 Russell K. Pachynski1*
Russell K. Pachynski1* Ping Wang1
Ping Wang1 Nicole Salazar2,3,4
Nicole Salazar2,3,4 Yayue Zheng2
Yayue Zheng2 Leona Nease2
Leona Nease2 Jesse Rosalez5Weng-In Leong6
Jesse Rosalez5Weng-In Leong6 Gurpal Virdi1
Gurpal Virdi1 Keith Rennier1
Keith Rennier1 Woo Jae Shin1
Woo Jae Shin1 Viet Nguyen4Eugene C. Butcher3,7
Viet Nguyen4Eugene C. Butcher3,7 Brian A. Zabel2
Brian A. Zabel2- 1Division of Oncology, Department of Medicine, Center for Human Immunology and Immunotherapy Programs, Washington University School of Medicine, St. Louis, MO, United States
- 2Department of Research and Development, Palo Alto Veterans Institute for Research, Palo Alto, CA, United States
- 3Department of Pathology, Stanford University, Stanford, CA, United States
- 4Department of Biology, San Francisco State University, San Francisco, CA, United States
- 5Department of Industrial and Systems Engineering, San José State University, San José, CA, United States
- 6Dynavax Technologies, Berkeley, CA, United States
- 7Laboratory of Immunology and Vascular Biology, VA Palo Alto Health Care Systems, Palo Alto, CA, United States
Infiltration of immune cells into the tumor microenvironment (TME) can regulate growth and survival of neoplastic cells, impacting tumorigenesis and tumor progression. Correlations between the number of effector immune cells present in a tumor and clinical outcomes in many human tumors, including breast, have been widely described. Current immunotherapies utilizing checkpoint inhibitors or co-stimulatory molecule agonists aim to activate effector immune cells. However, tumors often lack adequate effector cell numbers within the TME, resulting in suboptimal responses to these agents. Chemerin (RARRES2) is a leukocyte chemoattractant widely expressed in many tissues and is known to recruit innate leukocytes. CMKLR1 is a chemotactic cellular receptor for chemerin and is expressed on subsets of dendritic cells, NK cells, and macrophages. We have previously shown that chemerin acts as a tumor suppressive cytokine in mouse melanoma models by recruiting innate immune defenses into the TME. Chemerin/RARRES2 is down-regulated in many tumors, including breast, compared to normal tissue counterparts. Here, using a syngeneic orthotopic EMT6 breast carcinoma model, we show that forced overexpression of chemerin by tumor cells results in significant recruitment of NK cells and T cells within the TME. While chemerin secretion by EMT6 cells did not alter their phenotypic behavior in vitro, it did significantly suppress tumor growth in vivo. To define the cellular effectors required for this anti-tumor phenotype, we depleted NK cells or CD8+ T cells and found that either cell type is required for chemerin-dependent suppression of EMT6 tumor growth. Finally, we show significantly reduced levels of RARRES2 mRNA in human breast cancer samples compared to matched normal tissues. Thus, for the first time we have shown that increasing chemerin expression within the breast carcinoma TME can suppress growth by recruitment of NK and T cells, thereby supporting this approach as a promising immunotherapeutic strategy.
Introduction
Breast cancer is one of the most common malignancies with an estimated ~266,000 new cases in 2018, according to SEER estimates. The impact of infiltrating lymphocytes on breast cancer patient outcomes has been studied in several contexts, but in general is a favorable prognostic factor (1–3). The presence of pre-existing immune effectors cells within the tumor microenvironment (TME) within breast and other tumor types can not only predict response to traditional cytotoxic chemotherapy, but also immunotherapies (4–6). Compared with tumor types that are more responsive to checkpoint immunotherapy, however, there is a relative paucity of infiltrating lymphocytes in breast cancer (7). Thus, strategies to enhance recruitment of immune effector cells to the breast TME are highly desirable.
Chemerin (retinoic acid receptor responder 2; RARRES2) is a leukocyte chemoattractant initially discovered as being highly up-regulated in the skin by the synthetic retinoid tazarotene (8). Chemerin is widely expressed throughout tissues and has myriad roles including the chemoattraction of innate cells [e.g., NK cells, macrophages, dendritic cells (DCs)] (9–11), functioning as an important antimicrobial agent in the skin (12), and is able to induce angiogenesis in human endothelial cells (13), suggesting chemerin may be a key factor in early immune responses to infection, injury, and/or inflammation. Chemerin is initially secreted in an inactive pro-form, prochemerin, which is then cleaved by specific proteases to become bioactive (9, 10). Chemerin has three described serpentine cell membrane receptors: chemokine-like receptor 1 (CMKLR1; ChemR23), C-C chemokine receptor-like 2 (CCRL2), and G protein-coupled receptor 1 (GPR1) (9, 14, 15). CMKLR1 is a chemotactic cellular receptor, while, atypical chemoattractant receptor CCRL2 likely acts to sequester and concentrate chemerin at sites of CCRL2 expression, such as on activated endothelial cells (14–17). The function of GPR1 is poorly understood, though it is reported to be expressed in the CNS (18, 19).
Chemerin/RARRES2 has been studied in the context of several different tumor types, with its dysregulation dependent on the specific context. While we and others have reported on several tumor types where chemerin/RARRES2 is significantly down-regulated compared to normal tissue counterparts (e.g., melanoma, lung, prostate, liver, adrenal, etc.) (20–25), chemerin/RARRES2 has been shown to be up-regulated in fewer tumor types (e.g., mesothelioma, squamous oral cancers) (26–28). Several groups have correlated chemerin/RARRES2 expression levels in the TME with clinical outcomes, showing improved patient survival in those patients with higher expression levels (20–22, 24). Importantly, two of these studies also evaluated the tumor biopsies for infiltrating leukocytes, showing an increase and correlation between higher chemerin levels and infiltrating NK cells in those patients with improved overall survival (20, 21).
Our group was the first to show that in a mouse melanoma model, overexpression and secretion of chemerin protein by tumor cells increased total CD45+ tumor infiltrating leukocytes (TIL), resulting in significantly suppressed tumor growth. In this model, the effect was mediated by NK cells, as depletion via anti-asialo GM1 resulted in complete abrogation of chemerin's tumor suppressive effects (22). In contrast, T cells were dispensable, as RAG deficiency had no effect on the anti-melanoma effects of chemerin in vivo (22). Importantly, neither engineered chemerin expression nor incubation of mouse B16F0 melanoma cells with exogenous, recombinant chemerin affected in vitro growth or phenotype, suggesting chemerin's main anti-tumor activity was due primarily to its ability to recruit immune effector cells into the TME.
Here, we studied the effect of chemerin/RARRES2 overexpression using the transplantable orthotopic syngeneic EMT6 breast carcinoma model, which has been shown to be responsive to immunomodulation in a variety of settings (29–31). Utilizing a similar approach as in the B16 model, we engineered EMT6 tumor cells to express and secrete functional chemerin within the TME and then assessed the impact on tumor growth and TIL. Chemerin overexpression significantly suppressed tumor growth, which correlated with an increase in TIL. Depletion studies identified NK and CD8+ T cells as key effector leukocytes mediating chemerin's anti-tumor activity, suggesting an interplay between innate and adaptive arms. In human breast tissue, chemerin/RARRES2 RNA expression was significantly reduced in malignant samples compared to normal controls. Taken together, these data suggest that loss of chemerin/RARRES2 expression occurs in breast cancer during tumorigenesis, potentially as an immune evasion mechanism, and that restoring or enhancing chemerin levels within the TME may prove efficacious in increasing TIL, thereby slowing or reversing tumor progression in the clinic.
Materials and Methods
Microarray Analysis
Publicly available breast cancer studies were evaluated using the Oncomine database (www.oncomine.org), in which expression data has been curated using statistical methods and standardized normalization technique as previously described (32). The two largest breast cancer studies comparing normal to malignant tissues were selected: Curtis et al. (http://www.ebi.ac.uk/ega/studies/EGAS00000000083) (33) and TCGA (http://tcga-data.nci.nih.gov/tcga) (34). The Curtis dataset contains 1,992 breast carcinoma samples and 144 paired normal breast samples which were analyzed for the METABRIC project using the Illumina HumanHT-12 V3.0 R2 Array. The TCGA data included 532 invasive breast carcinomas and 61 paired normal breast tissue samples using level 2 (processed) data from the TCGA portal. The RARRES2 probe was selected for normal, invasive/infiltrating ductal carcinoma (IDC) and invasive/infiltrating lobular carcinoma (ILC) subsets, and gene expression (mRNA) data were shown as log2 transformed, median centered per array with p-values and fold change between subsets generate by Oncomine.
Mice and Cell Lines
All mice were used in experiments were purchased from The Jackson Laboratory. Wild type or Rag1 knockout (RAG KO; #003145, Rag1tm1Mom) (35) female BALB/c mice were used as indicated. Mice were maintained in the facilities at Washington University under the direction and guidelines of the Division of Comparative Medicine and used at approximately 9–12 weeks of age. All animal experiments were conducted in accordance with approved Washington University and National Institutes of Health Institutional Animal Care and Use Committee guidelines under an approved protocol (#20140232). The EMT6 mouse mammary carcinoma cell line was purchased from ATCC (CRL-2755). L1.2 cells transfected to express mouse CMKLR1 were a kind gift from BA Zabel. Cell lines were grown in complete media consisting of RPMI 1640 or DMEM supplemented with 10% FBS, sodium pyruvate, penicillin/streptomycin, and beta-mercaptoethanol, with or without appropriate antibiotics for selection. EMT6 cell lines (wild type and transduced) were serially tested for mycoplasma and found to be negative using the MycoProbe Mycoplasma Detection Kit (R&D Systems).
