 Alicia Lacoma1Lourdes Mateo2Ignacio Blanco3Maria J. Méndez4Carlos Rodrigo5
Alicia Lacoma1Lourdes Mateo2Ignacio Blanco3Maria J. Méndez4Carlos Rodrigo5 Irene Latorre1
Irene Latorre1 Raquel Villar-Hernandez1
Raquel Villar-Hernandez1 Jose Domínguez1†
Jose Domínguez1† Cristina Prat1*†
Cristina Prat1*†- 1Servei de Microbiologia, Hospital Universitari Germans Trias i Pujol, Institut d'Investigació Germans Trias i Pujol, Universitat Autònoma de Barcelona, CIBER Enfermedades Respiratorias, Barcelona, Spain
- 2Servei de Reumatologia, Hospital Universitari Germans Trias i Pujol, Institut d'Investigació Germans Trias i Pujol, Universitat Autònoma de Barcelona, Barcelona, Spain
- 3Clinical Genetics and Genetic Counseling Program, Hospital Universitari Germans Trias i Pujol, Institut d'Investigació Germans Trias i Pujol, Barcelona, Spain
- 4Servei de Pediatria, Hospital Universitari Germans Trias i Pujol, Institut d'Investigació GermansTrias i Pujol, Universitat Autònoma de Barcelona, Barcelona, Spain
- 5Servei de Pediatria, Hospital Universitari Vall d'Hebron, Vall d'Hebron Institut de Recerca, Facultat de Medicina, Unitat Docent Germans Trias i Pujol, Universitat Autònoma de Barcelona, Barcelona, Spain
Host susceptibility to respiratory tract infections (RTI) is dependent on both genetic and acquired risk factors. Repeated bacterial and viral RTI, such as pneumonia from encapsulated microorganisms, respiratory tract infections related to respiratory syncytial virus or influenza, and even the development of bronchiectasis and asthma, are often reported as the first symptom of primary immunodeficiencies. In the same way, neutropenia is a well-known risk factor for invasive aspergillosis, as well as lymphopenia for Pneumocystis, and mycobacterial infections. However, in the last decades a better knowledge of immune signaling networks and the introduction of next generation sequencing have increased the number and diversity of known inborn errors of immunity. On the other hand, the use of monoclonal antibodies targeting cytokines, such as tumor necrosis factor alpha has revealed new risk groups for infections, such as tuberculosis. The use of biological response modifiers has spread to almost all medical specialties, including inflammatory diseases and neoplasia, and are being used to target different signaling networks that may mirror some of the known immune deficiencies. From a clinical perspective, the individual contribution of genetics, and/or targeted treatments, to immune dysregulation is difficult to assess. The aim of this article is to review the known and newly described mechanisms of impaired immune signaling that predispose to RTI, including new insights into host genetics and the impact of biological response modifiers, and to summarize clinical recommendations regarding vaccines and prophylactic treatments in order to prevent infections.
Epidemiology and Pathogenesis of Respiratory Tract Infections
Acute and chronic respiratory tract infections (RTI) are one of the most frequent causes of infections and antimicrobial prescription, and the leading cause of death in developing countries (1, 2). Pneumonia accounts for 1.3 million deaths annually in children <5 years of age (3). In 2017, 1.6 million people died of tuberculosis (TB). Children (aged <15 years) accounted for 15% of total deaths, higher than their share of estimated cases, suggesting poorer access to diagnosis and treatment. About 1.7 billion people, 23% of the world's population, are estimated to have a latent TB infection (4). The control of latent TB, a stage in which a person is infected with Mycobacterium tuberculosis plays an important role in disease control, since dormant bacilli are a reservoir of potential TB cases (5). Viral acute RTI are estimated to cause 75% of acute diseases in children, and is the main reason for hospitalization worldwide (6). The annual prevalence in an otherwise healthy child is from 3 to 10 infections (7). Early and recurrent lower RTI are linked to a higher risk to develop asthma or bronchiectasis (8–10). However, bronchiectasis secondary to recurrent and severe infections alone have declined, with an increasing proportion of patients being recognized as having underlying conditions predisposing to its development (11).
Improvements in immunization programs and the wide availability of antimicrobials, have led to optimism for most of the devastating infectious diseases. Always without forgetting that alleviation of poverty is crucial, the combination of genetic versatility and ecological opportunism of the microbial world appears to have been under-estimated (12). Some emerging pathogens, such as Legionella, avian influenza, and coronavirus species were described in the past decades (13). Ethnic variations in the incidence of RTI have also been reported, suggesting genetic susceptibility to disease (14). Most children, on reaching 2 years of age, have been in contact with the most common respiratory viruses, such as respiratory syncytial virus (RSV), but while some develop a mild disease, others develop severe bronchiolitis (15). Influenza viruses cause mild to moderate respiratory illness in most people, but some develop fatal infections. The virulence factors encoded by viral genes can explain seasonal or geographical differences at a population level, but are unlikely to account for inter-individual clinical variability (16). TB outcome depends on the pathogen and extrinsic elements, as well as on host factors that are still unclear (17).
As regards bacteria, focusing on those species whose normal ecological niche is the airways, therapeutic decisions are a daily clinical challenge (18). The shift from commensalism to infection is shaped by host intrinsic (genetics) and extrinsic factors (for example, diet and exposure to cigarette smoke and environmental pollution) and by bacterial features that also contribute to inter-individual variability (19). Bacteria develop adaptive mechanisms (at genetic/phenotypic level) in order to survive in a hostile environment, such as the respiratory tract (20, 21). Whether pathogen virulence generates clinical symptoms depends on how well the immune system limits its impact. Recently, changes in gut and lung microbiome composition (dysbiosis) have also been related to dysfunctional immune modulation (22).
