 Kathryn S. Torok1*
Kathryn S. Torok1* Suzanne C. Li2,3
Suzanne C. Li2,3 Heidi M. Jacobe4
Heidi M. Jacobe4 Sarah F. Taber5,6
Sarah F. Taber5,6 Anne M. Stevens7,8
Anne M. Stevens7,8 Francesco Zulian9
Francesco Zulian9 Theresa T. Lu5,10,11*
Theresa T. Lu5,10,11*- 1Division of Pediatric Rheumatology, Department of Pediatrics, Childrens's Hospital of Pittsburgh, University of Pittsburgh, Pittsburgh, PA, United States
- 2Division of Pediatric Rheumatology, Department of Pediatrics, Hackensack University Medical Center, Hackensack, NJ, United States
- 3Hackensack Meridian School of Medicine at Seton Hall University, Clifton, NJ, United States
- 4Department of Dermatology, UT Southwestern Medical Center, Dallas, TX, United States
- 5Division of Pediatric Rheumatology, Department of Rheumatology, Hospital for Special Surgery, New York, NY, United States
- 6Department of Pediatrics, Weill Cornell Medicine, New York, NY, United States
- 7Division of Pediatric Rheumatology, Department of Pediatrics, University of Washington, Seattle, WA, United States
- 8Seattle Children's Research Institute, University of Washington, Seattle, WA, United States
- 9Pediatric Rheumatology Unit, Department of Woman's and Child's Health, University of Padua, Padua, Italy
- 10HSS Research Institute, Hospital for Special Surgery, New York, NY, United States
- 11Department of Microbiology and Immunology, Weill Cornell Medicine, New York, NY, United States
Localized scleroderma (LS) is a complex disease characterized by a mixture of inflammation and fibrosis of the skin that, especially in the pediatric population, also affects extracutaneous tissues ranging from muscle to the central nervous system. Although developmental origins have been hypothesized, evidence points to LS as a systemic autoimmune disorder, as there is a strong correlation to family history of autoimmune disease, the presence of shared HLA types with rheumatoid arthritis, high frequency of auto-antibodies, and elevated circulating chemokines and cytokines associated with T-helper cell, IFNγ, and other inflammatory pathways. This inflammatory phenotype of the peripheral blood is reflected in the skin via microarray, RNA Sequencing and tissue staining. Research is underway to identify the key players in the pathogenesis of LS, but close approximation of inflammatory lymphocytic and macrophage infiltrate with collagen and fibroblasts deposition supports the notion that LS is a disease of inflammatory driven fibrosis. The immune system is dynamic and undergoes changes during childhood, and we speculate on how the unique features of the immune system in childhood could potentially contribute to some of the differences in LS between children and adults. Interestingly, the immune phenotype in pediatric LS resembles to some extent the healthy adult cellular phenotype, possibly supporting accelerated maturation of the immune system in LS. We discuss future directions in better understanding the pathophysiology of and how to better treat pediatric LS.
Disease Manifestations and Unique Features of Localized Scleroderma (LS) In Childhood
Localized scleroderma (LS) is the most common form of pediatric scleroderma, a disease whose histologic pathology involves inflammation and fibrosis, similar to that of systemic sclerosis (SSc), although the clinical phenotypes are markedly different. Overall, the annual incidence in the US of LS collectively in adults and children is slighter higher than for systemic sclerosis (SSc) (2.7 vs. 1.9 per 100,000, respectively) (1, 2). A much larger difference is found for childhood onset of these diseases, however, with 34% of LS beginning in childhood (by age 18 years), compared to < <5% of SSc (by age 16 years) (2, 3).
The mean age of pediatric LS disease onset is 6.4–8.7 years, with the disease more prevalent in Caucasians (4). As is true for other autoimmune disease, females are more commonly affected than males (2.3 4:1 ratio) (4). This female preponderance is consistent over different ethnicities, as clinical centers with more homogeneous racial populations, such as Mexico (5), have reported similar ratios of female:male patients.
LS can present in several different patterns (subtypes) depending on depth and distribution of lesions, including circumscribed morphea (plaque lesions), linear scleroderma of the trunk/limb or head (band-like lesion), generalized morphea (multiple plaque lesions), pansclerotic morphea, or a combination of two or more of these subtypes (mixed morphea) (6). Besides the obvious difference in age of disease onset, there are several major differences between pediatric and adult onset disease. These include a different subtype predominance, higher frequency of deep tissue and extracutaneous involvement in pediatric disease, and longer disease duration in pediatric disease [reviewed in (4)]. These differences contribute to the higher frequency of serious morbidity in patients with pediatric compared to adult onset disease (4, 7), with morbidity including arthropathy, uveitis, facial hemiatrophy, seizures, and neuropathy (7, 8). Functional impairment has been reported in 30–38% of juvenile LS patients (9–11).
Twenty to 70% of juvenile LS patients have been reported to have extracutaneous involvement, with higher frequencies reported in prospective studies (7, 8, 11–15). The most common type of extracutaneous involvement is musculoskeletal, which includes joint, tendon, muscle, and bone issues. Joint and tendon issues include arthralgia, arthritis, joint contractures, and angulation defects, some of which require corrective surgeries (16). Muscle involvement includes myalgia, myositis, and muscle atrophy (8). Because the disease commonly begins before most children have undergone their major growth spurt, children are at risk for undergrowth of the affected side, which can lead to limited physical function, pain, and major disfigurement. Growth impairment is common, with studies reporting facial hemiatrophy in half the patients with linear scleroderma of the head, deformity from tissue atrophy and/or muscle bulk reduction in half, and a bone length difference in 15–18% of patients (15–17).
Central nervous system (CNS) involvement is a less common but notable extracutaneous manifestation. The overall frequency of CNS involvement in juvenile LS is ~5% but in patients with linear scleroderma of the head (LSh), it ranges between 44% (18) and 50% (19). Seizures, in particular, partial complex seizures, are the most common neurological symptom, followed by headache, hemiparesis, cranial nerve palsy, optic neuritis; and less commonly, neuropsychiatric disorders, deterioration of intelligence, and/or ischemic stroke (8). Temporal relationship between onset of the neurological symptoms and skin lesions is variable, with the majority of the patients having preceding scalp and facial lesions before CNS presentation, though approximately one-quarter of the cases can present with neurological manifestations (20) (21). Radiological and cerebrospinal fluid (CSF) laboratory findings in LSh patients further support that the disease affects the CNS, likely in an autoimmune manner. When brain imaging is performed in symptomatic patients, abnormalities, such as cortical and subcortical white matter lesions, atrophy, and calcinosis are common, with 34 of the 54 reported patients in one review (63%) found to have multiple or diffuse brain lesions on magnetic resonance imaging (MRI) (20). These brain lesions seem to be more epileptogenic than other autoimmune diseases, such as multiple sclerosis (20). Furthermore, analysis of CSF obtained via lumbar puncture reveals findings consistent with an inflammatory process in some LSh patients demonstrating oligoclonal bands, elevated IgG levels, and autoantibodies (22–24). Further evidence supporting CNS inflammation includes histologic findings of LSh brain biopsies, which demonstrate the same changes as seen in skin: chronic perivascular lymphocytic inflammation with some vessels showing intimal thickening and hyalinization (25).
Potential Pathogenic Etiologies
Genetics
Familial history of disease, either immediate or remote, is common for those with autoimmune diseases. For LS patients, 10–30% of patients reported having a family history of autoimmune disease, such as lupus and arthritis (26–30). Ten percent of LS patients have concurrent autoimmune diseases; in children, the most commonly identified diseases are vitiligo, alopecia areata, and juvenile rheumatoid arthritis (26).