EMT6 Clone Production
The full-length gene that encodes mouse active chemerin, mouse RARRES2, was inserted into the lentiviral transfer vector pCDH1-MSC1-EF1-Puro (System Biosciences) using the NheI and EcoRI restriction enzyme digestion sites. Empty vector pCDH1-MSC1-EF1-Puro was used to produce control lentivirus. 293T/17 cells were grown in DMEM complete media in 10 cm dishes for 16 h before transfected with packaging plasmid (Δ8.2), coat protein vector (pCMV-VSV-G) and transfer vector (pCDH-Puro-wt RARRES2 or pCDH-Puro Empty vector) by using the FuGENE® HD Transfection Reagent (Promega) according to the manufacturer's protocol. The culture supernatants containing lentiviruses were collected at 48 and 72 h post transfection. The collected media were centrifuged at 300 × g to remove cell debris and followed by filtration with 0.45 μM filters. Viral supernatants were either used immediately for cell transduction or stored at −80°C. To create EMT6 cell lines with constitutive chemerin expression or control vector, viral supernatants added with polybrene were used to infect wild type EMT6 cells. Starting 24 h infection, cells were selected with media containing 2 μg/ml puromycin for 3 days. Culture media containing puromycin was replaced daily. Monoclonal cell populations were obtained by limiting dilution.
In vitro Cell Line Evaluation
EMT6-pCDH-VEC or EMT6-pCDH-RARRES2 cells (1,000 cells/well) were plated in 96-well black walled plates (Corning). Cells were grown in a 5% CO2 humidified incubator at 37°C for the indicated days. On each day, alamar blue reagent (ThermoFisher Scientific) was added directly to each well, the plates were incubated at 37°C for 1–4 h and the fluorescence signal was measured according to the manufacturer's protocol. Data were shown as relative fluorescence values compared with that of day 0, which was normalized to 1. Control and chemerin-expressing EMT6 lines were plated at 200 k/ml/well in 24 well plates and evaluated for chemerin secretion by using a mouse chemerin ELISA (R&D Systems) on 48 h conditioned media. Surface marker expression of control and chemerin-expressing clones was evaluated by flow cytometry with indicated monoclonal antibodies and appropriate isotype controls (Biolegend). The functionality of secreted chemerin was tested using conditioned media from control and chemerin-expressing clones in chemotaxis assays. Briefly, 96 well HTS Transwell Permeable Supports with 5 μm pores (Corning) were used according the manufacturers protocol; 250 k mCMKRL1+ L1.2 cells/75 μl were placed in the top chamber and 240 μl of complete media +/− 3 nM recombinant, active mouse chemerin (R&D Systems), or conditioned media in the bottom chamber. Assays were left at 37°C for ~1–1.5 h. Migrated cells in the bottom chamber were counted and percent migration calculated.
Tumor Inoculation
To evaluate the effect of constitutive chemerin secretion on in vivo tumor growth, control or chemerin-expressing EMT6 breast tumor cells (0.5–1 × 106) were inoculated subcutaneously into 9–12 weeks old female BALB/c mice (JAX). Prior to inoculation, EMT6 lines were grown to ~60–80% confluence to ensure log-growth kinetics, and viability was tested using trypan blue and ensured to be ~>95% (or cells were not used). Tumor growth was measured every 2–4 d by calipers, and size was expressed either as the volume product of perpendicular length by width in square millimeters, or by tumor size as indicated by width × length (in square mm). Mice were euthanized when tumor size reached ~400 mm2 or when tumor sites ulcerated or at indicated time points for TIL analyses.
In vivo Leukocyte Depletion
Mice were injected i.p. with 100 μl of anti-asialo GM1 or control rabbit sera (Wako Chemicals) diluted 1:10 in PBS. Mice were treated with antibodies on day 1, day 0 and every 2–3 days after tumor inoculation. NK depletion efficiency was determined by staining blood cells collected from the venous sinus. Briefly, blood samples were isolated via retro-orbital bleed and washed once with PBS. After centrifugation at 300 × g for 5 min, cells were stained with CD45, CD3, and DX5 or its isotype control (Biolegend) and analyzed by FACS. For CD4+, CD8+ T cell depletion, mice were injected i.p. with 250 μg/500 μl PBS of anti-CD4 (clone GK1.5, BioXCell), anti-CD8β (Lyt 3.2) (clone 53-5.8, BioXCell) or both, and rat IgG (Sigma) for control. Antibodies were given weekly for 3 doses. Depletion efficiency was determined by staining blood cells collected via retro-orbital bleed with CD45, CD3, CD4, and CD8 antibodies (Biolegend) and analyzed by FACS.
Evaluation of Tumor Infiltrating Leukocytes
At indicated time points, whole subcutaneous tumors were resected en bloc including overlying skin and subcutaneous tissues. Tumors were then processed into single cell suspensions as previously described (22). Briefly, cells were counted using trypan blue, and samples were blocked with PBS/FBS containing 1% rat serum and Fc block (anti-CD16/32; Biolegend). Stained samples were analyzed on a BD Fortessa. For live/dead cell discrimination, AmCyan LIVE/DEAD Fixable Dead Cell Stain kit (Invitrogen) was used. Antibodies or appropriate isotype controls were purchased from Biolegend and FlowJo software (Tree Star) was used for analysis, with gating based on appropriate isotype control staining, and percentages expressed as shown of total live tumor cells or total live CD45+ cells, as indicated. FACS analyses was used to define the follow leukocyte subsets (all Live+CD45+): plasmacytoid DCs (pDCs; Lin-CD11cintB220hi), conventional DCs (cDCs; Lin- CD11chiB220low), CD4 (CD3+CD4+) T cells, CD8 (CD3+CD8a+) T cells, total T cells (CD3+CD4+CD8+), NK cells (CD3-DX5+), monocyte/macrophages (Lin-CD11b+GR1−), MDSCs (Lin-CD11b+GR1+), M1 (Lin-CD11chiF4/80+), and M2 (Lin-CD11clowF4/80+) macrophages, CD19+ B cells (CD3-CD19+). CD8+ T cell subsets were based on staining with CD44 and CD62L: naïve (CD44lowCD62Lhi), effector (CD44intCD62Llow), or memory (CD44hiCD62Llow).
Breast Tissue Microarrays
Tissue microarray (TMA) Breast Tissue FFPE sections were collected from the St. Louis Breast Tissue Registry (funded by The Department of Surgery at Washington University School of Medicine, St. Louis, MO) under IRB-approved institutional protocols. All patient information was de-identified prior to sharing with investigators. Data and tissue was obtained in accordance with the guidelines established by the Washington University Institutional Review Board (IRB #201102394) and WAIVER of Elements of Consent per 45 CFR 46.116 (d). Each TMA core was 5 μm thick and 2 mm in diameter. Normal and Tumor tissue was confirmed by a Board-Certified Pathologist (Dr. Marshall Poger) using a stained Hematoxylin and Eosin (H&E) section. Breast Tumor TMA section contained 37 IDC cases and 8 ILC cases. Normal TMA Section contained 45 cases of Terminal Ductal-Lobular Unit (TDLU) with 1 Tonsil and 4 Liver cores for control and TMA positioning.
Quantitative Real-Time PCR
De-identified, paired RNA samples of malignant or non-malignant human breast tissues were from the Siteman Cancer Center Tissue Procurement Core, collected under an IRB-approved research protocol (#201106191). Quantitative Real-Time PCR was carried out using the SYBR® Green master mix (Bio-Rad) with the real-time PCR primers for human chemerin and the housekeeping gene GAPDH (sequence listed below). Measurements were standardized to GAPDH using delta-delta Ct methods. RNA from human liver was the positive control for chemerin expression. RNA from RAJI cells was the negative control. Data were expressed as fold expression levels of negative control (RAJI, normalized to 1). Data shown are mean ± SEM of two independent experiments using identical starting RNA. Significant outliers identified by Grubbs' test were removed. The primers used for human RARRES2 have been previously described (36): Forward: 5′- TGGAAGAAACCCGAGTGCAAA-3′; Reverse: 5′-AGAACTTGGGTCTCTATGGGG-3′ Primers for human GAPDH: Forward: 5′- GAGTCAACGGTTTGGTCGTATTG-3′; Reverse: 5′- ATGTAGTTGAGGTCAATGAAGGGG-3′.
In situ Hybridization and Analysis
Manual chromogenic RNAScope (ACDBio) was performed using company protocols on TMA tissue sections to detect target RNA at single cell level. Tissue pre-treatment (Liver) included baking for 1 h at 60 degrees Celsius, deparaffinization using xylene and alcohol, RNAscope® Hydrogen Peroxide (ACD# 322335) treatment for 10 min at RT and protease treatment (RNAscope® Protease Plus ACD# 322331) for 30 min at 40 degrees Celsius using the HybEZ Oven. Pre-treatment of non-adherent cells (RAJI) included fixation by 10% NBF and dehydration in series of 50, 70, and 100% ethanol. Cells were treated with RNAscope® Hydrogen Peroxide for 10 min at RT (ACD# 322335) and treated with RNAscope® Protease III (ACD# 322337) for 30 min in 40 degrees Celsius using HybEZ oven. For all tissue sections and non-adherent cells, ACDBio pre-treatment protocol was used according to manufacturer's instructions. Detection of specific probe binding sites was with RNAScope 2.5 HD Reagent kit—brown from ACD (Cat. No. 322310). Single ISH detection for human RARRES2 (ACD Probe: 457921), Positive Control Probe (PPIB - ACD Probe: 313901) and Negative Control Probe (Dapb—ACD Probe: 310043) was performed manually using RNAscope® 2.5 HD Reagent Kit-Brown (ACD, 322310). Target probes were hybridized for 2 h at 40 degrees Celsius using HybEZ oven and a series of 6 amplification steps followed. A DAB-based chromogenic reagent was used to detect the brown signal for the RARRES2 probe expression. The experimental procedure followed the manufacturer's instructions for single plex assay. Positive staining was indicated by brown granular dots present in the nucleus and/or cytoplasm.
Quantitative analysis was completed using regions of interest (ROIs) and by random sampling. The ROIs for Normal and Tumor breast tissue were manually selected by a Board-Certified Pathologist (Dr. Marshall Poger) for imaging. Random sampling was done by numbering each core on the TMA section and using a random number generator to select which TMA core was to be selected for analysis. HALO Software by Indica Labs was used, specifically with the RNAScope ISH Module per recommendation by ACD, with user-defined thresholds. This module allowed the user to teach HALO software to recognize hematoxylin (blue) and positive signal (brown granular dots). Positive signal is reported by number of RNA copies. The Cytonuclear Module was used to teach HALO Software to recognize hematoxylin (blue) to identify nuclei. This generated a contrasted image allowing the user to count the number of nuclei in the region of interest. ISH module provided the user the number of RNA copies and the Cytonuclear module provided the user the number of cells. Thus, RNA copies per nuclei was determined allowing analysis to be normalized to each nuclei. Slides were imaged using a Nikon eclipse 50i microscope at 40x resolution. Three comparable regions of interest for tumor (IDC and ILC) and normal breast (TDLU) were subject to HALO Software for image analysis.