Immune Response to Respiratory Tract Infections
Respiratory immune responses are complex, and inborn errors can be present at any level. Essential pathways can be summarized as follows: Firstly, the pathogen has to be detected by host cells. This identification relies on a set of pathogen associated molecular profiles that bind to pattern recognition receptors (PRR). PRR can be found as transmembrane, cytosolic or extracellular components. Among PRRs, it is important to mention toll-like receptors (TLR), nucleotide-binding oligomerization domain-containing (NOD) receptor, NOD-like receptors (NLR), RIG-I-Like Receptors (RLR), and receptor CD14 because of their importance during respiratory infections (23). Depending on the PRR, different intracellular signaling pathways are activated (24). Most of the signaling pathways converge on signaling hubs, such as transcription nuclear factor κβ (NF-κβ), interferon regulatory factor families (IRF3, IRF7), and mitogen-activated protein kinase, leading to the induction of gene expression encoding adhesion molecules, pro-inflammatory cytokines, chemokines, and type I interferon, among others. NLRs directly trigger inflammasome assembly and caspase-1 activation, leading to interleukin (IL)-1β and IL-18 processing (25). Type III interferons, also termed IFN-λ, have been recently identified as regulators of immunity and homeostasis in the respiratory tract (26) during infections, as well as during chronic lung diseases, such as asthma and chronic obstructive pulmonary disease (COPD) (27). Alveolar macrophages and dendritic cells (DC) have an important role sensing microbes and thus activating lung epithelial cells and neutrophils. These are essential for the defense against bacteria, viruses, and Aspergillus (28, 29), as well as in the pathogenesis of acute lung injury. In a recent study, patterns of differentially expressed cellular genes shared by several respiratory pathogens were searched using transcriptomics (30). Most of the commonly up-regulated host genes were related to the innate immune response and/or apoptosis, with Toll-like, RIG-I-like, and NLR among the top 10 signalers. Some of the genes showed a high degree of interconnection and possible redundancy to respiratory viral and bacterial infections. The adaptive immune response requires the activation of antigen-specific T and B lymphocytes to trigger protective cellular and humoral responses. Most of the T lymphocyte subsets, along with B lymphocytes and DC, are essential for immune defense and/or regulation (31). In particular, the protective immunity against M. tuberculosis depends on CD4+ T-helper1 lymphocytes that mainly secrete interferon-gamma (IFN-γ), IL-2, and tumor necrosis factor alpha (TNF-α), which leads to macrophage activation, cytokine production, and bacterial control (32). HIV-revealed T-cell lymphopenia as a well-defined risk group for Pneumocystis jirovecii pneumonia (PJP), but also in other situations where CD4 lymphocyte count is lower, such as renal transplant recipients (33).
Genetic Susceptibility to Respiratory Tract Infections
The study of susceptibility to lower respiratory tract infections is complex, and requires different approaches. There are three main elements playing a role: host genetic background (in relation to lung tissue functionality and immune response), pathogen virulence determinants, and environmental factors.
Early life (children under 5 years of age) is a challenging period because pulmonary tissue and the immune system are still in a maturation process while being continuously exposed to airborne antigens (34). However, the occurrence of life-threatening bacterial/viral/fungal infection in an otherwise healthy individual deserves further immunological and genetic studies (35, 36). Complications during upper RTI include sinusitis and otitis media, and in the lower airways, pneumonia, bronchitis, as well as the development of bronchiectasis, interstitial lung diseases, organizing pneumonia, and hyperreactive airway diseases (37). Indeed, genetic susceptibility for the concomitant illnesses that predispose to RTI can also play a role, including congenital defects of the airways, familial congenital bronchiectasis or tracheobronchomegaly (11). As regards impaired mucociliary clearance, cystic fibrosis is the most common autosomal recessive disorder and primary cause of bronchiectasis in the developed world. Mutations are well-defined, but its severity is influenced by genes involving inflammatory and anti-inflammatory mediators (38, 39). Other disorders include ciliopathies and disorders of humoral immunity. Alpha 1-antitrypsin is a circulating serine protease inhibitor (serpin) made in the liver that plays an important role in modulating immunity, inflammation, apoptosis, and possibly cellular senescence programs and its deficiency is considered the genetic cause of COPD, but there are other genetic factors that may affect disease activity and outcomes, even in patients without this deficiency (27).
High-throughput whole genome sequencing technologies and novel bioinformatics tools are revealing the sequence and annotation of the complete human genome, as well as genome-wide maps of polymorphic microsatellite markers and single nucleotide polymorphisms (SNP). In order to characterize genetic susceptibility, two complementary approaches can be envisaged: whole genome association studies (WGAS) for the identification of variants with high population frequency but low impact at individual level in terms of risk of infection (although SNP identification can potentially be later included in healthcare planning protocols); and mechanistic studies for identifying disease-causing mutations with deleterious effects, related to a high risk of infection at individual level, although its frequency in general population is low. Many genetic variants have been associated with complex human diseases and traits, but often confer relatively small increases in risk (40). According to a recent review, there are more than 300 primary immunodeficiency disorders (PIDs), most of them monogenic conditions with Mendelian inheritance, that are mainly associated with crucial defects in adaptive immunity (31). Innate immune responses are largely redundant, with pleiotropic nature of some gene products (31), thus most of the defects can be potentially counterbalanced. According to the literature, there is another view suggesting that while patients with broad immunodeficiencies may present with one of their many infections, the phenotype of particular inborn errors of immunity is very narrow, with susceptibility to only one specific infection (36, 41, 42). A set of inborn errors affecting “primarily” innate immunity, exercise their effect on the adaptive immune response (41). The range and nature of infections depend on several factors. The improving recognition of immune dysregulation diseases, autoinflammatory disorders, and interferonopathies leads to changes in terminology. The annual report of the authoritative International Union of Immunological Societies (43) has categorized and listed (as of February 2017) 354 inborn errors of immunity, and those with a predominant RTI phenotype have been included in Table 1.
