 Victoria Klepsch
Victoria Klepsch Alexander R. Moschen2
Alexander R. Moschen2 Gottfried Baier
Gottfried Baier Natascha Hermann-Kleiter
Natascha Hermann-Kleiter
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol. , 14 May 2019
Sec. Mucosal Immunity
Volume 10 - 2019 | https://doi.org/10.3389/fimmu.2019.01070
This article is part of the Research Topic The Role of Physical and Biological Gut Barriers in Modulating Crosstalk between the Microbiota and the Immune System View all 16 articles
Gastrointestinal (GI) homeostasis is strongly dependent on nuclear receptor (NR) functions. They play a variety of roles ranging from nutrient uptake, sensing of microbial metabolites, regulation of epithelial intestinal cell integrity to shaping of the intestinal immune cell repertoire. Several NRs are associated with GI pathologies; therefore, systematic analysis of NR biology, the underlying molecular mechanisms, and regulation of target genes can be expected to help greatly in uncovering the course of GI diseases. Recently, an increasing number of NRs has been validated as potential drug targets for therapeutic intervention in patients with inflammatory bowel disease (IBD). Besides the classical glucocorticoids, especially PPARγ, VDR, or PXR-selective ligands are currently being tested with promising results in clinical IBD trials. Also, several pre-clinical animal studies are being performed with NRs. This review focuses on the complex biology of NRs and their context-dependent anti- or pro-inflammatory activities in the regulation of gastrointestinal barrier with special attention to NRs already pharmacologically targeted in clinic and pre-clinical IBD treatment regimens.
This review is based on the most recent advances in our understanding of the complex biology of nuclear receptors (NRs) within both the healthy and inflamed intestinal tract (Tables 1–3) and the emerging number of ligands successfully used in preclinical and clinical trials (Table 4) to target inflammation and treat inflammatory bowel disease (IBD) without focusing on intestinal infections (1–5). In general, NRs enable fine-tuning of cellular processes to environmental changes such as external milieu signals and the cell-intrinsic metabolic state and are therefore ideally suited as targets for therapeutic interventions (6–9). Within the gastrointestinal (GI) system, NRs are highly expressed and well-known sensors of nutrients, hormones, and specific host-bacterial metabolites (1–5). Gut physiology is regulated by several nuclear receptors such as ERβ (NR3B2), GR (NR3C1), FXR (NR1H), PPARγ (NR1C3), PXR (NR1I2), RARα (NR1B1), VDR (NR1I1), HNF4α (NR2A1), or NR2F6 (Ear2) which have been demonstrated to play fundamental roles in epithelial intestinal cell integrity and especially in shaping intestinal immune cell composition and function (Figure 1) (9–14).

Table 1. Steroidal nuclear receptors.
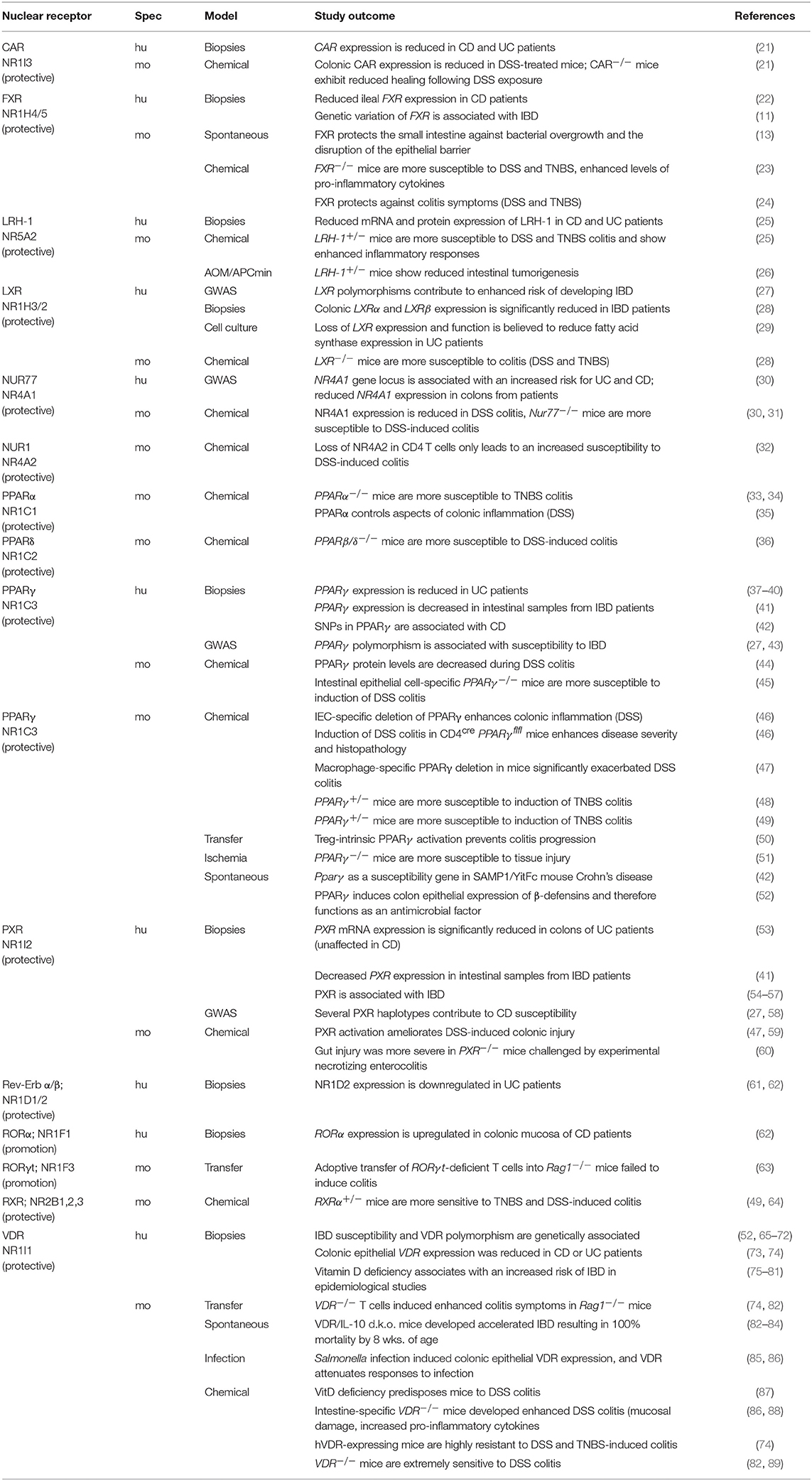
Table 2. Non-steroidal nuclear receptors.

Table 3. Orphan nuclear receptors.
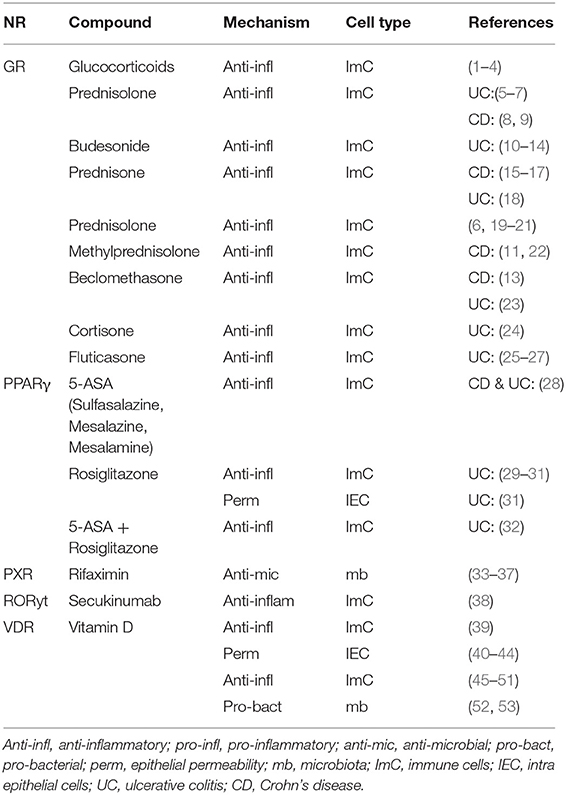
Table 4. Human IBD therapy—clinical trials.
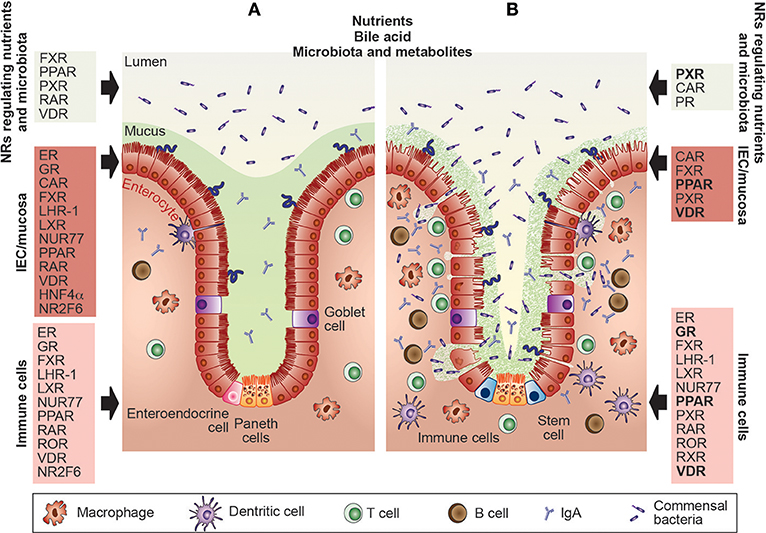
Figure 1. Nuclear receptors are essential for the maintenance of gut homeostasis and have already been targeted in IBD patients. (A) Within the healthy gastrointestinal system, nuclear receptors (NRs) such as FXR, PPAR, PXR, RAR, or VDR are well-known sensors of nutrients, toxic dietary products, specific host-bacterial metabolites, and bile acid. Intestinal barrier function and epithelial intestinal cell integrity are dependent on the appropriate function of the ER, GR, CAR, FXR, LHR-1, LXR, NUR77, PPAR, RAR, VDR, HNF4α, and NR2F6 which regulate mucus secretion, expression of tight junction proteins autophagy, circadian clock as well as goblet and paneth cell numbers. Also, NRs such as the ER, GR, FXR, LHR-1, LXR, NUR77, PPAR, RAR, ROR, VDR HNF4α, and NR2F6 contribute to gut homeostasis by shaping intestinal immune cell development, and the composition and effector functions of macrophages, dendritic cells, T and B cells. (B) The primary protective role of the NRs in the pathophysiology of inflammatory bowel diseases has been validated in pre-clinical animal models and clinical trials. NRs targeted by therapeutic drugs in IBD patients are GR, PPAR, PXR, and the VDR (highlighted in bold), NRs tested in preclinical mouse models are CAR, ER, FXR, LHR-1, LXR, NUR77, PPAR, PXR, RAR, ROR, and RXR; thus, novel concepts integrating NR, and gastrointestinal physiology have been integrated into the development of effective therapies. CAR, constitutive androstane receptor; ER, estrogen receptor; FXR, farnesoid X receptor; GR, glucocorticoid receptor; HNF4α, hepatocyte nuclear factor-4-alpha; IBD, inflammatory bowel disease; IECs, intestinal epithelial cells; LRH, liver-related homolog; LXR, liver X receptor; NR2F6, nuclear receptor subfamily 2 group F member 6; NR4A1/2, nuclear receptor subfamily 4 group A member 1/2 (NUR77, NUR1); PPAR, peroxisome proliferator-activated receptor; PXR, pregnane X receptor; RAR, retinoic acid receptor; RevErb, nuclear receptor subfamily 1, group D, member 1; ROR, RAR-related orphan receptor; RXR, retinoid X receptor; VDR, vitamin D receptor.
In humans, the NR family consists of 48 members and is, therefore, the most significant group of transcriptional regulators. It includes the receptors for steroid and thyroid hormones together with receptors for lipophilic vitamins and cholesterol metabolites (6, 7). The physiological ligands for approximately half of NRs are known, whereas the rest are classified as orphan receptors (8, 15–17). Members of the NR family are highly conserved; the modular domain structure consists of an activation domain (AF), the central DNA-binding-domain (DBD), the hinge region, the ligand-binding domain (LBD), and the activation function 2 (AF2) (15). NRs differ in their modes of action. In the classical steroid receptor signaling, for instance, the ligand (steroid) enters the cell to activate the receptor located in the cytoplasm. Due to the resulting conformational change, the receptor translocates to the nucleus and binds to its cognate nuclear receptor response element on the DNA within the target gene promoters thereby altering the transcription levels (18). However, other nuclear receptors, such as thyroid hormone receptors or the peroxisome proliferator-activated receptors (PPARs), are localized in the nucleus regardless of whether or not they are bound to a ligand and constitutively interact with DNA response elements (1). Of note, additional non-genomic functions of nuclear receptors in the cytosol have been firmly established such as the activation of cAMP, Ca2+ or the MAPK signaling cascade (17–19). The specificity of transcriptional activation by a given NR is achieved by the tissue-selective expression of co-repressors or co-activators as well as post-transcriptional modifications of both.
Expression analysis of biopsies from IBD patients as well as animal studies with NR ligands suggests a significant correlation between NR biology and IBD pathology (Tables 1–4) (Figure 1). Interestingly, the presence of NRs or their ligand agonists seems to be mainly protective during IBD (Tables 1–3). The NR superfamily is one of the primary classes of therapeutic drug targets for human disease (1, 2). How NRs regulate gut homeostasis in the complex interplay between intestinal epithelial cells, the immune system, and the microbiota is an active area of research.
Ligands targeting NRs in IBD, either being tested in clinical trials or already in use to treat IBD patients, are dexamethasone and methylprednisolone (targeting GR), rosiglitazone, pioglitazone, bezafibrate, and curcumin (targeting PPARγ), and 1,25-di-hydroxyvitamin, calcitriol, and cholecalciferol (targeting VDR) (3, 4) (Table 4).
Current strategies to treat IBD include anti-inflammatory drugs, immunosuppressives, biological agents, antibiotics, and changes in dietary habits in combination with pain medication (20). These treatment options help relieve symptoms and reduce the risks of recurrence and complications, but in most cases, only a subgroup of patients responds to the available therapies. Surgery is the last therapeutic possibility when there is loss of response and adverse side effects. In the context of an increasing number of IBD patients, new approaches to treatment are needed, and molecular targets such as NRs represent a promising avenue to pursue in a search for more effective drugs. This review focuses on the complex relationship between nutrition, inflammation and nuclear receptor biology within the GI and the emerging number of NR ligands used in IBD therapy.
Nutrient uptake and elimination of toxic dietary components or xenobiotics within gut epithelium are dependent on the dietary lipid-activated NRs such as CAR (NR1I3), FXR, PXR, and VDR. Also, glucose, fatty acid, triglycerides, and lipoprotein metabolism in intestinal epithelial cells (IECs) are regulated by the PPAR family (α, β, δ), whereas cholesterol transport and absorption and bile acid metabolism are dependent on LXR and LRH (NR5A2) (21).