Statistical Analysis
In vitro and in vivo tumor data was plotted using Prism software v7 and further analyzed with InStat (GraphPad Software). Differences between groups were evaluated by applying unpaired Student's t-test or non-parametric Mann-Whitney test, as indicated. Paired human RNA samples were evaluated by a paired student's t-test. p < 0.05 were considered significant.
Results
Reduced RARRES2 Expression in Human Tumors and Associated Poor Survival Outcome
We and others previously showed that chemerin/RARRES2 expression is commonly down-regulated in multiple tumor types, including breast cancer, compared to normal tissue controls RARRES2 (22). Our published expression analysis was limited to the publicly available GEO microarray datasets, thus, to confirm reproducibility we sought to further investigate chemerin expression in larger datasets. Here, we analyzed the two largest breast cancer datasets with data for RARRES2 that were curated within the Oncomine database (32). Chemerin/RARRES2 expression in both Curtis (Figure 1A) and TCGA (Figure 1B) datasets was significantly decreased by approximately 2.6- to 3.4-fold in tumor specimens compared to normal (33, 34). Subsequently, to examine the association between reduced RARRES2 expression and patient survival outcome, we analyzed two sets of mRNA microarray data with cohort sizes of 33 breast cancer patients and 135 early-stage breast cancer patients, respectively, and found that low chemerin levels significantly correlated with poor survival outcomes in both groups (Figures 1C,D). By in situ hybridization (ISH) comparing normal tissues to both invasive ductal carcinomas (IDC) as well as invasive lobular carcinoma (ILC)—the two most common histologic subtypes (37)—chemerin/RARRES2 was also down-regulated in the tumor samples (Figure 2). Thus, across multiple datasets and analytical expression methods, chemerin/RARRES2 was consistently down-regulated in malignant breast cancer samples vs. controls, and reduced chemerin/RARRES2 expression was correlated with poor survival outcome.
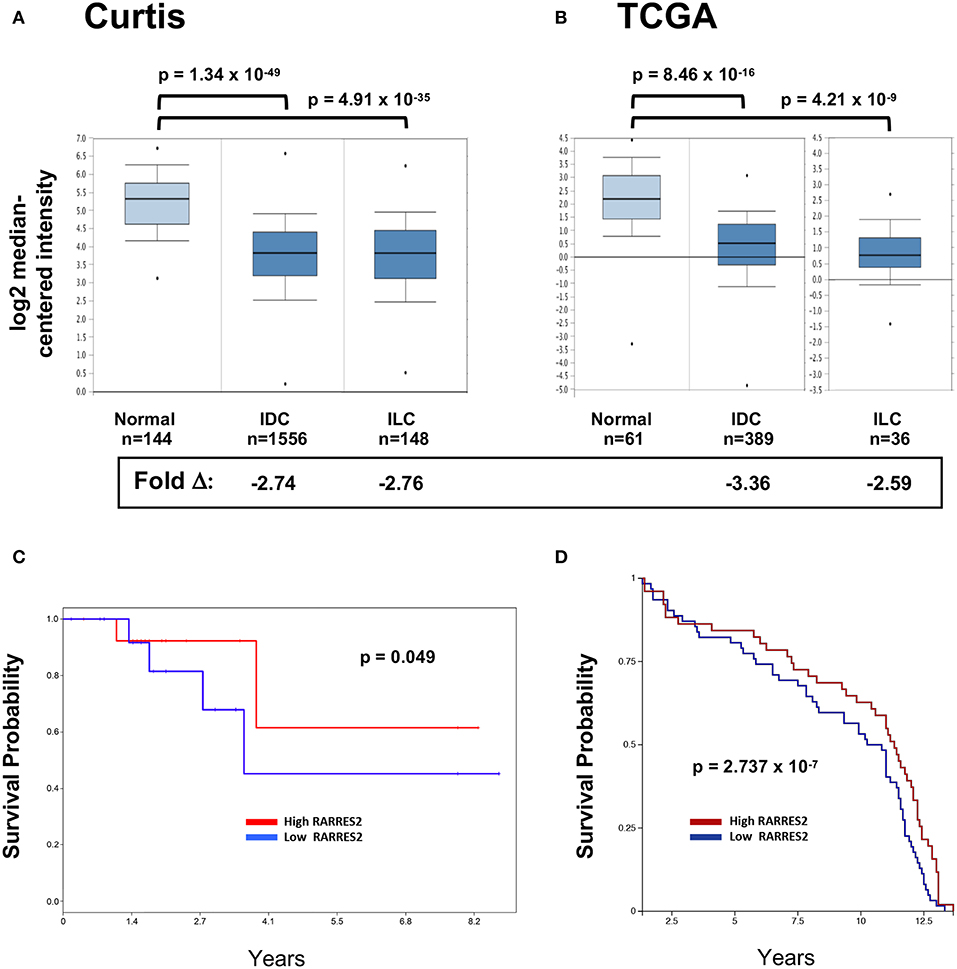
Figure 1. RARRES2 microarray expression in breast tissues. The two largest mRNA studies comparing normal and malignant breast tissues were selected in Oncomine (www.oncomine.org) for analysis. (A) Curtis, n = 2,136 total, (B) TCGA, n = 593 total. Both infiltrating ductal carcinoma (IDC) and infiltrating lobular carcinoma (ILC) subsets show significantly lower expression of RARRES2 mRNA when compared to normal breast tissue. RARRES2 probes were selected and relative expression by log2 median-centered intensity plotted for normal, IDC, and ILC subsets within each study. Oncomine calculated p-values and fold change compared with normal subset are shown. Down-regulation of chemerin in breast cancer can be associated with poor survival outcomes. (C) mRNA microarray data (accession # GSE6130-GPL887) from a cohort of 33 patients with breast cancer. mRNA microarray data was visualized using PROGgeneV2. The patients were stratified according to chemerin expression (divided at 50th percentile), and survival plotted for each group. Hazard ratio: 0.42 (0.18–1.00), p-value: 0.049, indicating that low chemerin levels significantly correlated with poor survival in this group. (D) mRNA microarray data (Caldas, Naderi Gene Exp 2007) from a cohort of 135 early-stage breast cancer, visualized using the UCSC Xena Browser. The patients were stratified according to chemerin expression (divided at 50th percentile), and survival plotted for each group. p-value: 2.737 × 10−7, indicating that low chemerin levels significantly correlated with poor survival in this group.
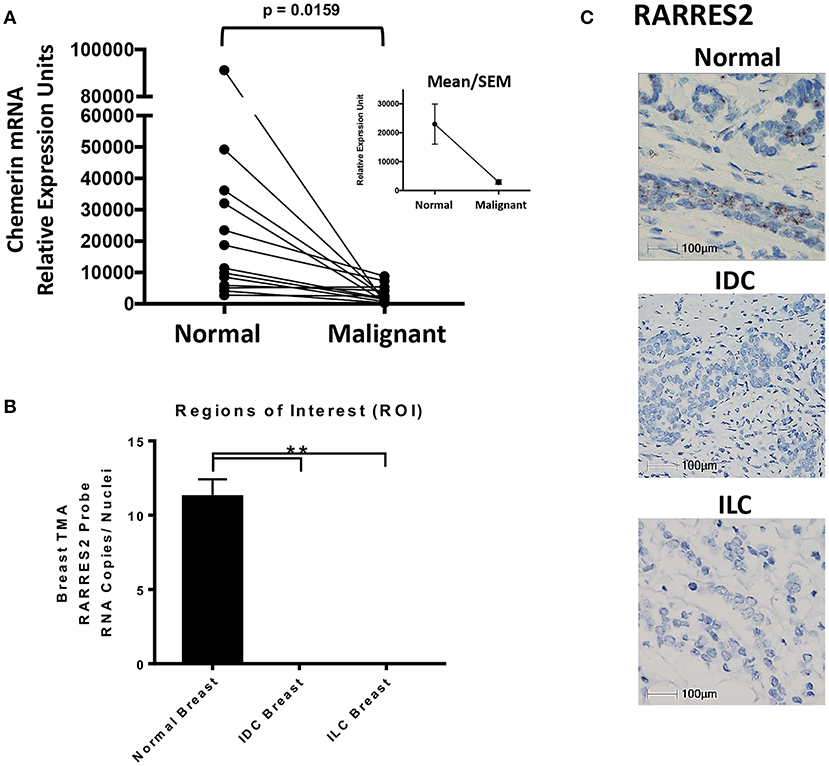
Figure 2. Chemerin RNA expression in human breast tissue. (A) Real-time quantitative PCR (RT-qPCR) of chemerin mRNA expression in matched normal and malignant breast tissue. RNA was isolated from paraffin embedded tissues and assessed for concentration, purity, and integrity. Chemerin expression was normalized to GAPDH loading control for each sample (**p = 0.0159 compared to normal breast tissue, n = 13 for each subset using two tailed paired t-test). Inset shows group means/SEM. (B) Quantified RARRES2 RNA expression in normal breast, IDC breast, and ILC breast tumor tissue microarrays (TMA) using RNAScope in-situ hybridization (ISH). Three comparable regions of interest (ROI) from each case of normal breast (n = 7) or tumor (IDC and ILC; n = 7 each) were subject to HALO Software for image analysis. The ROIs for normal and tumor breast tissue were manually selected for imaging/analysis. RNA expression is normalized to the number of nuclei in each image to determine RNA copies per nuclei. (**p = 0.0001 compared to normal breast using a one sample t-test). Results are representative of two TMAs containing (1) 45 normal cases and (2) 37 IDC cases and 8 ILC cases. (C) Representative ISH images for RARRES2 RNA expression in normal, IDC breast, and ILC breast tissue. Slides were imaged using a Nikon eclipse 50i microscope at 40X resolution; 100 mm bar shown. Positive staining is indicated by brown granular dots present in the cell nucleus and/or cytoplasm.
