 Dong Ho Park
Dong Ho Park Kip M. Connor
Kip M. Connor John D. Lambris
John D. Lambris- 1Department of Ophthalmology, School of Medicine, Kyungpook National University, Kyungpook National University Hospital, Daegu, South Korea
- 2Angiogenesis Laboratory, Department of Ophthalmology, Massachusetts Eye & Ear Infirmary, Boston, MA, United States
- 3Department of Ophthalmology, Harvard Medical School, Boston, MA, United States
- 4Department of Pathology and Laboratory Medicine, Perelman School of Medicine, University of Pennsylvania, Stellar Chance Laboratories, Philadelphia, PA, United States
Ocular inflammation is a defining feature of sight threating diseases and its dysregulation can catalyze and or propagate ocular neurodegenerative maladies such as age-related macular degeneration (AMD). The complement system, an intrinsic component of the innate immunity, has an integral role in maintaining immune-surveillance and homeostasis in the ocular microenvironment; however, overstimulation can drive ocular inflammatory diseases. The mechanism for complement disease propagation in AMD is not fully understood, although there is accumulating evidence showing that targeted modulation of complement-specific proteins has the potential to become a viable therapeutic approach. To date, a major focus of complement therapeutics has been on targeting the alternative complement system in AMD. Recent studies have outlined potential complement cascade inhibitors that might mitigate AMD disease progression. First-in-class complement inhibitors target the modulation of complement proteins C3, C5, factor B, factor D, and properdin. Herein, we will summarize ocular inflammation in the context of AMD disease progression, current clinical outcomes and complications of complement-mediated therapeutics. Given the need for additional therapeutic approaches for ocular inflammatory diseases, targeted complement modulation has emerged as a leading candidate for eliminating inflammation-driven ocular maladies.
Age-related macular degeneration (AMD) is the leading cause of blindness of the elderly in the Western world (1). Its prevalence is expected to rise as aging populations rise and ~288 million people will be affected by AMD by 2040 (2). The buildup of debris within retinal pigment epithelial (RPE) cells and under the RPE layer is considered a hallmark of AMD development and progression (3). Photoreceptors require a large membrane surface area for phototransduction, and have a large outer segment that must be continuously replaced. The RPE are specialized cells that phagocytize the shed outer segments of photoreceptors, and are the most active phagocytizing cells of the body (4). As aging progresses, it is thought that the machinery of these cells deteriorates, along with their capacity to degrade and recycle photoreceptor metabolic waste. During the lifetime of a person, the choroidal capillary area becomes thinner, and Bruch's membrane may also accumulate lipoprotein material from the choroidal capillaries, which adds to the incompletely digested material deposited by RPE cells. Collectively, this accumulating material is referred to as drusen, which is a major pathological feature of AMD.
Drusen size can predict the development of AMD. Accordingly, a new clinical classification scheme for AMD using drusen size and presence of pigmentary abnormalities has been introduced by the Beckman Initiative for Macular Research Classification Committee (5). In this scheme, individuals over 55 years of age with small (< 63 μm) drusen were considered to have normal aging. Early AMD is characterized by the presence of medium-sized drusen (≥63 and < 125 μm) with no pigmentary abnormalities, and is accompanied by a mild loss of vision. Intermediate AMD is characterized by larger drusen deposits (≥125 μm) and/or presence of pigmentary abnormalities, and is associated with a moderate loss of vision. Late/Advanced AMD is characterized by the presence of any choroidal neovascularization (CNV), known as neovascular AMD, or gross pigment abnormalities and cellular degeneration such as geographic atrophy (GA). Collectively, advanced AMD causes severe loss of vision and can lead to blindness. The incidence of late-stage AMD increases exponentially with age, with the two forms of late AMD, neovascular AMD and GA, occurring with roughly equal prevalence (2, 6). An estimated 1.22 million and 973,000 people in the United States have neovascular AMD and GA in at least one eye, respectively (7). As the population ages, the prevalence of late AMD is expected to rise from 9.6 million to 11.3 million in 2020 and 18.6 million in 2040 (2).
Currently, there is no effective prevention, individual risk estimation, or reliable prognostic evaluation available for the clinical management of AMD. Further, there are no therapeutics approved for the treatment of GA. The development of AMD depends on a complex interplay of risk factors such as age, genetics (8), and behavior including; smoking (9), diet (10), and sunlight exposure (11). Genetic variations in genes involved in the complement system, as well as others, are associated with risk for developing AMD, or risk of progression from early to late AMD (12). Overall, these findings suggest that AMD is a progressive neurodegenerative disease involving inflammation (13), and in particular an inflammatory immune response (14).
The Complement System is a Vital Component of Innate Immunity
The immune system is divided into two distinct types, the innate and adaptive systems. The complement system, as part of the innate immune system, plays an integral role in maintaining immune-surveillance and homeostasis in the ocular microenvironment (15, 16). The complement system consists of three systems classical, lectin, and alternative (17). The classical system is mediated by the binding of the complement component C1q of C1 protein to antigen–antibody complexes. The lectin system is activated by mannose-binding lectin recognition of the polysaccharide or glycoprotein motifs on the cell surface of damaged host and non-host cells (17, 18). Lastly, the alternative complement system is constitutively active through the spontaneous cleavage of an internal C3 thioester bond (17, 19) (Figure 1). The spontaneous hydrolysis of this internal thioester bond within the complement protein C3 forms C3(H2O), and while not cleaved, C3(H2O) can function in the same manner as C3b in a C3 convertase. This “tick-over” of C3 to C3 (H2O) enables it to complex with complement factor B, following by its cleavage by complement factor D, a serine protease, into Ba and Bb fragments. The resulting C3(H2O)Bb complex is a C3 convertase, which can efficiently cleave C3 into C3a and C3b. Factor B enables further downstream activation of the alternative system through its binding to C3b (17–21). These components form the C3 convertase enzyme, C3bBb, promoting the cleavage of C3 and creating a positive feedback loop (19, 21). Subsequently, C5 convertase is created by the combination of the C3 convertase with an additional C3b molecule. The C5 convertase then cleaves C5 into C5a and C5b (17–19). C5b recruits C6, − 7, −8, and−9, forming the membrane attack complex (MAC), which forms a pore in the cell membrane causing cell lysis and death, which can be down-regulated by complement inhibitors such as CD59 (22, 23). However, according to a previous immunohistochemical study from human donor eyes with drusen, the antibody against C5b-9 indicating MAC showed a restricted pattern, while the C3 or C5 antibody showed a diffuse pattern throughout the drusen and choroid (24). Thus, it is unclear if MAC formation is linked to AMD disease progression. It may be complement's upstream opsonization functions that are perturbed in this pathology.
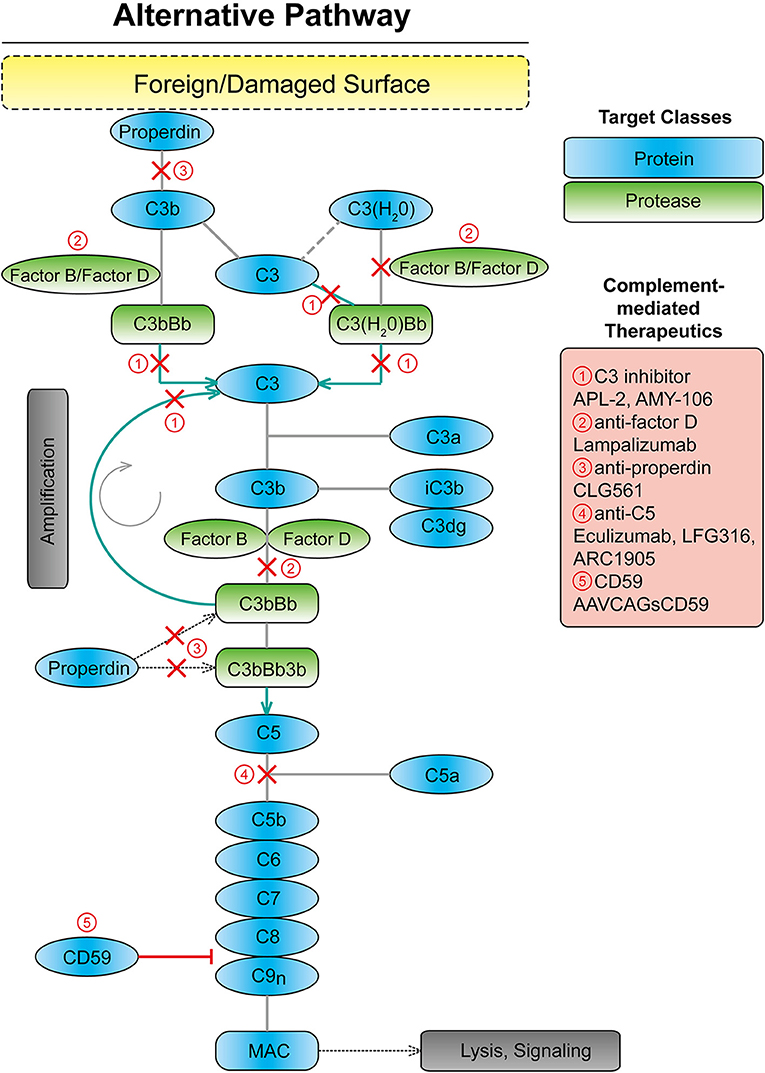
Figure 1. Scheme showing the alternative complement system with targets for complement mediated therapeutics. MAC, membrane attack complex.
Alternative Complement Cascade Dysfunction in Age-Related Macular Degeneration
Complement Factor H and Related Proteins
Over a lifetime, the photoreceptors and RPE cells of the retina encounter a number of innate immune activators, such that tight regulation of immunity is necessary to prevent deleterious inflammatory events. Dysregulation of the complement system may potentially drive ocular inflammation, which contributes to vision loss in AMD. Complement byproducts such as complement factor H (FH) in the drusen were the first indication that the complement system is involved in AMD progression (25). In 2005, genome-wide association studies (GWAS) suggested that the complement cascade was involved in AMD disease progression. Table 1 summarizes the genes for the alternative complement pathway that are known to be involved in AMD. Early GWAS and targeted sequencing studies identified a common single nucleotide polymorphism (SNP) in the CFH gene (rs1061170) that is associated with an increased risk of developing AMD (26–29). This SNP replaces a tyrosine residue with a histidine residue at position 402 (Y402H) (49). Heterozygous individuals and homozygotes for the Y402H polymorphism have a 2.3 and 5.2-fold increased risk of developing AMD, respectively (30). Approximately 30% of individuals of European descents are carriers of at least one of the Y402H risk alleles (30).