Furthermore, NRs such as ERβ, RARα, HNF4α, and NR2F6 regulate essential aspects of intestinal barrier functions such as mucus secretion, goblet and paneth cell numbers, autophagy and expression of tight junction proteins (Figures 1, 2) (11–14, 22).
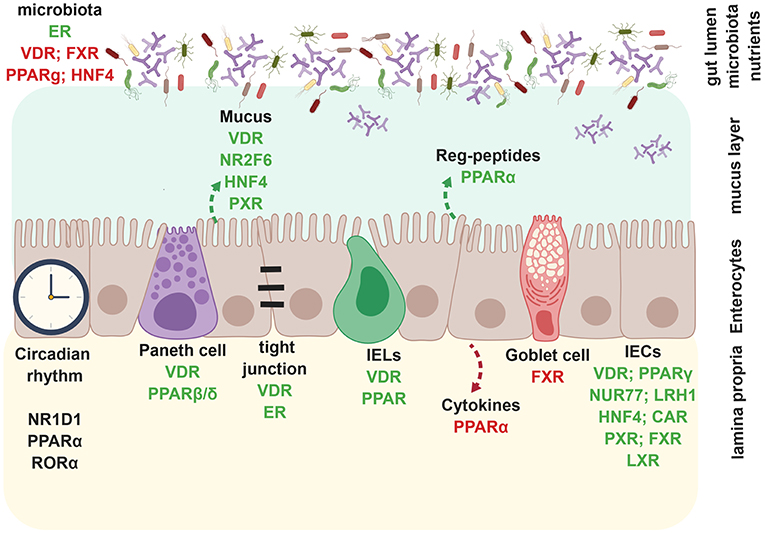
Figure 2. Nuclear receptors regulate intestinal epithelial barrier. During homeostasis, nuclear receptors such as VDR, FXR, PPARγ, HNF4α inhibit bacterial outgrowth whereas ER enhances microbiota richness. VDR, NR2F6, HNF4α, and PXR promote mucus secretion and epithelial barrier integrity, whereas VDR and ER directly enhance tight junctions. PPARα specifically promotes the production of anti-microbial Reg-peptides, beneficial for an intact barrier. Circadian rhythm in enterocytes is dependent on NR1D1, PPARα, and RORα. VDR and PPARβ/δ positively regulate paneth cell development. VDR and PPARα also promote CD8αα+ IELs. PPARα enhances expression of pro-inflammatory in enterocytes. FXR inhibits goblet cell development. VDR, PPARγ, NUR77, LRH-1, HNF4α, PXR, FXR, and LXR promote enterocyte development, whereas CAR is involved in wound healing of IECs. Created with BioRender.
Microbiota, and their metabolites such as butyrate, propionate, or indole, influence NR biology directly, functioning as ligands to target FXR, PPARγ, or PXR (Figure 1) (5). Depletion of butyrate-producing microbes by antibiotic treatment reduces epithelial signaling through PPARγ showing that microbiota-activated PPARγ signaling prevents the dysbiotic expansion of potential pathogens (23) (Figures 1, 2). However, FXR activation itself alters the intestinal microbiota and could provide opportunities for microbiome biomarker discovery or new approaches to engineering the human microbiome (24–26) (Figure 2). For detailed aspects of nuclear receptor and microbiota biology, we refer to a recent review by Duszka and Wahli (27).
Within the intestinal epithelium, NRs such as VDR, HNF4α, LXR, PPARγ, LRH1, and NR2F6 play protective roles in intestinal epithelial integrity (Figures 1, 2); decreased mRNAs have also been validated in intestinal samples from IBD patients (10, 12) (Table 1). In mice, deletion of the VDR increases mucosal injury that leads to high mortality in DSS-induced experimental colitis (10). In parallel, the activation of the farnesoid X receptor (FXR) prevents chemically-induced intestinal inflammation, improves colitis symptoms, inhibits epithelial permeability, and reduces goblet cell loss (13) (Figure 2). Intestinal steroidogenesis controls PPARγ expression in the colon, and this axis is impaired in ulcerative colitis (11).
The microbiota-NR axis influences not only metabolism of the intestinal epithelium, but also the components of the circadian clock; in particular, RORα (NR1F1) and RevErbα (NR1D1) influence corticosterone synthesis in IEC whereas PPAR and LXR families can alter the hepatic circadian clock (28, 29) (Figure 2).
NRs contribute especially to gut homeostasis by shaping intestinal immune cells; on one side, they are constantly challenged in the face of stimulatory signals from nutrients and gut microbiota, and on the other, they shape the composition of the microbiota themselves (Figures 1, 2) (5, 13, 19, 28, 29). Already the development of gut-associated lymphoid tissue is dependent on the expression of NRs like RORγt (NR1F3), which is required for the generation of lymphoid tissue inducer (LTi) cells and subsequent formation of Peyer's patches. As the amount of RORγ protein is reduced in the absence of the vitamin A metabolite retinoic acid (RA), this suggests that RAR directly controls the fetal development of intestinal secondary lymphoid organs (SLOs) as well as the fitness of the immune system in adulthood [recently reviewed (19)].
Macrophages expressing LXR, NR4A1 (NUR77), PPARγ, or RARα are essential for gut immune homeostasis (30–34) (Figure 3). Especially the reciprocal differentiation potential of naïve CD4+ T cells into either pro-inflammatory Th17 or tolerance-inducing regulatory T cells is dependent on several NRs such as RORγ, RORα, LXR, NR4A2 (NURR1), PPARγ, RAR, or VDR (35) (Figure 3). Whereas, RAR-related orphan receptors (RORγ and RORα) are key transcriptional activators, RAR, RXR, NUR77, PPARγ, LXR, GR, VDR, and ER contribute to anti-inflammatory effects (31, 32, 36–40) (Figure 3).
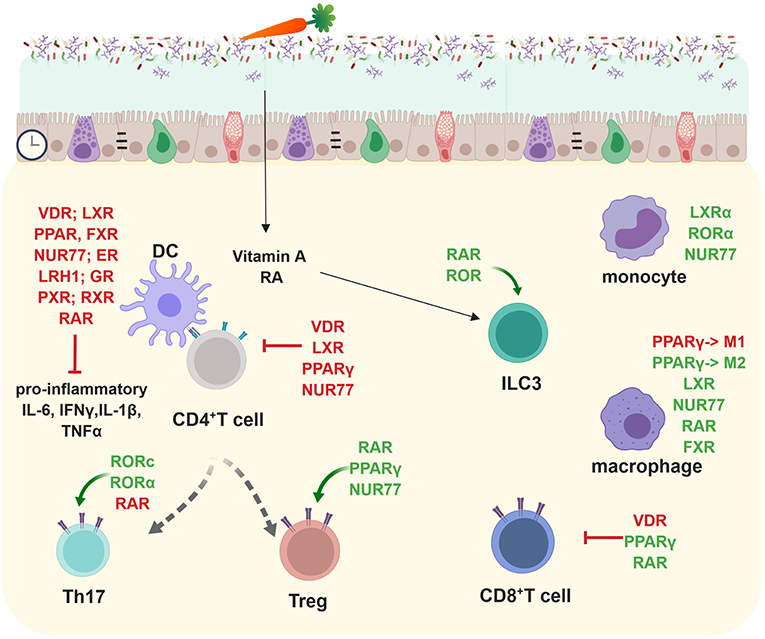
Figure 3. Nuclear receptors shape adaptive and innate immune responses in the lamina propria and subsequently gut homeostasis. Most NRs (VDR, LXR, PPAR, FXR, NUR77, ER, LRH-1, GR, PXR, RXR, and RAR) target pro-inflammatory cytokine production (IL-6, IFNγ, IL-1β, TNFα). RORc and RORα are important in promoting Th17 development whereas RAR, PPARγ, and NUR77 positively influence regulatory T cells. VDR, LXR, PPARγ, and NUR77 inhibit CD4+ T cell responses. RAR and ROR promote ILC3 differentiation. LXRα, RORα, and NUR77 positively influence monocytes. PPARγ inhibits M1 macrophages but promotes M2 macrophages which are important anti-inflammatory effector cytokine producers. LXR, NUR77, RAR, and FXR foster in parallel the anti-inflammatory responses of macrophages. VDR enhances inflammatory whereas PPARγ, and RAR suppresses pro-inflammatory CD8+ T cell responses. Created with BioRender.
The NR network in CD4 Th17 cells is highly complex. PPARγ suppresses Th17 differentiation by directly interfering with the silencing mediator of retinoid acid and thyroid hormone receptor (SMRT) clearance from the Rorc promoter. PPARγ activation suppresses not only the expression of Rorc but also subsequently that of Il17a, Il17f, Tnfa, Il22, Il21, Il23r, CCR6, and CCL20 (18). LXR reduces the expression of Rorγt, Il17a, Il17f, Il22, Il23r, and Ahr but does not affect that of Il21 and Rora. RAR suppresses Th17 differentiation through retinoic acid-mediated inhibition of Il23r, Il6r, and Irf4 expression and Smad2/3 phosphorylation via the TGFβ receptor pathway (43). NR2F6 directly binds to RORE sites within the Il17a locus and subsequently interferes with the transactivation of the Il17a promoter by RORc (41). It is still unclear how specificity is achieved despite highly homologous hormone response (HRE) DNA-binding core consensus sequences.
NRs, such as VDR or PPAR also regulate intestinal CD8 T cell responses (42–44). In parallel to the CD4 Th17 compartment, NR of the retinoic acid and retinoic acid-related family are key regulators of innate lymphoid cells (ILCs).
Innate lymphoid cells are tissue-resident immune cells that play essential roles in maintaining and protecting the gastrointestinal barrier against invading pathogens. In particular, ILCs themselves mediate immune responses but are controlled by dietary components and microbial metabolites. ILCs also directly regulate host metabolism and glucose tolerance (45). NR of the retinoic acid and retinoic acid-related family are key regulators of group ILC2 and 3 (46–48) (Figure 3). In a healthy state, ILC3 are responsible for mucosal homeostasis through the secretion of moderate amounts of IL-22, IL-17, and GM-CSF (49). Especially, regulation of gastrointestinal homeostasis and dysregulation by ILC3s result in overexpression of the IL-17, IFNγ, and IL-22 pro-inflammatory cytokines which can be seen in mice and IBD patients (50–57).
Thus, whereas on one side, microbiota and their metabolites shape the homeostasis of the gut immune system, innate and adaptive lymphocytes sequentially shape gut microbiota and lipid metabolism on the other. Thus, NRs occupy a center stage of gut immune homeostasis (27).
The two major forms of chronic inflammatory disorders within the gastrointestinal tract are Crohn's disease (CD) and ulcerative colitis (UC), characterized by clinical symptoms like severe diarrhea, pain, fatigue and weight loss (58). UC primarily affects the colon and rectum, whereas CD targets the small and large intestine, the mouth, esophagus, stomach, and the anus (59, 60). The etiology of the disease is multifactorial including genetic predisposition, the composition of gut microbiota, and environmental factors such as nutrition and antibiotic usage which subsequently can also alter immune responses (61). Intestinal barrier integrity is one of the most critical factors for a healthy GI tract, as an invasion of solutes, microorganisms and luminal antigens cause immune cell infiltration and inflammatory responses (62). Treatment options such as the corticosteroid prednisone or the anti-tumor necrosis factor-α antibody Infliximab suppress the immune system and relieve symptoms of patients.
Several members of the NR family have a protective role during disease progression, and their loss in different IBD animal models (DSS, TNBS, T cell transfer, or infection) leads to exacerbated colitis symptoms (Tables 1–3) (Figure 1). In human genome-wide association studies (GWAS), several nuclear receptor polymorphisms have been associated with IBD, and NR expression is mostly down-regulated in biopsies from UC and CD patient in comparison to healthy subjects (12, 63–73) (Tables 1–3). As IBD onset typically occurs in the second and third decade of life with a high number of patients progressing to relapse and chronic disease, an urgent need to develop new therapies with either low adverse side effects during long-term management or even curative potential is needed in the future.
The characterization of NRs that either promote or suppress intestinal inflammation has led to efficacious therapeutics for IBD. One classic anti-inflammatory drug, namely 5-ASAs, augments PPARγ expression and promotes its translocation from the cytoplasm to the nucleus resulting in activation of peroxisome-proliferator hormone response element-driven genes to suppress colitis activity (74, 75). Regarding combinatorial therapy, the treatment with 5-ASAs and rosiglitazone (PPARγ agonist) had a better therapeutic effect in UC than 5-ASA alone (76–78) (Table 4).
Regulating the glucocorticoid receptor (GR), glucocorticoids play an important role in inducing remission in IBD (79). Unfortunately, response rates are low and vary between 20 and 30% of patients showing resistance with the therapy also inducing common side effects (80) (Table 4).
There is strong evidence in support of vitamin D that targets the VDR having protective effects in IBD-related inflammatory responses (81). Treatment with vitamin D3 in mild to moderate CD patients significantly improved disease activity and quality of life after 24 weeks of treatment (Table 4) (82). More studies showed a positive effect of vitamin D supplementation in UC and CD patients (82–86). However, since the clinical efficacy and mechanism of action of vitamin D therapy are unclear, additional studies are necessary to fully explore its possible immunomodulatory and anti-inflammatory effects, also in relation to decreasing epithelial permeability and maintaining barrier integrity (87) (Tables 2B, 4).
Rifaximin, an intestine-specific human PXR agonist, appears to have more antimicrobial efficacy in the therapy of CD than traditional medications like metronidazole or ciprofloxacin; it decreases intestinal permeability and targets (NFκB), regulating anti-inflammatory effects in IBD patients (88–90) (Table 4).
Besides clinical data on nuclear receptor therapy targets, so far, several NRs have only been tested pre-clinically in animal models.
The estrogen receptors, members of the steroid hormone receptor family, play an essential role in the maintenance of colonic homeostasis. Understanding the biological effects of ERα and ERβ within the gut and the immune system are important for unraveling the gender-dependent differences in intestinal inflammatory diseases. Interestingly, estrogen levels impact the composition of gut microbiota itself (91); however, the composition of the microbiota influences the bioavailability of estrogen (92) (Figure 2). Males are at greater risk than females for developing ulcerative colitis (UC) and experiencing worse clinical progression, whereas females are more likely to develop CD (93–96). Especially ERβ is expressed abundantly in the colonic epithelium, where it regulates maintenance of colonic architecture, tight-junction formation, and barrier function (97, 98). ERα and ERβ gene expression levels are comparable between male and female UC colon samples, suggesting that sex-based differences in ER-mediated effects are most likely not caused by differences in gene expression (Table 1) (99).
Nevertheless, ERβ expression has been found markedly decreased in colonic mucosa of CD/UC patients with active disease; specifically, ERβ expression in female mice protected against DSS colitis, whereas it failed to protect male mice (100). Recently, Wendy A. Goodman et al. (93) reported that fundamental differences in ERα/ERβ signaling ratios impact colitis in males and females. Analysis of gene expression from inflamed colonic tissues identified alteration of typical estrogen-responsive genes such as Socs3, Ctsd, and Fos as being up-regulated in colon tissues of DSS-treated ERα-knockout male mice compared with ERα-knockout females. In line with these data, similar gene expression profiles of SOCS3, CTSD, and FOS were found in colonic biopsy specimens from male and female patients suffering from UC (93, 99) (Table 1).