Reduced Chemerin Expression in Human Invasive Breast Cancers
Next, we wanted to independently validate the findings of our public microarray analyses (Figure 1). We collected human breast tissues from two different sources and, using two different modalities, evaluated chemerin expression via measurement of RARRES2 mRNA. Matched total RNA from normal and malignant breast tissues (n = 13 patients with IDC) were obtained from the Siteman Cancer Center Tissue Procurement Core. De-identified frozen samples were collected under approved consents, pathologically reviewed, and processed into RNA per established protocols. RNA quantity and quality (i.e., RIN) was assured and validated primers for human RARRES2 (36) were used in real-time quantitative PCR. Expression of RARRES2 mRNA in malignant breast tissues was significantly reduced compared to patient matched, normal tissue (Figure 2). Group mean/SEM of individually matched samples (Figure 2A) are shown. Next, using samples collected from the St. Louis Breast Tissue Registry under IRB-approved institutional protocols, we then constructed normal (n = 45 cases) and malignant breast tissue microarrays (TMA) incorporating both IDC (n = 37 cases) and ILC (n = 8 cases). Utilizing ACDBio RNAscope, RNA in situ hybridization (ISH) was performed. RARRES2 was undetectable in both IDC and ILC samples compared to low but significant RARRES2 signal in normal tissues (Figure 2B). Duplicate TMA slides were used with positive and negative probes (ACDBio), in parallel with human liver (RARRES2-positive) and Raji cells (RARRES2-negative) as controls (Supplemental Figure 1). Representative images are shown in Figure 2C, with the majority of staining localized to epithelial components of the normal breast tissue. Taken together, our data confirms significant down-regulation of RARRES2 mRNA expression in both IDC and ILC compared to normal breast tissues.
Forced Expression of Chemerin by EMT6 Breast Carcinoma
After confirming down-regulation of RARRES2 mRNA in additional human studies, we then set out to favorably modulate chemerin expression in the EMT6 mammary carcinoma model. The EMT6 tumor line is a clonal isolate from a mouse mammary carcinoma that arose from an implanted hyperplastic alveolar nodule (38), and has been shown to be responsive to immunomodulation (31, 39, 40). In order to test our hypothesis that forced overexpression of chemerin by tumor cells would act to recruit anti-tumor leukocytes and suppress tumor growth, we used lentiviral transduction to introduce the mouse RARRES2 gene into EMT6 tumor cells. The pCDH1-MSC1-EF1-Puro (System Biosciences) vector was used to produce either control (empty vector) or RARRES2 viral particles for transduction. Control and chemerin-expressing EMT6 clonal lines were generated by limiting dilution plating. Evaluation of tumor-secreted chemerin was assessed by mouse chemerin ELISA (R&D Systems). Both wild type and control-transduced EMT6 lines showed no detectable chemerin by ELISA (not shown), while RARRES2-transduced clones showed significant production of secreted chemerin in the ng/ml range (Figure 3A). From these, two clones, one with low (LC) and one with high (HC) chemerin expression, were then selected for further evaluation. In order to determine if the tumor-secreted chemerin was functional and active, we utilized standard chemotaxis assays using 5 um pore transwell chambers. Conditioned media from both control and chemerin-expressing tumor lines was evaluated. The mouse pre-B lymphocyte cell line L1.2 engineered to express high levels of mouse CMKLR1 (10) was used to assess chemerin-dependent migration. Conditioned media from control transduced lines was unable to induce CMKLR1+ L1.2 cell chemotaxis, while conditioned media from chemerin-expressing tumor lines triggered robust migration comparable to recombinant, active chemerin (3 nM, R&D Systems). Chemotaxis of CMKRL1+ L1.2 cells in the HC clone was ~2-fold compared to the LC clone, in line with measured secreted chemerin levels (Figure 3B). In order to assess the effects of chemerin production and secretion on in vitro tumor cell proliferation, we utilized an alamar blue assay (ThermoFisher) and measured growth as a function of fluorescence signal over several days. There were no consistent differences between control or chemerin-expressing EMT6 clones (Figure 3C). Next, we looked at expression of several common surface markers involved in tumor-immune recognition (MHC class I, CD1d, PD-L1) as well as tumor cell migration and invasion (CD44) (41). While CMKLR1 has been reported to be expressed on some human tumors (42), we did not see detectable surface levels of CMKLR1 above isotype control, in line with our prior studies of the mouse melanoma line B16F0 (22). Figure 3D shows comparable phenotypic expression of these markers between control and chemerin-expressing tumor lines. These data show that transduction with RARRES2 and expression/secretion of chemerin by EMT6 tumor cells does not appear to meaningfully impact in vitro growth or the immunophenotype of key surface proteins, and that secreted chemerin is functionally active and can induce migration of CMKLR1+ cells.
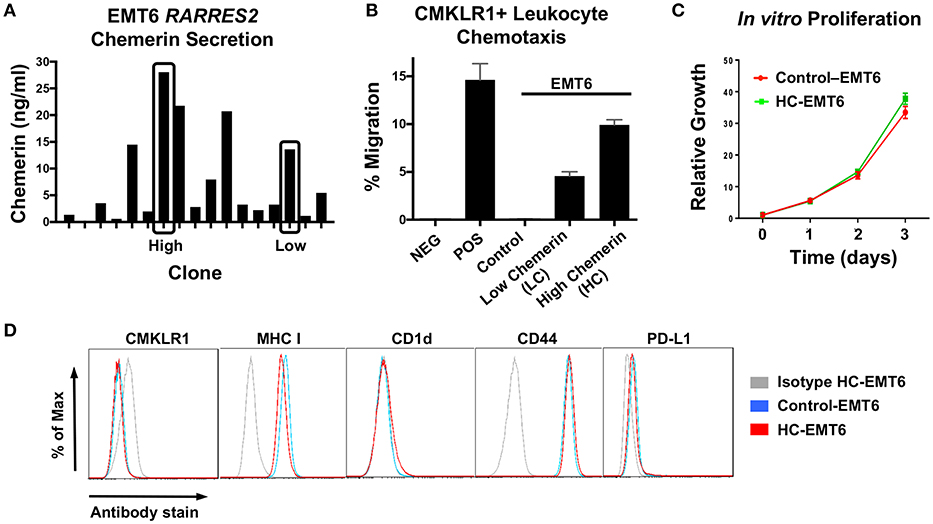
Figure 3. Evaluation of chemerin-expressing EMT6 tumor lines. The mouse breast carcinoma line EMT6 was used to study the impact of forced chemerin expression on tumor growth. Wild type EMT6 cells were transduced using the lentiviral transfer vector pCDH1-MSC1-EF1-Puro, with the full length mouse RARRES2 gene inserted (RARRES2) or not (control). (A) Clonal cell lines (n = 17) were generated and chemerin protein secretion in clonal conditioned media samples was quantified by ELISA. Clones with low (LC) and high chemerin (HC) secretion (indicated by boxes) were selected for subsequent in vitro and in vivo analysis. (B) Chemotaxis assays using CMKLR1+L1.2 transfectants and 5 micron transwell chambers were performed to confirm functionality of EMT6-secreted chemerin. Conditioned media was used from control and chemerin-expressing tumor lines (LC, HC). Media alone (NEG) and 3 nM recombinant mouse chemerin (POS; R&D Systems) were used as controls. The normalized “percent of input” migration is shown; mean/SEM plotted for duplicate wells for each condition. Representative proliferation and chemotaxis assays are shown, and were each performed several times prior to tumor inoculation in mice. (C) Control and high chemerin-expressing lines were evaluated for in vitro cell proliferation using an alamar blue assay; relative fluorescence values normalized to 1 on day 0 are shown. (D) Surface expression of CMKLR1, MHC class I, CD-1d, CD44, and PD-L1 was determined by FACS for control and high chemerin-expressing EMT6 tumor lines. For each marker indicated, the appropriate isotype control antibody (gray) is shown.
Chemerin Overexpression Suppresses EMT6 Tumor Growth in vivo
Given that chemerin-overexpression failed to impact EMT6 proliferation in vitro or expression of MHC class I, CD1d, CD44, or PD-L1, we next wanted to study the impact of chemerin expression in the TME on in vivo growth. Using WT female BALB/c recipients, control or chemerin-expressing EMT6 tumor cells were orthotopically implanted into the mammary fat pad as described (22). To determine if the level of chemerin secretion from transduced clones affected in vivo tumor growth, we implanted low-chemerin (LC) and high-chemerin (HC)-secreting clones. The in vivo growth of HC EMT6 tumors was significantly suppressed compared to LC- or control-EMT6 cells (Figure 4A), with some mice showing complete suppression of in vivo tumorigenesis. To confirm this was not an effect of clonality, we utilized completely independent, bulk transduced EMT6 tumor cell lines (i.e., polyclonal) and saw a similar significant reduction in in vivo tumor growth (Figures 4A,B). This might suggest that an adequate concentration gradient of chemerin within the TME needs to be established to recruit anti-tumor leukocytes and suppress tumor growth. Indeed, there was an approximately 2-fold increase in the total CD45+ tumor infiltrating leukocytes (TIL) relative to tumor cells in HC-EMT6 tumors compared to LC- or control-EMT6 tumors at time of euthanasia (Figures 4C,D). We next looked at the composition of infiltrating leukocyte subsets in the TME by flow cytometry and identified significant increases in the relative percentages of total T cells, CD4+ T cells, and NK cells among total CD45+ cells in HC-EMT6 tumors compared with controls by day 35 of tumor growth (Figures 4E,F; Supplemental Figure 2). CD8+ T cells were also enriched among the total CD45+ cells in the HC-EMT6 tumors by day 35, compared to day 14 (Figure 4G). However, no significant differences in percentages of total T cells, CD4+ or CD8+ T cell subsets, B cells, NK cells, cDCs, pDCs, MDSCs, or macrophages among CD45+ TILs were detected between the two groups at an earlier time point in tumor growth (day 14, not shown), potentially suggesting that sufficient time is needed to establish an adequate concentration gradient of chemerin and resultant chemoattraction of effector cells. No significant differences were seen in either CD4+ or CD8+ regulatory T cells (CD25+FoxP3+) between the groups at either early or late time points (Figures 4H,I). Taken together, these data show high-chemerin expression within the EMT6 TME results in significant tumor growth suppression and a favorable anti-tumor skewing of both NK cells and T cells, as a percentage of total TIL.