Few studies have examined HLA associations in LS. The largest to date was performed using participants from the Morphea in Adults and Children Cohort, which includes about 1/3 childhood-onset LS. In this case control study, HLA Class II genotyping and SSCP typing of HLA A, B, and C alleles was performed and associations between HLA-Class I and II alleles and LS as well as its subphenotypes was determined. Notably, there was only one common allele with adult SSc, DRB*04:04, implying LS and SSc are immunogenetically distinct. In contrast, the strongest associations were with DRB1*04:04 and HLA-B*37 (31). DRB1*04:04 is also strongly associated with risk for rheumatoid arthritis. Interestingly, population based studies examining the autoimmune profile of RA have identified increased risk (SIR) of LS in patients with RA. Conversely, studies have indicated there may be increased risk of RA in LS patients. Taken together, this implies that there may be common genetic susceptibility in LS and RA as well as other autoimmune disorders (27, 32). The strong association of LS with specific class-I HLA alleles supports the role of CD8 or natural killer cell associated immune responses in the pathogenesis of LS and implicate loss of tolerance to an unknown self-antigen (31).
Developmental Etiology
Several studies have confirmed that localized scleroderma follows the distribution pattern of Blaschko lines, invisible patterns in the skin, distinct from dermatomes, which is a normal embryonic pattern (Figure 1) (34, 35). This includes a retrospective chart review of 65 children with linear scleroderma, plotting skin lesions on standardized head and body charts and then comparing these clinical diagrams to a computer-assisted comparison, which demonstrated excellent correlation (35). Some dermatologic diseases which distribute along lines are believed to be evidence of genomic mosaicism, caused by a somatic mutation during embryogenesis that gives rise to an individual with two genetically distinct populations of cells (36). Whole exome sequencing has been used to prove that affected tissue from other conditions that follow Blaschko lines, including some types of epidermal nevi, contains somatic mutations not present in germline tissue from the same patient (37). Though these findings suggest that localized scleroderma is a form of cutaneous mosaicism, this theory has not yet been confirmed on a molecular basis.
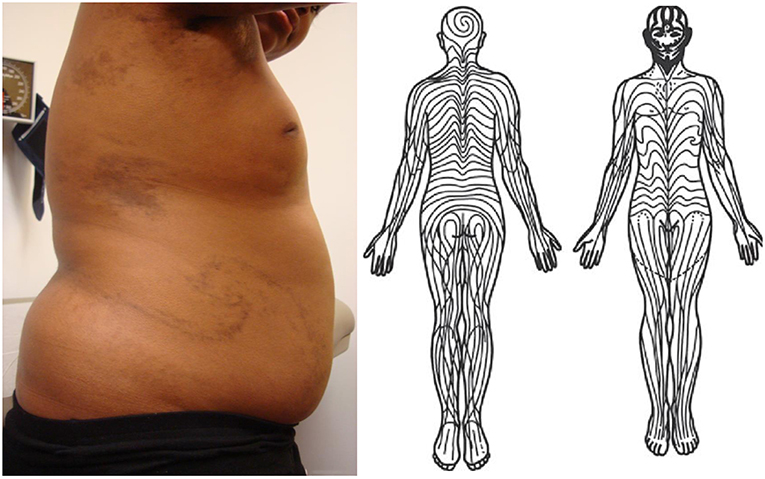
Figure 1. (Left) Swirling lines of Blaschko on patient's right trunk with pediatric-onset localized scleroderma. Note several other patches of morphea on trunk and axilla. She also had linear bands of fibrosis traveling down posterior aspects of bilateral arms and right greater than left leg following lines of Blaschko. Written informed consent was obtained for the clinical photograph. (Right) Diagram of lines of Blaschko. Without modification from Tenea (33).
A neuroectodermal origin has been postulated for CNS disease in juvenile LS. Given that face and brain parenchyma tissues derive from a common progenitor in the ectodermal site of the neural tube, an early mutation in the rostral area might cause both cerebral dysgenesis and progressive facial hemiatrophy (38). In support of this hypothesis are the coexistence of ipsilateral cutaneous and neurological lesions and reports of Sturg-Weber syndrome-like intracranial lesions and multiple hamartomas in patients with linear scleroderma of the face (39, 40).
Immune Etiology
LS as an Autoimmune Disease
Takehara and colleagues were among the first to recognize and publish the summary of immunologic findings to support localized scleroderma as an autoimmune disorder with serological supportive evidence being the presence of autoantibodies (autoAb), elevated circulating cytokines, their soluble receptors and soluble cell adhesion molecules (41). Further, examination of the histopathology of LS lesions shows close approximation of immune cell infiltrates with sclerosis in inflammatory lesions, further supporting the interplay between immune dysregulation and increased extracellular matrix components including type I collagen in LS (42). Specifically in regards to LSh, oligoclonal bands and elevated IgG in the CSF have been documented, further supporting an autoimmune phenomenon. Additionally, clinical response of skin, musculoskeletal, and neurological manifestation of LS to immunosuppressive agents, such as corticosteroids and methotrexate, further provides support for the autoimmune hypothesis (43–46). Here, we provide an updated summary of the literature that builds upon this foundation, summarize immune signature grouping or speculations of findings, as well as future directions to give a better understanding of the LS immunophenotype.
Histopathologic Evidence for Immune Involvement
In a cross-sectional study of 83 patients with localized scleroderma in which 101 biopsy specimens were examined the authors found that the microanatomical location and degree of inflammation and sclerosis were associated with the stage of evolution of the lesion as well as clinical disease manifestations. The authors categorized sclerosis patterns as top heavy (confined to the papillary dermis), bottom heavy (confined to the reticular dermis and beyond), and throughout (extending from the papillary dermis to subcutis and beyond). They also quantified the degree and location of inflammation and cell types present. Important observations included that all patterns of sclerosis (top, bottom, and throughout) were present in circumscribed, generalized, and linear subtypes. Interestingly, regardless of subtype the bottom heavy or throughout pattern of sclerosis was associated with increased risk of pain and functional limitation, implying that both the location of a lesion on the skin as well as the microanatomical location of the pathology are important in determining those at risk for more serious sequelae. In terms of inflammatory cell types, lymphocytes predominated with plasma cells as second most common. Surprisingly eosinophils were present in 21% of specimens. The microanatomical location of inflammation closely mirrored that of the pattern of sclerosis present in an individual specimen. In other words, sclerosis occurred in areas that were enriched in inflammatory cell infiltrate. This implies a link between inflammation and activation of fibroblasts in the surrounding dermis and subcutis. Taken together, these results support the use of microanatomical location of inflammation and sclerosis in assessing risk of pain and functional limitations in localized scleroderma as well as further linking immune dysregulation as a driver of sclerosis (42).
In addition to fibroblasts, the immune cells could potentially activate skin structures. The location of the inflammatory cell infiltrate in skin is typically in a perivascular, peri-eccrine and peri-neural distribution, with the interplay with the neurovascular bundles sometimes termed “lymphocytic neurovasculitis” (47). In CNS disease, chronic perivascular lymphocytic infiltrate with intimal thickening and hyalinization of the vessels are also found when brain biopsies are obtained (23–25). T lymphocytes were also identified in a brain biopsy (48). Together, the histopathology in multiple organs in LS further suggest an autoimmune phenomenon.