Experimental colitis studies in mice and rats have shown that ER has pre-clinical therapeutic implications (93, 101–103). Supraphysiological doses of 17β-estradiol have anti-inflammatory (in the DNB mouse colitis model) as well as pro-inflammatory (in the DSS mouse colitis model) effects demonstrating complex immunomodulation in female mice during intestinal inflammation (102). Additionally, studies in male rats and in the HLA-B27 transgenic rat IBD model demonstrated reduced colonic damage score with estradiol treatment during acute colonic injury (101, 103).
Glucocorticoids targeting the GR contribute to diverse biological processes including glucose metabolism, stress, or immune responses. Endogenous GCs are predominantly produced by the adrenal glands, but within the IEC the NR LRH-1 regulates extra-adrenal glucocorticoid synthesis in the intestine (104).
The GR is expressed in almost every cell in the body and is a multi-tasking transcription factor, changing its role and function from anti-inflammatory effects via direct gene suppression or activation to potential pro-inflammatory actions as well [reviewed in (105)]. Nevertheless, rapid non-genomic mechanisms of GC signaling have also been reported [reviewed in (105)].
A mechanism for glucocorticoid-mediated inhibition of immune responses is the interference with activities and modulation of key pro-inflammatory transcription factors, including NF-κB, activator protein 1 (AP-1), members of the signal transducer and activator of transcription (STAT), CCAT/enhancer-binding protein (C/EBP), and nuclear factor of activated T cells (NFAT) families (106, 107). Through GR-mediated transrepression, expression of pro-inflammatory cytokines and chemokines like IL-1α, IL-1β, and IL-8 are down-regulated (Figure 3). Additionally, the GR can directly activate suppressive inflammatory mediators like TGF-β and IL-10, inhibit T and B lymphocyte proliferation, and promote a tolerant macrophage profile (M2), altogether increasing its anti-inflammatory function (105, 108) (Figure 3). The signaling pathways of GR and PPARα, another nuclear receptor, can cooperate and increase the inhibition of cytokine gene expression to alleviate inflammation (109).
Within the immune system, glucocorticoids are circadian mediators (110) and regulate diurnal oscillations in T cell distribution by inducing IL-7R and CXCR4 (111) and regulate T cell responses in gastrointestinal Peyer's patches. Dexamethasone suppresses IL-23–mediated IL-22 production in human and mouse ILC3s (112).
GCs have a long history in IBD therapy and are well-known immune suppressants. Nevertheless, the expression of the GR itself does not appear to predict steroid treatment responses in IBD patients although conflicting data exist (Table 1). GCs are especially able to protect mice and men against TNF-induced inflammatory symptoms, and GR dimers control intestinal STAT1 and TNF-induced inflammation in mice (113, 114).
The role of the GR in IBD has been reviewed recently (115). Human trials demonstrated that standard systemic corticosteroids (cortisone, prednisone, methylprednisolone, fluticasone) are effective in inducing remission in UC by suppressing immune responses, and might be of benefit in CD (3) (Table 4). Therefore, glucocorticoids are still the mainstay for induction of clinical remission in cases of acute relapse of both CD and UC, and second-generation corticosteroids such as budesonide or beclomethasone have been developed. Whereas, budesonide induces remission in active ileal CD, it shows less efficacy in and does not prevent CD relapse (3, 116) (Table 4). Many preclinical studies were performed using different IBD animal models to investigate the complex cellular and molecular basis of glucocorticoid action at the interface between the endocrine, the immune, and the intestinal system. As a future perspective, screening assays for GR agonists are ongoing in order to develop new effective medications against acute inflammation (117).
The role of other steroid hormone receptors such as the androgen receptor (AR) and the progesterone receptor (PR) during colitis progression have been investigated only poorly. Pre-diagnostic circulating testosterone is associated with a lower risk of CD but not UC in women (95). Progesterone therapy decreases oxidative damage, characterized by decreased MDA, MPO, TNFα and caspase-3 activity, in the colonic mucosa (118).
Despite a wide range of pre-clinical IBD trials with compounds specifically targeting NR family members, the following NRs have not yet reached UC or CD patients in clinical trials.
The xenobiotic NR CAR can be regulated by xenobiotics and endobiotics but also by steroid hormones (119). One of its diverse metabolic functions (119) includes the clearance of xeno- and endobiotics such as toxic bilirubin (Table 2A) (120). Its expression in the intestine and the liver is dependent on the presence of microbiota (119, 121, 122).
CAR is expressed in the healthy intestinal epithelium, but the expression is reduced within intestinal mucosal biopsies from patients with UC and CD, or tissue from DSS mice (67, 123) (Figures 1, 2). In the pre-clinical DSS mouse model, especially wound healing of intestinal epithelial cells is reduced in Car-deficient mice whereas activation of CAR using a selective CAR agonist 3,3',5,5'-tetrachloro-1,4-bis(pyridyloxy)benzene (TCPOBOP) enhances mucosal healing (67) in mice (Figure 2). In a rat DSS colitis model, CAR agonists reduced the mRNA expression of several pro-inflammatory cytokines in a CAR-dependent manner; CAR inhibited apoptosis by inducing Gadd45b within an in vitro cell analysis (124). Therefore, CAR activation may also prove effective in patients with IBD.
FXR functions as an enterohepatic regulator of bile acid homeostasis and regulates especially lipid (125) and glucose metabolism (126), as well as inflammation (13). Fxr-deficient mice are more susceptible to IBD models such as TNBS or DSS due to enhanced expression of pro-inflammatory cytokines in innate immune cells (127) (Table 2A) (Figures 1, 3). Also monocytes and dendritic cells (DCs) are modulated by FXR and there is a decrease in epithelial expression of pro-inflammatory molecules both in vivo and in stimulated epithelial cultures after induction of FXR signaling, suggesting that the immunomodulation observed might be partly mediated through epithelial effects (13, 128) (Table 2A; Figure 2). Along with the FXR ligand, INT-747 represses the expression of various pro-inflammatory cytokines, chemokines and their receptors (13, 127). Colon inflammation in CD patients and rodent models of colitis is associated with reduced expression of FXR mRNA (Table 2A) (127). FXR also regulates gut barrier function due to its antibacterial growth effect (129) and its control of proliferating Lgr5+ intestinal stem cells (130) (Figure 2). Bile acids are well known natural ligands of FXR and regulate the protective activity of FXR in shielding the intestine from bacteria-induced damage and thereby maintaining a competent gut barrier and preventing the development of IBD (Table 2A) (13, 131).
Several pharmacological modulators of FXR activity have been tested in human clinical trials, but its role in IBD has so far only been investigated in pre-clinical mouse models (13). Fexaramine is an intestinal-specific FXR modulator which is potentially safer than systemic FXR agonists as it preferentially activates FXR target genes in the intestine (132), but its functional role has not yet been investigated in IBD models.
LRH-1 is mostly known for its regulatory role in cholesterol and bile acid homeostasis but has recently emerged as a key regulator of intestinal function. Unlike most of the other NRs, LRH-1 acts constitutively to drive the transcription of its target genes (133). Nevertheless, this atypical NR contains a well-ordered hormone-binding pocket, which binds signaling phospholipids including phosphoinositides (134, 135).
LRH-1 is expressed in intestinal crypts, where intestinal stem cells (ISCs) reside, and where it contributes to epithelial renewal by potentiating WNT/β-catenin signaling (136–138). GWAS meta-analyses of IBD patients found a significant association between LRH-1 and IBD (Table 2A). Subsequent analysis on IBD patients revealed a significant decrease in expression of LRH-1 and its transcriptional targets such as CYP11A1 and CYP11B1 in the affected tissues (69, 139). Both Lrh-1 haploinsufficiency and somatic deficiency of Lrh-1 in the intestinal epithelium rendered mice more susceptible to experimentally induced DSS or TNBS colitis (Table 1) (69). One pathway how LRH-1 limits inflammation involves the regulation of extra-adrenal glucocorticoid production in the gut (69, 133). Apart from the immune-regulatory action on local immune cells, glucocorticoids may also induce intestinal tight junction proteins and improve epithelial barrier function (Table 1; Figures 1, 2) (140). It is plausible that after hapten-induced mucosal inflammation, the cell cycle regulatory function of LRH-1 comes into play to promote mucosal renewal and regeneration (69). A recent study also underpins the human relevance, using humanized mouse intestinal organoids, a humanized in vivo IBD model, and human intestinal organoids (Table 2A). Thereby, Bayrer et al., uncovered an essential role for LRH-1 in intestinal epithelial homeostasis and cell survival, which mitigates inflammatory injury (135). As preliminary therapeutic results, the use of DLPC (dilauroyl phosphatidylcholine) as an extrinsic agonist ligand for LRH-1 has been reported to result in decreased colitis symptoms (69).
LXRs control lipid and glucose homeostasis and respond to physiological concentrations of sterols. Whereas, LXRα is mainly expressed in the liver, intestine, kidney, and immune cells, LXRβ is more ubiquitously expressed (141–143). Within the immune system, both LXRs are important anti-inflammatory transcription factors and physiological regulators of innate and adaptive immune responses, apoptosis, and phagocytosis (144).
Several LXR agonists are effective in pre-clinical models of diseases such as atherosclerosis or diabetes and are used as an anti-inflammatory agent (143, 145). Lxr-deficient mice are more susceptible to DSS colitis, show slower recovery and decreased survival (Table 2A). Expression of both LXRA and LXRB is significantly suppressed in the inflamed colon from both CD and UC patients compared with a non-inflamed colon (70), and LXR polymorphism has been linked to enhanced IBD risk (Table 2A) (146). While LXRα induces anti-inflammatory effects in innate immune cell populations (Figure 3), LXRβ has anti-inflammatory effects in colon epithelial cells (Figure 2). Addition of an LXR agonist GW3965 results in faster recovery and increased survival in pre-clinical mouse colitis models, making LXRs an exciting target to suppress inflammatory responses in IBD (70).
The orphan NR4A subfamily includes three members, which are expressed in a wide variety of tissues, especially innate and adaptive immune cells (147).
Although NR4A family members belong to the nuclear receptors superfamily, their activity is not considered to be regulated by physiological ligands, because their ligand-binding pockets are hidden by bulky amino acids, and their ligand-binding domains are constitutively active (148). Nevertheless, structurally diverse synthetic ligands for NR4A2 and NR4A3 have recently been identified (147). Genetic variants of the NUR77 gene locus are associated with increased risk for both UC and CD, and NUR77 expression is significantly reduced in colon tissues from patients with UC or CD and mice treated with DSS (Table 2A) (31, 149). Nur77-deficiency increases the susceptibility of mice to DSS and TNBS colitis and prevents intestinal recovery (31, 149) (Table 2A) (Figure 1). Mechanistically, NUR77 negatively regulate the TLR–IL-1R signaling axis (149). An independent study demonstrated that loss of Nur77 in mice leads to enhanced colon inflammation with larger numbers of infiltrating neutrophils, T-cells, and macrophages during DSS colitis. Nur77 overexpression dampens the pro-inflammatory state of both RAW macrophages and epithelial Caco-2 cells (Table 2A) (31).
The family member NR4A2 also regulates immune cell function and subsequently colitis, as deletion of NR4A2 in T cells attenuates induction of Tregs and causes aberrant induction of Th1 CD4+ T cells and subsequent exacerbation of colitis (Figure 3) (150). Treatment with cytosporone B (Csn-B), an agonist for Nur77, significantly attenuated excessive inflammatory response in mouse DSS colitis. Therefore, Nur77 has been suggested as a potential target for the prevention and treatment of IBD (149).
PPARs are involved in the control of energy metabolism, inflammation and immune responses activated by natural ligands such as fatty acids, eicosanoids, and phospholipids (151, 152). PPARγ is highly expressed in both IECs and immune cells. Impaired epithelial expression has been documented in preclinical animal models of IBD and UC patients (Tables 2A,B) (Figures 1, 2).
Expression and activity of PPARγ are directly induced by microbial metabolites such as butyrate or propionate (153, 154).
Direct targeting of the activity of PPARγ to enhance anti-inflammatory effects via its agonistic ligand rosiglitazone is efficacious in the treatment of mild to moderately active UC (78, 155, 156) (Table 4). In combination with the anti-inflammatory drug 5-ASA (5-Aminosalicylate and its generics), rosiglitazone achieves better therapeutic effects without causing side effects in UC patients (76). Therefore, rosiglitazone is the most widely used therapeutic agent in conjunction with PPARγ activation that directly leads to trans-repressing of several pro-inflammatory target genes such as NF-κB and signal transducers and activators of transcription (STATs) (77, 157–161) (Figure 3). Also, microbiota-activated PPARγ signaling prevents dysbiotic expansion of potential pathogens by reducing the bioavailability of respiratory electron acceptors in the lumen of the colon (23) (Figure 2). Accordingly, several preclinical studies in IBD animal models have been and are being performed to investigate the molecular mode of action of new compounds that target PPARγ to enhance its anti-inflammatory effects within the immune and the epithelial compartment (Tables 2A,B) (162).
In addition to PPARγ, the two other family members, PPARα (NR1C1) and PPARδ (NR1C2) are used for therapeutic IBD intervention. Each PPAR isotype has a specific expression pattern within the gut, but all of them mediate the effects of the microbiota (163). Agonistic ligands targeting PPARα such as dexamethasone (164), fenofibrate (165), palmitoylethanolamide (166), or bezafibrate (167) have anti-inflammatory effects in pre-clinical animal studies. The first-generation PPARα agonists, the fibrates, have however been hampered by drug-drug interaction issues, statin drop-in, and ill-designed cardiovascular intervention trials reviewed in Bougarne et al. (168). Ambiguous results were obtained targeting PPARδ with GW0742 or dietary punicic acid showing either anti-, pro-inflammatory or no effects in experimental mouse IBD models (Tables 2A,B) (165, 169, 170).
PXR protects the body from harmful foreign toxicants and endogenous toxic substances as it induces genes involved in drug transport and metabolism (171). Pregnane X receptor is primarily expressed in the liver and the intestine; the distribution and function of human PXR in the gastrointestinal system contribute to its emerging role as a modulator of inflammation and the intestinal mucosal barrier (Table 2B) (171, 172) (Figures 1, 2). In contrast to most other NRs, PXR has a wide spectrum of ligands such as drugs, endogenous ligands or products of the gut microflora (173–175). Despite high homology between the LBD and DBD domain of human and mouse PXR, species-specific responses to ligand activation (such as rifampicin, or pregnenolone-16α-carbonitrile) are surprisingly different making results of pre-clinical mouse studies less extrapolatable for human trials (171).