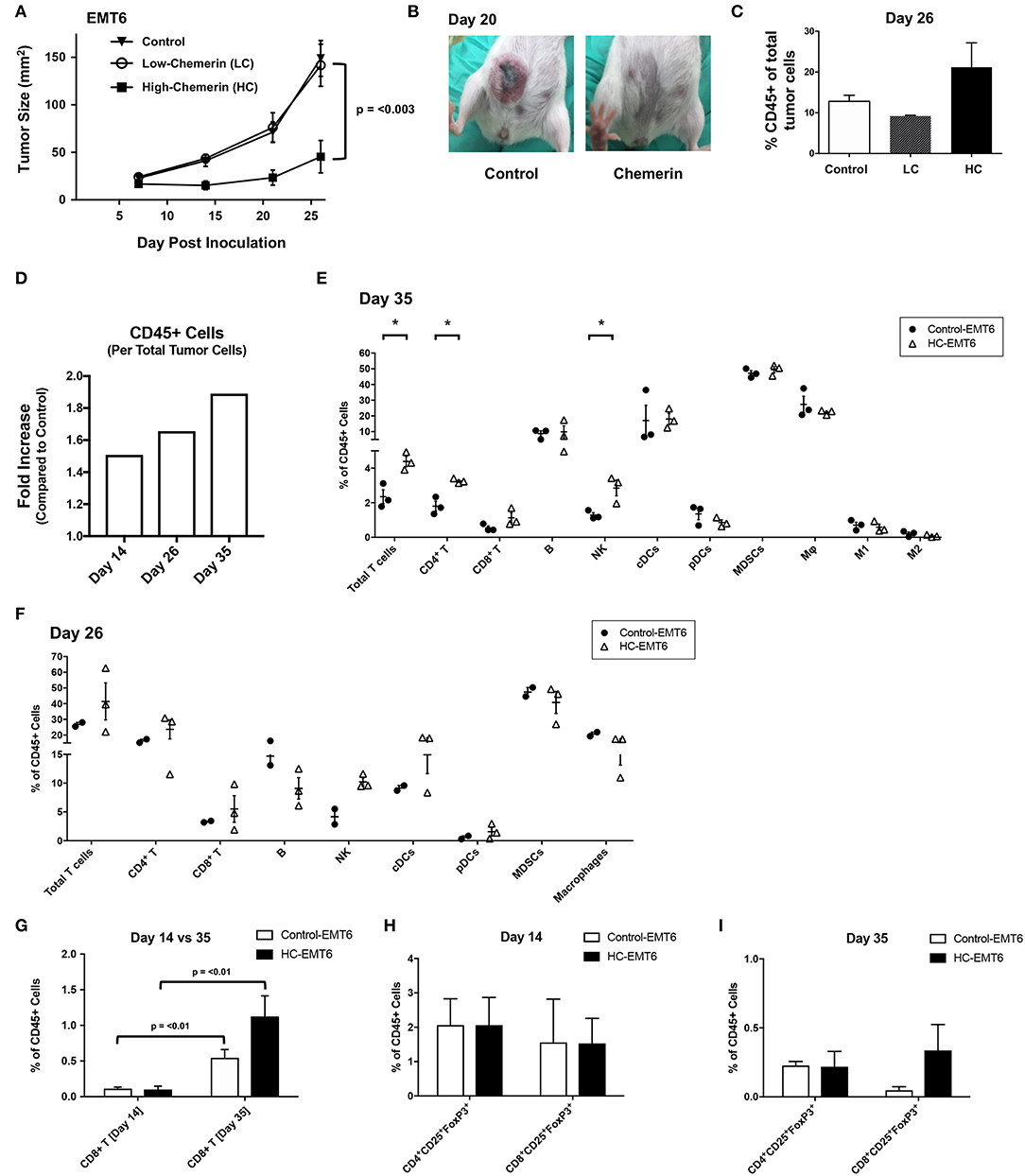
Figure 4. High chemerin expression by EMT6 tumors suppresses in vivo growth and results in altered TIL makeup. (A) High and low chemerin-expressing EMT6 clones were used in parallel with a control transduced EMT6 cells. 0.5–1 × 106 tumor cells were inoculated subcutaneously into 9–12 weeks old female BALB/c mice. Tumor growth was serially assessed using calipers. Mean/SEM shown with groups n = 10 per indicated line. Two-way ANOVA with Tukey's multiple comparisons test show no significant differences between control and LC groups, with p-value as indicated for differences compared to HC group. (B) Representative images of mice from control and chemerin-expressing groups showing significant in vivo suppression of tumorigenesis, 20 days post-inoculation. (C) Percent CD45 positive of total tumor cells by FACS analysis within the tumor microenvironment (TME) shown for (A) tumors (n = 2–3/group) resected at time of euthanasia (day 26). (D) Graph of fold increase showing CD45+ cells per total tumor cells, comparing HC-EMT6 tumors to control-EMT6 tumors resected at time of euthanasia (day 14, n = 6/group; day 26, n = 2–3/group; day 35, n = 3/group) and analyzed via FACS. Graph depicts two independent experiments; tumors collected on day 14 and day 35 are derived from the same experiment. (E,G–I) In a separate cohort of mice, we euthanized animals at pre-defined timepoints for TIL analysis. Of the mice that were initially inoculated; six mice per group were euthanized on day 14 for FACS analysis, and an additional three mice per group were euthanized on day 35 for further FACS analysis. Graphs show mean/SEM values; statistical significance (defined in Methods) was determined between groups using a 2-sided unpaired t-test. (E) FACS analysis of TIL from control-EMT6 or HC-EMT6 tumors (n = 3/group) resected on day 35. (F) FACS analysis of TIL from control-EMT6 or HC-EMT6 tumors resected on day 26 (n = 2–3/group). (G) FACS analysis of TIL from HC-EMT6 tumors (n = 6) resected on day 14 compared to TIL from HC-EMT6 tumors resected on day 35 (n = 3), specifically showing the CD8+ T cell population. (H) FACS analysis of TIL from control-EMT6 or HC-EMT6 tumors resected on day 14 (n = 6/group), specifically showing CD4+ and CD8+ regulatory T cell populations (CD25+FoxP3+). (I) FACS analysis of TIL from control-EMT6 or HC-EMT6 tumors resected on day 35 (n = 3/group), specifically showing CD4+ and CD8+ regulatory T cell populations (CD25+FoxP3+). *P < 0.05.
The Anti-Tumor Effects of Chemerin Are Mediated by NK Cells and T Cells
Our initial in vivo EMT6 tumor data identified a correlation among high-chemerin expression by EMT6 tumors, increased NK cells and T cells in the TIL population and suppressed tumor growth. (Figure 4). To further define the cellular mechanism of action of chemerin-dependent tumor growth suppression, we selectively depleted candidate lymphocyte subsets (or used genetically-modified subset-deficient animals) and evaluated HC-EMT6 tumor growth. We first used anti-asialo GM1 to deplete NK cells. Control and chemerin-expressing lines were inoculated into mice treated with control sera or anti-asialo GM1 (Wako Chemicals) sera. Anti-asialo GM1 treatment had no effect on control EMT6 tumor growth in vivo, while similar treatment resulted in the complete abrogation of tumor suppression in the chemerin-expressing tumors (Figure 5A). There were no significant differences noted between the growth of control-EMT6 tumors (+/- anti-asialo GM1) and NK cell-depleted chemerin-expressing tumors (Figure 5A). The extent of NK cell depletion was confirmed by analysis of peripheral blood prior to tumor inoculation (Figure 5B; Supplemental Figure 3A). Next, to explore the potential role of adaptive immunity in chemerin-dependent EMT6-tumor growth suppression, we used Rag1 KO mice, which lack mature T and B cells (35). Growth suppression by tumor-secreted chemerin was only seen in wild type mice and was completely abrogated in RAG KO mice (Figure 5C), suggesting a requirement of the adaptive immune response in this model. Given the lack of change in B cells and the significant increase in T cells in the TIL population in chemerin-expressing tumors, we then set out to define specific T cell subsets responsible for the chemerin-dependent anti-tumor effect. We used specific antibodies to deplete CD4+ and/or CD8+ T cells as indicated. Control antibody treatment did not affect suppression of tumor growth in chemerin-expressing tumors. However, depletion of CD8+ T cells in chemerin-expressing tumors—either alone or in combination with CD4+ T cell depletion—resulted in growth comparable to control tumors (Figure 5D). T cell subset depletion was confirmed by analysis of peripheral blood, which was essentially complete (Figure 5E; Supplemental Figure 3B). Interestingly, CD4+ T cell depletion alone in chemerin-expressing tumors resulted in improved tumor growth suppression (Figure 5D). Recently published data show that CD4+ T cell depletion in the EMT6 model results in a significant increase in CD45+ TIL, with a ~3-fold increase in IFNγ+CD8+ T cells in the draining lymph nodes compared to controls. CD4+ T cell depletion—as in our model—resulted in significantly reduced tumor growth, hypothesized to be due to a reduction in immunosuppressive regulatory CD4+ T cells (40). In line with this data, analysis of our control and T cell depleted cohorts showed a significant increase (~3-fold) in total CD45+ TIL only in the CD4+ T cell depleted mice (not shown). Taken together, these data suggest critical roles for both NK and CD8+ T cells in mediating chemerin tumor suppression.
