diverse protein families (18). All members of the IgSF contain at least one immunoglobulin or immunoglobulin-like domain and most members are type I transmembrane proteins with an extracellular domain (containing the Ig domain[s]), transmembrane domain and a cytoplasmic tail. The most well-known members include major histocompatibility complex (MHC) class I and II molecules and proteins of the T cell receptor (TCR) complex (18). Intercellular adhesion molecules (ICAMs), vascular cell adhesion molecules (VCAMs), MAdCAM-1 and activated leukocyte cell adhesion molecule (ALCAM), which are important in leukocyte trafficking events, also belong to this family of adhesion receptors (19–21). Of interest, MAdCAM-1 contains both Ig and mucin domains placing it as a member of both IgSF and mucin families (19).
Yet another subfamily of the IgSF is the nectin family which members mediate cell-cell adhesion in various tissues including endothelium, epithelium and neural tissue (9). The members can form homophilic interactions with each other or heterophilic interactions with other nectins or other ligands. They can also co-operate with cadherins to establish adherens junctions (9, 22). From an immunological point of view, interactions between nectins and immune modulatory receptors such as DNAM-1 (CD226) and TIGIT are of particular interest due to their involvement in regulation of effector cell function and their recently appreciated role in anti-tumor responses (23). Interestingly, the expression of nectins is also often up-regulated in various tumors (9).
Integrins
Integrins are large heterodimers consisting of α- and β-chains that together form the intact receptor in the plasma membrane. Integrins bind to a wide variety of ligands in the extracellular matrix, on the surface of other cells and also soluble proteins. Leukocytes express various integrins while the β1-integrins (α4β1), β2-integrins (αLβ2, αMβ2, αXβ2, αDβ2), and β7-integrins [α4β7 and αE(CD103)β7] playing particularly important roles in immune cells.
β2-integrins are the predominant integrins on leukocytes (24). The different integrins in this family have different expression in different leukocyte subpopulations (25, 26). LFA-1 (αLβ2, CD11a/CD18) is expressed in all leukocytes and is the predominant integrin in lymphocytes. Mac-1 (αMβ2, CD11b/CD18, CR3) dominates on myeloid leukocytes, especially neutrophils, but is also expressed in NK cells, B cells and some T cells, whilst αXβ2 (CD11c/CD18, CR4) is most abundant on myeloid dendritic cells (DCs). αDβ2 (CD11d/CD18) is the most recently discovered β2-integrin and is expressed on neutrophils, monocytes and NK cells. LFA-1-integrin is more distantly related than the other β2-integrin family members and has a more restricted ligand binding profile compared to the other β2-integrins. It mainly binds members of the ICAM-1-5 and JAM-1 families. In contrast, Mac-1-integrin has a very broad ligand repertoire, with more than 40 reported ligands including ICAM-1-4, iC3b, fibrinogen, fibronectin, factor X, heparin, polysaccharides, and even denatured proteins (26). αXβ2 and αDβ2 are more closely related to Mac-1 than to LFA-1 and have similar, but more restricted, ligand binding properties than Mac-1.
The extracellular domains of integrins are large and consist of several domains (27). In those integrins that contain it, the αI (or A) domain is the most important ligand binding domain, and mediates Mg2+-dependent ligand binding. The I-domain consists of a central β-sheet surrounded by seven α-helices. Ligand binding happens at the MIDAS-site which provides three surface loops to co-ordinate the Mg2+ ion, whilst a glutamate or aspartate from the ligand provides the fourth coordination position. This is referred to as the I-domain open conformation. In the closed conformation, the fourth coordination position is replaced by a water molecule – this induces structural changes in the I-domain e.g., rearrangement of the metal-co-ordinating residues and a 10Å shift in the α7-helix. Ligand binding shifts the equilibrium from the closed towards the open state (27).
The I-domain forms part of the ligand binding head domain of the integrin extracellular domain, which also contains the βI-domain (that has a similar structure as the αI domain) and the propeller domain in the α-chain (27). The I-domain sits on top of the propeller domain. Structural signals can be transduced through the integrin β-chain to induce conformational changes in the integrin head-domain or from the ligand-bound head into the cell (27).
In addition to I-domain conformational changes in the ligand-bound and unbound states, integrins can undergo much larger scale structural changes (27–29) (Figure 1). When the first integrin ectodomain was crystallized, it was a surprise that the integrin was found in a bent state, with both the α-chain and the β-chain being “bent at the knee” (genu), causing the ligand-binding head to turn down towards the legs of the integrin heterodimer. Indeed, integrins can be found in bent conformation, extended/closed conformation (where the integrin is extended but the I-domain is closed) as well as extended/open conformation (extended integrin/I-domain is in the open conformation) (28).
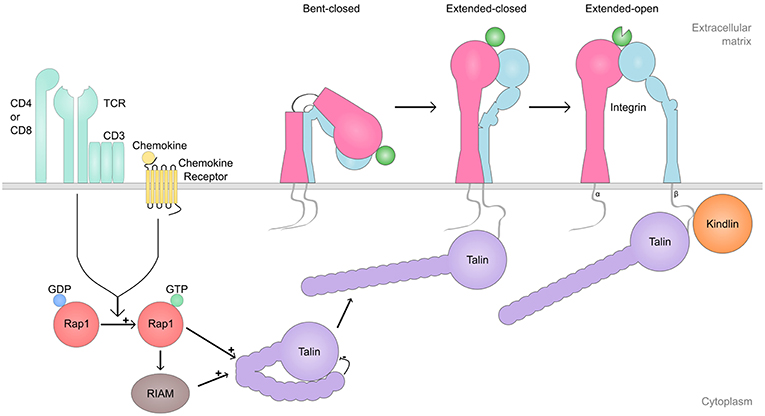
Figure 1. Integrin inside-out signaling. Shown is a simplified representation of the integrin inside-out process, which regulates integrin activation (i.e., integrin conformation switch from bent-closed or extended-closed to extended-open conformation). Cell signaling initiated by receptors such as chemokine receptors, T cell receptor (TCR), Toll-like receptors (TLR), and selectins, among others, trigger the switch from Rap1-GDP to Rap1-GTP which, either dependently of RIAM or not, activate Talin and enable its binding to the β-cytoplasmic tail of the integrin. Finally, Kindlin binds to the β-cytoplasmic tail of the integrin, and together with talin induces the separation of the cytoplasmic tails, and triggers the activation of the ligand-binding domain. The extended-open conformation of the integrin remains stable with Talin and Kindlin bound.
The transmembrane domains of integrins associate with each other at two motifs, maintaining the integrin in an inactive state (30). The intracellular domains are short and devoid of enzymatic activity. However, they are nevertheless important for regulating integrin function (see below). The cytoplasmic domains of the integrin β-chain are structurally related to each other with several important motifs that are essential for integrin regulation. In contrast, the cytoplasmic domains of the integrin α-chain are more diverse, presumably allowing different integrins to have different functional characteristics.
Inside the cell, these receptors link to the actin cytoskeleton through their cytoplasmic tails (31). In cells such as a fibroblast, they form large multiprotein complexes with intracellular molecules called focal adhesions, the composition of which have been determined by proteomic methods in recent years (31, 32). Integrins also participate in so called outside-in signaling, e.g., transmitting signals into cells through a variety of signaling pathways to change cell behavior (actin reorganization, cell migration, gene expression etc.) (33).
Integrin Regulation
Integrins are not constitutively active and able to bind ligands. Instead, their activity is regulated from inside the cell, through a process called inside-out signaling (24) (Figure 1). During this process, cell signaling initiated by other cell surface receptors, induce integrin activation. Receptors such as chemokine receptors, TCR, Toll-like receptors (TLRs), selectins as well as many other cell surface receptors, have been reported to induce integrin activation in immune cells. Integrins can also influence the activity status of other integrins, a process called integrin transregulation (34).
Ultimately, integrin activation in response to inside-out signaling is achieved by cytoplasmic factors that interact with the integrin β-chain cytoplasmic portion (Figure 1). Talin is the most well-known integrin activator. Talin binds to the membrane-proximal NPXY motif in the β-chain and is of fundamental importance for integrin activation. Talin binding to the integrin β-chain cytoplasmic domain destabilizes the transmembrane linkage between the α- and β-chain of the integrin, allowing integrin activation (30).
Kindlin is a more recently discovered integrin interaction partner which is nevertheless very important for integrin regulation (35–37). Kindlin binds to the membrane-distal NPXY-motif and a threonine-motif between the NPXY-motifs, but exactly how kindlin regulates integrin function remains incompletely understood. It has been suggested that talin is required for the conformational change of the integrin to the extended, intermediate affinity conformation, but that both talin and kindlin-3 are required for the induction of the high-affinity conformation (38, 39) (Figure 1). However, kindlin has also been reported to play a role in integrin clustering, stabilizing the integrin-ligand contact and strengthening cell adhesion (40), by recruiting downstream components such as actin, ILK, and paxillin (41–44).
The small GTPase Rap1 also plays a critical role in integrin activation (45, 46). The effects of Rap1 on integrin activation are at least in part dependent on the Rap1 interacting protein, RIAM (47). RIAM is a Rap1 effector molecule that is important for at least β2-integrin lymphocyte trafficking from blood into peripheral lymph nodes (pLNs) (45), but not for the regulation of all integrins (β3-integrin activation in platelets is independent of RIAM). However, for β2-integrins, the pathway seems to consist of Rap1/RIAM/Talin (45, 48). Very late antigen-4 (VLA-4) (α4β1, CD49d/CD29) -mediated adhesion is dependent on talin but only partly dependent on RIAM (45, 48) suggesting that also RIAM-independent Rap1/talin pathways exist. Recent studies seem to indicate that a RIAM-independent Rap1/talin pathway is of particular importance in cells that rely on quick integrin activation to function efficiently, such as neutrophils and platelets (49).
In addition to talin and kindlin, integrins interact with a multitude of cytoplasmic proteins, for example filamin A, Dok1 and 14-3-3 proteins (50) (Figure 2). Filamin A was previously thought to be a negative regulator of integrins, either by inhibiting talin binding (51) and/or by crosslinking integrin cytoplasmic domains (52). However, recent results indicate that it may instead be important for integrin-mediated shear flow adhesion and trafficking of immune cells in vivo (53). Integrin cytoplasmic domain phosphorylation has been reported for many integrins and plays a role in regulating interactions with cytoplasmic molecules and therefore further regulates integrin function (24).
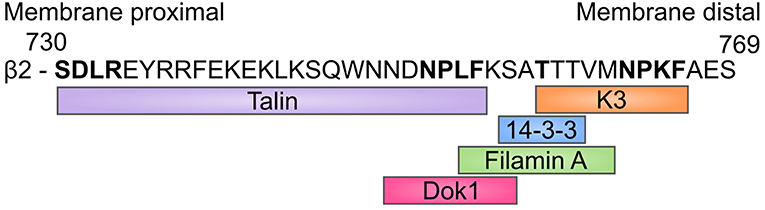
Figure 2. β2-integrin binding sites. Amino acid sequence of the β2-cytoplasmic tail where most of the main integrin binding proteins bind, and the sequences to which they bind. The amino acids highlighted in bold are of particular importance. 14-3-3 proteins only bind to Th758-phosphorylated integrin, whilst phosphorylation of this site inhibits Filamin A binding.
The Function of Integrins and Other Cell Adhesion Molecules in Immune Responses
Patients suffering from leukocyte adhesion deficiency type I (LAD-I) have lost or reduced expression of β2-integrins on their leukocytes, and these patients suffer from recurrent bacterial infections (54). Symptoms also include leukocytosis, periodontitis and delayed wound healing. In leukocyte adhesion deficiency type III (LAD-III), integrins are expressed but dysfunctional because kindlin-3 is mutated or absent, and these patients have similar symptoms as LAD-I patients (54). However, they also suffer from a Glanzmann-type bleeding disorder as kindlin-3 is required not only for β2-integrin-mediated leukocyte adhesion but also for β3-integrin-mediated platelet adhesion. These findings show that β2-integrins and their cytoplasmic regulators play fundamentally important roles in immunity (55). Studies with mice deficient for different β2-integrins have further revealed individual contributions to various leukocyte processes (56, 57).
Leukocytes traffic out of the blood stream into the lymph nodes, tissues or tumors by using the leukocyte adhesion cascade, which is regulated by sequential function of adhesion molecules (selectins, integrins, receptors of the IgSF) (58, 59). In brief, selectin-selectin ligand interactions lead to rolling of the leukocyte on endothelial cells, allowing activation of the cell by chemokines present on the endothelium. This leads to activation of integrins on the surface of the immune cell (15). LFA-1 and VLA-4 integrin activation by talin and kindlin allows firm interaction between the immune cell such as a T cell or a neutrophil and endothelial cells, which express integrin ligands such as ICAMs, VCAM-1, and MAdCAM (37, 38, 58, 60, 61). This is followed by cell spreading, Mac-1-mediated crawling (62), paracellular or transcellular extravasation, and migration into lymph nodes or tissues. In effector T cells, LFA-1 is up-regulated and constitutively activated, which contributes to the trafficking properties of these cells to peripheral tissues (63, 64). In tumors, several steps of the leukocyte trafficking process can be severely disrupted (discussed below).
Adhesion is important also in other immune cell interactions. LFA-1-ICAM-1 interaction, in particular, plays an essential role in the formation of the immunological synapse (IS) between a DC and a T cell (65–67). The structure of an IS is highly organized with key interacting molecules organized in distinct areas called supra-molecular activation complexes (SMACs) (68). The central region of the SMAC (cSMAC) is enriched in TCRs and associated molecules while LFA-1 and ICAM-1 are localized in the peripheral region of the SMAC (pSMAC) and large molecules such as CD45 and CD43 in the distal area of the SMAC (dSMAC). Also VLA-4 is localized at the pSMAC (69). Due to the crucial role for the stabilization of the immunological synapse, LFA-1 is important for T cell activation and proliferation (70, 71). In addition, talin and kindlin-3-mediated activation of LFA-1 has been shown to be important in T cell activation in vivo (71, 72). LFA-1 also provides a necessary co-stimulatory signal for T cells lowering the threshold for activation and proliferation following TCR engagement and promotes their IL-2 production (71, 73–75) In addition, LFA-1 has been reported to play a role in Th1/Th2 polarization, development and/or maintenance of Tregs and follicular T cells, and for generation of memory T cells (61, 76–79). Further, LFA-1-aided IS formation is important in the contact between cytotoxic CD8+ T cell/NK cell and the target cell such as a tumor cell and for the release of cytotoxic granules and target cell killing by CD8+ T cells and NK cells (61, 80, 81). Together, these studies show that LFA-1-ICAM-1 pathway plays a crucial role in T cell trafficking, activation and function and thus in the protection of the host from infections and cancer.
Mac-1 and αXβ2 are important receptors for complement iC3b thus mediating phagocytosis of complement-coated particles (82). Also neutrophil degranulation is dependent on Mac-1 integrin (56, 83). Therefore, Mac-1 is vital for the functionality of myeloid cells. However, interestingly, Mac-1 in myeloid cells can also function as a suppressor of immune responses (26) by e.g., inhibiting TLR-signaling in macrophages (84, 85). β2-integrins and the β2-integrin ligand ICAM-1 can also repress DC-mediated T cell activation (86–89) and Th17 differentiation (90, 91) and restrict DC trafficking from peripheral sites to lymph nodes (89, 92).
ICAM-1 is the main ligand for β2-integrins (24). It is expressed at basal levels on several cell types including fibroblasts, keratinocytes, endothelial cells, and leukocytes and its expression increases during inflammation due to inflammatory cytokines such as IFNγ, IL-1β, and TNFα (93, 94). In inflamed tissues, ICAM-1 expressed on endothelial cells binds to β2-integrins LFA-1 and Mac-1 on leukocytes and facilitates their transendothelial migration to the inflammation site. Given the role in mediating leukocyte migration, ICAM-1 up-regulation has been associated to various inflammatory, autoimmune and allergic diseases (94–96). Interestingly, ICAM-1 is also expressed in many types of tumors where it plays a dual role in tumor growth (discussed below).
VCAM-1 is predominantly expressed on endothelial cells and, similar to ICAM-1, its expression increases during inflammation due to inflammatory cytokines such as TNFα (97, 98). VCAM-1 is an important mediator of immune cell mediated rolling, adhesion and extravasation into inflamed tissues by binding to VLA-4 on leukocytes. Thus, VCAM-1 expression has been associated with several autoimmune disorders including rheumatoid arthritis and asthma. In addition, again similar to ICAM-1, VCAM-1 has been shown to play a dual role in cancer development (discussed below).
Integrins and integrin ligands therefore play crucial roles in several immune system functions relevant for tumor rejection, especially in immune cell migration and activation. Indeed, cell adhesion molecules have been shown to play both positive and negative roles in anti-tumor immunity.
Cold Tumors Often Display Dysregulated Expression of Cell Adhesion Molecules on the Tumor-Associated Vessels
Based on the immune landscape, tumors can be divided into inflamed or “hot” and non-inflamed or “cold” tumors. Hot tumors are characterized by transcripts encoding for various T cell-lineage markers, innate immune cell molecules, chemokines that regulate effector T cell recruitment and also for molecules mediating immune suppression such as PD-L1, Foxp3 and indoleamine-2,3-dioxygenase (IDO) (59, 99, 100). These tumors are highly infiltrated by tumor-infiltrating lymphocytes (TILs) but their function is inhibited due to immune suppressive tumor microenvironment. By contrast, cold tumors have low expression of the before-mentioned transcripts but instead express high levels of factors associated with angiogenesis, molecules involved in Notch and/or β-catenin pathway and serine protease inhibitors. Cold tumors are generally poorly infiltrated by TILs and effective T cell homing into the tumor remains a major obstacle for endogenous anti-tumor immunity and for the success of cancer immunotherapies. Migration of immune cells into tumors can be hampered by many factors, such as impaired chemokine expression at tumor sites, mismatch between chemokines expressed at the tumor site and the set of chemokine receptors being expressed on immune cells, fibrosis around the tumors, and abnormal tumor vasculature (101). Aberrant adhesion molecule expression on leukocytes and cancer cells enables immunoediting and evasion of immune system surveillance while aberrant expression of adhesion molecules on tumor-associated blood vessels can render the whole tumor mass inaccessible for the immune system.