PXR agonists reduce the mRNA expression of several pro-inflammatory cytokines in a PXR-dependent manner such as TNF-α and IL-1β in a rat DSS colitis model (124). PXR has been implicated in the pathogenesis of IBD, and its activator rifaximin (works only in humans, not in mice) has demonstrated efficacy in CD and UC (Table 2B). Antibiotic therapy with rifaximin, which was approved in 2004 for the treatment of traveler's diarrhea (176), was associated with the induction and maintenance of remission (90, 177). In CD patients, bile acid malabsorption is associated with deactivation of PXR (178). Despite the difference between human and mouse PXR species-specific responses, a lot of effort was made to test different ligands of PXR in mouse models of colitis utilizing its anti-microbial effects and its anti-inflammatory potential on immune cells as well as epithelial cells, helping the latter to maintain an intact epithelial barrier in the gut (Figure 3).
RARs usually form heterodimers and function as ligand-dependent transcription factors but also play extra-nuclear and non-genomic roles. The vitamin A metabolite all-trans-retinoic acid (atRA) acts as a ligand for RAR and is involved in the regulation of both the intestinal barrier function as well as immune homeostasis (19, 179, 180).
In general, the NRs of the RAR family play important pleiotropic roles in the regulation of innate immune cells such as dendritic cells, macrophages, and ILCs, and are especially important in the regulation of T cell homing to the gut as well as IgA class switching in B cells (Figure 3) (19). The pleiotropic roles of retinoic acid and RARs as modulators of the immune system, for example, induction of Th1, Th2, and Th17 responses together with the release of pro-inflammatory cytokines like IL-12 and Il-23 by DCs, have been reviewed in much detail recently and will therefore not be discussed here (19, 30).
The vitamin A metabolite retinoic acid (RA) can also enhance ILC3 responses in mice through multiple mechanisms, including direct binding to the Rorc or Il22 loci, promoting maturation of LTi-like ILC3s, and regulating ILC3 proliferation (47, 51, 181). In addition to promoting maintenance of the intestinal epithelium, during fetal development vitamin A and the metabolite RA control the size of secondary lymphoid tissues via LTi cells in mice, which can influence the efficiency of protection from viral infections later in life (47, 51, 182).
In mouse or rat colitis models Vitamin A inhibits the development of DSS colitis and colon cancer (183, 184) (Table 2B). Several pre-clinical studies in mice show anti-inflammatory effects of RAR ligands like ATRA (185–187), Neomangiferin (188), 13cis-retinoic acid (189), or RA (44, 190). Importantly, atRA supplementation reduced the tumor burden in a mouse model of colorectal cancer via enhancing protective CD8+ T cell responses highlighting the relevance of NRs as a potential therapeutic option to treat colon cancer patients (191) (Figure 3).
RORγ has a broad pattern of expression but is observed at very high levels within the thymus. There RORγ regulates thymocyte and lymphoid development but is also involved in the regulation of metabolism and the circadian rhythm (192). The recently de-orphanized RORγ is known to bind to sterols, with certain oxysterols having a very high affinity for this receptor. Synthetic inverse agonists of RORγ are effective in treating and preventing autoimmunity in mouse models and are beneficial in glucose and lipid metabolism (1). A cross-talk between RORγt+ ILCs and intestinal macrophages induces mucosal IL-22 production in Crohn's disease (193). In CD4+ Th17, the splice isoform RORγt controls the secretion of the cytokines IL-17a and IL-17f. Especially IL-17f has recently been identified as highly pathogenic in gut inflammation. Therefore, RORγt-expressing Th17 cells induce murine chronic intestinal inflammation (Table 2B) (194, 195). In parallel, delivery of IL-15 to CD4+ T cells in the colon downmodulates Foxp3 expression and enhances RORγt expression rapidly triggering IBD characterized by enhanced production of pro-inflammatory cytokines (such as interferon-γ, IL-6) and accumulation of Th1/Th17 cells (196) (Figure 3). Pharmacologic inhibition of RORγt via GSK805 provides therapeutic benefit in mouse models of intestinal inflammation and reduces the frequency of Th17 cells isolated from primary intestinal samples of individuals with inflammatory bowel disease (IBD) (197). In the course of IBD, RORα-dependent ILC3 functions are pivotal in mediating gut fibrosis, and they can offer an avenue for therapeutic intervention in Crohn's-like diseases (198).
RXRs have been implicated in a diversity of cellular processes. These pleiotropic effects originate from the ability of RXRs to dimerize with diverse NRs, which exert transcriptional control on specific aspects of cell biology, and the ability to stimulate transcriptional activation by RXR partner receptors (199). RXRs form heterodimers, either spontaneously or in a ligand-dependent manner, with NRs well known to play crucial roles in the regulation of intestinal homeostases such as VDR, PPAR, FXR, LXR, or CAR (199) (Figure 2). Especially the RXR/PPARγ heterodimers, which are permissive to activation by both PPARγ and RXR ligands, have been investigated in colitis models (200). Rxra+/− mice are highly sensitive to TNBS colitis and AOM/DSS colitis induction (Table 2B) (201). In the colon, the RXR ligand LG101305 is equally effective as PPARγ ligands in reducing intestinal inflammation during TNBS colitis. Also, rexinoids have a marked synergistic effect with PPARγ agonists on inflammation suggesting that co-administration of low doses of PPARγ and RXR agonists might be worth exploring in human IBD (200) (Table 4). Conventional full RXR agonists are known to show considerable adverse effects, but the partial RXR agonist, CBt-PMN, efficiently ameliorated the symptoms of colitis. This effect is attributed to the down-regulation of pro-inflammatory cytokines such as Tnf and Il6 in colon-infiltrating monocytes probably by the activation of PPARδ/RXR and Nur77/RXR heterodimers by CBt-PMN (202).
In the intestine, VDR signaling regulates microbial homeostasis, barrier integrity as well as immune cell distribution and function (Table 2B) (203). The immune system, in particular, is influenced by vitamin D3 via enhancement of chemotactic and phagocytic responses of macrophages and production of antimicrobial proteins, such as cathelicidin; it inhibits the surface expression of the MHC-II-complex antigen and costimulatory molecules and downregulates the production of many pro-inflammatory cytokines, such as IL-1, IL-6, IL-8, and TNF-α (204). Association studies showing a higher incidence of CD follow a “North-South gradient” and support animal and clinical data demonstrating an important role for vitamin D as a risk factor and potential therapeutic target in CD (205) (Table 2B).
IL-22 production is dependent on vitamin D and in the absence of vitamin D, mice develop a more severe enteric infection that takes longer to resolve (Figure 2) (206).
In contrast Chen et al., report that VDR KO mice have more interleukin-22 (IL-22)-producing ILCs and more antibacterial peptides than WT mice. The increased ILCs in the VDR KO mice was a cell-autonomous effect of VDR deficiency on ILC frequencies (207).
A double-blind, randomized placebo-controlled study of the effect of vitamin D supplementation over 3 months showed significantly increased 25(OH)D levels in patients in remission accompanied by maintenance of the intestinal permeability (86) (Table 4). Several therapeutic studies in experimental IBD animal models and patients have subsequently shown vitamin D to have therapeutic efficacy on epithelial permeability as well as anti-inflammatory properties, and the effects of different analogs have been summarized in a recent review (203) (Figure 3; Table 2B).
For two other non-steroidal nuclear receptors Rev-erb α/β (NR1D1/2) and RORα (NR1F1), data only from human patient biopsy is available. Whereas, NR1D2 expression in UC patients is downregulated (72), NR1F1 expression is upregulated in CD patients' colonic mucosa (208) (Table 2B).
The orphan NR HNF4α is considered to be an important actor in intestinal epithelial cell homeostasis and mucosal barrier integrity as this NR regulates proper intestinal epithelial cell differentiation (209–211), lipid metabolism (212), goblet cell maturation, epithelial junctions and Muc gene expression (Table 3) (12, 211, 213, 214) (Figures 1, 2). Its role in liver and intestinal inflammatory networks has recently been reviewed in detail elsewhere (215). In humans, HNF4A expression is strongly reduced in intestinal biopsies of UC and CD patients (12, 64) and a GWAS has identified HNF4A locus as a susceptibility gene for UC (216). Besides, a single-nucleotide polymorphism within the HNF4A locus has also been associated with UC and pediatric CD (12, 216–219). Two HNF4α isoforms P1 and P2 are expressed in different compartments in the colonic epithelium, interact with distinct sets of proteins, and regulate the expression of unique sets of target genes, and thus play distinct roles during pathological conditions such as colitis (Table 3) (220). Pre-clinical mouse and human association studies suggest a highly important role for this NR, but as an orphan NR family member, currently, no agonistic compounds targeting HNF4α are available for treatment options.
We were the first to unravel the role of the COUP-TF family member NR2F6 in the pathogenesis of IBD (14). In immune cells, NR2F6 inhibits CD4+ Th17 T cell responses and autoimmunity (38, 221) arnd suppresses CD4+ and CD8+ T cell-driven anti-tumor immunity (40, 222, 223). In the gut, NR2F6 directly protects the colonic intestinal epithelium and thus enhances gut barrier homeostasis. Nr2f6-deficient mice are highly susceptible to DSS-induced colitis; mechanistically, NR2F6 directly binds to a consensus sequence at −2 kb of murine and human MUC2 promoter and transactivates Muc2 expression. Loss of NR2F6, therefore, increases intestinal permeability and results in spontaneous late-onset colitis in Nr2f6-deficient mice (14) (Figures 1, 2). Beside this pre-clinical dataset in mice, several studies from the literature document reduced NR2F6 gene expression in patients with IBD (Table 3) (65, 72, 224, 225).
NRs and NR ligands control important gastrointestinal functions ranging from nutrient uptake, the composition of the microbiota and intestinal immune cells. Mechanistic studies have identified several NRs involved in the pathophysiology of IBD; therefore, novel concepts integrating NR and gastrointestinal physiology have been integrated into the successful development of effective drug therapies into the clinic.
Despite this expanding use of NR targeting as a therapeutic approach, there are many unknown issues about some classes of NRs, especially orphan NRs. In addition to the translation of the existing knowledge on NR biology, advances in current knowledge especially assessing dynamic NR regulation throughout disease progression should lead to the development of new drug targets for treating IBD. In this context, it should be mentioned that currently NRs are investigated not only with a focus on gastrointestinal diseases but also from a broader perspective. NRs such as the GR (dexamethasone), RXR (bexarotene and alitretinoin), PPARα (fibrates), and PPARγ (thiazolidinediones) have already been successfully targeted by approved drugs for treating autoimmunity, cancer, hyperlipidemia, or type 2 diabetes, respectively. Understanding the molecular mechanism of NRs in other human diseases will hopefully provide important insights into how to optimize NR-targeting therapies in IBD.
VK and NH-K wrote the manuscript. ARM, HT, and GB contributed to the colitis and nuclear receptor biology aspects and helped with writing the manuscript.