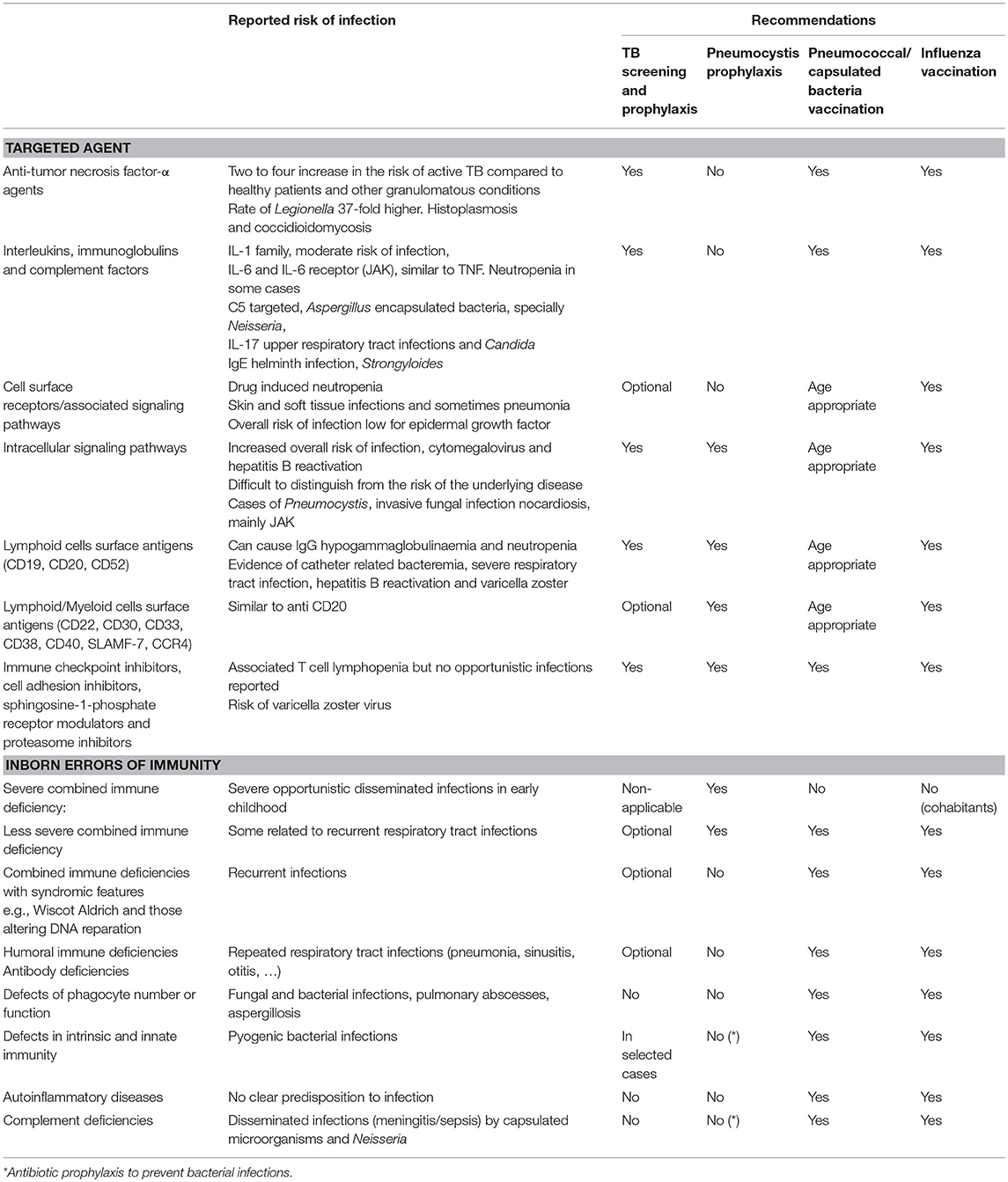
Table 1. Reported risk of infection and recommended prophylaxis according to functional classification of biologicals-based on ESCMID consensus document (44) and to categorization of inborn errors of immunity-based on International Union of Immunological Societies annual report (43).
Despite the limitations of molecular genetic studies in pulmonary infections, several associations have been described between SNPs and bacterial pneumonia and mycobacterial infections (14, 45). Polymorphisms affecting community-acquired pneumonia including, among others, those related to mannose-binding lectin and the IgG2 Fc gamma receptor II, and are discussed extensively elsewhere (14). The genetic contribution for the propensity to develop severe RSV infection was estimated to account for ~20% of the variance in RSV disease severity. Several studies have attempted to link candidate host SNPs to disease severity, mostly in chemokine receptors and PRRs (46, 47).
As regards TB, SNPs are frequently found in loci involving TLR-2, TNF-α, IL-12, and IFN-γ, and their corresponding receptors (45). Genetic variations in dendritic cell-specific ICAM-3 grabbing non-integrin have been linked with reduced risk of developing TB (48). Mendelian susceptibility to mycobacterial disease is a syndrome characterized by susceptibility to weakly virulent mycobacteria, including the attenuated vaccine Bacillus Calmette-Guerin (BCG) strain and non-tuberculous mycobacteria (NTM). Different gene mutations have been identified, most of which are related to IFN-γ-mediated immunity (49–51). Using exome and transcriptome sequencing, three rare loss-of-function variants have been recently characterized in theIFIH1 gene. These encode a RIG-I-like receptor involved in the sensing of viral RNA (52). The deficiency causes a primary immunodeficiency manifested in extreme susceptibility to common respiratory RNA viruses. Interestingly, human primary immunodeficiency disorders (PID) affecting T and B cells were not found to predispose to severe influenza. However, human IRF7 was shown to be essential for IFN-α/β- and IFN-λ-dependent protective immunity against primary influenza in vivo (53).
Impact of Biological Response Modifiers in Respiratory Tract Infections and Tuberculosis
Biological response modifiers (BRM) are substances that interact with and modify the host immune system by acting on a therapeutic target considered important in the pathogenic process of the disease. Monoclonal antibodies (mAbs) are now established as therapies for malignancies, transplant rejection, several immune disorders from most organ systems, and even infectious diseases (54). Safety problems related to immunomodulation and infection have been identified in some cases (55). The use of mAb indirectly provides insights into the function of the molecule to combat particular pathogens, increasing our knowledge of the immune system (56). A recent consensus document has reviewed the groups of drugs according to the targeted site of action, the expected impact on susceptibility to infection, the evidence of risk, and the recommendation of prevention strategies. It is also important to mention the influence of previous or concomitant therapies, underlying conditions, and the accumulative exposure to the agent (44). As regards lower RTI, treatment with BRM results in an increased risk is reported for pneumonia, influenza-related complications, TB and NTM, Pneumocystis, and fungal infections, such as histoplasmosis, taking into account the impact of geographical variations on incidence rates (57). The knowledge obtained from experience with the prescription of BRM may be particularly valuable for the understanding of some genetic inborn errors, as the type of infections acquired as a side effect may help to identify which genetic defects favors a similar infectious phenotype. With the current knowledge and because of pleiotropic effects, it is not feasible to show how biological agents actually mimic some inborn errors of immunity, but several parallelisms can be inferred. We provide a Table containing the list of BRM according to their functional classification, and inborn errors categorized according to common infectious phenotypes (Table 1). Data presented are extracted from the respective consensus documents, and lists the main RTI and preventive recommendations.
Current recommendations should be focused on rheumatic diseases because of the greater experience in follow-up time (more than 15 years) and number of patients treated. Biological therapies targeting TNF-α, T cells, B cells, and various cytokines (including IL-6 and IL-1) have become essential for the treatment of rheumatic diseases [mainly rheumatoid arthritis (RA), ankylosing spondylitis, and psoriatic arthritis], as well as other immune-mediated diseases. Moreover, additional drugs with novel targets, including those that inhibit IL-12–IL-23, IL-17α, or the Janus activating kinase system have been introduced more recently. Immunomodulation offered by biological and non-biological disease-modifying therapies and prednisone contributes greatly to the increased risks of opportunistic infections (OI) (58, 59). In Figure 1 we present the sites of action and associated risks of the most frequently prescribed BRM.