Autoantibodies and Clinical Associations in LS
Autoantibodies (autoAb) are commonly observed in individuals with LS which reflects activation of the immune system and auto-reactivity to self-antigens. Although not as specific with regard to organ manifestation or scleroderma subtype as seen in systemic sclerosis (SSc), autoAb may be helpful in associating to disease severity and/or depth of LS. When more classic SSc-associated autoAb, such as anti-centromere and anti-topoisomerase, are tested in LS patients, they occur in 3–18% of the LS subjects (4, 8, 26, 49, 50), none of which had or developed SSc in addition to their LS diagnosis. A recent study of the SSc line immunoassay (LIA) (Euroimmun, Germany) in pediatric LS subjects found similar percentages (centromere 14%, topoisomerase 10%, RNA Polymerase III 12%) and when compared to clinical parameters, correlated to deep tissue involvement, signified by joint contractures, muscle involvement and nerve entrapment (51). Interestingly, none of the SSc-antibodies associated with LS subtype designation (51).
A high proportion of pediatric LS patients are Anti-Nuclear Antibody (ANA) positive, ranging from 30 to 70% when tested by indirect immunofluorescence (4, 11, 26–29, 41, 52, 53). The frequency and clinical utility of autoantibodies in LS has been the subject of numerous studies, with varied results. The presence ANA in several studies corresponded to deeper disease involvement (beyond the subcutis) by associating with features, such as joint contractures, muscle atrophy and extremity shortening, but not disease subtype or age of onset (4, 26–29, 53, 54). A recent longitudinal cohort study supports a positive ANA at LS diagnosis to be predictive of likelihood for recurrence; therefore, ANA is likely promoting autoimmunity in some fashion or reflecting a more auto-reactive state (55). Other auto-antibodies which may be reflecting positive ANA in LS have been reported, most commonly, anti-histone antibody (AHA) and anti–single-stranded DNA antibody (ssDNA Ab). In those tested, a range of 10–50% of LS patients are positive for ssDNA and/or AHA (56–58) with both correlating to severity features, such as deep muscle involvement, joint contractures, and increased number of lesions (41, 57, 59), and is able to track with disease activity status in a subset of patients (60).
When evaluated, rheumatoid factor was present in 16- 29% of patients and associated with arthritis (29, 50). Other markers of immune activation, such as IgG, IgA, and IgM were found to be increased in patients with linear scleroderma, and deep and pansclerotic morphea (29). Elevation of other more typical laboratory parameters tested in connective tissue diseases, such as muscle enzymes, corresponded to deeper tissue involvement. Elevated creatine phosphokinase (CPK) and aldolase were associated with disease parameters including muscle atrophy and extremity shortening in a North American pediatric LS cohort, indicating muscle involvement (11).
Cytokine and Cellular Signatures in LS
The exact cellular signature of LS is still being investigated. In both adult and pediatric LS analyses, lymphocytes and their associated cytokine and chemokine populations are observed in both the blood and skin. Flow cytometry studies of the circulating cellular phenotype of LS (pediatric and adult) have shown a predominance of CD4+T helper cells along with decreased functional T regulatory cells (61–63). This decrease in T regulatory cells, possibly reflecting a more “permissive state,” was also seen in pediatric SSc without increases in other T cell populations (64). Furthermore, when comparing paired active to inactive PBMC phenotypes in LS, those with active disease states demonstrated much higher populations of IFNγ-expressing T cells (reflecting TH1 cells; CD4+ IFNγ + T cells) (63). An expanded study utilizing multiparameter mass cytometry by time-of-flight spectrometry (CyTOF) also supported increased IFNγ expression from CD4+T cells, as well as NK cell populations, in active LS PBMC samples (65).
T helper cells consist of 3 main types including TH1, TH2, and TH17. These cell types produce distinct interactive cytokine profiles. TH1 cells secrete IFN-γ and IL-2 and are stimulated by IL-12, whereas TH2 cells produce IL-4, IL-5, IL-6, IL-10, and IL-13 and are activated by IL-4. TH17 cells produce IL-17 A/F, IL-21, IL-22 and are propagated by IL-23, IL-6, and IL-1. While elevation of cytokines associated with all three TH lineages have been observed in LS, the pattern of expression is consistent with that of a TH1 predominance (Table 1). In peripheral blood, elevation of TH1 related cytokines, chemokines, and their receptors include: IL-2 (67, 69, 73), IL-12 (66, 67), TNFα (70), TGFβ (66, 67, 71, 72), MCP-1 (66), IFNγ related proteins, CXCL9 (monokine induced by gamma interferon [MIG]) (67, 76, 77), CXCL10 (Interferon gamma-induced protein 10 [IP-10]) (67, 76) and CXCL11 (Interferon-inducible T-cell alpha chemoattractant [I-TAC]), and IFNγ chemokine receptor (CXCR3). TH2 related cytokines IL-4 (69), IL-6 (69, 72), IL-13 (70), and TH17 related cytokines, IL-17A (66, 75), and IL-23 (75), were also reported as significantly elevated in peripheral blood of LS patients compared to controls. Peripheral blood levels of the IFNγ-related chemokines CXCL9 and CXCL10 also correlated to disease activity measures, such as clinical scores, the modified Localized scleroderma skin severity index (mLoSSI), and the Physician Global Assessment of disease activity (66, 67, 78) (Table 1), underscoring their potential as serological biomarkers of disease activity.
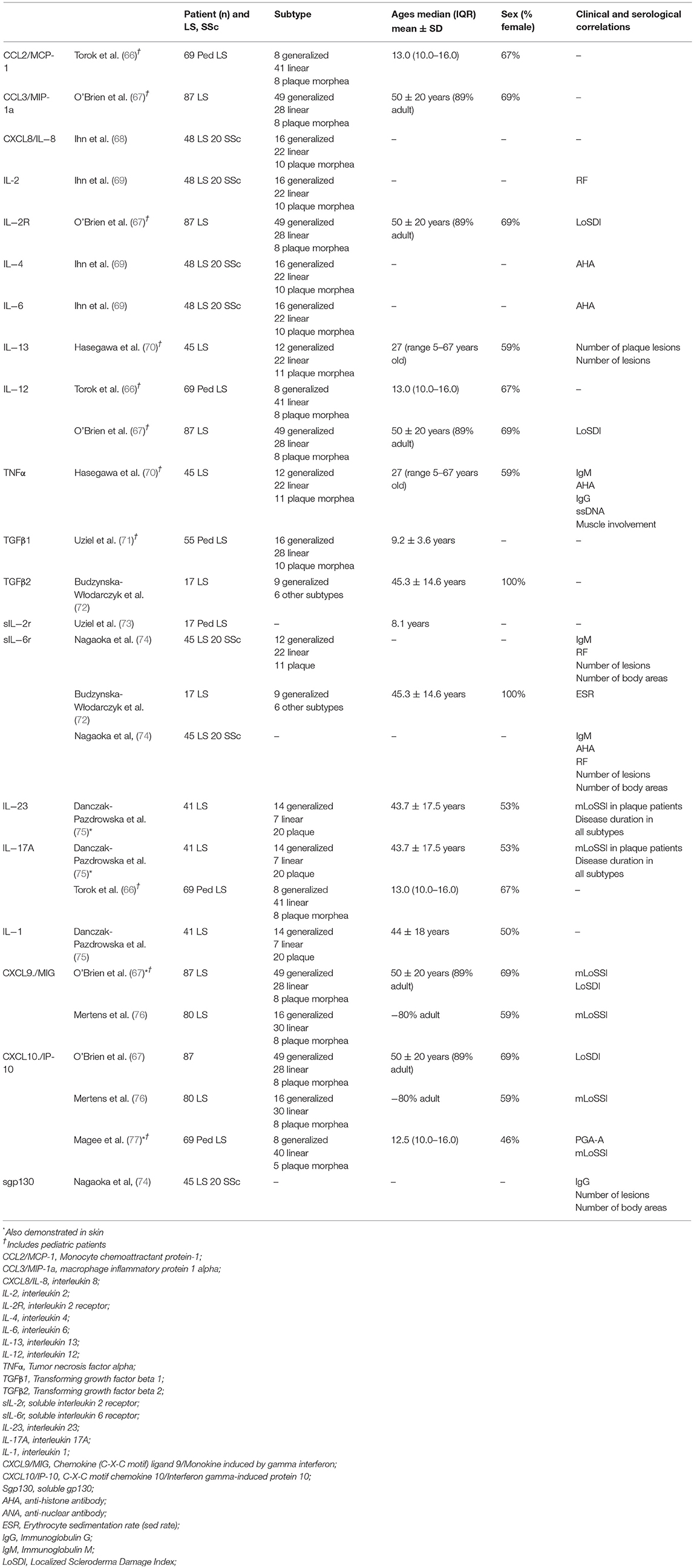
Table 1. Peripheral Blood Cytokine Profiles in LS: Elevated cytokines associate with T helper cell linages and correlate with other inflammatory disease indicators, such as activity scores and clinical laboratory tests.