The FWF Austrian Science Fund supported this work with the following grants P28694-B30 and P25044-B21.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
1. Kojetin DJ, Burris TP. REV-ERB and ROR nuclear receptors as drug targets. Nat Rev Drug Discov. (2014) 13:197–216. doi: 10.1038/nrd4100
2. Burris TP, Solt LA, Wang Y, Crumbley C, Banerjee S, Griffett K, et al. Nuclear receptors and their selective pharmacologic modulators. Pharmacol Rev. (2013) 65:710–78. doi: 10.1124/pr.112.006833
3. Ford AC, Bernstein CN, Khan KJ, Abreu MT, Marshall JK, Talley NJ, et al. Glucocorticosteroid therapy in inflammatory bowel disease: systematic review and meta-analysis. Am J Gastroenterol. (2011) 106:590–9. doi: 10.1038/ajg.2011.70
4. Laverny G, Penna G, Vetrano S, Correale C, Nebuloni M, Danese S, Adorini L. Efficacy of a potent and safe vitamin D receptor agonist for the treatment of inflammatory bowel disease. Immunol Lett. (2010) 131:49–58. doi: 10.1016/j.imlet.2010.03.006
5. Ranhotra SHL, Flannigan K, Brave M, Mukherjee S, Lukin J, Hirota A, et al. Xenobiotic receptor-mediated regulation of intestinal barrier function and innate immunity. Nucl Recept Res. (2016) 3:1–9. doi: 10.11131/2016/101199
6. Evans R. The steroid and thyroid hormone receptor superfamily. Science. (1988) 240:889–95. doi: 10.1126/science.3283939
7. Mangelsdorf DJ, Thummel C, Beato M, Herrlich P, Schütz G, Umesono K, et al. The nuclear receptor superfamily: the second decade. Cell. (1995) 83:835–9. doi: 10.1016/0092-8674(95)90199-X
8. Mangelsdorf DJ, Evans RM. The RXR heterodimers and orphan receptors. Cell. (1995) 83:841–50. doi: 10.1016/0092-8674(95)90200-7
9. Glass CK, Ogawa S. Combinatorial roles of nuclear receptors in inflammation and immunity. Nat Rev Immunol. (2006) 6:44–55. doi: 10.1038/nri1748
10. Li YC, Chen Y, Du J. Critical roles of intestinal epithelial vitamin D receptor signaling in controlling gut mucosal inflammation. J Steroid Biochem Mol Biol. (2015) 148:179–83. doi: 10.1016/j.jsbmb.2015.01.011
11. Bouguen G, Langlois A, Djouina M, Branche J, Koriche D, Dewaeles E, et al. Intestinal steroidogenesis controls PPARγ expression in the colon and is impaired during ulcerative colitis. Gut. (2015) 64:901–10. doi: 10.1136/gutjnl-2014-307618
12. Ahn S-H, Shah YM, Inoue J, Morimura K, Kim I, Yim S, et al. Hepatocyte nuclear factor 4alpha in the intestinal epithelial cells protects against inflammatory bowel disease. Inflamm Bowel Dis. (2008) 14:908–20. doi: 10.1002/ibd.20413
13. Gadaleta RM, van Erpecum KJ, Oldenburg B, Willemsen ECL, Renooij W, Murzilli S, et al. Farnesoid X receptor activation inhibits inflammation and preserves the intestinal barrier in inflammatory bowel disease. Gut. (2011) 60:463–72. doi: 10.1136/gut.2010.212159
14. Klepsch V, Gerner RR, Klepsch S, Olson WJ, Tilg H, Moschen AR, et al. Nuclear orphan receptor NR2F6 as a safeguard against experimental murine colitis. Gut. (2017) 67:gutjnl-2016-313466. doi: 10.1136/gutjnl-2016-313466
15. Giguere V. Orphan Nuclear Receptors: from Gene to Function*. Endocr Rev. (1999) 20:689–725. doi: 10.1210/er.20.5.689
16. Laudet V, Auwerx J, Gustafsson JA, Walter Wahli. Letter to the editor a unified nomenclature system for the nuclear receptor superfamily. Cell. (1999) 97:161–3. doi: 10.1016/S0092-8674(00)80726-6
17. Germain P, Staels B, Dacquet C, Spedding M, Laudet V. Overview of nomenclature of nuclear receptors. Pharmacol Rev. (2006) 58:685–704. doi: 10.1124/pr.58.4.2
19. Larange A, Cheroutre H. Retinoic acid and retinoic acid receptors as pleiotropic modulators of the immune system. Annu Rev Immunol. (2016) 34:369–94. doi: 10.1146/annurev-immunol-041015-055427
20. Triantafillidis JK, Merikas E, Georgopoulos F. Current and emerging drugs for the treatment of inflammatory bowel disease. Drug Des Devel Ther. (2011) 5:185–210. doi: 10.2147/DDDT.S11290
21. Francis GA, Fayard E, Picard F, Auwerx J. Nuclear receptors and the control of metabolism. Annu Rev Physiol. (2003) 65:261–311. doi: 10.1146/annurev.physiol.65.092101.142528
22. Jie D, Yunzi C, Yongyan S, Tianjing L, Yong C, Yue T, et al. 1,25-Dihydroxyvitamin D protects intestinal epithelial barrier by regulating the myosin light chain kinase signaling pathway. Inflamm Bowel Dis. (2015) 21:2495–506. doi: 10.1097/MIB.0000000000000526
23. Olsan EE, Byndloss AJ, Litvak Y, Tiffany CR, Faber F, Revzin A, et al. Microbiota-activated PPAR-γ signaling inhibits dysbiotic Enterobacteriaceae expansion. Science. (2017) 357:570–5. doi: 10.1126/science.aam9949
24. Parséus A, Sommer N, Sommer F, Caesar R, Molinaro A, Stahlman M, et al. Microbiota-induced obesity requires farnesoid X receptor. Gut. (2017) 66:429–37. doi: 10.1136/gutjnl-2015-310283
25. Yuan T, Jingwei C, Wei G, Robert G, Nichols IK, Jingtao Z, et al. Berberine directly affects the gut microbiota to promote intestinal farnesoid x receptor activation. Drug Metab Dispos. (2019) 47:86–93. doi: 10.1124/dmd.118.083691
26. Friedman ES, Li Y, Shen TCD, Jiang J, Chau L, Adorini L, et al. FXR-dependent modulation of the human small intestinal microbiome by the bile acid derivative obeticholic acid. Gastroenterology. (2018) 155:1741–52.e5. doi: 10.1053/j.gastro.2018.08.022
27. Duszka K, Wahli W. Enteric microbiota–gut–brain axis from the perspective of nuclear receptors. Int J Mol Sci. (2018) 19:2210. doi: 10.3390/ijms19082210
28. Mukherji A, Kobiita A, Ye T, Chambon P. Homeostasis in intestinal epithelium is orchestrated by the circadian clock and microbiota cues transduced by TLRs. Cell. (2013) 153:812–27. doi: 10.1016/j.cell.2013.04.020
29. Montagner A, Korecka A, Polizzi A, Lippi Y, Blum Y, Canlet C, et al. Hepatic circadian clock oscillators and nuclear receptors integrate microbiome-derived signals. Sci Rep. (2016) 6:20127. doi: 10.1038/srep20127
30. Erkelens MN, Mebius RE. Retinoic acid and immune homeostasis: a balancing act. Trends Immunol. (2017) 38:168–80. doi: 10.1016/j.it.2016.12.006
31. Hamers AJ, Van Dam L, Duarte JMT, Vos M, Marinković G, Van Tiel CM, et al. Deficiency of nuclear receptor Nur77 aggravates mouse experimental colitis by increased NFκB activity in macrophages. PLoS ONE. (2015) 10:e0133598. doi: 10.1371/journal.pone.0133598
32. Glass CK, Natoli G. Molecular control of activation and priming in macrophages. Nat Immunol. (2016) 17:26–33. doi: 10.1038/ni.3306
33. Hamers AAJ, Argmann C, Moerland PD, Koenis DS, Marinković G, Sokolović M, et al. Nur77-deficiency in bone marrow-derived macrophages modulates inflammatory responses, extracellular matrix homeostasis, phagocytosis and tolerance. BMC Genom. (2016) 17:162. doi: 10.1186/s12864-016-2469-9
34. Fiorucci S, Biagioli M, Zampella A, Distrutti E. Bile acids activated receptors regulate innate immunity. Front Immunol. (2018) 9:1853. doi: 10.3389/fimmu.2018.01853
35. Liu XK, Lin X, Gaffen SL. Crucial role for nuclear factor of activated T cells in T cell receptor-mediated regulation of human interleukin-17. J Biol Chem. (2004) 279:52762–71. doi: 10.1074/jbc.M405764200
36. Yang XO, Pappu BP, Nurieva R, Akimzhanov A, Kang HS, Chung Y, et al. T helper 17 lineage differentiation is programmed by orphan nuclear receptors ROR alpha and ROR gamma. Immunity. (2008) 28:29–39. doi: 10.1016/j.immuni.2007.11.016
37. Nolting J, Daniel C, Reuter S, Stuelten C, Li P, Sucov H, et al. Retinoic acid can enhance conversion of naive into regulatory T cells independently of secreted cytokines. J Exp Med. (2009) 206:2131–9. doi: 10.1084/jem.20090639
38. Hermann-Kleiter N, Gruber T, Lutz-Nicoladoni C, Thuille N, Fresser F, Labi V, et al. The nuclear orphan receptor NR2F6 suppresses lymphocyte activation and T helper 17-dependent autoimmunity. Immunity. (2008) 29:205–16. doi: 10.1016/j.immuni.2008.06.008
39. Klotz L, Burgdorf S, Dani I, Saijo K, Flossdorf J, Hucke S, et al. The nuclear receptor PPARγ selectively inhibits Th17 differentiation in a T cell–intrinsic fashion and suppresses CNS autoimmunity. J Exp Med. (2009) 206:2079–89. doi: 10.1084/jem.20082771
40. Hermann-Kleiter N, Klepsch V, Wallner S, Siegmund K, Klepsch S, Tuzlak S, et al. The nuclear orphan receptor NR2F6 Is a central checkpoint for cancer immune surveillance. Cell Rep. (2015) 12:2072–85. doi: 10.1016/j.celrep.2015.08.035
41. Hermann-Kleiter N, Meisel M, Fresser F, Thuille N, Müller M, Roth L, et al. Nuclear orphan receptor NR2F6 directly antagonizes NFAT and RORγt binding to the Il17a promoter. J Autoimmun. (2012) 39:428–40. doi: 10.1016/j.jaut.2012.07.007
42. Chen J, Bruce D, Cantorna MT. Vitamin D receptor expression controls proliferation of naïve CD8+ T cells and development of CD8 mediated gastrointestinal inflammation. BMC Immunol. (2014) 15:6. doi: 10.1186/1471-2172-15-6
43. Guri AJ, Mohapatra SK, Horne WT, Hontecillas R, Bassaganya-Riera J. The role of T cell PPAR gamma in mice with experimental inflammatory bowel disease. BMC Gastroenterol. (2010) 10:60. doi: 10.1186/1471-230X-10-60
44. Bai A, Lu N, Guo Y, Liu Z, Chen J, Peng Z. All-trans retinoic acid down-regulates inflammatory responses by shifting the Treg/Th17 profile in human ulcerative and murine colitis. J Leukoc Biol. (2009) 86:959–69. doi: 10.1189/jlb.0109006
45. Wilhelm C, Masouleh SK, Kazakov A. Metabolic regulation of innate lymphoid cell-mediated tissue protection-linking the nutritional state to barrier immunity. Front Immunol. (2017) 8:1742. doi: 10.3389/fimmu.2017.01742
46. Finke D, Esser C, Vonarbourg C, Kopfmann S, Diefenbach A, Kiss EA, et al. Natural aryl hydrocarbon receptor ligands control organogenesis of intestinal lymphoid follicles. Science. (2011) 334:1561–5. doi: 10.1126/science.1214914
47. Van De Pavert SA, Ferreira M, Domingues RG, Ribeiro H, Molenaar R, Moreira-Santos L, et al. Maternal retinoids control type 3 innate lymphoid cells and set the offspring immunity. Nature. (2014) 508:123–7. doi: 10.1038/nature13158
48. Eberl G, Marmon S, Sunshine MJ, Rennert PD, Choi Y, Littmann DR. An essential function for the nuclear receptor RORγt in the generation of fetal lymphoid tissue inducer cells. Nat Immunol. (2004) 5:64–73. doi: 10.1038/ni1022
49. Matthew R, Hepworth Laurel A., Monticelli Thomas C, Fung, et al. Innate lymphoid cells regulate CD4+ T cell responses to intestinal commensal bacteria. Nature. (2013) 498:113–7. doi: 10.1038/nature12240
50. Matthew R, Hepworth Thomas C, Fung Samuel H, Masur, et al. Group 3 innate lymphoid cells mediate intestinal selection of commensal. Science. (2015) 348:1031–5. doi: 10.1126/science.aaa4812
51. Arthur M, Aleksey C, Daigo H, Milena B, Sean PS, Yasmine B, et al. Microbiota-dependent crosstalk between macrophages and ILC3 promotes intestinal homeostasis. Science. (2014) 343:1–7. doi: 10.1126/science.1249288
52. Zeng B, Shi S, Ashworth G, Dong C, Liu J, Xing F. ILC3 function as a double-edged sword in inflammatory bowel diseases. Cell Death Dis. (2019) 10:315. doi: 10.1038/s41419-019-1540-2
53. A Eken AK, Singh PM, Treuting MO. IL-23R+ innate lymphoid cells induce colitis via interleukin-22- dependent mechanism. Mucosal Immunol. (2014) 7:143–54. doi: 10.1038/mi.2013.33
54. Alessandra G, Carolina V, Arancibia C, Myles P, Fleming P, Nigel R, et al. IL-23-responsive innate lymphoid cells are increased in inflammatory bowel disease: commentary. Inflamm Bowel Dis Monit. (2011) 12:74. doi: 10.1084/jem.20101712
55. Bauché D, Joyce-Shaikh B, Jain R, Grein J, Ku KS, Blumenschein WM, et al. LAG3 + regulatory T cells restrain interleukin-23-producing CX3CR1 + gut-resident macrophages during group 3 innate lymphoid cell-driven colitis. Immunity. (2018) 49:342–52.e5. doi: 10.1016/j.immuni.2018.07.007
56. Powell N, Lo JW, Biancheri P, Vossenkämper A, Pantazi E, Walker AW, et al. Interleukin 6 increases production of cytokines by colonic innate lymphoid cells in mice and patients with chronic intestinal inflammation. Gastroenterology. (2015) 149:456–67.e15. doi: 10.1053/j.gastro.2015.04.017
57. Buonocore S, Ahern PP, Uhlig HH, Ivanov II, Littman DR, Maloy KJ, et al. Group Innate lymphoid cells drive IL-23 dependent innate intestinal pathology. Nature. (2010) 464:1371–5. doi: 10.1038/nature08949
58. Podolsky DK. Inflammatory bowel disease. N Engl J Med. (2002) 347:417–29. doi: 10.1056/NEJMra020831
59. Baumgart DC, Sandborn WJ. Inflammatory bowel disease: clinical aspects and established and evolving therapies. Lancet. (2007) 369:1641–57. doi: 10.1016/S0140-6736(07)60751-X
60. Xavier RJ, Podolsky DK. Unravelling the pathogenesis of inflammatory bowel disease. Nature. (2007) 448:427–34. doi: 10.1038/nature06005
61. Molodecky N, Soon IS, Rabi DM, Ghali W, Ferris M, Chernoff G, et al. Increasing incidence and prevalence of the inflammatory bowel diseases with time, based on systematic review. Gastroenterology. (2012) 142:46–54.e42. doi: 10.1053/j.gastro.2011.10.001
62. Antoni L, Nuding S, Wehkamp J, Stange EF. Intestinal barrier in inflammatory bowel disease. World J Gastroenterol. (2014) 20:1165–79. doi: 10.3748/wjg.v20.i5.1165
63. Konstantinopoulos PA, Kominea A, Vandoros G, Sykiotis GP, Andricopoulos P, Varakis I, et al. Oestrogen receptor beta (ERβ) is abundantly expressed in normal colonic mucosa, but declines in colon adenocarcinoma paralleling the tumour's dedifferentiation. Eur J Cancer. (2003) 39:1251–8. doi: 10.1016/S0959-8049(03)00239-9
64. Darsigny M, Babeu JP, Dupuis AA, Furth EE, Seidman EG, Levy E, et al. Loss of hepatocyte-nuclear-factor-4a affects colonic ion transport and causes chronic inflammation resembling inflammatory bowel disease in mice. PLoS ONE. (2009) 4:2–11. doi: 10.1371/journal.pone.0007609
65. Lepage P, Häusler R, Spehlmann ME, Rehman A, Zvirbliene A, Begun A, et al. Twin study indicates loss of interaction between microbiota and mucosa of patients with ulcerative colitis. Gastroenterology. (2011) 141:227–36. doi: 10.1053/j.gastro.2011.04.011
66. Rogler G, Meinel A, Lingauer A, Michl J, Zietz B, Gross V, et al. Glucocorticoid receptors are down-regulated in inflamed colonic mucosa but not in peripheral blood mononuclear cells from patients with inflammatory bowel disease. Eur J Clin Invest. (1999) 29:330–6. doi: 10.1046/j.1365-2362.1999.00460.x
67. Hudson GM, Flannigan KL, Erickson SL, Vicentini F, Zamponi A, Hirota CL, et al. Constitutive androstane receptor regulates the intestinal mucosal response to injury. Br J Pharmacol. (2017) 172:1857–71. doi: 10.1111/bph.13787
68. Nijmeijer RM, Gadaleta RM, van Mil SWC, van Bodegraven AA, Crusius JBA, Dijkstra G, et al. Farnesoid X receptor (FXR) activation and FXR genetic variation in inflammatory bowel disease. PLoS ONE. (2011) 6:e23745. doi: 10.1371/journal.pone.0023745
69. Coste A, Dubuquoy L, Barnouin R, Annicotte JS, Magnier B, Notti M, et al. LRH-1-mediated glucocorticoid synthesis in enterocytes protects against inflammatory bowel disease. Proc Natl Acad Sci USA. (2007) 104:13098–103. doi: 10.1073/pnas.0702440104
70. Jakobsson T, Vedin LL, Hassan T, Venteclef N, Greco D, D'Amato M, et al. The oxysterol receptor LXRβ protects against DSS- and TNBS-induced colitis in mice. Mucosal Immunol. (2014) 7:1416–28. doi: 10.1038/mi.2014.31
71. Langmann T, Moehle C, Mauerer R, Scharl M, Liebisch G, Zahn A, et al. Loss of detoxification in inflammatory bowel disease: dysregulation of pregnane X receptor target genes. Gastroenterology. (2004) 127:26–40. doi: 10.1053/j.gastro.2004.04.019
72. Swan C, Duroudier NP, Campbell E, Zaitoun A, Hastings M, Dukes GE, et al. Identifying and testing candidate genetic polymorphisms in the irritable bowel syndrome (IBS): association with TNFSF15 and TNFalpha. Gut. (2013) 62:985–94. doi: 10.1136/gutjnl-2011-301213
73. Abreu MT, Kantorovich Y, Vasiliauskas EA, Gruntmanis U, Matuk R, Daigle K, et al. Measurement of vitamin D levels in inflammatory bowel disease patients reveals a subset of Crohn's disease patients with elevated 1,25-dihydroxyvitamin D and low bone mineral density. Gut. (2004) 53:1129–36. doi: 10.1136/gut.2003.036657
74. Desreumaux P1 RO. 5-aminosalicylates and colorectal cancer: preventive role in chronic inflammatory bowel disease? Gastroenterol Clin Biol. (2004) 28:509.