Indeed, down-regulation of adhesion molecules on tumor-associated endothelial cells, a process termed as endothelial anergy, is an effective mechanism utilized by tumors to prevent immune cell trafficking into the tumor site (102, 103). Down-regulation of several adhesion molecules such as ICAM-1/2, VCAM-1, E-selectin, P-selectin, and MAdCAM-1 has been reported in tumor-associated vessels in various human malignancies (104–109) which is at least partly due to high levels of angiogenic factors such as basic fibroblast growth factor (bFGF) and vascular endothelial growth factor (VEGF) in the tumor microenvironment (105, 110). It has been demonstrated both in vitro (111) and in vivo (112) that low adhesion molecule expression on the endothelial cells due to angiogenic factors leads to diminished leukocyte-vessel wall interactions. Further, diminished leukocyte interactions with tumor endothelium is caused by down-regulation of adhesion molecules on the endothelium and not by decreased expression of LFA-1, VLA-4, or L-selectin on leukocytes (110). From a clinical perspective, up-regulation of ICAM-1 in the tumor microenvironment have been shown to be related to favorable prognosis among patients with various cancers, suggesting an enhancement in cancer immunosurveillance (113–115). Indeed, an increase in TILs was observed in ICAM-1 positive gastric and colorectal cancers compared to ICAM-1 negative tumors (114–116). In addition, Tachimoro et al. showed that more human peripheral blood mononuclear cells (PBMCs) adhered to colon carcinoma (LM-H3) cells in vitro that were transfected with ICAM-1 compared to cells with an empty vector (117). PBMC-mediated cytotoxicity was also enhanced towards ICAM-1 expressing LM-H3 cells. Further, when injected into nude mice, a significant reduction was observed in subcutaneous tumor growth and ability to metastases liver with ICAM-1+ LM-H3 cells compared to ICAM-1 negative cells. In summary, expression levels of integrin ligands on tumor endothelium clearly influences anti-tumor immune responses by affecting immune cell infiltration into tumors.
Expression of Integrins and Other Cell Adhesion Molecules Affect T Cell Infiltration Into Tumors
T cells are important for the recognition of tumor-specific antigens and for the killing of malignant cells (118, 119). Thus T cells, particularly CD8+ T cells, have been demonstrated to be crucial in protecting the host from malignant tumor growth (120, 121). Indeed, CD8+ T cell infiltration and high CD8+ T cell/Treg ratio in the tumor microenvironment has been associated with favorable prognosis in different human malignancies (4, 122, 123). In order for CD8+ T cells to mediate tumor cell killing, they must first become into contact with the malignant cells. However, T cells often fail to infiltrate the tumor tissue, thereby causing a major obstacle for successful treatment of cancer patients with immunotherapy (99, 124). The mechanism of T cell homing to healthy and infected tissues is well-understood but this process may be significantly altered during cancer.
Murine studies have demonstrated that expression levels of both integrin ligands on endothelial cells and integrins on T cells influence T cell tumor infiltration. Fisher et al. showed that ICAM-1 deficiency or blockade decreased CD8+ T cell infiltration into melanoma (B16-OVA) or colon carcinoma tumors (CT26), respectively, demonstrating ICAM-1 expression affecting T cell infiltration into tumors at least in certain tumor models (125). Sartor et al. observed a suppression in tumor growth rate in mice inoculated with fibrosarcoma tumor cells expressing ICAM-1 compared to mice with ICAM-1 negative tumors suggesting an increase in T cell-mediated immunosurveillance in the presence of ICAM-1 (126). Interestingly, also the expression of αE (CD103) on tumor-infiltrating CD8+ T cells has been shown to increase CD8+ T cell trafficking into tumors and thus promoting anti-tumor immunity (127). αE+ CD8+ T cells had superior capacity to accumulate in tumors in humanized mouse models of breast cancer due to the specific binding of αE on CD8+ T cells to E-cadherin expressed on epithelial cancer cells.
In human patients, high expression of ICAM-1, VCAM-1, and MAdCAM-1 has been shown to correlate with higher density of CD8+ T cells in colorectal cancer (CRC) tumors and also with prolonged disease-free survival (128). Further, human hepatocellular carcinomas (HCC) are more heavily infiltrated with T cells compared to colorectal hepatic metastases (CHM) and that the T cell infiltration was associated with higher expression of ICAM-1 and VAP-1 on tumor-associated endothelial cells in HCC (106). Additionally, a higher proportion of tumor-infiltrating T cells in both tumor types expressed LFA-1 and VLA-4 compared to peripheral blood T cells. In addition, mainly anti-ICAM-1 and anti-VAP-1 and to a lesser extent anti-VCAM-1 mAbs inhibited HCC-derived T cell binding to tumor vascular endothelium in vitro suggesting that LFA-1/ICAM-1 and VAP-1 receptor/VAP-1 are crucial pathways mediating T cell recruitment into the tumor site in HCC. Correlation between levels of cell adhesion molecules (ICAM-1, E-selectin, P-selectin) and T cell infiltration levels has also been shown in melanoma, glioblastoma, Merkel cell carcinoma and squamous cell carcinoma (SCC) (108, 109, 129, 130). Interestingly, when SCC samples were treated with a TLR-7 agonist, imiquimod, tumor vessels up-regulated E-selectin expression causing an increase in CLA+ CD8+ T cell influx into the tumor, a decrease in Treg frequency and tumor regression. Together, both human and murine studies indicate that integrins and VAP-1 on T cells and integrin ligands on endothelial cells are of crucial importance for T cell infiltration into tumors.
Pre-clinical mouse studies indicate that integrins and integrin ligands may also have other effects on anti-tumor responses besides T cell recruitment into tumors, by influencing T cell priming and effector functions. Schmits et al. showed that LFA-1 deficient mice had defects in CD8+ T cell priming against tumor-specific antigens and thus, failed to reject immunogenic fibrosarcoma tumors (57). Mukai et al. further demonstrated that administration of anti-LFA-1 mAbs abrogated the efficacy of adoptive T cell therapy in mouse models of pulmonary and intracranial fibrosarcomas (131). By contrast, ICAM-1 deficiency or mAbs targeting ICAM-1, VCAM-1, or VLA-4, showed no inhibition on the efficacy of transferred T cells in the same mouse models. Interestingly, LFA-1 blockade and to a lesser extent ICAM-1 blockade caused a decrease in T cell IFNγ production in mixed tumor/T cell cultures but did not affect the level of T cell infiltration into the tumor. These results suggested that the LFA-1 pathway mainly affects T cell effector function but not migration to the tumor site. In addition, given that LFA-1 blockade but not ICAM-1 deficiency/blockade affected the anti-tumor efficacy of adoptive T cell therapy, LFA-1 interactions with other ligands such as ICAM-2 seem to be sufficient to preserve T cell effector function. Accordingly, adoptive T cell studies conducted by Blank et al. further demonstrated that CD8+ T cell infiltration into tumor site is not inhibited in ICAM-1 deficient mice (132). Rather, host ICAM-1 expression affected the priming of adoptively transferred tumor-antigen-specific CD8+ T cells leading to delayed tumor rejection in ICAM-1 deficient mice. In addition to priming, LFA-1 and integrin αE(CD103)β7 expressed on CD8+ T cells play important roles for CD8+ T cell cytotoxicity towards tumor cells expressing ICAM-1 and E-cadherin (133).
In addition to T cell migration into tumors, T cells must also be able to recirculate from the tumor site to draining lymph nodes in order to mount distant responses (134). Interestingly, Yanguas et al. showed that in a mouse model of melanoma, increased number of intra-tumorally injected tumor-specific T cells migrated into the draining lymph nodes in mice treated with anti-ICAM-1 or anti-LFA-1 mAbs compared to mice treated with control IgG (134). Further, activated T cells formed intra-tumoral clusters mediated by LFA-1/ICAM-1 interactions in mouse models of melanoma and breast cancer and similar T cell clusters were also visible in primary human melanoma. These results suggested that LFA-1/ICAM-1 pathway also mediates a mechanism to trap activated CD8+ T cells in the tumor tissue.
In summary, integrins, integrin ligands and other cell adhesion molecules expressed on T cells and endothelial cells mediate CD8+ T cell trafficking into tumors at least in some tumor models, but may also play crucial roles in T cell priming and effector functions, thereby affecting anti-tumor immunity in a multitude of ways.
Integrins and Other Cell Adhesion Molecules Affect DC Function During Anti-Tumor Immunity
DCs orchestrate immune responses and it is now widely appreciated that DCs also play a crucial role in regulating the host immune responses to cancer. Indeed, DCs have been found in various types of tumors both in humans and mice (2, 135–140). In the tumor microenvironment, many tumor cells die naturally or as a result of anti-cancer therapies such as chemotherapy and thus DCs often interact with dying tumor cells enabling them to acquire tumor antigens (141). In addition, immature DCs can also interact with live cells, including other immature or mature DCs and acquire tumor antigens by transferring parts of plasma membrane and intracellular proteins in a process termed “nibbling” (142). Further, immature DCs can also directly interact with live tumor cells and acquire tumor antigens by nibbling (143). Following antigen capture, DCs will ultimately become activated and migrate to the draining lymph node via the lymphatic vessels to present tumor-antigens to T and B cells (141) (Figure 3).
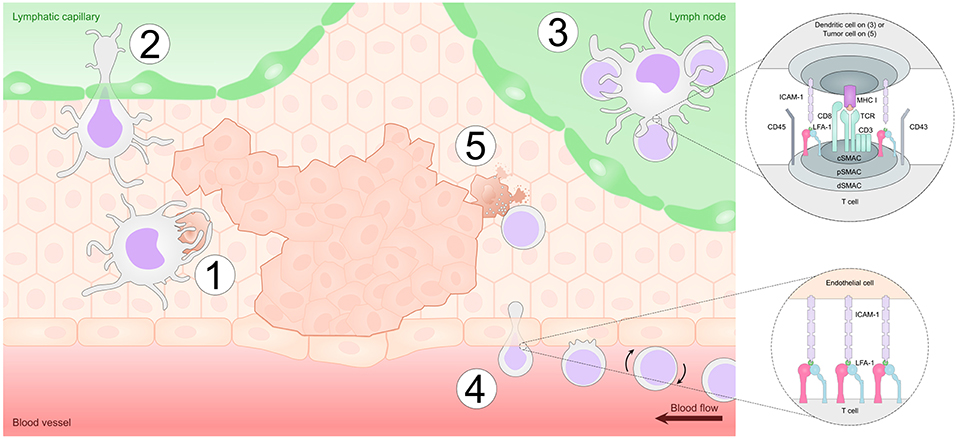
Figure 3. Integrins play a vital role in anti-tumor immunity. Dendritic cells (DCs) take up tumor antigens in the tumor microenvironment by phagocytosing dying tumor cells in a process mediated by adhesion molecules such as αvβ5 integrins (Step 1). DCs then enter the lymphatic vessels partly in an LFA-1/ICAM-1-dependent manner and migrate to the draining lymph node (Step 2). In the lymph node, DCs form an immunological synapse with CD8+ T cells in order to present the tumor antigen. LFA-1-ICAM interactions mediate adhesion in the immunological synapse and also provide an additional co-stimulatory signal to the T cells (Step 3). Once activated, T cells travel via the blood stream and enter the tumor site by interacting with adhesion molecules including E-selectin, ICAMs and VCAM-1 on endothelial cells in a process termed leukocyte adhesion cascade. This process is regulated by sequential expression of selectins (L-selectin) and integrins (LFA-1, VLA-4) on the migrating T cell (Step 4). Finally, after reaching the tumor microenvironment, CD8+ T cells form an immunological synapse with tumor cells and kill the malignant cells via the release of cytotoxic granules (Step 5).
Adhesion receptors on DCs are involved in many of the processes involved in DC-mediated anti-tumor responses. Several receptors such as αVβ5-integrin expressed on immature DCs are involved in the interaction with and phagocytosis of dying cells (144, 145). In addition, given that dying cells often become opsonized by complement component iC3b, DCs can also interact with dying tumor cells via the β2-integrins Mac-1 and αXβ2 (146, 147). However, as described above, β2-integrins often have anti-inflammatory effects in myeloid cells such as DCs, and these interactions lead to suppression of DC activation and, thus, tolerance. Further, since inflammation of various levels has often been associated with tumor development (148), and ICAM-1 expression is up-regulated in lymphatic vessels during inflammation (94), interaction between Mac-1 and ICAM-1 expressed on DCs and inflamed lymphatic endothelium, respectively, may lead to decreased ability of DCs to activate T cells (87). Therefore, integrins on DCs may be involved in the uptake of dying tumor cells via adhesion receptors such as αVβ5-integrin, and in the subsequent initiation of DC-mediated anti-tumor responses. However, β2-integrins expressed on DCs may instead be involved in suppressing DC function. How these processes influence the anti-tumor responses mediated by DCs in vivo is currently unclear.
Following tumor-antigen capture, DCs need to enter the lymphatic vessels in order to migrate to the draining lymph node to prime tumor-specific T cells. The role of integrins and other adhesion molecules in the migration of DCs to lymphatic vessels is currently under debate. Ma et al. elaborated anti-ICAM-1 or anti-LFA-1 mAbs effectively inhibiting antigen-bearing epidermal DCs from migrating to regional lymph nodes in vivo (149). Studies conducted by Xu et al. further demonstrated the importance of ICAM-1 expression on lymphatic endothelium for the migration of hapten-bearing Langerhans cells into the draining lymph node (150). In addition, Johnson et al. demonstrated the up-regulation of ICAM-1 and VCAM-1 on dermal lymphatic endothelial cells at the presence of inflammatory cytokines which then mediated DC trafficking into the lymph nodes via afferent lymphatic vessels (151). However, according to Grabbe et al., hapten-bearing DC migration from the blood to inflamed skin or from skin into the regional lymph node is similar in β2 (CD18) deficient mice compared to WT mice suggesting that β2-integrins are not required for DC migration into the regional lymph nodes (152). Furthermore, Morrison et al. reported that a mutation in β2 which disrupts the integrin/kindlin interaction and thereby renders the integrin inactive, resulted in increased rather than decreased DC migration to peripheral lymphoid organs through an effect on gene transcription in these cells (37). Accordingly, Podgrabinska et al. further showed that the rate of antigen-bearing DC migration into the lymph nodes was similar between ICAM-1 deficient mice and WT mice (87). Interestingly, according to Thompson et al. naïve tumor-specific CD8+ T cells can also become activated and gain effector cell phenotype directly at the tumor site, suggesting that cross-presenting DCs are also able to prime CD8+ T cells in the tumor. These results indicate that DC migration into lymph nodes may not even be completely necessary for DC-mediated anti-tumor responses (153). In conclusion, as the role of β2-integrins in DC migration into lymph nodes is currently unclear, whether β2-integrins on DCs influence DC migration and anti-tumor responses in humans is currently not known and requires further study.
In contrast to the unclear role of β2-integrins in DC biology and DC-mediated anti-tumor responses, the αE-integrins may promote DC anti-tumor responses. Several studies have shown that mouse DCs expressing the integrin αE (CD103) are superior in promoting anti-tumor T cell responses (154–157). Indeed, αE+ DCs are the main cell population carrying tumor-antigens into the draining lymph node and critical for effective anti-tumor CD8+ T cell priming both in vitro and in vivo. Further, increased tumor growth is observed in the absence of αE+ DCs in mouse models of cancer (154). In humans, high αE+/αE− ratio has been significantly associated with increased overall survival in various malignancies including breast cancer, head-neck squamous cell carcinoma and lung adenocarcinoma (154). However, the expression of CD141 (BDCA-3, thrombomodulin), rather than αE, has been thought to mark human DC population functionally equivalent to mouse αE+ DCs (158). Indeed, Jongbloed et al. and Bachem et al. demonstrated that CD141+ DCs were superior in their capacity to cross-present various antigens to CD8+ T cells compared to other DC populations (159, 160). Accordingly, a study assessing gene-signatures also associated high intra-tumoral levels of CD141+ DCs with better overall survival in melanoma patients (155). Increased disease-free survival among patients with aggressive triple-negative breast cancer was also associated with gene signature specific for CD141+ DCs (161).
In conclusion, although integrins play a key role in DC biology, the role of cell adhesion molecules in DC-mediated anti-tumor responses is still unclear and clearly requires further study, especially in human patients.
Integrins and Other Adhesion Molecules Affect the Development and Tumor-Infiltration of Regulatory Cells
Tregs, defined as CD4+CD25hiCD127lo or CD4+CD25+Foxp3+ in humans and mice, respectively, are critical in maintaining peripheral tolerance (162). Tregs are able to suppress the effector function and proliferation of various cell populations including T cells, B cells, NK cells, DCs and macrophages by secreting inhibitory molecules such as TGFβ and IL-10, by direct cell-cell contact or by indirect mechanism via antigen presenting cells (APCs) (162–167). For their suppressive function, it is crucial that Tregs are able to migrate to the site of inflammation and thus Tregs express high levels of cell adhesion molecules including ICAM-1, L-selectin, P-selectin, and VLA-4 (168). Interestingly, some adhesion molecules, particularly β2-integrins, also play an important role in Treg development and function (77, 169–171). Wang et al. showed that reduced CD18 (β2) expression in mouse Tregs disrupted the interactions between Tregs and DCs which led to poor Treg proliferation and decreased ability to produce TGFβ1 and thus decreased suppressive function in vitro compared to WT Tregs (169). In concordance with CD18−/− mice, also LFA-1−/− mice showed reduced Treg numbers in secondary lymphoid organs and decreased conversion rate of conventional CD4+ T cells into Tregs in the periphery (170). In addition, LFA-1 deficient Tregs failed to suppress CD4+ T effector cells in vitro and were unable to prevent disease development in experimental colitis model in vivo. However, on the contrary to CD18−/− mice, LFA-1−/− mice showed increase in Treg numbers in the thymus suggesting that in addition to LFA-1, other β2-integrin(s) also affects Treg development. Besides β2-integrins, also L-selectin and integrin αE (CD103) have been associated with Treg function. High expression of L-selectin has been shown to mark Tregs with more potent ability to inhibit graft-versus-host disease (GVHD) and bone marrow (BM) graft rejection in mice compared to L-selectinlo Tregs (172) and αE+ Tregs have been shown to be more effective in suppressing acute inflammatory reactions in the induced SCID colitis model and antigen-induced arthritis model compared to αE negative Tregs (173, 174).
Given that Tregs suppress the effector function of various immune cells, they can also inhibit immune responses against cancer cells and thus promote tumor growth (6). Further, the presence of Tregs in the tumor microenvironment can present a major obstacle for successful immunotherapy. Indeed, high number of tumor-infiltrating Tregs has been associated with poor prognosis in several malignancies (4, 175). Conditional depletion of Tregs has been shown to increase anti-tumor immunity in mouse models of cancer suggesting that targeting Tregs could be beneficial approach for cancer patients (176–178). However, the lack of Treg-specific cell surface markers presents a major challenge for this task. Further, targeting Tregs specifically in the tumor microenvironment in order to prevent harmful systemic immune reactions present even a greater challenge. Interestingly, Anz et al. discovered that integrin αE (CD103) is expressed at significantly higher levels in tumor-infiltrating Tregs in several mouse cancer models compared to other peripheral Tregs (90% in CT26-infiltrating Tregs vs. 20% in splenic Tregs) due to increased levels of TGFβ in the tumor microenvironment (179). In addition, αE+ Tregs displayed significantly more suppressive phenotype in vitro compared to αE negative Tregs. However, given the high expression also on anti-tumorigenic DCs and CD8+ T cells, αE seems to be an unsuitable target for cancer immunotherapy.