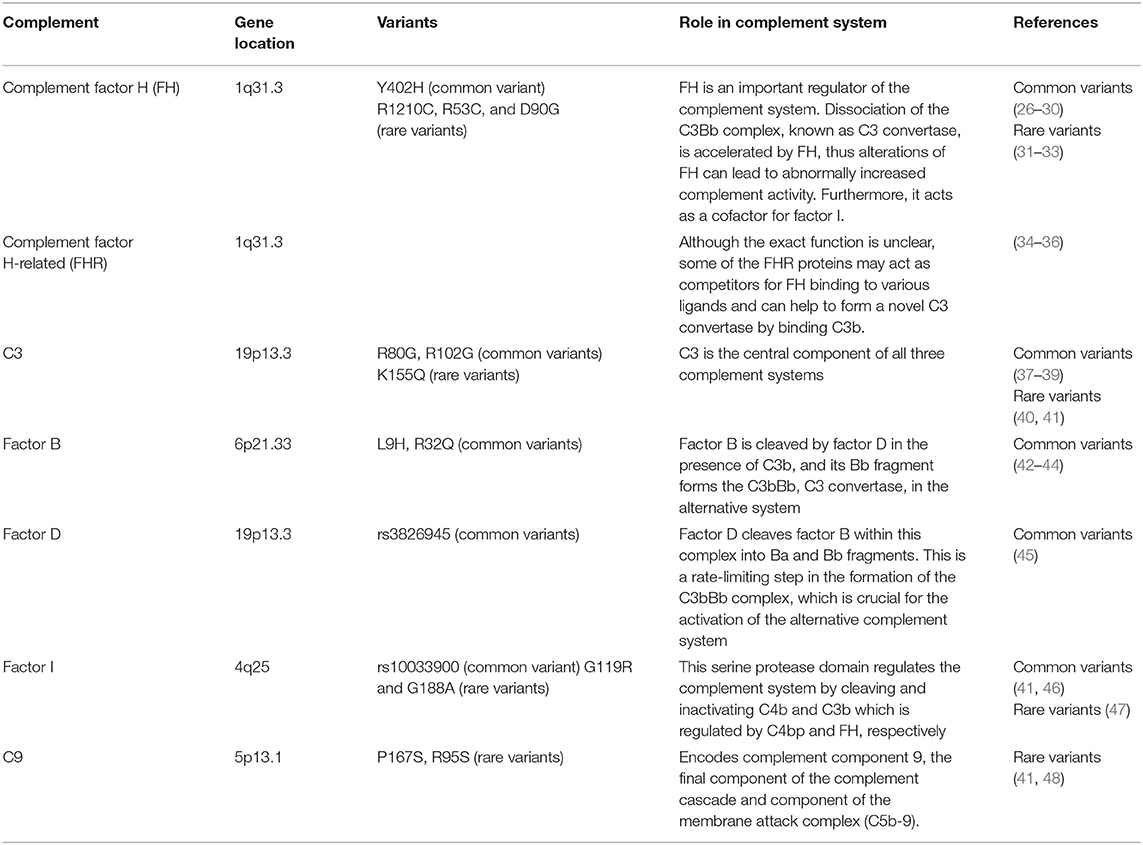
Table 1. Genes in the alternative complement pathway that are involved in age-related macular degeneration.
FH is an important regulator of the complement system (50). Increasingly, it has been found that complement activation is linked with AMD associated with genetic variants on chromosome 1 locus (1q32) that contains the CFH and CHFR genes, rather than chromosome 10 (10q31) around the ARMS2/HTRA1 genes (51). AMD patients with chromosome 1 mutations have significant complement over-activation in the extracellular matrix of the choriocapillaris, underlying Bruch's membrane. Complement activation and turnover have been identified in the choriocapillaris layer as well, and increased levels are found in individuals with AMD (52, 53). This is also found in donor eyes from individuals genetically at risk for AMD, but who do not yet have clinical manifestations of the disease (54). The activation of the alternative complement pathway is initiated by the formation of the C3bBb complex, also known as C3 convertase. Formation of this complex leads to the amplification of complement signaling and immune response. FH accelerates dissociation of this C3bBb complex, inhibiting alternative complement system activation. Furthermore, FH, as a cofactor, facilitates factor I-mediated C3b inactivation. FH is anchored to the extracellular matrix and the cell surface through interactions with glycosaminoglycans (GAGs) (55). The Y402H polymorphism does not alter the overall protein structure (56). However, the Y402H polymorphism disrupts binding of the complement control region to GAG chains in the Bruch's membrane (55). Because the Y402H variant has decreased binding affinity to numerous components of the damaged retina (57–59), the inhibitory effect of FH on the complement system is thought to be decreased. This could result in poorly controlled complement turnover and excessive chronic local inflammation. The FH and factor H-like protein (FHL-1), components of the alternative system, are capable of suppressing complement activation on the extracellular matrix (51). The shortened splice variant of FH and FHL-1 appears to prevail in the ECM in/around Bruch's membrane (60–63). Because FHL-1 only has the one GAG-mediated anchoring site in its CCP7 domain which anchors FHL-1 to Bruch's membrane and the intercapillary septa, this GAG-binding site is affected by the Y402H polymorphism. In contrast, FH has two anchoring sites and is not particularly affected by the Y402H polymorphism (51). This may explain why the Y402H polymorphism has a disproportionate effect on protein anchoring via GAGs.
Other rare variants in CFH SNPs have also been reported to affect AMD, including the R1210C, R53C and D90G polymorphisms (31, 32). R1210C is extremely rare, with a minor allele frequency of 0.0173 %, but has an even stronger association with AMD than Y402H, potentially by acting as a functionally null allele (31). The highly penetrant R1210C variant is associated with a 6-year earlier onset of AMD with drusen phenotype (33). Advanced age and decreased FH induced sub-RPE deposit formation leading to complement activation, which contributed to RPE damage and visual function impairment (64).
A number of other SNPs further downstream on chromosome 1 are associated with AMD, suggesting involvement of the five factor H-related (FHR) proteins in disease pathogenesis (34). Although the exact function of FHR proteins is unclear at present, some of them may bind competitively to ligands involved in FH binding and may facilitate formation of a novel C3 convertase by binding C3b (65). Furthermore, a haplotype of complete deletion of the genes for FHR-1 and FHR-3 was associated with a decreased risk of AMD in human studies (34–36).
C3
C3 is the central component of all three complement systems, and functional changes in C3 directly affect the downstream cascade (66). A common SNP, rs2230199 (R80G), is associated with increased risk of AMD (odds ratio 2.6) (37, 38). The R102G polymorphism of C3 results in reduced FH binding to the 102G variant. As FH acts as a cofactor for factor I-mediated cleavage of C3b, R102G polymorphism of C3 decreased the factor I-mediated cofactor activity. By extending the convertase lifetime, activation of the alternative complement system is enhanced (39). A single rare variant in C3, K155Q, is positively associated with AMD risk (odds ratio 3.8) (41). K155 in C3 is in close proximity to the FH binding site (40). In surface plasmon resonance experiments, binding of this C3 mutant to FH was reduced compared to wild type C3. The K155Q allele in C3 results in resistance to proteolytic inactivation by FH and CFI. Taken together, these findings implicate that loss of C3 functionality contributes to AMD pathogenesis (41). Increased plasma complement protein is also associated with advanced stages of AMD, further supporting the hypothesis that complement activation may be correlated with disease progression as well (67, 68). C3a was significantly increased in AMD patients, ranging from 4.6 to 87.2 % (69). Furthermore, C3a and C5a are markers of acute activation of the alternative complement system. Thus, the plasma concentrations of activation peptides including C3a and C5a are positively correlated biomarkers of AMD (69).
Factor B
Both haplotypes, L9H variant of factor B and the E318D variant in C2, as well as a variant in intron 10 of C2 and the R32Q variant of factor B, are considered highly protective against AMD development (odds ratio 0.45 and 0.36, respectively), although the protection is likely mediated by factor B mutations (42, 43). Factor B fragments are found in the drusen at similar levels to that of factor H, and convertase formation, which strengthens complement activation, is decreased by the R32Q mutation (44). Furthermore, studies in mouse models of complement dysregulation support a causative role for the complement cascade in multiple retinal pathologies. Factor B is up-regulated in the retinal detachment mouse model, and in the vitreous from patients with retinal detachment (70). In addition, factor B knockout mice are protected from retinal detachment associated photoreceptor death which further emphasizes a role for this system in photoreceptor degeneration.
Factor D
Factor D is one of the serine proteases that regulates the activation of the alternative complement system (71). Factor D becomes transiently active and is able to cleave factor B within this complex into Ba and Bb fragments. This is a rate limiting step in the formation of C3bBb which strengthens the signal and consequently activates the complement system (21, 71). A small case-control series showed that factor D gene SNP rs3826945 is positively associated with AMD risk (odds ratio 1.44) (45).
Factor I
The factor I gene spans 63 kb and contains 13 exons, the first 8 of which encode the heavy chain and the last 5 the light chain, which contains a serine protease domain (72). The serine protease domain is responsible for cleaving and inactivating C4b and C3b (73). Factor I-mediated C3b inactivation is assisted by FH. The FH acts as a cofactor for factor I-mediated cleavage of C3b (74), and thus accelerating breakdown of the alternative system C3 convertase, C3bBb. A common polymorphism near the factor I gene, rs10033900, has been described (46). A cohort study of 2,493 advanced AMD cases and controls showed that 7.8% of AMD cases compared to 2.3% of controls are carriers of rare missense factor I variants (odds ratio 3.6) (41). Another cohort study reported that two variants of factor I, G119R and G188A remained significant (47). The rare factor I G119R variant conferred a particularly high risk of AMD (odds ratio 22.2). Functional studies on the G119R variant showed that the secretion of the mutant protein was lower than that of the wild type protein, which led to a decrease in factor I-mediated cleavage of C3b.