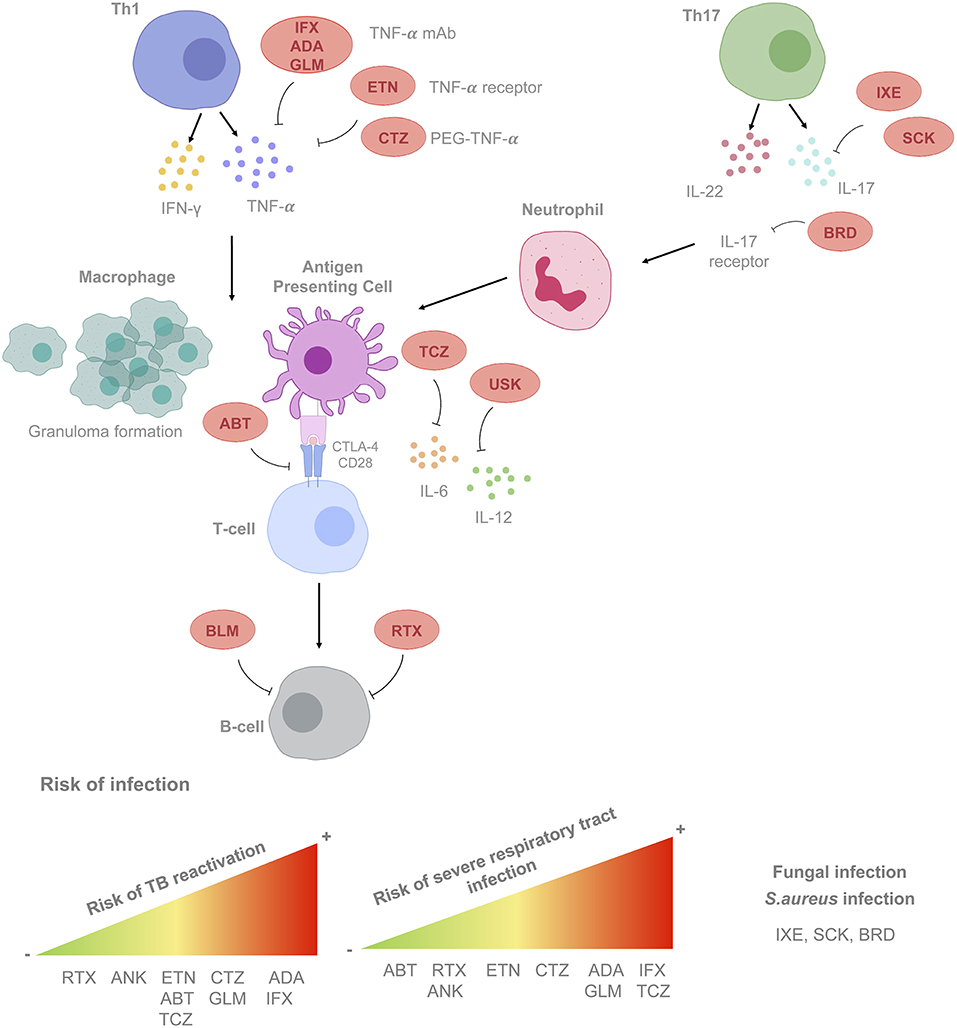
Figure 1. Mode of action of biological response modifiers (BRM) according to cell type, cytokine and/or receptor targeted. Risk of developing infections according to the BRM considered is also shown. List of BRM. Anti-tumor necrosis factor-α (TNF-α) agents. ADA, adalimumab; CTZ, certolizumab; GLM, golimumab; IFX, infliximab; ETN, etanercept. Anti-interleukins, immunoglobulins, and complement factors. Anti IL-1, anakinra ANK; Anti IL-6: TCZ, tocilizumab; Anti IL-17: SCK, secukinumab; IXE, ixekizumab; BRD, brodalumab. Anti-IL12/23: USK, ustekinumab. Cell surface receptors/associated signaling pathways agents. Anti-CD28: ABT, abatacept; B-cell activating factor (BAFF): BLM, belimumab. Lymphoid cells surface antigens. Anti-CD20: RTX, rituximab. mAb, monoclonal antibody; PEG, polyethylene glycol; TB, tuberculosis; LTBI, latent tuberculosis infection.
Two recent meta-analysis have calculated the relative risk of infection for rheumatic patients under biological treatment, with an odds ratio (OR) of 1.31–1.41 (60, 61). The absolute increase in the number of serious infections per 1,000 patients treated/year is six times higher than that observed with synthetic disease-modifying anti-rheumatic drugs (DMARDs). Different meta-analyses and national registries have confirmed the increase on the impact of any infections (20%), serious infections (40%), and TB (250%), associated with anti-TNF-α use (60). In addition, the risk of serious infections is highest during the first 6 months of therapy (62) (up to 4.5-fold risk), although, after 1 year this risk is no different from conventional DMARDs. Recurrent infections in RA are common. In a prospective observational cohort study, the baseline annual rate of a first serious infection was 4.6%. Additionally, 14% of this cohort experienced a recurrent episode/year during their follow-up, with the highest risk being within the first year (29%), and with respiratory infections being the most common (44% of all episodes) (63). Factors that have shown to be predictive of infection include, age, functional status, specific comorbidities (chronic renal/lung disease), corticosteroid treatment, number of previous DMARD, treatment failures, previous serious infections, and current treatment with anti-TNF-α inhibitors or non-biological DMARDs (64). Nevertheless, recent data suggest that patients having a serious infection and exposed to biological treatment have a significantly lower risk of sepsis and fatal outcome than patients treated with conventional DMARDs (62, 65). British and French national biological registries have reported OI rates of 200–270/100,000 in patients using anti-TNF-α therapies (66, 67). In particular, there is evidence of an increased risk of M. tuberculosis, herpes zoster, and Listeria infections. The overall incidence of OI is not significantly different considering drug classes; however, the rate of PJP is significantly higher in those patients using rituximab in comparison to anti-TNF-α therapy. The absolute risk of PJP is low, although corticosteroid exposure is a strong predictor. Current data do not support PJP prophylaxis for all rituximab users. However, it may be appropriate in certain high-risk individuals. Furthermore, rituximab-associated neutropenia and impaired antibody response is also well-described.