MDSCs and tumor-associated macrophages (TAMs) also represent cell populations capable of efficient suppression of natural anti-tumor immunity. MDSCs are a heterogeneous population of cells consisting of immature myeloid cells and myeloid progenitor cells (180). As other leukocytes, they are generated in the BM but during pathological conditions such infection or cancer they fail to differentiate into mature DCs, macrophages or granulocytes which leads to accumulation of immature myeloid cells with highly immunosuppressive phenotype (180, 181). MDSCs can be divided into different sub-populations in humans (CD11b+CD14−CD15+, CD11b+CD14−CD66b+ and CD11b+CD14+HLA-DR−/loCD15) and mice (CD11b+Ly6G+Ly6Clo and CD11b+Ly6G−Ly6Chi) based on their phenotypic differences but they all share myeloid origin and the ability to strongly suppress T and NK cell activity (180–182). MDSCs are recruited into the tumor site by the malignant cells by increasing the level of various soluble factors including IL-6, GM-CSF, TGFβ, VEGF and chemokines such as CCL2 and CCL5, in the tumor microenvironment (180, 183). After reaching the tumor site, MDSCs efficiently suppress the anti-tumor immunity by various mechanisms such as by depleting T cell nutrients such as L-arginine, by producing reactive oxygen species (ROS) and nitric oxide (NO) and by promoting the development of Tregs. Indeed, several studies have reported an increase in anti-tumor immunity following depletion or suppression of MDSCs in mouse models of cancer (184–186). Among human patients, an increase in MDSCs has been observed in the tumor tissue and peripheral blood in many cancer types (187–191). Further, an increase in MDSCs in the peripheral blood has been associated with poor prognosis in patients with various solid tumors (5, 192). Interestingly, Jin et al. reported VLA-4 being responsible for recruitment of circulating monocytes and macrophages into the tumor site (193). Later it was found that tumor growth was significantly suppressed in mice lacking activated form of VLA-4 and in WT mice treated with mAbs targeting VLA-4 (194). In addition, tumors derived from these mice had significantly reduced frequencies of total CD11b+Gr1+ myeloid cells but increased numbers of CD8+ T cells and mature (CD80+) DCs. In vitro IL-4 stimulated macrophages derived from mice lacking activated form of VLA-4 also showed decreased levels of Il10, Tgfb1, and Arg1 mRNAs but increased levels of mRNAs encoding for IFNγ and IL-12 compared to WT macrophages. These results suggested that VLA-4 regulates MDSC trafficking into the tumor site and also promotes myeloid cell polarization toward immune suppressive phenotype, inhibits anti-tumor immunity and thus promotes tumor growth.
Mac-1 is highly expressed in myeloid cells and plays a key role in various myeloid cell functions including migration, phagocytosis, and chemotaxis (195). In addition, given that mAbs targeting Mac-1 have been shown to decrease myeloid cell tissue infiltration and inflammation (196), targeting Mac-1 could also reduce the recruitment of suppressive myeloid cells into the tumor site. Indeed, Zhang et al. demonstrated that CD11b (αM-chain of Mac-1) deficiency reduced intestinal tumor growth in mice by reducing myeloid cell trafficking to the tumor microenvironment (197). Further, CD11b blockade also decreased myeloid cell recruitment into tumors in immune deficient mice bearing human squamous cell carcinoma xenografts and thus improved the anti-tumor responses to radiotherapy (198). However, as Mac-1 has been shown to play other roles in immunity than simply affecting cell recruitment, e.g., in immune suppression, it is possible that CD11b blockade also has other, as yet unrecognized effects on anti-tumor immunity.
Integrins and Other Cell Adhesion Molecules Also Have Cell Intrinsic Effects on Tumor Cells
Integrin expression on tumor cells themselves has been associated with tumor progression and metastasis by increasing the proliferation, survival, migration and invasion of the malignant cells (199). Integrin ligation can promote tumor cell survival by several mechanisms such as by inhibiting p53 and caspase-9 via αV (200) and by increasing anti-apoptotic proteins Bcl-2 and Bcl-XL via αVβ5 and αVβ3 (201). Particularly integrin αVβ3 has been associated with tumor progression in various human malignancies (202–204) and it co-operates with SRC oncogene to enhance anchorage-independent tumor growth and promotes lymph node metastases (205, 206). Other integrins have also been shown to co-operate with oncogenes including β4 which amplifies the signaling of ErbB2 to promote mammary tumorigenesis (207). In addition, integrins such as β3 may also function as markers for highly tumorigenic cancer stem cells (208).
Besides on tumor-associated vasculature, aberrant VCAM-1 expression has also been described on many types of tumor cells such as breast, renal and gastric carcinoma cells (97, 209). Up-regulation of VCAM-1 on malignant cells is associated with increased ability to metastasize and recruit tumor-associated monocytes and macrophages. Also ICAM-1 expression has been associated with the malignant potential of tumor cells and has thus been associated with metastases and poor prognosis in several cancers including melanoma, breast, lung and oral cancer (210–213). Given that MUC-1 which is often expressed on tumor cells (214) can also interact with ICAM-1, circulating cancer cells can adhere to endothelial cells which may represent the first step in metastases formation (215). MUC-1-ICAM-1 interaction can then induce cytokine secretion and ICAM-1 up-regulation in the malignant cells thus recruiting macrophages. Indeed, macrophage infiltration has been demonstrated to correlate with ICAM-1 expressing tumor cells in oral squamous cell carcinoma (213). Macrophage-derived cytokines can then further attract neutrophils which secrete proteases promoting extraluminal migration of the malignant cells (215). Further strengthening the role of ICAM-1 in tumor metastasis, tumor cell lines transfected with ICAM-1 showed enhanced invasive capacity and proliferation in vitro (211, 213) and ICAM-1 blockade have been shown to decrease tumor cell invasion in vitro (211, 216). In addition, an increase in serum levels of soluble ICAM-1 (sICAM-1) has been associated with disease progression, tumor aggressiveness and decreased survival in several malignancies such as melanoma, chronic B-lymphocytic leukemia, lymphoma, and CRC (217–220).
Given the specific expression on hematopoietic cells, LFA-1 plays a role in the development of hematological malignancies such as leukemias and lymphomas. Chronic lymphocytic leukemia (CLL) is the most common leukemia in the Western countries (221). It is characterized by clonal mature B cell accumulation in the BM, blood and other lymphoid tissues and advanced disease often manifests itself as lymphadenopathy, hepatomegaly, splenomegaly, BM failure, and recurrent infections. The malignant CLL cells are highly dependent on lymphoid microenvironment where they interact with and receive survival and proliferation signals from the accessory cells (222). In addition to chemokines, malignant cells require the expression of leukocyte adhesion molecules, particularly LFA-1 and VLA-4, in order to migrate into the lymphoid tissues. Interestingly, Montresor et al. reported differences in integrin signaling in human CLL cells compared to healthy B cells (223). Signaling molecules such as PIP5KC, RAC1, and CDC42 regulating integrin conformational change into the active form in normal lymphocytes showed either none or severely decreased regulatory role in CLL cells. By using live cell imaging, Till et al. further showed that in contrast to LFA-1 expressed on normal human B cells, LFA-1 expressed on CLL cells can be in its active conformation without chemokine induced clustering (224). In addition to aberrant integrin signaling, the expression levels of adhesion molecules on CLL cells usually differ from healthy B cells. Generally, the expression of various adhesion molecules including β2- and β1-integrins, CD54, CD62L, and CD44 is very low on peripheral blood and BM CLL cells compared to cells derived from healthy donors (225). Interestingly, Hartmann et al. demonstrated that CLL cells require the same integrins as healthy B cells in trafficking through the BM, spleen, and lymph nodes (226). On the contrary to normal human B cells, CLL cells were unable to arrest in ICAM-1 expressing endothelium in vitro and to migrate to lymph nodes of NOD/SCID mice in vivo due to low expression of LFA-1. In addition to low LFA-1 expression, VLA-4 expression was variable on CLL cells but yet significantly reduced compared to healthy B cells. Therefore, the ability of human CLL cells to migrate to the BM of NOD/SCID mice in vivo was significantly reduced compared to normal B cells. These results suggested that CLL cell migration to the BM and lymph nodes is decreased due to low expression of LFA-1 and VLA-4 thus causing an accumulation in the blood and spleen. Interestingly, the same study also reported that significantly higher expression of LFA-1 and VLA-4 was detected on CLL cells derived from high-risk patients with unfavorable cytogenetic abnormalities such as trisomy 12, deletion 17p or deletion 11q (226). Increased expression of integrins including LFA-1 and VLA-4 on CLL cells among patients with trisomy 12 has also been reported by others and the increase has been associated with up-regulation of molecules regulating integrin inside-out signaling such as RAP1B and RAPL and with enhanced ligand (ICAM-1, VCAM-1) binding and migration in vitro (227, 228). These results suggest that up-regulation of cell adhesion molecules, particularly LFA-1 and VLA-4, could increase CLL cell migration to lymphoid tissues where they would receive more proliferation and survival signals thus leading to more aggressive disease (226, 227). Indeed, higher expression of CD11a (LFA-1 α-chain) on CLL cells has been associated with increase in tumor burden (225) and higher expression of CD49d (VLA-4 α-chain) with disease progression and decreased overall survival (229–231).
Multiple myeloma (MM) is a cancer of the BM caused by malignant, terminally differentiated plasma cells (PCs) (232, 233). It is the second most common hematological malignancy in the world and its symptoms include increased calcium levels, renal insufficiency, anemia, and/or bone lesions. In most cases the malignant plasma cells also secrete monoclonal immunoglobulin proteins (M-proteins) that can be detected in blood and/or urine. Despite the recent improvements in MM therapies, MM is still largely considered to be an incurable disease. As in CLL (222), adhesion molecules play an important role in the interactions between MM cells and accessory cells in the BM thus promoting the proliferation and survival of the malignant cells (234). Indeed, high proportion of human myeloma cells have been shown to express various adhesion molecules including LFA-1, VLA-4, CD44, and ICAM-1 (235–237). LFA-1 expression on myeloma cells in particular has been associated with aggressive disease (236, 237) and to correlate with disease activity (235). Interestingly, by using a mouse MM cell line (5T33MMvt), Asosingh et al. demonstrated LFA-1 playing a role in homotypic cell-cell adhesion and cell proliferation of myeloma cells in vitro (238). Further, in contrast to LFA-1 negative cells, only LFA-1 expressing 5T33MMvt cells were able to cause disease in vivo. Finally, various adhesion molecules including LFA-1 and VLA-4 have also been associated with drug resistance in MM patients (239).
Anti-Tumor Therapy Targeting Cell Adhesion Molecules
Given that integrin function has been associated with tumor progression, there has been great interest in targeting integrins in the treatment of cancer. However, since integrins also play diverse roles in immunity and anti-tumor responses, blocking or enhancing the function of these molecules in vivo may be difficult.
The majority of drugs targeting integrins in the aim to treat cancer inhibit the function of αV- or β1-integrins (240, 241). Both integrin classes have been shown to be highly expressed in many human malignancies and antagonists targeting αV or β1 indeed suppressed tumor growth in preclinical mouse models (199, 202, 242–248). As a result, there have been multiple clinical trials targeting integrins either as a single therapy or in combination with conventional therapies in treating various human cancers (249–289) (Table 1). However, in contrast to the pre-clinical studies, the results from clinical trials were rather disappointing with αV or β1 antagonists generally showing either no or only week anti-tumor efficacy (Table 1). In one phase II clinical trial with advanced prostate cancer patients, intetumumab (CNTO 95), a mAb targeting multiple αV integrins, in fact resulted in decreased progression-free and overall survival compared to placebo (276). However, it could be possible to improve therapeutic responses of integrin targeting by combining it with other immunotherapies. Indeed, mouse studies conducted by Kwan et al. demonstrated that by combining a fusion protein, consisting of a mouse Fc domain and RGD-binding integrin targeting peptide, with albumin/IL-2 or anti-PD-1 immunotherapy it was possible to enhance anti-tumor immunity and tumor suppression (290).
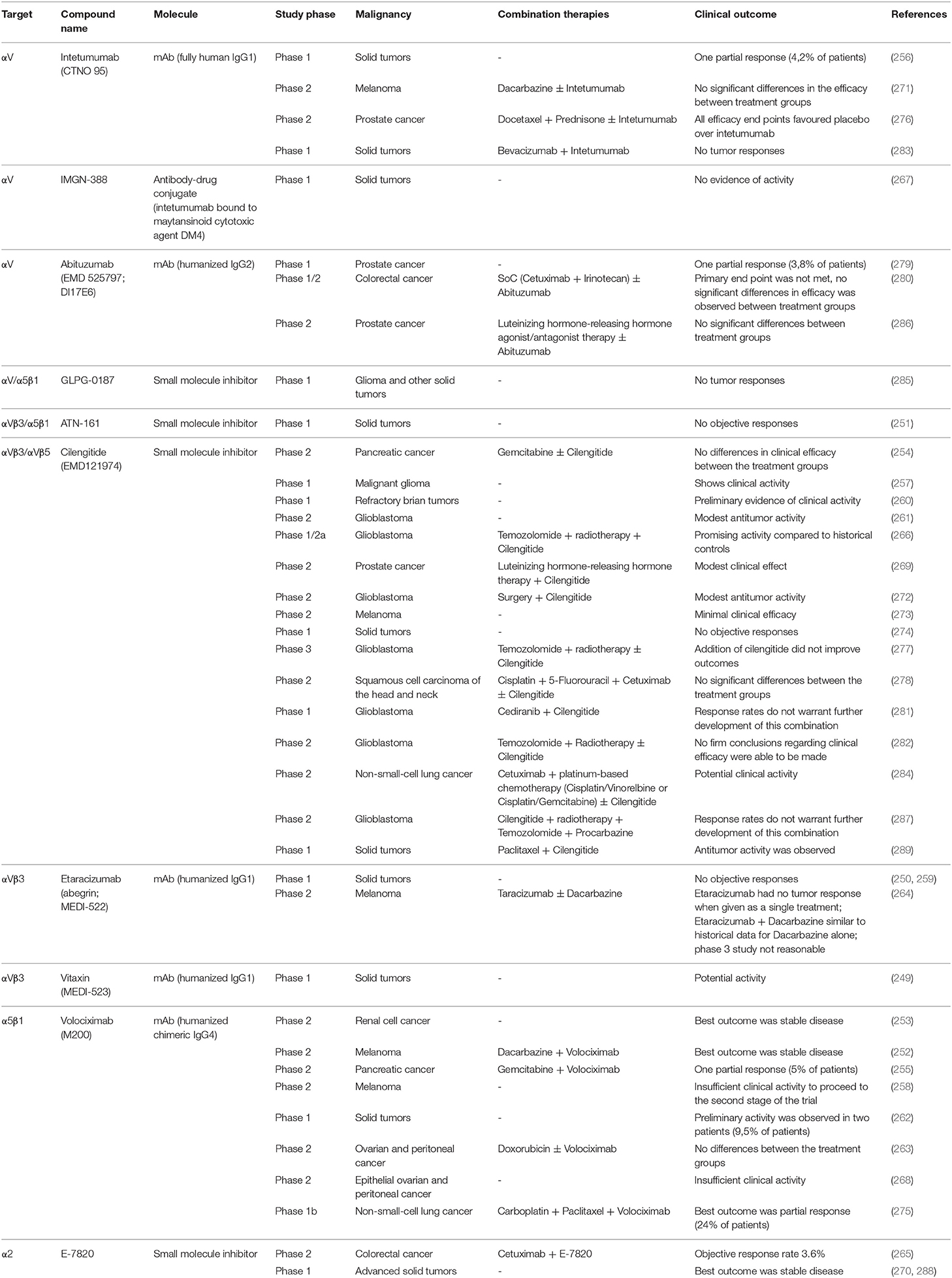
Table 1. Clinical trials targeting integrins.
Due to the specific expression on hematopoietic cells, β2-integrins have mainly been aimed to target to treat inflammatory diseases including liver fibrosis (291) and autoimmune diseases, including arthritis (292), and psoriasis (293). In pre-clinical studies, LFA-1 small-molecule antagonist (BMS-587101) inhibited LFA-1-mediated T cell adhesion to endothelial cells, Th1 cytokine production, and T cell proliferation in vitro and also inhibited inflammation in vivo (292). In the context of cancer, β2-integrins, mainly LFA-1, would be an attractive target to treat hematological cancers such as leukemias and lymphomas (294, 295). In addition, LFA-1 small molecule antagonists have also demonstrated anti-tumor efficacy against solid tumors in mice (296). However, given that LFA-1 promotes T cell activation and migration, blocking the function of LFA-1 may in fact increase the risk of malignancies and infections (297). Indeed, anti-LFA-1 therapy has been associated with rare but severe systemic adverse events such as immune-mediated thrombocytopenia and hemolytic anemia (298). Further, in 2009, after more than 45,000 psoriasis patients had been treated with efalizumab (humanized anti-CD11a mAb), the drug was withdrawn from the market due to three confirmed cases of fatal viral-based multifocal leukoencephalopathy (PML) (297, 299). One possibility to harness the anti-tumor efficacy of LFA-1 blockade without inducing severe adverse events would be to target LFA-1 specifically on tumor cells. Indeed, Cohen et al. demonstrated that by using bispecific antibody which simultaneously targets LFA-1 and a tumor specific antigen it was possible to specifically block LFA-1-mediated tumor cell adhesion without affecting immune responses in mice (294). In addition, it is also noteworthy that since activation of LFA-1 has been shown to regulate the activity of VLA-4, drugs targeting LFA-1 may also affect the function of other integrins (300).
Given the lack of anti-tumor efficacy and in the case of LFA-1 blockers, the severity of adverse events, it could be more feasible to target the integrin ligands on tumor vessels than the integrins themselves in order to enhance T cell infiltration to the tumor site. As discussed before, previously E-selectin negative tumor-associated vessels in human SCC samples up-regulated E-selectin following treatment with imiquimod (TLR-7 agonist) which resulted in CD8+ T cell influx into the tumor and tumor regression (129). Also systemic thermal therapy (STT) induced the activation of IL-6 trans-signaling causing up-regulation of ICAM-1 on tumor vascular endothelium and thus increased the homing of adoptively transferred CD8+ T cells into tumors in mouse models of cancer (125). Further, CpG-ODN (oligodeoxynucleotides (ODN) with cytocine-guanine-rich (CpG) motifs) vaccination caused up-regulation of ICAM-1 and VCAM-1 on tumor-associated blood vessel endothelia leading to strong tumor-infiltration of adoptively transferred tumor-specific T cells and tumor suppression in mouse model of pancreatic islet cell carcinoma (301). These results demonstrated that by increasing inflammatory signals in the tumor microenvironment it could be possible to enhance T cell infiltration into tumors and thus T-cell mediated tumor cell killing. The most straight forward way to increase inflammatory signals would be to administer inflammatory cytokines such as TNFα or IFNγ systemically (302). However, this could lead to severe adverse events as has been demonstrated with the systemic administration of TNFα which was associated with risk of septic shock syndrome leading to multi-organ failure (303). Delivery of inflammatory cytokines or other inflammatory stimuli directly to the tumor site could reduce the risk of side effects (302). Further, systemic administration of TNFα coupled to ACDCRGDCFCG-peptide, a ligand for αVβ3-integrin, has enabled targeting the tumor vasculature and inducing anti-tumor effects in tumor-bearing mice (304). Given that angiogenic factors have been shown to cause decrease in ICAM-1 and VCAM-1 expression on tumor associated vessels (105, 110), targeting angiogenesis could also increase T cell infiltration into tumors. Indeed, treatment with angiogenesis inhibitor anginex significantly up-regulated the expression of VCAM-1 and E-selectin on tumor blood vessel endothelial cells resulting in increase in tumor-infiltrating leukocytes and suppression of tumor growth in mouse models of cancer (305). Further, VEGF blockade significantly increased tumor infiltration of adoptively transferred T cells thus promoting tumor suppression in mice (306).