In LS skin, TH1/IFNγ related chemokines CXCL9 (67) and CXCL10 (77) were increased, while TH17 related cytokines IL-23 (75) and IL-17A (75) were decreased compared to healthy control skin. Both CXCL9 and CXCL10 were found to be present in the perivascular lymphocytic infiltrate of the papillary and reticular dermis (67, 77). Additionally, CXCL9 was found to stain in close approximation to both CD4+TH cells and macrophages (67), suggesting potential interaction between lymphocytes and macrophages utilizing IFNγ chemokine signaling (Figure 2). Overall, this may synergistically promote fibroblasts to increase collagen expression in LS, eventually causing increased collagen deposition, fibrosis, and a shift toward a later TH2 profile.
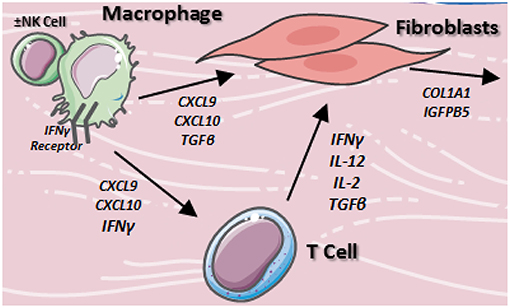
Figure 2. Proposed cellular interactions of macrophages, fibroblasts, and T cells in localized scleroderma. Resident macrophages stimulate T cells and fibroblasts via TH1/IFNγ associated cytokine network to produce inflammation and collagen accumulation in the skin.
In summary, in LS there is a TH1/IFNγ signature prevalent in the active or initial inflammatory stage of the disease and likely a more fibrotic TH2 signature follows in the collagenous stage of LS, which more closely resembles long term SSc disease profiles. These two states of inflammatory and fibrotic disease have unique profiles that reflect the clinical perception of disease. In pediatric patients, CXCL9, CXCL10, CXCL11, MIP-3β, IL-9, IL-2, and CCL-1 were elevated in the active state compared to the inactive state of disease (77, 78) as indicated by these cytokine profiles of the blood and skin. The exact relationship of the circulating cellular and cytokine/chemokine players to skin pathophysiology is still under study. One study focused on skin expression of select transcripts, and found increased mRNA levels of CCR7 and CCL5/RANTES in LS lesions (79). This supported the idea that lymphocytes are recruited from the blood to lesional skin. Investigation into skin-homing T cell profiles are underway in LS, with preliminary data showing that both skin homing CD8+(Tc)CCR10+ and CD4+(TH)CCR10+ T cells subsets produce inflammatory cytokine populations, including IFNγ, that were significantly increased in the active disease state compared to the inactive disease state (80). In adult SSc, IL-13+, and not IFNγ+, CD8+CCR10+ cells appear to be the skin homing cell type that propagates disease (81), emphasizing possible biological differences in scleroderma phenotypes: a more inflammatory, TH1/TC1 phenotype in LS and a more fibrotic, TH2/TC2 phenotype in SSc.
The LS Transcriptome
Investigation into the transcriptome of scleroderma, specifically SSc, has been successful at classifying samples based on genetic differences that could indicate or predict disease progression and potential future treatment plans. Adult SSc subjects were classified into 4 distinct genetic signatures based on overall microarray expression of the skin (82). These classifications include a diffuse-proliferation group composed of diffuse SSc patients, the inflammatory group containing diffuse SSc, limited SSc and LS (4 morphea subjects were included in the analyses), the limited group composed of limited SSc, and the normal-like group, which includes healthy controls along with a few SSc patients (82). All of the LS subjects included fell nicely into the inflammatory group, expressing primarily T-lymphocyte and IFNγ related genes (82), which corresponds to the peripheral blood and protein skin expression discussed above. Recent transcriptional analysis using RNA bulk sequencing of pediatric LS skin showed a distinct subset of patients expressing similar inflammatory genes including interferon-inducible chemokines, such as CXCL9, CXCL10, CXCL11, and IFNγ itself (83). Similar to SSc, subgroupings of LS patients are expected to be present as these inflammatory genes were more highly expressed in LS patients with more inflammatory or active lesions, with associated higher clinical activity scores, mLoSSI and PGA-A (83). This transcriptome classification system in SSc has been used more recently to predict patient response to therapy, such as the inflammatory subset showing better response to mycophenolate mofetil and the fibroproliferative group showing better response to stem cell transplant (84, 85). A methodologically similar classification of LS using immunophenotyping of the transcriptome could help to delineate immunological subtypes and determine therapeutic responses to disease.
Speculation: Differences in Immune System Function in Children and Adults
The greater prevalence of extracutaneous manifestations and autoimmunity in children than in adults suggests the possibility of some age-related differences in disease mechanisms that may potentially reflect differences in immune system function between children and adults. The immune system is relatively immature at birth, and many changes occur within the first year of life. Newborn dependency on antibodies from the maternal circulation and immune cells undergo phenotypic changes over the first few months of life including a robust thymic output of T cells that subsides over the first few years (86, 87). However, there are also changes from early childhood to adulthood. For example, in the peripheral blood, CD4 and CD8 T cell counts increase from childhood to adulthood while B cell counts drop (88). At the same time, regulatory T cell (87), monocyte (89) and NK cell (90) counts are higher in infancy or early childhood than in adulthood. Whether these differences in cell population reflect differential generation, trafficking, or survival is currently unclear. T and B cells are continuously trafficking between blood and secondary lymphoid organs, such as spleen and lymph nodes (91), and an increased proportion of B cells in the blood circulation at a young age could reflect relative reductions in B cell entry into lymph nodes, and potentially less exposure to an environment that supports the generation of a robust but also a well-regulated response. Further understanding how differences in immune cell numbers reflect differential immune system function could help to better understand disease.
In addition to differences in cell numbers, immune cells also differ in function in children and adults. Generally, early life is dominated by a more regulatory state that favors healing and repair (92). For innate immunity, NK cell phenotype is more consistent with cytokine producing rather than cytotoxic function early in post-natal life, with more mature phenotype during the first years of life (90). Responses to toll like receptor (TLR) stimulation are reduced during early months to years in Western countries (90, 92). At the same time, production of IL-10, a regulatory cytokine decreases during infancy to below adult levels, and then increases again until adult levels are reached (90). The functional implications of these alterations for different manifestations of scleroderma in childhood and adulthood are unclear, but these differences drive home the point that immunopathogenesis at different times of life may be different.