Pre-clinical and clinical evidence indicate that anti-TNF-α therapy (infliximab, adalimumab, golimumab, certolizumab pegol, and etanercept) is associated with a 2- to 4-fold increase in the risk of active tuberculosis and other granulomatous conditions. Risk seems to be lower for etanercept (68). Risk also depends on local TB prevalence: in the year 2000, Spanish investigators reported an estimated TB incidence of 1,893/100,000 person-years in patients with RA treated with infliximab (69). This rate is ~10- to 20-fold higher than the observed rate in naïve patients. These rates have decreased dramatically since the establishment of latent tuberculosis infection (LTBI) screening prior to biological therapy (67, 70). It is essential to rule out LTBI in such individuals in order to reduce the risk of active TB reactivation. Interferon-gamma release assays (IGRAs) are useful tools for LTBI diagnosis. They are more specific than the tuberculin skin test (TST) because they do not show cross-reactivity with BCG-vaccination or NTM sensitization (71–73). Moreover, these in-vitro assays incorporate a mitogen control that can detect the presence of anergy, common in patients on immunosuppressive therapy (74). However, the clinical performance of IGRAs is still controversial due to the variety of concomitant immunosuppressive drug-regimens used at the time of LTBI screening, population heterogeneity, and the severity of the disease itself (75). Therefore, the clinical accuracy of IGRAs seems to be differentially affected depending on the specific type of immune disorder. Crohn's disease and/or its concomitant drug-profile (such as azathioprine or high-dose corticosteroids) could negatively affect the clinical performance of IGRAs when compared with other immune-mediated diseases, such as psoriasis or inflammatory rheumatic diseases (76). Thus, it seems prudent and convenient to perform dual LTBI testing with TST and IGRAs (77). Patients with RA and underlying structural lung diseases are at increased risk of developing NTM infection (78), mostly Mycobacterium avium. In some countries, NTM infections are more common than TB after anti-TNF-α treatment. However, there are still no established recommendations as regards screening and prophylaxis (79). A baseline chest x-ray should be recommended prior to starting therapy, and in patients with chronic unexplained cough, further work-up should include chest computed tomography scans and culture of respiratory specimens.
Immunization strategies are recommended for all cases, regardless of whether the patient has PID or is receiving immunosuppressive treatment, and it is of importance to be vaccinated according to the national immunization routine schedules. For patients with anti-TNF-α treatment, pneumococcal and age-appropriate anti-viral vaccinations (i.e., influenza) should be administered (68). Immunization before and after BRM is well-established as regards inactivated vaccines, and precautions should be taken for live vaccines (57). However, even if response to vaccines is impaired in patients with PID (80), it may have an effect in patients receiving some BRM. This may be partially explained by the concept of trained immunity-based vaccines (81).
In conclusion, RTIs belong to the most common causes of infections in humans worldwide. The genetic contribution to severe RTIs may have been masked by other interventions (82). The inborn errors of innate immunity show us that the absence of a measurable immunological defect does not exclude an immunodeficiency (41). Further functional genetic studies are necessary in order to fully validate the impact of host genetics during lung infections. The knowledge obtained from experience with the prescription of BRM may be particularly valuable, as the infections acquired as a side effect may help to identify genetic defects with a similar infectious phenotype. In the meantime, recommendations based on biological rationale and clinical experience are mandatory in order to prevent re-emerging severe infections.
Author Contributions
CP organized the structure and supervised the manuscript elaboration, revised literature and wrote a part of every chapter. AL revised literature, wrote the sections related to the immune response to infection and part of genetics, and edited the manuscript. IL, RV-H, and JD revised and wrote the aspects related to tuberculosis, and JD also supervised the manuscript elaboration. LM revised and wrote the section regarding the impact of biological response modifiers specially related to rheumatologic diseases. LM, AL, and RV-H prepared the figure. MM and CR revised aspects related to immunodeficiencies, and impact of BRM in children. IB revised host genetic factors. All authors revised and approved the final version of the manuscript.
Funding
This research was supported by two grants from the Instituto de Salud Carlos III (PI 16/01912, PI 17/01139, and PI18/00411), integrated in the Plan Nacional de I + D + I and funded jointly by the ISCIII Subdirección General de Evaluación and the Fondo Europeo de Desarrollo Regional (FEDER). JD is a researcher from the Miguel Servet programme. CP was awarded by programa Germans Trias Sapiens Fundació Catalunya la Pedrera.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Ferkol T, Schraufnagel D. The global burden of respiratory disease. Ann Am Thorac Soc. (2014) 11:404–6. doi: 10.1513/AnnalsATS.201311-405PS
2. Mizgerd JP. Lung infection–a public health priority. PLoS Med. (2006) 3:e76. doi: 10.1371/journal.pmed.0030076
3. Walker CL, Rudan I, Liu L, Nair H, Theodoratou E, Bhutta ZA, et al. Global burden of childhood pneumonia and diarrhoea. Lancet. (2013) 381:1405–16. doi: 10.1016/S0140-6736(13)60222-6
4. World Health Organization. Global Tuberculosis Report 2018 (WHO/CDS/TB/2018.20). Geneva: World Health Organization.
5. Rustad TR, Sherrid AM, Minch KJ, Sherman DR. Hypoxia: a window into Mycobacterium tuberculosis latency. Cell Microbiol. (2009) 11:1151–9. doi: 10.1111/j.1462-5822.2009.01325.x
6. Nair H, Simoes EA, Rudan I, Gessner BD, Azziz-Baumgartner E, Zhang JS, et al. Global and regional burden of hospital admissions for severe acute lower respiratory infections in young children in 2010: a systematic analysis. Lancet. (2013) 381:1380–90. doi: 10.1016/S0140-6736(12)61901-1
7. Regamey N, Kaiser L, Roiha HL, Deffernez C, Kuehni CE, Latzin P, et al. Viral etiology of acute respiratory infections with cough in infancy: a community-based birth cohort study. Pediatr Infect Dis J. (2008) 27:100–5. doi: 10.1097/INF.0b013e31815922c8
8. Blanken MO, Rovers MM, Molenaar JM, Winkler-Seinstra PL, Meijer A, Kimpen JL, et al. Respiratory syncytial virus and recurrent wheeze in healthy preterm infants. N Engl J Med. (2013) 368:1791–9. doi: 10.1056/NEJMoa1211917
9. Henderson J, Hilliard TN, Sherriff A, Stalker D, Al Shammari N, Thomas HM. Hospitalization for RSV bronchiolitis before 12 months of age and subsequent asthma, atopy and wheeze: a longitudinal birth cohort study. Pediatr Allergy Immunol. (2005) 16:386–92. doi: 10.1111/j.1399-3038.2005.00298.x
10. Gern JE. Viral respiratory infection and the link to asthma. Pediatr Infect Dis J. (2008) 27(10 Suppl.):S97–103. doi: 10.1097/INF.0b013e318168b718
11. Gould CM, Freeman AF, Olivier KN. Genetic causes of bronchiectasis. Clin Chest Med. (2012) 33:249–63. doi: 10.1016/j.ccm.2012.03.002
12. McMichael A. Environmental and social influences on infectious diseases. In: Baquero F, Nombela C, Cassell G, Gutiérrez-Fuentes J, editors. Evolutionary Biology of Bacterial and Fungal Pathogens. Washington: American Society for Microbiology Press (2008). p. 31–8.