Complement 9
Complement component 9 (C9) is the most downstream component of the terminal complement complex cascade, acting as the terminal effector molecule of all three complement activation systems. Because the ultrastructure terminal complement complex is partially dependent on the amount of C9 molecules relative to C5b-8 molecules, a significant reduction in C9 can alter the terminal complement complex stoichiometry, reducing cytolytic activity (75, 76). Genotyping in 5,115 independent samples confirmed associations to AMD with a P167S allele in C9 (41). When compared to the controls, a two-fold increase was observed for this variant in the AMD cases. Another R95X variant was negatively associated with AMD risk in a small study (48), but has not been confirmed. Together, these findings suggest that the terminal complement pathway has a potential role in AMD pathogenesis.
Therapeutic Approaches and Clinical Trials Using Complement-Mediated Drugs
In addition to strong genetic and basic research studies supporting a pathological role for the complement system in AMD, many clinical trials for therapeutics targeting alternative complement cascade are at various stages or completed (Table 2). Clinical trials for eight drugs that target the alternative complement cascade include two targeting C3, one targeting factor D, one targeting properdin, three targeting C5, and one targeting CD59.
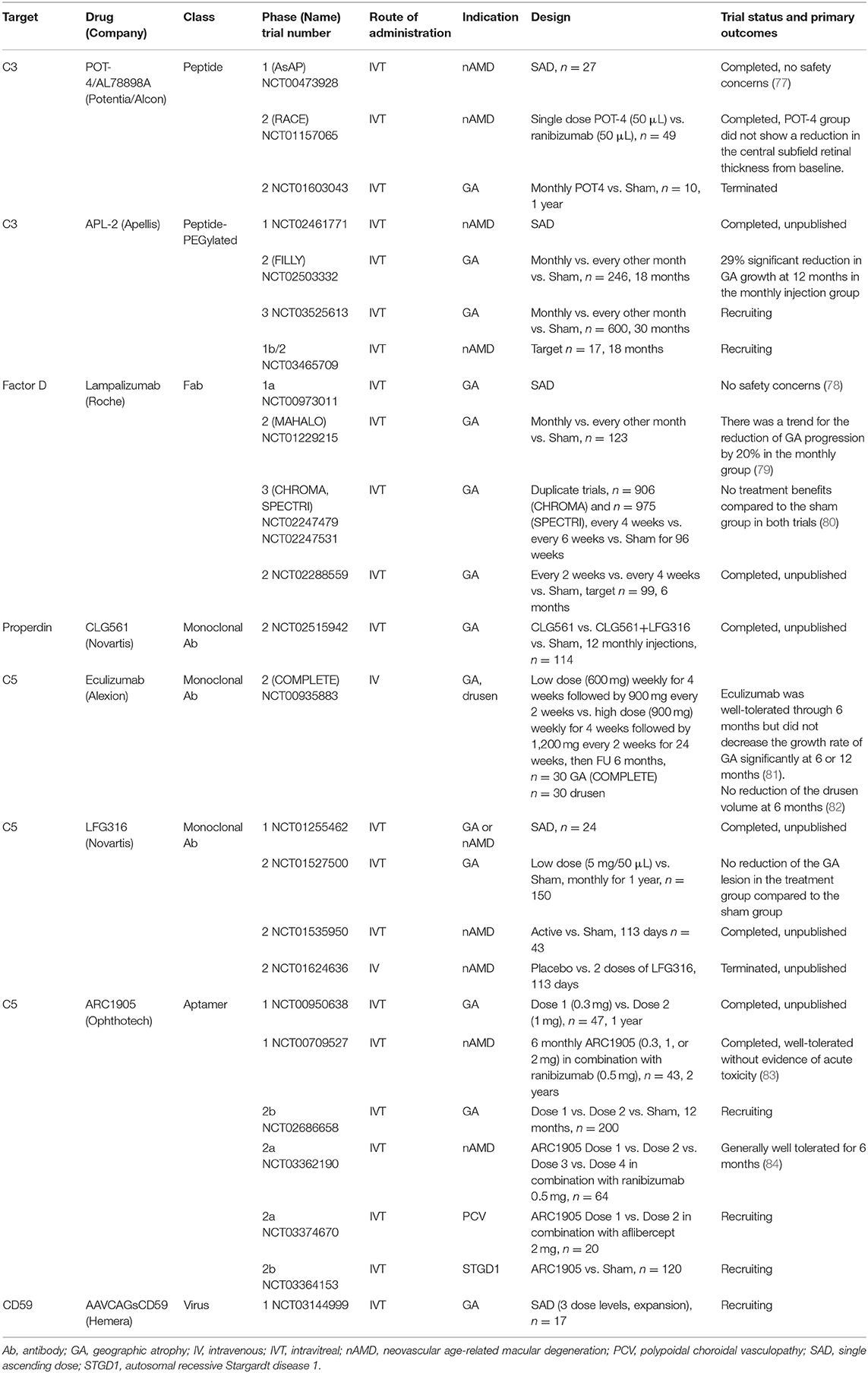
Table 2. Complement-mediated therapeutics in clinical trials for the treatment of age-related macular degeneration.
C3-Mediated Therapeutics
Inhibiting the C3 protein, including the C3 convertase, may be a desirable therapeutic approach. C3 modulates amplification of all initiation pathways, the generation of anaphylatoxins (C3a, C5a), and the membrane attack complex (MAC), while C3 inhibition attenuates these events. C3 functions as a central hub that mediates and controls the upstream activation and downstream effector functions of the complement cascade. Initial attempts in developing small molecules that inhibit conversion of C3 failed due to lack of potency and specificity (85).
In contrast, the peptidic inhibitor compstatin exerts its function via direct binding to native C3. Compstatin was discovered by screening phage-display libraries searching for C3b-binding peptides (86). Compstatin is a 13 amino acid cyclic peptide that inhibits complement activation by binding C3 and interfering with convertase formation and C3 cleavage (and the subsequent C3b opsonization, amplification, and generation of effectors) (85). Thus, compstatin could be a potent inhibitor, blocking all three complement systems (87).
POT-4 (Potentia Pharmaceuticals, Crestwood, KY), a compstatin derivative, is the first complement-specific drug that has entered clinical trials for the indication of AMD. A phase 1 clinical trial (AsAP, NCT00473928) using intravitreal POT-4 delivery was conducted in patients with neovascular AMD, and suggested that there were no safety concerns (77). Following that, a double-masked, randomized, multicenter phase 2 clinical trial for patients with neovascular AMD (RACE, NCT01157065) was performed. Forty-nine patients were randomized to receive either single POT-4 (50 μL) or single ranibizumab (Lucentis®, Roche AG, Basel, Switzerland) (50 μL) injections, and were followed up for 12 weeks. The primary outcome was a mean reduction from baseline in the central subfield retinal thickness at week 4. Compared to the ranibizumab group (199.9 μm), the POT-4 group showed no benefit (-12.1 μm) in the reduction of macular edema (88). For the indication of GA, a phase 2 clinical trial with 10 patients (NCT01603043) was terminated before completion, and product deposits was reported in four cases among seven patients (57.4%).
The observation of product precipitation, led to an attempt to increase its solubility by pegylation of POT-4 with a 40,000 Da PEG (termed APL-2, Apellis Pharmaceuticals Inc., Crestwood, KY); APL-2 is currently under clinical trials for patients with GA and neovascular AMD. A phase 2 clinical trial (FILLY, NCT02503332) evaluated the efficacy of APL-2 by comparing three groups: a monthly intravitreal APL-2 injection (15 mg/0.1 mL) group, every other month APL-2 injection (15 mg/0.1 mL) group, and the sham group. The monthly intravitreal APL-2 injection group (n = 84) showed a 29% significant reduction in GA growth at 12 months compared to the sham group (n = 80) (89). The every other month APL-2 injection group (n = 78) showed a 20% reduction in GA growth, though this decrease was not statistically significant. Best-corrected visual acuity (BCVA) did not differ among the three groups. Of concern was that individuals receiving APL-2 had an 8 and 18% conversion rate to neovascular AMD in the every other month and monthly injection groups, respectively (90). Conversion to neovascular AMD appeared to be dose dependent based on monthly and bi-monthly injections. A possible explanation is that PEG can induce CNV in a mouse model by upregulating complement proteins in the RPE-choroid tissue or that a complement inhibition-mediated shift of pro-inflammatory M1 to pro-angiogenic M2 macrophages as part of the repair process, may lead to small exudations (91, 92). Endophthalmitis occurred in two out of 86 patients (1.3%) and in one out of 79 patients (2.3%) in the every other month and monthly injection groups, respectively (93). At present, a new 30-month, Phase 3, multicenter clinical trial for GA is recruiting subjects (NCT03525613) (94). Patients will be randomized to monthly and every other month injections for both APL-2 (15 mg/0.1 mL) and sham groups. Additionally, a phase 2 clinical trial for patients with neovascular AMD has been initiated, and is currently recruiting subjects (NCT03465709) (95).
In the meantime, a novel non-pegylated C3 inhibitor is under development (termed AMY-106, Amyndas Pharmaceuticals) and has shown prolonged residence in ocular tissues at C3-saturating levels, extending over 3 months after a single intravitreal injection in cynomolgus monkeys (96). The increased bioavailability of this C3 inhibitor, indicates its clinical potential for ocular indications associated with C3 convertase dysregulation (e.g., AMD).
Factor D-Mediated Therapeutics
FCFD4514S (Lampalizumab)
As a rate limiting enzyme, factor D cleaves factor B to generate C3 convertase and is a pivotal activator of the alternative complement system (21, 97). FCFD4514S (lampalizumab, Roche AG, Basel, Switzerland), a Fab fragment of humanized IgG murine anti-factor D antibody, was developed to block factor D-mediated formation of the C3 convertase (98). A phase 1 trial (NCT00973011) of intravitreal lampalizumab injection in patients with GA found that this modality is safe and well-tolerated, and there were no adverse events (78). This was followed by the phase 2 MAHALO study. “The phase 2 clinical trial (MAHALO, NCT01229215) was a multicenter, randomized, controlled study that evaluated intravitreal delivery of lampalizumab (10 mg/100 μL) administered monthly (n = 42) and every other month (n = 41) vs. sham control (n = 40) in patients with GA. The primary endpoint was the mean change in the lesion area from baseline to month 18 as measured by fundus autofluorescence (79).” The results showed a trend for a reduction in GA progression of 20% in the lampalizumab group, but there was no significant difference between the lampalizumab and sham groups (79). Interestingly, in a subgroup of factor I risk-allele carriers (57% of the patients), monthly treatment showed a 44% reduction in GA area progression compared to the sham control (95% CI, 15 to 73%). “The MAHALO study demonstrated an acceptable safety profile during the 18-month treatment period. The MAHALO study concluded that modulation of the complement system can affect the progression of GA, which is supported by human genetic studies for which various genetic variants in the alternative complement system increase the AMD risk (79).”