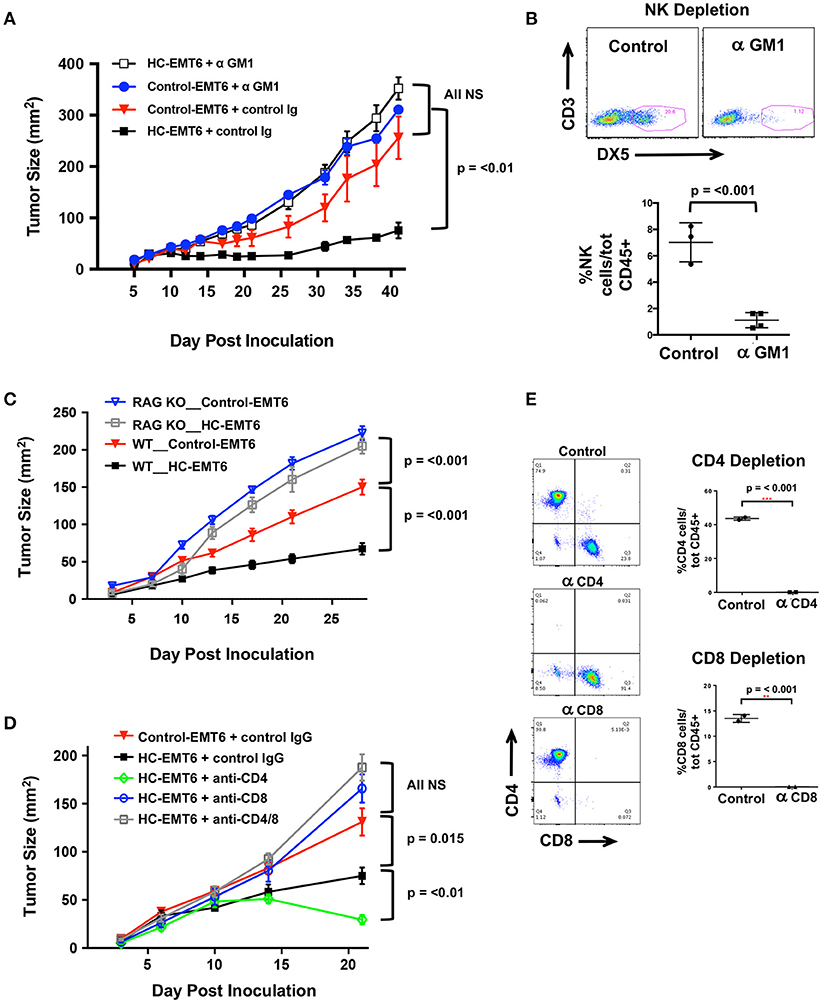
Figure 5. Depletion of NK, T cells abolishes chemerin-induced tumor suppression. (A) Control or high chemerin-expressing (HC) EMT6 cells were inoculated in wild type BALB/c mice (n = 4–5/group). NK cell depletion was accomplished using anti-asialo GM1 (control rabbit sera was used as a negative control) (Wako Chemicals). (B) Depletion of blood NK cells was confirmed in each experiment at either day −1 or day 0 (time of inoculation). (C) Control and chemerin-expressing lines were inoculated in both wild type (WT; n = 10/group) and Rag-1 knockout (RAG KO, Jackson Labs; n = 7/group). (D) Antibody depletion of T cell subsets (CD4, CD8) was accomplished using i.p., injection of 250 ug/500 ul PBS of anti-CD4 (clone GK1.5, BioXCell), anti-CD8β (Lyt 3.2) (clone 53-5.8, BioXCell) or both, or control rat IgG (Sigma). Control or depleting antibodies were used in both control and chemerin-expressing (HC) tumors as indicated (n = 5–6/group). (E) Depletion of blood T cell subsets was confirmed in each experiment at either day−1 or day 0 (time of inoculation). Graphs show mean/SEM from representative experiments with similar results (n = 4 experiments for NK depletion, n = 2 experiments each for T cell depletion, RAG KO studies). P-values are indicated from 2-tailed unpaired t-tests between indicated groups at time of euthanization.
Discussion
Chemerin is a multifunctional protein with wide tissue expression and myriad roles in host defense, implicated in antibiosis, angiogenesis, as well as chemoattraction of leukocytes (43). Several groups have described its dysregulation in the context of tumorigenesis, with the majority—but not all—showing decreased chemerin/RARRES2 expression within malignant tissues (20–22, 42, 44–48). Our group was the first to show tumor suppression via therapeutic modulation of chemerin in a mouse tumor model, with now several studies confirming the role of chemerin as a tumor suppressor in various settings (22, 45, 46, 48, 49). Importantly, two independent studies showed not only improved patient survival but also increased immune effector cell infiltrates in tumor samples with higher chemerin expression (20, 21). Our prior studies in the B16F0 mouse melanoma model showed increases in tumor-infiltrating NK and T cells with forced overexpression of chemerin by tumor cells, with suppression mediated by NK cells in that model (22). This led us to hypothesize that chemerin may play a key role in tumor immune surveillance and, further, that malignant tissues may selectively down-regulate chemerin/RARRES2 as a means of immune escape (Figure 6).
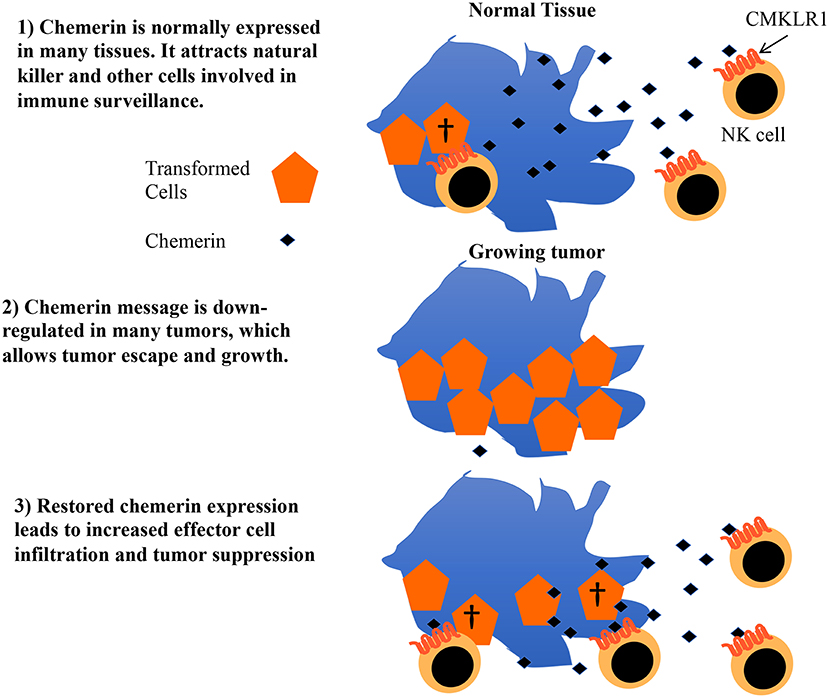
Figure 6. Proposed model for role of chemerin in tumor immune surveillance and suppression. Chemerin/RARRES2 is down-regulated in breast and other tumors compared to their normal tissue counterparts. Data from our mouse tumor models show that forced over-expression of chemerin within the tumor microenvironment (TME) results in an increase in tumor-infiltrating leukocyte effector subsets (NK cells, CD8+ T cells) that are required to suppress tumor growth. Strategies focused on augmenting chemerin in the TME may represent an attractive strategy to increase effector cell composition within the tumor and potentially favorably impact the effect of existing immunotherapies such as checkpoint inhibitors in otherwise treatment-refractory tumors.
Human breast cancers have variable levels of infiltrating immune cells, with ER/PR+HER2- subtypes typically showing the lowest (2). Breast cancer subtypes with high TILs may also show higher expression of checkpoint molecules such as PD-1 and CTLA-4 (50), which may play a role in higher response rates to checkpoint inhibitors in these tumor subtypes (e.g., ER/PR-HER2-, HER2+) (51). Decreased levels of TIL have been described in metastatic breast tumors compared to matched primary tumors (52), suggesting a role for immune escape in breast cancer progression. Thus, strategies to increase TIL and improve immunosurveillance in breast cancer are attractive from a therapeutic standpoint.
Here, we present—for the first time to our knowledge—studies focused on the expression and role of chemerin/RARRES2 in human breast tissues and a mouse model of breast cancer. Using the fully immune competent mouse EMT6 breast tumor model, we have shown that overexpression and secretion of chemerin by tumor cells significantly suppressed tumor growth in vivo. As in our melanoma model, chemerin appears to have no significant effect on tumor intrinsic proliferation or phenotype in vitro, though this may be a function of specific tumor types as well as the presence or absence of chemerin receptors on tumor cells, as others have shown direct effects of chemerin on tumor cells (45, 46, 48, 49). RARRES2-transduced EMT6 clones with lower expression of chemerin grew similarly to control cells in vivo, suggesting that in this model adequate expression and secretion of chemerin within the TME is necessary to successfully establish the concentration gradient necessary to recruit leukocytes.
The EMT6 mouse tumor model has recently been shown to recapitulate an “immune excluded” tumor phenotype with exclusion of CD8+ T cells from the tumor parenchyma, often seen in human tumors such as urothelial cancers (31). Importantly, we found meaningful increases in both NK and T cells within chemerin-expressing tumors compared to controls, similar to our findings in the melanoma model. Depletion studies indicate important roles for NK and CD8+ T cells in mediating the tumor suppressive effects of chemerin in this model, not surprisingly as supportive roles of NK cells in T cell function and the adaptive immune response are well described (53–56). Though chemerin does not seem to directly recruit CD8+ T cells via CMKLR1 interactions in this model, there is compelling evidence in the literature to suggest that NK cells mediate various functions that enhance CD8+ T cell cytolytic activity; for example, NK cells have been shown to moderate CD8+ T cell priming during influenza A viral infection and activate CD8+ T cell anti-tumor activity in the YAC-1 mouse lymphoma model (57, 58). Other studies have found that intratumor NK cell recruitment induces further leukocyte infiltration into the tumor (59), together articulating the point that chemerin may not need to act directly on CD8+ T cells to play a role in chemerin-dependent tumor growth inhibition. Additionally, ongoing studies include the impact of chemerin expression on the establishment of immune memory as well as the development of metastatic disease in this model.
Our de novo studies of human breast tissues using two independent cohorts of normal, IDC, and ILC samples across two assay platforms confirm large publicly available microarray datasets showing RARRES2 is significantly down-regulated in breast malignancies. Additionally, analysis of two mRNA microarray datasets showed that reduced chemerin levels significantly correlated with poor survival outcomes. In our in vivo experiments, we did not directly assess the effects of chemerin down-regulation/silencing during tumorigenesis in the EMT6 model. Rather, we focused on studying the potential therapeutic activity of restoring and/or overexpressing chemerin in the TME. Additional tumor studies are needed in animals with spontaneous carcinomas to determine whether chemerin down-regulation in the TME correlates with poor survival and thus models the clinical results we described in Figures 1, 2. Given the variability within and across tumor types, evaluation of chemerin/RARRES2 and receptor expression will be important prior to pursuing human translational studies. Importantly, recently published data provides a mechanistic link between chemerin and PTEN expression and function in hepatocellular carcinoma (48), suggesting chemerin may have other tumor suppressive mechanisms of action in addition to the recruitment of immune effector cells into the TME. Taken together our data elucidate mechanistic insights into the role of chemerin in breast tumor suppression and provide rationale for translational studies in human breast cancer.