Among therapies involving adoptively transferred cells, chimeric antigen receptor (CAR) T cell therapy has shown promise in cancer treatment due to measurable responses in several clinical trials across various malignancies (307–310). Given that CAR T cells are usually constructed by joining an antigen-recognition moiety such as single chain antibody (scFv) to TCR/CD3 complex and by adding costimulatory domain such as CD28 or CD137 to improve cell survival and proliferation, CAR T cell therapy has several advantages to conventional T cell therapy such as higher antigen binding affinity and independency of MHC expression on tumor cells (311). However, the major obstacle for CAR T cell therapy is often the inability of the transferred cells to migrate and extravasate into the tumor (312). One way to overcome this obstacle could be to generate a CAR T cell with specificity to tumor-associated vasculature instead of the tumor cells themselves (313). Fu et al. constructed a CAR T cell containing a peptide sequence for echistatin (T-eCAR) which has a high affinity to the integrin highly expressed on tumor vasculature endothelium, αVβ3 (313). Indeed, T-eCAR cells efficiently lysed αVβ3 expressing human umbilical vein endothelial cells and tumor cells in vitro. Further, these cells led to extensive bleeding in the tumor tissue and thus tumor growth suppression while sparing normal tissues in mice. In addition to CAR T cells, integrins expressed on the tumor vasculature could also be utilized in order to target drug-delivering nanoparticles to the tumor tissue more efficiently (199). Murphy et al. demonstrated that doxorubicin containing, αVβ3-targeted lipid nanoparticles effectively targeted tumor vessels leading to their apoptosis in mouse models of cancer (314). Moreover, αVβ3-targeted nanoparticles showed anti-metastatic effects in spontaneous metastases models without causing adverse events associated with systemic administration of free doxorubicin.
It may also be possible to enhance the anti-tumor response of adoptively transferred cells by increasing LFA-1 function in the cells. Simply “locking” LFA-1 in an active state (through mutation) is unlikely to be successful, as T cells expressing such mutated integrins are actually deficient in T cell migration, which requires dynamic modulation of integrin activity (315). Instead, indirect approaches may be required. Indeed, mutation of a phosphorylation site in VLA-4 leads to increased LFA-1 activity (through integrin transregulation) and enhanced tumor immunity (316). Also, modulating LFA-1 activation status through engineering of ALCAM increases tumor rejection of brain tumors (317).
Conclusions and Future Perspectives
Taken together, adhesion molecules play vital roles in the function of the immune system both in health and disease. During cancer development, adhesion molecules, particularly integrins, mediate crucial functions in nearly every step of the anti-tumor response including in tumor antigen uptake, activation of tumor-specific T cells, leukocyte trafficking into the tumor site and tumor cell killing. However, malignant cells can also utilize cell adhesion molecule pathways to promote tumor growth. Expression of various integrins on tumor cells promotes tumor cell proliferation, survival and metastases while increased secretion of angiogenic molecules causes down-regulation of adhesion molecules on tumor-associated blood vessels and thus prevents immune effector cell infiltration into the tumor. Tumor cells also recruit regulatory cells such as Tregs and MDSCs which express high levels of integrins enabling them to reach the tumor site. The main cell adhesion molecule-mediated events promoting tumor growth are listed in Table 2.

Table 2. Adhesion molecule-mediated events promoting tumor growth.
Immunotherapy including immune checkpoint blockade and CAR T cells has revolutionized the field of cancer therapy during the last decades. Given the various roles in tumor development, integrins also seem like promising targets for cancer therapy. However, clinical trials targeting integrins directly on malignant cells have shown disappointing results with low therapeutic efficacy. Rather, increasing the expression or function of β2-integrins on immune cells or their ligands on tumor-associated blood vessels and enhancing anti-tumor responses may represent a more efficient approach. In addition, considering that still a considerable number of patients do not benefit from current forms of immunotherapy due to the inability of T cells to access the tumor microenvironment, enhancing β2-integrin function could open a possibility to overcome this impediment. For this and also to prevent harmful treatment-related adverse events, it is vital to fully understand the various functions of β2-integrins and how their expression and function are regulated.
Author Contributions
All authors contributed to writing the manuscript, with HH and SF playing leading roles in manuscript planning and writing. ML and CG wrote part of the text. ML made the figures.
Funding
This study was funded by the Academy of Finland, Sigrid Juselius Foundation, University of Helsinki, Magnus Ehrnrooth Foundation, Liv och Hälsa Foundation, HiLIFE (all to SF).
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Grivennikov SI, Greten FR, Karin M. Immunity, inflammation, and cancer. Cell. (2010) 140:883–99. doi: 10.1016/j.cell.2010.01.025
2. Fridman WH, Pagès F, Sautès-Fridman C, Galon J. The immune contexture in human tumours: impact on clinical outcome. Nat Rev Cancer. (2012) 12:298. doi: 10.1038/nrc3245
3. Barnes TA, Amir E. HYPE or HOPE: the prognostic value of infiltrating immune cells in cancer. Br J Cancer. (2017) 117:451. doi: 10.1038/bjc.2017.220
4. Shang B, Liu Y, Jiang S-J, Liu Y. Prognostic value of tumor-infiltrating FoxP3+ regulatory T cells in cancers: a systematic review and meta-analysis. Sci Rep. (2015) 5:15179. doi: 10.1038/srep15179
5. Zhang S, Ma X, Zhu C, Liu L, Wang G, Yuan X. The role of myeloid-derived suppressor cells in patients with solid tumors: a meta-analysis. PLoS ONE. (2016) 11:e0164514. doi: 10.1371/journal.pone.0164514
6. Tanaka A, Sakaguchi S. Regulatory T cells in cancer immunotherapy. Cell Res. (2017) 27:109–18. doi: 10.1038/cr.2016.151
7. Schreiber RD, Old LJ, Smyth MJ. Cancer immunoediting: integrating immunity's roles in cancer suppression and promotion. Science. (2011) 331:1565–70. doi: 10.1126/science.1203486
8. Mittal D, Gubin MM, Schreiber RD, Smyth MJ. New insights into cancer immunoediting and its three component phases–elimination, equilibrium and escape. Curr Opin Immunol. (2014) 27:16–25. doi: 10.1016/j.coi.2014.01.004
9. Samanta D, Almo SC. Nectin family of cell-adhesion molecules: structural and molecular aspects of function and specificity. Cell Mol Life Sci. (2015) 72:645–58. doi: 10.1007/s00018-014-1763-4
10. Jalkanen S, Karikoski M, Mercier N, Koskinen K, Henttinen T, Elima K, et al. The oxidase activity of vascular adhesion protein-1 (VAP-1) induces endothelial E- and P-selectins and leukocyte binding. (2007) 110:1864–70. doi: 10.1182/blood-2007-01-069674
11. Sperandio M. Selectins and glycosyltransferases in leukocyte rolling in vivo. FEBS Journal. (2006) 273:4377–89. doi: 10.1111/j.1742-4658.2006.05437.x
12. Patel SD, Chen CP, Bahna F, Honig B, Shapiro L. Cadherin-mediated cell-cell adhesion: sticking together as a family. Curr Opin Struct Biol. (2003) 13:690–8. doi: 10.1016/j.sbi.2003.10.007
13. Kappelmayer J, Nagy B Jr . The interaction of selectins and PSGL-1 as a key component in thrombus formation and cancer progression. Hindawi BioMed Res Int. (2017) 2017:6138145. doi: 10.1155/2017/6138145
14. Mcever RP, Zu C. Rolling cell adhesion. Annu Rev Cell Dev Biol. (2010) 26:363–96. doi: 10.1146/annurev.cellbio.042308.113238
15. Tadayuki Yago NZ, Liang Z, Charles S, Abrams MRP. Selectins and chemokines use shared and distinct signals to activate β2 integrins in neutrophils. Blood Adv. (2018) 2:731–44. doi: 10.1182/bloodadvances.2017015602
16. Perez-Vilar J, Hill RL. The structure and assembly of secreted mucins. J Biol Chem. (1999) 274:31751–4. doi: 10.1074/jbc.274.45.31751
17. Kufe DW. Mucins in cancer: function, prognosis and therapy. Nat Rev Cancer. (2009) 9:874–85. doi: 10.1038/nrc2761
18. Wai Wong C, Dye DE, Coombe DR. The role of immunoglobulin superfamily cell adhesion molecules in cancer metastasis. J Int J Cell Biol. (2012) 2012:340296. doi: 10.1155/2012/340296
19. Briskin MJ, Mcevoy LM, Butcher EC. MAdCAM-1 has homology to immunoglobulin and mucin-like adhesion receptors and to IgA1. Nature. (1993) 363:461. doi: 10.1038/363461a0
20. Cayrol R, Wosik K, Berard JL, Dodelet-Devillers A, Ifergan I, Kebir H, et al. Activated leukocyte cell adhesion molecule promotes leukocyte trafficking into the central nervous system. Nat Immunol. (2007) 9:137. doi: 10.1038/ni1551
21. Muller WA. Getting leukocytes to the site of inflammation. Vet Pathol. (2013) 50:7–22. doi: 10.1177/0300985812469883
22. Tachibana K, Nakanishi H, Mandai K, Ozaki K, Ikeda W, Yamamoto Y, et al. Two cell adhesion molecules, nectin and cadherin, interact through their cytoplasmic domain-associated proteins. J Cell Biol. (2000) 150:1161–76. doi: 10.1083/jcb.150.5.1161
23. Dougall WC, Kurtulus S, Smyth MJ, Anderson AC. TIGIT and CD96: new checkpoint receptor targets for cancer. Immunotherapy. (2017) 276:112–20. doi: 10.1111/imr.12518
24. Gahmberg CG, Fagerholm SC, Nurmi SM, Chavakis T, Marchesan S, Gronholm M. Regulation of integrin activity and signalling. Biochim Biophys Acta. (2009) 1790:431–44. doi: 10.1016/j.bbagen.2009.03.007
25. Tan SM. The leucocyte beta2 (CD18) integrins: the structure, functional regulation and signalling properties. Biosci Rep. (2012) 32:241–69. doi: 10.1042/BSR20110101
26. Schittenhelm L, Hilkens CM, Morrison VL. beta2 integrins as regulators of dendritic cell, monocyte, and macrophage function. Front Immunol. (2017) 8:1866. doi: 10.3389/fimmu.2017.01866
27. Arnaout MA. Biology and structure of leukocyte beta 2 integrins and their role in inflammation. F1000Res. (2016) 5:2433. doi: 10.12688/f1000research.9415.1
28. Nishida N, Xie C, Shimaoka M, Cheng Y, Walz T, Springer TA. Activation of leukocyte beta2 integrins by conversion from bent to extended conformations. Immunity. (2006) 25:583–94. doi: 10.1016/j.immuni.2006.07.016
29. Li J, Springer TA. Integrin extension enables ultrasensitive regulation by cytoskeletal force. Proc Natl Acad Sci USA. (2017) 114:4685–90. doi: 10.1073/pnas.1704171114
30. Kim M, Carman CV, Springer TA. Bidirectional transmembrane signaling by cytoplasmic domain separation in integrins. Science. (2003) 301:1720–5. doi: 10.1126/science.1084174
31. Winograd-Katz SE, Fassler R, Geiger B, Legate KR. The integrin adhesome: from genes and proteins to human disease. Nat Rev Mol Cell Biol. (2014) 15:273–88. doi: 10.1038/nrm3769
32. Horton ER, Humphries JD, James J, Jones MC, Askari JA, Humphries MJ. The integrin adhesome network at a glance. J Cell Sci. (2016) 129:4159–63. doi: 10.1242/jcs.192054
33. Humphries JD, Chastney MR, Askari JA, Humphries MJ. Signal transduction via integrin adhesion complexes. Curr Opin Cell Biol. (2018) 56:14–21. doi: 10.1016/j.ceb.2018.08.004
34. Rose DM, Liu S, Woodside DG, Han J, Schlaepfer DD, Ginsberg MH. Paxillin binding to the alpha 4 integrin subunit stimulates LFA-1 (integrin alpha L beta 2)-dependent T cell migration by augmenting the activation of focal adhesion kinase/proline-rich tyrosine kinase-2. J Immunol. (2003) 170:5912–8. doi: 10.4049/jimmunol.170.12.5912
35. Moser M, Nieswandt B, Ussar S, Pozgajova M, Fässler R. Kindlin-3 is essential for integrin activation and platelet aggregation. Nat Med. (2008) 14:325. doi: 10.1038/nm1722
36. Moser M, Bauer M, Schmid S, Ruppert R, Schmidt S, Sixt M, et al. Kindlin-3 is required for β2 integrin–mediated leukocyte adhesion to endothelial cells. Nat Med. (2009) 15:300. doi: 10.1038/nm.1921
37. Morrison VL, Macpherson M, Savinko T, San Lek H, Prescott A, Fagerholm SC. The β2 integrin–kindlin-3 interaction is essential for T-cell homing but dispensable for T-cell activation in vivo. (2013) 122:1428–36. doi: 10.1182/blood-2013-02-484998
38. Lefort CT, Rossaint J, Moser M, Petrich BG, Zarbock A, Monkley SJ, et al. Distinct roles for talin-1 and kindlin-3 in LFA-1 extension and affinity regulation. Blood. (2012) 119:4275–82. doi: 10.1182/blood-2011-08-373118
39. Sun Z, Costell M, Fassler R. Integrin activation by talin, kindlin and mechanical forces. Nat Cell Biol. (2019) 21:25–31. doi: 10.1038/s41556-018-0234-9
40. Ye F, Petrich BG, Anekal P, Lefort CT, Kasirer-Friede A, Shattil SJ, et al. The mechanism of kindlin-mediated activation of integrin alphaIIbbeta3. Curr Biol. (2013) 23:2288–95. doi: 10.1016/j.cub.2013.09.050
41. Huet-Calderwood C, Brahme NN, Kumar N, Stiegler AL, Raghavan S, Boggon TJ, et al. Differences in binding to the ILK complex determines kindlin isoform adhesion localization and integrin activation. J Cell Sci. (2014) 127:4308–21. doi: 10.1242/jcs.155879
42. Bledzka K, Bialkowska K, Sossey-Alaoui K, Vaynberg J, Pluskota E, Qin J, et al. Kindlin-2 directly binds actin and regulates integrin outside-in signaling. J Cell Biol. (2016) 213:97–108. doi: 10.1083/jcb.201501006
43. Theodosiou M, Widmaier M, Bottcher RT, Rognoni E, Veelders M, Bharadwaj M, et al. Kindlin-2 cooperates with talin to activate integrins and induces cell spreading by directly binding paxillin. Elife. (2016) 5:e10130. doi: 10.7554/eLife.10130
44. Gao J, Huang M, Lai J, Mao K, Sun P, Cao Z, et al. Kindlin supports platelet integrin alphaIIbbeta3 activation by interacting with paxillin. J Cell Sci. (2017) 130:3764–75. doi: 10.1242/jcs.205641
45. Su W, Wynne J, Pinheiro EM, Strazza M, Mor A, Montenont E, et al. Rap1 and its effector RIAM are required for lymphocyte trafficking. Blood. (2015) 126:2695–703. doi: 10.1182/blood-2015-05-644104
46. Stefanini L, Lee RH, Paul DS, O'shaughnessy EC, Ghalloussi D, Jones CI, et al. Functional redundancy between RAP1 isoforms in murine platelet production and function. Blood. (2018) 132:1951–62. doi: 10.1182/blood-2018-03-838714
47. Lafuente EM, Van Puijenbroek AA, Krause M, Carman CV, Freeman GJ, Berezovskaya A, et al. RIAM, an Ena/VASP and profilin ligand, interacts with Rap1-GTP and mediates Rap1-induced adhesion. Dev Cell. (2004) 7:585–95. doi: 10.1016/j.devcel.2004.07.021
48. Klapproth S, Sperandio M, Pinheiro EM, Prunster M, Soehnlein O, Gertler FB, et al. Loss of the Rap1 effector RIAM results in leukocyte adhesion deficiency due to impaired beta2 integrin function in mice. Blood. (2015) 126:2704–12. doi: 10.1182/blood-2015-05-647453
49. Bromberger T, Klapproth S, Rohwedder I, Zhu L, Mittmann L, Reichel CA, et al. Direct Rap1/Talin1 interaction regulates platelet and neutrophil integrin activity in mice. Blood. (2018) 2004:846766. doi: 10.1182/blood-2018-04-846766
50. Morse EM, Brahme NN, Calderwood DA. Integrin cytoplasmic tail interactions. Biochemistry. (2014) 53:810–20. doi: 10.1021/bi401596q
51. Kiema T, Lad Y, Jiang P, Oxley CL, Baldassarre M, Wegener KL, et al. The molecular basis of filamin binding to integrins and competition with talin. Mol Cell. (2006) 21:337–47. doi: 10.1016/j.molcel.2006.01.011
52. Liu J, Das M, Yang J, Ithychanda SS, Yakubenko VP, Plow EF, et al. Structural mechanism of integrin inactivation by filamin. Nat Struct Mol Biol. (2015) 22:383–9. doi: 10.1038/nsmb.2999
53. Savinko T, Guenther C, Uotila LM, Llort Asens M, Yao S, Tojkander S, et al. Filamin A Is required for optimal T cell integrin-mediated force transmission, flow adhesion, and T cell trafficking. J Immunol. (2018) 200:3109–16. doi: 10.4049/jimmunol.1700913
54. Etzioni A. Genetic etiologies of leukocyte adhesion defects. Curr Opin Immunol. (2009) 21:481–6. doi: 10.1016/j.coi.2009.07.005
55. Fagerholm SC, Guenther C, Llort Asens M, Savinko T, Uotila LM. Beta2-Integrins and interacting proteins in leukocyte trafficking, immune suppression, and immunodeficiency disease. Front Immunol. (2019) 10:254. doi: 10.3389/fimmu.2019.00254
56. Coxon A, Rieu P, Barkalow FJ, Askari S, Sharpe AH, Von Andrian UH, et al. A novel role for the beta 2 integrin CD11b/CD18 in neutrophil apoptosis: a homeostatic mechanism in inflammation. Immunity. (1996) 5:653–66. doi: 10.1016/S1074-7613(00)80278-2
57. Schmits R, Kundig TM, Baker DM, Shumaker G, Simard JJ, Duncan G, et al. LFA-1-deficient mice show normal CTL responses to virus but fail to reject immunogenic tumor. J Exp Med. (1996) 183:1415–26. doi: 10.1084/jem.183.4.1415
58. Ley K, Laudanna C, Cybulsky MI, Nourshargh S. Getting to the site of inflammation: the leukocyte adhesion cascade updated. Nat Rev Immunol. (2007) 7:678–89. doi: 10.1038/nri2156
59. Bellone M, Calcinotto A. Ways to enhance lymphocyte trafficking into tumors and fitness of tumor infiltrating lymphocytes. Front Oncol. (2013) 3:231. doi: 10.3389/fonc.2013.00231
60. Manevich-Mendelson E, Feigelson SW, Pasvolsky R, Aker M, Grabovsky V, Shulman Z, et al. Loss of Kindlin-3 in LAD-III eliminates LFA-1 but not VLA-4 adhesiveness developed under shear flow conditions. Blood. (2009) 114:2344–53. doi: 10.1182/blood-2009-04-218636
61. Walling BL, Kim M. LFA-1 in T cell migration and differentiation. Front Immunol. (2018) 9:952. doi: 10.3389/fimmu.2018.00952
62. Phillipson M, Heit B, Colarusso P, Liu L, Ballantyne CM, Kubes P. Intraluminal crawling of neutrophils to emigration sites: a molecularly distinct process from adhesion in the recruitment cascade. J Exp Med. (2006) 203:2569–75. doi: 10.1084/jem.20060925
63. Shulman Z, Cohen SJ, Roediger B, Kalchenko V, Jain R, Grabovsky V, et al. Transendothelial migration of lymphocytes mediated by intraendothelial vesicle stores rather than by extracellular chemokine depots. Nat Immunol. (2011) 13:67–76. doi: 10.1038/ni.2173
64. MacIver NJ, Michalek RD, Rathmell JC. Metabolic regulation of T lymphocytes. Ann Rev Immunol. (2013) 31:259–83. doi: 10.1146/annurev-immunol-032712-095956
65. Grakoui A, Bromley SK, Sumen C, Davis MM, Shaw AS, Allen PM, et al. The immunological synapse: a molecular machine controlling T cell activation. Science. (1999) 285:221–7. doi: 10.1126/science.285.5425.221
66. Dustin ML. Cell adhesion molecules and actin cytoskeleton at immune synapses and kinapses. Curr Opin Cell Biol. (2007) 19:529–33. doi: 10.1016/j.ceb.2007.08.003
67. Comrie WA, Li S, Boyle S, Burkhardt JK. The dendritic cell cytoskeleton promotes T cell adhesion and activation by constraining ICAM-1 mobility. J Cell Biol. (2015) 208:457–73. doi: 10.1083/jcb.201406120
68. Huppa JB, Davis MM. T-cell-antigen recognition and the immunological synapse. Nat Rev Immunol. (2003) 3:973. doi: 10.1038/nri1245
69. Mittelbrunn M, Molina A, Escribese MM, Yanez-Mo M, Escudero E, Ursa A, et al. VLA-4 integrin concentrates at the peripheral supramolecular activation complex of the immune synapse and drives T helper 1 responses. Proc Natl Acad Sci USA. (2004) 101:11058–63. doi: 10.1073/pnas.0307927101
70. Kandula S, Abraham C. LFA-1 on CD4+ T cells is required for optimal antigen-dependent activation in vivo. J Immunol. (2004) 173:4443–51. doi: 10.4049/jimmunol.173.7.4443
71. Morrison VL, Uotila LM, Llort Asens M, Savinko T, Fagerholm SC. Optimal T cell activation and B cell antibody responses in vivo require the interaction between leukocyte function-associated antigen-1 and kindlin-3. J Immunol. (2015) 195:105–15. doi: 10.4049/jimmunol.1402741
72. Wernimont SA, Wiemer AJ, Bennin DA, Monkley SJ, Ludwig T, Critchley DR, et al. Contact-dependent T cell activation and T cell stopping require talin1. J Immunol. (2011) 187:6256–67. doi: 10.4049/jimmunol.1102028
73. Abraham C, Griffith J, Miller J. The dependence for leukocyte function-associated antigen-1/ICAM-1 interactions in T cell activation cannot be overcome by expression of high density tcr ligand. J Immunol. (1999) 162:4399–405.