Childhood is also a time of immune repertoire development for lymphocytes, whereby lymphocytes that are naïve will be exposed to antigen, and antigen- experienced lymphocytes will constitute a larger proportion of the body's lymphocytes (87). Exposure over infancy and childhood through the gut, skin, and respiratory tract to different microbes, food, and the environment is thought to contribute to shaping and increasing the memory T cell compartment and protective antibodies. While the immune repertoire is modulated with intercurrent infections, even in the absence of frank infections, these exposures help to generate memory cells and antibodies that can potentially cross react enough with potential pathogens to protect the child later on (87). In addition to the antigenic exposure, the diversity of an individual's immune repertoire is also shaped by stochastic events. The selection of the exact T and B cells that will dominate in the response to antigen exposure has a large element of randomness, as reflected in the different immune repertoires in twins (93). During this period of high expansion of the immune repertoire, T and B cells that are cross-reactive to self and pathogenic could incidentally be generated.
Given these differences in immune function in children and adults, it is interesting to consider that the biomarkers of disease seen in pediatric LS compared to healthy pediatric controls—the reduced regulatory T cell, increased TH1, TH2, and TH17 cells, and increased innate cell activation—resemble the pattern seen in a healthy adult immune system. We speculate that, while autoreactivity could contribute to pathogenesis, the cell distribution and activity in pediatric LS may actually signal an accelerated development of the immune system. This would be complementary but distinct from “aging” of the immune system and generation of senescent pro-inflammatory cells (94), but an acceleration of normal transition to a more permissive immune system that, perhaps in addition to the increased antigenic exposure during childhood, contributes to pathology in children.
Future Directions for Research
While treatment strategies effective for most patients have been identified, there is a major need for additional treatment options and strategies. Thirty percent of jLS patients may fail to respond to initial standard immunosuppressive treatment (95), and 15–53% of patients can relapse following treatment (15, 28, 95). Active disease can persist for decades (53, 96). Failure to achieve remission and relapsing disease are both associated with poorer outcome (97).
Identifying optimal treatment strategies for jLS will require comparative effectiveness studies; the feasibility of this approach was demonstrated by a recent pilot study of three standardized methotrexate based regimens (13). Because treatment is focused on controlling inflammation, sensitive monitoring of disease activity is essential for conducting such trials. A recent study identified specific lesion features for tracking disease activity that are likely to improve the sensitivity and specificity of existing clinical measures (14). Future work may lead to development of a weighted clinical activity measure to further improve our ability to identify relative differences in treatment efficacies. The identification of biomarkers that facilitate monitoring activity level and/or help identify response to specific treatments will enable us to work toward personalized medicine for these patients.
Further understanding disease pathophysiology will aid in the development of new therapeutic approaches. Insight into the exact nature of the immune dysfunction and how it contributes to skin fibrosis and the extracutaneous manifestations may help to better target the immune system to treat the disease. Understanding how the unique aspects of the immune system of childhood intersect with the disease process can further provide insight into disease etiology and perhaps teach us how to best reduce the immune dysfunction, or at the very least, repair the damage.
Author Contributions
KT, SL, HJ, ST, AS, FZ, and TL conceived of and wrote the manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work was supported by NIH R01AI079178, Scleroderma Foundation, St. Giles Foundation, and the Lupus Research Alliance (TL).
References
1. Mayes MD, Lacey JVJr, Beebe-Dimmer J, Gillespie BW, Cooper B, Laing TJ, et al. Prevalence, incidence, survival, and disease characteristics of systemic sclerosis in a large US population. Arthritis Rheum. (2003) 48:2246–55. doi: 10.1002/art.11073
2. Peterson L, Nelson A, Su W, Mason T, O'Fallon W, Gabriel S. The epidemiology of morphea (localized scleroderma) in Olmstead County 1960–1993. J Rheumatol. (1997) 24:73–80.
3. Scalapino K, Arkaschaisri T, Lucas M, Fertig N, Helfrich D, Londino AJ, et al. Childhood onset systemic sclerosis: classification, clinical and serologic features, and survival in comparison with adult onset disease. J Rheumatol. (2006) 33:1004–13.
4. Li SC. Scleroderma in children and adolescents: localized scleroderma and systemic sclerosis. Pediatr Clin North Am. (2018) 65:757–81. doi: 10.1016/j.pcl.2018.04.002
5. Guevara-Gutierrez E, Yinh-Lao J, Garcia-Gutierrez P, Tlacuilo-Parra A. Frequency of antinuclear antibodies in mestizo Mexican children with morphea. Clin. Rheumatol. (2010) 29:1055–9. doi: 10.1007/s10067-010-1515-2
6. Laxer R, Zulian F. Localized scleroderma. Curr Opin Rheumatol. (2006) 18:606–13. doi: 10.1097/01.bor.0000245727.40630.c3
7. Ardalan K, Zigler CK, Torok KS. Predictors of longitudinal quality of life in juvenile localized scleroderma. Arthritis Care Res. (2017) 69:1082–7. doi: 10.1002/acr.23101
8. Zulian F, Vallongo C, Woo P, Russo R, Ruperto N, Harper J, et al. Localized scleroderma in childhood is not just a skin disease. Arthritis Rheum. (2005) 52:2873–81. doi: 10.1002/art.21264
9. Piram M, McCuaig CC, Saint-Cyr C, Marcoux D, Hatami A, Haddad E, et al. Short- and long-term outcome of linear morphoea in children. Br J Dermatol. (2013) 169:1265–71. doi: 10.1111/bjd.12606
10. Weibel L, Laguda B, Atherton D, Harper JI. Misdiagnosis and delay in referral of children with localized scleroderma. Br J Dermatol. (2011) 165:1308–13. doi: 10.1111/j.1365-2133.2011.10600.x
11. Wu EY, Li SC, Torok KS, Virkud Y, Fuhlbrigge R, Rabinovich CE. Baseline description of the juvenile localized scleroderma subgroup from the childhood arthritis and rheumatology research alliance legacy registry. ACR Open Rheumatol In press. (2019) 1:119–124. doi: 10.1002/acr2.1019
12. Condie D, Grabell D, Jacobe H. Comparison of outcomes in adults with pediatric-onset morphea and those with adult-onset morphea: a cross-sectional study from the morphea in adults and children cohort. Arthritis Rheumatol. (2014) 66:3496–504. doi: 10.1002/art.38853
13. Li S, Torok K, Hong S, Ferguson P, Rabinovich C, Becker M, et al. (2018a). Working Towards Comparative Effectiveness Studies for Moderate to Severe Juvenile Localized Scleroderma: Results of a 1-Year Pilot Study of Three Methotrexate-Based Consensus Treatment Plans. Pediatric Rheumatol Online J. 16(Suppl 1):42. doi: 10.1186/s12969-018-0252-y
14. Li SC, Li X, Pope E, Stewart K, Higgins GC, Rabinovich CE, et al. New Features for Measuring Disease Activity in Pediatric Localized Scleroderma. J Rheumatol in press. (2018b) 45:1680–8: doi: 10.3899/jrheum.171381
15. Weibel L, Sampaio MC, Visentin MT, Howell KJ, Woo P, Harper JI. Evaluation of methotrexate and corticosteroids for the treatment of localized scleroderma (morphoea) in children. Br J Dermatol. (2006) 155:1013–20. doi: 10.1111/j.1365-2133.2006.07497.x
16. Schoch J, Schoch B, Davis D. Orthopedic complications of linear morphea: Implications for early interdisciplinary care. Pediatric Dermatol. (2018) 35:43–6. doi: 10.1111/pde.13336
17. Tollefson M, Witman P. En coup de sabre morphea and parry-romberg syndrome: a retrospective review of 54 patients. J Am Acad Dermatol. (2007) 56:257–63. doi: 10.1016/j.jaad.2006.10.959
18. Blaszczyk M, Krolicki L, Krasu M, Glinska O, Jablonska S. Progressive facial hemiatrophy: central nervous system involvement and relationship with scleroderma en coup de sabre. J Rheumatol. (2003) 30:1997–2004.