Ethics Statement
This study was carried out in compliance with the ethics policies of Washington University School of Medicine. All animal studies were carried out under institutional IACUC-approved protocol, and human tissues were obtained from the St. Louis Breast Tissue Registry (funded by The Department of Surgery at Washington University School of Medicine, St. Louis, MO) in accordance with the guidelines established by the Washington University Institutional Review Board (IRB #201102394) and WAIVER of Elements of Consent per 45 CFR 46.116 (d). All patient information was de-identified prior to sharing with investigators. All of the human research activities and all activities of the IRBs designated in the Washington University (WU) Federal Wide Assurance (FWA), regardless of sponsorship, are guided by the ethical principles in The Belmont Report: Ethical Principles and Guidelines for the Protection of Human Subjects Research of the National Commission for the Protection of Human Subjects of Biomedical and Behavioral Research.
Author Contributions
RP, EB, and BZ conceived and designed experiments. RP, W-IL, PW, GV, KR, JR, YZ, LN, and NS performed experiments. RP, PW, GV, KR, WS, VN, and BZ analyzed data. RP, PW, WS, GV, KR, and BZ wrote the manuscript.
Funding
This work is supported by NIH/NIAID R01 CA169354 (EB, RP), American Cancer Society MSRG 125078-MRSG-13-244-01-LIB, California Breast Cancer Research Program, ASCO YIA, and the Kimmel Foundation (RP). This work was also supported by the NIH (R01AI079320 to BZ; R01CA169354 and R01GM037734 to EB), and the Emerson Collective Cancer Research Fund (BZ and EB). This work was supported in part by Merit Review Award Number I01 BX004115 from the United States (U.S.) Department of Veterans Affairs Biomedical Laboratory R&D (BLRD) Service to BZ. NS was supported by a diversity supplement from the NIH (3R01CA169354-03S1), and JR was supported by an SJSU NIH-MARC fellowship from the NIGMS Grant #4T34GM008253-30.
Conflict of Interest Statement
W-IL is currently employed by Dynavax Technologies, Berkeley, CA; no experiments, data-sharing or analyses, or other work were performed at or in collaboration with Dynavax.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We are grateful to Dr. Marshall Poger for his help with pathologic review and analyses of human tissues. We would like to thank Kayla Corman and Christina Mizerny for administrative support. We kindly thank Tom Walsh and the St. Louis Breast Tissue Registry for production of human breast tissue microarrays. We thank the Alvin J. Siteman Cancer Center at Washington University School of Medicine and Barnes-Jewish Hospital in St. Louis, MO. and the Institute of Clinical and Translational Sciences (ICTS) at Washington University in St. Louis, for the use of the Tissue Procurement Core, which provided human breast tissue RNA. The Siteman Cancer Center is supported in part by an NCI Cancer Center Support Grant #P30 CA091842 and the ICTS is funded by the National Institutes of Health's NCATS Clinical and Translational Science Award (CTSA) program grant #UL1 TR002345.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2019.00983/full#supplementary-material
References
1. Stanton SE, Adams S, Disis ML. Variation in the incidence and magnitude of tumor-infiltrating lymphocytes in breast cancer subtypes: a systematic review. JAMA Oncol. (2016) 2:1354–60. doi: 10.1001/jamaoncol.2016.1061
2. Stanton SE, Disis ML. Clinical significance of tumor-infiltrating lymphocytes in breast cancer. J Immunother Cancer. (2016) 4:59. doi: 10.1186/s40425-016-0165-6
3. Xu T, He BS, Liu XX, Hu XX, Lin K, Pan YQ, et al. The predictive and prognostic role of stromal tumor-infiltrating lymphocytes in HER2-positive breast cancer with trastuzumab-based treatment: a meta-analysis and systematic review. J Cancer. (2017) 8:3838–48. doi: 10.7150/jca.21051
4. Tumeh PC, Harview CL, Yearley JH, Shintaku IP, Taylor EJ, Robert L, et al. PD-1 blockade induces responses by inhibiting adaptive immune resistance. Nature. (2014) 515:568–71. doi: 10.1038/nature13954
5. Denkert C, von Minckwitz G, Brase JC, Sinn BV, Gade S, Kronenwett R, et al. Tumor-infiltrating lymphocytes and response to neoadjuvant chemotherapy with or without carboplatin in human epidermal growth factor receptor 2-positive and triple-negative primary breast cancers. J Clin Oncol. (2015) 33:983–91. doi: 10.1200/JCO.2014.58.1967
6. Daud AI, Loo K, Pauli ML, Sanchez-Rodriguez R, Sandoval PM, Taravati K, et al. Tumor immune profiling predicts response to anti-PD-1 therapy in human melanoma. J Clin Invest. (2016) 126:3447–52. doi: 10.1172/JCI87324
7. Vonderheide RH, Domchek SM, Clark AS. Immunotherapy for breast cancer: what are we missing? Clin Cancer Res. (2017) 23:2640–6. doi: 10.1158/1078-0432.CCR-16-2569
8. Nagpal S, Patel S, Jacobe H, DiSepio D, Ghosn C, Malhotra M, et al. Tazarotene-induced gene 2 (TIG2), a novel retinoid-responsive gene in skin. J Invest Dermatol. (1997) 109:91–5.
9. Wittamer V, Franssen J-D, Vulcano M, Mirjolet J-F, Le Poul E, Migeotte I, et al. Specific recruitment of antigen-presenting cells by chemerin, a novel processed ligand from human inflammatory fluids. J Exp Med. (2003) 198:977–85. doi: 10.1084/jem.20030382
10. Zabel BA, Allen SJ, Kulig P, Allen JA, Cichy J, Handel TM, et al. Chemerin activation by serine proteases of the coagulation, fibrinolytic, and inflammatory cascades. J Biol Chem. (2005) 280:34661–6. doi: 10.1074/jbc.M504868200
11. Parolini S, Santoro A, Marcenaro E, Luini W, Massardi L, Facchetti F, et al. The role of chemerin in the colocalization of NK and dendritic cell subsets into inflamed tissues. Blood. (2007) 109:3625–32. doi: 10.1182/blood-2006-08-038844
12. Banas M, Zabieglo K, Kasetty G, Kapinska-Mrowiecka M, Borowczyk J, Drukala J, et al. Chemerin is an antimicrobial agent in human epidermis. PLoS ONE. (2013) 8:e58709. doi: 10.1371/journal.pone.0058709
13. Kaur J, Adya R, Tan BK, Chen J, Randeva HS. Identification of chemerin receptor (ChemR23) in human endothelial cells: chemerin-induced endothelial angiogenesis. Biochem Biophys Res Commun. (2010) 391:1762–8. doi: 10.1016/j.bbrc.2009.12.150
14. Zabel BA, Nakae S, Zúñiga L, Kim J-Y, Ohyama T, Alt C, et al. Mast cell–expressed orphan receptor CCRL2 binds chemerin and is required for optimal induction of IgE-mediated passive cutaneous anaphylaxis. J Exp Med. (2008) 205:2207–20. doi: 10.1084/jem.20080300
15. Yoshimura T, Oppenheim JJ. Chemokine-like receptor 1 (CMKLR1) and chemokine (C-C motif) receptor-like 2 (CCRL2); two multifunctional receptors with unusual properties. Exp Cell Res. (2011) 317:674–84. doi: 10.1016/j.yexcr.2010.10.023
16. Monnier J, Lewén S, O'Hara E, Huang K, Tu H, Butcher EC, et al. Expression, regulation and function of atypical chemerin receptor CCRL2 on endothelial cells. J Immunol. (2012) 189:956–67. doi: 10.4049/jimmunol.1102871
17. Mazzotti C, Gagliostro V, Bosisio D, Del Prete A, Tiberio L, Thelen M, et al. The atypical receptor CCRL2 (C-C Chemokine Receptor-Like 2) does not act as a decoy receptor in endothelial cells. Front Immunol. (2017) 8:1233. doi: 10.3389/fimmu.2017.01233
18. Bondue B, Wittamer V, Parmentier M. Chemerin and its receptors in leukocyte trafficking, inflammation and metabolism. Cytokine Growth Factor Rev. (2011) 22:331–8. doi: 10.1016/j.cytogfr.2011.11.004
19. De Henau O, Degroot GN, Imbault V, Robert V, De Poorter C, McHeik S, et al. Signaling properties of chemerin receptors CMKLR1, GPR1 and CCRL2. PLoS ONE. (2016) 11:e0164179. doi: 10.1371/journal.pone.0164179
20. Lin W, Chen YL, Jiang L, Chen JK. Reduced expression of chemerin is associated with a poor prognosis and a lowed infiltration of both dendritic cells and natural killer cells in human hepatocellular carcinoma. Clin Lab. (2011) 57:879–85.