74. Abraham C, Miller J. Molecular mechanisms of IL-2 gene regulation following costimulation through LFA-1. J Immunol. (2001) 167:5193–201. doi: 10.4049/jimmunol.167.9.5193
75. Wang Y, Li D, Nurieva R, Yang J, Sen M, Carreno R, et al. LFA-1 affinity regulation is necessary for the activation and proliferation of naive T cells. J Biol Chem. (2009) 284:12645–53. doi: 10.1074/jbc.M807207200
76. Rogers PR, Croft M. CD28, Ox-40, LFA-1, and CD4 modulation of Th1/Th2 differentiation is directly dependent on the dose of antigen. J Immunol. (2000) 164:2955–63. doi: 10.4049/jimmunol.164.6.2955
77. Marski M, Kandula S, Turner JR, Abraham C. CD18 is required for optimal development and function of CD4+CD25+ T regulatory cells. J Immunol. (2005) 175:7889–97. doi: 10.4049/jimmunol.175.12.7889
78. Meli AP, Fontes G, Avery DT, Leddon SA, Tam M, Elliot M, et al. The integrin LFA-1 controls T follicular helper cell generation and maintenance. Immunity. (2016) 45:831–46. doi: 10.1016/j.immuni.2016.09.018
79. Capece T, Walling BL, Lim K, Kim KD, Bae S, Chung HL, et al. A novel intracellular pool of LFA-1 is critical for asymmetric CD8(+) T cell activation and differentiation. J Cell Biol. (2017) 216:3817–29. doi: 10.1083/jcb.201609072
80. Anikeeva N, Somersalo K, Sims TN, Thomas VK, Dustin ML, Sykulev Y. Distinct role of lymphocyte function-associated antigen-1 in mediating effective cytolytic activity by cytotoxic T lymphocytes. Proc Natl Acad Sci USA. (2005) 102:6437–42. doi: 10.1073/pnas.0502467102
81. Osman MS, Burshtyn DN, Kane KP. Activating Ly-49 receptors regulate LFA-1-mediated adhesion by NK cells. J Immunol. (2007) 178:1261–7. doi: 10.4049/jimmunol.178.3.1261
82. Dupuy AG, Caron E. Integrin-dependent phagocytosis: spreading from microadhesion to new concepts. J Cell Sci. (2008) 121:1773–83. doi: 10.1242/jcs.018036
83. Anderson KE, Boyle KB, Davidson K, Chessa TA, Kulkarni S, Jarvis GE, et al. CD18-dependent activation of the neutrophil NADPH oxidase during phagocytosis of Escherichia coli or Staphylococcus aureus is regulated by class III but not class I or II PI3Ks. Blood. (2008) 112:5202–11. doi: 10.1182/blood-2008-04-149450
84. Han C, Jin J, Xu S, Liu H, Li N, Cao X. Integrin CD11b negatively regulates TLR-triggered inflammatory responses by activating Syk and promoting degradation of MyD88 and TRIF via Cbl-b. Nat Immunol. (2010) 11:734–42. doi: 10.1038/ni.1908
85. Wang L, Gordon RA, Huynh L, Su X, Park Min KH, Han J, et al. Indirect inhibition of Toll-like receptor and type I interferon responses by ITAM-coupled receptors and integrins. Immunity. (2010) 32:518–30. doi: 10.1016/j.immuni.2010.03.014
86. Varga G, Balkow S, Wild MK, Stadtbaeumer A, Krummen M, Rothoeft T, et al. Active MAC-1 (CD11b/CD18) on DCs inhibits full T-cell activation. Blood. (2007) 109:661–9. doi: 10.1182/blood-2005-12-023044
87. Podgrabinska S, Kamalu O, Mayer L, Shimaoka M, Snoeck H, Randolph GJ, et al. Inflamed lymphatic endothelium suppresses dendritic cell maturation and function via Mac-1/ICAM-1-dependent mechanism. J Immunol. (2009) 183:1767–79. doi: 10.4049/jimmunol.0802167
88. Balkow S, Heinz S, Schmidbauer P, Kolanus W, Holzmann B, Grabbe S, et al. LFA-1 activity state on dendritic cells regulates contact duration with T cells and promotes T-cell priming. Blood. (2010) 116:1885–94. doi: 10.1182/blood-2009-05-224428
89. Morrison VL, James MJ, Grzes K, Cook P, Glass DG, Savinko T, et al. Loss of beta2-integrin-mediated cytoskeletal linkage reprogrammes dendritic cells to a mature migratory phenotype. Nat Commun. (2014) 5:5359. doi: 10.1038/ncomms6359
90. Ehirchiou D, Xiong Y, Xu G, Chen W, Shi Y, Zhang L. CD11b facilitates the development of peripheral tolerance by suppressing Th17 differentiation. J Exp Med. (2007) 204:1519–24. doi: 10.1084/jem.20062292
91. Nowatzky J, Manches O, Khan SA, Godefroy E, Bhardwaj N. Modulation of human Th17 cell responses through complement receptor 3 (CD11b/CD18) ligation on monocyte-derived dendritic cells. J Autoimmun. (2018) 92:57–66. doi: 10.1016/j.jaut.2018.05.005
92. Savinko TS, Morrison VL, Uotila LM, Wolff CH, Alenius HT, Fagerholm SC. Functional Beta2-integrins restrict skin inflammation in vivo. J Invest Dermatol. (2015) 135:2249–57. doi: 10.1038/jid.2015.164
93. Springer TA. Adhesion receptors of the immune system. Nature. (1990) 346:425–34. doi: 10.1038/346425a0
94. Roebuck KA, Finnegan A. Regulation of intercellular adhesion molecule-1 (CD54) gene expression. J Leukoc Biol. (1999) 66:876–88. doi: 10.1002/jlb.66.6.876
95. Wegner CD, Gundel RH, Reilly P, Haynes N, Letts LG, Rothlein R. Intercellular adhesion molecule-1 (ICAM-1) in the pathogenesis of asthma. Science. (1990) 247:456–9. doi: 10.1126/science.1967851
96. Borchers AT, Shimoda S, Bowlus C, Keen CL, Gershwin ME. Lymphocyte recruitment and homing to the liver in primary biliary cirrhosis and primary sclerosing cholangitis. Semin Immunopathol. (2009) 31:309–22. doi: 10.1007/s00281-009-0167-2
97. Schlesinger M, Bendas G. Vascular cell adhesion molecule-1 (VCAM-1)–an increasing insight into its role in tumorigenicity and metastasis. Int J Cancer. (2015) 136:2504–14. doi: 10.1002/ijc.28927
98. Kong D-H, Kim YK, Kim MR, Jang JH, Lee S. Emerging roles of vascular cell adhesion molecule-1 (VCAM-1) in immunological disorders and cancer. Int J Mol Sci. (2018) 19:1057. doi: 10.3390/ijms19041057
99. Harlin H, Meng Y, Peterson AC, Zha Y, Tretiakova M, Slingluff C, et al. Chemokine expression in melanoma metastases associated with CD8+ T-cell recruitment. Cancer Res. (2009) 69:3077–85. doi: 10.1158/0008-5472.CAN-08-2281
100. Gajewski TF, Fuertes M, Spaapen R, Zheng Y, Kline J. Molecular profiling to identify relevant immune resistance mechanisms in the tumor microenvironment. Curr Opin Immunol. (2011) 23:286–92. doi: 10.1016/j.coi.2010.11.013
101. Vignali D, Kallikourdis M. Improving homing in T cell therapy. Cytokine Growth Factor Rev. (2017) 36:107–16. doi: 10.1016/j.cytogfr.2017.06.009
102. Wu AA, Drake V, Huang H-S, Chiu S, Zheng L. Reprogramming the tumor microenvironment: tumor-induced immunosuppressive factors paralyze T cells. Oncoimmunology. (2015) 4:e1016700. doi: 10.1080/2162402X.2015.1016700
103. Klein D. The tumor vascular endothelium as decision maker in cancer therapy. (2018) 8:367. doi: 10.3389/fonc.2018.00367
104. Piali L, Fichtel A, Terpe HJ, Imhof BA, Gisler RH. Endothelial vascular cell adhesion molecule 1 expression is suppressed by melanoma and carcinoma. J Exp Med. (1995) 181:811–6. doi: 10.1084/jem.181.2.811
105. Griffioen AW, Damen CA, Martinotti S, Blijham GH, Groenewegen G. Endothelial intercellular adhesion molecule-1 expression is suppressed in human malignancies: the role of angiogenic factors. Cancer Res. (1996) 56:1111–7.
106. Yoong KF, Mcnab G, Hubscher SG, Adams DH. Vascular adhesion protein-1 and ICAM-1 support the adhesion of tumor-infiltrating lymphocytes to tumor endothelium in human hepatocellular carcinoma. J Immunol. (1998) 160:3978–88.
107. Enarsson K, Johnsson E, Lindholm C, Lundgren A, Pan-Hammarstrom Q, Stromberg E, et al. Differential mechanisms for T lymphocyte recruitment in normal and neoplastic human gastric mucosa. Clin Immunol. (2006) 118:24–34. doi: 10.1016/j.clim.2005.08.001
108. Weishaupt C, Munoz KN, Buzney E, Kupper TS, Fuhlbrigge RC. T-cell distribution and adhesion receptor expression in metastatic melanoma. Clin Cancer Res. (2007) 13:2549–56. doi: 10.1158/1078-0432.CCR-06-2450
109. Afanasiev OK, Nagase K, Simonson W, Vandeven N, Blom A, Koelle DM, et al. Vascular E-selectin expression correlates with CD8 lymphocyte infiltration and improved outcome in Merkel cell carcinoma. J Invest Dermatol. (2013) 133:2065–73. doi: 10.1038/jid.2013.36
110. Dirkx AE, Oude Egbrink MG, Kuijpers MJ, Van Der Niet ST, Heijnen VV, Bouma-Ter Steege JC, et al. Tumor angiogenesis modulates leukocyte-vessel wall interactions in vivo by reducing endothelial adhesion molecule expression. Cancer Res. (2003) 63:2322–9.
111. Griffioen AW, Relou IaM, Gallardo Torres HI, Damen CA, Martinotti S, Groenewegen GJA, et al. The angiogenic factor bFGF impairs leukocyte adhesion and rolling under flow conditions. (1998) 2:235–43. doi: 10.1023/A:1009237324501
112. Tromp SC, Oude Egbrink MGA, Dings RPM, Van Velzen S, Slaaf DW, Hillen HFP, et al. Tumor angiogenesis factors reduce leukocyte adhesion in vivo. Int Immunol. (2000) 12:671–6. doi: 10.1093/intimm/12.5.671
113. Ogawa Y, Hirakawa K, Nakata B, Fujihara T, Sawada T, Kato Y, et al. Expression of intercellular adhesion molecule-1 in invasive breast cancer reflects low growth potential, negative lymph node involvement, and good prognosis. Clin Cancer Res. (1998) 4:31–6.
114. Fujihara T, Yashiro M, Inoue T, Sawada T, Kato Y, Ohira M, et al. Decrease in ICAM-1 expression on gastric cancer cells is correlated with lymph node metastasis. Gastric Cancer. (1999) 2:221–5. doi: 10.1007/s101200050067
115. Maeda K, Kang SM, Sawada T, Nishiguchi Y, Yashiro M, Ogawa Y, et al. Expression of intercellular adhesion molecule-1 and prognosis in colorectal cancer. Oncol Rep. (2002) 9:511–4. doi: 10.3892/or.9.3.511
116. Koyama S, Ebihara T, Fukao K. Expression of intercellular adhesion molecule 1 (ICAM-1) during the development of invasion and/or metastasis of gastric carcinoma. J Cancer Res Clin Oncol. (1992) 118:609–14. doi: 10.1007/BF01211806
117. Tachimori A, Yamada N, Sakate Y, Yashiro M, Maeda K, Ohira M, et al. Up regulation of ICAM-1 gene expression inhibits tumour growth and liver metastasis in colorectal carcinoma. Eur J Cancer. (2005) 41:1802–10. doi: 10.1016/j.ejca.2005.04.036
118. Van der Bruggen P, Traversari C, Chomez P, Lurquin C, De Plaen E, Van Den Eynde B, et al. A gene encoding an antigen recognized by cytolytic T lymphocytes on a human melanoma. Science. (1991) 254:1643–7. doi: 10.1126/science.1840703
119. Robbins PF, Kawakami Y. Human tumor antigens recognized by T cells. Curr Opin Immunol. (1996) 8:628–36. doi: 10.1016/S0952-7915(96)80078-1
120. Shrikant P, Mescher MF. Control of syngeneic tumor growth by activation of CD8+ T cells: efficacy is limited by migration away from the site and induction of nonresponsiveness. J Immunol. (1999) 162:2858–66.