19. Moko SB, Mistry Y, Blandin de Chalain TM. Parry-Romberg syndrome: intracranial MRI appearances. J Cranio Maxillo Facial Surgery. (2003) 31:321–4. doi: 10.1016/S1010-5182(03)00028-3
20. Kister I, Inglese M, Laxer RM, Herbert J. Neurologic manifestations of localized scleroderma: a case report and literature review. Neurology. (2008) 71:1538–45. doi: 10.1212/01.wnl.0000334474.88923.e3
21. Sartori S, Martini G, Calderone M, Patrizi A, Gobbi G, Zulian F. Severe epilepsy preceding by four months the onset of scleroderma en coup de sabre. Clin Exp Rheumatol. (2009) 27:64–7.
22. De Somer L, Morren MA, Muller PC, Despontin K, Jansen K, Lagae L, et al. Overlap between linear scleroderma, progressive facial hemiatrophy and immune-inflammatory encephalitis in a paediatric cohort. Eur J Pediatr. (2015) 174:1247–54. doi: 10.1007/s00431-015-2532-6
23. Luer W, Jockel D, Henze T, Schipper HI. Progressive inflammatory lesions of the brain parenchyma in localized scleroderma of the head. J Neurol. (1990) 237:379–81. doi: 10.1007/BF00315664
24. Sathornsumetee S, Schanberg L, Rabinovich E, Lewis D Jr Weisleder P. Parry-Romberg syndrome with fatal brain stem involvement. J Pediatr. (2005) 146:429–31. doi: 10.1016/j.jpeds.2004.10.026
25. Stone J, Franks AJ, Guthrie JA, Johnson MH. Scleroderma “en coup de sabre”: pathological evidence of intracerebral inflammation. J Neurol Neurosurg Psychiatr. (2001) 70:382–5. doi: 10.1136/jnnp.70.3.382
26. Christen-Zaech S, Hakim MD, Afsar FS, Paller AS. Pediatric morphea (localized scleroderma): review of 136 patients. J Am Acad Dermatol. (2008) 59:385–96. doi: 10.1016/j.jaad.2008.05.005
27. Leitenberger JJ, Cayce RL, Haley RW, Adams-Huet B, Bergstresser PR, Jacobe HT. Distinct autoimmune syndromes in morphea: a review of 245 adult and pediatric cases. Arch Dermatol. (2009) 145:545–50. doi: 10.1001/archdermatol.2009.79
28. Pequet MS, Holland KE, Zhao S, Drolet BA, Galbraith SS, Siegel DH, et al. Risk factors for morphoea disease severity: a retrospective review of 114 paediatric patients. Br J Dermatol. (2014) 170:895–900. doi: 10.1111/bjd.12758
29. Zulian F, Athreya BH, Laxer R, Nelson AM, Feitosa de Oliveira SK, Punaro MG, et al. Juvenile localized scleroderma: clinical and epidemiological features in 750 children. An international study. Rheumatology. (2006) 45:614–20. doi: 10.1093/rheumatology/kei251
30. el-Azhary RA, Aponte CC, Nelson AM, Weaver AL, Homburger HA. Antihistone antibodies in linear scleroderma variants. Int. J. Dermatol. (2006) 45:1296–9. doi: 10.1111/j.1365-4632.2006.02891.x
31. Jacobe H, Ahn C, Arnett FC, Reveille JD. Major histocompatibility complex class I and class II alleles may confer susceptibility to or protection against morphea: findings from the morphea in adults and children cohort. Arthritis Rheumatol. (2014) 66:3170–7. doi: 10.1002/art.38814
32. Hemminki K, Li X, Sundquist J, Sundquist K. Familial associations of rheumatoid arthritis with autoimmune diseases and related conditions. Arthritis Rheum. (2009) 60:661–8. doi: 10.1002/art.24328
34. Jue MS, Kim MH, Ko JY, Lee CW. Digital image processing for the acquisition of graphic similarity of the distributional patterns between cutaneous lesions of linear scleroderma and Blaschko's lines. J Dermatol. (2011) 38:778–83. doi: 10.1111/j.1346-8138.2010.01162.x
35. Weibel L, Harper JI. Linear morphoea follows blaschko's lines. Br J Dermatol. (2008) 159:175–81. doi: 10.1111/j.1365-2133.2008.08647.x
36. Molho-Pessach V, Schaffer JV. Blaschko lines and other patterns of cutaneous mosaicism. Clin Dermatol. (2011) 29:205–25. doi: 10.1016/j.clindermatol.2010.09.012
37. Levinsohn JL, Tian LC, Boyden LM, McNiff JM, Narayan D, Loring ES, et al. Whole-exome sequencing reveals somatic mutations in HRAS and KRAS, which cause nevus sebaceus. J Invest Dermatol. (2013) 133:827–30. doi: 10.1038/jid.2012.379
38. Dupont S, Catala M, Hasboun D, Semah F, Baulac M. Progressive facial hemiatrophy and epilepsy: a common underlying dysgenetic mechanism. Neurology. (1997) 48:1013–8. doi: 10.1212/WNL.48.4.1013
39. Chung MH, Sum J, Morrell MJ, Horoupian DS. Intracerebral involvement in scleroderma en coup de sabre: report of a case with neuropathologic findings. Ann Neurol. (1995) 37:679–81. doi: 10.1002/ana.410370519
40. Derex L, Isnard H, Revol M. Progressive facial hemiatrophy with multiple benign tumors and hamartomas. Neuropediatrics. (1995) 26:306–9. doi: 10.1055/s-2007-979779
41. Takehara K, Sato S. Localized scleroderma is an autoimmune disorder. Rheumatology. (2005) 44:274–9. doi: 10.1093/rheumatology/keh487
42. Walker D, Susa JS, Currimbhoy S, Jacobe H. Histopathological changes in morphea and their clinical correlates: results from the morphea in adults and children cohort V. J Am Acad Dermatol. (2017) 76:1124–30. doi: 10.1016/j.jaad.2016.12.020
43. Goldberg-Stern HT, deGrauw Passo M, Ball WS, Jr. Parry-Romberg syndrome: follow-up imaging during suppressive therapy. Neuroradiology. (1997) 39:873–6. doi: 10.1007/s002340050525
44. Holland KE, Steffes B, Nocton JJ, Schwabe MJ, Jacobson RD, Drolet BA. Linear scleroderma en coup de sabre with associated neurologic abnormalities. Pediatrics. (2006) 117:e132–6. doi: 10.1542/peds.2005-0470
45. Torok KS, Arkachaisri T. Methotrexate and corticosteroids in the treatment of localized scleroderma: a standardized prospective longitudinal single-center study. J Rheumatol. (2012) 39:286–94. doi: 10.3899/jrheum.110210
46. Zulian F, Martini G, Vallongo C, Vittadello F, Falcini F, Patrizi A, et al. Methotrexate treatment in juvenile localized scleroderma: a randomized, double-blind, placebo-controlled trial. Arthritis Rheum. (2011) 63:1998–2006. doi: 10.1002/art.30264
47. Pensler JM, Murphy GF, Mulliken JB. Clinical and ultrastructural studies of Romberg's hemifacial atrophy. Plast Reconstr Surg. (1990) 85:669–74; discussion 675–66. doi: 10.1097/00006534-199005000-00001
48. Doolittle DA, Lehman VT, Schwartz KM, Wong-Kisiel LC, Lehman JS, Tollefson MM. CNS imaging findings associated with Parry-Romberg syndrome and en coup de sabre: correlation to dermatologic and neurologic abnormalities. Neuroradiology. (2015) 57:21–34. doi: 10.1007/s00234-014-1448-6
49. Kreuter A, Wischnewski J, Terras S, Altmeyer P, Stucker M, Gambichler T. Coexistence of lichen sclerosus and morphea: a retrospective analysis of 472 patients with localized scleroderma from a German tertiary referral center. J Am Acad Dermatol. (2012) 67:1157–62. doi: 10.1016/j.jaad.2012.04.003
50. Vancheeswaran R, Black CM, David J, Hasson N, Harper J, Atherton D, et al. Childhood-onset scleroderma: is it different from adult-onset disease. Arthritis Rheum. (1996) 39:1041–9. doi: 10.1002/art.1780390624
51. Porter A, Mirizio E, Fritzler MJ, Brown R, Choi M, Schollaert-Fitch K, et al. Autoantibody testing in pediatric localized scleroderma (LS). Arthritis Rheumatol. (2018) 70(suppl 10). Available online at: https://acrabstracts.org/abstract/autoantibody-testing-in-pediatric-localized-scleroderma-ls/
52. Kreuter A, Tigges C, Gaifullina R, Kirschke J, Altmeyer P, Gambichler T. Pulsed high-dose corticosteroids combined with low-dose methotrexate treatment in patients with refractory generalized extragenital lichen sclerosus. Arch Dermatol. (2009) 145:1303–8. doi: 10.1001/archdermatol.2009.235
53. Marzano AV, Menni S, Parodi A, Borghi A, Fuligni A, Fabbri P, et al. Localized scleroderma in adults and children. clinical and laboratory investigations on 239 cases. Eur J Dermatol. (2003) 13:171–6.