75. Rousseaux C, Lefebvre B, Dubuquoy L, Lefebvre P, Romano O, Auwerx J, et al. Intestinal antiinflammatory effect of 5-aminosalicylic acid is dependent on peroxisome proliferator–activated receptor-γ. J Exp Med. (2005) 201:1205–15. doi: 10.1084/jem.20041948
76. Liang HL, Ouyang Q. A clinical trial of combined use of rosiglitazone and 5-aminosalicylate for ulcerative colitis. World J Gastroenterol. (2008) 14:114–9. doi: 10.3748/wjg.14.114
77. Lewis JD, Lichtenstein GR, Deren JJ, Sands BE, Hanauer SB, Katz J, et al. Rosiglitazone for active ulcerative colitis: a randomized placebo-controlled trial. Gastroenterology. (2008) 134:688–95. doi: 10.1053/j.gastro.2007.12.012
78. Lewis J, Lichtenstein G, Stein R, Deren J, Judge T, Fogt F, et al. An open-label trial of the PPAR-gamma ligand rosiglitazone for active ulcerative colitis. Am J Gastroenterol. (2001) 96:3323–8. doi: 10.1111/j.1572-0241.2001.05333.x
79. Ali Rezaie, M Ellen K, Eric IB, Anne MG, Anthony RO, Hillary S, et al. Budesonide for induction of remission in Crohn's disease. Cochrane Database Syst Rev. (2015) 3:CD000296. doi: 10.1002/14651858.CD000296.pub4
80. Faubion W, Loftus EV, Harmsen WS, Zinsmeister AR, Sandborn WJ. The natural history of corticosteroid therapy for inflammatory bowel disease: a population-based study. Gastroenterology. (2001) 121:255–60. doi: 10.1053/gast.2001.26279
81. Reich KM, Fedorak RN, Madsen K, Kroeker KI. Vitamin D improves inflammatory bowel disease outcomes: basic science and clinical review. World J Gastroenterol. (2014) 20:4934–47. doi: 10.3748/wjg.v20.i17.4934
82. Yang L, Weaver V, Smith JP, Bingaman S, Hartman TJ, Cantorna MT. Therapeutic effect of Vitamin D supplementation in a pilot study of crohn's patients. Clin Transl Gastroenterol. (2013) 4:e33. doi: 10.1038/ctg.2013.1
83. Miheller P, Muzes G, Hritz I, Lakatos G, Pregun I, Lakatos PL, et al. Comparison of the effects of 1,25 dihydroxyvitamin D and 25 hydroxyvitamin D on bone pathology and disease activity in Crohn's disease patients. Inflamm Bowel Dis. (2009) 15:1656–62. doi: 10.1002/ibd.20947
84. Ananthakrishnan AN, Cagan A, Gainer VS, Cai T, Cheng SC, Savova G, et al. Normalization of plasma 25-hydroxy vitamin D is associated with reduced risk of surgery in Crohn's disease. Inflamm Bowel Dis. (2013) 19:1921–7. doi: 10.1097/MIB.0b013e3182902ad9
85. Pappa HM, Mitchell PD, Jiang H, Kassiff S, Filip-Dhima R, DiFabio D, et al. Maintenance of optimal Vitamin D status in children and adolescents with inflammatory bowel disease: a randomized clinical trial comparing two regimens. J Clin Endocrinol Metab. (2014) 99:3408–17. doi: 10.1210/jc.2013-4218
86. Raftery T, Martineau AR, Greiller CL, Ghosh S, McNamara D, Bennett K, et al. Effects of vitamin D supplementation on intestinal permeability, cathelicidin and disease markers in Crohn's disease: results from a randomised double-blind placebo-controlled study. United Eur Gastroenterol J. (2015) 3:294–302. doi: 10.1177/2050640615572176
87. Limketkai BN, Bechtold ML, Nguyen DL. Vitamin D and the pathogenesis of inflammatory bowel disease. Curr Gastroenterol Rep. (2016) 18:52. doi: 10.1007/s11894-016-0526-9
88. Morgan RA, Dudley ME, Wunderlich JR, Hughes MS, Yang JC, Sherry RM, et al. Cancer regression in patients after transfer of genetically engineered lymphocytes. Science. (2006) 314:126–9. doi: 10.1126/science.1129003
89. Lauritano EC, Gabrielli M, Scarpellini E, Ojetti V, Roccarina D, Villita A, et al. Antibiotic therapy in small intestinal bacterial overgrowth: rifaximin versus metronidazole. Eur Rev Med Pharmacol Sci. (2009) 13:111–6.
90. Shafran I, Burgunder P. Adjunctive antibiotic therapy with rifaximin may help reduce Crohn's disease activity. Dig Dis Sci. (2010) 55:1079–84. doi: 10.1007/s10620-009-1111-y
91. Moreno-Indias I, Sánchez-Alcoholado L, Sánchez-Garrido MÁ, Martín-Núñez GM, Pérez-Jiménez F, Tena-Sempere M, et al. Neonatal androgen exposure causes persistent gut microbiota dysbiosis related to metabolic disease in adult female rats. Endocrinology. (2016) 157:4888–98. doi: 10.1210/en.2016-1317
92. Littman DR, Pamer EG. Role of the commensal microbiota in normal and pathogenic host immune responses. Cell Host Microbe. (2011) 10:311–23. doi: 10.1016/j.chom.2011.10.004
93. Goodman WA, Havran HL, Quereshy HA, Kuang S, De Salvo C, Pizarro TT. Estrogen receptor α Loss-of-function protects female mice from DSS-induced experimental colitis. Cmgh. (2018) 5:630–3.e1. doi: 10.1016/j.jcmgh.2017.12.003
94. Krege JH, Hodgin JB, Couse JF, Enmark E, Warner M, Mahler JF, et al. Generation and reproductive phenotypes of mice lacking estrogen receptor beta. Proc Natl Acad Sci USA. (1998) 95:15677–82. doi: 10.1073/pnas.95.26.15677
95. Khalili H, Ananthakrishnan AN, Konijeti GG, Higuchi LM, Fuchs CS, Richter JM, et al. Endogenous levels of circulating androgens and risk of Crohn's disease and ulcerative colitis among women: a nested case-control study from the nurses' health study cohorts. Inflamm Bowel Dis. (2015) 21:1378–85. doi: 10.1097/MIB.0000000000000385
96. Rubtsova K, Marrack P, Rubtsov AV. Sexual dimorphism in autoimmunity. J Clin Invest. (2015) 125:2187–93. doi: 10.1172/JCI78082
97. Langen ML, Hotte N, Dieleman L, Albert E, Mulder C, Madsen KL, et al. Estrogen receptor-B signaling modulates epithelial barrier function. Am J Physiol. (2011) 300:G621–6. doi: 10.1152/ajpgi.00274.2010
98. Wada-Hiraike O, Imamov O, Hiraike H, Hultenby K, Schwend T, Omoto Y, et al. Role of estrogen receptor beta in colonic epithelium. Proc Natl Acad Sci USA. (2006) 103:2959–64. doi: 10.1073/pnas.0511271103
99. De Simone V, Matteoli G. Estrogen-mediated effects underlie gender bias in inflammatory bowel disease. CMGH. (2018) 5:638–9 e1. doi: 10.1016/j.jcmgh.2018.01.017
100. Pierdominici M, Maselli A, Varano B, Barbati C, Cesaro P, Spada C, et al. Linking estrogen receptor β expression with inflammatory bowel disease activity. Oncotarget. (2015) 6:40443–51. doi: 10.18632/oncotarget.6217
101. Harnish DC, Albert LM, Leathurby Y, Eckert AM, Ciarletta A, Kasaian M, et al. Beneficial effects of estrogen treatment in the HLA-B27 transgenic rat model of inflammatory bowel disease. Am J Physiol Gastrointest Liver Physiol. (2004) 286:G118–25. doi: 10.1152/ajpgi.00024.2003
102. Verdu EF, Deng Y, Bercik P, Collins SM. Modulatory effects of estrogen in two murine models of experimental colitis. Am J Physiol Gastrointest Liver Physiol. (2002) 283:G27–36. doi: 10.1152/ajpgi.00460.2001
103. Günal O, Oktar B, Ozçinar E, Sungur M, Arbak S, Yegen B. Estradiol treatment ameliorates acetic acid-induced gastric and colonic injuries in rats. Inflammation. (2003) 27:351–9. doi: 10.1023/B:IFLA.0000006703.53427.da
104. Mueller M, Cima I, Noti M, Fuhrer A, Jakob S, Dubuquoy L, et al. The nuclear receptor LRH-1 critically regulates extra-adrenal glucocorticoid synthesis in the intestine. J Exp Med. (2006) 203:2057–62. doi: 10.1084/jem.20060357
105. Desmet SJ, De Bosscher K. Glucocorticoid receptors: finding the middle ground. J Clin Invest. (2017) 127:1136–45. doi: 10.1172/JCI88886
106. Cain DW, Cidlowski JA. Immune regulation by glucocorticoids. Nat Rev Immunol. (2017) 17:233–47. doi: 10.1038/nri.2017.1
107. Ratman D, Vanden Berghe W, Dejager L, Libert C, Tavernier J, Beck IM, et al. How glucocorticoid receptors modulate the activity of other transcription factors: a scope beyond tethering. Mol Cell Endocrinol. (2013) 380:41–54. doi: 10.1016/j.mce.2012.12.014
108. Liberman AC, Budziñski ML, Sokn C, Gobbini RP, Steininger A, Arzt E. Regulatory and mechanistic actions of glucocorticoids on T and inflammatory cells. Front Endocrinol (Lausanne). (2018) 9:1–14. doi: 10.3389/fendo.2018.00235
109. Bougarne N, Paumelle R, Caron S, Hennuyer N, Mansouri R, Gervois P, et al. PPARalpha blocks glucocorticoid receptor alpha-mediated transactivation but cooperates with the activated glucocorticoid receptor alpha for transrepression on NF-kB. Proc Natl Acad Sci USA. (2009) 106:7397–402. doi: 10.1073/pnas.0806742106
110. So AY-L, Bernal TU, Pillsbury ML, Yamamoto KR, Feldman BJ. Glucocorticoid regulation of the circadian clock modulates glucose homeostasis. Proc Natl Acad Sci USA. (2009) 106:17582–7. doi: 10.1073/pnas.0909733106
111. Shimba A, Cui G, Tani-ichi S, Ogawa M, Abe S, Okazaki F, et al. Glucocorticoids drive diurnal oscillations in T cell distribution and responses by inducing interleukin-7 receptor and CXCR4. Immunity. (2018) 48:286–8 e6. doi: 10.1016/j.immuni.2018.01.004
112. Seshadri S, Pope RL, Zenewicz LA. Glucocorticoids inhibit group 3 innate lymphocyte IL-22 production. J Immunol. (2018) 201:1267–74. doi: 10.4049/jimmunol.1800484
113. Ballegeer M, Vandenbroucke RE, Libert C, Ballegeer M, Van Looveren K, Timmermans S, et al. Glucocorticoid receptor dimers control intestinal STAT1 and TNF-induced inflammation in mice Graphical abstract find the latest version: glucocorticoid receptor dimers control intestinal STAT1 and TNF-induced inflammation in mice. J Clin Invest. (2018) 128:3265–79. doi: 10.1172/JCI96636
114. Van Hauwermeiren F, Vandenbroucke RE, Grine L, Lodens S, Van Wonterghem E, De Rycke R, et al. TNFR1-induced lethal inflammation is mediated by goblet and Paneth cell dysfunction. Mucosal Immunol. (2015) 8:828–40. doi: 10.1038/mi.2014.112
115. Dubois-Camacho K, Ottum PA, Franco-Muñoz D, De La Fuente M, Torres-Riquelme A, Díaz-Jiménez D, et al. Glucocorticosteroid therapy in inflammatory bowel diseases: from clinical practice to molecular biology. World J Gastroenterol. (2017) 23:6628–38. doi: 10.3748/wjg.v23.i36.6628
116. De Cassan C, Fiorino G, Danese S. Second-generation corticosteroids for the treatment of Crohn's disease and ulcerative colitis: more effective and less side effects? Dig Dis. (2012) 30:368–75. doi: 10.1159/000338128
117. Souffriau J, Eggermont M, Van Ryckeghem S, Van Looveren K, Van Wyngene L, Van Hamme E, et al. A screening assay for selective dimerizing glucocorticoid receptor agonists and modulators (SEDIGRAM) that are effective against acute inflammation. Sci Rep. (2018) 8:1–13. doi: 10.1038/s41598-018-31150-w
118. Karatepe O, Altiok M, Battal M, Kamali G, Ahu Kemik, Aydin T, et al. The effect of progesterone in the prevention of the chemically induced experimental colitis in rats. Acta Cir Bras. (2012) 27:23–9. doi: 10.1590/S0102-86502012000100005
119. Wei P, Zhang J, Egan-ha M, Liang S, Moore DD, Egan-Hafley M, et al. The nuclear receptor CAR mediates speci ® c xenobiotic induction of drug metabolism. Nature. (2000) 407:6–9. doi: 10.1038/35038112
120. Huang W, Zhang J, Chua SS, Qatanani M, Han Y, Granata R, et al. Induction of bilirubin clearance by the constitutive androstane receptor (CAR). Proc Natl Acad Sci USA. (2003) 100:4156–61. doi: 10.1073/pnas.0630614100