However, the subsequent following phase 3 trials showed conflicting results. Two similarly designed phase 3 double-masked, randomized, sham-controlled clinical trials (CHROMA, NCT02247479 and SPECTRI, NCT02247531) enrolled participants at 275 sites in 23 countries (99, 100). “Participants were aged 50 years or older, with bilateral GA and no prior or active CNV in either eye. Participants were randomized 2:1:2:1 to receive 10 mg of intravitreal lampalizumab every 4 weeks, a sham procedure every 4 weeks, 10 mg of lampalizumab every 6 weeks, or a sham procedure every 6 weeks, through 96 weeks. The primary endpoint was the mean change in the GA area at 48 weeks by fundus autofluorescence (80).” In contrast to the favorable results from the phase 2 study, the phase 3 study CHROMA (n = 906) showed both dose arms of the monthly and every 6-week injections did not show any significant efficacy compared to the sham group (80). The SPECTRI study (n = 975) also demonstrated no treatment benefit of lampalizumab. No benefit of lampalizumab was observed across prespecified subgroups, including by factor I, noted in the earlier phase 2 study. It was noted that development of new CNV in patients with bilateral GA occurred in < 2% of the study or fellow eyes. This finding is consistent with observational studies, which reported conversion rates of 2% at 2 years and 11% at 4 years in patients with bilateral GA and no baseline CNV (101). CHROMA and SPECTRI, which are the largest GA complement studies conducted to date, concluded that lampalizumab did not reduce GA enlargement vs. sham over 48 weeks of treatment. The limitation of these studies comes from the selection criteria of enrolled patients which excluded patients with smaller or larger lesions, unilateral GA, autofluorescence patterns except for banded or diffuse, current or prior CNV, GA from causes other than AMD, or earlier stages. Recent diffusion studies with enriched Bruch's membrane from human donor eyes over a wide age-range demonstrated that there is distinct selectivity in the permeability of Bruch's membrane to complement proteins with size and glycosylation being the important determinants (102). Factor D can penetrate Bruch's membrane suggesting a possible reason as to why the clinical trials with lampalizumab may have failed. Furthermore, an in vivo study with mice reported that the absence of both factor H and factor D pushes the complement system identifying a factor D bypass mechanism that is likely always present but only clinically germane in association with factor H dysfunction (103).
Properdin-Mediated Therapeutics
C3 convertase (C3bBb) is unstable, and is stabilized by the binding of properdin. Stabilized C3bBb can cleave more C3, thus generating a feedback loop (17, 19). Therefore, it is hypothesized that use of an anti-properdin antibody should destabilize the C3 convertase and block the feedback loop. An anti-properdin monoclonal antibody (CLG561, Novartis International AG, Basel, Switzerland) was developed by Novartis, and a phase 2 clinical trial (NCT02515942) was completed. The purpose of that study was to evaluate the safety and efficacy of 12 (every 28 days) intravitreal injections of CLG561 as a monotherapy and as a combination therapy with LFG316 (anti-C5, discussed below) compared to the sham in subjects with GA. However, to date the results remain unpublished (104).
C5-Mediated Therapeutics
Cleavage of C5 generates C5a and the initiation of MAC, which are key mediators of the terminal complement pathway and complement activation (18). C5a is immunomodulatory, while the MAC initiates cell lysis. Histopathologic specimens of human dry AMD lesions strongly stain for C5 and MAC at the key sites of pathology (24). C5 inhibitors do not affect the formation of upstream complement components such as C3b, which are important in host defense mechanisms. It was suggested that by inhibiting C5-mediated inflammatory and MAC activities, a therapeutic benefit may be achieved in both dry and wet AMD while sparing the immunoprotective functions of the complement system, however, this has not yet been proven in the clinic (105). Three anti-C5 antibodies are in clinical trials: eculizumab (Soliris®; Alexion Pharmaceuticals, Cheshire, CT, US), LFG316 (Novartis Pharma AG, Basel, Switzerland), and ARC1905 (Zimura®, Ophthotech Corp., Princeton, NJ, US).
Eculizumab
Eculizumab is a humanized monoclonal antibody derived from the murine anti-human C5 antibody m5G1. This antibody specifically binds the terminal complement protein C5, thereby inhibiting cleavage to C5a and C5b during complement activation and preventing MAC formation. The United States Food and Drug Administration (FDA) and European Medicines Agency (EMA) approved eculizumab for the treatment of two rare genetic deficiencies of complement inhibition, atypical hemolytic uremic syndrome and paroxysmal nocturnal hemoglobinuria (106). Recently, the FDA/EMA approved eculizumab for the treatment of adult patients with generalized myasthenia gravis, who are anti-acetyl-choline receptor antibody positive (107).
A phase 2 clinical trial (COMPLETE, NCT00935883) was the first prospective, randomized, placebo-controlled investigation of complement inhibition for the treatment of AMD (108). This trial was conducted to evaluate the effect of intravenous injection of eculizumab, a systemic C5 inhibitor, on the expansion of GA in patients with AMD (81). “Patients were randomized 2:1 to receive intravenous eculizumab or placebo over 6 months. In the eculizumab treatment arm, the first 10 patients received a low-dose regimen of 600 mg weekly for 4 weeks followed by 900 mg every 2 weeks until week 24, and the next 10 patients received a high-dose regimen of 900 mg weekly for 4 weeks followed by 1200 mg every 2 weeks until week 24. At 26 weeks, the GA area increased by 0.19 ± 0.12 and 0.18 ± 0.15 mm in the eculizumab and placebo groups, respectively (P = 0.96). At 52 weeks, the GA area increased by 0.37 ± 0.22 mm in the eculizumab group and by 0.37 ± 0.21 mm in the placebo group (P = 0.93). None of the eyes converted to neovascular AMD (81).” The lack of any trend toward therapeutic efficacy with eculizumab led the study authors to conclude that eculizumab was well-tolerated through 6 months but did not decrease the growth rate of GA significantly. Furthermore, the effect of eculizumab treatment on the drusen volume was also evaluated (82). The mean drusen cube root volumes were 0.49 and 0.47 mm (P = 0.64) at baseline and 0.51 and 0.42 mm (P = 0.17) at 26 weeks in the eculizumab and placebo groups, respectively, which showed no beneficial effect of eculizumab in reducing the drusen volume compared to the placebo.
There are several limitations in the COMPLETE clinical trial, including the route of administration. The rationale for using a systemic drug in that clinical trial was based on the belief that complement activation in the choroid played an important role in the progression of GA. Furthermore, eculizumab already has been approved for systemic delivery for patients with paroxysmal nocturnal hemoglobinuria (109) and atypical hemolytic uremic syndrome (110); thus, systemic delivery of eculizumab was possible without additional safety studies. Although systemic complement inhibitors are successfully used for other systemic diseases including paroxysmal nocturnal hemoglobinuria, the amount of drug delivered systemically could be inadequate to penetrate the blood retinal barrier and affect the pathological progression of GA. Another possible explanation could be that the clinical trials focused on the wrong targets. Eculizumab for GA is no longer listed among Alexion's pipeline therapeutics (111).
LFG316
LFG316 is a human monoclonal C5 antibody. A phase 2 clinical trial (NCT01535950) to evaluate the efficacy of intravitreal LFG316 in patients with neovascular AMD was completed though the results remain unpublished (112). Another phase 2 clinical trial (NCT01527500) for 150 patients with GA was completed. However, the intravitreal LFG316 treatment group (5 mg/50 μL) did not have reduced GA lesions compared to the sham group over 1 year of follow-up (113). A phase 2 clinical trial (NCT01624636) evaluating the safety and tolerability of intravenous LFG316 injection was terminated before completion (114).
ARC1905
ARC1905 is an intravitreal anti-C5 aptamer. Results of the phase 1 study (NCT00709527) were presented at the Association for Research in Vision and Ophthalmology 2010 Annual Meeting (83). “Forty-three patients with subfoveal neovascular AMD received six monthly administrations of ARC1905 (0.3, 1, or 2 mg) in combination with ranibizumab (0.5 mg). The BCVA in the study eye was 20/63 to 20/200. It did not show any dose-limiting toxicity during 6 months. The mean change in BCVA at week 24 was an increase of +13.6, +11.7, and +15.3 letters at the doses of 0.3, 1, and 2 mg, respectively. Furthermore, 46%, 47%, and 60% of patients gained 3 or more lines of visual acuity at the doses of 0.3, 1, and 2 mg, respectively. The mean change in center point thickness by optical coherence tomography was−150 μm.” The phase 1 study showed that the combined therapy of C5 and VEGF inhibition was well-tolerated without any toxicity issue. An additional phase 1 trial (NCT00950638), which was conducted to evaluate the safety and tolerability of ARC1905 in patients with GA, was completed though the results have not been published (115).
A phase 2 clinical trial for patients with GA started recruiting (NCT02686658) (116). Furthermore, 64 patients with neovascular AMD were enrolled in the randomized, dose-ranging, open-label, multicenter phase 2a safety trial (NCT03362190) of ARC1905 in combination with ranibizumab (117). Ophthotech announced that after 6 months of treatment, the ARC1905 combination therapy was generally well-tolerated in neovascular AMD. It is interesting that though this clinical trial, with a small sample size, was not designed to detect a significant difference in efficacy, 60% of patients who had received monthly ARC1905 (2 mg) in combination with ranibizumab (0.5 mg) gained greater than or equal to three lines of vision, or 15 Early Treatment of Diabetic Retinopathy Study letters, defined as a significant visual gain (84).