13. Morens DM, Folkers GK, Fauci AS. The challenge of emerging and re-emerging infectious diseases. Nature. (2004) 430:242–9. doi: 10.1038/nature02759
14. Affandi JS, Price P, Waterer G. Can immunogenetics illuminate the diverse manifestations of respiratory infections? Ther Adv Respir Dis. (2010) 4:161–76. doi: 10.1177/1753465810371484
15. Lambert L, Sagfors AM, Openshaw PJ, Culley FJ. Immunity to RSV in early-Life. Front Immunol. (2014) 5:466. doi: 10.3389/fimmu.2014.00466
16. Ciancanelli MJ, Abel L, Zhang SY, Casanova JL. Host genetics of severe influenza: from mouse Mx1 to human IRF7. Curr Opin Immunol. (2016) 38:109–20. doi: 10.1016/j.coi.2015.12.002
17. Bastos HN, Osorio NS, Gagneux S, Comas I, Saraiva M. The Troika host-pathogen-extrinsic factors in tuberculosis: modulating inflammation and clinical outcomes. Front Immunol. (2017) 8:1948. doi: 10.3389/fimmu.2017.01948
18. Cookson W, Cox MJ, Moffatt MF. New opportunities for managing acute and chronic lung infections. Nat Rev Microbiol. (2018) 16:111–20. doi: 10.1038/nrmicro.2017.122
19. Sen R, Nayak L, De RK. A review on host-pathogen interactions: classification and prediction. Eur J Clin Microbiol Infect Dis. (2016) 35:1581–99. doi: 10.1007/s10096-016-2716-7
20. Cullen L, McClean S. Bacterial adaptation during chronic respiratory infections. Pathogens. (2015) 4:66–89. doi: 10.3390/pathogens4010066
21. Prat C, Lacoma A. Bacteria in the respiratory tract-how to treat? Or do not treat? Int J Infect Dis. (2016) 51:113–22. doi: 10.1016/j.ijid.2016.09.005
22. Budden KF, Gellatly SL, Wood DL, Cooper MA, Morrison M, Hugenholtz P, et al. Emerging pathogenic links between microbiota and the gut-lung axis. Nat Rev Microbiol. (2017) 15:55–63. doi: 10.1038/nrmicro.2016.142
23. Mizgerd JP. Acute lower respiratory tract infection. N Engl J Med. (2008) 358:716–27. doi: 10.1056/NEJMra074111
24. Hancock RE, Nijnik A, Philpott DJ. Modulating immunity as a therapy for bacterial infections. Nat Rev Microbiol. (2012) 10:243–54. doi: 10.1038/nrmicro2745
25. Franchi L, Munoz-Planillo R, Nunez G. Sensing and reacting to microbes through the inflammasomes. Nat Immunol. (2012) 13:325–32. doi: 10.1038/ni.2231
26. Andreakos E, Salagianni M, Galani IE, Koltsida O. Interferon-lambdas: front-line guardians of immunity and homeostasis in the respiratory tract. Front Immunol. (2017) 8:1232. doi: 10.3389/fimmu.2017.01232
27. Egli A, Mandal J, Schumann DM, Roth M, Thomas B, Lorne Tyrrell D, et al. IFNLambda3/4 locus polymorphisms and IFNLambda3 circulating levels are associated with COPD severity and outcomes. BMC Pulm Med. (2018) 18:51. doi: 10.1186/s12890-018-0616-6
28. Dagenais TR, Keller NP. Pathogenesis of Aspergillus fumigatus in invasive aspergillosis. Clin Microbiol Rev. (2009) 22:447–65. doi: 10.1128/CMR.00055-08
29. Camp JV, Jonsson CB. A role for neutrophils in viral respiratory disease. Front Immunol. (2017) 8:550. doi: 10.3389/fimmu.2017.00550
30. Martinez I, Oliveros JC, Cuesta I, de la Barrera J, Ausina V, Casals C, et al. Apoptosis, toll-like, RIG-I-like and NOD-like receptors are pathways jointly induced by diverse respiratory bacterial and viral pathogens. Front Microbiol. (2017) 8:276. doi: 10.3389/fmicb.2017.00276
31. Fischer A, Rausell A. What do primary immunodeficiencies tell us about the essentiality/redundancy of immune responses? Semin Immunol. (2018) 36:13–6. doi: 10.1016/j.smim.2017.12.001
32. O'Garra A, Redford PS, McNab FW, Bloom CI, Wilkinson RJ, Berry MP. The immune response in tuberculosis. Annu Rev Immunol. (2013) 31:475–527. doi: 10.1146/annurev-immunol-032712-095939
33. Brunot V, Pernin V, Chartier C, Garrigue V, Vetromile F, Szwarc I, et al. An epidemic of Pneumocystis jiroveci pneumonia in a renal transplantation center: role of T-cell lymphopenia. Transplant Proc. (2012) 44:2818–20. doi: 10.1016/j.transproceed.2012.09.089
34. Lambert L, Culley FJ. Innate immunity to respiratory infection in early life. Front Immunol. (2017) 8:1570. doi: 10.3389/fimmu.2017.01570
35. Casanova JL, Abel L. The human model: a genetic dissection of immunity to infection in natural conditions. Nat Rev Immunol. (2004) 4:55–66. doi: 10.1038/nri1264
36. Casanova JL, Abel L. Human genetics of infectious diseases: Unique insights into immunological redundancy. Semin Immunol. (2018) 36:1–12. doi: 10.1016/j.smim.2017.12.008
37. Yazdani R, Abolhassani H, Asgardoon M, Shaghaghi M, Modaresi M, Azizi G, et al. Infectious and noninfectious pulmonary complications in patients with primary immunodeficiency disorders. J Investig Allergol Clin Immunol. (2017) 27:213–24. doi: 10.18176/jiaci.0166
38. Cutting GR. Modifier genes in Mendelian disorders: the example of cystic fibrosis. Ann N Y Acad Sci. (2010) 1214:57–69. doi: 10.1111/j.1749-6632.2010.05879.x
39. Corvol H, Blackman SM, Boelle PY, Gallins PJ, Pace RG, Stonebraker JR, et al. Genome-wide association meta-analysis identifies five modifier loci of lung disease severity in cystic fibrosis. Nat Commun. (2015) 6:8382. doi: 10.1038/ncomms9382
40. Manolio TA, Collins FS, Cox NJ, Goldstein DB, Hindorff LA, Hunter DJ, et al. Finding the missing heritability of complex diseases. Nature. (2009) 461:747–53. doi: 10.1038/nature08494
41. Bucciol G, Moens L, Bosch B, Bossuyt X, Casanova JL, Puel A, et al. Lessons learned from the study of human inborn errors of innate immunity. J Allergy Clin Immunol. (2019) 143:507–27. doi: 10.1016/j.jaci.2018.07.013