54. Kreuter A. Localized scleroderma. Dermatol Ther. (2012) 25:135–47. doi: 10.1111/j.1529-8019.2012.01479.x
55. Kurzinski K, Zigler CK, Torok KS. Prediction of disease relapse in a cohort of juvenile localized scleroderma patients. Br J Dermatol. (2018) doi: 10.1111/bjd.17312
56. Dharamsi JW, Victor S, Aguwa N, Ahn C, Arnett F, Mayes MD, et al. Morphea in adults and children cohort III: nested case-control study–the clinical significance of autoantibodies in morphea. JAMA Dermatol. (2013) 149:1159–65. doi: 10.1001/jamadermatol.2013.4207
57. Falanga V, Medsger TAJr, Reichlin M, Rodnan GP. Linear scleroderma. clinical spectrum, prognosis, and laboratory abnormalities. Ann Intern Med. (1986) 104:849–57. doi: 10.7326/0003-4819-104-6-849
58. Sato S, Ihn H, Kikuchi K, Takehara K. Antihistone antibodies in systemic sclerosis. Assoc Pulmonary Fibrosis Arthritis Rheum. (1994) 37:391–4. doi: 10.1002/art.1780370313
59. Arkachaisri T, Fertig N, Pino S, Medsger TAJr. Serum autoantibodies and their clinical associations in patients with childhood- and adult-onset linear scleroderma. a single-center study. J Rheumatol. (2008) 35:2439–44. doi: 10.3899/jrheum.080098
60. Arkachaisri T, Pino S. Localized scleroderma severity index and global assessments: a pilot study of outcome instruments. J Rheumatol. (2008) 35:650–7.
61. Inoshita T, Whiteside TL, Rodnan GP, Taylor FH. Abnormalities of T lymphocyte subsets in patients with progressive systemic sclerosis (PSS, scleroderma). J Lab Clin Med. (1981) 97:264–77.
62. Keystone EC, Lau C, Gladman DD, Wilkinson S, Lee P, Shore A. Immunoregulatory T cell subpopulations in patients with scleroderma using monoclonal antibodies. Clin Exp Immunol. (1982)48:443–8.
63. Mirizio E, Marathi A, Hershey N, Ross C, Schollaert K, Salgadgo CM, et al. Identifying the signature immune phenotypes present in pediatric localized scleroderma. J Invest Dermatol. (2018) 139:715–8. doi: 10.1016/j.jid.2018.09.025
64. Reiff A, Weinberg KI, Triche T, Masinsin B, Mahadeo KM, Lin CH, et al. T lymphocyte abnormalities in juvenile systemic sclerosis patients. Clin Immunol. (2013) 149:146–55. doi: 10.1016/j.clim.2013.07.005
65. Mirizio E, Mellins ED, Macaubas C, Maecker HT, Gartner F, Konnikova L, et al. Multiparameter mass cytometry by time-of-flight spectrometry (CyTOF) phenotyping in pediatric localized scleroderma [abstract]. Arthritis Rheumatol. (2018a) 70(suppl 10). Available online at: https://acrabstracts.org/abstract/multiparameter-mass-cytometry-by-time-of-flight-spectrometry-cytof-phenotyping-in-pediatric-localized-scleroderma/
66. Torok KS, Kurzinski K, Kelsey C, Yabes J, Magee K, Vallejo AN, et al. Peripheral blood cytokine and chemokine profiles in juvenile localized scleroderma: T-helper cell-associated cytokine profiles. Semin Arthritis Rheum. (2015) 45:284–93. doi: 10.1016/j.semarthrit.2015.06.006
67. O'Brien JC, Rainwater YB, Malviya N, Cyrus N, Auer-Hackenberg L, Hynan LS, et al. Transcriptional and cytokine profiles identify CXCL9 as a biomarker of disease activity in morphea. J Invest Dermatol. (2017) 137:1663–70. doi: 10.1016/j.jid.2017.04.008
68. Ihn H, Sato S, Fujimoto M, Kikuchi K, Takehara K. Demonstration of interleukin 8 in serum samples of patients with localized scleroderma. Arch Dermatol. (1994) 130:1327–8.
69. Ihn H, Sato S, Fujimoto M, Kikuchi K, Takehara K. Demonstration of interleukin-2, interleukin-4 and interleukin-6 in sera from patients with localized scleroderma. Arch Dermatol Res. (1995) 287:193–7. doi: 10.1007/BF01262331
70. Hasegawa M, Sato S, Nagaoka T, Fujimoto M, Takehara K. Serum levels of tumor necrosis factor and interleukin-13 are elevated in patients with localized scleroderma. Dermatology. (2003) 207:141–7. doi: 10.1159/000071783
71. Uziel Y, Feldman BM, Krafchik BR, Laxer RM, Yeung RS. Increased serum levels of TGFbeta1 in children with localized scleroderma. Pediatr Rheumatol Online J. (2007) 5:22. doi: 10.1186/1546-0096-5-22
72. Budzynska-Wlodarczyk J, Michalska-Jakubus MM, Kowal M, Krasowska D. Evaluation of serum concentrations of the selected cytokines in patients with localized scleroderma. Postepy Dermatol Alergol. (2016) 33:47–51. doi: 10.5114/pdia.2015.48044
73. Uziel Y, Krafchik BR, Feldman B, Silverman ED, Rubin LA, Laxer RM. Serum levels of soluble interleukin-2 receptor. a marker of disease activity in localized scleroderma. Arthritis Rheum. (1994) 37:898–901. doi: 10.1002/art.1780370618
74. Nagaoka T, Sato S, Hasegawa M, Ihn H, Takehara K. Serum levels of soluble interleukin 6 receptor and soluble gp130 are elevated in patients with localized scleroderma. J Rheumatol. (2000) 27:1917–21.