121. Lundin A, Bok CM, Aronsson L, Björkholm B, Gustafsson JÅ, Pott S, et al. Gut flora, toll-like receptors and nuclear receptors: a tripartite communication that tunes innate immunity in large intestine. Cell Microbiol. (2008) 10:1093–103. doi: 10.1111/j.1462-5822.2007.01108.x
122. Björkholm B, Bok CM, Lundin A, Rafter J, Hibberd ML, Pettersson S. Intestinal microbiota regulate xenobiotic metabolism in the liver. PLoS ONE. (2009) 4:e6958. doi: 10.1371/journal.pone.0006958
123. Martin P, Riley R, Back DJ, Owen A. Comparison of the induction profile for drug disposition proteins by typical nuclear receptor activators in human hepatic and intestinal cells. Br J Pharmacol. (2008) 153:805–19. doi: 10.1038/sj.bjp.0707601
124. Uehara D, Tojim H, Kakizaki S, Yamazaki Y, Horiguchi N, Takizawa D, et al. Constitutive androstane receptor and pregnane X receptor cooperatively ameliorate DSS-induced colitis. Dig Liver Dis. (2019) 51:226–35. doi: 10.1016/j.dld.2018.10.008
125. Sinal CJ, Tohkin M, Miyata M, Ward JM, Lambert G, Gonzalez FJ. Targeted disruption of the nuclear receptor FXR/BAR impairs bile acid and lipid homeostasis. Cell. (2000) 102:731–44. doi: 10.1016/S0092-8674(00)00062-3
126. Ma K, Saha PK, Chan L, Moore DD. Farnesoid X receptor is essential for normal glucose homeostasis. J Clin Invest. (2006) 116:1102. doi: 10.1172/JCI25604
127. Vavassori P, Mencarelli A, Renga B, Distrutti E, Fiorucci S. The bile acid receptor FXR is a modulator of intestinal innate immunity. J Immunol. (2009) 183:6251–61. doi: 10.4049/jimmunol.0803978
128. Wildenberg ME, Van Den Brink GR. FXR activation inhibits inflammation and preserves the intestinal barrier in IBD. Gut. (2011) 60:432–3. doi: 10.1136/gut.2010.233304
129. Inagaki T, Moschetta A, Lee Y-K, Peng L, Zhao G, Downes M, et al. Regulation of antibacterial defense in the small intestine by the nuclear bile acid receptor. Proc Natl Acad Sci USA. (2006) 103:3920–5. doi: 10.1073/pnas.0509592103
130. Fu T, Coulter S, Yoshihara E, Oh TG, Fang S, Cayabyab F, et al. FXR regulates intestinal cancer stem cell proliferation. Cell. (2019) 176:1098–112 e18. doi: 10.1016/j.cell.2019.01.036
131. Ding L, Yang L, Wang Z, Huang W. Bile acid nuclear receptor FXR and digestive system diseases. Acta Pharm Sin B. (2015) 5:135–44. doi: 10.1016/j.apsb.2015.01.004
132. Nicolaou KC, Evans RM, Roecker AJ, Hughes R, Downesc M, Pfefferkorn JA. Discovery and optimization of non-steroidal FXR agonists from natural product-like libraries. Org Biomol Chem. (2003) 1:908–20. doi: 10.1002/chin.200324210
133. Fernandez-Marcos PJ, Auwerx J, Schoonjans K. Emerging actions of the nuclear receptor LRH-1 in the gut. Biochim Biophys Acta Mol Basis Dis. (2011) 1812:947–55. doi: 10.1016/j.bbadis.2010.12.010
134. Krylova IN, Sablin EP, Moore J, Xu RX, Waitt GM, MacKay JA, et al. Structural analyses reveal phosphatidyl inositols as ligands for the NR5 orphan receptors SF-1 and LRH-1. Cell. (2005) 120:343–55. doi: 10.1016/j.cell.2005.01.024
135. Bayrer JR, Wang H, Nattiv R, Suzawa M, Escusa HS, Fletterick RJ, et al. LRH-1 mitigates intestinal inflammatory disease by maintaining epithelial homeostasis and cell survival. bioRxiv. (2018) 9:1–10. doi: 10.1038/s41467-018-06137-w
136. Botrugno OA, Fayard E, Annicotte JS, Haby C, Brennan T, Wendling O, et al. Synergy between LRH-1 and β-catenin Induces G1 cyclin-mediated cell proliferation. Mol Cell. (2004) 15:499–509. doi: 10.1016/j.molcel.2004.07.009
137. Yumoto F, Nguyen P, Sablin EP, Baxter JD, Webb P, Fletterick RJ. Structural basis of coactivation of liver receptor homolog-1 by -catenin. Proc Natl Acad Sci USA. (2012) 109:143–8. doi: 10.1073/pnas.1117036108
138. Schoonjans K, Dubuquoy L, Mebis J, Fayard E, Wendling O, Haby C, et al. Liver receptor homolog 1 contributes to intestinal tumor formation through effects on cell cycle and inflammation. Proc Natl Acad Sci USA. (2005) 102:2058–62. doi: 10.1073/pnas.0409756102
139. Jostins L, Ripke S, Weersma RK, Duerr RH, McGovern DP, Hui KY, et al. Host-microbe interactions have shaped the genetic architecture of inflammatory bowel disease. Nature. (2012) 491:119–24. doi: 10.1038/nature11582
140. Boivin MA, Ye D, Kennedy JC, Al-Sadi R, Shepela C, Ma TY. Mechanism of glucocorticoid regulation of the intestinal tight junction barrier. (2006) 292:590–8. doi: 10.1152/ajpgi.00252.2006
141. Janowski BA, Willy PJ, Devi TR, Falck JR, Mangelsdorf DJ. An oxysterol signalling pathway mediated by the nuclear receptor LXRα. Nature. (1996) 383:728–31. doi: 10.1038/383728a0
142. Willy PJ, Mangelsdorf DJ. Unique requirements for retinoid-dependent transcriptional activation by the orphan receptor LXR. Genes Dev. (1997) 11:289–98. doi: 10.1101/gad.11.3.289
143. Lo Sasso G, Murzilli S, Salvatore L, D'Errico I, Petruzzelli M, Conca P, et al. Intestinal specific LXR activation stimulates reverse cholesterol transport and protects from atherosclerosis. Cell Metab. (2010) 12:187–93. doi: 10.1016/j.cmet.2010.07.002
144. Steffensen KR, Jakobsson T, Gustafsson JÅ. Targeting liver X receptors in inflammation. Expert Opin Ther Targets. (2013) 17:977–90. doi: 10.1517/14728222.2013.806490
145. Kratzer A, Buchebner M, Pfeifer T, Becker TM, Uray G, Miyazaki M, et al. Synthetic LXR agonist attenuates plaque formation in apoE-/- mice without inducing liver steatosis and hypertriglyceridemia. J Lipid Res. (2009) 50:312–26. doi: 10.1194/jlr.M800376-JLR200
146. Andersen V, Christensen J, Ernst A, Jacobsen BA, Tjønneland A, Krarup HB, et al. Polymorphisms in NF-κB, PXR, LXR, PPARγ and risk of infammatory bowel disease. World J Gastroenterol. (2011) 17:197–206. doi: 10.3748/wjg.v17.i2.197
147. Safe S, Jin UH, Morpurgo B, Abudayyeh A, Singh M, Tjalkens RB. Nuclear receptor 4A (NR4A) family - Orphans no more. J Steroid Biochem Mol Biol. (2016) 157:48–60. doi: 10.1016/j.jsbmb.2015.04.016
148. Wang Z, Benoit G, Liu J, Prasad S, Aarnisalo P, Liu X, et al. Structure and function of Nurr1 identifies a class of ligand- independent nuclear receptors. Nature. (2003) 423:555–60. doi: 10.1038/nature01645
149. Wu H, Li XM, Wang JR, Gan WJ, Jiang FQ, Liu Y, et al. NUR77 exerts a protective effect against inflammatory bowel disease by negatively regulating the TRAF6/TLR-IL-1R signalling axis. J Pathol. (2016) 238:457–69. doi: 10.1002/path.4670
150. Sekiya T, Kashiwagi I, Inoue N, Morita R, Hori S, Waldmann H, et al. The nuclear orphan receptor Nr4a2 induces Foxp3 and regulates differentiation of CD4+ T cells. Nat Commun. (2011) 2:269. doi: 10.1038/ncomms1272
151. Desvergne B, Wahli W. Peroxisome proliferator-activated receptors: nuclear control of metabolism. Endocr Rev. (1999) 20:649–88. doi: 10.1210/edrv.20.5.0380
152. Green S, Wahli W. Peroxisome proliferator-activated receptors: finding the orphan a home. Mol Cell Endocrinol. (1994) 100:149–53. doi: 10.1016/0303-7207(94)90294-1
153. Schwab M, Reynders V, Loitsch S, Steinhilber D, Stein J, Schröder O. Involvement of different nuclear hormone receptors in butyrate-mediated inhibition of inducible NFκB signalling. Mol Immunol. (2007) 44:3625–32. doi: 10.1016/j.molimm.2007.04.010
154. Nepelska M, De Wouters T, Jacouton E, Béguet-Crespel F, Lapaque N, Doré J, et al. Commensal gut bacteria modulate phosphorylation-dependent PPARλ transcriptional activity in human intestinal epithelial cells. Sci Rep. (2017) 7:1–13. doi: 10.1038/srep43199
155. Balzola F, Bernstein C, Ho GT, Lees C. Immunoregulatory actions of epithelial cell PPAR gamma at the colonic mucosa of mice with experimental inflammatory bowel disease: commentary. Inflamm Bowel Dis Monit. (2010) 11:31. doi: 10.1371/journal.pone.0010215
156. Pedersen G, Brynskov J. Topical rosiglitazone treatment improves ulcerative colitis by restoring peroxisome proliferator-activated receptor-gamma activity: commentary. Am J Gastroenterol. (2010) 11:82–3. doi: 10.1038/ajg.2009.749
157. Iacucci M, De Silva S, Ghosh S. Mesalazine in inflammatory bowel disease: a trendy topic once again? Can J Gastroenterol. (2010) 24:127–33. doi: 10.1155/2010/586092
158. Kornbluth A, Sachar DB. Ulcerative colitis practice guidelines in adults (update): American College of Gastroenterology, Practice Parameters Committee. Am J Gastroenterol. (2004) 99:1371–85. doi: 10.1111/j.1572-0241.2004.40036.x
159. Allgayer H. Review article: mechanisms of action of mesalazine in preventing colorectal carcinoma in inflammatory bowel disease. Aliment Pharmacol Ther. (2003) 18:10–4. doi: 10.1046/j.1365-2036.18.s2.1.x
160. Suzuki Y, Iida M, Ito H, Nishino H, Ohmori T, Arai T, et al. 2.4 g mesalamine (Asacol 400 mg tablet) once daily is as effective as three times daily in maintenance of remission in ulcerative colitis. Inflamm Bowel Dis. (2017) 23:822–32. doi: 10.1097/MIB.0000000000001065
161. Nagahori M, Kochi S, Hanai H, Yamamoto T, Nakamura S, Omuro S, et al. Real life results in using 5-ASA for maintaining mild to moderate UC patients in Japan, a multi-center study, OPTIMUM Study. BMC Gastroenterol. (2017) 17:47. doi: 10.1186/s12876-017-0604-y
162. Nozu T, Miyagishi S, Nozu R, Takakusaki K, Okumura T. Pioglitazone improves visceral sensation and colonic permeability in a rat model of irritable bowel syndrome. J Pharmacol Sci. (2019) 139:46–9. doi: 10.1016/j.jphs.2018.11.006
163. Braissant O, Foufelle F, Scotto C, Dauca M, Wahli W. Differential expression of peroxisome proliferator-activated receptors (PPARs): tissue distribution of PPAR-a, -b, and -y in the adult rat. Endocrinology. (1996) 137:354–66. doi: 10.1210/endo.137.1.8536636
164. Riccardi L, Mazzon E, Bruscoli S, Esposito E, Crisafulli C, Di PR, et al. PPAR-alpha modulates the anti-inflammatory effect of Glucocorticoids in a model of inflammatory bowel disease in mice. Shock. (2009) 31:308–16. doi: 10.1097/SHK.0b013e31818339e7
165. Lee JW, Bajwa PJ, Carson MJ, Jeske DR, Cong Y, Elson CO, et al. Fenofibrate represses interleukin-17 and interferon-g expression and improves colitis in interleukin-10-deficient mice. Gastroenterology. (2007) 133:108–23. doi: 10.1053/j.gastro.2007.03.113
166. Esposito G, Capoccia E, Turco F, Palumbo I, Lu J, Steardo A, et al. Palmitoylethanolamide improves colon inflammation through an enteric glia/toll like receptor 4-dependent PPAR-α activation. Gut. (2014) 63:1300–12. doi: 10.1136/gutjnl-2013-305005
167. Tanaka T, Kohno H, Yoshitani S, Takashima S, Okumura A, Murakami A, et al. Ligands for peroxisome proliferator-activated receptors alpha and gamma inhibit chemically induced colitis and formation of aberrant crypt foci in rats1. Cancer Res. (2001) 61:2424–8.