121. Durgeau A, Virk Y, Corgnac S, Mami-Chouaib F. Recent advances in targeting CD8 T-cell immunity for more effective cancer immunotherapy. Front Immunol. (2018) 9:14. doi: 10.3389/fimmu.2018.00014
122. Sato E, Olson SH, Ahn J, Bundy B, Nishikawa H, Qian F, et al. Intraepithelial CD8+ tumor-infiltrating lymphocytes and a high CD8+/regulatory T cell ratio are associated with favorable prognosis in ovarian cancer. Proc Natl Acad Sci USA. (2005) 102:18538–43. doi: 10.1073/pnas.0509182102
123. Ino Y, Yamazaki-Itoh R, Shimada K, Iwasaki M, Kosuge T, Kanai Y, et al. Immune cell infiltration as an indicator of the immune microenvironment of pancreatic cancer. Br J Cancer. (2013) 108:914–23. doi: 10.1038/bjc.2013.32
124. Melero I, Rouzaut A, Motz GT, Coukos G. T-cell and NK-cell infiltration into solid tumors: a key limiting factor for efficacious cancer immunotherapy. Cancer Discov. (2014) 4:522–6. doi: 10.1158/2159-8290.CD-13-0985
125. Fisher DT, Chen Q, Skitzki JJ, Muhitch JB, Zhou L, Appenheimer MM, et al. IL-6 trans-signaling licenses mouse and human tumor microvascular gateways for trafficking of cytotoxic T cells. J Clin Invest. (2011) 121:3846–59. doi: 10.1172/JCI44952
126. Sartor WM, Kyprianou N, Fabian DF, Lefor AT. Enhanced expression of ICAM-1 in a murine fibrosarcoma reduces tumor growth rate. J Surg Res. (1995) 59:66–74. doi: 10.1006/jsre.1995.1133
127. Wu T-C, Xu K, Banchereau R, Marches F, Yu CI, Martinek J, et al. Reprogramming tumor-infiltrating dendritic cells for CD103+ CD8+ mucosal T-cell differentiation and breast cancer rejection. Cancer Immunol Res. (2014) 2:487–500. doi: 10.1158/2326-6066.CIR-13-0217
128. Mlecnik B, Tosolini M, Charoentong P, Kirilovsky A, Bindea G, Berger A, et al. Biomolecular network reconstruction identifies T-cell homing factors associated with survival in colorectal cancer. Gastroenterology. (2010) 138:1429–40. doi: 10.1053/j.gastro.2009.10.057
129. Clark RA, Huang SJ, Murphy GF, Mollet IG, Hijnen D, Muthukuru M, et al. Human squamous cell carcinomas evade the immune response by down-regulation of vascular E-selectin and recruitment of regulatory T cells. J Exp Med. (2008) 205:2221–34. doi: 10.1084/jem.20071190
130. Lohr J, Ratliff T, Huppertz A, Ge Y, Dictus C, Ahmadi R, et al. Effector T-cell infiltration positively impacts survival of glioblastoma patients and is impaired by tumor-derived TGF-beta. Clin Cancer Res. (2011) 17:4296–308. doi: 10.1158/1078-0432.CCR-10-2557
131. Mukai S, Kagamu H, Shu S, Plautz GE. Critical role of CD11a (LFA-1) in therapeutic efficacy of systemically transferred antitumor effector T cells. Cell Immunol. (1999) 192:122–32. doi: 10.1006/cimm.1998.1439
132. Blank C, Brown I, Kacha AK, Markiewicz MA, Gajewski TF. ICAM-1 contributes to but is not essential for tumor antigen cross-priming and CD8+ T cell-mediated tumor rejection in vivo. J Immunol. (2005) 174:3416–20. doi: 10.4049/jimmunol.174.6.3416
133. Le Floc'h A, Jalil A, Vergnon I, Le Maux Chansac B, Lazar V, Bismuth G, et al. Alpha E beta 7 integrin interaction with E-cadherin promotes antitumor CTL activity by triggering lytic granule polarization and exocytosis. J Exp Med. (2007) 204:559–70. doi: 10.1084/jem.20061524
134. Yanguas A, Garasa S, Teijeira Á, Aubá C, Melero I, Rouzaut A. ICAM-1-LFA-1 dependent CD8+ T-lymphocyte aggregation in tumor tissue prevents recirculation to draining lymph nodes. Front Immunol. (2018) 9:2084. doi: 10.3389/fimmu.2018.02084
135. Bell D, Chomarat P, Broyles D, Netto G, Harb GM, Lebecque S, et al. In breast carcinoma tissue, immature dendritic cells reside within the tumor, whereas mature dendritic cells are located in peritumoral areas. J Exp Med. (1999) 190:1417–26. doi: 10.1084/jem.190.10.1417
136. Aspord C, Pedroza-Gonzalez A, Gallegos M, Tindle S, Burton EC, Su D, et al. Breast cancer instructs dendritic cells to prime interleukin 13-secreting CD4+ T cells that facilitate tumor development. J Exp Med. (2007) 204:1037–47. doi: 10.1084/jem.20061120
137. Okita Y, Tanaka H, Ohira M, Muguruma K, Kubo N, Watanabe M, et al. Role of tumor-infiltrating CD11b+ antigen-presenting cells in the progression of gastric cancer. J Surg Res. (2014) 186:192–200. doi: 10.1016/j.jss.2013.08.024
138. Nakahara T, Oba J, Shimomura C, Kido-Nakahara M, Furue M. Early Tumor-infiltrating dendritic cells change their characteristics drastically in association with murine melanoma progression. J Investig Dermatol. (2016) 136:146–53. doi: 10.1038/JID.2015.359
139. Jang J-E, Hajdu CH, Liot C, Miller G, Dustin ML, Bar-Sagi D. Crosstalk between regulatory T cells and tumor-associated dendritic cells negates anti-tumor immunity in pancreatic cancer. Cell Rep. (2017) 20:558–71. doi: 10.1016/j.celrep.2017.06.062
140. Hotblack A, Holler A, Piapi A, Ward S, Stauss HJ, Bennett CL. Tumor-resident dendritic cells and macrophages modulate the accumulation of TCR-engineered T cells in melanoma. Mol Ther. (2018) 26:1471–81. doi: 10.1016/j.ymthe.2018.03.011
141. Dhodapkar MV, Dhodapkar KM, Palucka AK. Interactions of tumor cells with dendritic cells: balancing immunity and tolerance. Cell Death Differentiation. (2008) 15:39–50. doi: 10.1038/sj.cdd.4402247
142. Harshyne LA, Watkins SC, Gambotto A, Barratt-Boyes SM. Dendritic cells acquire antigens from live cells for cross-presentation to CTL. J Immunol Mar. (2001) 166:3717–23. doi: 10.4049/jimmunol.166.6.3717
143. Harshyne LA, Zimmer MI, Watkins SC, Barratt-Boyes SM. A role for class A scavenger receptor in dendritic cell nibbling from live cells. J Immunol. (2003) 170:2302–9. doi: 10.4049/jimmunol.170.5.2302
144. Albert ML, Pearce SFA, Francisco LM, Sauter B, Roy P, Silverstein RL, et al. Immature dendritic cells phagocytose apoptotic cells via αvβ5 and CD36, and cross-present antigens to cytotoxic T lymphocytes. J Exp Med. (1998) 188:1359–68. doi: 10.1084/jem.188.7.1359
145. Akakura S, Singh S, Spataro M, Akakura R, Kim J-I, Albert ML, et al. The opsonin MFG-E8 is a ligand for the αvβ5 integrin and triggers DOCK180-dependent Rac1 activation for the phagocytosis of apoptotic cells. Exp Cell Res. (2004) 292:403–16. doi: 10.1016/j.yexcr.2003.09.011
146. Verbovetski I, Bychkov H, Trahtemberg U, Shapira I, Hareuveni M, Ben-Tal O, et al. Opsonization of apoptotic cells by autologous iC3b facilitates clearance by immature dendritic cells, down-regulates DR and CD86, and up-regulates CC chemokine receptor 7. J Exp Med. (2002) 196:1553–61. doi: 10.1084/jem.20020263
147. Škoberne M, Somersan S, Almodovar W, Truong T, Petrova K, Henson PM, et al. The apoptotic-cell receptor CR3, but not αvβ5, is a regulator of human dendritic-cell immunostimulatory function. Blood J. (2006) 108:947–55. doi: 10.1182/blood-2005-12-4812
148. Hanahan D, Weinberg R. Hallmarks of cancer: the next generation. Cell. (2011) 144:646–74. doi: 10.1016/j.cell.2011.02.013
149. Ma J, Wang J-H, Guo Y-J, Sy M-S, Bigby M. In vivo treatment with Anti-ICAM-1 and anti-LFA-1 antibodies inhibits contact sensitization-induced migration of epidermal langerhans cells to regional lymph nodes. Cell Immunol. (1994) 158:389–99. doi: 10.1006/cimm.1994.1285
150. Xu H, Guan H, Zu G, Bullard D, Hanson J, Slater M, et al. The role of ICAM-1 molecule in the migration of Langerhans cells in the skin and regional lymph node. Eur J Immunol. (2001) 31:3085–93. doi: 10.1002/1521-4141(2001010)31:10<3085::AID-IMMU3085>3.0.CO;2-B
151. Johnson LA, Clasper S, Holt AP, Lalor PF, Baban D, Jackson DG. An inflammation-induced mechanism for leukocyte transmigration across lymphatic vessel endothelium. J Exp Med. (2006) 203:2763–77. doi: 10.1084/jem.20051759
152. Grabbe S, Varga G, Beissert S, Steinert M, Pendl G, Seeliger S, et al. Beta2 integrins are required for skin homing of primed T cells but not for priming naive T cells. J Clin Investig. (2002) 109:183–92. doi: 10.1172/JCI0211703
153. Thompson ED, Enriquez HL, Fu Y-X, Engelhard VH. Tumor masses support naive T cell infiltration, activation, and differentiation into effectors. J Exp Med. (2010) 207:1791–804. doi: 10.1084/jem.20092454
154. Broz M, Binnewies M, Boldajipour B, Nelson A, Pollack J, Erle D, et al. Dissecting the tumor myeloid compartment reveals rare activating antigen-presenting cells critical for T cell immunity. Cancer Cell. (2014) 26:638–52. doi: 10.1016/j.ccell.2014.09.007
155. Roberts EW, Broz ML, Binnewies M, Headley MB, Nelson AE, Wolf DM, et al. Critical role for CD103(+)/CD141(+) dendritic cells bearing CCR7 for tumor antigen trafficking and priming of T cell immunity in melanoma. Cancer Cell. (2016) 30:324–36. doi: 10.1016/j.ccell.2016.06.003
156. Salmon H, Idoyaga J, Rahman A, Leboeuf M, Remark R, Jordan S, et al. Expansion and activation of CD103(+) dendritic cell progenitors at the tumor site enhances tumor responses to therapeutic PD-L1 and BRAF inhibition. Immunity. (2016) 44:924–38. doi: 10.1016/j.immuni.2016.03.012
157. Böttcher JP, Sousa REC. The role of type 1 conventional dendritic cells in cancer immunity. Trends Cancer. (2018) 4:784–92. doi: 10.1016/j.trecan.2018.09.001
158. Haniffa M, Shin A, Bigley V, Mcgovern N, Teo P, See P, et al. Human tissues contain CD141hi cross-presenting dendritic cells with functional homology to mouse CD103+ nonlymphoid dendritic cells. Immunity. (2012) 37:60–73. doi: 10.1016/j.immuni.2012.04.012
159. Bachem A, Güttler S, Hartung E, Ebstein F, Schaefer M, Tannert A, et al. Superior antigen cross-presentation and XCR1 expression define human CD11c+CD141+ cells as homologues of mouse CD8+ dendritic cells. (2010) 207:1273–1281. doi: 10.1084/jem.20100348
160. Jongbloed SL, Kassianos AJ, Mcdonald KJ, Clark GJ, Ju X, Angel CE, et al. Human CD141+ (BDCA-3)+ dendritic cells (DCs) represent a unique myeloid DC subset that cross-presents necrotic cell antigens. J Exp Med. (2010) 207:1247–60. doi: 10.1084/jem.20092140
161. Michea P, Noël F, Zakine E, Czerwinska U, Sirven P, Abouzid O, et al. Adjustment of dendritic cells to the breast-cancer microenvironment is subset specific. Nat Immunol. (2018) 19:885–97. doi: 10.1038/s41590-018-0145-8
162. Sakaguchi S, Yamaguchi T, Nomura T, Ono M. Regulatory T cells and immune tolerance. Cell. (2008) 133:775–87. doi: 10.1016/j.cell.2008.05.009
163. Cottrez F, Hurst SD, Coffman RL, Groux H. T regulatory cells 1 inhibit a Th2-specific response in vivo. J Immunol. (2000) 165:4848–53. doi: 10.4049/jimmunol.165.9.4848
164. Nakamura K, Kitani A, Strober W. Cell contact-dependent immunosuppression by CD4(+)CD25(+) regulatory T cells is mediated by cell surface-bound transforming growth factor beta. J Exp Med. (2001) 194:629–44. doi: 10.1084/jem.194.5.629
165. Grossman WJ, Verbsky JW, Barchet W, Colonna M, Atkinson JP, Ley TJ. Human T regulatory cells can use the perforin pathway to cause autologous target cell death. Immunity. (2004) 21:589–601. doi: 10.1016/j.immuni.2004.09.002
166. Wrzesinski SH, Wan YY, Flavell RA. Transforming growth factor-beta and the immune response: implications for anticancer therapy. Clin Cancer Res. (2007) 13:5262–70. doi: 10.1158/1078-0432.CCR-07-1157
167. Li M, Lin J, Wang Z, He S, Ma X, Li D. Oxidized low-density lipoprotein-induced proinflammatory cytokine response in macrophages are suppressed by CD4CD25(+)Foxp3(+) regulatory T cells through downregulating toll like receptor 2-mediated activation of NF-kappaB. Cell Physiol Biochem. (2010) 25:649–56. doi: 10.1159/000315084
168. Kohm AP, Carpentier PA, Anger HA, Miller SD. Cutting edge: CD4+CD25+ regulatory T cells suppress antigen-specific autoreactive immune responses and central nervous system inflammation during active experimental autoimmune encephalomyelitis. J Immunol. (2002) 169:4712–6. doi: 10.4049/jimmunol.169.9.4712
169. Wang H, Peters T, Sindrilaru A, Kess D, Oreshkova T, Yu XZ, et al. TGF-beta-dependent suppressive function of Tregs requires wild-type levels of CD18 in a mouse model of psoriasis. J Clin Invest. (2008) 118:2629–39. doi: 10.1172/JCI34916
170. Wohler J, Bullard D, Schoeb T, Barnum S. LFA-1 is critical for regulatory T cell homeostasis and function. Mol Immunol. (2009) 46:2424–8. doi: 10.1016/j.molimm.2009.04.004
171. Haasken S, Auger JL, Binstadt BA. Absence of β2 integrins impairs regulatory T cells and exacerbates CD4+T cell-dependent autoimmune carditis. J Exp Med. (2011) 187:2702–10. doi: 10.4049/jimmunol.1000967
172. Taylor PA, Panoskaltsis-Mortari A, Swedin JM, Lucas PJ, Gress RE, Levine BL, et al. L-Selectin(hi) but not the L-selectin(lo) CD4+25+ T-regulatory cells are potent inhibitors of GVHD and BM graft rejection. Blood. (2004) 104:3804–12. doi: 10.1182/blood-2004-05-1850
173. Lehmann J, Huehn J, De La Rosa M, Maszyna F, Kretschmer U, Krenn V, et al. Expression of the integrin alpha Ebeta 7 identifies unique subsets of CD25+ as well as CD25- regulatory T cells. Proc Natl Acad Sci USA. (2002) 99:13031–6. doi: 10.1073/pnas.192162899
174. Huehn J, Siegmund K, Lehmann JC, Siewert C, Haubold U, Feuerer M, et al. Developmental stage, phenotype, and migration distinguish naive- and effector/memory-like CD4+ regulatory T cells. J Exp Med. (2004) 199:303–13. doi: 10.1084/jem.20031562
175. Curiel TJ, Coukos G, Zou L, Alvarez X, Cheng P, Mottram P, et al. Specific recruitment of regulatory T cells in ovarian carcinoma fosters immune privilege and predicts reduced survival. Nat Med. (2004) 10:942–9. doi: 10.1038/nm1093
176. Klages K, Mayer CT, Lahl K, Loddenkemper C, Teng MW, Ngiow SF, et al. Selective depletion of Foxp3+ regulatory T cells improves effective therapeutic vaccination against established melanoma. Cancer Res. (2010) 70:7788–99. doi: 10.1158/0008-5472.CAN-10-1736
177. Teng MW, Ngiow SF, Von Scheidt B, Mclaughlin N, Sparwasser T, Smyth MJ. Conditional regulatory T-cell depletion releases adaptive immunity preventing carcinogenesis and suppressing established tumor growth. Cancer Res. (2010) 70:7800–9. doi: 10.1158/0008-5472.CAN-10-1681
178. Marabelle A, Kohrt H, Sagiv-Barfi I, Ajami B, Axtell RC, Zhou G, et al. Depleting tumor-specific Tregs at a single site eradicates disseminated tumors. J Clin Invest. (2013) 123:2447–63. doi: 10.1172/JCI64859
179. Anz D, Mueller W, Golic M, Kunz WG, Rapp M, Koelzer VH, et al. CD103 is a hallmark of tumor-infiltrating regulatory T cells. Int J Cancer. (2011) 129:2417–26. doi: 10.1002/ijc.25902
180. Gabrilovich DI, Nagaraj S. Myeloid-derived suppressor cells as regulators of the immune system. Nat Rev Immunol. (2009) 9:162–74. doi: 10.1038/nri2506
181. Najjar YG, Finke JH. Clinical perspectives on targeting of myeloid derived suppressor cells in the treatment of cancer. Front Oncol. (2013) 3:49. doi: 10.3389/fonc.2013.00049
182. Bronte V, Brandau S, Chen SH, Colombo MP, Frey AB, Greten TF, et al. Recommendations for myeloid-derived suppressor cell nomenclature and characterization standards. Nat Commun. (2016) 7:12150. doi: 10.1038/ncomms12150
183. Kumar V, Patel S, Tcyganov E, Gabrilovich DI. The nature of myeloid-derived suppressor cells in the tumor microenvironment. Trends Immunol. (2016) 37:208–20. doi: 10.1016/j.it.2016.01.004
184. Suzuki E, Kapoor V, Jassar AS, Kaiser LR, Albelda SM. Gemcitabine selectively eliminates splenic Gr-1+/CD11b+ myeloid suppressor cells in tumor-bearing animals and enhances antitumor immune activity. Clin Cancer Res. (2005) 11:6713–21. doi: 10.1158/1078-0432.CCR-05-0883
185. Serafini P, Meckel K, Kelso M, Noonan K, Califano J, Koch W, et al. Phosphodiesterase-5 inhibition augments endogenous antitumor immunity by reducing myeloid-derived suppressor cell function. J Exp Med. (2006) 203:2691–702. doi: 10.1084/jem.20061104
186. Wang G, Lu X, Dey P, Deng P, Wu CC, Jiang S, et al. Targeting YAP-dependent MDSC infiltration impairs tumor progression. Cancer Discov. (2016) 6:80–95. doi: 10.1158/2159-8290.CD-15-0224
187. Almand B, Clark JI, Nikitina E, Van Beynen J, English NR, Knight SC, et al. Increased production of immature myeloid cells in cancer patients: a mechanism of immunosuppression in cancer. J Immunol. (2001) 166:678–89. doi: 10.4049/jimmunol.166.1.678
188. Ochoa AC, Zea AH, Hernandez C, Rodriguez PC. Arginase, prostaglandins, and myeloid-derived suppressor cells in renal cell carcinoma. Clin Cancer Res. (2007) 13:721s−6s. doi: 10.1158/1078-0432.CCR-06-2197
189. Diaz-Montero CM, Salem ML, Nishimura MI, Garrett-Mayer E, Cole DJ, Montero AJ. Increased circulating myeloid-derived suppressor cells correlate with clinical cancer stage, metastatic tumor burden, and doxorubicin-cyclophosphamide chemotherapy. Cancer Immunol Immunother. (2009) 58:49–59. doi: 10.1007/s00262-008-0523-4
190. Eruslanov E, Neuberger M, Daurkin I, Perrin GQ, Algood C, Dahm P, et al. Circulating and tumor-infiltrating myeloid cell subsets in patients with bladder cancer. Int J Cancer. (2012) 130:1109–19. doi: 10.1002/ijc.26123
191. Yu J, Du W, Yan F, Wang Y, Li H, Cao S, et al. Myeloid-derived suppressor cells suppress antitumor immune responses through IDO expression and correlate with lymph node metastasis in patients with breast cancer. J Immunol. (2013) 190:3783–97. doi: 10.4049/jimmunol.1201449
192. Weide B, Martens A, Zelba H, Stutz C, Derhovanessian E, Di Giacomo AM, et al. Myeloid-derived suppressor cells predict survival of patients with advanced melanoma: comparison with regulatory T cells and NY-ESO-1- or melan-A-specific T cells. Clin Cancer Res. (2014) 20:1601–9. doi: 10.1158/1078-0432.CCR-13-2508
193. Jin H, Su J, Garmy-Susini B, Kleeman J, Varner J. Integrin alpha4beta1 promotes monocyte trafficking and angiogenesis in tumors. Cancer Res. (2006) 66:2146–52. doi: 10.1158/0008-5472.CAN-05-2704
194. Foubert P, Kaneda MM, Varner JA. PI3Kgamma activates integrin alpha4 and promotes immune suppressive myeloid cell polarization during tumor progression. Cancer Immunol Res. (2017) 5:957–68. doi: 10.1158/2326-6066.CIR-17-0143
195. Arnaout M. Structure and function of the leukocyte adhesion molecules CD11/CD18. Blood. (1990) 75:1037–50.
196. Palmen MJ, Dijkstra CD, Van Der Ende MB, Peña AS, Van Rees EP. Anti-CD11b/CD18 antibodies reduce inflammation in acute colitis in rats. Clin Exp Immunol. (1995) 101:351–6. doi: 10.1111/j.1365-2249.1995.tb08363.x
197. Zhang QQ, Hu XW, Liu YL, Ye ZJ, Gui YH, Zhou DL, et al. CD11b deficiency suppresses intestinal tumor growth by reducing myeloid cell recruitment. Sci Rep. (2015) 5:15948. doi: 10.1038/srep15948
198. Ahn GO, Tseng D, Liao CH, Dorie MJ, Czechowicz A, Brown JM. Inhibition of Mac-1 (CD11b/CD18) enhances tumor response to radiation by reducing myeloid cell recruitment. Proc Natl Acad Sci USA. (2010) 107:8363–8. doi: 10.1073/pnas.0911378107
199. Desgrosellier JS, Cheresh DA. Integrins in cancer: biological implications and therapeutic opportunities. Nat Rev Cancer. (2010) 10:9–22. doi: 10.1038/nrc2748
200. Bao W, Strömblad S. Integrin alphav-mediated inactivation of p53 controls a MEK1-dependent melanoma cell survival pathway in three-dimensional collagen. J Cell Biol. (2004) 167:745–56. doi: 10.1083/jcb.200404018
201. Uhm JH, Dooley NP, Kyritsis AP, Rao JS, Gladson CL. Vitronectin, a glioma-derived extracellular matrix protein, protects tumor cells from apoptotic death. Clin Cancer Res. (1999) 5:1587–94.