A phase 2a open-label trial to assess the safety of ARC1905 in combination with aflibercept (Eylea®, Bayer AG, Leverkusen, Germany) 2 mg, in patients with idiopathic polypoidal choroidal vasculopathy has started recruiting (NCT03374670) (118). Furthermore, a phase 2b randomized, double-masked, controlled trial to evaluate the safety and efficacy of ARC1905 compared to sham in subjects with autosomal recessive Stargardt disease 1 is recruiting (NCT03364153) (119).
CD59-Mediated Therapeutics
Soluble and cell-bound regulators of complement including CD59 help to protect healthy host tissue from self-recognition and serve to prevent activation of a complement response (17). However, damaged or diseased host cells can down-regulate membrane-bound complement inhibitors which enables targeted clearance. An imbalance between complement recognition and initiation on healthy host cells can lead to unregulated complement activation, opsonization, and/or subsequent cellular damage. Moreover, accumulation of MAC on cell surfaces leads to cell damage and death, associated with several clinical findings observed in AMD. CD59 functions by binding the C5b678 terminal complement protein complex and preventing the incorporation of the multiple C9 molecules required to complete the formation of a pore in the cell membrane. Normal cells within the human body produce a surface protein, CD59, which blocks formation of the MAC (22, 23). In mouse models with retinal pathologies including retinal detachment (70) and oxygen-induced retinopathy (120, 121), CD59 expression was suppressed compared to the control mice. The above animal studies showed that the presence of CD59 can have a dichotomous role. Cd59 down-regulation can either be protective in pathologies involving dividing cells (e.g., vascular endothelial cells in neovascular disease), or can lead to neurodegeneration when Cd59 down-regulation enables complement to target non-dividing cells of the central nervous system (e.g., photoreceptor cell). In AMD, the complement cascade is thought to be upregulated and it has been postulated that targeting MAC formation can protect against self-cell death. AAVCAGsCD59 (Hemera, QC, Canada), an ocular gene therapy product that is delivered intravitreally, causes normal retinal cells to increase the expression of a soluble form of CD59 (sCD59). This soluble recombinant CD59 is designed to protect retinal cells by inhibiting MAC formation. Adeno-associated virus (AAV) serotype 2 was used because it has been shown to be safe for use in humans and is generally considered less immunogenic than adenovirus vectors (122). A phase 1 trial (NCT03144999) was initiated to evaluate drug safety after a single injection of AAVCAGsCD59 administered in an office setting for patients with GA (123). Regarding the frequency of injections, the biggest advantage of this gene therapeutic approach is that it requires only one injection compared to other drugs with monthly or bimonthly intravitreal injections. This could be beneficial in minimizing potential endophthalmitis, which is a risk that can arise from multiple injections.
Treatment Challenges
Vascular endothelial growth factor (VEGF) drives ocular neovascularization, and anti-VEGF therapies are highly effective in the treatment of neovascular AMD. Although these drugs are highly effective, they require frequent intraocular injections, and are costly, reducing patient compliance. Furthermore, anti-VEGF therapies are not effective in treating GA.
A significant body of work in animal models, genetic studies and clinical trials suggests an important but complex role for the complement system in AMD, including GA. However, therapies targeting the alternative complement cascade have thus far had only modest therapeutic effects in GA and neovascular AMD. The reasons for these unexpected outcomes have not been fully elucidated, but are likely due the disease stages treated, and in some cases, insufficient drug delivery. The complement system may be more relevant in the earlier stages of the disease, before clinical pathologies such as GA, CNV and decreased visual acuity develop. Drug delivery may also have been problematic in some studies such as intravenous eculizumab injection in the COMPLETE study. Intravitreal injections may be the most popular, but there are issues with continued dosing and getting to the right locations within the eye (i.e., monthly injections). Gene therapy has made great strides recently, and sub-retinal injections of AAV-delivered therapeutics are now FDA approved for other ocular indications such as inherited retinal dystrophy (i.e., Voretigene neparvovec, Luxterna, Spark Therapeutics Inc.) (124), which could show the beneficial mode of delivery of the future complement therapeutics. In addition, an emerging hypothesis points toward a dual role for complement in the progression of age-related and degenerative diseases, which are often driven by accumulating debris (125). However, a previous study using a primate model with early-onset macular degeneration, which develop drusen in < 2 years after birth, showed that intravitreal C3 inhibitor compstatin injection for 6 months resulted in drusen disappearance (126). The “fitness” of the cascade, largely defined by the complotype of polymorphisms/mutations in complement genes (127), is likely of high importance in these chronic, slowly developing disorders.
Conclusion
Abundant evidence from pathological as well as genetic studies has contributed to a breakthrough in our understanding of the role of the complement system in the pathogenesis of AMD. Thus, local inhibition of complement activation has been considered a promising approach for treating both forms of late AMD. In light of the probable role of the complement system in development of AMD, many clinical trials investigating the effect of complement inhibitors have been conducted or are in progress. The results of clinical trials, in which often only a subgroup of patients responded favorably, has shown that careful stratification of indications and patient cohorts will be critical to identify patients that may benefit from complement-mediated therapies. In this aspect, genetic and/or clinical diagnostic tools will be important. Continued research, including studies on the initial development of late AMD and the subsequent impairment of visual function, will be crucial to further understand the complement pathophysiology in AMD, and to identify additional potential therapeutic targets for complement modulation. Furthermore, the conceptual diversity of complement-mediated therapeutics could allow for accessible indications, treatment options, costs, and clinical availability. Thus, the results of ongoing clinical trials are eagerly awaited, with the hope of developing additional therapeutic modalities for this increasingly common malady.
Author Contributions
All authors listed have made a substantial, direct and intellectual contribution to the work, and approved it for publication.
Funding
This study was supported by the National Institute of Health/National Eye Institute: Grant R01EY027303 and R01EY029269 (to KC); and AI068730 (to JL) and the American Macular Degeneration Foundation Prevention Award (KC). Special thanks go to Department of Ophthalmology, Harvard University, and Massachusetts Eye and Ear Infirmary for supporting this research (KC). DP is financially supported by the Basic Science Research Program of the National Research Foundation of Korea (NRF), funded by the Ministry of Education (NRF-2017R1D1A1B03027966), and the Korea Health Technology R&D Project of the Korea Health Industry Development Institute (KHIDI), funded by the Ministry of Health & Welfare, Republic of Korea (HI16C1501).
Conflict of Interest Statement
JL declares that he is the founder of Amyndas Pharmaceuticals, is named as an inventor on patents or patent applications describing the therapeutic use of complement inhibitors (some of which are being developed by Amyndas Pharmaceuticals) and is the inventor of the compstatin analog licensed to Apellis Pharmaceuticals termed 4(1MeW)7 W (also known as POT-4 and APL-1) and pegylated derivatives such as APL-2.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Gehrs KM, Anderson DH, Johnson LV, Hageman GS. Age-related macular degeneration–emerging pathogenetic and therapeutic concepts. Ann Med. (2006) 38:450–71. doi: 10.1080/07853890600946724
2. Wong WL, Su X, Li X, Cheung CM, Klein R, Cheng CY, et al. Global prevalence of age-related macular degeneration and disease burden projection for 2020 and 2040: a systematic review and meta-analysis. Lancet Glob Health. (2014) 2:e106–16. doi: 10.1016/S2214-109X(13)70145-1
3. Sparrow JR, Hicks D, Hamel CP. The retinal pigment epithelium in health and disease. Curr Mol Med. (2010) 10:802–23. doi: 10.2174/156652410793937813
4. Kevany BM, Palczewski K. Phagocytosis of retinal rod and cone photoreceptors. Physiology. (2010) 25:8–15. doi: 10.1152/physiol.00038.2009
5. Ferris FL III, Wilkinson CP, Bird A, Chakravarthy U, Chew E, Csaky K, et al. Clinical classification of age-related macular degeneration. Ophthalmology. (2013) 120:844–51. doi: 10.1016/j.ophtha.2012.10.036
6. Rudnicka AR, Jarrar Z, Wormald R, Cook DG, Fletcher A, Owen CG. Age and gender variations in age-related macular degeneration prevalence in populations of European ancestry: a meta-analysis. Ophthalmology. (2012) 119:571–80. doi: 10.1016/j.ophtha.2011.09.027
7. Friedman DS, O'Colmain BJ, Munoz B, Tomany SC, McCarty C, de Jong PT, et al. Prevalence of age-related macular degeneration in the United States. Arch Ophthalmol. (2004) 122:564–72. doi: 10.1001/archopht.122.4.564
8. Chakravarthy U, Wong TY, Fletcher A, Piault E, Evans C, Zlateva G, et al. Clinical risk factors for age-related macular degeneration: a systematic review and meta-analysis. BMC Ophthalmol. (2010) 10:31. doi: 10.1186/1471-2415-10-31
9. Tomany SC, Wang JJ, Van Leeuwen R, Klein R, Mitchell P, Vingerling JR, et al. Risk factors for incident age-related macular degeneration: pooled findings from 3 continents. Ophthalmology. (2004) 111:1280–7. doi: 10.1016/j.ophtha.2003.11.010
10. Clemons TE, Milton RC, Klein R, Seddon JM, Ferris FL III, Age-Related Eye Disease Study Research Group. Risk factors for the incidence of Advanced Age-Related Macular Degeneration in the Age-Related Eye Disease Study (AREDS) AREDS report no. 19. Ophthalmology. (2005) 112:533–9. doi: 10.1016/j.ophtha.2004.10.047
11. Sui GY, Liu GC, Liu GY, Gao YY, Deng Y, Wang WY, et al. Is sunlight exposure a risk factor for age-related macular degeneration? A systematic review and meta-analysis. Br J Ophthalmol. (2013) 97:389–94. doi: 10.1136/bjophthalmol-2012-302281
12. Seddon JM, Reynolds R, Maller J, Fagerness JA, Daly MJ, Rosner B. Prediction model for prevalence and incidence of advanced age-related macular degeneration based on genetic, demographic, and environmental variables. Invest Ophthalmol Vis Sci. (2009) 50:2044–53. doi: 10.1167/iovs.08-3064
13. Cascella R, Ragazzo M, Strafella C, Missiroli F, Borgiani P, Angelucci F, et al. Age-related macular degeneration: insights into inflammatory genes. J Ophthalmol. (2014) 2014:582842. doi: 10.1155/2014/582842
14. Whitcup SM, Sodhi A, Atkinson JP, Holers VM, Sinha D, Rohrer B, et al. The role of the immune response in age-related macular degeneration. Int J Inflamm. (2013) 2013:348092. doi: 10.1155/2013/348092
15. Markiewski MM, Lambris JD. The role of complement in inflammatory diseases from behind the scenes into the spotlight. Am J Pathol. (2007) 171:715–27. doi: 10.2353/ajpath.2007.070166
16. Anderson DH, Radeke MJ, Gallo NB, Chapin EA, Johnson PT, Curletti CR, et al. The pivotal role of the complement system in aging and age-related macular degeneration: hypothesis re-visited. Prog Retin Eye Res. (2010) 29:95–112. doi: 10.1016/j.preteyeres.2009.11.003
17. Ricklin D, Hajishengallis G, Yang K, Lambris JD. Complement: a key system for immune surveillance and homeostasis. Nat Immunol. (2010) 11:785–97. doi: 10.1038/ni.1923
18. Walport MJ. Complement. First of two parts. N Engl J Med. (2001) 344:1058–66. doi: 10.1056/NEJM200104053441406
19. Harboe M, Mollnes TE. The alternative complement pathway revisited. J Cell Mol Med. (2008) 12:1074–84. doi: 10.1111/j.1582-4934.2008.00350.x
20. Walport MJ. Complement. Second of two parts. N Engl J Med. (2001) 344:1140–4. doi: 10.1056/NEJM200104123441506
21. Forneris F, Ricklin D, Wu J, Tzekou A, Wallace RS, Lambris JD, et al. Structures of C3b in complex with factors B and D give insight into complement convertase formation. Science. (2010) 330:1816–20. doi: 10.1126/science.1195821
22. Hamilton KK, Ji Z, Rollins S, Stewart BH, Sims PJ. Regulatory control of the terminal complement proteins at the surface of human endothelial cells: neutralization of a C5b-9 inhibitor by antibody to CD59. Blood. (1990) 76:2572–7.