42. Casanova JL. Severe infectious diseases of childhood as monogenic inborn errors of immunity. Proc Natl Acad Sci USA. (2015) 112:E7128–37. doi: 10.1073/pnas.1521651112
43. Picard C, Bobby Gaspar H, Al-Herz W, Bousfiha A, Casanova JL, Chatila T, et al. International Union of Immunological Societies: 2017 Primary Immunodeficiency Diseases Committee report on inborn errors of immunity. J Clin Immunol. (2018) 38:96–128. doi: 10.1007/s10875-017-0464-9
44. Fernandez-Ruiz M, Meije Y, Manuel O, Akan H, Carratala J, Aguado JM, et al. ESCMID Study Group for Infections in Compromised Hosts (ESGICH) Consensus document on the safety of targeted and biological therapies: an infectious diseases perspective (Introduction). Clin Microbiol Infect. (2018) 24 Suppl. 2:S2–9. doi: 10.1016/j.cmi.2018.01.029
45. van Tong H, Velavan TP, Thye T, Meyer CG. Human genetic factors in tuberculosis: an update. Trop Med Int Health. (2017) 22:1063–71. doi: 10.1111/tmi.12923
46. Amanatidou V, Apostolakis S, Spandidos DA. Genetic diversity of the host and severe respiratory syncytial virus-induced lower respiratory tract infection. Pediatr Infect Dis J. (2009) 28:135–40. doi: 10.1097/INF.0b013e31818c8d17
47. Janssen R, Bont L, Siezen CL, Hodemaekers HM, Ermers MJ, Doornbos G, et al. Genetic susceptibility to respiratory syncytial virus bronchiolitis is predominantly associated with innate immune genes. J Infect Dis. (2007) 196:826–34. doi: 10.1086/520886
48. Barreiro LB, Neyrolles O, Babb CL, Tailleux L, Quach H, McElreavey K, et al. Promoter variation in the DC-SIGN-encoding gene CD209 is associated with tuberculosis. PLoS Med. (2006) 3:e20. doi: 10.1371/journal.pmed.0030020
49. Bustamante J, Arias AA, Vogt G, Picard C, Galicia LB, Prando C, et al. Germline CYBB mutations that selectively affect macrophages in kindreds with X-linked predisposition to tuberculous mycobacterial disease. Nat Immunol. (2011) 12:213–21. doi: 10.1038/ni.1992
50. Schurr E. The contribution of host genetics to tuberculosis pathogenesis. Kekkaku. (2011) 86:17–28.
51. Boisson-Dupuis S, Ramirez-Alejo N, Li Z, Patin E, Rao G, Kerner G, et al. Tuberculosis and impaired IL-23-dependent IFN-gamma immunity in humans homozygous for a common TYK2 missense variant. Sci Immunol. (2018) 3:eaau8714. doi: 10.1126/sciimmunol.aau8714
52. Asgari S, Schlapbach LJ, Anchisi S, Hammer C, Bartha I, Junier T, et al. Severe viral respiratory infections in children with IFIH1 loss-of-function mutations. Proc Natl Acad Sci USA. (2017) 114:8342–7. doi: 10.1073/pnas.1704259114
53. Ciancanelli MJ, Huang SX, Luthra P, Garner H, Itan Y, Volpi S, et al. Life-threatening influenza and impaired interferon amplification in human IRF7 deficiency. Science. (2015) 348:448–53. doi: 10.1126/science.aaa1578
54. Kaplon H, Reichert JM. Antibodies to watch in 2018. MAbs. (2018) 10:183–203. doi: 10.1080/19420862.2018.1415671
55. Hansel TT, Kropshofer H, Singer T, Mitchell JA, George AJ. The safety and side effects of monoclonal antibodies. Nat Rev Drug Discov. (2010) 9:325–38. doi: 10.1038/nrd3003
56. Davis BP, Ballas ZK. Biologic response modifiers: Indications, implications, and insights. J Allergy Clin Immunol. (2017) 139:1445–56. doi: 10.1016/j.jaci.2017.02.013
57. Davies HD, Committee On Infectious D. Infectious complications with the use of biologic response modifiers in infants and children. Pediatrics. (2016) 138:e2016120. doi: 10.1542/peds.2016-1209
58. Fisher MC, Greenberg JD. Assessing infection risk with biologic agents in RA: methodological challenges. Nat Rev Rheumatol. (2009) 5:288–91. doi: 10.1038/nrrheum.2009.51
59. Woodrick RS, Ruderman EM. Safety of biologic therapy in rheumatoid arthritis. Nat Rev Rheumatol. (2011) 7:639–52. doi: 10.1038/nrrheum.2011.145
60. Minozzi S, Bonovas S, Lytras T, Pecoraro V, Gonzalez-Lorenzo M, Bastiampillai AJ, et al. Risk of infections using anti-TNF agents in rheumatoid arthritis, psoriatic arthritis, and ankylosing spondylitis: a systematic review and meta-analysis. Expert Opin Drug Saf. (2016) 15(sup1):11–34. doi: 10.1080/14740338.2016.1240783
61. Singh JA, Cameron C, Noorbaloochi S, Cullis T, Tucker M, Christensen R, et al. Risk of serious infection in biological treatment of patients with rheumatoid arthritis: a systematic review and meta-analysis. Lancet. (2015) 386:258–65. doi: 10.1016/S0140-6736(14)61704-9
62. Galloway JB, Hyrich KL, Mercer LK, Dixon WG, Fu B, Ustianowski AP, et al. Anti-TNF therapy is associated with an increased risk of serious infections in patients with rheumatoid arthritis especially in the first 6 months of treatment: updated results from the British Society for Rheumatology Biologics Register with special emphasis on risks in the elderly. Rheumatology (Oxford). (2011) 50:124–31. doi: 10.1093/rheumatology/keq242
63. Subesinghe S, Rutherford AI, Byng-Maddick R, Leanne Hyrich K, Benjamin Galloway J. Recurrent serious infections in patients with rheumatoid arthritis-results from the British Society for Rheumatology Biologics Register. Rheumatology (Oxford). (2018) 57:651–5. doi: 10.1093/rheumatology/kex469
64. Zink A, Manger B, Kaufmann J, Eisterhues C, Krause A, Listing J, et al. Evaluation of the RABBIT Risk Score for serious infections. Ann Rheum Dis. (2014) 73:1673–6. doi: 10.1136/annrheumdis-2013-203341