202. Brooks PC, Strömblad S, Klemke R, Visscher D, Sarkar FH, Cheresh DA. Antiintegrin alpha v beta 3 blocks human breast cancer growth and angiogenesis in human skin. J Clin Investig. (1995) 96:1815–22. doi: 10.1172/JCI118227
203. Takayama S, Ishii S, Ikeda T, Masamura S, Doi M, Kitajima M. The relationship between bone metastasis from human breast cancer and integrin αvβ3 expression. Anticancer Res. (2005) 25:79–83.
204. McCabe NP, De S, Vasanji A, Brainard J, Byzova TV. Prostate cancer specific integrin alphavbeta3 modulates bone metastatic growth and tissue remodeling. Oncogene. (2007) 26:6238–43. doi: 10.1038/sj.onc.1210429
205. Huveneers S, Van Den Bout I, Sonneveld P, Sancho A, Sonnenberg A, Danen EHJ. Integrin αvβ3 controls activity and oncogenic potential of primed c-Src. Cancer Res. (2007) 67:2693–700. doi: 10.1158/0008-5472.CAN-06-3654
206. Desgrosellier JS, Barnes LA, Shields DJ, Huang M, Lau SK, Prévost N, et al. An integrin alpha(v)beta(3)-c-Src oncogenic unit promotes anchorage-independence and tumor progression. Nat Med. (2009) 15:1163–9. doi: 10.1038/nm.2009
207. Guo W, Pylayeva Y, Pepe A, Yoshioka T, Muller WJ, Inghirami G, et al. β4 integrin amplifies ErbB2 signaling to promote mammary tumorigenesis. Cell. (2006) 126:489–502. doi: 10.1016/j.cell.2006.05.047
208. Vaillant F, Asselin-Labat M-L, Shackleton M, Forrest NC, Lindeman GJ, Visvader JE. The mammary progenitor marker CD61/β3 integrin identifies cancer stem cells in mouse models of mammary tumorigenesis. Cancer Res. (2008) 68:7711–7. doi: 10.1158/0008-5472.CAN-08-1949
209. Wu TC. The role of vascular cell adhesion molecule-1 in tumor immune evasion. Cancer Res. (2007) 67:6003–6. doi: 10.1158/0008-5472.CAN-07-1543
210. Johnson JP, Stade BG, Holzmann B, Schwable W, Riethmuller G. De novo expression of intercellular-adhesion molecule 1 in melanoma correlates with increased risk of metastasis. Proc Natl Acad Sci USA. (1989) 86:641–4. doi: 10.1073/pnas.86.2.641
211. Lin YC, Shun CT, Wu MS, Chen CC. A novel anticancer effect of thalidomide: inhibition of intercellular adhesion molecule-1-mediated cell invasion and metastasis through suppression of nuclear factor-kappaB. Clin Cancer Res. (2006) 12:7165–73. doi: 10.1158/1078-0432.CCR-06-1393
212. Schroder C, Witzel I, Muller V, Krenkel S, Wirtz RM, Janicke F, et al. Prognostic value of intercellular adhesion molecule (ICAM)-1 expression in breast cancer. J Cancer Res Clin Oncol. (2011) 137:1193–201. doi: 10.1007/s00432-011-0984-2
213. Usami Y, Ishida K, Sato S, Kishino M, Kiryu M, Ogawa Y, et al. Intercellular adhesion molecule-1 (ICAM-1) expression correlates with oral cancer progression and induces macrophage/cancer cell adhesion. Int J Cancer. (2013) 133:568–78. doi: 10.1002/ijc.28066
214. Nath S, Mukherjee P. MUC1: a multifaceted oncoprotein with a key role in cancer progression. Trends Mol Med. (2014) 20:332–42. doi: 10.1016/j.molmed.2014.02.007
215. Roland CL, Harken AH, Sarr MG, Barnett CCJr. ICAM-1 expression determines malignant potential of cancer. Surgery. (2007) 141:705–7. doi: 10.1016/j.surg.2007.01.016
216. Yu JA, Sadaria MR, Meng X, Mitra S, Ao L, Fullerton DA, et al. Lung cancer cell invasion and expression of intercellular adhesion molecule-1 (ICAM-1) are attenuated by secretory phospholipase A2 inhibition. J Thoracic Cardiovasc Surg. (2012) 143:405–11. doi: 10.1016/j.jtcvs.2011.10.026
217. Harning R, Mainolfi E, Bystryn JC, Henn M, Merluzzi VJ, Rothlein R. Serum levels of circulating intercellular adhesion molecule 1 in human malignant melanoma. Cancer Res. (1991) 51:5003–5.
218. Christiansen I, Gidlof C, Wallgren AC, Simonsson B, Totterman TH. Serum levels of soluble intercellular adhesion molecule 1 are increased in chronic B-lymphocytic leukemia and correlate with clinical stage and prognostic markers. Blood. (1994) 84:3010–6.
219. Abdelrazik N, Fouda M, Zaghloul MHE, Abbas D. Serum level of intercellular adhesion molecule-1 in children with malignant lymphoma. Med Principles Pract. (2008) 17:233–8. doi: 10.1159/000117798
220. Toiyama Y, Miki C, Inoue Y, Okugawa Y, Koike Y, Yokoe T, et al. Soluble intercellular adhesion molecule-1 as a prognostic marker for stage II colorectal cancer patients. Ann Surg Oncol. (2008) 15:1617–24. doi: 10.1245/s10434-008-9874-5
221. Fabbri G, Dalla-Favera R. The molecular pathogenesis of chronic lymphocytic leukaemia. Nat Rev Cancer. (2016) 16:145–62. doi: 10.1038/nrc.2016.8
222. Burger JA, Gribben JG. The microenvironment in chronic lymphocytic leukemia (CLL) and other B cell malignancies: insight into disease biology and new targeted therapies. Semin Cancer Biol. (2014) 24:71–81. doi: 10.1016/j.semcancer.2013.08.011
223. Montresor A, Bolomini-Vittori M, Simon SI, Rigo A, Vinante F, Laudanna C. Comparative analysis of normal versus CLL B-lymphocytes reveals patient-specific variability in signaling mechanisms controlling LFA-1 activation by chemokines. Cancer Res. (2009) 69:9281–90. doi: 10.1158/0008-5472.CAN-09-2009
224. Till KJ, Harris RJ, Linford A, Spiller DG, Zuzel M, Cawley JC. Cell motility in chronic lymphocytic leukemia: defective rap1 and αLβ2 activation by chemokine. Cancer Res. (2008) 68:8429–36. doi: 10.1158/0008-5472.CAN-08-1758
225. Lucio P, Faria M, Pinto A, Da Silva M, Correia Junior M, Da Costa R, et al. Expression of adhesion molecules in chronic B-cell lymphoproliferative disorders. Haematologica. (1998) 83:104–11.
226. Hartmann TN, Grabovsky V, Wang W, Desch P, Rubenzer G, Wollner S, et al. Circulating B-cell chronic lymphocytic leukemia cells display impaired migration to lymph nodes and bone marrow. Cancer Res. (2009) 69:3121–30. doi: 10.1158/0008-5472.CAN-08-4136
227. Riches JC, O'donovan CJ, Kingdon SJ, Mcclanahan F, Clear AJ, Neuberg DS, et al. Trisomy 12 chronic lymphocytic leukemia cells exhibit upregulation of integrin signaling that is modulated by NOTCH1 mutations. Blood. (2014) 123:4101–10. doi: 10.1182/blood-2014-01-552307
228. Hutterer E, Asslaber D, Caldana C, Krenn PW, Zucchetto A, Gattei V, et al. CD18 (ITGB2) expression in chronic lymphocytic leukaemia is regulated by DNA methylation-dependent and -independent mechanisms. Br J Haematol. (2015) 169:286–9. doi: 10.1111/bjh.13188
229. Gattei V, Bulian P, Del Principe MI, Zucchetto A, Maurillo L, Buccisano F, et al. Relevance of CD49d protein expression as overall survival and progressive disease prognosticator in chronic lymphocytic leukemia. Blood. (2008) 111:865–73. doi: 10.1182/blood-2007-05-092486
230. Rossi D, Zucchetto A, Rossi FM, Capello D, Cerri M, Deambrogi C, et al. CD49d expression is an independent risk factor of progressive disease in early stage chronic lymphocytic leukemia. Haematologica. (2008) 93:1575–9. doi: 10.3324/haematol.13103
231. Shanafelt TD, Geyer SM, Bone ND, Tschumper RC, Witzig TE, Nowakowski GS, et al. CD49d expression is an independent predictor of overall survival in patients with chronic lymphocytic leukaemia: a prognostic parameter with therapeutic potential. Br J Haematol. (2008) 140:537–46. doi: 10.1111/j.1365-2141.2007.06965.x
232. Röllig C, Knop S, Bornhäuser M. Multiple myeloma. Lancet. (2015) 385:2197–208. doi: 10.1016/S0140-6736(14)60493-1
233. Kumar SK, Rajkumar V, Kyle RA, Van Duin M, Sonneveld P, Mateos M-V, et al. Multiple myeloma. Nat Rev Dis Primers. (2017) 3:17046. doi: 10.1038/nrdp.2017.46
234. Guillerey C, Nakamura K, Vuckovic S, Hill GR, Smyth MJJC, Sciences ML. Immune responses in multiple myeloma: role of the natural immune surveillance and potential of immunotherapies. Cell Mol Life Sci. (2016) 73:1569–89. doi: 10.1007/s00018-016-2135-z
235. Ahsmann E, Lokhorst H, Dekker A, Bloem A. Lymphocyte function-associated antigen-1 expression on plasma cells correlates with tumor growth in multiple myeloma. Blood. (1992) 79:2068–75.
236. Vacca A, Loreto MD, Ribatti D, Stefano RD, Gadaleta-Caldarola G, Iodice G, et al. Bone marrow of patients with active multiple myeloma: angiogenesis and plasma cell adhesion molecules LFA-1, VLA-4, LAM-1, and CD44. Am J Hematol. (1995) 50:9–14. doi: 10.1002/ajh.2830500103
237. Tatsumi T, Shimazaki C, Goto H, Araki S, Sudo Y, Yamagata N, et al. Expression of adhesion molecules on myeloma cells. Jpn J Cancer Res. (1996) 87:837–42. doi: 10.1111/j.1349-7006.1996.tb02108.x
238. Asosingh K, Vankerkhove V, Van Riet I, Van Camp B, Vanderkerken K. Selective in vivo growth of lymphocyte function- associated antigen-1–positive murine myeloma cells: Involvement of function-associated antigen-1–mediated homotypic cell-cell adhesion. Exp Hemat. (2003) 31:48–55. doi: 10.1016/S0301-472X(02)00970-0
239. Di Marzo L, Desantis V, Solimando AG, Ruggieri S, Annese T, Nico B, et al. Microenvironment drug resistance in multiple myeloma: emerging new players. Oncotarget. (2016) 7:60698–711. doi: 10.18632/oncotarget.10849
240. Raab-Westphal S, Marshall JF, Goodman SL. Integrins as therapeutic targets: successes and cancers. Cancers. (2017) 9:110. doi: 10.3390/cancers9090110
241. Schnittert J, Bansal R, Storm G, Prakash J. Integrins in wound healing, fibrosis and tumor stroma: High potential targets for therapeutics and drug delivery. Advanced Drug Delivery Rev. (2018) 129:37–53. doi: 10.1016/j.addr.2018.01.020
242. Feldman LE, Shin KC, Natale RB, Todd RF. β1 integrin expression on human small cell lung cancer cells. Cancer Res. (1991) 51:1065–70.
243. Hood JD, Cheresh DA. Role of integrins in cell invasion and migration. Nat Rev Cancer. (2002) 2:91. doi: 10.1038/nrc727
244. Mulgrew K, Kinneer K, Yao X-T, Ward BK, Damschroder MM, Walsh B, et al. Direct targeting of αvβ3 integrin on tumor cells with a monoclonal antibody, Abegrin™. (2006). Mol Cancer Therapeut. 5:3122–9. doi: 10.1158/1535-7163.MCT-06-0356
245. Bhaskar V, Zhang D, Fox M, Seto P, Wong MH, Wales PE, et al. A function blocking anti-mouse integrin α5β1 antibody inhibits angiogenesis and impedes tumor growth in vivo. J Transl Med. (2007) 5:61. doi: 10.1186/1479-5876-5-61
246. Sawada K, Mitra AK, Radjabi AR, Bhaskar V, Kistner EO, Tretiakova M, et al. Loss of E-cadherin promotes ovarian cancer metastasis via alpha 5-integrin, which is a therapeutic target. Cancer Res. (2008) 68:2329–39. doi: 10.1158/0008-5472.CAN-07-5167
247. Weis SM, Cheresh DA. αV integrins in angiogenesis and cancer. Cold Spring Harbor Perspect Med. (2011) 1:a006478. doi: 10.1101/cshperspect.a006478
248. dos Santos PB, Zanetti JS, Ribeiro-Silva A, Beltrão EIC. Beta 1 integrin predicts survival in breast cancer: a clinicopathological and immunohistochemical study. Diagn Pathol. (2012) 7:104. doi: 10.1186/1746-1596-7-104
249. Gutheil JC, Campbell TN, Pierce PR, Watkins JD, Huse WD, Bodkin DJ, et al. Targeted antiangiogenic therapy for cancer using Vitaxin: a humanized monoclonal antibody to the integrin alphavbeta3. Clin Cancer Res. (2000) 6:3056–61.