23. Rollins SA, Sims PJ. The complement-inhibitory activity of CD59 resides in its capacity to block incorporation of C9 into membrane C5b-9. J Immunol. (1990) 144:3478–83.
24. Anderson DH, Mullins RF, Hageman GS, Johnson LV. A role for local inflammation in the formation of drusen in the aging eye. Am J Ophthalmol. (2002) 134:411–31. doi: 10.1016/S0002-9394(02)01624-0
25. Hageman GS, Luthert PJ, Victor Chong NH, Johnson LV, Anderson DH, Mullins RF. An integrated hypothesis that considers drusen as biomarkers of immune-mediated processes at the RPE-Bruch's membrane interface in aging and age-related macular degeneration. Prog Retin Eye Res. (2001) 20:705–32. doi: 10.1016/S1350-9462(01)00010-6
26. Edwards AO, Ritter R III, Abel KJ, Manning A, Panhuysen C, Farrer LA. Complement factor H polymorphism and age-related macular degeneration. Science. (2005) 308:421–4. doi: 10.1126/science.1110189
27. Hageman GS, Anderson DH, Johnson LV, Hancox LS, Taiber AJ, Hardisty LI, et al. A common haplotype in the complement regulatory gene factor H (HF1/CFH) predisposes individuals to age-related macular degeneration. Proc Natl Acad Sci USA. (2005) 102:7227–32. doi: 10.1073/pnas.0501536102
28. Haines JL, Hauser MA, Schmidt S, Scott WK, Olson LM, Gallins P, et al. Complement factor H variant increases the risk of age-related macular degeneration. Science. (2005) 308:419–21. doi: 10.1126/science.1110359
29. Klein RJ, Zeiss C, Chew EY, Tsai JY, Sackler RS, Haynes C, et al. Complement factor H polymorphism in age-related macular degeneration. Science. (2005) 308:385–9. doi: 10.1126/science.1109557
30. Sofat R, Casas JP, Webster AR, Bird AC, Mann SS, Yates JR, et al. Complement factor H genetic variant and age-related macular degeneration: effect size, modifiers and relationship to disease subtype. Int J Epidemiol. (2012) 41:250–62. doi: 10.1093/ije/dyr204
31. Raychaudhuri S, Iartchouk O, Chin K, Tan PL, Tai AK, Ripke S, et al. A rare penetrant mutation in CFH confers high risk of age-related macular degeneration. Nat Genet. (2011) 43:1232–6. doi: 10.1038/ng.976
32. Yu Y, Triebwasser MP, Wong EK, Schramm EC, Thomas B, Reynolds R, et al. Whole-exome sequencing identifies rare, functional CFH variants in families with macular degeneration. Hum Mol Genet. (2014) 23:5283–93. doi: 10.1093/hmg/ddu226
33. Ferrara D, Seddon JM. Phenotypic Characterization of Complement Factor H R1210C Rare Genetic Variant in Age-Related Macular Degeneration. JAMA Ophthalmol. (2015) 133:785–91. doi: 10.1001/jamaophthalmol.2015.0814
34. Hageman GS, Hancox LS, Taiber AJ, Gehrs KM, Anderson DH, Johnson LV, et al. Extended haplotypes in the complement factor H (CFH) and CFH-related (CFHR) family of genes protect against age-related macular degeneration: Characterization, ethnic distribution and evolutionary implications. Ann Med. (2006) 38:592–604. doi: 10.1080/07853890601097030
35. Hughes AE, Orr N, Esfandiary H, Diaz-Torres M, Goodship T, Chakravarthy U. A common CFH haplotype, with deletion of CFHR1 and CFHR3, is associated with lower risk of age-related macular degeneration. Nat Genet. (2006) 38:1173–7. doi: 10.1038/ng1890
36. Fritsche LG, Lauer N, Hartmann A, Stippa S, Keilhauer CN, Oppermann M, et al. An imbalance of human complement regulatory proteins CFHR1, CFHR3, and factor H influences risk for age-related macular degeneration (AMD). Hum Mol Genet. (2010) 19:4694–704. doi: 10.1093/hmg/ddq399
37. Maller JB, Fagerness JA, Reynolds RC, Neale BM, Daly MJ, Seddon JM. Variation in complement factor 3 is associated with risk of age-related macular degeneration. Nat Genet. (2007) 39:1200–1. doi: 10.1038/ng2131
38. Yates JR, Sepp T, Matharu BK, Khan JC, Thurlby DA, Shahid H, et al. Complement C3 variant and the risk of age-related macular degeneration. N Engl J Med. (2007) 357:553–61. doi: 10.1056/NEJMoa072618
39. Heurich M, Martinez-Barricarte R, Francis NJ, Roberts DL, Rodriguez de Cordoba S, Morgan BP, et al. Common polymorphisms in C3, factor B, and factor H collaborate to determine systemic complement activity and disease risk. Proc Natl Acad Sci USA. (2011) 108:8761–6. doi: 10.1073/pnas.1019338108
40. Wu J, Wu YQ, Ricklin D, Janssen BJ, Lambris JD, Gros P. Structure of complement fragment C3b-factor H and implications for host protection by complement regulators. Nat Immunol. (2009) 10:728–33. doi: 10.1038/ni.1755
41. Seddon JM, Yu Y, Miller EC, Reynolds R, Tan PL, Gowrisankar S, et al. Rare variants in CFI, C3 and C9 are associated with high risk of advanced age-related macular degeneration. Nat Genet. (2013) 45:1366–70. doi: 10.1038/ng.2741
42. Gold B, Merriam JE, Zernant J, Hancox LS, Taiber AJ, Gehrs K, et al. Variation in factor B (BF) and complement component 2 (C2) genes is associated with age-related macular degeneration. Nat Genet. (2006) 38:458–62. doi: 10.1038/ng1750
43. Maller J, George S, Purcell S, Fagerness J, Altshuler D, Daly MJ, et al. Common variation in three genes, including a noncoding variant in CFH, strongly influences risk of age-related macular degeneration. Nat Genet. (2006) 38:1055–9. doi: 10.1038/ng1873
44. Montes T, Tortajada A, Morgan BP, Rodriguez de Cordoba S, Harris CL. Functional basis of protection against age-related macular degeneration conferred by a common polymorphism in complement factor B. Proc Natl Acad Sci USA. (2009) 106:4366–71. doi: 10.1073/pnas.0812584106
45. Stanton CM, Yates JR, den Hollander AI, Seddon JM, Swaroop A, Stambolian D, et al. Complement factor D in age-related macular degeneration. Invest Ophthalmol Vis Sci. (2011) 52:8828–34. doi: 10.1167/iovs.11-7933
46. Fagerness JA, Maller JB, Neale BM, Reynolds RC, Daly MJ, Seddon JM. Variation near complement factor I is associated with risk of advanced AMD. Eur J Hum Genet. (2009) 17:100–4. doi: 10.1038/ejhg.2008.140
47. van de Ven JP, Nilsson SC, Tan PL, Buitendijk GH, Ristau T, Mohlin FC, et al. A functional variant in the CFI gene confers a high risk of age-related macular degeneration. Nat Genet. (2013) 45:813–7. doi: 10.1038/ng.2640
48. Nishiguchi KM, Yasuma TR, Tomida D, Nakamura M, Ishikawa K, Kikuchi M, et al. C9-R95X polymorphism in patients with neovascular age-related macular degeneration. Invest Ophthalmol Vis Sci. (2012) 53:508–12. doi: 10.1167/iovs.11-8425
49. Day AJ, Willis AC, Ripoche J, Sim RB. Sequence polymorphism of human complement factor H. Immunogenetics. (1988) 27:211–4.