65. Richter A, Listing J, Schneider M, Klopsch T, Kapelle A, Kaufmann J, et al. Impact of treatment with biologic DMARDs on the risk of sepsis or mortality after serious infection in patients with rheumatoid arthritis. Ann Rheum Dis. (2016) 75:1667–73. doi: 10.1136/annrheumdis-2015-207838
66. Mariette X, Gottenberg JE, Ravaud P, Combe B. Registries in rheumatoid arthritis and autoimmune diseases: data from the French registries. Rheumatology (Oxford). (2011) 50:222–9. doi: 10.1093/rheumatology/keq368
67. Rutherford AI, Patarata E, Subesinghe S, Hyrich KL, Galloway JB. Opportunistic infections in rheumatoid arthritis patients exposed to biologic therapy: results from the British Society for Rheumatology Biologics Register for Rheumatoid Arthritis. Rheumatology (Oxford). (2018) 57:997–1001. doi: 10.1093/rheumatology/key023
68. Baddley JW, Cantini F, Goletti D, Gomez-Reino JJ, Mylonakis E, San-Juan R, et al. ESCMID Study Group for Infections in Compromised Hosts (ESGICH) Consensus Document on the safety of targeted and biological therapies: an infectious diseases perspective (Soluble immune effector molecules [I]: anti-tumor necrosis factor-alpha agents). Clin Microbiol Infect. (2018) 24 Suppl. 2:S10–20. doi: 10.1016/j.cmi.2017.12.025
69. Gomez-Reino JJ, Carmona L, Valverde VR, Mola EM, Montero MD, Group B. Treatment of rheumatoid arthritis with tumor necrosis factor inhibitors may predispose to significant increase in tuberculosis risk: a multicenter active-surveillance report. Arthritis Rheum. (2003) 48:2122–7. doi: 10.1002/art.11137
70. Carmona L, Gomez-Reino JJ, Rodriguez-Valverde V, Montero D, Pascual-Gomez E, Mola EM, et al. Effectiveness of recommendations to prevent reactivation of latent tuberculosis infection in patients treated with tumor necrosis factor antagonists. Arthritis Rheum. (2005) 52:1766–72. doi: 10.1002/art.21043
71. Dominguez J, Ruiz-Manzano J, De Souza-Galvao M, Latorre I, Mila C, Blanco S, et al. Comparison of two commercially available gamma interferon blood tests for immunodiagnosis of tuberculosis. Clin Vaccine Immunol. (2008) 15:168–71. doi: 10.1128/CVI.00364-07
72. Ferrara G, Losi M, D'Amico R, Roversi P, Piro R, Meacci M, et al. Use in routine clinical practice of two commercial blood tests for diagnosis of infection with Mycobacterium tuberculosis: a prospective study. Lancet. (2006) 367:1328–34. doi: 10.1016/S0140-6736(06)68579-6
73. Latorre I, De Souza-Galvao M, Ruiz-Manzano J, Lacoma A, Prat C, Altet N, et al. Evaluating the non-tuberculous mycobacteria effect in the tuberculosis infection diagnosis. Eur Respir J. (2010) 35:338–42. doi: 10.1183/09031936.00196608
74. Dominguez J, Latorre I. Role of the T-cell interferon-gamma release assays in preventing reactivation of latent tuberculosis infection in immunosuppressed patients in treatment with anti-TNF agents. J Crohns Colitis. (2008) 2:250–4. doi: 10.1016/j.crohns.2008.05.007
75. Sester M, van Leth F, Bruchfeld J, Bumbacea D, Cirillo DM, Dilektasli AG, et al. Risk assessment of tuberculosis in immunocompromised patients. A TBNET study. Am J Respir Crit Care Med. (2014) 190:1168–76. doi: 10.1164/rccm.201405-0967OC
76. Latorre I, Minguez S, Carrascosa JM, Naves J, Villar-Hernandez R, Muriel B, et al. Immune-mediated inflammatory diseases differently affect IGRAs' accuracy for latent tuberculosis infection diagnosis in clinical practice. PLoS One. (2017) 12:e0189202. doi: 10.1371/journal.pone.0189202
77. Santin M, Garcia-Garcia JM, Rigau D, Altet N, Anibarro L, Casas I, et al. Executive summary of the guidelines for the use of interferon-gamma release assays in the diagnosis of tuberculosis infection. Enferm Infecc Microbiol Clin. (2016) 34:304–8. doi: 10.1016/j.eimc.2015.11.021
78. Winthrop KL, Chang E, Yamashita S, Iademarco MF, LoBue PA. Nontuberculous mycobacteria infections and anti-tumor necrosis factor-alpha therapy. Emerg Infect Dis. (2009) 15:1556–61. doi: 10.3201/eid1510.090310
79. Winthrop KL, Iseman M. Bedfellows: mycobacteria and rheumatoid arthritis in the era of biologic therapy. Nat Rev Rheumatol. (2013) 9:524–31. doi: 10.1038/nrrheum.2013.82
80. Mieves JF, Wittke K, Freitag H, Volk HD, Scheibenbogen C, Hanitsch LG. Influenza vaccination in patients with common variable immunodeficiency (CVID). Curr Allergy Asthma Rep. (2017) 17:78. doi: 10.1007/s11882-017-0749-3
81. Sanchez-Ramon S, Conejero L, Netea MG, Sancho D, Palomares O, Subiza JL. Trained immunity-based vaccines: a new paradigm for the development of broad-spectrum anti-infectious formulations. Front Immunol. (2018) 9:2936. doi: 10.3389/fimmu.2018.02936
Keywords: immunogenetics, biological response modifiers, respiratory tract infections, primary immunodeficiencies, inborn errors
Citation: Lacoma A, Mateo L, Blanco I, Méndez MJ, Rodrigo C, Latorre I, Villar-Hernandez R, Domínguez J and Prat C (2019) Impact of Host Genetics and Biological Response Modifiers on Respiratory Tract Infections. Front. Immunol. 10:1013. doi: 10.3389/fimmu.2019.01013
Received: 16 July 2018; Accepted: 23 April 2019;
Published: 07 May 2019.
Edited by:
Ian Marriott, University of North Carolina at Charlotte, United StatesReviewed by:
András N. Spaan, University Medical Center Utrecht, NetherlandsJesús Gonzalo-Asensio, University of Zaragoza, Spain
Copyright © 2019 Lacoma, Mateo, Blanco, Méndez, Rodrigo, Latorre, Villar-Hernandez, Domínguez and Prat. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Cristina Prat, Y3ByYXQuZ2VybWFuc3RyaWFzQGdlbmNhdC5jYXQ=
†These authors are co-senior authors of this study