250. McNeel DG, Eickhoff J, Lee FT, King DM, Alberti D, Thomas JP, et al. Phase I trial of a monoclonal antibody specific for alphavbeta3 integrin (MEDI-522) in patients with advanced malignancies, including an assessment of effect on tumor perfusion. Clin Cancer Res. (2005) 11:7851–60. doi: 10.1158/1078-0432.CCR-05-0262
251. Cianfrocca ME, Kimmel KA, Gallo J, Cardoso T, Brown MM, Hudes G, et al. Phase 1 trial of the antiangiogenic peptide ATN-161 (Ac-PHSCN-NH(2)), a beta integrin antagonist, in patients with solid tumours. Br J Cancer. (2006) 94:1621–6. doi: 10.1038/sj.bjc.6603171
252. Cranmer LD, Bedikian AY, Ribas A, O'day S, Forero-Torres A, Yazji S, et al. Phase II study of volociximab (M200), an α5β1 anti-integrin antibody in metastatic melanoma. J Clin Oncol. (2006) 24:8011. doi: 10.1200/jco.2006.24.18_suppl.8011
253. Figlin RA, Kondagunta GV, Yazji S, Motzer RJ, Bukowski RM. Phase II study of volociximab (M200), an α5β1 anti-integrin antibody in refractory metastatic clear cell renal cell cancer (RCC). J Clin Oncol. (2006) 24:4535. doi: 10.1200/jco.2006.24.18_suppl.4535
254. Friess H, Langrehr JM, Oettle H, Raedle J, Niedergethmann M, Dittrich C, et al. A randomized multi-center phase II trial of the angiogenesis inhibitor Cilengitide (EMD 121974) and gemcitabine compared with gemcitabine alone in advanced unresectable pancreatic cancer. BMC Cancer. (2006) 6:285. doi: 10.1186/1471-2407-6-285
255. Evans T, Ramanathan RK, Yazji S, Glynne-Jones R, Anthoney A, Berlin J, et al. Final results from cohort 1 of a phase II study of volociximab, an anti-α5β1 integrin antibody, in combination with gemcitabine (GEM) in patients (pts) with metastatic pancreatic cancer (MPC). J Clin Oncol. (2007) 25:4549. doi: 10.1200/jco.2007.25.18_suppl.4549
256. Mullamitha SA, Ton NC, Parker GJ, Jackson A, Julyan PJ, Roberts C, et al. Phase I evaluation of a fully human anti-alphav integrin monoclonal antibody (CNTO 95) in patients with advanced solid tumors. Clin Cancer Res. (2007) 13:2128–35. doi: 10.1158/1078-0432.CCR-06-2779
257. Nabors LB, Mikkelsen T, Rosenfeld SS, Hochberg F, Akella NS, Fisher JD, et al. Phase I and correlative biology study of cilengitide in patients with recurrent malignant glioma. J Clin Oncol. (2007) 25:1651–7. doi: 10.1200/JCO.2006.06.6514
258. Barton J. A multicenter phase II study of volociximab in patients with relapsed metastatic melanoma. J Clin Oncol. (2008) 26:9051. doi: 10.1200/jco.2008.26.15_suppl.9051
259. Delbaldo C, Raymond E, Vera K, Hammershaimb L, Kaucic K, Lozahic S, et al. Phase I and pharmacokinetic study of etaracizumab (Abegrin), a humanized monoclonal antibody against alphavbeta3 integrin receptor, in patients with advanced solid tumors. Invest New Drugs. (2008) 26:35–43. doi: 10.1007/s10637-007-9077-0
260. MacDonald TJ, Stewart CF, Kocak M, Goldman S, Ellenbogen RG, Phillips P, et al. Phase I clinical trial of cilengitide in children with refractory brain tumors: pediatric brain tumor consortium study PBTC-012. J Clin Oncol. (2008) 26:919–24. doi: 10.1200/JCO.2007.14.1812
261. Reardon DA, Fink KL, Mikkelsen T, Cloughesy TF, O'neill A, Plotkin S, et al. Randomized phase II study of cilengitide, an integrin-targeting arginine-glycine-aspartic acid peptide, in recurrent glioblastoma multiforme. J Clin Oncol. (2008) 26:5610–7. doi: 10.1200/JCO.2008.16.7510
262. Ricart AD, Tolcher AW, Liu G, Holen K, Schwartz G, Albertini M, et al. Volociximab, a chimeric monoclonal antibody that specifically binds alpha5beta1 integrin: a phase I, pharmacokinetic, and biological correlative study. Clin Cancer Res. (2008) 14:7924–9. doi: 10.1158/1078-0432.CCR-08-0378
263. Vergote IB, Colombo N, Kutarska E, Campo JD, Pippitt C, Casado A, et al. Phase II study comparing volociximab (an antiangiogenic antibody) and pegylated liposomal doxorubicin (PLD) with PLD alone in recurrent ovarian or primary peritoneal cancer. J Clin Oncol. (2009) 27:5560. doi: 10.1200/jco.2009.27.15s.5560
264. Hersey P, Sosman J, O'day S, Richards J, Bedikian A, Gonzalez R, et al. A randomized phase 2 study of etaracizumab, a monoclonal antibody against integrin alpha(v)beta(3), + or - dacarbazine in patients with stage IV metastatic melanoma. Cancer. (2010) 116:1526–34. doi: 10.1002/cncr.24821
265. Sawyer MB, Iqbal S, Lenz H, Lima CSR, Rossignol DP, Krivelevich I, et al. Phase II study of E7820 in combination with cetuximab in subjects (pts) with metastatic and refractory colorectal cancer (CRC). (2010) 28:3537–3537. doi: 10.1200/jco.2010.28.15_suppl.3537
266. Stupp R, Hegi ME, Neyns B, Goldbrunner R, Schlegel U, Clement PM, et al. Phase I/IIa study of cilengitide and temozolomide with concomitant radiotherapy followed by cilengitide and temozolomide maintenance therapy in patients with newly diagnosed glioblastoma. J Clin Oncol. (2010) 28:2712–8. doi: 10.1200/JCO.2009.26.6650
267. Thompson DS, Patnaik A, Bendell JC, Papadopoulos K, Infante JR, Mastico RA, et al. A phase I dose-escalation study of IMGN388 in patients with solid tumors. J Clin Oncol. (2010) 28:3058. doi: 10.1200/jco.2010.28.15_suppl.3058
268. Bell-McGuinn KM, Matthews CM, Ho SN, Barve M, Gilbert L, Penson RT, et al. A phase II, single-arm study of the anti-alpha5beta1 integrin antibody volociximab as monotherapy in patients with platinum-resistant advanced epithelial ovarian or primary peritoneal cancer. Gynecol Oncol. (2011) 121:273–9. doi: 10.1016/j.ygyno.2010.12.362
269. Bradley DA, Daignault S, Ryan CJ, Dipaola RS, Cooney KA, Smith DC, et al. Cilengitide (EMD 121974, NSC 707544) in asymptomatic metastatic castration resistant prostate cancer patients: a randomized phase II trial by the prostate cancer clinical trials consortium. Invest New Drugs. (2011) 29:1432–40. doi: 10.1007/s10637-010-9420-8
270. Mita M, Kelly KR, Mita A, Ricart AD, Romero O, Tolcher A, et al. Phase I study of E7820, an oral inhibitor of integrin alpha-2 expression with antiangiogenic properties, in patients with advanced malignancies. Clin Cancer Res. (2011) 17:193–200. doi: 10.1158/1078-0432.CCR-10-0010
271. O'Day S, Pavlick A, Loquai C, Lawson D, Gutzmer R, Richards J, et al. A randomised, phase II study of intetumumab, an anti-alphav-integrin mAb, alone and with dacarbazine in stage IV melanoma. Br J Cancer. (2011) 105:346–52. doi: 10.1038/bjc.2011.183
272. Gilbert MR, Kuhn J, Lamborn KR, Lieberman F, Wen PY, Mehta M, et al. Cilengitide in patients with recurrent glioblastoma: the results of NABTC 03-02, a phase II trial with measures of treatment delivery. J Neuro Oncol. (2012) 106:147–53. doi: 10.1007/s11060-011-0650-1
273. Kim KB, Prieto V, Joseph RW, Diwan AH, Gallick GE, Papadopoulos NE, et al. A randomized phase II study of cilengitide (EMD 121974) in patients with metastatic melanoma. Melanoma Res. (2012) 22:294–301. doi: 10.1097/CMR.0b013e32835312e4
274. O'Donnell PH, Undevia SD, Stadler WM, Karrison TM, Nicholas MK, Janisch L, et al. A phase I study of continuous infusion cilengitide in patients with solid tumors. Invest New Drugs. (2012) 30:604–10. doi: 10.1007/s10637-010-9537-9
275. Besse B, Tsao LC, Chao DT, Fang Y, Soria JC, Almokadem S, et al. Phase Ib safety and pharmacokinetic study of volociximab, an anti-alpha5beta1 integrin antibody, in combination with carboplatin and paclitaxel in advanced non-small-cell lung cancer. Ann Oncol. (2013) 24:90–6. doi: 10.1093/annonc/mds281
276. Heidenreich A, Rawal SK, Szkarlat K, Bogdanova N, Dirix L, Stenzl A, et al. A randomized, double-blind, multicenter, phase 2 study of a human monoclonal antibody to human alphanu integrins (intetumumab) in combination with docetaxel and prednisone for the first-line treatment of patients with metastatic castration-resistant prostate cancer. Ann Oncol. (2013) 24:329–36. doi: 10.1093/annonc/mds505
277. Stupp R, Hegi ME, Gorlia T, Erridge SC, Perry J, Hong YK, et al. Cilengitide combined with standard treatment for patients with newly diagnosed glioblastoma with methylated MGMT promoter (CENTRIC EORTC 26071-22072 study): a multicentre, randomised, open-label, phase 3 trial. Lancet Oncol. (2014) 15:1100–8. doi: 10.1016/S1470-2045(14)70379-1
278. Vermorken JB, Peyrade F, Krauss J, Mesia R, Remenar E, Gauler TC, et al. Cisplatin, 5-fluorouracil, and cetuximab (PFE) with or without cilengitide in recurrent/metastatic squamous cell carcinoma of the head and neck: results of the randomized phase I/II ADVANTAGE trial (phase II part). Ann Oncol. (2014) 25:682–8. doi: 10.1093/annonc/mdu003
279. Wirth M, Heidenreich A, Gschwend JE, Gil T, Zastrow S, Laniado M, et al. A multicenter phase 1 study of EMD 525797 (DI17E6), a novel humanized monoclonal antibody targeting alphav integrins, in progressive castration-resistant prostate cancer with bone metastases after chemotherapy. Eur Urol. (2014) 65:897–904. doi: 10.1016/j.eururo.2013.05.051
280. Elez E, Kocakova I, Hohler T, Martens UM, Bokemeyer C, Van Cutsem E, et al. Abituzumab combined with cetuximab plus irinotecan versus cetuximab plus irinotecan alone for patients with KRAS wild-type metastatic colorectal cancer: the randomised phase I/II POSEIDON trial. Ann Oncol. (2015) 26:132–40. doi: 10.1093/annonc/mdu474
281. Gerstner ER, Ye X, Duda DG, Levine MA, Mikkelsen T, Kaley TJ, et al. A phase I study of cediranib in combination with cilengitide in patients with recurrent glioblastoma. Neuro Oncol. (2015) 17:1386–92. doi: 10.1093/neuonc/nov085
282. Nabors LB, Fink KL, Mikkelsen T, Grujicic D, Tarnawski R, Nam DH, et al. Two cilengitide regimens in combination with standard treatment for patients with newly diagnosed glioblastoma and unmethylated MGMT gene promoter: results of the open-label, controlled, randomized phase II CORE study. Neuro Oncol. (2015) 17:708–17. doi: 10.1093/neuonc/nou356
283. Uronis HE, Jia J, Bendell JC, Howard L, Ready NA, Lee PH, et al. A Phase I/biomarker study of bevacizumab in combination with CNTO 95 in patients with advanced solid tumors. Cancer Chemother Pharmacol. (2015) 75:343–52. doi: 10.1007/s00280-014-2647-x
284. Vansteenkiste J, Barlesi F, Waller CF, Bennouna J, Gridelli C, Goekkurt E, et al. Cilengitide combined with cetuximab and platinum-based chemotherapy as first-line treatment in advanced non-small-cell lung cancer (NSCLC) patients: results of an open-label, randomized, controlled phase II study (CERTO). Ann Oncol. (2015) 26:1734–40. doi: 10.1093/annonc/mdv219
285. Cirkel GA, Kerklaan BM, Vanhoutte F, Van Der Aa A, Lorenzon G, Namour F, et al. A dose escalating phase I study of GLPG0187, a broad spectrum integrin receptor antagonist, in adult patients with progressive high-grade glioma and other advanced solid malignancies. Investigational New Drugs. (2016) 34:184–92. doi: 10.1007/s10637-015-0320-9
286. Hussain M, Le Moulec S, Gimmi C, Bruns R, Straub J, Miller K. Differential effect on bone lesions of targeting integrins: randomized phase ii trial of abituzumab in patients with metastatic castration-resistant prostate cancer. Clin Cancer Res. (2016) 22:3192–200. doi: 10.1158/1078-0432.CCR-15-2512
287. Khasraw M, Lee A, Mccowatt S, Kerestes Z, Buyse ME, Back M, et al. Cilengitide with metronomic temozolomide, procarbazine, and standard radiotherapy in patients with glioblastoma and unmethylated MGMT gene promoter in ExCentric, an open-label phase II trial. J Neurooncol. (2016) 128:163–71. doi: 10.1007/s11060-016-2094-0
288. Milojkovic Kerklaan B, Slater S, Flynn M, Greystoke A, Witteveen PO, Megui-Roelvink M, et al. A phase I, dose escalation, pharmacodynamic, pharmacokinetic, and food-effect study of alpha2 integrin inhibitor E7820 in patients with advanced solid tumors. Invest New Drugs. (2016) 34:329–37. doi: 10.1007/s10637-016-0344-9
289. Haddad T, Qin R, Lupu R, Satele D, Eadens M, Goetz MP, et al. A phase I study of cilengitide and paclitaxel in patients with advanced solid tumors. Cancer Chemother Pharmacol. (2017) 79:1221–7. doi: 10.1007/s00280-017-3322-9
290. Kwan BH, Zhu EF, Tzeng A, Sugito HR, Eltahir AA, Ma B, et al. Integrin-targeted cancer immunotherapy elicits protective adaptive immune responses. J Exp Med. (2017) 214:1679–90. doi: 10.1084/jem.20160831
291. Dold S, Laschke MW, Lavasani S, Menger MD, Thorlacius H. Cholestatic liver damage is mediated by lymphocyte function antigen-1–dependent recruitment of leukocytes. Surgery. (2008) 144:385–93. doi: 10.1016/j.surg.2008.05.010
292. Suchard SJ, Stetsko DK, Davis PM, Skala S, Potin D, Launay M, et al. An LFA-1 (αLβ2) small-molecule antagonist reduces inflammation and joint destruction in murine models of arthritis. J Immunol. (2010) 184:3917–26. doi: 10.4049/jimmunol.0901095
293. Frampton JE, Plosker GL. Efalizumab. Am J Clin Dermatol. (2009) 10:51–72. doi: 10.2165/0128071-200910010-00009
294. Cohen S, Haimovich J, Hollander N. Anti-idiotype × anti-LFA-1 bispecific antibodies inhibit metastasis of B cell lymphoma. J Immunol. (2003) 170:2695–701. doi: 10.4049/jimmunol.170.5.2695
295. Reina M, Espel E. Role of LFA-1 and ICAM-1 in cancer. Cancers. (2017) 9:153. doi: 10.3390/cancers9110153
296. San Sebastián E, Zimmerman T, Zubia A, Vara Y, Martin E, Sirockin F, et al. Design, synthesis, and functional evaluation of leukocyte function associated antigen-1 antagonists in early and late stages of cancer development. J Med Chem. (2013) 56:735–47. doi: 10.1021/jm3016848
297. Seminara NM, Gelfand JM. Assessing long-term drug safety: lessons (re) learned from raptiva. Semin Cutaneous Med Surg. (2010) 29:16–9. doi: 10.1016/j.sder.2010.01.001
298. Scheinfeld N. Efalizumab: a review of events reported during clinical trials and side effects. Expert Opin Drug Saf. (2006) 5:197–209. doi: 10.1517/14740338.5.2.197
299. Talamonti M, Spallone G, Di Stefani A, Costanzo A, Chimenti S. Efalizumab. Expert Opinion Drug Saf. (2011) 10:239–51. doi: 10.1517/14740338.2011.524925
300. Grönholm M, Jahan F, Bryushkova EA, Madhavan S, Aglialoro F, Soto Hinojosa L, et al. LFA-1 integrin antibodies inhibit leukocyte α4β1-mediated adhesion by intracellular signaling. Blood. (2016) 128:1270–81. doi: 10.1182/blood-2016-03-705160
301. Garbi N, Arnold B, Gordon S, Hämmerling GJ, Ganss R. CpG motifs as proinflammatory factors render autochthonous tumors permissive for infiltration and destruction. J Immunol. (2004) 172:5861–9. doi: 10.4049/jimmunol.172.10.5861
302. Peske JD, Woods AB, Engelhard VH. Control of CD8 T-cell infiltration into tumors by vasculature and microenvironment. Adv Cancer Res. (2015) 128:263–307. doi: 10.1016/bs.acr.2015.05.001
303. Lejeune FJ, Lienard D, Matter M, Ruegg C. Efficiency of recombinant human TNF in human cancer therapy. Cancer Immun. (2006) 6:6.
304. Curnis F, Gasparri A, Sacchi A, Longhi R, Corti A. Coupling tumor necrosis factor-α with αV integrin ligands improves its antineoplastic activity. Cancer Res. (2004) 64:565–71. doi: 10.1158/0008-5472.CAN-03-1753
305. Dirkx AE, oude Egbrink MG, Castermans K, van der Schaft DW, Thijssen VL, Dings RP, et al. Anti-angiogenesis therapy can overcome endothelial cell anergy and promote leukocyte-endothelium interactions and infiltration in tumors. FASEB J. (2006) 20:621–30. doi: 10.1096/fj.05-4493com
306. Shrimali RK, Yu Z, Theoret MR, Chinnasamy D, Restifo NP, Rosenberg SA. Antiangiogenic agents can increase lymphocyte infiltration into tumor and enhance the effectiveness of adoptive immunotherapy of cancer. Cancer Res. (2010) 70:6171–80. doi: 10.1158/0008-5472.CAN-10-0153
307. Kochenderfer JN, Wilson WH, Janik JE, Dudley ME, Stetler-Stevenson M, Feldman SA, et al. Eradication of B-lineage cells and regression of lymphoma in a patient treated with autologous T cells genetically engineered to recognize CD19. (2010) 116:4099–102. doi: 10.1182/blood-2010-04-281931
308. Parkhurst MR, Yang JC, Langan RC, Dudley ME, Nathan D-aN, et al. T cells targeting carcinoembryonic antigen can mediate regression of metastatic colorectal cancer but induce severe transient colitis. Mol Ther. (2011) 19:620–6. doi: 10.1038/mt.2010.272
309. Porter DL, Levine BL, Kalos M, Bagg A, June CH. Chimeric antigen receptor–modified t cells in chronic lymphoid leukemia. N Engl J Med. (2011) 365:725–33. doi: 10.1056/NEJMoa1103849
310. Robbins PF, Morgan RA, Feldman SA, Yang JC, Sherry RM, Dudley ME, et al. Tumor regression in patients with metastatic synovial cell sarcoma and melanoma using genetically engineered lymphocytes reactive with NY-ESO-1. J Clin Oncol. (2011) 29:917–24. doi: 10.1200/JCO.2010.32.2537
311. Sadelain M, Brentjens R, Rivière I. The promise and potential pitfalls of chimeric antigen receptors. Curr Opin Immunol. (2009) 21:215–23. doi: 10.1016/j.coi.2009.02.009
312. Kosti P, Maher J, Arnold JN. Perspectives on chimeric antigen receptor T-cell immunotherapy for solid tumors. Front Immunol. (2018) 9:1104. doi: 10.3389/fimmu.2018.01104
313. Fu X, Rivera A, Tao L, Zhang X. Genetically modified T cells targeting neovasculature efficiently destroy tumor blood vessels, shrink established solid tumors and increase nanoparticle delivery. Int J Cancer. (2013) 133:2483–92. doi: 10.1002/ijc.28269
314. Murphy EA, Majeti BK, Barnes LA, Makale M, Weis SM, Lutu-Fuga K, et al. Nanoparticle-mediated drug delivery to tumor vasculature suppresses metastasis. Proc Natl Acad Sci USA. (2008) 105:9343–8. doi: 10.1073/pnas.0803728105
315. Semmrich M, Smith A, Feterowski C, Beer S, Engelhardt B, Busch DH, et al. Importance of integrin LFA-1 deactivation for the generation of immune responses. J Exp Med. (2005) 201:1987–98. doi: 10.1084/jem.20041850
316. Cantor JM, Rose DM, Slepak M, Ginsberg MH. Fine-tuning tumor immunity with integrin trans-regulation. Cancer Immunol Res. (2015) 3:661–7. doi: 10.1158/2326-6066.CIR-13-0226
Keywords: cell adhesion, integrin, LFA-1, ICAM-1, VCAM-1, immunotherapy, dendritic cell (DC)
Citation: Harjunpää H, Llort Asens M, Guenther C and Fagerholm SC (2019) Cell Adhesion Molecules and Their Roles and Regulation in the Immune and Tumor Microenvironment. Front. Immunol. 10:1078. doi: 10.3389/fimmu.2019.01078
Received: 18 January 2019; Accepted: 29 April 2019;
Published: 22 May 2019.
Edited by:
Toshiyuki Murai, Osaka University, JapanReviewed by:
David Calderwood, Yale University, United StatesFrancisco Sanchez-Madrid, Autonomous University of Madrid, Spain
Copyright © 2019 Harjunpää, Llort Asens, Guenther and Fagerholm. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Susanna C. Fagerholm, c3VzYW5uYS5mYWdlcmhvbG0mI3gwMDA0MDtoZWxzaW5raS5maQ==