75. Danczak-Pazdrowska A, Kowalczyk M, Szramka-Pawlak B, Gornowicz-Porowska J, Szewczyk A, Silny W, et al. Interleukin-17A and interleukin-23 in morphea. Arch Med Sci. (2012) 8:1089–95. doi: 10.5114/aoms.2012.32421
76. Mertens JSEM, de Jong GJ, Pandit AM, Seyger MB. Regarding “transcriptional and cytokine profiles identify CXCL9 as a biomarker of disease activity in morphea”. J Invest Dermatol. (2018) 138:1212–5. doi: 10.1016/j.jid.2017.11.032
77. Magee KE, Kelsey CE, Kurzinski KL, Ho J, Mlakar LR, Feghali-Bostwick CA, et al. Interferon-gamma inducible protein-10 as a potential biomarker in localized scleroderma. Arthritis Res Ther. (2013) 15:R188. doi: 10.1186/ar4378
78. Torok KS, Mi Q, Mirizio E, Schollaert-Fitch K, Fritzler M, Fritzler MJ. Chemokine ligand 9 (CXCL9) [monokine induced by gamma interferon (MIG)] as a predictor of active disease status in localized scleroderma [abstract]. Arthritis Rheumatol. (2017) 69(suppl 10). Available online at: https://acrabstracts.org/abstract/chemokine-ligand-9-cxcl9-monokine-induced-by-gamma-interferon-mig-as-a-predictor-of-active-disease-status-in-localized-scleroderma/
79. Gambichler T, Skrygan M, Labanski AA, Kolios AG, Altmeyer P, Kreuter A. Significantly increased CCL5/RANTES and CCR7 mRNA levels in localized scleroderma. Regul Pept. (2011) 170:4–6. doi: 10.1016/j.regpep.2011.05.003
80. Macaubas C, Mirizio E, Schollaert-Fitch K, Mellins ED, Torok KS. (2017). Nterferon gamma (IFN-γ) Subpopulations in skin homing T cells of localized scleroderma [abstract]. Arthritis Rheumatol. 69(suppl 10). Available online at: https://acrabstracts.org/abstract/interferon-gamma-ifn-%ce%b3-subpopulations-in-skin-homing-t-cells-of-localized-scleroderma/
81. Li G, Larregina AT, Domsic RT, Stolz DB, Medsger TAJr, Lafyatis R, et al. Skin-resident effector memory CD8(+)CD28(-) T cells exhibit a profibrotic phenotype in patients with systemic sclerosis. J Invest Dermatol. (2017) 137:1042–50. doi: 10.1016/j.jid.2016.11.037
82. Milano A, Pendergrass SA, Sargent JL, George LK, McCalmont TH, Connolly MK, et al. Molecular subsets in the gene expression signatures of scleroderma skin. PLoS ONE. (2008) 3:e2696. doi: 10.1371/journal.pone.0002696
83. Mirizio E, Mandal R, Yan Q, Horne W, Schollaert-Fitch K, Torok KS. Genetic signatures from RNA sequencing of pediatric localized scleroderma (LS) skin. Arthritis Rheumatol. (2018b) 70 (suppl 10). Available online at: https://acrabstracts.org/abstract/genetic-signatures-from-rna-sequencing-of-pediatric-localized-scleroderma-ls-skin/
84. Franks J, Martyanov V, Wood TA, Croord L, Keyes-Elstein L, Furst DE, et al. Machine learning classification of peripheral blood gene expression identifies a subset of patients with systemic sclerosis most likely to show clinical improvement in response to hematopoietic stem cell transplant. Arthritis Rheumatol. (2018) 90(Suppl. 10). Available online at: https://acrabstracts.org/abstract/machine-learning-classification-of-peripheral-blood-geneexpression-identifies-a-subset-of-patients-with-systemic-sclerosis-mostlikely-to-show-clinical-improvement-in-response-to-hematopoietic-stem-c/
85. Hinchcliff M, Huang CC, Wood TA, Matthew Mahoney J, Martyanov V, Bhattacharyya S, et al. Molecular signatures in skin associated with clinical improvement during mycophenolate treatment in systemic sclerosis. J Invest Dermatol. (2013) 133:1979–89. doi: 10.1038/jid.2013.130
86. Olin A, Henckel E, Chen Y, Lakshmikanth T, Pou C, Mikes J, et al. Stereotypic immune system development in newborn children. Cell. (2018) 174:1277–92.e1214. doi: 10.1016/j.cell.2018.06.045
87. Simon AK, Hollander GA, McMichael A. Evolution of the immune system in humans from infancy to old age. Proc Biol Sci. (2015) 282:20143085. doi: 10.1098/rspb.2014.3085
88. Valiathan R, Ashman M, Asthana D. Effects of ageing on the immune system: infants to elderly. Scand J Immunol. (2016) 83:255–66. doi: 10.1111/sji.12413
89. Adeli K, Raizman JE, Chen Y, Higgins V, Nieuwesteeg M, Abdelhaleem M, et al. Complex biological profile of hematologic markers across pediatric, adult, and geriatric ages: establishment of robust pediatric and adult reference intervals on the basis of the Canadian health measures survey. Clin Chem. (2015) 61:1075–86. doi: 10.1373/clinchem.2015.240531
90. Georgountzou A, Papadopoulos NG. Postnatal innate immune development: from birth to adulthood. Front Immunol. (2017) 8:957. doi: 10.3389/fimmu.2017.00957
91. Young AJ. The physiology of lymphocyte migration through the single lymph node in vivo. Semin Immunol. (1999) 11:73–83. doi: 10.1006/smim.1999.0163
92. Kollmann TR, Kampmann B, Mazmanian SK, Marchant A, Levy O. Protecting the newborn and young infant from infectious diseases: lessons from immune ontogeny. Immunity. (2017) 46:350–63. doi: 10.1016/j.immuni.2017.03.009
93. Hawes GE, Struyk L, van den Elsen PJ. Differential usage of T cell receptor V gene segments in CD4+ and CD8+ subsets of T lymphocytes in monozygotic twins. J Immunol. (1993) 150:2033–45.
94. Vallejo AN. Immune remodeling: lessons from repertoire alterations during chronological aging and in immune-mediated disease. Trends Mol Med. (2007) 13:94–102. doi: 10.1016/j.molmed.2007.01.005
95. Zulian F, Vallongo C, Patrizi A, Belloni-Fortina A, Cutrone M, Alessio M, et al. A long-term follow-up study of methotrexate in juvenile localized scleroderma (morphea). J Am Acad Dermatol. (2012) 67:1151–6. doi: 10.1016/j.jaad.2012.03.036
96. Saxton-Daniels S, Jacobe HT. An evaluation of long-term outcomes in adults with pediatric-onset morphea. Arch Dermatol. (2010) 146:1044–5. doi: 10.1001/archdermatol.2010.239
Keywords: localized scleroderma, morphea, pediatric rheumatology, immunophenotype, disease etiology, autoimmune disease, skin, fibrosis
Citation: Torok KS, Li SC, Jacobe HM, Taber SF, Stevens AM, Zulian F and Lu TT (2019) Immunopathogenesis of Pediatric Localized Scleroderma. Front. Immunol. 10:908. doi: 10.3389/fimmu.2019.00908
Received: 05 February 2019; Accepted: 09 April 2019;
Published: 30 April 2019.
Edited by:
Randy Q. Cron, University of Alabama at Birmingham, United StatesReviewed by:
Richard M. Silver, Medical University of South Carolina, United StatesAnn Marie Reed, Duke University School of Medicine, United States
Copyright © 2019 Torok, Li, Jacobe, Taber, Stevens, Zulian and Lu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Kathryn S. Torok, a2F0aHJ5bi50b3Jva0BjaHAuZWR1
Theresa T. Lu, bHV0QGhzcy5lZHU=