21. Zhao S, Li C, Ye Y-b, Peng F, Chen Q. Expression of chemerin correlates with a favorable prognosis in patients with non-small cell lung cancer. Lab Med. (2011) 42:553–7. doi: 10.1309/lmww79nits6zadpt
22. Pachynski RK, Zabel BA, Kohrt HE, Tejeda NM, Monnier J, Swanson CD, et al. The chemoattractant chemerin suppresses melanoma by recruiting natural killer cell antitumor defenses. J Exp Med. (2012) 209:1427–35. doi: 10.1084/jem.20112124
23. Alholle A, Brini AT, Gharanei S, Vaiyapuri S, Arrigoni E, Dallol A, et al. Functional epigenetic approach identifies frequently methylated genes in Ewing sarcoma. Epigenetics. (2013) 8:1198–204. doi: 10.4161/epi.26266
24. Liu-Chittenden Y, Patel D, Gaskins K, Giordano TJ, Assie G, Bertherat J, et al. Serum RARRES2 is a prognostic marker in patients with adrenocortical carcinoma. J Clin Endocrinol Metab. (2016) 101:3345–52. doi: 10.1210/jc.2016-1781
25. Lin PC, Giannopoulou EG, Park K, Mosquera JM, Sboner A, Tewari AK, et al. Epigenomic alterations in localized and advanced prostate cancer. Neoplasia. (2013) 15:373–83. doi: 10.1593/neo.122146
26. Mohr S, Bottin MC, Lannes B, Neuville A, Bellocq JP, Keith G, et al. Microdissection, mRNA amplification and microarray: a study of pleural mesothelial and malignant mesothelioma cells. Biochimie. (2004) 86:13–9. doi: 10.1016/j.biochi.2003.11.008
27. Wang N, Wang QJ, Feng YY, Shang W, Cai M. Overexpression of chemerin was associated with tumor angiogenesis and poor clinical outcome in squamous cell carcinoma of the oral tongue. Clin Oral Investig. (2014) 18:997–1004. doi: 10.1007/s00784-013-1046-8
28. Ghallab NA, Shaker OG. Serum and salivary levels of chemerin and MMP-9 in oral squamous cell carcinoma and oral premalignant lesions. Clin Oral Investig. (2017) 21:937–47. doi: 10.1007/s00784-016-1846-8
29. Shi W, Siemann DW. Augmented antitumor effects of radiation therapy by 4-1BB antibody (BMS-469492) treatment. Anticancer Res. (2006) 26:3445–53
30. Gorczynski RM, Chen Z, Erin N, Khatri I, Podnos A. Comparison of immunity in mice cured of primary/metastatic growth of EMT6 or 4THM breast cancer by chemotherapy or immunotherapy. PLoS ONE. (2014) 9:e113597. doi: 10.1371/journal.pone.0113597
31. Mariathasan S, Turley SJ, Nickles D, Castiglioni A, Yuen K, Wang Y, et al. TGFbeta attenuates tumour response to PD-L1 blockade by contributing to exclusion of T cells. Nature. (2018) 554:544–8. doi: 10.1038/nature25501
32. Rhodes DR, Yu J, Shanker K, Deshpande N, Varambally R, Ghosh D, et al. ONCOMINE: a cancer microarray database and integrated data-mining platform. Neoplasia. (2004) 6:1–6. doi: 10.1016/S1476-5586(04)80047-2
33. Curtis C, Shah SP, Chin SF, Turashvili G, Rueda OM, Dunning MJ, et al. The genomic and transcriptomic architecture of 2,000 breast tumours reveals novel subgroups. Nature. (2012) 486:346–52. doi: 10.1038/nature10983
34. Cancer Genome Atlas Network. Comprehensive molecular portraits of human breast tumours. Nature. (2012) 490, 61–70. doi: 10.1038/nature11412
35. Mombaerts P, Iacomini J, Johnson RS, Herrup K, Tonegawa S, Papaioannou VE. RAG-1-deficient mice have no mature B and T lymphocytes. Cell. (1992) 68:869–77.
36. Goralski KB, McCarthy TC, Hanniman EA, Zabel BA, Butcher EC, Parlee SD, et al. Chemerin, a novel adipokine that regulates adipogenesis and adipocyte metabolism. J Biol Chem. (2007) 282:28175–88. doi: 10.1074/jbc.M700793200
37. Makki J. Diversity of breast carcinoma: histological subtypes and clinical relevance. Clin Med Insights Pathol. (2015) 8:23–31. doi: 10.4137/CPath.S31563
38. Rockwell SC, Kallman RF, Fajardo LF. Characteristics of a serially transplanted mouse mammary tumor and its tissue-culture-adapted derivative. J Natl Cancer Inst. (1972) 49:735–49.
39. McAdam AJ, Felcher A, Woods ML, Pulaski BA, Hutter EK, Frelinger JG, et al. Transfection of transforming growth factor-beta producing tumor EMT6 with interleukin-2 elicits tumor rejection and tumor reactive cytotoxic T-lymphocytes. J Immunother Emphasis Tumor Immunol. (1994) 15:155–64.
40. Curry A, Khatri I, Kos O, Zhu F, Gorczynski R. Importance of CD200 expression by tumor or host cells to regulation of immunotherapy in a mouse breast cancer model. PLoS ONE. (2017) 12:e0171586. doi: 10.1371/journal.pone.0171586
41. Senbanjo LT, Chellaiah MA. CD44: A multifunctional cell surface adhesion receptor is a regulator of progression and metastasis of cancer cells. Front Cell Dev Biol. (2017) 5:18. doi: 10.3389/fcell.2017.00018
42. Kumar JD, Kandola S, Tiszlavicz L, Reisz Z, Dockray GJ, Varro A. The role of chemerin and ChemR23 in stimulating the invasion of squamous oesophageal cancer cells. Br J Cancer. (2016) 114:1152–9. doi: 10.1038/bjc.2016.93
43. Zabel BA, Kwitniewski M, Banas M, Zabieglo K, Murzyn K, Cichy J. Chemerin regulation and role in host defense. Am J Clin Exp Immunol. (2014) 3:1–19.
44. Cai Q, Huang Z, Qi L, Wang T, Shen Y, Huang J. Tazarotene-induced gene 2 is associated with poor survival in non-small cell lung cancer. Oncol Lett. (2016) 12:2680–5. doi: 10.3892/ol.2016.5025
45. Lin Y, Yang X, Liu W, Li B, Yin W, Shi Y, et al. Chemerin has a protective role in hepatocellular carcinoma by inhibiting the expression of IL-6 and GM-CSF and MDSC accumulation. Oncogene. (2017) 36:3599–608. doi: 10.1038/onc.2016.516
46. Liu-Chittenden Y, Jain M, Gaskins K, Wang S, Merino MJ, Kotian S, et al. RARRES2 functions as a tumor suppressor by promoting β-catenin phosphorylation/degradation and inhibiting p38 phosphorylation in adrenocortical carcinoma. Oncogene. (2017) 36:3541–52. doi: 10.1038/onc.2016.497
47. Zhang J, Zhou J, Tang X, Zhou L-Y, Zhai L-L, Vanessa ME-D, et al. Reduced expression of chemerin is associated with poor clinical outcome in acute myeloid leukemia. Oncotarget. (2017) 8:92536–44. doi: 10.18632/oncotarget.21440
48. Li JJ, Yin HK, Guan DX, Zhao JS, Feng YX, Deng YZ, et al. Chemerin suppresses hepatocellular carcinoma metastasis through CMKLR1-PTEN-Akt axis. Br J Cancer. (2018) 118:1337–48. doi: 10.1038/s41416-018-0077-y
49. Unver N, Yilmaz G, Canpinar H, Guc D, Esendagli G. Regulatory role of chemerin in lung tumorigenesis and cancer-associated inflammation. Acta Med. (2018) 49, 16–25.
50. Solinas C, Garaud S, De Silva P, Boisson A, Van den Eynden G, de Wind A, et al. Immune checkpoint molecules on tumor-infiltrating lymphocytes and their association with tertiary lymphoid structures in human breast cancer. Front Immunol. 8:1412. doi: 10.3389/fimmu.2017.01412
51. Luen SJ, Savas P, Fox SB, Salgado R, Loi S. Tumour-infiltrating lymphocytes and the emerging role of immunotherapy in breast cancer. Pathology. (2017) 49:141–55. doi: 10.1016/j.pathol.2016.10.010
52. Ogiya R, Niikura N, Kumaki N, Bianchini G, Kitano S, Iwamoto T, et al. Comparison of tumor-infiltrating lymphocytes between primary and metastatic tumors in breast cancer patients. Cancer Sci. (2016) 107:1730–5. doi: 10.1111/cas.13101
53. Trambley J, Bingaman AW, Lin A, Elwood ET, Waitze SY, Ha J, et al. Asialo GM1(+) CD8(+) T cells play a critical role in costimulation blockade-resistant allograft rejection. J Clin Invest. (1999) 104:1715–22. doi: 10.1172/JCI8082
54. Kelly JM, Darcy PK, Markby JL, Godfrey DI, Takeda K, Yagita H, et al. Induction of tumor-specific T cell memory by NK cell-mediated tumor rejection. Nat Immunol. (2002) 3:83–90. doi: 10.1038/ni746
55. Byrne P, McGuirk P, Todryk S, Mills KH. Depletion of NK cells results in disseminating lethal infection with Bordetella pertussis associated with a reduction of antigen-specific Th1 and enhancement of Th2, but not Tr1 cells. Eur J Immunol. (2004) 34:2579–88. doi: 10.1002/eji.200425092
56. Nishikado H, Mukai K, Kawano Y, Minegishi Y, Karasuyama H. NK cell-depleting anti-asialo GM1 antibody exhibits a lethal off-target effect on basophils in vivo. J Immunol. (2011) 186:5766–71. doi: 10.4049/jimmunol.1100370
57. Ge MQ, Ho AWS, Tang Y, Wong KHS, Chua BYL, Gasser S, et al. NK cells regulate CD8+ T cell priming and dendritic cell migration during influenza A infection by IFN-γ and perforin-dependent mechanisms. J Immunol. (2012) 189:2099–109. doi: 10.4049/jimmunol.1103474
58. Fan Z, Yu P, Wang Y, Wang Y, Fu ML, Liu W, et al. NK-cell activation by LIGHT triggers tumor-specific CD8+ T-cell immunity to reject established tumors. Blood. (2005) 107:1342–51. doi: 10.1182/blood-2005-08-3485
Keywords: chemerin, RARRES2, breast cancer, leukocyte trafficking, immunotherapy, NK cells, T cells
Citation: Pachynski RK, Wang P, Salazar N, Zheng Y, Nease L, Rosalez J, Leong W-I, Virdi G, Rennier K, Shin WJ, Nguyen V, Butcher EC and Zabel BA (2019) Chemerin Suppresses Breast Cancer Growth by Recruiting Immune Effector Cells Into the Tumor Microenvironment. Front. Immunol. 10:983. doi: 10.3389/fimmu.2019.00983
Received: 06 August 2018; Accepted: 16 April 2019;
Published: 08 May 2019.
Edited by:
Annalisa Del Prete, University of Brescia, ItalyReviewed by:
Cinzia Fionda, Sapienza University of Rome, ItalySantos Mañes, Spanish National Research Council (CSIC), Spain
Anna Mondino, San Raffaele Hospital (IRCCS), Italy
Copyright © 2019 Pachynski, Wang, Salazar, Zheng, Nease, Rosalez, Leong, Virdi, Rennier, Shin, Nguyen, Butcher and Zabel. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Russell K. Pachynski, rkpachynski@wustl.edu