50. Forneris F, Wu J, Xue X, Ricklin D, Lin Z, Sfyroera G, et al. Regulators of complement activity mediate inhibitory mechanisms through a common C3b-binding mode. EMBO J. (2016) 35:1133–49. doi: 10.15252/embj.201593673
51. Clark SJ, Bishop PN. The eye as a complement dysregulation hotspot. Semin Immunopathol. (2018) 40:65–74. doi: 10.1007/s00281-017-0649-6
52. Mullins RF, Schoo DP, Sohn EH, Flamme-Wiese MJ, Workamelahu G, Johnston RM, et al. The membrane attack complex in aging human choriocapillaris: relationship to macular degeneration and choroidal thinning. Am J Pathol. (2014) 184:3142–53. doi: 10.1016/j.ajpath.2014.07.017
53. Whitmore SS, Sohn EH, Chirco KR, Drack AV, Stone EM, Tucker BA, et al. Complement activation and choriocapillaris loss in early AMD: implications for pathophysiology and therapy. Prog Retin Eye Res. (2015) 45:1–29. doi: 10.1016/j.preteyeres.2014.11.005
54. Keenan TD, Toso M, Pappas C, Nichols L, Bishop PN, Hageman GS. Assessment of proteins associated with complement activation and inflammation in maculae of human donors homozygous risk at chromosome 1 CFH-to-F13B. Invest Ophthalmol Vis Sci. (2015) 56:4870–9. doi: 10.1167/iovs.15-17009
55. Clark SJ, Bishop PN, Day AJ. Complement factor H and age-related macular degeneration: the role of glycosaminoglycan recognition in disease pathology. Biochem Soc Trans. (2010) 38:1342–8. doi: 10.1042/BST0381342
56. Herbert AP, Deakin JA, Schmidt CQ, Blaum BS, Egan C, Ferreira VP, et al. Structure shows that a glycosaminoglycan and protein recognition site in factor H is perturbed by age-related macular degeneration-linked single nucleotide polymorphism. J Biol Chem. (2007) 282:18960–8. doi: 10.1074/jbc.M609636200
57. Laine M, Jarva H, Seitsonen S, Haapasalo K, Lehtinen MJ, Lindeman N, et al. Y402H polymorphism of complement factor H affects binding affinity to C-reactive protein. J Immunol. (2007) 178:3831–6. doi: 10.4049/jimmunol.178.6.3831
58. Sjoberg AP, Trouw LA, Clark SJ, Sjolander J, Heinegard D, Sim RB, et al. The factor H variant associated with age-related macular degeneration (His-384) and the non-disease-associated form bind differentially to C-reactive protein, fibromodulin, DNA, and necrotic cells. J Biol Chem. (2007) 282:10894–900. doi: 10.1074/jbc.M610256200
59. Clark SJ, Perveen R, Hakobyan S, Morgan BP, Sim RB, Bishop PN, et al. Impaired binding of the age-related macular degeneration-associated complement factor H 402H allotype to Bruch's membrane in human retina. J Biol Chem. (2010) 285:30192–202. doi: 10.1074/jbc.M110.103986
60. Clark SJ, Bishop PN, Day AJ. The proteoglycan glycomatrix: a sugar microenvironment essential for complement regulation. Front Immunol. (2013) 4:412. doi: 10.3389/fimmu.2013.00412
61. Clark SJ, Ridge LA, Herbert AP, Hakobyan S, Mulloy B, Lennon R, et al. Tissue-specific host recognition by complement factor H is mediated by differential activities of its glycosaminoglycan-binding regions. J Immunol. (2013) 190:2049–57. doi: 10.4049/jimmunol.1201751
62. Langford-Smith A, Keenan TD, Clark SJ, Bishop PN, Day AJ. The role of complement in age-related macular degeneration: heparan sulphate, a ZIP code for complement factor H? J Innate Immun. (2014) 6:407–16. doi: 10.1159/000356513
63. Langford-Smith A, Day AJ, Bishop PN, Clark SJ. Complementing the sugar code: role of GAGs and sialic acid in complement regulation. Front Immunol. (2015) 6:25. doi: 10.3389/fimmu.2015.00025
64. Toomey CB, Kelly U, Saban DR, Bowes Rickman C. Regulation of age-related macular degeneration-like pathology by complement factor H. Proc Natl Acad Sci USA. (2015) 112:E3040–9. doi: 10.1073/pnas.1424391112
65. Hebecker M, Jozsi M. Factor H-related protein 4 activates complement by serving as a platform for the assembly of alternative pathway C3 convertase via its interaction with C3b protein. J Biol Chem. (2012) 287:19528–36. doi: 10.1074/jbc.M112.364471
66. Ricklin D, Reis ES, Mastellos DC, Gros P, Lambris JD. Complement component C3 - The “Swiss Army Knife” of innate immunity and host defense. Immunol Rev. (2016) 274:33–58. doi: 10.1111/imr.12500
67. Hecker LA, Edwards AO, Ryu E, Tosakulwong N, Baratz KH, Brown WL, et al. Genetic control of the alternative pathway of complement in humans and age-related macular degeneration. Hum Mol Genet. (2010) 19:209–15. doi: 10.1093/hmg/ddp472
68. Kijlstra A, Berendschot TT. Age-related macular degeneration: a complementopathy? Ophthalmic Res. (2015) 54:64–73. doi: 10.1159/000432401
69. Scholl HP, Charbel Issa P, Walier M, Janzer S, Pollok-Kopp B, Borncke F, et al. Systemic complement activation in age-related macular degeneration. PLoS ONE. (2008) 3:e2593. doi: 10.1371/journal.pone.0002593
70. Sweigard JH, Matsumoto H, Smith KE, Kim LA, Paschalis EI, Okonuki Y, et al. Inhibition of the alternative complement pathway preserves photoreceptors after retinal injury. Sci Transl Med 7 (297), 297ra116. doi: 10.1126/scitranslmed.aab1482
71. Volanakis JE, Narayana SV. Complement factor D, a novel serine protease. Protein Sci. (1996) 5:553–64. doi: 10.1002/pro.5560050401
72. Vyse TJ, Bates GP, Walport MJ, Morley BJ. The organization of the human complement factor I gene (IF): a member of the serine protease gene family. Genomics. (1994) 24:90–8. doi: 10.1006/geno.1994.1585
73. Catterall CF, Lyons A, Sim RB, Day AJ, Harris TJ. Characterization of primary amino acid sequence of human complement control protein factor I from an analysis of cDNA clones. Biochem J. (1987) 242:849–56.
74. Xue X, Wu J, Ricklin D, Forneris F, Di Crescenzio P, Schmidt CQ, et al. Regulator-dependent mechanisms of C3b processing by factor I allow differentiation of immune responses. Nat Struct Mol Biol. (2017) 24:643–51. doi: 10.1038/nsmb.3427
75. Harriman GR, Esser AF, Podack ER, Wunderlich AC, Braude AI, Lint TF, et al. The role of C9 in complement-mediated killing of Neisseria. J Immunol. (1981) 127:2386–90.
76. Podack ER, Tschoop J, Muller-Eberhard HJ. Molecular organization of C9 within the mem$$brane attack complex of complement. Induction of circular C9 polymerization by the C5b-8 assembly. J Exp Med. (1982) 156:268–82.
77. Kaushal S, Grossi F, Francois C, Slakter J, ASaP Study Group. Complement C3 inhibitor POT-4: clinical safety of intravitreal administration. Invest Ophthalmol Vis Sci. (2009) 50:1. Available online at: https://iovs.arvojournals.org/article.aspx?articleid=2367196
78. Do DV, Pieramici DJ, van Lookeren Campagne M, Beres T, Friesenhahn M, Zhang Y, et al. A phase ia dose-escalation study of the anti-factor D monoclonal antibody fragment FCFD4514S in patients with geographic atrophy. Retina. (2014) 34:313–20. doi: 10.1097/IAE.0b013e3182979ddd
79. Yaspan BL, Williams DF, Holz FG, Regillo CD, Li Z, Dressen A, et al. Targeting factor D of the alternative complement pathway reduces geographic atrophy progression secondary to age-related macular degeneration. Sci Transl Med. (2017) 9:aaf1443. doi: 10.1126/scitranslmed.aaf1443
80. Holz FG, Sadda SR, Busbee B, Chew EY, Mitchell P, Tufail A, et al. Efficacy and safety of lampalizumab for geographic atrophy due to age-related macular degeneration: chroma and spectri phase 3 randomized clinical trials. JAMA Ophthalmol. (2018) 136:666–77. doi: 10.1001/jamaophthalmol.2018.1544
81. Yehoshua Z, de Amorim Garcia Filho CA, Nunes RP, Gregori G, Penha FM, Moshfeghi AA, et al. Systemic complement inhibition with eculizumab for geographic atrophy in age-related macular degeneration: the COMPLETE study. Ophthalmology. (2014) 121:693–701. doi: 10.1016/j.ophtha.2013.09.044
82. Garcia Filho CA, Yehoshua Z, Gregori G, Nunes RP, Penha FM, Moshfeghi AA, et al. Change in drusen volume as a novel clinical trial endpoint for the study of complement inhibition in age-related macular degeneration. Ophthalmic Surg Lasers Imaging Retina. (2014) 45:18–31. doi: 10.3928/23258160-20131217-01
83. Cousins SW, Ophthotech Study Group. Targeting Complement Factor 5 in Combination with Vascular Endothelial Growth Factor (VEGF) Inhibition for Neovascular Age Related Macular Degeneration (AMD): results of a phase 1 study. Invest Ophthalmol Visual Sci. (2010) 51:1. Available online at: https://iovs.arvojournals.org/article.aspx?articleid=2369585
84. Ophthotech. Ophthotech Announces Results from Phase 2a Safety Trial of Zimura® in Combination with Lucentis® in Wet Age-Related Macular Degeneration. (2018). Available: https://www.businesswire.com/news/home/20181112005203/en/Ophthotech-Announces-Results-Phase-2a-Safety-Trial (accessed November 25, 2018).
85. Ricklin D, Lambris JD. Compstatin: a complement inhibitor on its way to clinical application. Adv Exp Med Biol. (2008) 632:273–92.
86. Sahu A, Kay BK, Lambris JD. Inhibition of human complement by a C3-binding peptide isolated from a phage-displayed random peptide library. J Immunol. (1996) 157:884–91.
87. Mastellos DC, Yancopoulou D, Kokkinos P, Huber-Lang M, Hajishengallis G, Biglarnia AR, et al. Compstatin: a C3-targeted complement inhibitor reaching its prime for bedside intervention. Eur J Clin Invest. (2015) 45:423–40. doi: 10.1111/eci.12419
88. Alcon. Evaluation of AL-78898A in Exudative Age-Related Macular Degeneration. (2013). Available online at: https://ClinicalTrials.gov/show/NCT01157065 (accessed November 19, 2018).
89. Apellis Pharmaceuticals Inc. Study of APL-2 Therapy in Patients Geographic Atrophy. (2017). Available online at: https://ClinicalTrials.gov/show/NCT02503332 (accessed November 18, 2018).