168. Bougarne N, Weyers B, Desmet SJ, Deckers J, Ray DW, Staels B, et al. Molecular actions of PPARα in lipid metabolism and inflammation. Endocr Rev. (2018) 39:760–802. doi: 10.1210/er.2018-00064
169. Hollingshead HE, Morimura K, Adachi M, Kennett MJ, Billin AN, Willson TM, et al. PPARβ/δ protects against experimental colitis through a ligand-independent mechanism. Dig Dis Sci. (2007) 52:2912–9. doi: 10.1007/s10620-006-9644-9
170. Bassaganya-Riera J, DiGuardo M, Climent M, Vives C, Carbo A, Jouni ZE, et al. Activation of PPARγ and δ by dietary punicic acid ameliorates intestinal inflammation in mice. Br J Nutr. (2011) 106:878–86. doi: 10.1017/S0007114511001188
171. Cheng J, Shah YM, Gonzalez FJ. Pregnane X receptor as a target for treatment of inflammatory bowel disorders. Trends Pharmacol Sci. (2012) 33:323–30. doi: 10.1016/j.tips.2012.03.003
172. Pavek P, Dvorak Z. Xenobiotic-induced transcriptional regulation of xenobiotic metabolizing enzymes of the cytochrome P450 superfamily in human extrahepatic tissues. Curr Drug Metab. (2008) 9:129–43. doi: 10.2174/138920008783571774
173. di Masi A, DeMarinisa E, Ascenzi P, Marinoa M. Nuclear receptors CAR and PXR: molecular, functional, and biomedical aspects. Mol Aspects Med. (2009) 30:297–343. doi: 10.1016/j.mam.2009.04.002
174. Venkatesh M, Mukherjee S, Wang H, Li H, Sun K, Benechet AP, et al. Symbiotic bacterial metabolites regulate gastrointestinal barrier function via the xenobiotic sensor PXR and toll-like receptor 4. Immunity. (2014) 41:296–310. doi: 10.1016/j.immuni.2014.06.014
175. Pavek P. Pregnane X receptor (PXR)-mediated gene repression and cross-talk of PXR with other nuclear receptors via coactivator interactions. Front Pharmacol. (2016) 7:1–16. doi: 10.3389/fphar.2016.00456
176. Laustsen G, Wimmett L. 2004 drug approval highlights FDA update. Nurse Pract. (2005) 30:14–29. doi: 10.1097/00006205-200502000-00004
177. Yang J, Lee HR, Low K, Chatterjee S, Pimentel M. Rifaximin versus other antibiotics in the primary treatment and retreatment of bacterial overgrowth in IBS. Dig Dis Sci. (2008) 53:169–74. doi: 10.1007/s10620-007-9839-8
178. Iwamoto J, Saito Y, Honda A, Miyazaki T, Ikegami T, Matsuzaki Y. Bile acid malabsorption deactivates pregnane X receptor in patients with crohn's disease. Inflamm Bowel Dis. (2013) 19:1278–84. doi: 10.1097/MIB.0b013e318281f423
179. Li Y, Gao Y, Cui T, Yang T, Liu L, Li T, et al. Retinoic acid facilitates toll-like receptor 4 expression to improve intestinal barrier function through retinoic acid receptor beta. Cell Physiol Biochem. (2017) 42:1390–406. doi: 10.1159/000479203
180. Chanchevalap S, Nandan MO, Merlin D, Yang VW. All-trans retinoic acid inhibits proliferation of intestinal epithelial cells by inhibiting expression of the gene encoding Krüppel-like factor 5. FEBS Lett. (2004) 578:99–105. doi: 10.1016/j.febslet.2004.10.079
181. Mielke LA, Jones SA, Raverdeau M, Higgs R, Stefanska A, Groom JR, et al. Retinoic acid expression associates with enhanced IL-22 production by γδ T cells and innate lymphoid cells and attenuation of intestinal inflammation. J Exp Med. (2013) 210:1117–24. doi: 10.1084/jem.20121588
182. Sonnenberg GF, Artis D. Innate lymphoid cells in the initiation, regulation and resolution of inflammation. Nat Med. (2015) 21:698–708. doi: 10.1038/nm.3892
183. Okayasu I, Hana K, Nemoto N, Yoshida T, Saegusa M, Yokota-nakatsuma A, et al. Vitamin A inhibits development of dextran sulfate sodium-induced colitis and colon cancer in a mouse model. Biomed Res Int. (2016) 2016:4874809. doi: 10.1155/2016/4874809
184. Reifen R, Nur T, Ghebermeskel K, Zaiger G, Urizky R, Pines M. Vitamin A deficiency exacerbates inflammation in a rat model of colitis through activation of nuclear factor- kB and collagen formation. J Nutr. (2002) 9:2743–7. doi: 10.1093/jn/132.9.2743
185. Bai A, Lu N, Zeng H, Li Z, Zhou X, Chen J, et al. All-trans retinoic acid ameliorates trinitrobenzene sulfonic acid-induced colitis by shifting Th1 to Th2 profile. J Interferon Cytokine Res. (2010) 30:399–406. doi: 10.1089/jir.2009.0028
186. Hong K, Zhang Y, Guo Y, Xie J, Wang J, He X, et al. All-trans retinoic acid attenuates experimental colitis through inhibition of NF-κB signaling. Immunol Lett. (2014) 162:34–40. doi: 10.1016/j.imlet.2014.06.011
187. Conway TF, Hammer L, Furtado S, Mathiowitz E, Nicoletti F, Mangano K, et al. Oral delivery of particulate transforming growth factor beta 1 and all-trans retinoic acid reduces gut inflammation in murine models of inflammatory bowel disease. J Crohn's Colitis. (2015) 9:647–58. doi: 10.1093/ecco-jcc/jjv089
188. Lim SM, Kang GD, Jeong JJ, Choi HS, Kim DH. Neomangiferin modulates the Th17/Treg balance and ameliorates colitis in mice. Phytomedicine. (2016) 23:131–40. doi: 10.1016/j.phymed.2016.01.002
189. Frey-Wagner I, Fischbeck A, Cee A, Leonardi I, Gruber S, Becker E, et al. Effects of retinoids in mouse models of colitis: benefit or danger to the gastrointestinal tract? Inflamm Bowel Dis. (2013) 19:2356–65. doi: 10.1097/MIB.0b013e31829cf1fc
190. Menning A, Loddenkemper C, Westendorf AM, Szilagyi B, Buer J, Siewert C, et al. Retinoic acid-induced gut tropism improves the protective capacity of Treg in acute but not in chronic gut inflammation. Eur J Immunol. (2010) 40:2539–48. doi: 10.1002/eji.200939938
191. Bhattacharya N, Yuan R, Prestwood TR, Penny HL, DiMaio MA, Reticker-Flynn NE, et al. Normalizing microbiota-induced retinoic acid deficiency stimulates protective CD8+T cell-mediated immunity in colorectal cancer. Immunity. (2016) 45:641–55. doi: 10.1016/j.immuni.2016.08.008
192. Sun Z. Requirement for RORgamma in thymocyte survival and lymphoid organ development. Science. (2000) 288:2369–73. doi: 10.1126/science.288.5475.2369
193. Mizuno S, Mikami Y, Kamada N, Handa T, Hayashi A, Sato T, et al. Cross-talk between RORγt+ innate lymphoid cells and intestinal macrophages induces mucosal IL-22 production in Crohn's disease. Inflamm Bowel Dis. (2014) 20:1426–34. doi: 10.1097/MIB.0000000000000105
194. Leppkes M, Becker C, Ivanov II, Hirth S, Wirtz S, Neufert C, et al. RORgamma-expressing Th17 cells induce murine chronic intestinal inflammation via redundant effects of IL-17A and IL-17F. Gastroenterology. (2009) 136:257–67. doi: 10.1053/j.gastro.2008.10.018
195. Tang C, Kakuta S, Shimizu K, Kadoki M, Kamiya T, Shimazu T, et al. Suppression of IL-17F, but not of IL-17A, provides protection against colitis by inducing Tregcells through modification of the intestinal microbiota. Nat Immunol. (2018) 19:755–65. doi: 10.1038/s41590-018-0134-y
196. Tosiek MJ, Fiette L, El Daker S, Eberl G, Freitas AA. IL-15-dependent balance between Foxp3 and RORγt expression impacts inflammatory bowel disease. Nat Commun. (2016) 7:1–11. doi: 10.1038/ncomms10888
197. Withers DR, Hepworth MR, Wang X, Mackley EC, Halford EE, Dutton EE, et al. Transient inhibition of ROR-γt therapeutically limits intestinal inflammation by reducing TH17 cells and preserving group 3 innate lymphoid cells. Nat Med. (2016) 22:319–23. doi: 10.1038/nm.4046
198. Lo BC, Gold MJ, Hughes MR, Antignano F, Valdez Y, Zaph C, et al. The orphan nuclear receptor RORα and group 3 innate lymphoid cells drive fibrosis in a mouse model of Crohn's disease. Inflamm Bowel Dis. (2016) 1:eaaf8864. doi: 10.1126/sciimmunol.aaf8864
199. Lefebvre P, Benomar Y, Staels B. Retinoid X receptors: common heterodimerization partners with distinct functions. Trends Endocrinol Metab. (2010) 11:676–83. doi: 10.1016/j.tem.2010.06.009
200. Desreumaux P, Dubuquoy L, Nutten S, Peuchmaur M, Englaro W, Schoonjans K, et al. Attenuation of colon inflammation through activators of the retinoid X receptor (Rxr)/peroxisome proliferator–activated receptor γ (Pparγ) heterodimer. J Exp Med. (2001) 193:827–38. doi: 10.1084/jem.193.7.827
201. Knackstedt R, Shaoli S, Moseley V, Wargovich M. The importance of the retinoid X receptor alpha in modulating inflammatory signaling in acute murine colitis. Dig Dis Sci. (2014) 59:753–9. doi: 10.1007/s10620-013-2902-8
202. Onuki M, Watanabe M, Ishihara N, Suzuki K, Takizawa K, Hirota M, et al. A partial agonist for retinoid X receptor mitigates experimental colitis. Int Immunol. (2018) 31:251–62. doi: 10.1093/intimm/dxy089
203. Ardesia M, Ferlazzo G, Fries W, Ardesia M, Ferlazzo G, Fries W. Vitamin D and inflammatory bowel disease. Biomed Res Int. (2015) 2015:1–16. doi: 10.1155/2015/470805
204. Sun J. Vitamin D and mucosal immune function. Curr Opin Gastroenterol. (2010) 26:591–5. doi: 10.1097/MOG.0b013e32833d4b9f
205. Hughes DJ, McManus R, Neary P, O'Morain C, O'Sullivan M. Common variation in the vitamin D receptor gene and risk of inflammatory bowel disease in an Irish case–control study. Eur J Gastroenterol Hepatol. (2011) 23:807–12. doi: 10.1097/MEG.0b013e328349283e
206. Lin YD, Arora J, Diehl K, Bora SA, Cantorna MT. Vitamin D is required for ILC3 derived IL-22 and protection from citrobacter rodentium infection. Front Immunol. (2019) 10:1. doi: 10.3389/fimmu.2019.00001
207. Chen J, Waddell A, Lin YD, Cantorna MT. Dysbiosis caused by vitamin D receptor deficiency confers colonization resistance to Citrobacter rodentium through modulation of innate lymphoid cells. Mucosal Immunol. (2015) 8:618–26. doi: 10.1038/mi.2014.94
208. Palmieri O, Mazzoccoli G, Bossa F, Maglietta R, Palumbo O, Ancona N, et al. Systematic analysis of circadian genes using genome-wide cDNA microarrays in the inflammatory bowel disease transcriptome. Chronobiol Int. (2015) 32:903–16. doi: 10.3109/07420528.2015.1050726
209. Babeu J-P, Darsigny M, Lussier CR, Boudreau F. Hepatocyte nuclear factor 4α contributes to an intestinal epithelial phenotype in vitro and plays a partial role in mouse intestinal epithelium differentiation. Am J Physiol Liver Physiol. (2009) 297:G124–34. doi: 10.1152/ajpgi.90690.2008
210. Cattin A-L, Le Beyec J, Barreau F, Saint-Just S, Houllier A, Gonzalez FJ, et al. Hepatocyte nuclear factor 4, a key factor for homeostasis, cell architecture, and barrier function of the adult intestinal epithelium. Mol Cell Biol. (2009) 29:6294–308. doi: 10.1128/MCB.00939-09
211. Lussier CR, Babeu J, Auclair BA, Perreault N, Boudreau F. Hepatocyte nuclear factor-4a promotes differentiation of intestinal epithelial cells in a coculture system. Am J Clin Nutr. (2008) 4:418–28. doi: 10.1152/ajpgi.00418.2007
212. Archer A, Sauvaget D, Chauffeton V, Bouchet P-E, Chambaz J, Pinçon-Raymond M, et al. Intestinal apolipoprotein A-IV gene transcription is controlled by two hormone-responsive elements: a role for hepatic nuclear factor-4 isoforms. Mol Endocrinol. (2005) 19:2320–34. doi: 10.1210/me.2004-0462
213. Battle M, Konopka G, Parviz F, Gaggl AL, Yang C, Sladek FM, et al. Hepatocyte nuclear factor 4alpha orchestrates expression of cell adhesion proteins during the epithelial transformation of the developing liver. Proc Natl Acad Sci USA. (2006) 103:8419–24. doi: 10.1073/pnas.0600246103
214. Chiba H, Gotoh T, Kojima T, Satohisa S, Kikuchi K, Osanai M, et al. Hepatocyte nuclear factor (HNF)-4α triggers formation of functional tight junctions and establishment of polarized epithelial morphology in F9 embryonal carcinoma cells. Exp Cell Res. (2003) 286:288–97. doi: 10.1016/S0014-4827(03)00116-2
215. Babeu JP, Boudreau F. Hepatocyte nuclear factor 4-alpha involvement in liver and intestinal inflammatory networks. World J Gastroenterol. (2014) 20:22–30. doi: 10.3748/wjg.v20.i1.22
216. Barrett J, Lee J, Lees C, Prescott N, Anderson C, Phillips A, et al. Genome-wide association study of ulcerative colitis identifies three new susceptibility loci, including the HNF4A region. Nat Genet. (2009) 41:1330–4. doi: 10.1038/ng.483
217. Marcil V, Sinnett D, Seidman E, Gendron F, Beaulieu J, Levy E. Association between genetic variants in the HNF4a gene and childhood-onset of crohn's disease. (2016) 13:556–65. doi: 10.1038/gene.2012.37
218. Chellappa K, Jankova L, Schnabl JM, Pan S, Brelivet Y, Fung CL-S, et al. Src tyrosine kinase phosphorylation of nuclear receptor HNF4 correlates with isoform-specific loss of HNF4 in human colon cancer. Proc Natl Acad Sci USA. (2012) 109:2302–7. doi: 10.1073/pnas.1106799109
219. Tanaka T, Jiang S, Hotta H, Takano K, Iwanari H, Sumi K, et al. Dysregulated expression of P1 and P2 promoter-driven hepatocyte nuclear factor-4α in the pathogenesis of human cancer. J Pathol. (2006) 208:662–72. doi: 10.1002/path.1928
220. Chellappa K, Deol P, Evans JR, Vuong LM, Chen G, Briançon N, et al. Opposing roles of nuclear receptor HNF4α isoforms in colitis and colitis-associated colon cancer. Elife. (2016) 5:1–25. doi: 10.7554/eLife.10903
221. Hermann-Kleiter N, Baier G. Orphan nuclear receptor NR2F6 acts as an essential gatekeeper of Th17 CD4 + T cell effector functions. Cell Commun Signal. (2014) 12:38. doi: 10.1186/1478-811X-12-38
222. Klepsch V, Hermann-Kleiter N, Baier G. Beyond CTLA-4 and PD-1: orphan nuclear receptor NR2F6 as T cell signaling switch and emerging target in cancer immunotherapy. Immunol Lett. (2016) 178:31–6. doi: 10.1016/j.imlet.2016.03.007
223. Klepsch V, Hermann-Kleiter N, Do-Dinh P, Jakic B, Offermann A, Efremova M, et al. Nuclear receptor NR2F6 inhibition potentiates responses to PD-L1/PD-1 cancer immune checkpoint blockade. Nat Commun. (2018) 9:1538. doi: 10.1038/s41467-018-04004-2
224. Olsen J, Gerds T, Seidelin J, Csillag C, Bjerrum J, Troelsen J, et al. Diagnosis of ulcerative colitis before onset of inflammation by multivariate modeling of genome-wide gene expression data. Inflamm Bowel Dis. (2009) 15:1032–8. doi: 10.1002/ibd.20879
225. Bjerrum JT, Hansen M, Olsen J, Nielsen OH. Genome-wide gene expression analysis of mucosal colonic biopsies and isolated colonocytes suggests a continuous inflammatory state in the lamina propria of patients with quiescent ulcerative colitis. Inflamm Bowel Dis. (2010) 16:999–1007. doi: 10.1002/ibd.21142
Keywords: nuclear receptor, intestinal barrier homeostasis, immune system, microbiota, inflammatory bowel disease
Citation: Klepsch V, Moschen AR, Tilg H, Baier G and Hermann-Kleiter N (2019) Nuclear Receptors Regulate Intestinal Inflammation in the Context of IBD. Front. Immunol. 10:1070. doi: 10.3389/fimmu.2019.01070
Received: 05 March 2019; Accepted: 26 April 2019;
Published: 14 May 2019.
Edited by:
Julien Diana, Institut National de la Santé et de la Recherche Médicale (INSERM), FranceReviewed by:
Carlo Riccardi, University of Perugia, ItalyCopyright © 2019 Klepsch, Moschen, Tilg, Baier and Hermann-Kleiter. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Natascha Hermann-Kleiter, bmF0YXNjaGEua2xlaXRlckBpLW1lZC5hYy5hdA==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.