90. Apellis Pharmaceuticals Inc. FILLY - Phase 2 Study of APL-2 in Geographic Atrophy. (2018). Available online at: http://investors.apellis.com/static-files/b5d132fc-e22a-441c-9cc0-62c694beff43 (accessed January 14, 2019).
91. Lyzogubov VV, Tytarenko RG, Liu J, Bora NS, Bora PS. Polyethylene glycol (PEG)-induced mouse model of choroidal neovascularization. J Biol Chem. (2011) 286:16229–37. doi: 10.1074/jbc.M110.204701
92. Cao X, Shen D, Patel MM, Tuo J, Johnson TM, Olsen TW, Chan CC. Macrophage polarization in the maculae of age-related macular degeneration: a pilot study. Pathol Int. (2011) 61:528–35. doi: 10.1111/j.1440-1827.2011.02695.x
93. Apellis Pharmaceuticals Inc. APL-2 Geogrphaic Atrophy Preliminary 18-month Results. (2018). Available online at: http://investors.apellis.com/static-files/96d866e8-627e-4948-ba43-5dc72ffe5078 (accessed January 14, 2019).
94. Apellis Pharmaceuticals Inc. Study to Compare the Efficacy and Safety of Intravitreal APL-2 Therapy With Sham Injections in Patients With Geographic Atrophy (GA) Secondary to Age-Related Macular Degeneration (AMD). (2018). Available online at: https://ClinicalTrials.gov/show/NCT03525613 (accessed November 18, 2018).
95. Apellis Pharmaceuticals Inc. APL-2 in Neovascular AMD. (2018). Available online at: https://ClinicalTrials.gov/show/NCT03465709 (accessed November 18, 2018).
96. Amyndas Pharmaceuticals Inc. Amyndas' Priority Clinical Programs. (2019). Available online at: http://amyndas.com/research-focus/ (accessed February 13, 2019).
97. Holers VM. The spectrum of complement alternative pathway-mediated diseases. Immunol Rev. (2008) 223:300–16. doi: 10.1111/j.1600-065X.2008.00641.x
98. Katschke KJ Jr, Wu P, Ganesan R, Kelley RF, Mathieu MA, et al. Inhibiting alternative pathway complement activation by targeting the factor D exosite. J Biol Chem. (2012) 287:12886–92. doi: 10.1074/jbc.M112.345082
99. Roche. A Study Investigating the Efficacy and Safety of Lampalizumab Intravitreal Injections in Participants With Geographic Atrophy Secondary to Age-Related Macular Degeneration. (2018). Available online at: https://ClinicalTrials.gov/show/NCT02247479 (accessed November 19, 2018).
100. Roche. A Study Investigating the Safety and Efficacy of Lampalizumab Intravitreal Injections in Participants With Geographic Atrophy Secondary to Age-Related Macular Degeneration. (2018). Available online at: https://ClinicalTrials.gov/show/NCT02247531 (accessed November 19, 2018).
101. Sunness JS, Gonzalez-Baron J, Bressler NM, Hawkins B, Applegate CA. The development of choroidal neovascularization in eyes with the geographic atrophy form of age-related macular degeneration. Ophthalmology. (1999) 106:910–9. doi: 10.1016/S0161-6420(99)00509-6
102. Clark SJ, McHarg S, Tilakaratna V, Brace N, Bishop PN. Bruch's membrane compartmentalizes complement regulation in the eye with implications for therapeutic design in age-related macular degeneration. Front Immunol. (2017) 8:1778. doi: 10.3389/fimmu.2017.01778
103. Irmscher S, Doring N, Halder LD, Jo EAH, Kopka I, Dunker C, et al. Kallikrein Cleaves C3 and activates complement. J Innate Immun. (2018) 10:94–105. doi: 10.1159/000484257
104. Novartis. CLG561 Proof-of-Concept Study as a Monotherapy and in Combination With LFG316 in Subjects With Geographic Atrophy (GA). (2017). Available online at: https://ClinicalTrials.gov/show/NCT02515942 (accessed November 19, 2018).
105. Mastellos DC, Reis ES, Ricklin D, Smith RJ, Lambris JD. Complement C3-targeted therapy: replacing long-held assertions with evidence-based discovery. Trends Immunol. (2017) 38:383–94. doi: 10.1016/j.it.2017.03.003
106. Mastellos DC, Reis ES, Yancopoulou D, Risitano AM, Lambris JD. Expanding complement therapeutics for the treatment of paroxysmal nocturnal hemoglobinuria. Semin Hematol. (2018) 55:167–75. doi: 10.1053/j.seminhematol.2018.02.002
107. Howard JF Jr, Utsugisawa K, Benatar M, Murai H, Barohn RJ, et al. Safety and efficacy of eculizumab in anti-acetylcholine receptor antibody-positive refractory generalised myasthenia gravis (REGAIN): a phase 3, randomised, double-blind, placebo-controlled, multicentre study. Lancet Neurol. (2017) 16:976–86. doi: 10.1016/S1474-4422(17)30369-1
108. Alexion. Complement Inhibition With Eculizumab for the Treatment of Non-Exudative Macular Degeneration (AMD). (2017). Available online at: https://ClinicalTrials.gov/show/NCT00935883 (accessed November 25, 2018).
109. Hillmen P, Young NS, Schubert J, Brodsky RA, Socie G, Muus P, et al. The complement inhibitor eculizumab in paroxysmal nocturnal hemoglobinuria. N Engl J Med. (2006) 355:1233–43. doi: 10.1056/NEJMoa061648
110. Maga TK, Nishimura CJ, Weaver AE, Frees KL, Smith RJ. Mutations in alternative pathway complement proteins in American patients with atypical hemolytic uremic syndrome. Hum Mutat. (2010) 31:E1445–60. doi: 10.1002/humu.21256
111. Alexion. Alexion Research and Development: Pipeline. (2018). Available online at: http://alxn.com/research-development/pipeline (accessed November 23, 2018).
112. Novartis. Safety and Efficacy of Intravitreal LFG316 in Wet Age Related Macular Degeneration (AMD). (2016). Available online at: https://ClinicalTrials.gov/show/NCT01535950 (accessed November 25, 2018).
113. Novartis. Intravitreal LFG316 in Patients With Age-related Macular Degeneration (AMD). (2016). Available online at: https://ClinicalTrials.gov/show/NCT01527500 (accessed November 19, 2018).
114. Novartis. Safety and Tolerability of Intravenous LFG316 in Wet Age-related Macular Degeneration (AMD). (2016). Available online at: https://ClinicalTrials.gov/show/NCT01624636 (accessed November 25, 2018).
115. Ophthotech. A Study of ARC1905 (Anti-C5 Aptamer) in Subjects With Dry Age-related Macular Degeneration. (2017). Available online at: https://ClinicalTrials.gov/show/NCT00950638 (accessed November 25, 2018).
116. Ophthotech. Zimura in Subjects With Geographic Atrophy Secondary to Dry Age-Related Macular Degeneration. (2018). Available online at: https://ClinicalTrials.gov/show/NCT02686658 (accessed November 21, 2018).
117. Ophthotech. ZIMURA in Combination With LUCENTIS in Patients With Neovascular Age Related Macular Degeneration (NVAMD). (2018). Available online at: https://ClinicalTrials.gov/show/NCT03362190 (accessed November 21, 2018).
118. Ophthotech. ZIMURA in Combination With Eylea in Patients With Idiopathic Polypoidal Choroidal Vasculopathy (IPCV). (2018). Available online at: https://ClinicalTrials.gov/show/NCT03374670 (accessed November 21, 2018).
119. Ophthotech. Zimura Compared to Sham in Patients With Autosomal Recessive Stargardt Disease (STGD1). (2018). Available online at: https://ClinicalTrials.gov/show/NCT03364153 (accessed November 21, 2018).
120. Sweigard JH, Yanai R, Gaissert P, Saint-Geniez M, Kataoka K, Thanos A, et al. The alternative complement pathway regulates pathological angiogenesis in the retina. FASEB J. (2014) 28:3171–82. doi: 10.1096/fj.14-251041
121. Kim C, Smith KE, Castillejos A, Diaz-Aguilar D, Saint-Geniez M, Connor KM. The alternative complement pathway aids in vascular regression during the early stages of a murine model of proliferative retinopathy. FASEB J. (2016) 30:1300–5. doi: 10.1096/fj.15-280834
122. Cashman SM, Ramo K, Kumar-Singh R. A non membrane-targeted human soluble CD59 attenuates choroidal neovascularization in a model of age related macular degeneration. PLoS ONE. (2011) 6:e19078. doi: 10.1371/journal.pone.0019078
123. Hemera. Treatment of Advanced Dry Age Related Macular Degeneration With AAVCAGsCD59. (2017). Available online at: https://ClinicalTrials.gov/show/NCT03144999 (accessed November 19, 2018).
124. Spark Therapeutics Inc. Luxturna. (2018). Available online at: https://luxturna.com/ (accessed March 26, 2019).
125. Ricklin D, Lambris JD. New milestones ahead in complement-targeted therapy. Semin Immunol. (2016) 28:208–22. doi: 10.1016/j.smim.2016.06.001
126. Chi ZL, Yoshida T, Lambris JD, Iwata T. Suppression of drusen formation by compstatin, a peptide inhibitor of complement C3 activation, on cynomolgus monkey with early-onset macular degeneration. Adv Exp Med Biol. (2010) 703:127–35. doi: 10.1007/978-1-4419-5635-4_9
Keywords: age-related macular degeneration, complement system, immune modulation, ocular inflammation, therapeutics
Citation: Park DH, Connor KM and Lambris JD (2019) The Challenges and Promise of Complement Therapeutics for Ocular Diseases. Front. Immunol. 10:1007. doi: 10.3389/fimmu.2019.01007
Received: 18 February 2019; Accepted: 18 April 2019;
Published: 15 May 2019.
Edited by:
Robert Braidwood Sim, University of Oxford, United KingdomReviewed by:
Michael Kirschfink, Universität Heidelberg, GermanySimon John Clark, University of Manchester, United Kingdom
Copyright © 2019 Park, Connor and Lambris. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Kip M. Connor, S2lwJiN4MDAwNUY7Q29ubm9yQG1lZWkuaGFydmFyZC5lZHU=